

Abstract
Osteoarthritis (OA) is a prevalent disease characterized by chronic joint degeneration and low-grade localized inflammation. There is no available treatment to delay OA progression. We report that in human primary articular chondrocytes, erythromycin, a well-known macrolide antibiotic, had the ability to inhibit pro-inflammatory cytokine Interleukin 1β (IL-1β)-induced catabolic gene expression and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation. Furthermore, erythromycin inhibited monosodium iodoacetate (MIA) -induced joint inflammation and cartilage matrix destruction in mice, an arthritis model that reflects the inflammatory and cartilage matrix loss aspects of OA. EM900, an erythromycin-derivative lacking antibiotic function, had the same activity as erythromycin in vitro and in vivo, indicating distinct anti-inflammatory and antibiotic properties. Using an antibody against erythromycin, we found erythromycin was present on chondrocytes in a dose-dependent manner. The association of erythromycin with chondrocytes was diminished in ghrelin receptor null chondrocytes, and administration of the ghrelin ligand prevented the association of erythromycin with chondrocytes. Importantly, the anti-inflammatory activity of erythromycin was diminished in ghrelin receptor null chondrocytes. Moreover, erythromycin could not exert its chondroprotective effect in ghrelin receptor null mice, and the loss of ghrelin receptor further augmented joint damage upon MIA-injection. Therefore, our study identified a novel pharmacological mechanism for how erythromycin exerts its chondroprotective effect. This mechanism entails ghrelin receptor signaling, which is necessary for alleviating inflammation and joint destruction.
Keywords: Erythromycin, ghrelin receptor, osteoarthritis, chondrocyte, cartilage, synovitis
Graphical Abstract
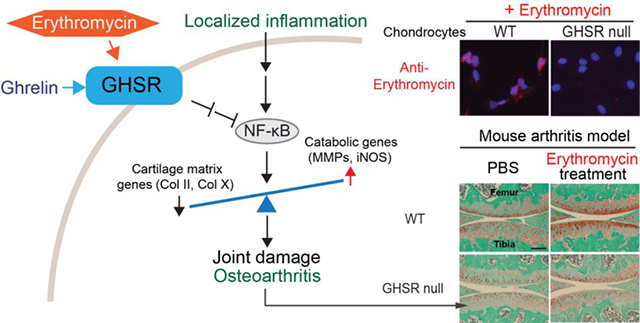
1. Introduction
Osteoarthritis (OA) is a prevalent disease characterized by chronic joint degeneration, including articular cartilage loss, synovitis and osteophyte formation [1–5]. The cause of OA is metabolic imbalance and mechanical stress, which is coupled with inflammation, leading to a catabolic shift in cartilage homeostasis. Increased macrophage activity has been reported as a biomarker for human OA [6], and antagonizing pro-inflammatory cytokine Interleukin 1β (IL-1β) directly in the joint protects cartilage matrix in post-traumatic experimental OA, indicating a pivotal role of inflammation in OA pathogenesis [7]. IL-1β activity is mediated by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway, which upon activation induces the expression of cartilage matrix-degrading enzymes such as matrix metalloproteinases (MMPs) and subsequent matrix degradation [8].
Recently, certain antibiotics used to treat bacterial infections have been shown to alter the response of chondrocytes toward inflammatory stimuli. Tetracycline and doxycycline chelate zinc at the active sites of MMPs and inhibit experimental OA and rheumatoid arthritis [9–14]; however, reduction in human joint pain and swelling was limited [15, 16]. The macrolide class of antibiotics, including erythromycin (EM) and azithromycin, has also been found to be anti-inflammatory. In patients with diffuse panbronchiolitis, they inhibit macrophage activation and inflammation in the lung [17–19]. Similarly, EM inhibited aseptic loosening after bone implants [20, 21]. Our group recently showed that EM inhibits inflammatory stimuli IL-1β and Lipopolysaccharides (LPS)-induced catabolic events in bovine articular chondrocytes [22]. However, the mechanism underlying this activity remains poorly understood.
In the current study, we discovered that the activity of EM is mediated by the ghrelin receptor. Ghrelin is a 28 amino acid peptide that is best known as the “hunger hormone,” as it increases appetite and controls blood sugar levels [23–26]. The ghrelin receptor is also known as the Growth Hormone Secretagogue Receptor (GHSR1a, or abbreviated as GHSR) [27]. While a truncated variant GHSR1b also exists, it does not bind to ghrelin; therefore, ghrelin signals through its binding to GHSR1a [28]. In chondrocytes, ghrelin has been found to control fatty acid uptake and inhibit inflammation when overexpressed [29–31]. However, these are gain-of-function experiments, and it is still not known whether ghrelin signaling is truly necessary for joint maintenance under disease conditions.
We identified a novel functional link between EM and ghrelin signaling in chondrocytes, since the loss of the ghrelin receptor abolished EM-mediated chondro-protection and resulted in more cartilage destruction in vivo. Administration of ghrelin to chondrocytes had a similar inhibitory activity as EM toward pro-inflammatory cytokine IL-1β-induced catabolic gene expression. This activity was diminished in ghrelin receptor null cells, suggesting that EM and ghrelin both act through the ghrelin receptor.
2. Materials and Methods
2.1. Experimental animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committees at Tufts University. C57/B6 mice were used. The ghrelin receptor null mouse (i.e. GHSR null, in C57/B6 background) was a generous gift from Dr. Jeffrey Zigman, University of Texas Southwestern Medical Center [26]. Mice were caged in groups under the standard conditions with a 12-hour light/dark cycle, fed with a standard chow diet and identified by ear tags. Tail biopsies were used for PCR-based genotyping based on the established method with primers (5’ to 3’): Forward: CCACTGCACGTCTCTCCCTATTT and reverse for WT: CGGTCTCCACCCTTCATTACTTTA or Rev for null: AAGAGCTACAGGAAGGCAGGTCA [26]. Mice were euthanized by carbon dioxide overdose followed by cervical translocation or by decapitation under inhalation anesthesia.
2.2. Isolation of normal and OA human cartilage specimens for RT-PCR analysis
Normal and OA human cartilage specimens were obtained as previously described [32], with additional samples over time. Samples of severe OA that showed no presence of articular cartilage were excluded. Articular cartilage slices of normal and OA specimens were excised from the tibial plateau of cadaveric joints (National Disease Research Interchange and Articular Engineering, age/sex: 84/M, 75/M, 65/F, 49/F). Human OA tibial plateau was obtained from patients undergoing total knee replacement surgery at Tufts Medical Center (age/sex: 53/F, 63/F, 65/F, 73/F, 72/M). All cartilage samples were de-identified before we received them and would have otherwise been discarded. Sample collection protocols were reviewed by the Institutional Review Board (IRB) at Tufts University and classified as exempt. Cartilage explants from cadavers were refrigerated for no longer than 36hrs postmortem before being used for chondrocyte isolation or gene expression analysis. Cartilage from knee replacement surgeries were obtained within 1hr after the excision. Due to the avascular nature of articular cartilage, chondrocytes in excised cartilage specimens remain alive for long periods of time, and have been used postmortem for culturing and gene expression analysis [33–37]. Each specimen was divided into two groups. The first group was subjected to histological analysis to confirm the integrity of the articular cartilage as previously reported [32]. The second group was subjected to gene expression analysis by RT-PCR.
2.3. In vitro culture of primary normal human articular chondrocytes
Primary normal human articular chondrocytes (nHACs) were purchased from Lonza (Walkersville, MD), or isolated from cadaveric joints retrieved within 36hrs post-mortem (National Disease Research Interchange). Experiments were performed using chondrocytes from three donors: age/sex: 49/M, 38/M, and 84/M). Before use, nHACs were re-differentiated per Lonza’s instructions. Briefly, nHACs were encapsulated in 1.2% alginate beads (Sigma, St. Louis, MO) at a density of 8×105 cells/ml and cultured in chondrocyte differentiation media (CDM) containing TGF-β1 (Lonza) supplemented with ascorbic acid for 2 to 4 weeks. Alginate beads were dissolved in 55mM citric acid (Sigma) supplemented with 10mM HEPES (Thermo Fisher Scientific, Waltham, MA) to collect re-differentiated nHACs. Cell viability was confirmed by 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma). Usually more than 80% cell viability was obtained. Re-differentiation of chondrocytes was confirmed by elevated Collagen II (Col-II) mRNA expression and alcian blue (Thermo Fisher Scientific) staining before proceeding. Once re-differentiation was confirmed, cells were treated with EM (Sigma), EM900 (a 12-membered macrolide and EM derivative manufactured in Drs. Sunazuka and Ömura laboratories) and ghrelin (Phoenix Pharmaceuticals, Burlingame, CA) overnight, and then subjected to IL-1β (1ng/ml) (Peprotech, Rocky Hill, NJ) treatment. For RT-qPCR analysis of EM, EM900 and ghrelin-treatment, cells were lysed after 3 days of IL-1β treatment. For RT-qPCR analysis of MIA-treated chondrocytes in vitro, cells were analyzed after 2 days of MIA (Thermo Fisher Scientific) treatment. All experiments were performed at least three times.
