

Abstract
Abacavir (ABC) is a guanosine nucleoside reverse transcriptase inhibitor (NRTI) with potent antiretroviral activity. Since NRTIs exhibit tissue-specific inhibition of mitochondrial DNA (mtDNA) synthesis, the ability of ABC to inhibit mtDNA synthesis in vivo was evaluated. Inbred wild-type (WT) and transgenic mice (TG) treated with ABC (3.125 mg/d p. o., 35 days) were used to define mitochondrial oxidative stress and cardiac function. Chosen TGs exhibited overexpression of HIV-1 viral proteins (NL4–3Δgag/pol, non-replication competent), hemizygous depletion or overexpression of mitochondrial superoxide dismutase (SOD2+/− knock-out (KO) or MnSOD OX, respectively), overexpression of mitochondrially targeted catalase (MCAT), or double “knockout” deletion of aldehyde dehydrogenase activity (ALDH2 KO). Impact on mtDNA synthesis was assessed by comparing changes in mtDNA abundance between ABC-treated and vehicle-treated WTs and TGs. No changes in mtDNA abundance occurred from ABC treatment in any mice, suggesting no inhibition of mtDNA synthesis. Left ventricle (LV) mass and LV end-diastolic dimension (LVEDD) were defined echocardiographically and remained unchanged as well. These results indicate that treatment with ABC has no visible cardiotoxicity in these adult mice exposed for 5 weeks compared to findings with other antiretroviral NRTI studies and support some claims for its relative safety.
Keywords: ABC, NRTIs, Mitochondrial DNA (mtDNA), ECHO, Drug safety, Antiretrovirals
Introduction
Combinations with nucleoside analogs (NRTIs) and protease inhibitors as antiretroviral treatment regimens (HAART) have effectively reduced morbidity and mortality associated with HIV infection. Treatment with NRTIs, however, can result in serious dose-limiting toxicities in addition to HIV-related complications. Peripheral neuropathy is an often-associated side effect of 2′, 3′-dideoxyinosine (ddI), 2′, 3′-dideoxycytidine (ddC), and 2′, 3′-didehydrothymidine (D4T). In addition, cardiomyopathy can occur from use of 3′-deoxy-3′-azidothymidine (AZT) and has been documented in murine models by us previously [1–5]. The shared toxic mechanisms of NRTIs appear to result from the inhibition of polymerase γ (pol γ), the mitochondrial (mt-) DNA polymerase [6–9]. Phosphorylated nucleoside analogs particularly (5′-triphosphates) are potent inhibitors of pol γ and may interfere with normal mtDNA biosynthesis.
Abacavir (ABC) is a carbocyclic 2′-deoxyguanosine NRTI. The antiviral activity of abacavir results from its intracellular metabolism to a 5′-triphosphate anabolite (carbovir triphosphate, CBV-TP) [10]. CBV-TP competes with endogenous nucleotide 2′-deoxyguanosine triphosphate (dGTP) for incorporation into the nucleic acid chain and leads to DNA chain termination [11, 12]. CBV lacks mitochondrial toxicity in vitro in CEM cells [13]. However, in studies with HepG2 cells, CBV strongly impaired hepatocyte proliferation and increased lactate and lipid production, but not mtDNA depletion [14] giving a mixed picture of mitochondrial dysfunction compared to other NRTIs [15, 16].
The potential toxicity of ABC is unknown, but increased cardiovascular risk has been suggested [17]. To address this important question experimentally, we investigated ABC effects on murine cardiac function by echocardiographic analysis and on mtDNA replication (mtDNA abundance) in “2 × 2” studies using transgenic mice (TG) and wild-type littermates. The selected TGs either exhibit HIV-1 viral protein overexpression, or important pharmacological activity/metabolism with various NRTIs, including ABC. Results indicate neither mitochondrial toxicity nor change in cardiac function following ABC treatment.
