

Abstract
Chronic unresolved inflammation is the primary determinant of cardiovascular disease. Precise mechanisms that define the genesis of unresolved inflammation in heart failure with preserved ejection fraction (HFpEF) are of interest due to the obesity epidemic. To examine the obesity phenotype and its direct/indirect consequences, multiple approaches were employed using lipoxin receptor (abbreviated as ALX) dysfunction mouse model. Indirect calorimetry analyses revealed that deletion of ALX dysregulated energy metabolism driving toward age-related obesity. Heart function data suggest that obesity-prone ALX deficient mice had impaired myocardium strain. Comprehensive measurement of chemokines, extracellular matrix, and arrhythmogenic arrays confirmed dysregulation of multiple ion channels gene expression with amplified inflammatory chemokines and cytokines response at the age of 4 months compared with WT counterparts. Quantitative analyses of leukocytes demonstrated an increase of proinflammatory Ly6ChiCCR2+ macrophages in the spleen and heart at a steady-state resulting in an inflamed splenocardiac axis. Signs of subtle inflammation were marked with cardiorenal, endothelial defects with decreased CD31 and eNOS and an increased iNOS and COX2 expression. Thus, ALX receptor deficiency serves as an experimental model that defines multiple cellular and molecular mechanisms in HFpEF that could be a target for the development of HFpEF therapy in cardiovascular medicine.
Keywords: Obesogenic aging, non-resolving inflammation, inflammatory macrophage, kidney function
Introduction
Heart failure (HF) is a multi-morbidity clinical syndrome and recently has been classified into two main types; heart failure with preserved ejection fraction (HFpEF) and heart failure with reduced ejection fraction (HFrEF) (1). In recent years, with an increased incidence of patients with metabolic dysregulation, obesity, aging, and hypertension are associated with the risk of HFpEF syndrome (2–5). Epidemiological data indicate that due to the obesity epidemic, HFpEF poses as a global health problem that is relatively higher than ischemic cardiomyopathy due to differential diagnostic criteria (6),(7). Moreover, >50% of patients with clinical HF syndrome have HFpEF which is commonly associated with physical limitations, immune, and metabolic dysregulation that impair life quality (8). There are a total of 7 million HFpEF patients, of them 80% present an obesity phenotype (9) and two-thirds of HFpEF patients develop arrhythmia either before, after, or during the diagnostic progression (10),(11). In the majority of HFpEF patients, aging is a common, universal, and inevitable factor that overlaps with obesity. Thereby, the impact of age-related obesity on cardiac health remains of interest (12),(13). Obesity and aging are the prime triggers of low-grade, remote, and suboptimal inflammation at multiple levels including hemodynamic, metabolic, inflammatory, and hormonal perturbations (14). Given the HFpEF prevalence, it is unclear whether leukocytes trigger the non-resolving, suboptimal inflammation, or chronic inflammatory coexisting conditions serve as a major contributor to HFpEF pathophysiological phenotype that includes endothelial inflammation and kidney dysfunction (15).
Many experimental animal models such as systemic hypertension rat (SHR), Zucker rat, db/ob genetically altered mice are proposed to mimic the HFpEF syndrome to describe its molecular and cellular pathways; however, there is limited success in developing a comprehensive HFpEF model in cardiovascular medicine (16). To date, no precise treatment or genetically modified model is available to mimic aging, non-resolving chronic inflammation along with cardiorenal syndrome as a multi-factor risk entity to study HFpEF (17). Our previous report indicates that ALX/FPR2 receptor (synonym FPR2; referred to as ALX in the text) is essential for cardiometabolic health and ALX dysfunction drives obesity and cardiorenal inflammation in aging mice (18). This leukocyte expressed ALX receptor is essential for action of inflammation-resolution entity such as lipoxinA4, aspirin-trigger lipoxins, and resolving D1 (19–21). In this study, we tested whether the age-associated alteration in leukocyte profiling due to ALX receptor deficiency can qualify as an experimental model of HFpEF based on multi-organ and systems biology criteria. We demonstrate that leukocyte directed immune dysregulation in progressive obesogenic phenotype in spleen and heart. Sustained inflammation with amplified endothelial dysfunction of heart and kidney are primary outcomes as signs of age-related cardiomyopathy of obesity and HFpEF.
Methods
Animal compliance
Animal monitoring was conducted according to the “Guide for the Care and Use of Laboratory Animals” (8th Edition. 2011), and AVMA Guidelines for the Euthanasia of Animals: (2013 Edition) and were approved by the Institutional Animal Care and Use Committees at the University of Alabama at Birmingham, and University of South Florida, Tampa, USA.
Study design of obesogenic aging
The C57BL/6J (WT) mice were procured from the Jackson laboratory and ALX/FPR2−/− (ALX−/−) male mice were obtained originally from Idorsia Pharmaceuticals Ltd (Switzerland, formerly known as Actelion) and in-breed for future experiments. Mice were randomized to groups at the age of 2 and 4 months of age, maintained on standard lab chow diet and used to investigate age-related obesogenic phenotype. The age of 4 months old is considered middle-aged mice based on our previous life span measurement in ALX −/− mice (18). Two sets of age groups were compared with WT age-matched controls for molecular, cellular, and histological experiments. To comply with a systems biology approach, tissue organs such as spleen, heart, and kidney from all four groups were used to define the role of ALX receptor in obesity and aging-induced suboptimal inflammation in the progression of cardiovascular disease (Figure 1A; study design).
Figure 1. ALX−/− mice onsets early age obesity and energy imbalance.
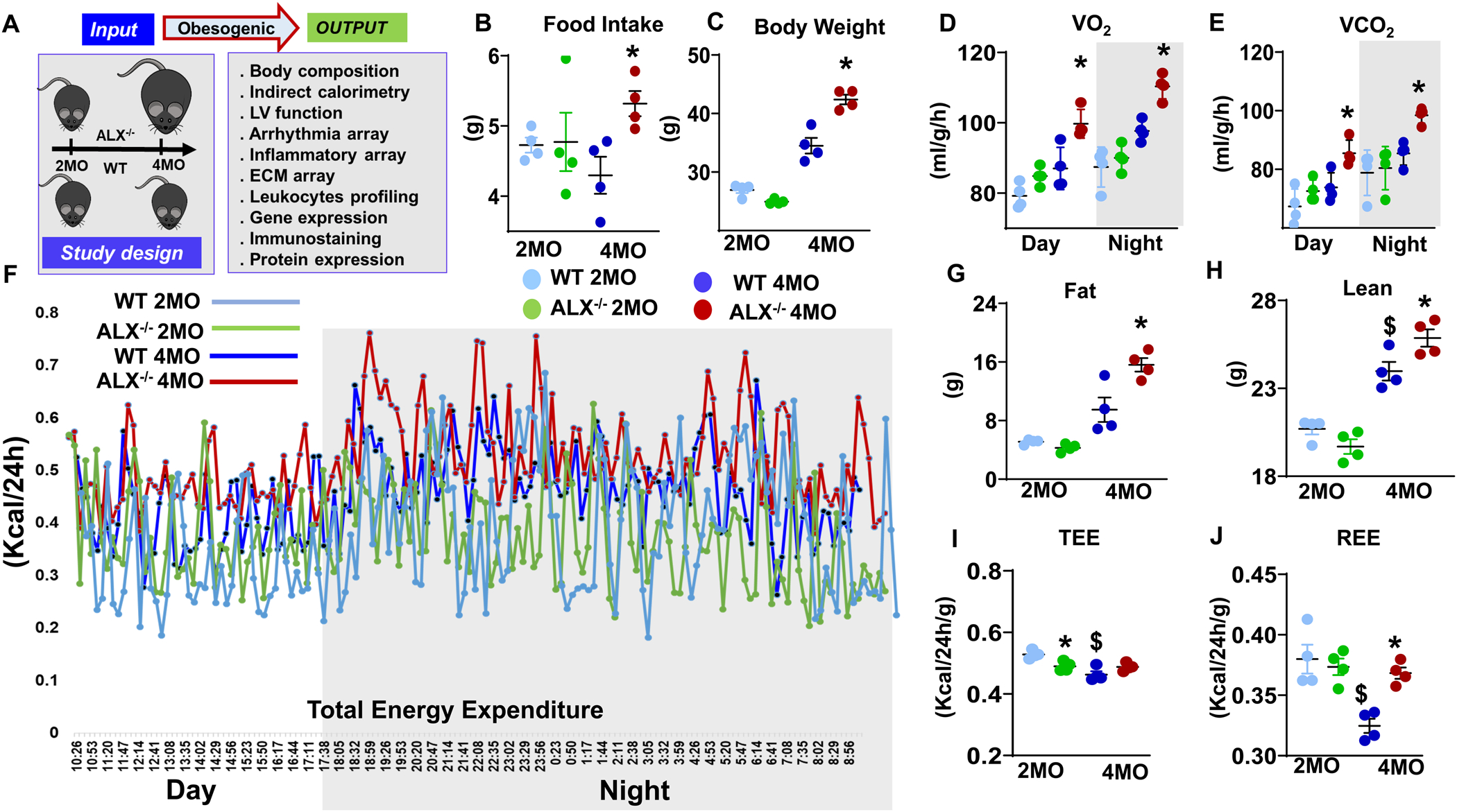
(A) Study design summarizing input and output parameters of obesogenic aging in WT and ALX−/− mice. (B, C) Dot plots showing food intake and body weight changes, (D, E) Indirect calorimetry measurement of VO2 and VCO2 during day- and night-time, (F) Line kinetics of the total energy expenditure (TEE) at day and night time, (G, H) Quantitative magnetic resonance imaging (QMRI) analysis of change in fat and lean mass, (I, J) Dot plot graph representing indirect calorimetry measurements of total energy expenditure (TEE), resting energy expenditure (REE) in WT and ALX−/− mice at the age of 2 and 4 months. *p<0.05 vs respective 2 months ALX−/− group; $ p<0.05 vs respective 2 months WT group; n= 4 mice/group; Values are means ± SEM.
Food intake measurements using indirect calorimetry measurements
The exact measurement of food intake is a key component in the analysis of energy metabolism. The WT and ALX−/− mice of 2 and 4 months of age were separated into single cages 10 days before to assure a smooth transition and to avoid single cage-related isolation stress. Food was placed in the cage of the mice after the first acclimation day and measured daily for 5 days. Food intake was measured as food consumed per gram/day (22). Indirect calorimetry was performed using the doubly-labeled water method. An isotope-elimination technique used to measure the metabolic rate of small free-living animals, which are released in the field between two-time points to measure O2 and CO2 consumption (ml/h). The energy expenditure is calculated based on the amount of O2 consumed and CO2 produced. Total energy expenditure (TEE, kcal/h, and kcal/24h), resting energy expenditure (REE, kcal/hr and kcal/24h) were exported for four groups of WT and ALX−/− mice at 2 and 4 months-old. Quantitative graphs were plotted and results distinguished per day/night for precise analysis (23),(24).