2.4. In vitro culture of murine articular chondrocytes
Murine femoral and tibial chondrocytes were isolated based on a previously published protocol [38]. Briefly, cartilage pieces from the femoral head, femoral condyle and tibial plateau were isolated and digested in collagenase D (Thermo Fisher Scientific) to remove residual soft tissues and to isolate chondrocytes. WT and GHSR null chondrocytes at passage 1 were seeded in a well of a 24-well plate in DMEM/F-12 (Thermo Fisher Scientific) supplemented with 10% FBS (Thermo Fisher Scientific). For analyzing the presence of EM in chondrocytes, cells were seeded in a well of an 8-wells chamber (Thermo Fisher Scientific) before being treated with EM (Sigma) or co-treated with ghrelin (Phoenix Pharmaceuticals) in serum-free media for 1 hour. For RT-qPCR analysis, cells were seeded in wells of a 24-wells plate. After 24 hours, EM or ghrelin were added to the cells, followed by IL-1β (1ng/ml) (Peprotech) treatment on the next day for an additional 3 days. All experiments were performed at least three times.
2.5. Explant culture of human articular cartilage
For ex vivo culturing of OA cartilage slices, OA cartilage pieces were prepared from OA donors as described above (72/M, 69/M, 73/F, 67/M, 54/F, 53/F, 60/F). Cartilage slices were cut into 2×3×2mm and cultured in the presence or absence of ghrelin (100nM), EM or EM900 (10μM) for 3 weeks with media changes every 3 days. Explant cultures were performed in duplicate per group and repeated for at least three donors.
2.6. Monosodium iodoacetate (MIA)-induced articular cartilage degeneration model
Adult male mice (18–22 weeks) were anesthetized by isoflurane/O2 inhalation. EM or EM900 at 40μg/g body weight/day was delivered by intraperitoneal injection using a 28G½” needle. PBS was delivered as a control. 5μL of 10μg/μL MIA dissolved in PBS was injected intra-articularly using a 30G½” needle. PBS was injected to contralateral knees as a control. EM or EM900 was administered every day until mice were euthanized at 7 days post injection of MIA. For each experiment, at least n=4 mice were used.
2.7. Cytotoxicity assay of erythromycin (EM) and EM900
Chondrocytes were seeded on tissue culture dishes and treated with EM and EM900 (0–100μM) for 4 days before being subjected to Live/Dead Cell Viability/Cytotoxicity assay (Thermo Fisher Scientific) according to the manufacturer’s instruction. The ratios of dead cells/total number of cells were determined.
2.8. RT-qPCR
Total RNA was isolated using the RNeasy mini kit (Qiagen, Germantown, MD) and reverse transcribed using the M-MLV reverse transcriptase (Thermo Fisher Scientific). Quantitative PCR was carried out using iTaq universal SYBR Green supermix (Bio-Rad, Hercules, CA) on the iQ5 Real Time Detection System (Bio-Rad). TATA-binding protein (TBP) served as a reference gene. The sequences for PCR primers are: human primers: TBP, forward 5’-GGTGCTAAAGTCAGAGCAGAA-3’, reverse 5’-CAAGGGTACATGAGAGCCATTA-3’; Ghrelin, for, 5’-AAGTGATCGCCCACAAGCCTTACT-3’, reverse, 5’-TGTACAACAGTCGTGGGAGTTGCT-3’; Aggrecan (ACAN), forward, 5’-AAGGTCTCTCTCTCTCAGCCACCT-3’, reverse, 5’-TGGCTGAGGGAATGGACACTGAAA-3’; Collagen II (COL-II), forward, 5’-TTCATCCCACCCTCTCACAGTT-3’, reverse, 5’-CCTCTGCCTTGACCCGAA-3’; Collagen IX (COL-IX), forward, 5’-TCGATGTGCTGTCTCTGGAGTGAT-3’, reverse, 5’-CTTTGCTTTCACATAGAAGCGCTCA-3’; Collagen X (COL-X), forward, 5’- ACCCAGCATAACTTGGAAACAGGT-3’, reverse, 5’- TCACTTGAATGGGAGGCACAAGGT-3’; IL-1β, forward, 5’-AACAGGCTGCTCTGGGATTCTCTT-3’, reverse, 5’- ATTTCACTGGCGAGCTCAGGTACT-3’; TNF⍺ (tumor necrosis factor alpha), forward, 5’-AGGACGAACATCCAACCTTCCCAA-3’, reverse, 5’- TTTGAGCCAGAAGAGGTTGAGGGT-3’; iNOS (inducible nitric oxide synthase), forward, 5’- GCGTTACTCCACCAACAATGGCAA-3’, reverse, 5’- ATAGCGGATGAGCTGAGCATTCCA-3’; MMP1, forward, 5’- AGTGACTGGGAAACCAGATGCTGA-3’, reverse, 5’- TCAGTGAGGACAAACTGAGCCACA-3’; MMP3, forward, 5’- AGGCAAGACAGCAAGGCATAGAGA-3’, reverse, 5’- ACAAGGTTCATGCTGGTGTCCTCA-3’; MMP9, forward, 5’- GACCTGGGCAGATTCCAAA-3’, reverse, 5’-GGCAAGTCTTCCGAGTAGTTT-3’; MMP13, forward, 5’-ACAAGTAGTTCCAAAGGCTACAA-3’, reverse, 5’- ATAGGAAACATGAGTGCTCCAG-3’. Mouse primers: TBP, forward, 5’- CTACCGTGAATCTTGGCTGTAA-3’, reverse, 5’- GTTGTCCGTGGCTCTCTTATT-3’; MMP13, forward, 5’-TCTGGGCTCTGAATGCTTATG-3’, reverse, 5’-GCTCAGTCTCTTCACCTCTTT-3’.
2.9. NF-κB transactivation assay
70ng of a six-copy NF-κB element-driven luciferase reporter construct and 10ng of Renilla luciferase internal control were transiently co-transfected into nHACs using X-tremeGENE HP (Roche, Indianapolis, IN). EM (10μM), EM900 (10μM) or ghrelin (10nM) was added at the time of transfection. At 24 hours after the transfection, chondrocytes were treated with 1ng/ml IL-1β for an additional 16 hours. Luciferase assay was conducted using the Dual Luciferase Assay kit (Promega, Madison, WI). Luminescence was detected using a microplate luminometer (Perkin Elmer, 1450 Microbeta TriLux). NF-κB luciferase activity was normalized to Renilla luciferase activity. Fold induction of normalized luciferase activity was determined based on the control sample as a baseline. At least three independent experiments in triplicate were performed.
2.10. Serum response element (SRE) reporter assay
100ng of a SRE luciferase reporter construct (gift from Dr. Alan Kopin, Tufts Medical Center) and 30ng of Renilla luciferase internal control were transiently co-transfected into nHACs using X-tremeGENE HP (Roche). At 24 hours after the transfection, cells were treated with EM (10μM), EM900 (10μM) or ghrelin (10nM) in serum-free media for additional 18 hours. Luciferase assay was conducted using the Dual Luciferase Assay kit (Promega). Luminescence was detected using a microplate luminometer (Perkin Elmer, 1450 Microbeta TriLux). SRE luciferase activity was normalized to Renilla luciferase activity. Fold induction of normalized luciferase activity was determined based on the control sample as a baseline. At least three independent experiments in triplicate were performed.
2.11. Immunocytochemistry (ICC) and histological analysis
For ICC analysis, chondrocytes were fixed with ice-chilled ethanol (VWR, Radnor, PA) for 15 minutes. Cells were blocked with goat serum (Thermo Fisher Scientific) for 1 hour and incubated with primary sheep anti-EM antibody (Abcam, ab123983, Cambridge, MA) followed by incubation with Alexa594 or fluorescein-conjugated secondary goat anti-sheep antibody (Thermo Fisher Scientific). Nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole, Thermo Fisher Scientific) staining. Five different views under the microscope were randomly selected from each well for quantification. Experiments were performed in triplicates.
For histological analysis of joint tissues, samples were fixed in 1% PFA (VWR, Radnor, PA), then decalcified in 0.33M EDTA (VWR) 10 days for mouse knee joints and overnight for human cartilage specimens. Decalcified samples were dehydrated in 25, 50, 75, 100 and 100% ethanol/PBS and xylene twice for each 1hr. Samples were embedded in paraffin (Paraplast plus, McCormick Scientific, St. Louis, MO). Mouse knee joints and human cartilage specimens were sectioned at 5μm thickness.
