

Abstract
Once regarded merely as a bland lipid storage disease consequence of aging, atherosclerosis is currently considered a slow and continuous inflammatory process (partially controllable by treatment) with complex etiology involving a multitude of genetic and environmental risk factors which ultimately result in the formation of the plaque. The vascular endothelium, a monolayer of endothelial cells (ECs), is an important regulatory "organ" critical for cardiovascular homeostasis in health which also contributes significantly to the pathomechanisms of several disease states, including atherosclerosis. Over the years, there has been evidence highlighting the central role of endoplasmic reticulum (ER) in the maintenance of endothelial function and perturbations in ER biology have been proposed to adversely affect a diverse range of endothelial functions. Of particular interest is the evidence that under certain pathophysiological circumstances, abnormal ER ultrastructure correlates with altered ER function and signaling and can contribute to cell injury and apoptosis. Therefore, the ultrastructural traits of ER membranes can have important implications not only for their functional bearings but also for the etiology and pathophysiology of diverse human disorders. With regard to atherosclerosis, the focus of ER research has been centered on the molecular signals originated from the ER to manage conditions of stress, leaving the fine structure of this organelle an almost unexplored (but promising) area of studies. There is, also, increasing evidence that mitochondrial dysfunction plays a critical role in promoting cell apoptosis, inflammation, and oxidative stress, thereby contributing to atheroma growth. It is within this context that the present study has been undertaken to investigate the microscopic architecture of ECs in human atherosclerosis and to determine whether the potential structural abnormalities of ER and mitochondria may play a central pathogenic role in atherogenesis or may merely reflect the condition of a tissue whose integrity has already been disturbed or destroyed. For this purpose, transmission electron microscopy (TEM) remains a powerful technique that can not only provide information about the ultrastructural state of cell organelles but also allow the correlation between different subcellular alterations indicative of a certain pathophysiological condition and cellular response. The present study expands the spectrum of ultrastructural defects known to exist in human atherosclerosis and suggests that ER alterations may be of great importance in the pathogenesis of the disease. The architectural changes of ER may be considered early pathological events that precede any overt histologic abnormalities in the vascular endothelium and its subcellular organelles, primarily the mitochondrial pool.
Keywords: atherosclerosis, electron microscopy, endoplasmic reticulum, endothelial cells, mitochondria
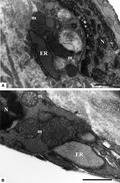
Dilation of ER is a common ultrastructural feature found in the ECs of human atherosclerotic plaque. Adjacent mitochondria also show alterations in their structural organization.
1. INTRODUCTION
Cardiovascular diseases (CVD) are the leading cause of death in the European Union (EU) accounting for around 2 million deaths per year (Townsend et al. 2016). The term “CVD” refers to a broad group of medical problems affecting the circulatory system that often result from atherosclerosis, a slow, complex and continuous process involving numerous interconnected cellular and acellular processes that lead to the deposition of fatty substances, cholesterol, cellular waste products, and calcium in the innermost layer of the artery wall (tunica intima) (Chroni et al. 2011; Rafieian‐Kopaei et al. 2014). The intima consists of a lining of endothelial cells (ECs) that rests on a thin layer of connective tissue and serves as the contact surface with blood (Sandoo et al. 2010; Mazurek et al. 2017). Due to its strategic position, the endothelium is the main regulator of vascular homeostasis and exerts many vasoprotective, anti‐inflammatory, and antithrombotic effects (Widmer & Lerman 2014; Gimbrone & García‐Cardeña 2016). However, in different pathological contexts, such as atherosclerosis, the anatomical integrity of the ECs is severely damaged, cell junctions are disrupted, and cell ultrastructure undergoes profound changes creating areas of vascular damage in which plaques build up (Cai & Harrison 2000; Davignon & Ganz 2004; Favero et al 2014; Sun et al 2019). Endothelial injury has been demonstrated to result not only from oxidative and nitroxidative stress but also from conditions that affect the ER whose disturbances can influence different cell signaling cascades, dynamics, and biosynthetic pathways (Cimellaro et al. 2016; Luchetti et al. 2017; Maamoun et al. 2019). In this regard, multiple risk factors for endothelial dysfunction have been shown to have also an impact on ER biology. Gargalovic and co‐workers are among the first to directly link perturbations of ER homeostasis to EC dysfunction (Gargalovic et al. 2006; Battson et al. 2017). The researchers demonstrated that chemically induced ER stress triggers a strong inflammatory response in human aortic ECs that can be abolished upon silencing of UPR mediators (ATF4 or XBP1), thus suggesting the important role of ER in promoting vascular inflammation. Another mechanism by which ER alterations have been shown to cause direct damage to ECs is via oxidative stress. Interestingly, it has been proven that generation of ROS during conditions of ER stress can occur through different ways including the interplay between the ER and mitochondria. Although these previous results have been later confirmed and extended in cell culture experiments and animal model studies, to date, very few reports have investigated the causal role of ER in endothelial dysfunction during human atherosclerosis, probably due to the difficulty of obtaining human vascular tissues. Atherosclerotic plaques have been found to express markers of chronic ER stress that may evoke or promote inflammation and atherothrombotic events throughout the induction of oxidative stress and the perturbation of calcium storage leading to functional changes of ECs and apoptosis (Dong et al. 2010; Chistiakov et al. 2014). There is also some evidence that ER stress transducers are activated in the atherosusceptible endothelium of swine aorta where they promote cell survival at low levels of expression becoming also capable to induce EC apoptosis under a prolonged and sustained activation status (Civelek et al. 2009; Zeng et al. 2009). Contrary to the breakthroughs in understanding the molecular biology of ER, progress on the fine structure of this organelle and how it modifies in response to changes in cellular environment has been slower. This study begins to fill this research gap by examining the anatomical state of ER in the ECs of human atherosclerotic plaques. Here, it has been also hypothesized that alterations or defects in ER ultrastructure may also be present in the unaffected aortic tissue adjacent to the lesion. The obtained results indicate that in vascular ECs, ER damage might be one of the earliest event in the process of atherosclerotic plaque formation appearing even before any other histopathologic abnormality of the blood vessel wall also preceeding the structural remodeling of mitochondria.