Quantitative Magnetic Resonance Imaging
The body composition of WT and ALX−/− mice in each group was differentiated in fat mass, lean mass, and water content. Body weight was measured at designated time points of 2 and 4 months, and graphs were plotted as content per gram (25).
Echocardiography and ECG measurements
Heart function echocardiography of WT and ALX−/− mice were performed using Vevo 3100 equipped with transducer MX-400 (18–38 MHz) (VisualSonics Inc., Toronto, Canada) as described previously (26),(18). In brief, mice were anesthetized using 1– 2% isoflurane with a 100% oxygen mix and placed on the temperature-regulated platform (36–37 °C) on supine position. Three color-coded recording electrodes were subdermally inserted into both upper limbs and the left lower limb. The ambient room temperature was monitored using a temperature controller (LA CROSSE) technology. Mice that had an average heartbeat below 400 beats/min were excluded from the study. The recording electrodes were connected to an ix-228/S data acquisition unit (iWorx Systems). Signals were filtered between 3 and 500 Hz. The input range was within 5 mV. Signals were also digitized with 16-bit precision at a sampling rate of 1,000 samples/s. The ECG signal was continuously recorded for 10 min for baseline analysis. Only data from recordings of stable ECG signals were included. Data were then analyzed with Lab chart 8 software (AD Instruments) (27).
Flow cytometry
Single mononuclear cells were isolated from the heart and spleen of WT and ALX−/− mice at 4 months of age due to distinct obesity phenotype as previously described (28, 29). Data were acquired on BDTM LSRII Flow Cytometer and analyzed with FlowJo software, version 7.6.3. For the absolute cell counts, e-beads (123count™ eBeads -Thermo Fischer Scientific) were used as per manufacturers’ instruction. The absolute count has been normalized to the weight of the organ tissue (heart and spleen).
Left ventricle (LV) and endothelial vessel immunostaining using confocal microscopy
The LV and kidney sections from each group were fixed using 4% paraformaldehyde and permeated using 0.1% Triton X-100. The tissues were blocked for 1 h in 10% goat serum. For endothelial cells staining LV and kidney tissues were incubated with CD31 (14–0311081, 1:100, ebiosciences) and F4/80 (macrophages detection in the LV (ab16911,100, Abcam) antibodies overnight. Sections were washed three times with 1XPBS and Alexa 555-labeled anti-rabbit secondary antibody (Molecular Probes, A21422) and wheat germ agglutinin (WGA)-647 for LV and WGA-488 for kidney (W32466, 1:1000; Molecular Probes) was added to sections for 60 min, followed by Hoechst staining for 5 min. All the stained sections were washed with 1X PBS three times and mounted with anti-fade and acquired using Nikon A1 confocal microscope (18).
RealTime2 profiler ECM, inflammatory, and arrhythmogenic PCR array and network analysis
RNA was isolated from LV samples using trizol method. cDNA synthesis was performed using RT2 First Strand Kit (330401, Qiagen) according to manufacturer’s instruction. Real-Time RT2-PCR gene array for inflammatory cytokines and receptors (Qiagen, PAMM-011E), ECM and adhesion molecules (Qiagen, PAMM-013E) and customized arrhythmogenic genes (Qiagen, CAPM14006E were performed to quantify gene expression levels as per manufacturer’s protocol, on ABI7900HT. Gene levels were normalized by using the geometric mean of housekeeping genes control (hypoxanthine phosphoribosyltransferase (Hprt-1), actin and GAPDH). The results were reported as 2−ΔCt (ΔΔCt) values. Network analysis was done using STRING: functional protein association networks analysis database (18).
LV and kidney RNA isolation and real-time quantitative PCR
RNA was extracted from LV and kidney tissues using the trizol method. For cDNA, 2 μg of RNA was used and synthesis was done SuperScript Vilo cDNA Synthesis Kit (11754250, Thermo Fisher Scientific). Quantitative PCR for eNOS (Mm00443258_m1), iNOS (Mm01336189_m1), CD31 (Mm00485172_m1), FFAR-1 (Mm01324470_m1), FFAR-2 (Mm00506686_m1), FFAR-3 (Mm01176615_g1), FFAR-4 (Mm00477214_m1), and COX-2 (Mm00478374_m1) genes were performed using TaqMan (Applied Biosystems, CA, USA) as previously reported (10). Gene expression was normalized with hypoxanthine phosphoribosyltransferase-1 (HPRT-1). The results were reported as 2^−ΔCT. RT-PCR experiments were performed in technical duplicates with n = 4 mice per group (18),(28).
Western Blotting
Immunoblotting was done using 10–15 μg of LV tissues from 4 months old WT and ALX−/− mice. Briefly, LV lysates were prepared using radio-immunoprecipitation assay (RIPA) lysis buffer (Sigma) and protease inhibitor cocktail (Roche GmbH, Germany). The proteins were electrophoresed. The blots were probed with primary CD31 antibody (Cat# ab124432, Abcam, 1/1000), eNOS (Cat# ab76198, Abcam, 1/1000), peNOS (Cat# ab184154, Abcam, 1/1000), COX-2 (Cat# ab15191, Abcam, 1/1000), iNOS (Cat#ab15323, Abcam),1:1000 or GAPDH (cat# ab9485, Abcam; 1/5000) kept overnight at 4 °C followed by respective secondary antibody (Biorad) as described previously. The proteins were detected using the Femto chemiluminescence detection system (Pierce Chemical, Rockford, IL, USA). After exposure, blot membrane image were acquired using myECL™ Imager (Thermo Scientific, USA). Densitometry data were normalized to GAPDH or total eNOS expression using Image J software (NIH, USA) (21).
Glomerular filtration rate (GFR)
GFR measurement in WT and ALX−/− for 4–5 mice per group was performed at the age of 4 months. In brief, mice were anesthetized using 1.5–2% isoflurane, and then a small portion of hair was removed from the dorsal side of each mouse. GFR was monitored using a solution of 15 mg/mL of FITC-sinistrin dissolved in 0.9% sterile saline that was injected intravenously. Then, mice were returned to their respective cages for the next 2 h. Data were analyzed using elimination kinetics as described previously (30).
Statistical analysis
Data are expressed as mean ± SEM. At least 4–8 mice/group is used for each experiment. Statistical analyses were performed using Graphpad Prism 8. Analysis of variance One-way ANOVA or two-way ANOVA followed by Newman–Keuls posthoc test was used for multiple comparisons between 2- and 4-months WT and ALX−/− groups respectively. p < 0.05 was considered as statistically significant.
Results
Dysfunction of ALX receptor provoked an age-related obesogenic phenotype
To understand the age-related obesogenic phenotype in ALX−/− mice and whether the dysfunction of ALX receptor can advance obesity-mediated different abnormalities from an early age, we applied comprehensive and systems biology approaches summarized in the study design (Figure 1A). For phenotype characterization, the body fat mass and food intake were determined in WT and ALX−/− groups at the age of 2 and 4 months without high-fat diet intervention. Food intake results suggested that ALX−/− mice developed hyperphagia at early age of 4 months with a significant increase in food intake (24%, p<0.05) with increased body mass (23%, p<0.05) compared to the age-matched WT mice (Figure 1B, C). The results highlighted a crucial role of the ALX receptor in controlling age-related hyperphagic behavior and body fat mass.
Dysregulation of ALX receptor impaired total and resting energy expenditure
Since ALX−/− mice developed an age-related obesogenic phenotype, the energy dynamics of hyperphagia-induced obesity was determined using indirect calorimetry. The volume of O2 (VO2) and CO2 (VCO2) consumption is calculated in both WT and ALX−/− mice at the age of 2 and 4 months that outlined the total and resting energy expenditure (TEE and REE). Compared to WT mice, VO2 consumption increased to 14% in day and 12% at night-time while VCO2 increased up to 15% at both day and night in ALX−/− mice at the age of 4 months (Figure 1D, E). Likewise, total Energy Expenditure (TEE) was higher in ALX−/− mice at 2 months (green lines) which further amplified at 4 months (red lines) compared with age-matched WT mice (Figure 1F). The ALX−/− mice show dysregulated TEE, which is a known primary mechanism underlying the obese and lean phenotypes (31), which were further confirmed by measurements of body composition in the same mice and at the same time point by quantitative magnetic resonance imaging (QMRI). Compared with WT groups, ALX−/− mice increased body mass with higher fat mass (64%) with a proportional increase in lean mass (7%) at 4 months age (Figure 1G, H), with no difference in the water content.
To understand whether the age-associated increase in TEE (Figure1F) in ALX−/− mice (4 months) is due to the metabolic activity of the lean mass or consequence of genotype effect, we normalized TEE and REE to the lean mass that reflects the metabolic energy of the fat mass independent of lean mass. ALX−/− mice showed 8% decrease in TEE ratio compared with WT mice (Figure 1I) indicating metabolic dysregulation began at the early age of 2 months, in the absence of weight gain. However, no change was observed in REE at 2 months in ALX−/− mice compared with WT mice (Figure 1J). Thus, in ALX−/− mice energy dysregulation is the primary and early cause of obesity-mediated metabolic defects.
At 4 months of age, REE was increased in ALX−/− mice by 13% without affecting TEE compared with the WT group at the respective age indicative of higher consumption of basal energy relative to lean mass development (Figure 1I, J). On the other hand, as expected with normal development, age-matched WT mice decreased their TEE by 12% and REE by 15% when normalized with respective lean mass gain compared with ALX−/− (Figure 1I, J). Energy hemodynamics data suggest that ALX−/− mice dysregulated total and resting energy expenditure that contributed to metabolic abnormalities in the obesogenic phenotype at an early age.