Sections were stained with 0.1% Safranin O (Sigma) and counterstained with Hematoxylin (Sigma) and Fast green (Sigma). Cartilage integrity was evaluated by the established OARSI scoring system that is based on loss of Safranin O staining, as described previously [32, 39]. Severity of synovitis was scored by the established method [40, 41]. This scoring system has the following three criteria: enlargement of the synovial cell layer (0–3 points), cellularity of resident cells (0–3 points) and inflammatory infiltration (0–3 points). When the points are totaled, a full score of 9 points would indicate severe synovitis with lower scores indicating less severe synovitis. Three to four sections per mouse knee joint or human cartilage slices within 100μm intervals were evaluated and scored. All scorings were performed blinded.
For ghrelin receptor (GHSR) immunohistochemistry, heat-induced antigen retrieval in 10mM citric acid buffer at pH 6.0 was performed for 10min by steam. Primary GHS-R1a antibody (Santa Cruz, F-16, Dallas, Texas) and secondary biotin-conjugated horse anti-goat antibody (Vector labs, Burlingame, CA) were used. For chromogenic staining, the Vectastain ABC Elite kit (VWR, Radnor, PA) was used according to the manufacture’s instruction. Nuclei were counterstained with methyl green (Sigma).
2.12. Glucose consumption assay
Glucose consumption assays were performed as done previously [42]. Cells were seeded overnight and then treated with MIA for 2 days. OA and nHACs at passage 1 were seeded and grown for 2 days. Conditioned media were harvested at the end of culture and glucose concentration was determined using Glucose (HK) assay kit (Sigma) according to the manufacturer’s instruction. Briefly, 3μL of media samples and glucose standards were incubated with 250μL assay reagent for 15 minutes and the absorbance at 340nm was measured using a plate reader (Bio-Rad Benchmark Plus). Total glucose consumption amount was calculated by subtracting the total glucose amount of each sample from the initial glucose amount. Total cell numbers were used to for normalization. For each treatment, technical duplicates were performed, with triplicate biological repeats.
2.13. Intracellullar ATP assay
Intracellular ATP assays were performed as done previously [42]. Cells were seeded overnight and then treated with MIA for 2 days. OA and nHACs at passage 1 were seeded and grown for 2 days. At the end of culture, cells were lysed in the lysis buffer (200mM Tris pH.7.5, 2M NaCl, 20mM EDTA and 0.2% Triton X-100) for 5 minutes on ice, and supernatants were collected for ATP assay using ATP Determination kit (Thermo Fisher Scientific) according to the manufacturer’s instruction. Each sample and ATP standards were incubated with reaction solutions for 2 minutes and luminescence was detected using a microplate luminometer (Perkin Elmer, 1450 Microbeta TriLux). Total cell numbers were used for normalization. For each treatment, technical duplicates were performed, with triplicate biological repeats.
2.14. Reactive oxygen species (ROS) assay
ROS assays were performed as done previously [42]. Cells were seeded overnight and then treated with MIA for 2 days. OA and nHACs at passage 1 were seeded and grown for 2 days. ROS was detected using Image-iT Live Green Reactive Oxygen Species Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s instruction. Cells were washed with HBSS/Ca/Mg and incubated with 25μM carboxy-H2DCFDA (2′,7′-Dichlorodihydrofluorescein diacetate) for 30 minutes at 37˚C. ROS production was measured at 495/529nm (excitation/emission) using a plate reader (FlexStation 3, Molecular devices). ROS production was normalized by signal of Hoechst 33342 measured at 350/461nm (excitation/emission). For each treatment, technical duplicates were performed, with triplicate biological repeats.
2.15. Light microscopy
Bright field images were taken using an Olympus IX-71 microscope and Olympus DP70 digital cameras. The optical parameters and camera exposure time were kept constant between samples of the same experiment.
2.16. Statistical analysis
Parametric data were reported as mean ± standard deviation (SD) and statistical analysis was performed using a Student’s t-test, Dunnett’s test or a one-way analysis of variance followed by a post-hoc Tukey test with p-values of <0.05 considered significant. Non-parametric data (OARSI scores and synovitis scores) were reported as a box plot, where the median, the maximum and minimum data values were indicated; and statistical analysis was performed using the Kruskal-Wallis test followed by Mann-Whitney U test with p-values of <0.05 considered significant. All statistical analyses were conducted on Prism (Graphpad).
3. Results
3.1. EM inhibits pro-inflammatory cytokine IL-1β-induced catabolic gene expression and NF-κB activity in chondrocytes
To evaluate EM activity on human articular chondrocytes under inflammatory conditions, we treated primary normal human articular chondrocytes (nHACs) with EM and IL-1β. A “live-dead” assay was first performed on nHACs to determine the concentrations of EM to be used. We tested 1, 10 and 100μM of EM, which are in the range of systemic plasma concentrations achieved with typical therapeutic activity of EM in humans [43]. The level of cell death overall was minimal (1–4%), although higher EM concentrations caused slightly more cell death (Fig. 1A). As expected, IL-1β reduced cartilage matrix collagen II (Col-II) mRNA expression, and induced hypertrophic marker collagen X (Col-X), as well as catabolic genes iNOS and MMPs expression (Fig. 1B). EM treatment alone did not alter the expression of these genes. However, when chondrocytes were pre-treated with EM, the induction of hypertrophic and catabolic genes Col-X, iNOS and MMPs by IL-1β was strongly reduced, and Col-II expression restored (Fig. 1B). This result suggests that EM itself inhibits the response to pro-inflammatory stimuli in human chondrocytes.
Fig. 1. Erythromycin (EM) inhibits the catabolic activity of IL-1β on primary human articular chondrocytes (nHACs).

(A) Live-dead assay on chondrocytes treated with EM (1, 10 and 100μM) for 4 days. Percentage of dead cells/total cells was quantified. (B) RT-qPCR analysis of Col-II, Col-X, iNOS, MMP1, MMP9 and MMP13 on nHACs treated with EM (0, 1, and 10μM) and IL-1β (1ng/mL). TBP served as a reference gene for normalization. (C) NF-κB transactivation assay. nHACs were transiently transfected with an NF-κB reporter and treated with EM (10μM) and IL-1β (1ng/ml). A Renilla luciferase construct was co-transfected as an internal control for normalization. Data were reported as mean ± SD and analyzed by Dunnett’s test (B) and an unpaired t-test (C). At least three independent experiments were performed. * p<0.05.
To further investigate the underlying mechanism, we evaluated activation of NF-κB, the key mediator of IL-1β signaling [32, 44]. nHACs were transfected with an NF-κB luciferase reporter, which has NF-κB binding sites and serves as a readout for NF-κB transcriptional activity after NF-κB nuclear translocation [32, 45] (Fig. 1C). EM alone did not have any effects on the activity of this luciferase reporter construct. In contrast, IL-1β clearly induced NF-κB reporter activity, but co-treatment with EM significantly reduced IL-1β-induced luciferase activity (Fig. 1C). Thus, EM inhibits the ability of chondrocytes to activate NF-κB signaling and induce catabolic gene expression under inflammatory conditions.
3.2. Ghrelin receptor (GHSR) mediates the activity of erythromycin (EM) to antagonize IL-1β
To further understand EM activity on chondrocytes, we searched the literature for proteins that interact with EM. Interestingly, classic in vitro binding assays showed that a derivative of EM could bind to the ghrelin receptor [46–48] and ghrelin signaling inhibited inflammation [30, 31]. Thus, we hypothesized that ghrelin signaling might mediate the activity of EM. This hypothesis was tested through the use of the mouse strain lacking the ghrelin receptor GHSR, i.e. GHSR null mice. Immunohistochemistry (IHC) analysis confirmed that GHSR is expressed in articular chondrocytes and is absent in the GHSR null mice (Fig. 2A). Subsequently, primary chondrocytes from WT and GHSR null mice were used to determine whether EM requires GHSR to inhibit IL-1β activity. EM was capable of inhibiting IL-1β-induced MMP13 expression in WT cells, but not in GHSR null cells (Fig. 2B), suggesting that EM requires the ghrelin receptor for this activity. Next, we evaluated whether GHSR mediates the activity of EM on chondrocytes. In our immunocytochemistry analysis, EM was associated with chondrocytes in a dose-dependent manner (Fig. 2C). Furthermore, this binding was diminished in chondrocytes from the GHSR null mice (Fig. 2D), suggesting that the association of EM to chondrocytes is mediated by the GHSR. However, it is not clear whether EM and ghrelin compete for the same site on the ghrelin receptor.