2. MATERIALS AND METHODS
2.1. Patients
Surgical specimens of human ascending aorta (at the midascending tract) and their respective adjacent regions were obtained from six male and six female (n = 12) nondiabetic patients (mean age, 63.6 years) who had been operated on for aortic atherosclerotic aneurysms. The aortas were opened longitudinally and washed with phosphate‐buffered saline (PBS), pH 7.2. The grossly normal aortic areas and those regions with atherosclerotic lesions were identified macroscopically. Only the regions of the arterial wall involved in the atherosclerotic process have been processed for electron microscopic analysis. All patients gave written informed consent. Control specimens were obtained from age‐ and ethnicity‐matched healthy subjects at autopsy (six female patient and six male patient; mean age, 64 years), after obtaining informed consent from close relatives. All autopsies were performed within 24 hr of death. Control subjects had no history or pathomorphological evidence of hypertension, diabetes, or hypercholesterolemia. All procedures to collect samples were performed according to the recommendations of the institutional Ethics Committee.
2.2. Light microscopy
Specimens of healthy and atherosclerotic human aorta were fixed by immersion in Bouin's fixative, dehydrated in ascending ethanol concentrations, cleared in xylene, and subsequently embedded in paraffin (Singhal et al. 2016). Four‐micrometer transverse sections were stained with hematoxylin–eosin (H&E).
2.3. Electron microscopy
Immediately after collection, all specimens were fixed in 3% glutaraldehyde solution in 0.1 M phosphate buffer (pH 7.4) for 2 hr at 4°C, postfixed in osmium tetroxide (1%) for 2 hr, dehydrated in a graded series of acetone solutions, and then progressively embedded in acetone/resin with final embedment in pure resin (Araldite ‐ Fluka). Ultrathin sections (60‐90 nm in thickness) were cut with a diamond knife, mounted on copper grids (G300 Cu), contrasted using both aqueous uranyl acetate and lead citrate (Reynolds 1963), and then examined with a Jeol JEM 1400‐Plus electron microscope operating at 80 kV.
2.4. Data analysis
For quantification of ER lumen and mitochondrial size, at least two grids were prepared for each sample in the diseased (n = 12) and control (n = 12) groups. Images have been randomly captured, and 100 ER profiles and mitochondria have been analyzed for each grid. ER and mitochondria were identified based on morphology and sizes in each section. The diameter of ER and mitochondria was measured in EM images using ImageJ software (NIH). Statistical comparisons between two groups of data were performed using two‐tailed unpaired Student's t tests. p‐values <.05 have been considered significant. Data have been presented as mean ± SEM.
3. RESULTS
Firstly, the aortic wall specimens derived from the operated patients have been evaluated on the basis of their gross anatomy, histopathologically analyzed and then classified according to the American Heart Association Committee on Vascular Lesions (AHA) guidelines (Stary et al. 1995). Subsequently, a comparative analysis of ECs of healthy and atherosclerotic human aorta has been conducted by using an electron microscopic approach. Healthy human aorta characteristically consisted of a continuous layer of flat ECs whose nuclei bulged into the lumen (black arrowhead) and a tunica media made up of concentrically arranged elastic lamellae that appeared in cross section as wavy bands (asterisks) interspersed with layers of SMCs (Figure 1a). The aortic wall adjacent to atheromatous areas exhibited only a slight fibrointimal thickening (black arrow). Fatty accumulation was not present. An intact endothelial monolayer was observed to cover the lumen of these regions (Figure 1b). The atherosclerotic plaques analyzed in this study were histologically characterized by the presence of a necrotic core (nc) of acellular amorphous material and frequent cholesterol crystals (cc) separated from the lumen by a thick fibrous cap (fc) (Figure 1c). These lesions have been considered as type IV‐V lesions, atheromas. Ultrastructurally, the atherosclerotic plaques consisted of a cap of fibrous and avascular connective tissue (Figure 2a), and a pool of amorphous and ill‐defined material (the necrotic core) made up of lipid‐laden foam cells, extracellular lipids, cell debris, inflammatory cells, calcium, and cholesterol clefts (Figure 2b‐d, Figure S1). ER from control tissues appeared as an interconnected network of flattened, membrane‐enclosed tubules that extended throughout the cytoplasm, studded externally with ribosomes. Its luminal content was moderately electron dense and amorphous. Mitochondria were spherical or sausage‐shaped structures with a smooth outer membrane and a highly folded inner membrane. Mitochondria typically presented an inner compartment (the matrix) with low electron density in which lamellar or tubular cristae can be clearly distinguished (Figure 3a,b). With respect to their shape, size, and arrangement, ER and mitochondria showed a remarkable degree of constancy for all the analyzed specimens independently of their location within the cell and the orientation of the ultrathin sections. TEM analysis revealed that in the endothelium of atherosclerotic patients, the ER clearly differed from its normal counterpart. Diseased ECs contained dilated cisternae of the ER filled with a moderate electrondense material. The dilated ER profiles displayed fewer attached ribosomes, and in some cases, large stretches of these dilations almost completely lacked ribosomes (Figure 4). The mean ER diameter in diseased ECs was conspicuously wider than that of healthy cells (Figure 5). Gross alterations in shape and size accompanied by a remodeling of the inner membrane have been also documented in the mitochondria of lesional ECs (Figure 6). Mitochondria appeared swollen and/or enlarged with a greater circularity and an average size significantly larger than control group (Figure 7). They showed an aberrant architectural remodeling and acquired a variety of abnormal phenotypes with loss of their normal internal arrangement. Cristae were reduced in number and/or detached from the inner boundary membrane to form blind‐ending tubules within the matrix or circular structures located peripherally (Figure 8). Alterations in mitochondrial morphology were present not only in the perinuclear region but also at the periphery of the cell where breakage of mitochondrial membranes have been documented (Figure 9). In these ECs, nuclei displayed areas of nucleoplasm rarefaction (Figure 10a). ECs became intensely vacuolated with multiple, well‐defined clear vacuoles in their cytoplasm (Figure 10b). In advanced stages of degeneration, cell structure was dramatically altered. Dilated ER and enlarged mitochondria can be still recognized (Figure 10c). In the aortic tissue adjacent to the lesion, EM showed a mixed population of ECs: the first with a regular phenotype, normal mitochondria, and a well‐developed ER (Figure 11a); the second exhibiting evident alterations of ER including dilation of the cisternae and loss of ribosomes from the surface (Figure 11b). ER membranes also contained a floccular material. In these cells, mitochondria did not exhibit the profound ultrastructural defects observed in the diseased samples (Figure 11c,d).