Deficiency of ALX receptor reduced myocardium strain and impaired electrocardiogram in obesogenic aging
Obesity is the primary determinant in HFpEF and myocardium pathology. Compared to age-matched WT mice, ALX−/− mice increased end-diastolic volume (EDV, 25%; p<0.05) (Figure 2A), end-systolic volume (ESV, 60%; p<0.05) at 4 months (Figure 2B) with a decrease in global longitudinal strain (GLS, 11%; p<0.05) at 2 months of age determined using high-resolution echocardiography (Figure 2C). No significant change was observed in radial circumferential strain between both groups (Figure 2D). LV cardiac morphology assessed by measuring myocyte area cross-sections using WGA-Hoechst staining. At 4 months of age, ALX−/− mice myocardium revealed an increase in the surface area of myocyte, compared with WT controls (Figure 2E). The observed enlargement of cardiomyocyte may compress the non-myocyte cells like endothelial and residential macrophages that may affect the electrical cardiac conduction. Besides myocardium strain dysfunction and hemodynamic load, obesity has additionally adverse effects on heart function that includes arrhythmia. Electrocardiogram (ECG) analyses revealed abnormal electrical conductance in ALX−/− mice compared with WT mice. ECG analysis suggest that ALX−/− mice have shorter atrial depolarization (P wave; 20%) compared with lean WT mice at 4 months (Figure 2F). Compared with WT group in ALX−/− mice, R wave increased at 2 and 4 months by 49% and 66% respectively (Figure 2G), while at 4 months Q and ST waves were decreased by 120% and 41% respectively (Figure 2H, I). The observed electrical variations are suggestive of perturbed and pathological ventricle depolarization in ALX−/− mice. Defective ECG pattern illustrated as one heartbeat from ECG analyses of WT and ALX−/− mice (Figure 2J). Thus, obesity-prone ALX−/− mice impaired myocardium with defective electrical conduction.
Figure 2. Multiple levels of heart dysfunction following deletion of ALX receptor.
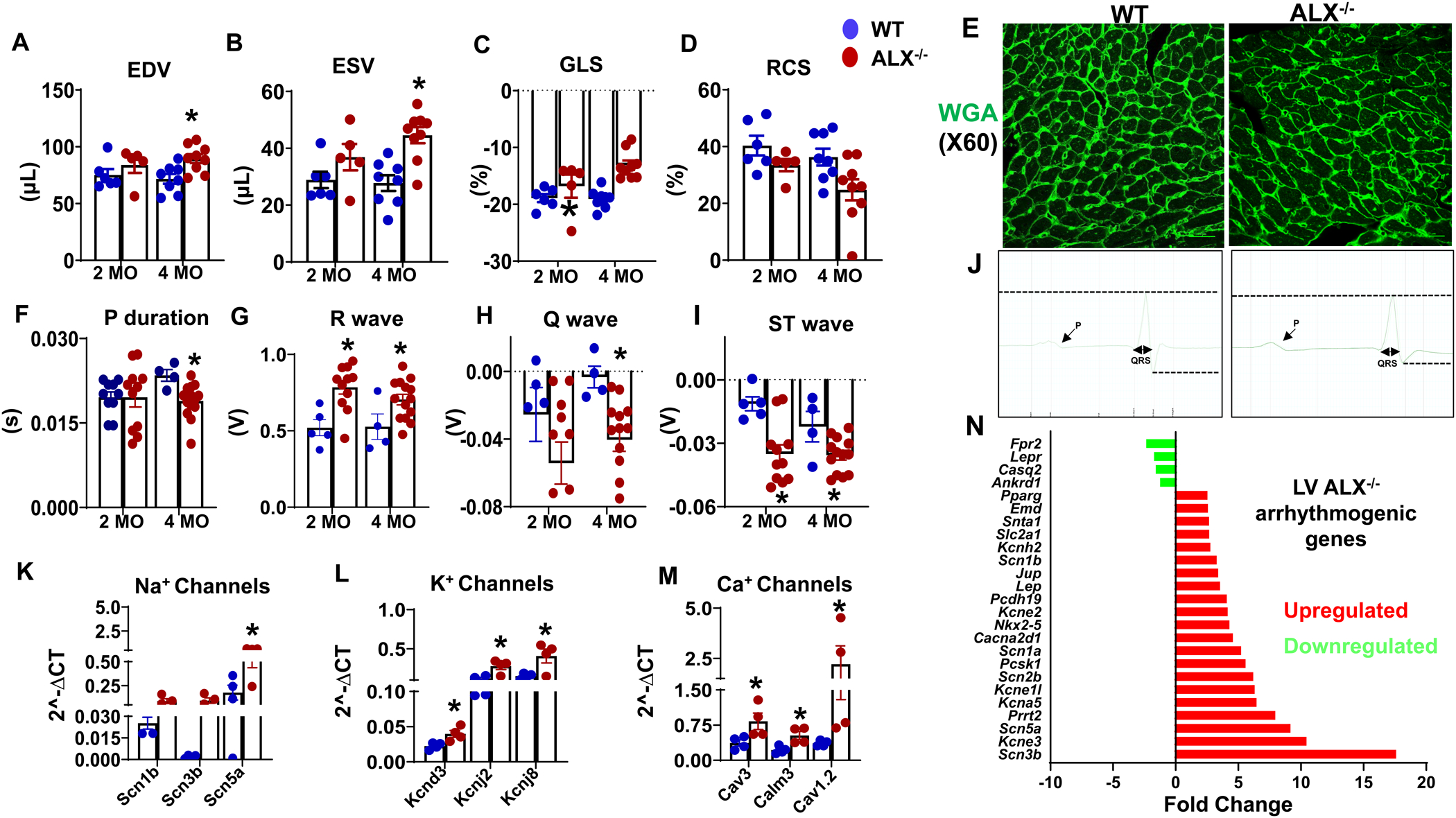
Echocardiography parameters presenting (A) end-diastolic volume (EDV), (B) end-systolic volume (ESV), (C) global longitudinal strain (GLS), (D) radial circumferential strain (RCS) measured in WT and ALX−/− group at the age of 2 and 4 months (n=4–8 mice/group). (E) Representative immunofluorescence images of LV stained with wheat germ agglutinin (WGA-green) showing changes in cardiomyocyte area in WT and ALX−/− mice at the age of 4 months. Images are representative of 4–5 sections, n =4 mice/group. Magnification, 60x. Scale bars, 25μm. ECG parameters presenting (F) P duration, (G) R wave, (H) Q wave, (I) ST wave, and (J) images of one ECG beat of WT and ALX−/− mice at the age of 4 months. Dotted lines indicate differences between ALX−/− and WT in QRS complex. mRNA expression with increase of (K) sodium (Na2+) channels, (L) potassium (K+) channels, (M) calcium (Ca2+) channels, (N) Fold change of arrhythmia gene expression analyzed using customized array in LV of ALX−/− mice at 4 months of age. The fold change is calculated by normalizing expression of respective genes to WT. mRNA expression of genes normalized to the geometric mean of HPRT, GAPDH, and actin; *p<0.05 vs respective 4 months WT group; n= 4–6 mice/group; Values are means ± SEM.
Insufficiency of ALX−/− intensified arrhythmogenic gene expression profile in obesogenic mice
Observations with humans and mice suggest that obesity has a direct impact on the electrical conductivity of the heart (32). To understand the impact of obesity in ALX−/− mice, arrhythmia gene expression was determined using an array including 84 customized genes of gating and trafficking channels in 4 months age mice. Globally, out of 84 genes, 8 genes were significantly upregulated, and 4 genes expression were downregulated in ALX−/− mice compared with WT mice. Thus, dysregulation of ion channels gene expression suggests an arrhythmogenic effect and supports defective ECG functional data in obesogenic mice. An increase in mRNA expression of voltage-gated Na channels, specifically Nav1.5, which is encoded by the gene SCNA5 (3 fold, p<0.05) was noticed in ALX−/− mice compared with WT mice. Higher expression of SCN1B and SCN3B were observed in ALX−/− mice when compared with WT mice (3.53 fold and 52 fold) (Figure 2K). These genes code for the entry of INa which controls the initial phase of the cardiac action potential (33). Genes that regulate active K+ channel physiology have significant effects on membrane action potential (34). Higher expression in mRNA level of the potassium inwardly-rectifying channel, subfamily J, member 2 and 8 was observed (KCNJ2, 2.31 fold KCNJ8, 2.68 fold, p<0.05) which encode for the channel subunits composed of Kir2.1 and Kir 6.1 respectively in ALX−/− mice compared with WT mice. Also, a marked increase in potassium voltage-gated channel, shal-related family, member 3 (Kcnd3) that code for Kv4.3 (1.76 fold, p<0.05) (Figure 2L) was observed in ALX−/− mice compared with WT mice counterparts. The upregulation in mRNA expression of calmodulin 3 (2.25 fold, p<0.05), Cav1.2 (2.40 fold, p<0.05) and Cav 3 (6.10 fold, p<0.05) in ALX−/− mice indicated modulation of calcium sensor stimuli compared with age-matched WT mice (Figure 2M). Amplified L-type Ca2+ channels expression explains the pathological increase in QRS amplitude in ALX−/− mice (Figure 2N). Thus, lack of ALX−/− in mice developed obesity and further dysregulated the cluster of Na+, K+, and Ca+ channels expression with ECG defects and impaired myocardium strain.
Lack of ALX receptor expanded CCR2+ population in spleen
Next, to determine whether age-related obesity impacts the immune cell profile with no-injury, we investigated the leukocyte phenotypes in spleen and heart using flow cytometry. In the spleen, ALX−/− mice expressed higher Ly6ChiCCR2+ macrophage population compared with WT (124±6 vs 55±13 cells/mg of tissue, p<0.05) suggestive of the inflamed spleen in steady-state (Figure 3A–C). A similar trend was observed in the heart of ALX−/− mice (24±12 vs 7±1 cells/mg) compared with WT mice (Figure 3D–F). Quantitative immune profiling in the heart demonstrated the abundance of inflamed macrophages in ALX−/− mice compared with WT mice. There was non-significant increase in Ly6CloCCR2+ and Ly6CloCCR2− resident macrophages in ALX−/− mice compared to WT mice in the heart (Figure 3D–H). As expected, in the steady myocardium neutrophils (Ly6G+) population was limited in WT mice, but was higher in ALX−/− mice compared with WT mice (1.2%±0.2 vs 0.6%±0.1, p<0.05) exemplified in comprehensive doughnut chart (Figure 3I). The observed neutrophil expansion could be explained by the higher Ly6ChiCCR2+ macrophages population observed in the spleen. Thus, dysfunction of ALX receptor drives inflammatory splenic leukocytes activation suggestive of low-grade and suboptimal inflammation.
Figure 3. Deletion of ALX receptor-activated inflamed immune population in splenocardiac manner.