Fig. 2. The anti-inflammatory activity of EM in chondrocytes is mediated by the ghrelin receptor GHSR.

(A) Immunohistochemical (IHC) analysis using an anti-GHSR antibody (anti-GHS-R1a) on WT and GHSR null mouse knee joints. Rectangles denote areas magnified. Arrows and arrowhead indicate GHSR positive and negative cells, respectively. Scale bar = 200μm. (B) Relative MMP13 mRNA analysis on WT and GHSR null chondrocytes treated with EM (0, 10, and 25μM) and IL-1β (1ng/ml). TBP served as a loading control. (C) Immunocytochemistry (ICC) analysis with anti-EM antibody (red) on WT chondrocytes treated with different doses of EM (0, 1, 10, and 25μM) for 1hr. Arrows indicate cells with EM staining. Percentage of EM-positive cells was calculated. (D) ICC analysis with EM presence on WT and GHSR null chondrocytes treated with EM (10μM) for 1hr. Percentage of EM-positive cells was calculated. Data were reported as mean ± SD and analyzed by Dunnett’s test (B and C) and an unpaired t-test (D). At least three independent experiments were performed. * p<0.05. n.s: not significant.
3.3. Ghrelin exhibits a similar activity as EM for inhibiting IL-1β-induced catabolic gene expression and NF-κB activation
Since we found that the ghrelin receptor mediates the activity of EM on chondrocytes under IL1β treatment, we further hypothesized that the natural ligand for the ghrelin receptor, ghrelin, would have a similar activity. Therefore, normal primary human articular chondrocytes (nHACs) were treated with IL-1β in the absence or presence of ghrelin and the response to IL-1β was assayed. While ghrelin treatment did not reduce IL-1β-induced downregulation of Col-II mRNA expression, it inhibited IL-1β-induced Col-X expression as well as those of catabolic genes iNOS and MMPs (Fig. 3A). In addition, ghrelin strongly inhibited IL-1β-induced NF-κB luciferase activity (Fig. 3B). Interestingly, ghrelin alone did not exert a significant effect on the expression of any of the genes or on NF-κB luciferase activity, suggesting that like EM, ghrelin specifically reduced catabolic activities in response to inflammatory stimuli. However, since EM, but not ghrelin, enhanced Col-II mRNA expression, but did not inhibit Col-X expression under IL-1β treatment, it suggests that ghrelin has a different activity as compared to EM in terms of cartilage matrix gene expression. Using a serum-response element (SRE) reporter, which is an established reporter that serves as a readout of GHSR signaling as a G-protein coupled receptor [49, 50], we found that both ghrelin and EM indeed activated GHSR (Fig. 3C), suggesting that both ghrelin and EM are agonists at the ghrelin receptor.
Fig. 3. Ghrelin signaling is necessary and sufficient to inhibit IL-1β-induced catabolic gene expression and NF-κB activation in chondrocytes.

(A) RT-qPCR analysis of Col-II, Col-X, iNOS, MMP1, MMP9 and MMP13 on nHACs treated with ghrelin (0, 10 and 100nM) and IL-1β (1ng/mL). TBP served as a reference gene. (B) NF-κB transactivation assay. nHACs were transiently transfected with NF-κB reporter construct for 24 hours and then treated with ghrelin (10nM) and IL-1β (1ng/mL). A Renilla luciferase construct was co-transfected as an internal control for normalization. (C) Serum Response Element (SRE) luciferase reporter assay. nHACs were transiently transfected with the SRE reporter construct for 24 hours and then treated with ghrelin (10nM) or EM (10μM) for 16 hours. A Renilla luciferase construct was co-transfected as an internal control for normalization. Data are presented as fold activation relative to untreated samples. (D) RT-qPCR analysis of MMP13 on WT and GHSR null mouse chondrocytes treated with ghrelin (0, 10, 100nM) and IL-1β (1ng/mL). TBP served as a reference gene. (E) ICC analysis with an anti-EM antibody on WT chondrocytes co-treated with EM (10μM) and different concentrations of ghrelin (1, 10 and 100nM). Arrows indicate cells with EM staining. Percentage of EM-positive cells was calculated. Data were reported as mean ± SD and analyzed by Dunnett’s test (A, D and E) and an unpaired t-test (B and C). At least three independent experiments were performed. * p<0.05. n.s: not significant.
Next, we determined whether ghrelin requires its receptor to inhibit IL-1β activity by using primary chondrocytes from WT and GHSR null mice. We found that ghrelin inhibited IL-1β-induced MMP13 expression in WT cells, but not in GHSR null cells (Fig. 3D). This result suggests that ghrelin, like EM, requires the ghrelin receptor to exert this function. We reasoned that if ghrelin and EM are both agonists at the ghrelin receptor, they would be competitors for the binding of this receptor. Thus, we treated mouse chondrocytes with EM and ghrelin simultaneously. Indeed, increasing concentrations of ghrelin led to a progressive decrease in the percentage of cells with EM presence in our immunocytochemistry analysis, thus further confirming that the ghrelin receptor is used by both EM and ghrelin (Fig. 3E). It is worth noting however, that the association of EM with chondrocytes could be overcome by a much lower concentration of ghrelin, suggesting that ghrelin is a much more potent competitor (Fig. 3E).
3.4. GHSR mediates the chondroprotective activity of EM in vivo
To determine whether GHSR is necessary to mediate the chondroprotective effect of EM in vivo, we subjected WT and GHSR null mice to intra-articular injection of monosodium iodoacetate (MIA). This model is well-established for recapitulating joint destruction and inflammation [51–56]. MIA is a specific inhibitor of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a key enzyme in the glycolysis pathway. Since articular cartilage is highly glycolytic as it adapts to its avascular environment [33, 34, 57], inhibition of glycolysis causes energy imbalance and cell death. As a result, there is a surge of inflammation. As MIA is cleared in the body, inflammation subsequently subsides to a large extent. However, as the case of injury-induced OA, already damaged cartilage continues to slowly disintegrate and enters the chronic phase of OA [51, 53–56]. To further validate the MIA model, we compared normal and human OA chondrocytes, and found OA chondrocytes to have reduced glucose uptake and ATP production as well as increased reactive oxygen species (ROS) production (Fig. 4A). These metabolic changes were also found in chondrocytes treated with MIA, suggesting MIA has caused oxidative stress as in human OA (Fig. 4B). Gene expression analysis indicated that MIA treatment increased pro-inflammatory cytokine IL-1β and TNF⍺ expression as well as hypertrophic chondrocyte marker Col-X and catabolic gene MMP13 expression in human articular chondrocytes (Fig. 4C). There were no differences in joint health between WT and GHSR null littermates under the control condition (PBS-injected), suggesting that the lack of GHSR does not affect the joint in the absence of OA (Fig. 4D). However, upon MIA treatment, GHSR null mice exhibited much more cartilage matrix loss than their WT littermates (Fig. 4D), suggesting that endogenous GHSR is required for preventing MIA-induced articular cartilage degeneration. We next determined whether GHSR mediates the chondro-protective activity of EM in vivo by subjecting WT and GHSR null mice to intraperitoneal injection of EM, in addition to intra-articular injection of MIA. In WT mice, EM dramatically restored MIA-induced loss of safranin O staining, whereas EM could not restore matrix in GHSR null littermates (Fig. 4E). Similarly, EM could not rescue MIA-induced synovitis in the GHSR null joint as compared to WT (Fig. 4F). These data strongly demonstrate that GHSR mediates the chondroprotective activity of EM in vivo.
Fig. 4. The Ghrelin receptor is required for EM to inhibit cartilage matrix loss and synovitis in vivo.