FIGURE 1.
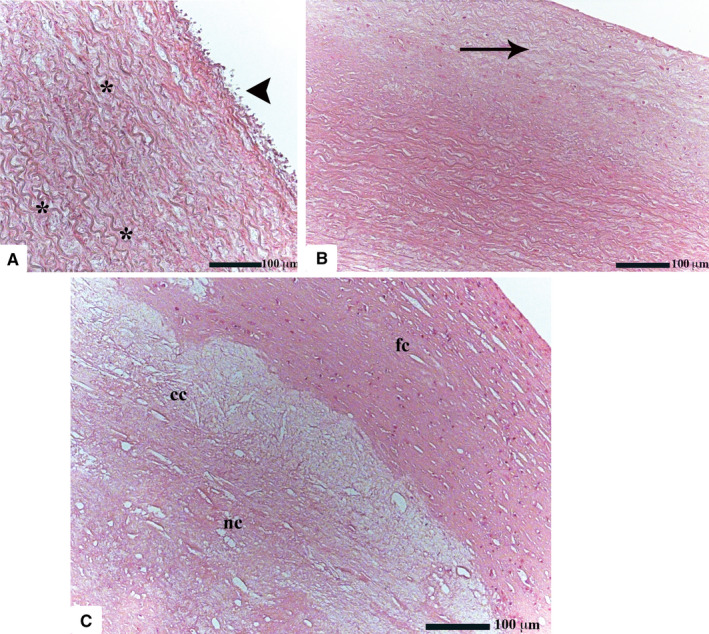
Histological features of healthy and atherosclerotic human aorta (H&E; original magnification ×50). Healthy ECs are very thin and flat (a). Their nuclei are frequently found to protrude into the lumen of the vessel (black arrowhead). In the tunica media, the characteristically wavy elastic fibers are entwined among collagenous fibers and SMCs. The aortic tissue adjacent to the lesion maintains a regular architecture showing only a mild intimal hyperplasia (black arrow) (b). Atherosclerotic plaques consist of a lipid‐rich necrotic core (nc) which is separated from the vascular lumen by a well‐developed fibrous cap (fc). Cholesterol crystals (cc), which appear as spindle‐shaped structures, can be also recognized (c)
FIGURE 2.
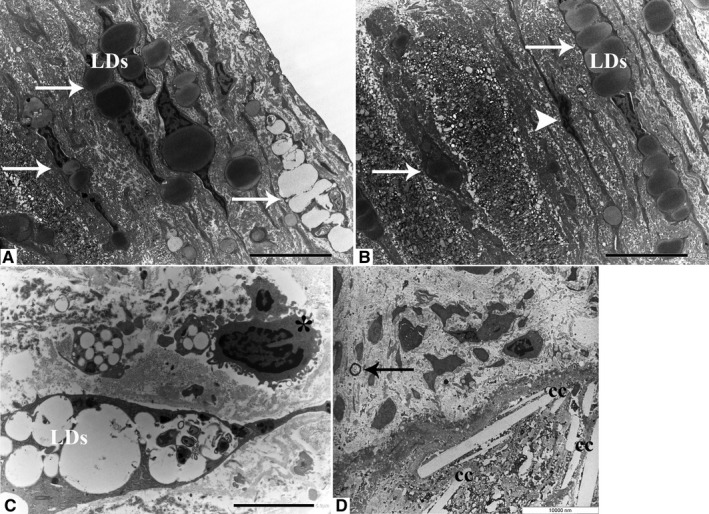
Ultrastructural properties of atherosclerotic plaques. The fibrous cap is composed mainly of connective tissue and foam cells filled with cytosolic lipid droplets (white arrows) (a, original magnification ×1200). The necrotic core contains SMCs (white arrowhead), foam cells (white arrows), and amorphous material (b, original magnification ×1200). Unlike SMCs that usually possess an elongated shape, macrophages appear as round cells with a central nucleus and abundant cytoplasmic protrusions. Nucleoli are typically not seen (asterisk) (c, original magnification ×2500). Ovoid microcalcifications (black arrow) and cholesterol clefts (cc) are present in the deep portion of the plaque (d, original magnification ×3150). LDs: lipid droplets
FIGURE 3.
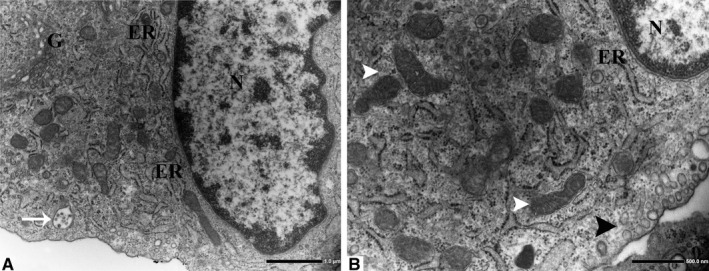
Subcellular ultrastructure of healthy ECs (a, original magnification ×8000). ER forms a labyrinth of interconnected flattened tubes studded on their outer surface (the cytosolic side) with numerous ribosomes. Mitochondria appear as cylindrical structures with an outer membrane that separates the organelle from the cytoplasm and an inner membrane with folded cristae. Notice the cristae projecting into the matrix (white arrowheads) (b, original magnification ×15000). Pinocytic vesicles (black arrowhead), free ribosomes, and multivesicular bodies (white arrow) are also present in the healthy endothelium. ER, endoplasmic reticulum; G, Golgi apparatus
FIGURE 4.
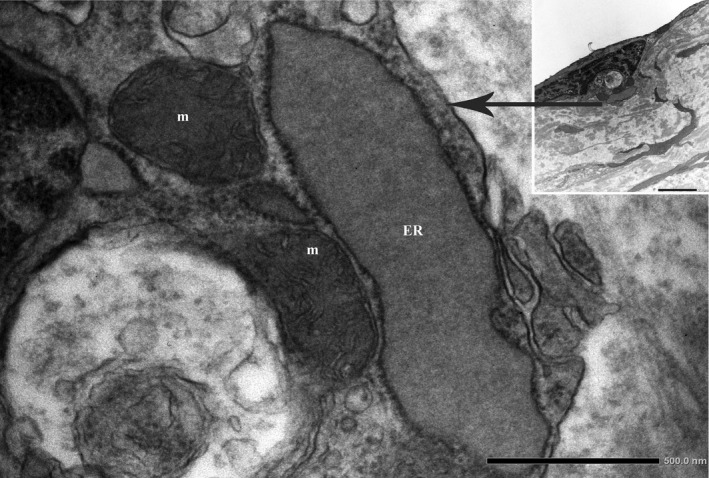
Pathologic dilation of ER in the ECs of human atherosclerotic plaque. The electron micrograph shows a higher magnification of the boxed area (Original magnification ×25000 and ×6000, respectively). ER, endoplasmic reticulum; m, mitochondria
FIGURE 5.