Materials and Methods
Mice and Genotyping
TG mice were from experiments in the senior investigator’s laboratory (Table 1). Hemizygous HIV-1 TG mice (from Paul Klotman) [18] provided a model of HIV viral protein expression to determine viral impact on cardiac function or mitochondrial biogenesis alone or combined with ABC treatment. Originally on FVB/n background, this TG line was bred congenically to C57/BL6 (and given the trivial name “MSB”). SOD2+/− knock-outs (KO) (from Brian Day and colleagues at the National Jewish Medical Research Center, Denver CO.) [19] provided a model to determine whether oxidative events are related to ABC treatment. In tandem, SOD2-OX TGs (from Ye-Shih Ho and colleagues at Wayne State University, Detroit, MI) [20] provided a model of protection against oxidative stress. MCAT TGs (from Peter Rabinovitch and colleagues at the University of Washington, Seattle, WA.) [21] provided an alternative model of protection from oxidative injury. ALDH2 KOs (from Jonathan Stamler at The Cleveland Clinic, Cleveland, OH.) [22] provided a model with altered ABC metabolism. It was hypothesized that ALDH2 absence could decrease ABC-4’-COOH, but possibly could increase intermediates that may result in cardiac toxicity. TGs were either developed on a C57BL/6 background or bred congenically to C57BL/6 (10 generations).
Table 1.
Transgenic mice
| Mouse | Target gene | Backgrounda | Source (Reference) |
|---|---|---|---|
| MSB | HIV-1 hemizygous NL4–3Δgag/pol overexpressed | Originally FVB/n, Bred congenically to C57/BL6 | Paul Klotman [18] |
| MnSOD OX | Superoxide dismutase overexpressed | C57/BL6 | Ye-Shih Ho [20] |
| SOD2+/− KO | Superoxide dismutase Hemizygous (±) “knockout” | C57/BL6 | Brian Day [19] |
| MCAT | Mitochondrially targeted catalase, overexpressed | C57/BL6 | Peter Rabinovitch [21] |
| ALDH2 KO | Aldehyde dehydrogenase double (−/−) “knockout” | C57/BL6 | Jonathan Stamler [22] |
Mouse strain; “wild-type” (WT) littermates used had same background
ABC Treatment Protocols
All procedures complied with Emory Institutional Animal Care and Use Committee and NIH guidelines. ABC (Ziagen, GlaxoSmithKline, Research Triangle Park, NC) was purchased from the pharmacy. WT and TG littermates (both genders) were age-matched (8–12 weeks) at the start of ABC or vehicle treatment. Standard rodent chow and water were provided ad libitum in a 12 h light: dark, humidity and temperature controlled environment at Emory. ABC was administered daily by gavage in doses that resemble those used in humans on a mg/kg/d basis to remain clinically relevant. Mice received vehicle control (0.9% saline) or vehicle containing ABC (3.125 mg/d). At day 35, echocardiographic measurements were made, animals terminated, heart samples retrieved and stored at −80°C for extraction of mitochondrial DNA (mtDNA).
mtDNA and Nuclear DNA (nDNA) Quantitation in Heart Tissue Using Real Time PCR
Methods employed were modifications of those from others [23] as employed by us in the past [4, 5]. Total DNA was extracted from heart tissue (~10 mg wet weight) using a MagNA Pure System and reagents (Roche Life Sciences, Indianapolis, IN). DNA sequences for primers and probes used for quantitation of mitochondrial and nuclear DNA were described [4]. Real-Time PCR was performed in duplicate for each amplicon. Amplification was performed using LC 480 (Roche). Standard DNA curves for quantitation of the LC products were employed. PCR products of mtDNA and nDNA were quantified using the corresponding external standard.
Echocardiography (ECHO) of TG Mice
ECHO was performed prior to termination. Left ventricular (LV) mass and LV end-diastolic dimension (LVEDD) were quantified and normalized as before [24].
Experimental Analysis and Statistics
Real-time PCR data were expressed as the ratio of mean values for mtDNA to the mean values of nDNA × 10−3. Resultant values were expressed as mean ± standard error, normalized to untreated WT mean (set at 1.0). Statistical analysis was performed on the resultant mtDNA abundance ratios using a one-way ANOVA (non-parametric) and Newman-Keuls test with a value of P < 0.05 considered statistically significant. Echocardiographic determinations from all groups were compared by 1-way ANOVA [24].