(A) Analysis of total glucose consumption, intracellular ATP levels and reactive oxygen species (ROS) levels at day 2 of culture in healthy and OA human articular chondrocytes. (B) Analysis of total glucose consumption, intracellular ATP levels and reactive oxygen species (ROS) levels at day 2 of culture in nHACs after MIA treatment. (C) RT-qPCR analysis of aggrecan (agg), Col-II, Col-X, IL-1β, TNF⍺ and MMP13 on nHACs treated with MIA (0, 0.1 and 1μM) for 2 days. TBP served as a reference gene for normalization. (D) Safranin O staining analysis of WT and GHSR null mouse knee joints. MIA (50μg/joint) was intra-articularly injected to knee joints at day 0, and samples were harvested at day 7. The degree of cartilage matrix loss was examined according to the OARSI scoring system. (E) Safranin O staining analysis of WT and GHSR null mouse knee joints injected with MIA. MIA was intra-articularly injected to knee joints at day 0. EM (PBS as control) was IP-injected at 50μg/g into the mice daily until sample harvest at day 7. The degree of cartilage matrix loss was examined according to the OARSI scoring system. (F) Synovitis analysis on sections from the medial tibial plateau of WT and GHSR null mice knees injected with EM. MIA was intra-articularly injected to knee joints at day 0. EM (PBS as control) was IP-injected into the mice daily until sample harvest at day 7. Arrowheads indicate thickening of the synovial layers and increased cellularity of resident cells. M = Meniscus. T = Tibia. F=Femur. Synovitis was scored according to established synovitis scoring protocols. Scale bar = 200μm. For A, data were reported as mean ± SD and analyzed by an unpaired t-test. For B and C, data were reported as mean ± SD and analyzed by Dunnett’s test. Comparisons were made between MIA treatments over untreated controls for each gene. For D-F, data were reported as a box plot, where the median, as well as the maximum and the minimum data points were indicated. Data from D-F were analyzed by Kruskal-Wallis statistical test followed by Mann-Whitney U test. * p<0.05. n.s: not significant.
3.5. EM900, a macrolide devoid of antibiotic function, has a similar activity to EM and ghrelin
One concern of antibiotic use in disease treatment is antibiotic resistance. To determine whether the anti-inflammatory activity of EM is separate from the macrolide’s antibiotic function, we used EM900 in in vitro and in vivo experiments. EM900 is a 12 membered EM derivative synthesized to be devoid of antibiotic activities, but has demonstrated anti-inflammatory activities in airway epithelial cells [58–60]. In our “live-dead” assay, chondrocytes showed good viability when treated with 1 and 10μM of EM900 (Fig. 5A), which were the concentrations subsequently used for the functional analysis of EM900. When EM900 was administered in pro-inflammatory cytokine IL-1β-treated chondrocytes, a reduction of catabolic gene MMP13 expression was observed, suggesting that EM900 inhibited the catabolic activity of IL-1β (Fig. 5B). Furthermore, the NF-κB reporter activity induced by IL-1β was also inhibited by EM900 (Fig. 5C), indicating that EM900 inhibits NF-κB activation. To determine whether EM900 also activates the SRE luciferase reporter that can be activated by ghrelin and EM, we administered EM900 to human primary articular chondrocytes transfected with this reporter. Similar to ghrelin and EM, EM900 also led to the activation of SRE reporter (Fig. 5D).
Fig. 5. EM900, a non-antibiotic EM derivative, inhibits IL-1β-induced MMP13 expression and NF-κB activation, and maintains joint health in MIA-injected knees.

(A) Live-dead assay on chondrocytes treated with EM900 (0, 1, 10 and 100μM) for 4 days. Percentage of dead cells/total cells was quantified. (B) RT-qPCR analysis of MMP13 mRNA on nHACs treated with EM900 (1, 10 and 25μM) overnight followed by IL-1β (1ng/mL) treatment for 3 days. TBP served as a reference gene. (C) NF-κB transactivation assay. nHACs were transiently transfected with the NF-κB reporter construct for 24 hours and then treated with EM900 (1μM) and IL-1β (1ng/mL). A Renilla luciferase construct was co-transfected as an internal control for normalization. (D) Serum Response Element (SRE) luciferase reporter assay as a readout of ghrelin receptor signaling. nHACs were transiently transfected with the SRE reporter construct for 24 hours and then treated with EM900 (10μM) and IL-1β (1ng/mL). A Renilla luciferase construct was co-transfected as an internal control for normalization. Data were presented as “fold activation” compared to untreated samples. (E) Safranin O staining images for cartilage matrix analysis of WT mouse knee joints. MIA was intra-articularly injected to knee joints at day 0. EM900 was IP-delivered daily from the day of MIA injection. Samples were collected for analysis 7 days later. The degree of cartilage matrix loss was scored according to the OARSI scoring system. Scale bar = 200μm. M = Meniscus. T = Tibia. F = Femur. (F) Synovitis analysis on sections from the medial tibial plateau of the WT mice knees injected with EM900. MIA was intra-articularly injected into knee joints at day 0. EM900 (PBS as control) was IP-injected into the mice daily until sample harvest at day 7. Arrowheads indicate thickening of the synovial layers and increased cellularity of resident cells. Synovitis was scored according to established synovitis scoring protocols. For A to D, data were reported as mean ± SD and analyzed by Dunnett’s test (B) and an unpaired t-test (C, D). For E and F, data were reported in a box plot, where the median, as well as the maximum and the minimum data points were indicated. Data from E and F were analyzed by Kruskal-Wallis statistical test followed by Mann-Whitney U test. * p<0.05.
To determine whether EM900 has a protective effect in vivo, we subjected normal mice to MIA-induced inflammatory osteoarthritis, and intraperitoneally administered EM900. Histological analyses showed significant level of cartilage matrix loss in MIA-treated knee joints, but much less matrix loss when EM900 was also injected, which was subsequently quantified using the OARSI scoring system (Fig. 5E). Consistently, EM900 also significantly reduced MIA-induced synovitis induced by MIA (Fig. 5F). While it is not clear whether the ghrelin receptor is required to mediate the activity of EM900, our results demonstrate that EM900 has an anti-inflammatory and joint protective activity similar to EM in isolated human chondrocytes as well as in an inflammatory OA animal model, and suggests that this activity is antibiotic-independent.
3.6. EM, EM900 and ghrelin enhance cartilage matrix levels in human OA cartilage
Although EM, EM900 and ghrelin have demonstrated inhibitory activities of IL-1β in the in vitro cultures of primary human chondrocytes, whether they could promote cartilage matrix levels in human cartilage under diseased conditions was not known. Thus, we subjected OA cartilage specimens to EM and ghrelin treatment. As expected, these OA cartilage samples also showed reduced expression of cartilage matrix genes aggrecan and collagen IX and had increased expression of catabolic genes MMPs and IL-1β (Fig. 6A). Significantly, ghrelin expression was reduced in OA cartilage specimens (Fig. 6A). When administered in ex vivo-cultured OA articular cartilage explants, EM, EM900 and ghrelin all led to significantly increased safranin O staining as compared to PBS control samples (Fig. 5B–5D), suggesting that EM, EM900, as well as ghrelin indeed enhance cartilage matrix levels even in advanced OA.
Fig. 6. EM and ghrelin enhance cartilage matrix levels in human OA cartilage.

(A) RT-qPCR analysis of Agg, Col-II, Col-IX, MMP13, iNOS, IL-1β and ghrelin in human normal cartilage from 4 healthy donors and OA cartilage from 5 OA donors. TBP served as a reference gene. (B) Safranin O staining analysis on sections of OA articular cartilage explants after 3 weeks of culturing with 10μM EM. Images of explants from donor 54/F (age/sex) are shown. (C) Safranin O staining analysis on sections of OA articular cartilage explants after 3 weeks of culturing with 10μM EM900. Images of explants from donor 54/F (age/sex) are shown. (D) Safranin O staining analysis on sections of OA articular cartilage explants after 3 weeks of culturing with 100nM ghrelin. Images of explants from donor 53/F (age/sex) are shown. For each experiment, duplicate technical repeats were analyzed. Experiments were repeated three times using samples from three different OA donors. As the extent of cartilage matrix loss and OA severity of the donor samples differed among experiments, cellularity and matrix levels varied, which is evident from the differences in control samples used for EM, EM900 and ghrelin experiments. Scale bar = 100μm. SZ = superficial zone. DZ = deep zone.
4. Discussion
This study provides a key mechanism for the pharmacological activity of erythromycin (EM). We report that the ghrelin receptor is an essential mediator of EM chondroprotective activity, demonstrating that both ghrelin and EM can activate the ghrelin receptor and inhibit pro-inflammatory cytokine IL-1β-induced NF-κB activation and catabolic gene expression. Furthermore, the ghrelin receptor is required to resist synovitis and joint destruction in EM-treated mice in vivo, thus playing an essential role for joint maintenance under pathological conditions.