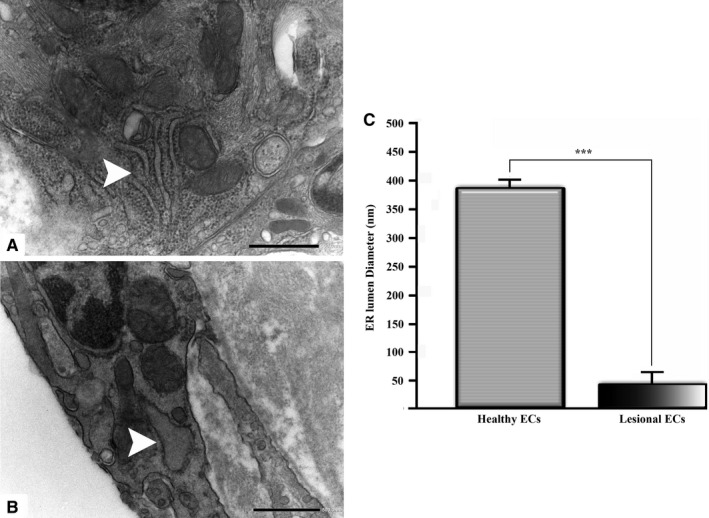
The diameter of the ER lumen has been obtained from EM images and compared between healthy (a) and lesional (b) ECs. Data are presented as mean ± S.E.M. ***denote p < .001 (c). (a, b original magnification ×20000)
FIGURE 6.
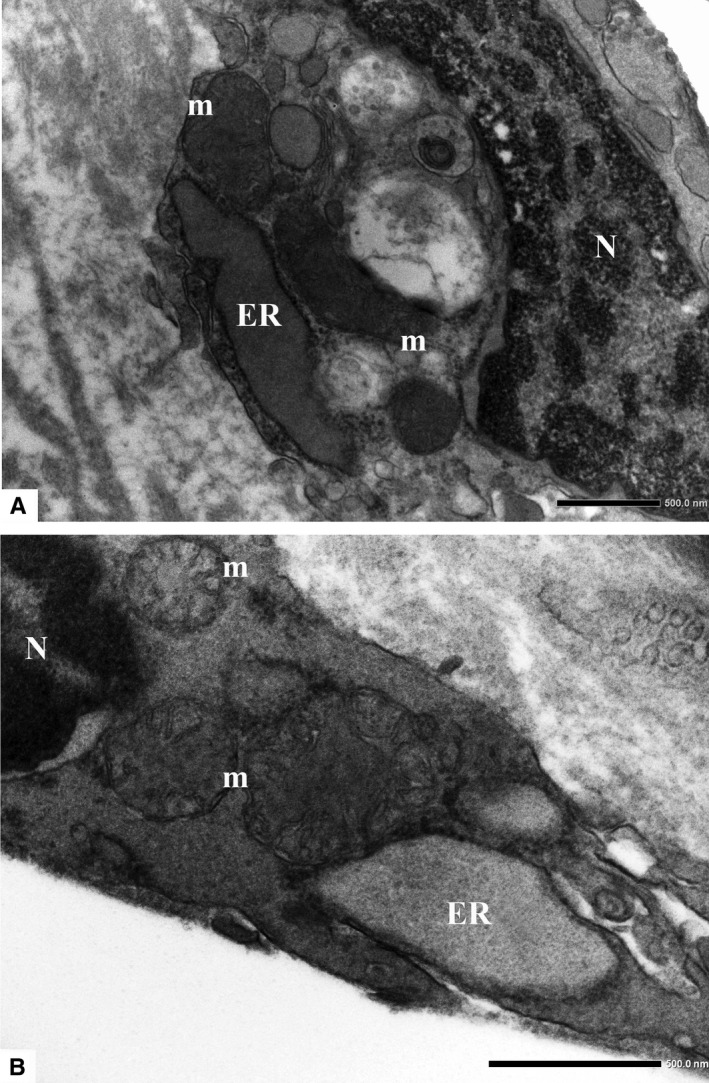
Dilation of ER is a common ultrastructural feature found in the ECs of human atherosclerotic plaque. Adjacent mitochondria also show alterations in their structural organization. (a, original magnification ×15000; b, original magnification ×25000). ER, endoplasmic reticulum; m, mitochondria; and N, nucleus
FIGURE 7.
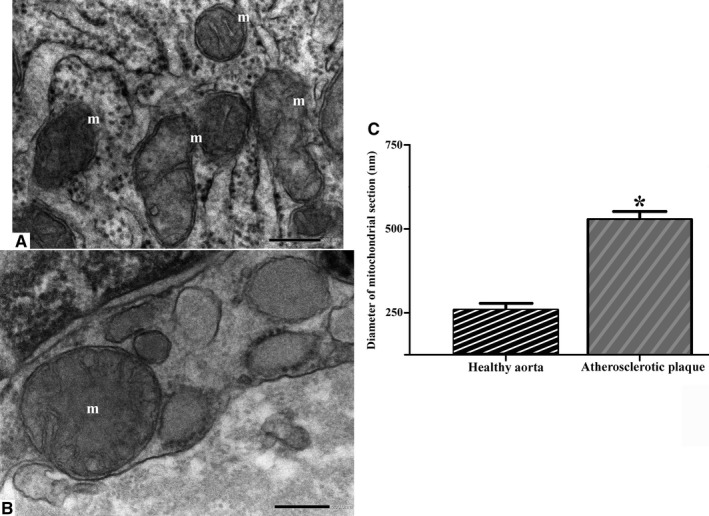
Compared to controls (a), the average diameter of diseased mitochondria is significantly increased (b) (a, b original magnification ×40000). The asterisk in (c) indicates statistical significant differences (*p < .05). m, mitochondria
FIGURE 8.