Results
General
All mice maintained normal body weights and growth, and normal levels of activity throughout the study (8–12 weeks old mice at initiation and 5 weeks treatment).
mtDNA Abundance in Cardiac Tissues
To determine whether 5 weeks of ABC treatment depleted cardiac mtDNA, mtDNA abundance (expressed as a ratio of mtDNA/nDNA) was assessed using DNA extracts from hearts. Results showed no change in mtDNA abundance among any of the vehicle-treated mice (including MSB, MnSOD OX, SOD2+/− KO, MCAT, and ALDH2 KO) compared to their respective vehicle-treated WTs (Fig. 1). As expected, ABC had no effect on mtDNA abundance in hearts from MnSOD OX and MCAT TGs, suggesting these TGs provide protection from potential oxidative stress related to ABC treatment (Fig. 1). A surprising finding was that ABC treatment also had no effect on mtDNA abundance in SOD2+/− KO, which should have been susceptible to oxidative stress. This result suggests that oxidative stress is not associated with ABC treatment and is therefore unique from other NRTI treatments (e.g. AZT). ALDH2 KOs, likewise, were found to have no change in mtDNA abundance following ABC treatment. The absence of ALDH2 activity appears to have had no critical impact on ABC metabolism, or at least did not render these TGs susceptible to changes in mtDNA abundance.
Fig. 1.
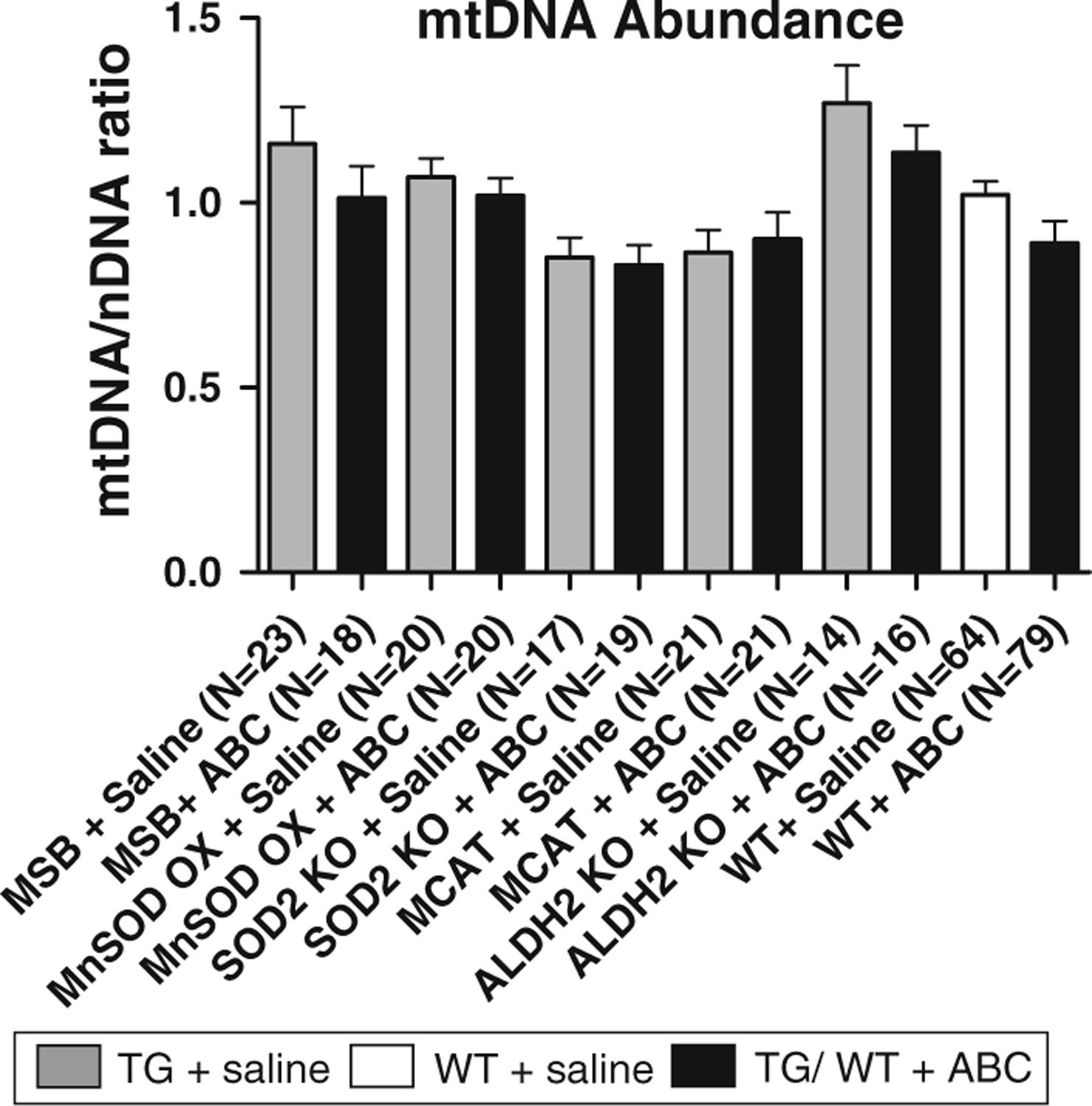
Cardiac mtDNA abundance in TGs and WTs following ABC treatment. Total DNA was extracted from cardiac tissues isolated from TGs and WTs treated with abacavir (ABC) or vehicle (saline) alone for 35 days. mtDNA abundance was assessed using a ratio of mtDNA/nDNA as determined by real-time PCR amplification. Cardiac mtDNA abundance remained unchanged in all TGs (with or without ABC) compared to WT littermates (1-way ANOVA, all comparisons had P > 0.05)
Cardiac Function in TGs and WT Following ABC