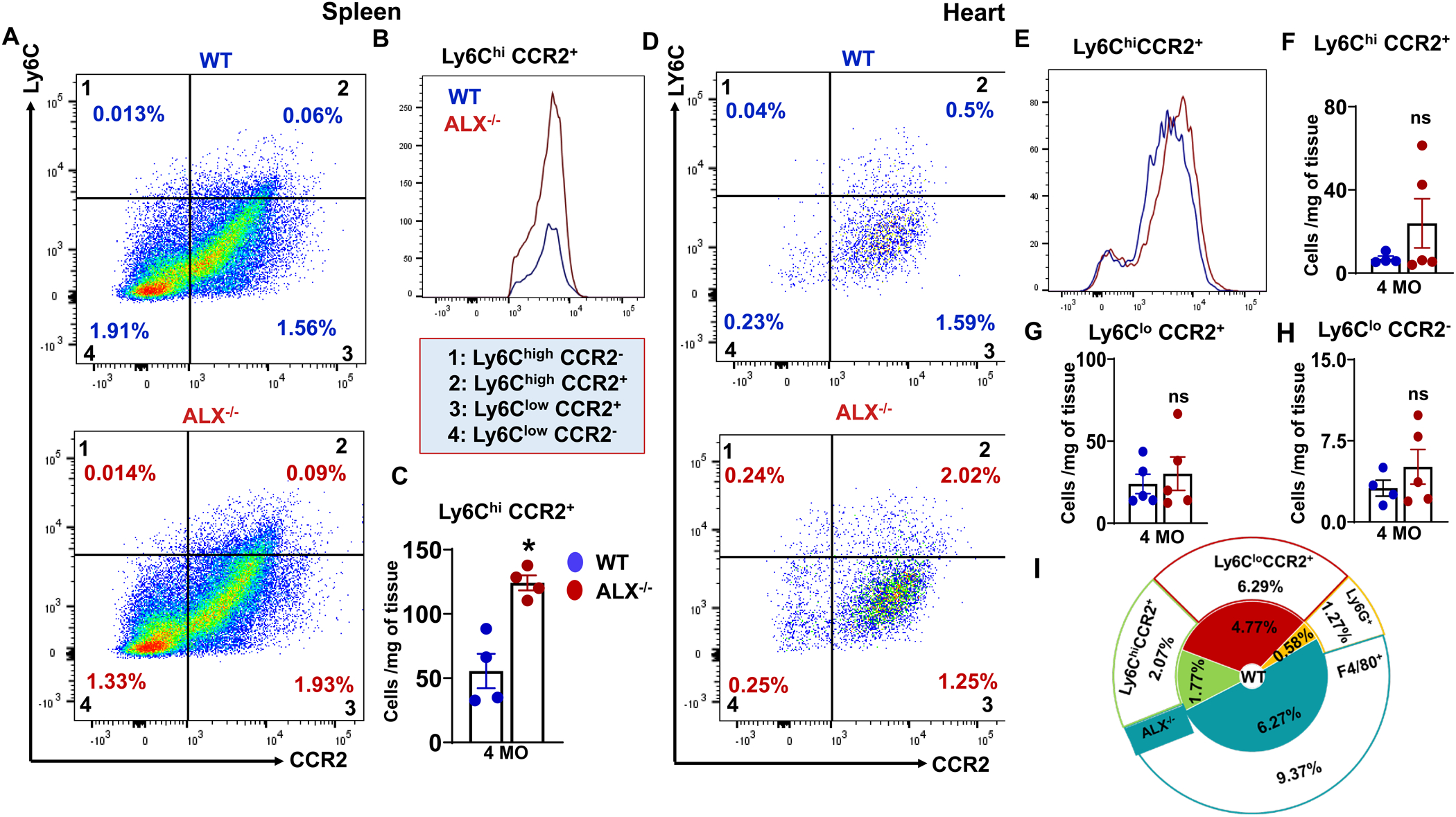
(A) Representative flow cytometry pseudoplots of the activated macrophages (Ly6C/CCR2) in the spleen of ALX−/− and WT mice at the age of 4 months. (B) Representative overlay of Ly6ChiCCR2+ expression in spleen of WT and ALX−/− mice. (C) Bar graph showing Ly6ChiCCR2+ population in the spleen of WT and ALX−/− mice at the age of 4 months. (D) Representative flow cytometry pseudoplots of activated macrophages (Ly6ChiCCR2+) in the heart of ALX−/− and WT mice at the age of 4 months. (E) Representative overlay of Ly6ChiCCR2+ expression in heart of WT and ALX−/− mice. Bar graph showing (F) Ly6ChiCCR2+, (G) Ly6CloCCR2+, (H) Ly6CloCCR2− population in the heart of WT and ALX−/− mice at the age of 4 months. (I) Summary of doughnut plots representing different percentages of the major cell population analyzed in both groups in the heart. Each color indicates the same cell population percentage in ALX−/− and WT groups at the age of 4 months. *p<0.05 vs WT; n= 4–5 mice/group; Values are means ± SEM. The cell population is represented as the absolute number of cells per mg of tissue.
Defective ALX receptor over primed chemokines in the heart
Activation of the innate immune cell indicated an inflammatory state due to insufficiency of the ALX receptor. To determine the leukocyte-directed inflammatory milieu, 84 gene RT2-PCR profiler inflammation and cytokines gene array were screened as altered levels of pro-inflammatory cytokines are also associated with prolongation of interval action potential (AP) in patients with inflammatory diseases (35). Activated splenic leukocyte in spleen and heart stimulated trafficking and migration supporting chemokines, especially the CCL (Ccl2, Ccl3, Ccl4, Ccl6, Ccl7, Ccl20, Ccl24) and TNF-α superfamily genes clusters (Tnf, Tnfsf10, Tnfsf13, Tnfsf13b, Tnfsf4, Mif, Osm, Cd40lg) family compared with age-matched WT controls (Figure 4A–D). A significant change in ECM deposition and degradation genes expression was observed in ALX−/− mice compared with WT control group (Figure 4E and F). ECM deposition genes (Itgax, Lama1, Col2a1, Cntn1, Ctnna2) were significantly upregulated in ALX−/− mice compared to WT mice suggestive of activated fibroblasts (Figure 4E). Likewise, the mRNA expression of ECM degradation genes (MMP10, 14, and 15) were increased compared with WT controls (Figure 4F) suggestive of an imbalance between degradation and deposition of ECM. Genes that regulate cell adhesion molecules (Pcdh19, Tgfβ3, Prrt2, Emd, Cpt1a, Ctnna3) were amplified suggestive of immune activation (Figure 4G), demonstrating the presence of dynamic relation between non-immune cells secreting ECM and leukocyte directed chemokines cluster in the heart of ALX−/− mice. Thus, amplified splenic leukocytes activated cytokines and perturbed extracellular matrix (ECM) homeostasis in the myocardium.
Figure 4. Age-related obesogenic phenotype dysregulated cytokines, chemokines, and extracellular matrix genes.
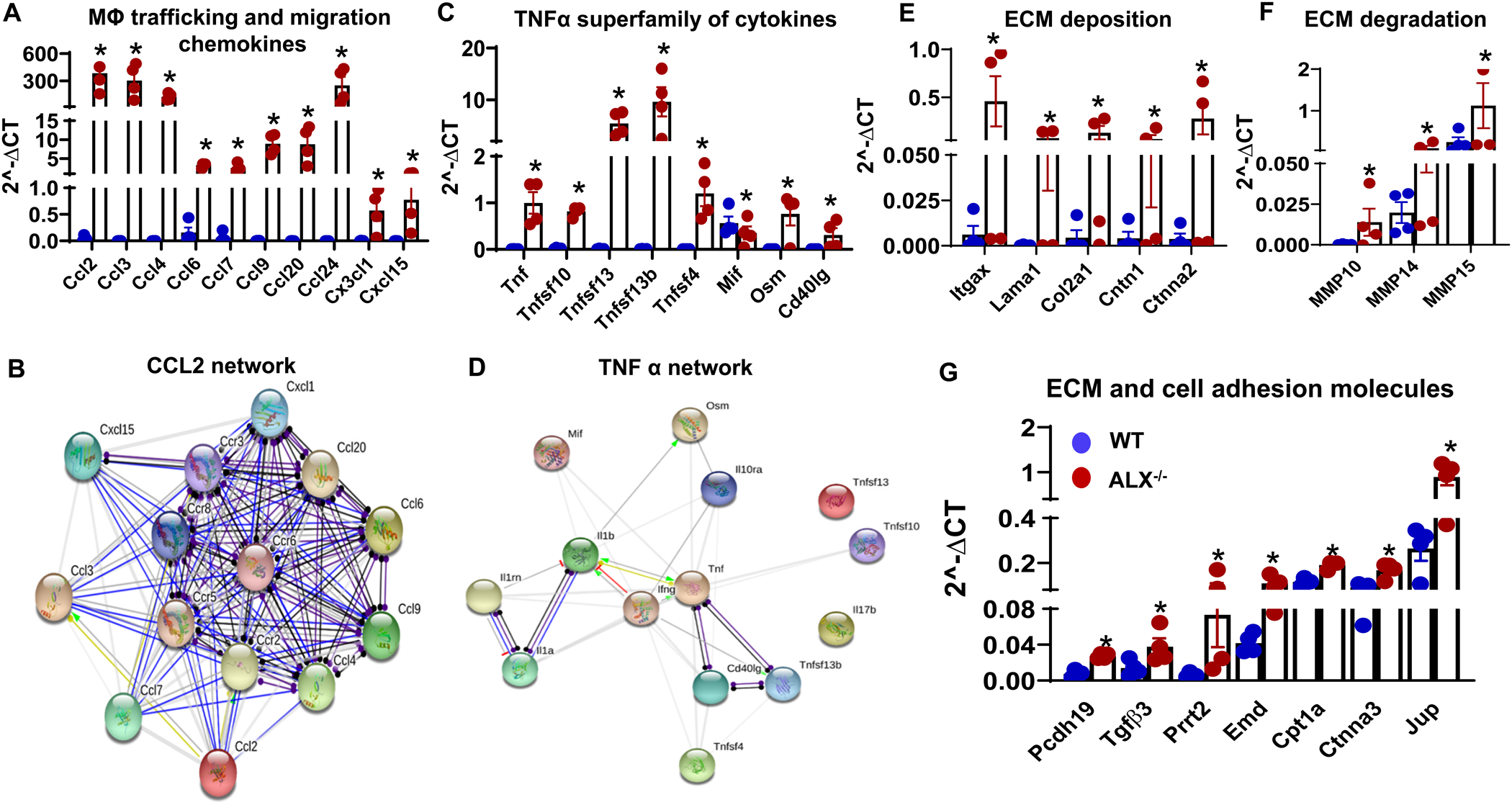
(A) mRNA expression of macrophage trafficking and migration chemokines, (B) CCL2 oriented protein-protein interaction network generated through STRING, (C) mRNA expression of TNFα superfamily cytokines, (D) TNFα superfamily oriented protein-protein interaction network generated through STRING. mRNA expression of (E) extracellular matrix (ECM) deposition genes, (F) ECM degradation genes, and (G) ECM and cell adhesion molecules genes. mRNA expression of genes normalized to the geometric mean of HPRT, GAPDH, and actin. *p<0.05 vs WT; n= 4 mice/group; values are means ± SEM.