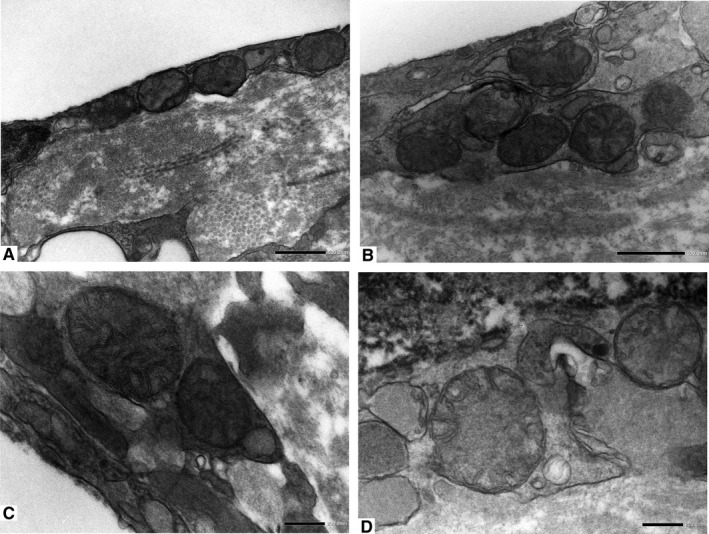
Ultrastructural documentation of the mitochondrial defects found in association with ER dilation in the ECs of human atherosclerotic plaque. The enlarged mitochondria show derangement of cristae and tend to exhibit a high degree of roundness. Internal “circular” cristae are located peripherally in contact with the inner membrane. (a, original magnification ×15000; b, original magnification ×20000; c and d, original magnification ×40000)
FIGURE 9.
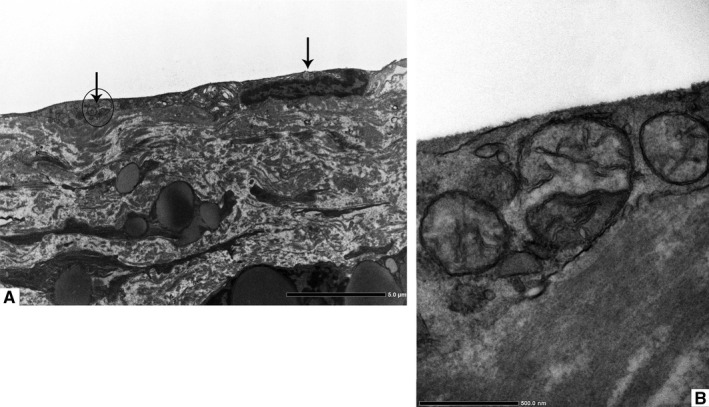
Aberrant mitochondria (black arrows) are present around the perinuclear region and scattered throughout the periphery (a, original magnification ×2500). High magnification image of the region marked with the circle in figure (a) is shown in figure (b). The presence of mitochondria with broken membranes has been also documented (b, original magnification ×25000)
FIGURE 10.
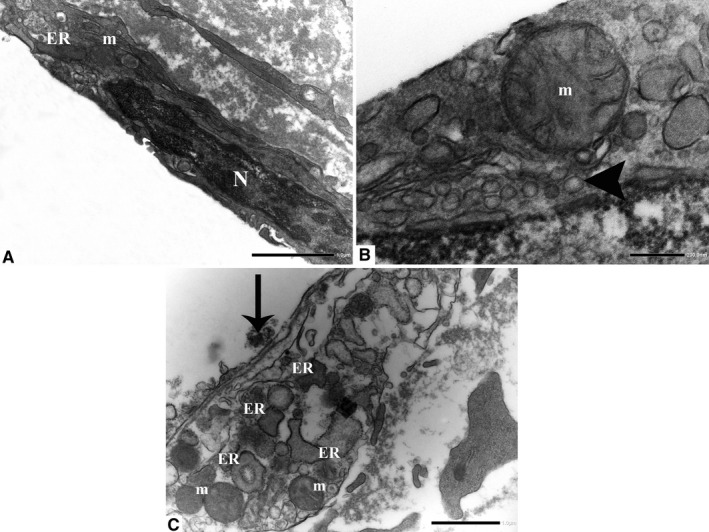
Endothelial cells with dilated ER membranes and altered mitochondria display cytoplasmic vacuolation (arrowhead). The nuclei frequently show areas of rarefaction and darker chromatin. An EC in an advanced stage of degeneration: Cell outlines are practically completely lost. Note the presence of dilated ER and altered mitochondria. ER: endoplasmic reticulum; m: mitochondria; and N: nucleus. (a, original magnification ×12000; b, original magnification ×40000; c, original magnification ×10000)
FIGURE 11.