Cardiac function was assessed for each group of mice. LV mass and LVEDD were derived from direct echocardiographic measurements of cavity and wall thickness in each mouse to define effects of ABC and/or genetic manipulation on LV. Results showed LV mass was unchanged in all vehicle-treated TGs (including MSB, MnSOD OX, SOD2+/− KO, MCAT, and ALDH2 KO) compared to vehicle-treated WTs (Fig. 2a). Similarly, ABC had no effect on LV mass in the MnSOD OX and MCAT TGs, as expected (Fig. 2a). Surprisingly, SOD2+/− KOs were also found to have no change in LV mass following ABC, further supporting the hypothesis that oxidative stress is not associated with ABC treatment (Fig. 2a). In contrast, SOD2+/− KO previously was shown to have increased LV mass following AZT treatment for 5 weeks [3]. Any potential changes in LV mass due to the absence of ALDH2 activity was also disproven with results here that showed no change in ALDH2 KOs treated with ABC compared to WTs (Fig. 2a).
Fig. 2.
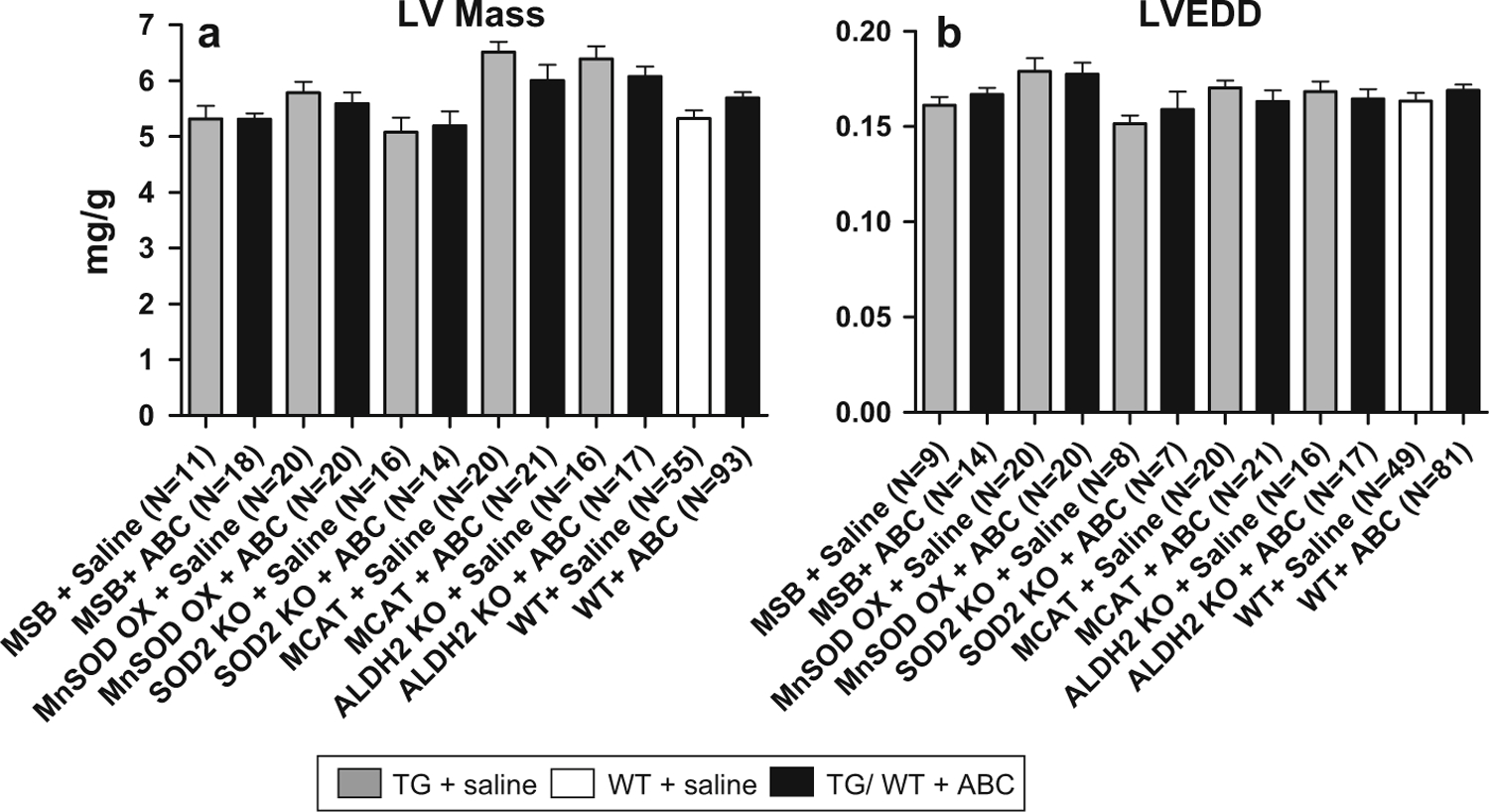
Quantitative analysis of ECHO images. LV mass was determined from ECHO images captured just prior to termination. Data were normalized to body weight (mg/g) and plotted as mean ± SEM. a ABC treatment had no effect on LV mass in WT and all TG models compared to vehicle-treated WTs (1-way ANOVA, all comparisons had P > 0.05). b LVEDD also remained unchanged in TGs and WTs following ABC treatment (1-way ANOVA, all comparisons had P > 0.05)
LVEDD also remained unchanged (with or without ABC treatment) in WTs and all TGs, even SOD2+/− KO and ALDH2 KO (Fig. 2b). These results further support the conclusion that ABC treatment for 5 weeks has no detectable cardiotoxicity and suggests that oxidative stress is not associated with ABC treatment.
Discussion
Mitochondrial side effects limit therapeutic efficacy and clinical options in HIV/AIDS. Mitochondrial toxicity is associated with many NRTIs. It came to clinical awareness with the reports of skeletal muscle and heart toxicity following AZT treatment [25, 26]. It has been hypothesized that mitochondrial toxicity is related to disruption of mtDNA replication and biogenesis [8, 27, 28]. Studies by our group and by others supported this hypothesis, in vivo [1, 2, 5, 29], but clinical data are less convincing [30, 31].