ALX deficiency-induced myocardium endothelial dysfunction
Multiple evidence suggests that classical Ly6ChiCCR2+ macrophages immune activation contribute to cardiac dysfunction and impair endothelial function (36). Expansion of splenic Ly6ChiCCR2+ macrophage primed CCL2 cluster chemokines in the heart, which led to the foundation to determine whether ALX deletion contributed to endothelial dysfunction. Thus, we determined the expression of endothelial markers in response to ALX deletion. First, at the level of blood vessel structure, obesity-prone ALX−/− mice showed lower expression of CD31 (red color) on the basement membrane of tunica intima with the absence of positive F4/80 (green color) macrophages. In contrast, WT mice vessel indicated the presence of CD31, determined using immunostaining as F4/80+ macrophages surrounding the vessel and CD31 expression on-base membrane of the vessel (Figure 5A). Second, in the myocardium, LV mid-cavity showed (WGA purple color) high CD31 expression supporting the presence of endothelial cells with less F4/80 positive cells (macrophages) in WT when compared to the ALX−/− mice exhibiting an inverse profile (Figure 5A). These data demonstrated that ALX receptor dysfunction impacted capillary density in ALX−/− mice. We further validated the molecular basis of endothelial dysfunction mRNA and protein expression of CD31, eNOS, and total eNOS in the heart (left ventricle) that were quantitated using RT-PCR. In agreement with immunohistochemistry results, the reduced levels of CD31 protein and mRNA expression (0.50 and 0.84 fold, p<0.05) were observed in ALX−/− mice compared with WT mice (Figure 5B, C, D). Paired with these data, a significant decrease in the ratio of peNOS/total eNOS protein expression along with lower gene expression of eNOS (0.44 and 0.83 fold, p<0.05) were observed in ALX−/− mice compared with age-matched WT control (Figure 5 E, F, G), indicative of significant endothelial dysfunction since CD31/peNOS pathway is altered.
Figure 5. The absence of ALX set off the myocardium endothelial dysfunction.
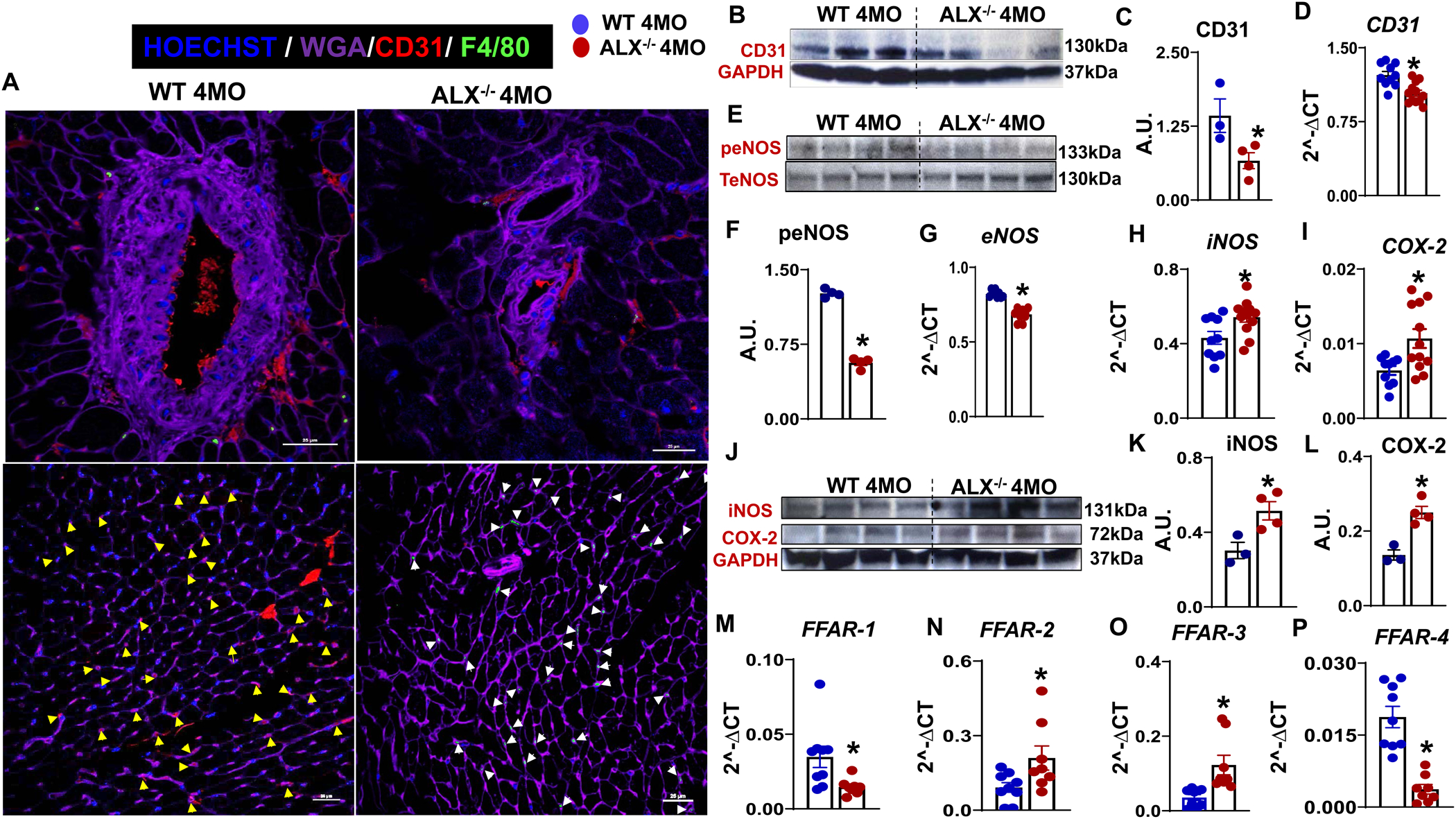
(A) LV immunofluorescence images showing a decrease in CD31 (red) expression in ALX−/− mice. The upper panel images are of CD31(red) and F4/80 (green) cells around vessels which showing ALX−/− vessels have the low number of macrophages and reduced CD31 staining. The lower panel shows LV without a vessel. Yellow arrow denotes CD31 staining and white arrow denotes F4/80 staining. WGA (purple) is used for marking cell boundaries. Nuclei are stained with Hoechst (blue). Images are representative of 4–5 sections, n =4 mice/group. Magnification, 60x. Scale bars, 25μm, (B) Immunoblot of CD31 LV protein expressions, (C) Densitometry analysis of CD31, (D) mRNA expression of CD31, (E) Immunoblot of phospho (S1177) eNOS and total eNOS protein expression, (F) Densitometry analysis of phospho (S1177) eNOS/TeNOS, mRNA expression of (G) eNOS (H) iNOS (I) COX-2 (J) Immunoblot of iNOS, COX-2 and GAPDH protein expression. Densitometry analysis of (K) iNOS, (L) COX-2, and (M-P) mRNA expression of FFAR-1, −2,−3, and −4. The gene and protein expression are measured in LV of WT and ALX−/− mice at the age of 4 months. Protein expression is normalized to GAPDH and mRNA expression is normalized to HPRT-1. *, p<0.05 vs WT. n= 3–4 mice/group. values are means ± SEM.
Parallel to the eNOS/NO pathway, COX2 and iNOS gene expression was determined in the heart since the LV environment is inflamed due to age-related obesity in ALX−/− mice. Higher expression of COX2 (1.66 fold, p<0.05) and iNOS (1.25 fold, p<0.05) were observed in ALX−/− mice compared with WT mice (Figure 5H, I). At the protein level, both COX2 and iNOS expression were mirrored with the observed mRNA gene expression results (Figure 5J, K, L) suggestive of chronic inflammation related to endothelial dysfunction. Taken all together, inflamed spleen feed-forward the inflammatory leukocytes to heart with signs of impaired endothelial dysfunction. To validate the metabolic dysregulation, fatty acids receptors (FFAR1 to FFAR4) gene expression were determined. Importantly, FFAR2 (2.29-fold, p<0.05) and FFAR3 (3.45 fold, p<0.05) were increased in ALX−/− mice compared with WT mice (Figure 5M, N). A decrease in FFAR1 (0.42 fold, p<0.05) (Figure 5O) and FFAR4 (0.19 fold, p<0.05) (Figure 5P) were observed in ALX−/− mice when compared with WT mice. Thus, our results showed that activated splenic leukocytes in the steady-state leads to endothelial dysfunction in the heart. This is an indirect impact of resolution receptor dysfunction and source of non-resolving inflammation due to obesity with profound alteration in immune metabolism.
Obesity increased hyper-filtration and kidney dysfunction in ALX−/− mice
In the obesogenic setting, the heart and kidney coordinate the vicious cycle of pathophysiological stress and dysfunction of one feed-forward the maladaptive response to others (37). Since ALX−/− mice displayed an age-related obesity phenotype with signs of inflamed myocardium and defects in ECG, we determined whether obesity of ALX−/− mice impacts kidney function. Kidney functional data suggest higher glomerular filtration rate (GFR) (1.22 fold, p<0.05) as a sign of hyperfiltration in ALX−/− group compared with WT group (Figure 6A) with no change in t1/2 (Figure 6B). These findings are in agreement with a human study that ranks obesity as a major confounding factor in the glomerular hyper-filtration (38). Further, to understand the molecular mechanism behind the observed glomerular hyper-filtration, we determined markers of endothelial function (CD31) because endothelial integrity serves as a pivotal barrier in the control of permeability using immunofluorescence-staining (Figure 6C, D, E). Histological data suggest a significant decrease of CD31+ (red color) in the medulla, especially in the tublo-interstitial area in ALX−/− mice compared with WT (Figure 6E). In the cortex zone, the total absence of CD31 positive cells was observed in ALX−/− mice compared with WT control (Figure 6C, D). Few endothelial cells were observed inside and surrounding the vessel in ALX−/− mice compared to WT mice (Figure 6E). Histological results further were validated by the evaluation of the molecular markers which revealed a decline of eNOS (0.35 fold, p<0.05) and CD31 (0.72 fold, p<0.05) gene expression (Figure 6F, G) in ALX−/− mice when compared with age-matched WT mice. Thus, the observed capillary rarefaction in the kidney is accompanied by an increase of COX-2 expression (3 fold, p<0.05) (Figure 6H) with limited change in iNOS (Figure 6I) in ALX−/− mice compared with WT mice. Thus, obesogenic aging in ALX−/− mice intensified renal dysfunction with suggestive signs of a remote systemic inflamed network, and age-related kidney endothelial dysfunction.