Beneficial effects of ghrelin on OA cartilage has only recently been reported. In 2017, Zou et al. found that the synovial fluid of knee OA patients contained lower levels of ghrelin [61]. In the same year, Qu et al. showed that ectopic intraperitoneal injection of ghrelin could reduce surgery-induced mouse knee cartilage damage and inflammation [31]. In 2018, Liu et al. also demonstrated that supplementing ghrelin in culture media inhibited IL-1β-induced catabolic changes in human cartilage [30]. Our study is consistent with these recent findings, but we have also significantly deepened the understanding on the role of ghrelin in OA in additional ways. First, we definitely demonstrate that ghrelin signaling is critical for joint health and resistance to inflammation. Past reports focused on ectopic ghrelin treatment and did not indicate whether ghrelin signaling is required for the maintenance of the joint [30, 31]. Our study demonstrated that ghrelin signaling is essential for resisting inflammatory cytokine-induced MMP expression in vitro and delaying cartilage destruction in vivo. Second, we showed that ghrelin enhances cartilage matrix levels in human OA cartilage, even in the absence of exogenous pro-inflammatory cytokine. This has important implications for the potential use of ghrelin or its agonists to treat existing OA and halt cartilage destruction. Third, we extended our analysis in vivo to the analysis of the synovium, which plays an important role in inflammation of the joint, rather than being restricted to cartilage analysis. It is worth noting that while ghrelin administration would increase feeding and body weight [62, 63], mice lacking ghrelin have the same weight as the wild type under normal feeding conditions [64, 65]. On the other hand, mice lacking the receptor for ghrelin showed sexual dimorphism in this aspect: male mice did not exhibit any difference in body weight differences when fed with the standard chow, but female mice did [26]. Since we only used male mice, it allays the concern of body weight-associated OA in our study.
One important discovery of our study is the identification of a pharmacological mechanism for the macrolide antibiotic erythromycin (EM) in promoting joint health. EM is a commonly used antibiotic that directly binds bacterial ribosomes to inhibit protein synthesis [66]. Although EM-like macrolide antibiotics have anti-inflammatory activities in the lung and bone in an antibiotic-independent manner, its chondroprotective effect in the joint has only been described in our recent study [17–22]. However, in all these systems, how EM exerts its effect on eukaryotic cells had not been elucidated. EM was previously shown to be capable of binding to the motilin receptor [67, 68]. The natural ligand of the motilin receptor is motilin, a peptide that stimulates GI motility [69]. Interestingly, in the mouse, motilin and its receptors are pseudogenes [70, 71]. Since our in vivo experiments indicate a joint protective effect of EM, we reasoned that the motilin receptor could not be mediating the activity of EM in the mouse [22]. The ghrelin receptor and the motilin receptor share 50% identity, and both ligands have very similar roles in promoting gastric emptying and GI motility [69, 72, 73]. While an EM derivative binds to the ghrelin receptor, it is still not known whether EM itself can directly bind to the receptor. Nevertheless, our subsequent experiments indicated that ghrelin can compete with EM in ghrelin receptor association, and that the effect of EM is lost when the ghrelin receptor is not present. Thus, our data strongly suggest that EM functionally acts through the ghrelin receptor to exert its activity in the mouse. However, this does not exclude the motilin receptor as a mediator of the activity of EM in species where motilin signaling is present, as in humans [70]. While the binding affinity of EM to the ghrelin receptor in vivo and in vitro is now known, our data indicate that the association of EM with chondrocytes can be outcompeted by a much lower concentration of ghrelin, suggesting that it is not likely that EM will inhibit the activity of endogenous ghrelin (Fig. 3F). Furthermore, in the case of OA, when ghrelin concentration is already lower in the synovial fluid [61], additional EM might stimulate the ghrelin receptor to exert its beneficial effect. However, in order to select the most appropriate macrolide for OA therapy, other EM-like antibiotics, such as azithromycin or clarithromycin, would also need to be investigated for similar effects on the joint as EM,
Systemic administration of macrolide antibiotics may raise the concern of antibiotic resistance. However, this concern may be allayed by intra-articular injection of antibiotics or by administration of non-antibiotic macrolides such as EM900. One limitation of this study is that we have not demonstrated whether EM900 acts through the ghrelin receptor to exert its activity, even though we have shown this is the case for EM. Another limitation is that only one mouse model for joint inflammation and destruction was tested. Since there are many causes of OA, it will be important to evaluate the role of these reagents in other models, such as the Destabilization of the Medial Meniscus (DMM), meniscectomy, or obesity-induced OA model [74–78].
Future studies will also involve the delivering of EM into OA joints after OA has initiated for a period of time so that its therapeutic potential can be evaluated. The only study on EM in human OA was restricted to a single clinical trial combining EM with acetaminophen [79]. While joint effusion and pain was reduced [79], changes in joint structure were not assessed and erythromycin efficacy as a monotherapy was also not determined [79]. Our study has characterized the structural changes of the joint and uncovered a new mechanism for EM to act as an anti-inflammatory reagent. While ghrelin has been reported to alleviate pain in the carrageenan intraplantar injection model [80], it remains unclear whether such parallels exist in the knee joint. Thus, future experiments would be designed to determine whether EM and ghrelin can provide pain relief in arthritic joints, to thoroughly explore the potential of these molecules as OA therapeutics.
Acknowledgments
We thank Dr. Jeffrey Zigman (University of Texas Southwestern Medical Center) for providing the ghrelin receptor null mice, Dr. Gail Sonenshein (Tufts University) for providing the NF-κB luciferase reporter construct, and Dr. Alan Kopin (Tufts Medical Center) for providing the SRE luciferase reporter construct. We thank Judith Hollander and Yuxi Chen (Tufts University Sackler School of Graduate Medical Sciences) for helpful discussions. This work has been supported by grants to LZ from the NIH (AR054611, AR069278), and a grant from the Tufts Clinical Translational Science Institute (UL1TR001064).
Footnotes
Conflict of interest
None of the authors have any conflict of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation --- United States, 2007–2009, MMWR Morb Mortal Wkly Rep, 59 (2010) 1261–1265. [PubMed] [Google Scholar]
- [2].Bitton R, The economic burden of osteoarthritis, Am J Manag Care, 15 (2009) S230–235. [PubMed] [Google Scholar]
- [3].Hochberg MC, Osteoarthritis year 2012 in review: clinical, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 20 (2012) 1465–1469. [DOI] [PubMed] [Google Scholar]
- [4].Rainbow R, Ren W, Zeng L, Inflammation and Joint Tissue Interactions in OA: Implications for Potential Therapeutic Approaches, Arthritis, 2012 (2012) 741582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lories RJ, Luyten FP, The bone-cartilage unit in osteoarthritis, Nature reviews. Rheumatology, 7 (2011) 43–49. [DOI] [PubMed] [Google Scholar]
- [6].Daghestani HN, Pieper CF, Kraus VB, Soluble macrophage biomarkers indicate inflammatory phenotypes in patients with knee osteoarthritis, Arthritis & rheumatology, 67 (2015) 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Furman BD, Mangiapani DS, Zeitler E, Bailey KN, Horne PH, Huebner JL, Kraus VB, Guilak F, Olson SA, Targeting pro-inflammatory cytokines following joint injury: acute intra-articular inhibition of interleukin-1 following knee injury prevents post-traumatic arthritis, Arthritis research & therapy, 16 (2014) R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goldring MB, Otero M, Plumb DA, Dragomir C, Favero M, El Hachem K, Hashimoto K, Roach HI, Olivotto E, Borzi RM, Marcu KB, Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis, European cells & materials, 21 (2011) 202–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Krakauer T, Buckley M, Doxycycline is anti-inflammatory and inhibits staphylococcal exotoxin-induced cytokines and chemokines, Antimicrobial agents and chemotherapy, 47 (2003) 3630–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Duivenvoorden WC, Hirte HW, Singh G, Use of tetracycline as an inhibitor of matrix metalloproteinase activity secreted by human bone-metastasizing cancer cells, Invasion & metastasis, 17 (1997) 312–322. [PubMed] [Google Scholar]