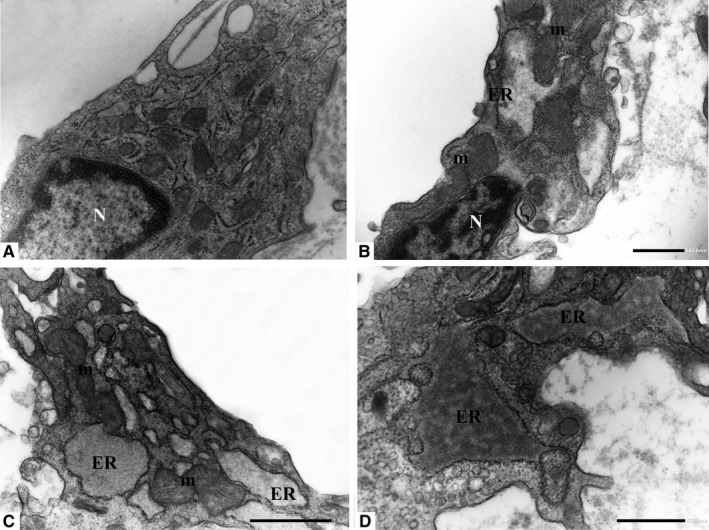
Ultrastructure of ECs in the area adjacent to the lesion. The first type possesses a regular arrangement with normal mitochondria and ER membranes (a). The second type shows evident dilation of ER that also contains a floccular material. In these cells, mitochondrial structure is preserved (b–d). ER, endoplasmic reticulum; m, mitochondria; and N, nucleus. (a, original magnification ×15000; b, original magnification ×15000; c, original magnification ×12000; d, original magnification ×20000)
4. DISCUSSION
Atherosclerosis is a chronic, progressive disease with multiple risk factors and pathological mechanisms responsible for the alterations of the structural and functional properties of the artery wall and, finally, for the evolution of the plaque (Lusis 2000; Robinson et al. 2009). As the major regulator of vascular homeostasis, the endothelium acts as guardian of the blood vessel integrity sensing external changes in the environment and responding, either directly or indirectly, by transmitting redox signals to neighboring cells in a paracrine fashion (Arnal et al. 2010; Fliser 2010; Giannotta et al. 2013). The endothelium controls the delicate balance between vasodilators and vasoconstrictors, stimulates or inhibits SMC migration and proliferation depending on the conditions, and mediates thrombosis and fibrinolysis in part through the production of coagulation factors and anticoagulant substances (Casscells 1992; Gertler & Abbott 1992; Pearson & Vanhoutte 1993; Jeremy et al. 1999; Pearson 1999; Hinsbergh 2012; Li et al. 2018). Under certain stressful situations, however, the endothelium deteriorates and the capacity to perform these tasks becomes severely compromised. The resulting imbalance leads to a condition called “endothelial dysfunction” which represents the primum movens of the atherosclerotic process (Rubanyi 1993; Hadi et al. 2005; Park & Park 2015). In recent years, there have been a lot of studies highlighting the importance of ER in the maintenance of endothelial function (Tabas et al. 2009; Stacchiotti et al. 2013; Galan et al. 2014). As is well known, the role of the ER tubules is not only restricted to protein synthesis, protein trafficking, and lipid synthesis but also extends to the control of the calcium ion concentration within the cell and to the supply of lipid bilayers to the nascent autophagosome (Krebs et al. 2015; McCaffrey & Braakman 2016; Ge et al. 2017; Jacquemyn et al. 2017; Marchi et al. 2018). Besides its physiological relevance, ER has been also increasingly implicated as a central contributor to the pathogenesis of a wide range of human diseases (Yoshida 2007; Lin et al. 2008). In general, almost any type of condition that affects the protein‐folding capacity of ER is able to induce ER stress. To rectify and restore the homeostasis of this compartment, cells engage a sophisticated surveillance pathway, the unfolded protein response (UPR), that requires the cooperation of different stress sensor proteins known to perform both anti‐ and pro‐apoptotic functions (depending on the stimulus), (Schröder & Kaufman 2005; Amen et al. 2019). It is therefore not surprising that, under some circumstances, the ER can exhibit a certain degree of heterogeneity and can influence cellular behavior and responses. As mentioned above, ER stress has been implicated in diverse pathological states, such as liver disease, neurodegeneration, and cancer. The most compelling evidence confirming the importance of ER stress in disease development and progression has been obtained from animal and human studies of type 2 diabetes mellitus demonstrating that pancreatic β‐cell failure driven by ER stress is among the fundamental causes of the disease (Eizirik et al. 2008; Dara et al. 2011; Fonseca et al. 2011; Malhi & Kaufman 2011; Back & Kaufman 2012; Papa 2012; Yadav et al. 2014; Remondelli & Renna 2017; Sprenkle et al. 2017; Lebeaupin et al. 2018; Lin et al 2019). In the context of atherosclerosis, the majority of researches have focused on the molecular mechanisms that govern ER stress signaling, leaving many uncertainties and doubts regarding the structural state of the ER within the spatial context of the cell. It has been found that ER stress sensor proteins (i.e., IRE1α, ATF6α, and XBP1) are upregulated in the endothelium of normal swine aorta at site of atherosusceptibility (Civelek et al. 2009). Similarly, artificial shear stress has been reported to promote endothelial ER stress via induction of GRP78, one of the UPR downstream effectors (Feaver et al. 2008). There is also some evidence that exposure of cultured ECs to disturbed flow profiles triggers the activation of XBP1 that may act in either a positive or a negative fashion. XBP1 is known to have a cytoprotective effect against disturbed flow. However, excessive and/or chronic XBP1 activation adversely affects cell physiology thereby promoting endothelial dysfunction and atherosclerosis (Zeng et al. 2009). Other possible athero‐relevant factors that are also implicated in the pathologic activation of ER stress in ECs include homocysteine and modified forms of LDL that act through different mechanisms involving the induction of cell death programs, the imbalance of ER calcium metabolism, and the stimulation of inflammatory cytokine production (Outinen et al. 1999; Zhang et al. 2001; Zhou et al. 2004). The UPR activation can be also triggered in ECs by oxidized 1‐palmitoyl‐2‐arachidonyl‐sn‐3‐glycero‐phosphorylcholine (oxPAPC), an oxidized phospholipid abundant in atherosclerotic lesions. Although with phenotypic differences between humans, an in vivo evidence has also demonstrated that oxidized phospholipid‐rich areas of human lesional endothelium show evidence of ER stress activation (Gargalovic et al. 2006; Romanoski et al. 2010). These observations are consistent with the notion that ER stress may promote plaque formation by initially inducing cell dysfunction, leading to endothelial apoptosis at later stage (Tabas 2010). A well‐documented ultrastructural sign of ER stress is the pronounced dilation of the ER lumen as demonstrated in yeast and mammalian cells that increase their ER volume as an adapatative response to manage the elevated levels of luminal constituents under stress conditions (Raposo et al. 1995; Schönthal 2012; Welihinda et al. 1999). Similarly, it has been demonstrated by histology in myocardial cells that ER develops morphological changes (which are indicative of ER overload) in order to counteract unfavorable environmental stimuli, such as hypoxia, oxidative stress, and enhanced protein synthesis (Hotamisligil 2010a, 2010b; Mei et al. 2015; Zhang et al. 2017). To date these ultrastructural changes have not been documented in human atherosclerosis nor it is known if the potential re‐organization of ER takes place in the