Figure 6. Obesity in ALX−/− mice advances kidney and endothelium dysfunction at the age of 4 months.
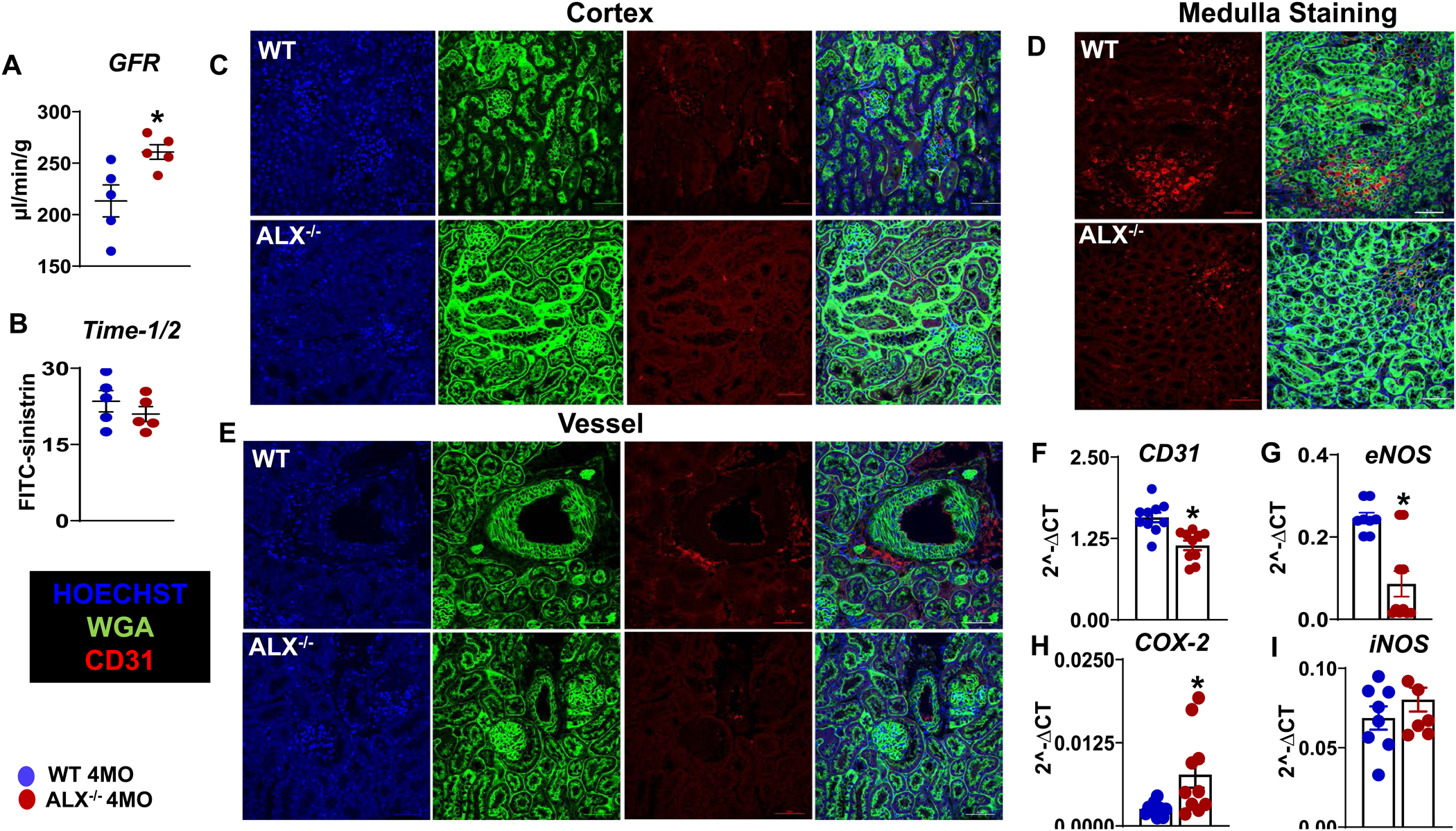
Bar graphs presenting (A) glomerular filtration rate (GFR) and (B) half time of injected FITC-sinistrin in the kidney of WT and ALX−/− mice. Representative immunofluorescence images of (C) cortex, (D) vessel, and (E) medulla stained with WGA (green)-CD31(red) in kidney of ALX−/− and WT mice. Images are representative of 4–5 sections, n =4 mice/group. Magnification, 60x. Scale bars, 50μm. Representative graph of mRNA gene expression in the kidney of (F) CD31, (G) eNOS, (H) COX-2, (I) iNOS in the WT and ALX−/− mice. mRNA expression is normalized to HPRT-1. *p<0.05 vs WT; n= 6 mice/group; values are means ± SEM.
Discussion
An obesogenic phenotype and non-ischemic heart dysfunction are known to impair immune function (39). Every aspect of the immune response including presentation of CCR2, complex MHCII, immune cell differentiation, and leukocytes class switching activity in wound healing intimately links with cell metabolism and resolution of inflammation (40–42). Previous reports suggest that endogenous/exogenous/agonist-based action of the ALX receptor facilitates the resolution of inflammation, anti-inflammatory action, and cardiac repair, in contrast, the dysfunction of ALX receptor leads to non-resolving inflammation with lower survival (43–45) (18),(21, 46). Here, we present that obesity-prone ALX−/− mice drives; (i) dysregulation of energy metabolism thereby developing an obesogenic phenotype leading to structural remodeling, perturbed electrical changes, and weakened myocardium strain of the heart; (ii) activation of splenic CCR2+ macrophages that over prime CCL cluster of chemokines with the initiation of arrhythmogenic gene expression in the left ventricle; and (iii) age-related cardiorenal and endothelial dysfunction. Thus, our multiple complementary approaches confirm the critical role of ALX receptor in cardiometabolic health and thus ALX deficiency could qualify as an experimental model of HFpEF.
First, at the phenotypic level, the role of the ALX receptor was defined in energy balance, structural, functional, and electrical remodeling. Our previous study indicates that ALX is essential for survival in obesity-prone male mice and to resolve inflammation (18). Compared to male, female ALX−/− mice are resistant to spontaneous obesity phenotype contrast to humans because menopause-related estrogen decline and obesity might contribute to HFpEF onset (47). The dysfunction of ALX in mice develops an obesogenic phenotype from 4 months of age due to bulimia. Based on calorimetry data, the likely explanation is that uneven energy dynamics provide the obesity-related pathophysiological structural remodeling that occurs within the heart as an adaptation to the increase of body mass in mice/humans. Obese patients and mice undergo dilation of the left ventricle with signs of disturbed electrical conduction (48) (49). In ALX−/− mice, heart structural remodeling and electrical disruption co-exist with signs of diastolic dysfunction without major defects in contractility at an early age. Recent reports identified that the impairment of the metabolic activity is linked with the alteration of cardiac electrophysiology (50), which led us to explore the ion channel expressions in the absence of a resolution sensor.
Second, at the molecular level, important findings are amplified Na+, K+, and Ca2+ ion channels gene expression following the deletion of ALX. The increase of Kcnj2 and Kcnj5 favor more towards the risks of re-entrant ventricle arrhythmias (51). However, SN5a gain-of-function study defines the cause of ventricular arrhythmias and early-onset atrial fibrillation (33). In multiple studies using the myocardial infarction model, it is apparent that inhibition of re-expressed T-type Ca2+ like Cav 3 channels had a beneficial role in limiting infarct size, and ventricular remodeling (52). Dysregulated ions channels data supports our findings and align with the ECG impairment marked by the increase in QRS duration in obesity-prone ALX null mice. Hence, maintenance of Na2+ and ca2+ homeostasis is crucial for electrical stability and heart function homeostasis (53, 54). However, it remains to be resolved if the upregulation of these genes is a cause or a consequence of functional arrhythmias in the heart of ALX null mice. Prescribed changes presented in the manuscript are a snapshot of an arrhythmogenic profile at early age suggestive of critical role in sudden cardiac death of ALX null mice (18).
Third, the innovative outcome is the cellular activation of splenic leukocytes; dysregulation of lipids and proteins that could particularly compromise the integrated metabolic response of macrophages (42, 55). Dysfunction of ALX receptor in mice primes over activation of chemokines and disruption of matrix homeostasis as the primary signs of suboptimal inflamed state reflected by the increase of Ly6ChiCCR2+ population in the spleen and to some extent in the heart. Clinical and experimental data support that suboptimal inflammation often manifests in endothelial dysfunction as the beginning of atherogenesis that progresses to atherosclerosis with time (56),(57). This outcome bridges the gap between ALX receptor defects that drives an obesogenic phenotype related to non-resolving inflammation and endothelial dysfunction, which is of high relevance to cardiomyopathy of obesity. The cardio-renal endothelium dysfunction indicates a part of direct (feed-forward) or indirect (feed-backward) consequences of age-related ALX dysfunction. The declining expression of the endothelial markers such as endothelial nitric oxide synthase in both myocardium and kidney of ALX null mice drives activation of induced nitric oxide synthase expression along with cyclooxygenase expression suggestive of suboptimal inflammation in steady-state. Furthermore, data are of translational significance as the endothelium-dependent dysfunction is a prime determinant in clinical studies when free fatty acids are experimentally increased in metabolic syndrome subjects (58). Recent crystal structure data of ALX/FPR2 indicate that this sensor has an unusual site compared to pro-inflammatory receptors that can accommodate proresolving mediators such as lipoxin A4 and resolvin D1 (59). This novel proresolving structure within the receptor offers a basis for structure-based drug design for disorders that are at epidemic proportions in the Western world, these could include resolvin D1 and lipoxin A4 mimetics (42). In contrast, pharmacological inhibition of ALX post-ischemia, dysregulated leukocyte trafficking in spleen and heart with signs of non-resolving inflammation (60). Presented data along with previously published reports underscore that the suboptimal and remote inflammation is a consequence of impaired biosynthesis of resolution bioactive lipids and either absence through enzyme or receptor dysfunction leading to the impairment of immune responsive metabolism (18), (42, 61).