While HLA-specific ABC-associated hypersensitivity reaction syndrome (HRS) has been established as a clinical side effect [32, 33], the possibility of mitochondrial toxicity for this NRTI is not defined. Present studies in WT mice underscored ABC safety. This is based on absence of cardiac mitochondrial toxicity after 5 weeks of treatment. In some ways, these results supported earlier in vitro data in which CBV had no effect on CEM and HepG2 cell mtDNA [13, 14].
Recent reports suggest increased rates of coronary heart disease (CHD) among HIV-infected patients. As survival increases, cardiovascular disease is an important cause of morbidity and mortality in this population [34], but mechanism(s) specific to this population remain elusive. Traditional risk factors play a role, but non-traditional factors including direct effects of HIV and side effects of antiretroviral drugs impact clinical outcomes [35]. We employed an established murine model (MSB) [36] to assess the combined effects of “HIV” and ABC treatment. Results demonstrated neither cardiac mitochondrial toxicity nor altered cardiac function from ABC. Recently, we found that the combination of “HIV” (MSB model) and tenofovir (an NRTI) treatment resulted in a tissue-specific mitochondrial toxicity [37]. In contrast, results here suggest ABC’s safety in combination with HIV-1 with respect to cardiac mitochondrial changes.
Oxidative stress and mtDNA depletion are integral mechanisms of mitochondrial toxicity [38–44] and cardiovascular diseases. TG models of mitochondrial oxidative stress are useful tools to explore the role of ROS related to NRTIs [45]. In these studies, we employed several relevant TG models to determine whether oxidative events are related to ABC treatment. TG models included those that enhanced mitochondrial ROS (such as SOD2+/− KO) or protected the heart from it (SOD-OX or mCAT) and which were previously found to be susceptible or resistant to oxidative stress following AZT treatment, respectively [3]. ABC treatment had no effect on genetically engineered susceptibility to or protection from oxidative injury.