- [11].Greenwald RA, Moak SA, Ramamurthy NS, Golub LM, Tetracyclines suppress matrix metalloproteinase activity in adjuvant arthritis and in combination with flurbiprofen, ameliorate bone damage, The Journal of rheumatology, 19 (1992) 927–938. [PubMed] [Google Scholar]
- [12].Greenwald RA, The road forward: the scientific basis for tetracycline treatment of arthritic disorders, Pharmacological research : the official journal of the Italian Pharmacological Society, 64 (2011) 610–613. [DOI] [PubMed] [Google Scholar]
- [13].de Bri E, Lei W, Svensson O, Chowdhury M, Moak SA, Greenwald RA, Effect of an inhibitor of matrix metalloproteinases on spontaneous osteoarthritis in guinea pigs, Adv Dent Res, 12 (1998) 82–85. [DOI] [PubMed] [Google Scholar]
- [14].Brandt KD, Modification by oral doxycycline administration of articular cartilage breakdown in osteoarthritis, The Journal of rheumatology. Supplement, 43 (1995) 149–151. [PubMed] [Google Scholar]
- [15].da Costa BR, Nuesch E, Reichenbach S, Juni P, Rutjes AW, Doxycycline for osteoarthritis of the knee or hip, The Cochrane database of systematic reviews, 11 (2012) CD007323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Otterness IG, Brandt KD, Le Graverand MP, Mazzuca SA, Urinary TIINE concentrations in a randomized controlled trial of doxycycline in knee osteoarthritis: implications of the lack of association between TIINE levels and joint space narrowing, Arthritis and rheumatism, 56 (2007) 3644–3649. [DOI] [PubMed] [Google Scholar]
- [17].Kwiatkowska B, Maslinska M, Macrolide therapy in chronic inflammatory diseases, Mediators of inflammation, 2012 (2012) 636157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aminov RI, Biotic acts of antibiotics, Frontiers in microbiology, 4 (2013) 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Desaki M, Okazaki H, Sunazuka T, Omura S, Yamamoto K, Takizawa H, Molecular mechanisms of anti-inflammatory action of erythromycin in human bronchial epithelial cells: possible role in the signaling pathway that regulates nuclear factor-kappaB activation, Antimicrobial agents and chemotherapy, 48 (2004) 1581–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ren W, Blasier R, Peng X, Shi T, Wooley PH, Markel D, Effect of oral erythromycin therapy in patients with aseptic loosening of joint prostheses, Bone, 44 (2009) 671–677. [DOI] [PubMed] [Google Scholar]
- [21].Ren W, Li XH, Chen BD, Wooley PH, Erythromycin inhibits wear debris-induced osteoclastogenesis by modulation of murine macrophage NF-kappaB activity, Journal of orthopaedic research : official publication of the Orthopaedic Research Society, 22 (2004) 21–29. [DOI] [PubMed] [Google Scholar]
- [22].Uchimura T, Foote AT, Markel DC, Ren W, Zeng L, The Chondroprotective Role of Erythromycin in a Murine Joint Destruction Model, Cartilage, 7 (2016) 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kojima M, Kangawa K, Ghrelin: structure and function, Physiological reviews, 85 (2005) 495–522. [DOI] [PubMed] [Google Scholar]
- [24].Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K, Ghrelin is a growth-hormone-releasing acylated peptide from stomach, Nature, 402 (1999) 656–660. [DOI] [PubMed] [Google Scholar]
- [25].Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, Bhattacharya S, Carpenter R, Grossman AB, Korbonits M, The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans, The Journal of clinical endocrinology and metabolism, 87 (2002) 2988. [DOI] [PubMed] [Google Scholar]
- [26].Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, Jones JE, Deysher AE, Waxman AR, White RD, Williams TD, Lachey JL, Seeley RJ, Lowell BB, Elmquist JK, Mice lacking ghrelin receptors resist the development of diet-induced obesity, The Journal of clinical investigation, 115 (2005) 3564–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Albarran-Zeckler RG, Smith RG, The ghrelin receptors (GHS-R1a and GHS-R1b), Endocrine development, 25 (2013) 5–15. [DOI] [PubMed] [Google Scholar]
- [28].Leung PK, Chow KB, Lau PN, Chu KM, Chan CB, Cheng CH, Wise H, The truncated ghrelin receptor polypeptide (GHS-R1b) acts as a dominant-negative mutant of the ghrelin receptor, Cell Signal, 19 (2007) 1011–1022. [DOI] [PubMed] [Google Scholar]
- [29].Caminos JE, Gualillo O, Lago F, Otero M, Blanco M, Gallego R, Garcia-Caballero T, Goldring MB, Casanueva FF, Gomez-Reino JJ, Dieguez C, The endogenous growth hormone secretagogue (ghrelin) is synthesized and secreted by chondrocytes, Endocrinology, 146 (2005) 1285–1292. [DOI] [PubMed] [Google Scholar]
- [30].Liu J, Cao L, Gao X, Chen Z, Guo S, He Z, Qian Y, Yu Y, Wang G, Ghrelin prevents articular cartilage matrix destruction in human chondrocytes, Biomed Pharmacother, 98 (2018) 651–655. [DOI] [PubMed] [Google Scholar]
- [31].Qu R, Chen X, Wang W, Qiu C, Ban M, Guo L, Vasilev K, Chen J, Li W, Zhao Y, Ghrelin protects against osteoarthritis through interplay with Akt and NF-kappaB signaling pathways, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 32 (2018) 1044–1058. [DOI] [PubMed] [Google Scholar]
- [32].Uchimura T, Foote AT, Smith EL, Matzkin EG, Zeng L, Insulin-Like Growth Factor II (IGF-II) Inhibits IL-1beta-Induced Cartilage Matrix Loss and Promotes Cartilage Integrity in Experimental Osteoarthritis, Journal of cellular biochemistry, 116 (2015) 2858–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jackson A, Gu W, Transport Properties of Cartilaginous Tissues, Curr Rheumatol Rev, 5 (2009) 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang Y, Wei L, Zeng L, He D, Wei X, Nutrition and degeneration of articular cartilage, Knee Surg Sports Traumatol Arthrosc, 21 (2013) 1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Alibegovic A, Balazic J, Petrovic D, Hribar G, Blagus R, Drobnic M, Viability of human articular chondrocytes harvested postmortem: changes with time and temperature of in vitro culture conditions, Journal of forensic sciences, 59 (2014) 522–528. [DOI] [PubMed] [Google Scholar]
- [36].Li Y, Wang Y, Chubinskaya S, Schoeberl B, Florine E, Kopesky P, Grodzinsky AJ, Effects of insulin-like growth factor-1 and dexamethasone on cytokine-challenged cartilage: relevance to post-traumatic osteoarthritis, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 23 (2015) 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kuettner KE, Cole AA, Cartilage degeneration in different human joints, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 13 (2005) 93–103. [DOI] [PubMed] [Google Scholar]
- [38].Gosset M, Berenbaum F, Thirion S, Jacques C, Primary culture and phenotyping of murine chondrocytes, Nature protocols, 3 (2008) 1253–1260. [DOI] [PubMed] [Google Scholar]
- [39].Glasson SS, Chambers MG, Van Den Berg WB, Little CB, The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 18 Suppl 3 (2010) S17–23. [DOI] [PubMed] [Google Scholar]
- [40].Krenn V, Morawietz L, Burmester GR, Kinne RW, Mueller-Ladner U, Muller B, Haupl T, Synovitis score: discrimination between chronic low-grade and high-grade synovitis, Histopathology, 49 (2006) 358–364. [DOI] [PubMed] [Google Scholar]
- [41].Matsukura Y, Muneta T, Tsuji K, Miyatake K, Yamada J, Abula K, Koga H, Tomita M, Sekiya I, Mouse synovial mesenchymal stem cells increase in yield with knee inflammation, Journal of orthopaedic research : official publication of the Orthopaedic Research Society, 33 (2015) 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Uchimura T, Hollander JM, Nakamura DS, Liu Z, Rosen CJ, Georgakoudi I, Zeng L, An essential role for IGF2 in cartilage development and glucose metabolism during postnatal long bone growth, Development, 144 (2017) 3533–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Colburn WA, Di Santo AR, Gibaldi M, Pharmacokinetics of erythromycin on repetitive dosing, Journal of clinical pharmacology, 17 (1977) 592–600. [DOI] [PubMed] [Google Scholar]
- [44].Goldring MB, Berenbaum F, The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide, Clinical orthopaedics and related research, (2004) S37–46. [DOI] [PubMed] [Google Scholar]
- [45].Gibson AL, Hui Mingalone CK, Foote AT, Uchimura T, Zhang M, Zeng L, Wnt7a Inhibits IL-1beta Induced Catabolic Gene Expression and Prevents Articular Cartilage Damage in Experimental Osteoarthritis, Scientific reports, 7 (2017) 41823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Asakawa A, Akio I, Ohinata K, Fujimiya M, Meguid MM, Yoshikawa M, EM574, a motilide, has an orexigenic activity with affinity for growth-hormone secretagogue receptor, Journal of gastroenterology and hepatology, 18 (2003) 881–882. [DOI] [PubMed] [Google Scholar]