earlier stage of the disease or later, when other structural and functional changes develop. The results of the present study reveal new findings relevant to the mechanisms of atherosclerotic plaque formation demonstrating that ER dilation accompanied by alterations of mitochondrial structure and cytoplasm vacuolation are present in the ECs covering the plaque. More importantly, it has been demonstrated that ER alterations occur as apparent early changes in the aortic tissue adjacent to the lesion exhibiting only slight fibrointimal thickening that may indicate the initial proliferative stage of atherosclerosis. There is no similar description with an electron microscope in the field of atherosclerosis research. Much of the current knowledge relates to the macrophages residing into the plaque and concentrates largely on the ability of ER stress to influence lipid metabolism, inflammation, and apoptotic responses (Hotamisligil 2010a, 2010b). Likewise, it has been shown that cultured SMCs can undergo programmed cell death when exposed to excess free cholesterol, through a mechanisms involving both ER‐ and mitochondria‐based signals (Kedi et al. 2009). ER dilation has been demonstrated to occur in a mouse model of neonatal hypoxic‐ischemic brain injury where it exerts a dualistic role and serves as a protective mechanism in the early phase of injury, switching to a terminal maladaptive phase if the stress condition persists and the compensatory UPR response fails. At this late stage, also mitochondria undergo striking structural changes and a “peppered” chromatin pattern appears (Chavez‐Valdez et al. 2016). The importance of ER in neonatal brain injury has also been highlighted in another paper by Martin and co‐workers demonstrating that neurodegeneration presents a specific temporal pattern of events in which ER dilation, Golgi‐derived vacuolation, and mitochondrial swelling are a sequence of successive ultrastructural changes that finally result in neuronal apoptosis (Martin et al. 2000). In line with these previous observations, the findings reported here of prominent ER dilation in the ECs covering the plaque add strength to the importance of ER in human atherosclerosis. These data are in line with the idea that perturbation of ER ultrastructure, probably as a consequence of ER stress, is initiated early in the disease course and may reflect the enormous capacity of ECs to buffer the stress throughout the enlargement of ER compartment. Alterations of ER membranes seem to precede other morphological changes of the vascular endothelium and its subcellular organelles, mainly the mitochondria. To date, aberrant changes of mitochondria have been only documented in the leukocytes of patients with atherosclerosis and in the SMCs of human coronary artery (Sazonova et al. 2017; Murray et al. 1968). Although much remains to be learned mechanistically, the electron microscopic results described here expand the spectrum of ultrastructural abnormalities known to exist in human atherosclerotic tissues and provide novel perceptions about the different pathophysiologic mechanisms responsible for atherosclerosis. The ultrastructural pathology of ER might have a profound impact on EC function, resilience, and survival and might represent an important step in the development of cell dysfunction contributing directly to the disturbance of the vascular homeostasis and thus to the initiation (not only the progression) of the disease. It is tempting to speculate that endothelial ER might act as a promising therapeutic target to improve endothelial function and to prevent atherosclerosis.
CONFLICT OF INTEREST
The author declares that there is no conflict of interest.
Supporting information
Fig S1
ACKNOWLEDGMENTS
The author would like to thank Dr Alfonso Sciangula (Department of Cardiovascular Surgery, Cardiac Surgery Unit, S. Anna Hospital Catanzaro, Italy) who kindly provided surgical specimens.
Perrotta I. The microscopic anatomy of endothelial cells in human atherosclerosis: Focus on ER and mitochondria. J. Anat. 2020;237:1015–1025. 10.1111/joa.13281
Funding information
This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors.
REFERENCES
- Amen, O.M. , Sarker, S.D. , Ghildyal, R. and Arya, A. (2019) Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity, and cardiac dysfunction: Therapeutic and molecular approach. Frontiers in Pharmacology, 10, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnal, J.F. , Fontaine, C. , Billon‐Galés, A. , Favre, J. , Laurell, H. , Lenfant, F. and Gourdy, P. (2010) Estrogen receptors and endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology, 30, 1506–1512. [DOI] [PubMed] [Google Scholar]
- Back, S.H. and Kaufman, R.J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annual Review of Biochemitry, 85, 767–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battson, M.L. , Lee, D.M. and Gentile, C.L. (2017) Endoplasmic reticulum stress and the development of endothelial dysfunction. American Journal of Physiology‐Heart and Circulatory Physiology, 312, H355–H367. [DOI] [PubMed] [Google Scholar]
- Cai, H. and Harrison, D.G. (2000) Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ Res, 87, 840–844. [DOI] [PubMed] [Google Scholar]
- Casscells, W. (1992) Migration of smooth muscle and endothelial cells. Critical events in restenosis. Circulation, 86, 723–729. [DOI] [PubMed] [Google Scholar]
- Chavez‐Valdez, R. , Flock, D.L. , Martin, L.J. and Northington, F.J. (2016) Endoplasmic reticulum pathology and stress response in neurons precede programmed necrosis after neonatal hypoxia‐ischemia. International Journal of Developmental Neuroscience, 48, 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov, D.A. , Sobenin, I.A. , Orekhov, A.N. and Bobryshev, Y.V. (2014) Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. BioMed Research International, 2014, 610140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chroni, A. , Leondaritis, G. and Karlsson, H. (2011) Lipids and lipoproteins in atherosclerosis. Journal of Lipids, 2011, 160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimellaro, A. , Perticone, M. , Fiorentino, T.V. Sciacqua, A. and Hribal, M.l. (2016) Role of endoplasmic reticulum stress in endothelial dysfunction. Nutrition, Metabolism and Cardiovascular Diseases, 26, 863–871. [DOI] [PubMed] [Google Scholar]
- Civelek, M. , Manduchi, E. , Riley, R.J. , Stoeckert, C.J. and Davies, P.F. (2009) Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circulation Research, 105, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dara, L. , Ji, C. and Kaplowitz, N. (2011) The contribution of endoplasmic reticulum stress to liver diseases. Hepatology, 53, 1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon, J. and Ganz, P. (2004) Role of endothelial dysfunction in atherosclerosis. Circulation, 109, III27–III32. [DOI] [PubMed] [Google Scholar]