ABC is extensively metabolized by the liver via two pathways [12]. We focused here on ABC metabolism through alcohol dehydrogenase to its inactive form (carboxylate) as a potential manipulation of drug toxicity. ALDH2 can effectively process ABC intermediate of alcohol dehydrogenase to ABC-4′-COOH [46]. It was hypothesized that ALDH2 absence could decrease ABC-4′-COOH, but possibly could increase intermediates that may result in cardiac toxicity. Results demonstrated that even in the absence of ALDH2 activity, ABC treatment resulted in no cardiac mitochondrial toxicity.
Together, the set of TG models and WT littermates establish in vivo evidence for the lack of cardiac mitochondrial toxicity associated with ABC under the experimental conditions tested. Granted, the findings in the current study do not rule out the possibility that higher daily doses or cumulative doses resulting with chronic treatment with human equivalent levels of ABC have not been determined. Results here support the safety of ABC within 5 weeks of treatment and suggest that ABC has little potential for risk of developing cardiac mitochondrial toxicity (short-term) compared to other NRTIs (e.g., AZT).
Acknowledgments
Studies were supported by R01 HL79867 and HL59798 to WL; JK is a recipient of K01 DK78513.
Abbreviations
- ABC
Abacavir
- ALDH2 KO
Aldehyde dehydrogenase double knockout
- AZT
3′-deoxy-3′-azidothymidine, zidovudine
- CBV/CBV-TP
Carbovir; carbovir triphosphate
- ddI
2′,3′-dideoxyinosine, didanosine
- LV
Left ventricle
- LVEDD
Left ventricle end-diastolic dimension
- MSB
HIV TG, NL4–3Δgag/pol
- MCAT
Mitochondrially targeted catalase
- MnSOD OX
Superoxide dismutase overexpressed
- mtDNA
Mitochondrial DNA
- NRTI
Nucleoside reverse transcriptase inhibitors; nucleoside analogs
- SOD2+/− KO
Hemizygous superoxide dismutase knockout
- TG
Transgenic mice
- WT
Wild-type
References
- 1.Kohler JJ, Hosseini SH, Cucoranu I, Hoying-Brandt A, Green E, Johnson D, et al. (2009). Murine cardiac mtDNA: Effects of transgenic manipulation of nucleoside phosphorylation. Laboratory Investigation, 89, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohler JJ, Hosseini SH, Green E, Hoying-Brandt A, Cucoranu I, Haase CP, et al. (2008). Cardiac-targeted transgenic mutant mitochondrial enzymes: mtDNA defects, antiretroviral toxicity and cardiomyopathy. Cardiovascular Toxicology, 8, 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohler JJ, Cucoranu I, Fields E, Green E, He S, Hoying A, et al. (2009). Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Laboratory Investigation, 89, 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hosseini SH, Kohler JJ, Haase CP, Tioleco N, Stuart T, Keebaugh E, et al. (2007). Targeted transgenic overexpression of mitochondrial thymidine kinase (TK2) alters mitochondrial DNA (mtDNA) and mitochondrial polypeptide abundance: Transgenic TK2, mtDNA, and antiretrovirals. American Journal of Pathology, 170, 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, et al. (2007). Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Laboratory Investigation, 87, 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cote HC (2007). Mechanisms of antiretroviral therapy-induced mitochondrial dysfunction. Current Opinion in HIV and AIDS, 2, 253–260. [DOI] [PubMed] [Google Scholar]
- 7.Kakuda TN (2000). Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clinical Therapeutics, 22, 685–708. [DOI] [PubMed] [Google Scholar]
- 8.Lewis W, Day BJ, & Copeland WC (2003). Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nature Reviews Drug Discovery, 2, 812–822. [DOI] [PubMed] [Google Scholar]
- 9.Parker WB, & Cheng YC (1994). Mitochondrial toxicity of NRTI analogs. Journal of NIH Research, 6, 57–61. [Google Scholar]
- 10.Stein DS, & Moore KH (2001). Phosphorylation of nucleoside analog antiretrovirals: A review for clinicians. Pharmacotherapy, 21, 11–34. [DOI] [PubMed] [Google Scholar]
- 11.Faletto MB, Miller WH, Garvey EP, St Clair MH, Daluge SM, & Good SS (1997). Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrobial Agents and Chemotherapy, 41, 1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuen GJ, Weller S, & Pakes GE (2008). A review of the pharmacokinetics of abacavir. Clinical Pharmacokinetics, 47, 351–371. [DOI] [PubMed] [Google Scholar]