Limitations
The presented results has some limitations. Current study focused on the genesis of non-resolving inflammation using monogenic mouse model, however in clinical setting the multi-organ remote inflammation originates from polygenic sources such as imbalanced nutrient intake/energy expenditure, altered sleep/wake up cycle, lack of exercise or antecedent/genetic predisposition. Due to spontaneous obesity, the development of insulin resistance, hyperglycemia, and diabetes that suggest possible role of neuro-endocrine system is understudied.
Conclusion
The deletion of ALX receptor triggers non-resolving inflammation appears to have a multi-dimensional role extending from cardiometabolic disorder to cardiorenal endothelial dysfunction. These profiles are different arms to one common syndrome, HFpEF in obesogenic aging. Data could be of interest in identifying novel mechanism(s) of action in cardiovascular medicine and target(s) for HFpEF pathobiology.
Figure 7. Summary of HFpEF model presenting splenocardiac and cardiorenal inflammation in obesity-prone ALX-deficient mice.
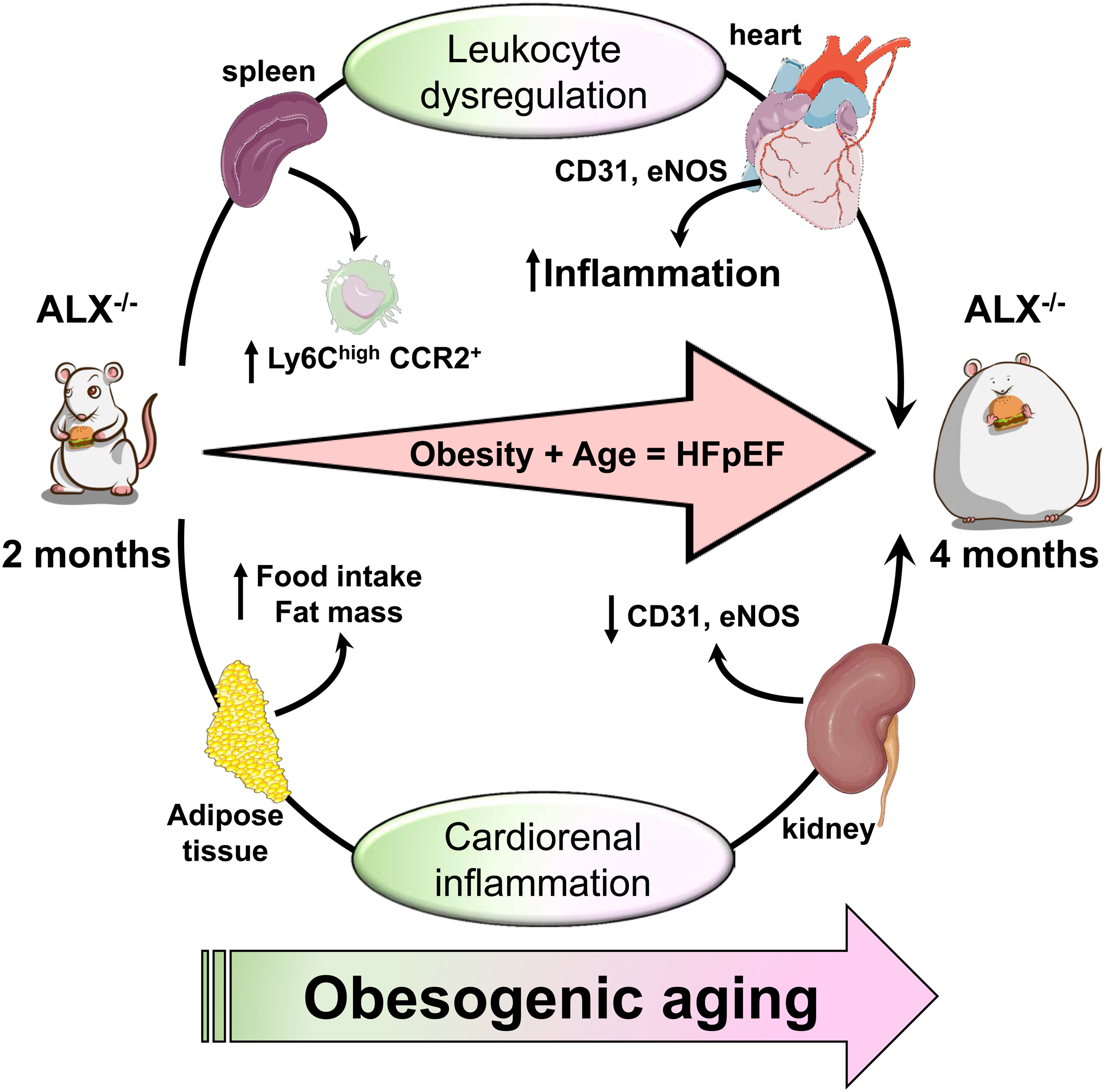
Obesity prone ALX null mice develops HFpEF phenotypes with leukocyte-directed suboptimal inflammation and dysregulation of energy dynamics with endothelial dysfunction.
Acknowledgement
Authors acknowledge the funding support from the National Institutes of Health (HL132989 and HL144788) to G.V.H., AT008375 to S.R.S., GM038765 to C.N.S. We acknowledge support from the UAB-UCSD O’Brien Core Center for Acute Kidney Injury Research for this project. Authors acknowledge Idorsia (formerly known as Actelion) Pharmaceuticals Ltd, Switzerland for providing the mice and Servier Medical Art gallery that used to illustrate figures in the manuscript.
Non standard abbreviations
- ALX receptor
lipoxin receptor
- Ca2+ channel
calcium channel
- CD31
cluster of differentiation 31
- COX-2
cyclooxygenase-2
- ECG
electrocardiogram
- ECM
extracellular matrix
- EDV
end diastolic volume
- ESV
end systolic volume
- eNOS
endothelial nitric oxide synthase
- FPR2
formyl peptide receptor 2 (synonym as ALX)
- GFR
glomerular filtration rate
- GLS
global longitudinal strain
- HFpEF
heart failure with preserved ejection fraction
- iNOS
inducible nitric oxide synthase
- K+ channel
potassium channel
- Na2+ channel
sodium channel
- peNOS
phosphorylated eNOS
- QMRI
quantitative magnetic resonance imaging
- REE
resting energy expenditure
- RCS
radial circumferential strain
- TEE
total energy expenditure
- VCO2
volume of CO2 consumption
- VO2
volume of O2 consumption
- WGA
wheat gram agglutinin
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Dunlay SM, Roger VL, and Redfield MM (2017) Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 14, 591–602 [DOI] [PubMed] [Google Scholar]
- 2.Packer M, and Kitzman DW (2018) Obesity-Related Heart Failure With a Preserved Ejection Fraction: The Mechanistic Rationale for Combining Inhibitors of Aldosterone, Neprilysin, and Sodium-Glucose Cotransporter-2. JACC Heart Fail 6, 633–639 [DOI] [PubMed] [Google Scholar]
- 3.Altara R, Giordano M, Nordén ES, Cataliotti A, Kurdi M, Bajestani SN, and Booz GW (2017) Targeting Obesity and Diabetes to Treat Heart Failure with Preserved Ejection Fraction. Front Endocrinol (Lausanne) 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zannad F, and Rossignol P (2018) Cardiorenal Syndrome Revisited. Circulation 138, 929–944 [DOI] [PubMed] [Google Scholar]
- 5.Yanovski SZ, and Yanovski JA (2011) Obesity prevalence in the United States--up, down, or sideways? N Engl J Med 364, 987–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pieske B, Tschöpe C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, Lancellotti P, Melenovsky V, Morris DA, Nagel E, Pieske-Kraigher E, Ponikowski P, Solomon SD, Vasan RS, Rutten FH, Voors AA, Ruschitzka F, Paulus WJ, Seferovic P, and Filippatos G (2019) How to diagnose heart failure with preserved ejection fraction: the HFA–PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). European Heart Journal 40, 3297–3317 [DOI] [PubMed] [Google Scholar]
- 7.Kjeldsen SE, von Lueder TG, Smiseth OA, Wachtell K, Mistry N, Westheim AS, Hopper I, Julius S, Pitt B, Reid CM, Devereux RB, and Zannad F (2019) Medical Therapies for Heart Failure With Preserved Ejection Fraction. Hypertension, Hypertensionaha11914057 [DOI] [PubMed] [Google Scholar]
- 8.Alpert CM, Smith MA, Hummel SL, and Hummel EK (2017) Symptom burden in heart failure: assessment, impact on outcomes, and management. Heart Fail Rev 22, 25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haass M, Kitzman DW, Anand IS, Miller A, Zile MR, Massie BM, and Carson PE (2011) Body mass index and adverse cardiovascular outcomes in heart failure patients with preserved ejection fraction: results from the Irbesartan in Heart Failure with Preserved Ejection Fraction (I-PRESERVE) trial. Circ Heart Fail 4, 324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly JP, Mentz RJ, Mebazaa A, Voors AA, Butler J, Roessig L, Fiuzat M, Zannad F, Pitt B, O’Connor CM, and Lam CSP (2015) Patient selection in heart failure with preserved ejection fraction clinical trials. J Am Coll Cardiol 65, 1668–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masarone D, Limongelli G, Rubino M, Valente F, Vastarella R, Ammendola E, Gravino R, Verrengia M, Salerno G, and Pacileo G (2017) Management of Arrhythmias in Heart Failure. J Cardiovasc Dev Dis 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Upadhya B, and Kitzman DW (2017) Heart Failure with Preserved Ejection Fraction in Older Adults. Heart Fail Clin 13, 485–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savji N, Meijers WC, Bartz TM, Bhambhani V, Cushman M, Nayor M, Kizer JR, Sarma A, Blaha MJ, Gansevoort RT, Gardin JM, Hillege HL, Ji F, Kop WJ, Lau ES, Lee DS, Sadreyev R, van Gilst WH, Wang TJ, Zanni MV, Vasan RS, Allen NB, Psaty BM, van der Harst P, Levy D, Larson M, Shah SJ, de Boer RA, Gottdiener JS, and Ho JE (2018) The Association of Obesity and Cardiometabolic Traits With Incident HFpEF and HFrEF. JACC Heart Fail 6, 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frasca D, Blomberg BB, and Paganelli R (2017) Aging, Obesity, and Inflammatory Age-Related Diseases. Front Immunol 8, 1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Redfield MM (2017) Heart Failure with Preserved Ejection Fraction. N Engl J Med 376, 897. [DOI] [PubMed] [Google Scholar]
- 16.Barandiaran Aizpurua A, Schroen B, van Bilsen M, and van Empel V (2018) Targeted HFpEF therapy based on matchmaking of human and animal models. Am J Physiol Heart Circ Physiol 315, H1670–h1683 [DOI] [PubMed] [Google Scholar]
- 17.Carbone S, Mauro AG, Toldo S, and Abbate A (2018) Diet-Induced Obesity HFpEF Murine Models. JACC Basic Transl Sci 3, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tourki B, Kain V, Pullen AB, Norris PC, Patel N, Arora P, Leroy X, Serhan CN, and Halade GV (2019) Lack of resolution sensor drives age-related cardiometabolic and cardiorenal defects and impedes inflammation-resolution in heart failure. Molecular Metabolism 31, 138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kain V, Liu F, Kozlovskaya V, Ingle KA, Bolisetty S, Agarwal A, Khedkar S, Prabhu SD, Kharlampieva E, and Halade GV (2017) Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Scientific reports 7, 9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiore S, Maddox JF, Perez HD, and Serhan CN (1994) Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. The Journal of experimental medicine 180, 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M, and Halade GV (2015) Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol 84, 24–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Smith DL Jr., Keating KD, Allison DB, and Nagy TR (2014) Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity (Silver Spring) 22, 2147–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alberts P, Johansson BG, and McArthur RA (2006) Characterization of energy expenditure in rodents by indirect calorimetry. Curr Protoc Neurosci Chapter 9, Unit9.23D [DOI] [PubMed] [Google Scholar]
- 24.Meyer CW, Reitmeir P, and Tschop MH (2015) Exploration of Energy Metabolism in the Mouse Using Indirect Calorimetry: Measurement of Daily Energy Expenditure (DEE) and Basal Metabolic Rate (BMR). Curr Protoc Mouse Biol 5, 205–222 [DOI] [PubMed] [Google Scholar]