- [47].Matsuura B, Dong M, Naik S, Miller LJ, Onji M, Differential contributions of motilin receptor extracellular domains for peptide and non-peptidyl agonist binding and activity, The Journal of biological chemistry, 281 (2006) 12390–12396. [DOI] [PubMed] [Google Scholar]
- [48].Nunoi H, Matsuura B, Utsunomiya S, Ueda T, Miyake T, Furukawa S, Kumagi T, Ikeda Y, Abe M, Hiasa Y, Onji M, A relationship between motilin and growth hormone secretagogue receptors, Regulatory peptides, 176 (2012) 28–35. [DOI] [PubMed] [Google Scholar]
- [49].Pugliese-Pires PN, Fortin JP, Arthur T, Latronico AC, Mendonca BB, Villares SM, Arnhold IJ, Kopin AS, Jorge AA, Novel inactivating mutations in the GH secretagogue receptor gene in patients with constitutional delay of growth and puberty, European journal of endocrinology / European Federation of Endocrine Societies, 165 (2011) 233–241. [DOI] [PubMed] [Google Scholar]
- [50].Liu G, Fortin JP, Beinborn M, Kopin AS, Four missense mutations in the ghrelin receptor result in distinct pharmacological abnormalities, The Journal of pharmacology and experimental therapeutics, 322 (2007) 1036–1043. [DOI] [PubMed] [Google Scholar]
- [51].Takahashi I, Matsuzaki T, Kuroki H, Hoso M, Induction of osteoarthritis by injecting monosodium iodoacetate into the patellofemoral joint of an experimental rat model, PloS one, 13 (2018) e0196625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Udo M, Muneta T, Tsuji K, Ozeki N, Nakagawa Y, Ohara T, Saito R, Yanagisawa K, Koga H, Sekiya I, Monoiodoacetic acid induces arthritis and synovitis in rats in a dose- and time-dependent manner: proposed model-specific scoring systems, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 24 (2016) 1284–1291. [DOI] [PubMed] [Google Scholar]
- [53].Guingamp C, Gegout-Pottie P, Philippe L, Terlain B, Netter P, Gillet P, Mono-iodoacetate-induced experimental osteoarthritis: a dose-response study of loss of mobility, morphology, and biochemistry, Arthritis and rheumatism, 40 (1997) 1670–1679. [DOI] [PubMed] [Google Scholar]
- [54].Guzman RE, Evans MG, Bove S, Morenko B, Kilgore K, Mono-iodoacetate-induced histologic changes in subchondral bone and articular cartilage of rat femorotibial joints: an animal model of osteoarthritis, Toxicologic pathology, 31 (2003) 619–624. [DOI] [PubMed] [Google Scholar]
- [55].Haslauer CM, Proffen BL, Johnson VM, Hill A, Murray MM, Gene expression of catabolic inflammatory cytokines peak before anabolic inflammatory cytokines after ACL injury in a preclinical model, Journal of inflammation, 11 (2014) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Johnson K, Svensson CI, Etten DV, Ghosh SS, Murphy AN, Powell HC, Terkeltaub R, Mediation of spontaneous knee osteoarthritis by progressive chondrocyte ATP depletion in Hartley guinea pigs, Arthritis and rheumatism, 50 (2004) 1216–1225. [DOI] [PubMed] [Google Scholar]
- [57].Lee RB, Urban JP, Evidence for a negative Pasteur effect in articular cartilage, The Biochemical journal, 321 ( Pt 1) (1997) 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Otsu K, Ishinaga H, Suzuki S, Sugawara A, Sunazuka T, Omura S, Jono H, Takeuchi K, Effects of a novel nonantibiotic macrolide, EM900, on cytokine and mucin gene expression in a human airway epithelial cell line, Pharmacology, 88 (2011) 327–332. [DOI] [PubMed] [Google Scholar]
- [59].Sugawara A, Sueki A, Hirose T, Nagai K, Gouda H, Hirono S, Shima H, Akagawa KS, Omura S, Sunazuka T, Novel 12-membered non-antibiotic macrolides from erythromycin A; EM900 series as novel leads for anti-inflammatory and/or immunomodulatory agents, Bioorganic & medicinal chemistry letters, 21 (2011) 3373–3376. [DOI] [PubMed] [Google Scholar]
- [60].Sugawara A, Sueki A, Hirose T, Shima H, Akagawa KS, Omura S, Sunazuka T, Novel 12-membered non-antibiotic macrolides, EM900 series with anti-inflammatory and/or immunomodulatory activity; synthesis, structure-activity relationships and in vivo study, The Journal of antibiotics, 65 (2012) 487–490. [DOI] [PubMed] [Google Scholar]
- [61].Zou YC, Deng HY, Mao Z, Zhao C, Huang J, Liu G, Decreased synovial fluid ghrelin levels are linked with disease severity in primary knee osteoarthritis patients and are increased following laser therapy, Clin Chim Acta, 470 (2017) 64–69. [DOI] [PubMed] [Google Scholar]
- [62].Tschop M, Smiley DL, Heiman ML, Ghrelin induces adiposity in rodents, Nature, 407 (2000) 908–913. [DOI] [PubMed] [Google Scholar]
- [63].Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S, A role for ghrelin in the central regulation of feeding, Nature, 409 (2001) 194–198. [DOI] [PubMed] [Google Scholar]
- [64].Sun Y, Ahmed S, Smith RG, Deletion of ghrelin impairs neither growth nor appetite, Molecular and cellular biology, 23 (2003) 7973–7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, Moncrieffe M, Thabet K, Cox HJ, Yancopoulos GD, Wiegand SJ, Sleeman MW, Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 8227–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Preston DA, Microbiological aspects of erythromycin, Pediatr Infect Dis, 5 (1986) 120–123. [DOI] [PubMed] [Google Scholar]
- [67].Feighner SD, Tan CP, McKee KK, Palyha OC, Hreniuk DL, Pong SS, Austin CP, Figueroa D, MacNeil D, Cascieri MA, Nargund R, Bakshi R, Abramovitz M, Stocco R, Kargman S, O’Neill G, Van Der Ploeg LH, Evans J, Patchett AA, Smith RG, Howard AD, Receptor for motilin identified in the human gastrointestinal system, Science, 284 (1999) 2184–2188. [DOI] [PubMed] [Google Scholar]
- [68].Broad J, Sanger GJ, The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin, British journal of pharmacology, 168 (2013) 1859–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].De Smet B, Mitselos A, Depoortere I, Motilin and ghrelin as prokinetic drug targets, Pharmacology & therapeutics, 123 (2009) 207–223. [DOI] [PubMed] [Google Scholar]
- [70].He J, Irwin DM, Chen R, Zhang YP, Stepwise loss of motilin and its specific receptor genes in rodents, Journal of molecular endocrinology, 44 (2010) 37–44. [DOI] [PubMed] [Google Scholar]
- [71].Sanger GJ, Holbrook JD, Andrews PL, The translational value of rodent gastrointestinal functions: a cautionary tale, Trends in pharmacological sciences, 32 (2011) 402–409. [DOI] [PubMed] [Google Scholar]
- [72].Holst B, Holliday ND, Bach A, Elling CE, Cox HM, Schwartz TW, Common structural basis for constitutive activity of the ghrelin receptor family, The Journal of biological chemistry, 279 (2004) 53806–53817. [DOI] [PubMed] [Google Scholar]
- [73].Brown JC, Cook MA, Dryburgh JR, Motilin, a gastric motor activity stimulating polypeptide: the complete amino acid sequence, Canadian journal of biochemistry, 51 (1973) 533–537. [DOI] [PubMed] [Google Scholar]
- [74].Sniekers YH, Weinans H, Bierma-Zeinstra SM, van Leeuwen JP, van Osch GJ, Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment - a systematic approach, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 16 (2008) 533–541. [DOI] [PubMed] [Google Scholar]
- [75].Utomo L, Eijgenraam SM, Meuffels DE, Bierma-Zeinstra SMA, Bastiaansen-Jenniskens YM, van Osch G, Meniscal extrusion and degeneration during the course of osteoarthritis in the Murine collagenase-induced osteoarthritis model, Journal of orthopaedic research : official publication of the Orthopaedic Research Society, 36 (2018) 2416–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Driban JB, Sitler MR, Barbe MF, Balasubramanian E, Is osteoarthritis a heterogeneous disease that can be stratified into subsets?, Clin Rheumatol, 29 (2010) 123–131. [DOI] [PubMed] [Google Scholar]
- [77].Glasson SS, Blanchet TJ, Morris EA, The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse, Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society, 15 (2007) 1061–1069. [DOI] [PubMed] [Google Scholar]
- [78].Berenbaum F, Eymard F, Houard X, Osteoarthritis, inflammation and obesity, Current opinion in rheumatology, 25 (2013) 114–118. [DOI] [PubMed] [Google Scholar]
- [79].Sadreddini S, Noshad H, Molaeefard M, Moloudi R, Ardalan MR, Ghojazadeh M, A double blind, randomized, placebo controlled study to evaluate the efficacy of erythromycin in patients with knee effusion due to osteoarthritis, International journal of rheumatic diseases, 12 (2009) 44–51. [DOI] [PubMed] [Google Scholar]
- [80].Sibilia V, Pagani F, Mrak E, Dieci E, Tulipano G, Ferrucci F, Pharmacological characterization of the ghrelin receptor mediating its inhibitory action on inflammatory pain in rats, Amino acids, 43 (2012) 1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]