- 13.Parker WB, Shaddix SC, Vince R, & Bennett LL Jr. (1997). Lack of mitochondrial toxicity in CEM cells treated with carbovir. Antiviral Research, 34, 131–136. [DOI] [PubMed] [Google Scholar]
- 14.Venhoff N, Setzer B, Melkaoui K, & Walker UA (2007). Mitochondrial toxicity of tenofovir, emtricitabine and abacavir alone and in combination with additional nucleoside reverse transcriptase inhibitors. Antiviral Therapy, 12, 1075–1085. [PubMed] [Google Scholar]
- 15.Lewis W, Griniuviene B, Tankersley KO, Levine ES, Montione R, Engelman L, et al. (1997). Depletion of mitochondrial DNA, destruction of mitochondria, and accumulation of lipid droplets result from fialuridine treatment in woodchucks (Marmota monax). Laboratory Investigation, 76, 77–87. [PubMed] [Google Scholar]
- 16.Lewis W, Levine ES, Griniuviene B, Tankersley KO, Colacino JM, Sommadossi JP, et al. (1996). Fialuridine and its metabolites inhibit DNA polymerase gamma at sites of multiple adjacent analog incorporation, decrease mtDNA abundance, and cause mitochondrial structural defects in cultured hepatoblasts. Proceedings of the National Academy of Sciences of the United States of America, 93, 3592–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kristoffersen US, Kofoed K, Kronborg G, Benfield T, Kjaer A, & Lebech AM (2009). Changes in biomarkers of cardiovascular risk after a switch to abacavir in HIV-1-infected individuals receiving combination antiretroviral therapy. HIV Medicine, 10, 627–633. [DOI] [PubMed] [Google Scholar]
- 18.Dickie P, Felser J, Eckhaus M, Bryant J, Silver J, Marinos N, et al. (1991). HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology, 185, 109–119. [DOI] [PubMed] [Google Scholar]
- 19.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, et al. (1998). A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nature Genetics, 18, 159–163. [DOI] [PubMed] [Google Scholar]
- 20.Ho YS, Magnenat JL, Gargano M, & Cao J (1998). The nature of antioxidant defense mechanisms: A lesson from transgenic studies. Environmental Health Perspectives, 106(Suppl 5), 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. (2005). Extension of murine life span by overexpression of catalase targeted to mitochondria. Science, 308, 1909–1911. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, et al. (2000). Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Letters, 476, 306–311. [DOI] [PubMed] [Google Scholar]
- 23.Cote HC, Yip B, Asselin JJ, Chan JW, Hogg RS, Harrigan PR, et al. (2003). Mitochondrial: Nuclear DNA ratios in peripheral blood cells from human immunodeficiency virus (HIV)-infected patients who received selected HIV antiretroviral drug regimens. Journal of Infectious Diseases, 187, 1972–1976. [DOI] [PubMed] [Google Scholar]
- 24.Lewis W, Haase CP, Raidel SM, Russ RB, Sutliff RL, Hoit BD, et al. (2001). Combined antiretroviral therapy causes cardiomyopathy and elevates plasma lactate in transgenic AIDS mice. Laboratory Investigation, 81, 1527–1536. [DOI] [PubMed] [Google Scholar]
- 25.Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, & Griffin JL (1990). Mitochondrial myopathy caused by long-term zidovudine therapy. New England Journal of Medicine, 322, 1098–1105. [DOI] [PubMed] [Google Scholar]
- 26.Lewis W, & Currie PF (2008). HIV/AIDS and the Cardiovascular System In Fuster V, O’Rourke RA, Walsh RA, & Poole-Wilson P (Eds.), Hurst’s the heart (pp. 2118–2131). New York: McGraw Hill Medical. [Google Scholar]
- 27.Lewis W, & Dalakas MC (1995). Mitochondrial toxicity of antiviral drugs. Nature Medicine, 1, 417–422. [DOI] [PubMed] [Google Scholar]