- 25.Halade GV, Ma Y, Ramirez TA, Zhang J, Dai Q, Hensler JG, Lopez EF, Ghasemi O, Jin YF, and Lindsey ML (2013) Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am J Physiol Heart Circ Physiol 305, H1830–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halade GV, Kain V, and Ingle KA (2018) Heart functional and structural compendium of cardiosplenic and cardiorenal networks in acute and chronic heart failure pathology. Am J Physiol Heart Circ Physiol 314, H255–h267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kain V, Ingle KA, Kachman M, Baum H, Shanmugam G, Rajasekaran NS, Young ME, and Halade GV (2018) Excess omega-6 fatty acids influx in aging drives metabolic dysregulation, electrocardiographic alterations, and low-grade chronic inflammation. Am J Physiol Heart Circ Physiol 314, H160–h169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halade GV, Kain V, Black LM, Prabhu SD, and Ingle KA (2016) Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging (Albany NY) 8, 2611–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halade GV, Norris PC, Kain V, Serhan CN, and Ingle KA (2018) Splenic leukocytes define the resolution of inflammation in heart failure. Sci Signal 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushita K, Saritas T, Eiwaz MB, McClellan N, Coe I, Zhu W, Ferdaus MZ, Sakai LY, McCormick JA, and Hutchens MP (2020) The acute kidney injury to chronic kidney disease transition in a mouse model of acute cardiorenal syndrome emphasizes the role of inflammation. Kidney Int 97, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler AA, and Kozak LP (2010) A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes 59, 323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aromolaran AS, and Boutjdir M (2017) Cardiac Ion Channel Regulation in Obesity and the Metabolic Syndrome: Relevance to Long QT Syndrome and Atrial Fibrillation. Front Physiol 8, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lieve KV, Verkerk AO, Podliesna S, van der Werf C, Tanck MW, Hofman N, van Bergen PF, Beekman L, Bezzina CR, Wilde AAM, and Lodder EM (2017) Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int J Cardiol 236, 187–193 [DOI] [PubMed] [Google Scholar]
- 34.Nelson MT, Patlak JB, Worley JF, and Standen NB (1990) Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol 259, C3–18 [DOI] [PubMed] [Google Scholar]
- 35.Lazzerini PE, Laghi-Pasini F, Bertolozzi I, Morozzi G, Lorenzini S, Simpatico A, Selvi E, Bacarelli MR, Finizola F, Vanni F, Lazaro D, Aromolaran A, El Sherif N, Boutjdir M, and Capecchi PL (2017) Systemic inflammation as a novel QT-prolonging risk factor in patients with torsades de pointes. Heart 103, 1821–1829 [DOI] [PubMed] [Google Scholar]
- 36.Kalucka J, Bierhansl L, Wielockx B, Carmeliet P, and Eelen G (2017) Interaction of endothelial cells with macrophages-linking molecular and metabolic signaling. Pflugers Arch 469, 473–483 [DOI] [PubMed] [Google Scholar]
- 37.House AA, Wanner C, Sarnak MJ, Piña IL, McIntyre CW, Komenda P, Kasiske BL, Deswal A, deFilippi CR, Cleland JGF, Anker SD, Herzog CA, Cheung M, Wheeler DC, Winkelmayer WC, McCullough PA, Abu-Alfa AK, Amann K, Aonuma K, Appel LJ, Baigent C, Bakris GL, Banerjee D, Boletis JN, Bozkurt B, Butler J, Chan CT, Costanzo MR, Dubin RF, Filippatos G, Gikonyo BM, Gikonyo DK, Hajjar RJ, Iseki K, Ishii H, Knoll GA, Lenihan CR, Lentine KL, Lerma EV, Macedo E, Mark PB, Noiri E, Palazzuoli A, Pecoits-Filho R, Pitt B, Rigatto C, Rossignol P, Setoguchi S, Sood MM, Störk S, Suri RS, Szummer K, Tang SCW, Tangri N, Thompson A, Vijayaraghavan K, Walsh M, Wang AY-M, and Weir MR (2019) Heart failure in chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International 95, 1304–1317 [DOI] [PubMed] [Google Scholar]
- 38.Ogna A, Forni Ogna V, Bochud M, Guessous I, Paccaud F, Burnier M, and Wuerzner G (2016) Association between obesity and glomerular hyperfiltration: the confounding effect of smoking and sodium and protein intakes. Eur J Nutr 55, 1089–1097 [DOI] [PubMed] [Google Scholar]
- 39.de Heredia FP, Gomez-Martinez S, and Marcos A (2012) Obesity, inflammation and the immune system. Proc Nutr Soc 71, 332–338 [DOI] [PubMed] [Google Scholar]
- 40.Odegaard JI, and Chawla A (2013) The immune system as a sensor of the metabolic state. Immunity 38, 644–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stunault MI, Bories G, Guinamard RR, and Ivanov S (2018) Metabolism Plays a Key Role during Macrophage Activation. Mediators Inflamm 2018, 2426138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, and Serhan CN (2002) Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med 8, 1296–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halade GV, Kain V, and Serhan CN (2018) Immune responsive resolvin D1 programs myocardial infarction-induced cardiorenal syndrome in heart failure. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 32, 3717–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corminboeuf O, and Leroy X (2015) FPR2/ALXR Agonists and the Resolution of Inflammation. Journal of Medicinal Chemistry 58, 537–559 [DOI] [PubMed] [Google Scholar]
- 46.Ge Y, Zhang S, Wang J, Xia F, Wan JB, Lu J, and Ye RD (2020) Dual modulation of formyl peptide receptor 2 by aspirin-triggered lipoxin contributes to its anti-inflammatory activity. FASEB journal: : official publication of the Federation of American Societies for Experimental Biology; [DOI] [PubMed] [Google Scholar]
- 47.Sabbatini AR, and Kararigas G (2020) Menopause-Related Estrogen Decrease and the Pathogenesis of HFpEF: JACC Review Topic of the Week. J Am Coll Cardiol 75, 1074–1082 [DOI] [PubMed] [Google Scholar]
- 48.Alex L, Russo I, Holoborodko V, and Frangogiannis NG (2018) Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 315, H934–h949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lakdawala NK, and Givertz MM (2010) Dilated cardiomyopathy with conduction disease and arrhythmia. Circulation 122, 527–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barth AS, and Tomaselli GF (2009) Cardiac metabolism and arrhythmias. Circ Arrhythm Electrophysiol 2, 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dehghani-Samani A, Madreseh-Ghahfarokhi S, and Dehghani-Samani A (2019) Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias. Acta Cardiol Sin 35, 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandmann S, Claas R, Cleutjens JP, Daemen MJ, and Unger T (2001) Calcium channel blockade limits cardiac remodeling and improves cardiac function in myocardial infarction-induced heart failure in rats. J Cardiovasc Pharmacol 37, 64–77 [DOI] [PubMed] [Google Scholar]
- 53.Gorski PA, Ceholski DK, and Hajjar RJ (2015) Altered myocardial calcium cycling and energetics in heart failure--a rational approach for disease treatment. Cell Metab 21, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, and Hill JA (2010) Electrophysiological remodeling in heart failure. J Mol Cell Cardiol 48, 619–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Remmerie A, and Scott CL (2018) Macrophages and lipid metabolism. Cell Immunol 330, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castellon X, and Bogdanova V (2016) Chronic Inflammatory Diseases and Endothelial Dysfunction. Aging Dis 7, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Premer C, Kanelidis AJ, Hare JM, and Schulman IH (2019) Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. Mayo Clin Proc Innov Qual Outcomes 3, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tripathy D, Mohanty P, Dhindsa S, Syed T, Ghanim H, Aljada A, and Dandona P (2003) Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 52, 2882–2887 [DOI] [PubMed] [Google Scholar]
- 59.Zhuang Y, Liu H, Edward Zhou X, Kumar Verma R, de Waal PW, Jang W, Xu TH, Wang L, Meng X, Zhao G, Kang Y, Melcher K, Fan H, Lambert NA, Eric Xu H, and Zhang C (2020) Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nature communications 11, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kain V, Jadapalli JK, Tourki B, and Halade GV (2019) Inhibition of FPR2 impaired leukocytes recruitment and elicited non-resolving inflammation in acute heart failure. Pharmacol Res 146, 104295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halade GV, Kain V, Tourki B, and Jadapalli JK (2019) Lipoxygenase drives lipidomic and metabolic reprogramming in ischemic heart failure. Metabolism 96, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]