- 28.Martin AM, Hammond E, Nolan D, Pace C, Den Boer M, Taylor L, et al. (2003). Accumulation of mitochondrial DNA mutations in human immunodeficiency virus-infected patients treated with nucleoside-analogue reverse-transcriptase inhibitors. American Journal of Human Genetics, 72, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis W, Kohler JJ, Hosseini SH, Haase CP, Copeland WC, Bienstock RJ, et al. (2006). Antiretroviral nucleosides, deoxynucleotide carrier and mitochondrial DNA: Evidence supporting the DNA pol gamma hypothesis. Aids, 20, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Negredo E, Miro O, Rodriguez-Santiago B, Garrabou G, Estany C, Masabeu A, et al. (2009). Improvement of mitochondrial toxicity in patients receiving a nucleoside reversetranscriptase inhibitor-sparing strategy: Results from the Multicenter Study with Nevirapine and Kaletra (MULTINEKA). Clinical Infectious Diseases, 49, 892–900. [DOI] [PubMed] [Google Scholar]
- 31.Van Dyke RB, Wang L, & Williams PL (2008). Toxicities associated with dual nucleoside reverse-transcriptase inhibitor regimens in HIV-infected children. Journal of Infectious Diseases, 198, 1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hetherington S, McGuirk S, Powell G, Cutrell A, Naderer O, Spreen B, et al. (2001). Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clinical Therapeutics, 23, 1603–1614. [DOI] [PubMed] [Google Scholar]
- 33.Martin AM, Nolan D, Gaudieri S, Almeida CA, Nolan R, James I, et al. (2004). Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proceedings of the National Academy of Sciences of the United States of America, 101, 4180–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grinspoon SK, Grunfeld C, Kotler DP, Currier JS, Lundgren JD, Dube MP, et al. (2008). State of the science conference: Initiative to decrease cardiovascular risk and increase quality of care for patients living with HIV/AIDS: Executive summary. Circulation, 118, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis W (2000). Cardiomyopathy in AIDS: A pathophysiological perspective. Progress in Cardiovascular Diseases, 43, 151–170. [DOI] [PubMed] [Google Scholar]
- 36.Lewis W (2001). AIDS cardiomyopathy: Physiological, molecular, and biochemical studies in the transgenic mouse. Annals of the New York Academy of Sciences, 946, 46–56. [PubMed] [Google Scholar]
- 37.Kohler JJ, Hosseini SH, Hoying-Brandt A, Green E, Johnson DM, Russ R, et al. (2009). Tenofovir renal toxicity targets mitochondria of renal proximal tubules. Laboratory Investigation, 89, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohler JJ, & Lewis W (2007). A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environmental and Molecular Mutagenesis, 48, 166–172. [DOI] [PubMed] [Google Scholar]
- 39.Szabados E, Fischer GM, Toth K, Csete B, Nemeti B, Trombitas K, et al. (1999). Role of reactive oxygen species and poly-ADP-ribose polymerase in the development of AZT-induced cardiomyopathy in rat. Free Radical Biology and Medicine, 26, 309–317. [DOI] [PubMed] [Google Scholar]
- 40.Bialkowska A, Bialkowski K, Gerschenson M, Diwan BA, Jones AB, Olivero OA, et al. (2000). Oxidative DNA damage in fetal tissues after transplacental exposure to 3′-azido-3′-deoxythymidine (AZT). Carcinogenesis, 21, 1059–1062. [DOI] [PubMed] [Google Scholar]
- 41.Caron M, Auclairt M, Vissian A, Vigouroux C, & Capeau J (2008). Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antiviral Therapy, 13, 27–38. [PubMed] [Google Scholar]
- 42.de la Asuncion JG, Del Olmo ML, Gomez-Cambronero LG, Sastre J, Pallardo FV, & Vina J (2004). AZT induces oxidative damage to cardiac mitochondria: Protective effect of vitamins C and E. Life Science, 76, 47–56. [DOI] [PubMed] [Google Scholar]
- 43.Dunge A, Chakraborti AK, & Singh S (2004). Mechanistic explanation to the variable degradation behaviour of stavudine and zidovudine under hydrolytic, oxidative and photolytic conditions. Journal of Pharmaceutical and Biomedical Analysis, 35, 965–970. [DOI] [PubMed] [Google Scholar]
- 44.Iwamoto T, Hiraku Y, Oikawa S, Mizutani H, Kojima M, & Kawanishi S (2003). Oxidative DNA damage induced by photodegradation products of 3(′)-azido-3(′)-deoxythymidine. Archives of Biochemistry and Biophysics, 416, 155–163. [DOI] [PubMed] [Google Scholar]
- 45.Wallace DC (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annual Review of Genetics, 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh JS, Reese MJ, & Thurmond LM (2002). The metabolic activation of abacavir by human liver cytosol and expressed human alcohol dehydrogenase isozymes. Chemicobiological Interactions, 142, 135–154. [DOI] [PubMed] [Google Scholar]