- Dong, Y. , Zhang, M. , Wang, S. , Liang, B. , Zhao, Z. , Liu, C. et al (2010) Activation of AMP‐activated protein kinase inhibits oxidized LDL‐triggered endoplasmic reticulum stress in vivo. Diabetes, 59, 1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik, D.L. , Cardozo, A.K. and Cnop, M. (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews, 29, 42–61. [DOI] [PubMed] [Google Scholar]
- Favero, G. , Paganelli, C. , Buffoli, B. , Rodella, L.F. and Rezzani, R. (2014) Endothelium and its alterations in cardiovascular diseases: Life style intervention. BioMed Research International, 2014, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feaver, R.E. , Hastings, N.E. , Pryor, A. and Blackman, B.R. (2008) GRP78 upregulation by atheroprone shear stress via p38‐, α2β1‐dependent mechanism in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, 1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliser, D. (2010) Perspectives in renal disease progression: The endothelium as a treatment target in chronic kidney disease. Journal of Nephrology, 23, 369–376. [PubMed] [Google Scholar]
- Fonseca, S.G. , Gromada, J. and Urano, F. (2011) Endoplasmic reticulum stress and pancreatic beta cell death. Trends Endocrinology Metabolism, 22, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán, M. , Kassan, M. , Kadowitz, P.J. , Trebak, M. , Belmadani, S. and Matrougui, K. (2014) Mechanism of endoplasmic reticulum stress‐induced vascular endothelial dysfunction. Biochimica et Biophysica Acta (BBA) ‐ Molecular Cell Research, 1843, 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargalovic, P.S. , Gharavi, N.M. , Clark, M.J. , Pagnon, J. , Yang, W.‐P. , He, A. et al (2006) The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 26, 2490–2496. [DOI] [PubMed] [Google Scholar]
- Ge, L. , Zhang, M. , Kenny, S.J. , Liu, D. , Maeda, M. , Saito, K. et al (2017) Remodeling of ER‐exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Reports, 18, 1586–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler, J.P. and Abbott, W.M. (1992) Prothrombotic and fibrinolytic function of normal and perturbed endothelium. Journal of Surgical Research, 52, 89–95. [DOI] [PubMed] [Google Scholar]
- Giannotta, M. , Trani, M. and Dejana, E. (2013) VE‐cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Developmental Cell, 26, 441–454. [DOI] [PubMed] [Google Scholar]
- Gimbrone, M.A. Jr and García‐Cardeña, G (2016) Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circulation Research, 118, 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadi, H.A. , Carr, C.S. and Al, S.J. (2005) Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vascular Health Risk Management, 1, 183–198. [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil, G.S. (2010a) Endoplasmic reticulum stress and atherosclerosis. Nature Medicine, 16, 396–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil, G.S. (2010b) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell, 140, 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemyn, J. , Cascalho, A. and Goodchild, R.E. (2017) The ins and outs of endoplasmic reticulum‐controlled lipid biosynthesis. EMBO Reports, 18, 1905–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremy, J.Y. , Rowe, D. , Emsley, A.M. and Newby, A.C. (1999) Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovascular Research, 43, 580–594. [DOI] [PubMed] [Google Scholar]
- Kedi, X. , Ming, Y. , Yongping, W. , Yi, Y. and Xiaoxiang, Z. (2009) Free cholesterol overloading induced smooth muscle cells death and activated both ER‐ and mitochondrial‐dependent death pathway. Atherosclerosis, 207, 123–130. [DOI] [PubMed] [Google Scholar]
- Krebs, J. , Agellon, L.B. and Michalak, M. (2015) Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochemical and Biophysical Research Communications, 460, 114–121. [DOI] [PubMed] [Google Scholar]
- Lebeaupin, C. , Vallée, D. , Hazari, Y. , Hetz, C. , Chevet, E. and Bailly‐Maitre, B. (2018) Endoplasmic reticulum stress signalling and the pathogenesis of non‐alcoholic fatty liver disease. Journal of Hepatology, 69, 927–947. [DOI] [PubMed] [Google Scholar]
- Li, M. , Qian, M. , Kyler, K. and Xu, J. (2018) Endothelial‐vascular smooth muscle cells interactions in atherosclerosis. Frontiers in Cardiovascular Medicine, 5, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J.H. , Walter, P. and Yen, T.S.B. (2008) Endoplasmic reticulum stress in disease pathogenesis. Annual Review of Pathology: Mechanisms of Disease, 3, 399–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. , Jiang, M. , Chen, W. , Zhao, T. and Wei, Y. (2019) Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomedicine & Pharmacotherapy, 118, 109249. [DOI] [PubMed] [Google Scholar]
- Luchetti, F. , Crinelli, R. , Cesarini, E. , Canonico, B. , Guidi, L. , Zerbinati, C. (2017) Endothelial cells, endoplasmic reticulum stress and oxysterols. Redox Biology, 13, 581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis, A.J. (2000) Atherosclerosis. Nature, 407, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maamoun, H. , Abdelsalam, S.S. , Zeidan, A. et al (2019) Endoplasmic reticulum stress: A critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. International Journal of Molecular Sciences, 20, 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi, H. and Kaufman, R.J. (2011) Endoplasmic reticulum stress in liver disease. Journal of Hepatology, 54, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi, S. , Patergnani, S. , Missiroli, S. , Morciano, G. , Rimessi, A. , Wieckowski, M.R et al (2018) Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium, 69, 62–72. [DOI] [PubMed] [Google Scholar]
- Martin, L.J. , Brambrink, A.M. , Price, A.C. , Kaiser, A. , Agnew, D.M. , Ichord, R.N. et al (2000) Neuronal death in newborn striatum after hypoxia‐ischemia is necrosis and evolves with oxidative stress. Neurobiology of Disease, 7, 169–191. [DOI] [PubMed] [Google Scholar]
- Mazurek, R. , Dave, J.M. , Chandran, R.R. , Misra, A. , Sheikh, A.Q. and Greif, D.M. (2017) Vascular cells in blood vessel wall development and disease. Advances in Pharmacology, 78, 323–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey, K. and Braakman, I. (2016) Protein quality control at the endoplasmic reticulum. Essays in Biochemistry, 60, 227–235. [DOI] [PubMed] [Google Scholar]
- Mei, Y. , Thompson, M.D. , Cohen, R.A. and Tong, X. (2015) Autophagy and oxidative stress in cardiovascular diseases. Biochimica et Biophysica Acta (BBA) ‐ Molecular Basis of Disease, 1852, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M. , Schrodtab, G.R. and Berga, H.F. (1968) Mitochondrial alterations in human and experimental atherosclerosis. Biochemical Medicine, 2, 118–135. [Google Scholar]
- Outinen, P.A. , Sood, S.K. , Pfeifer, S.I. , Pamidi, S. , Podor, T.J. , Li, J. et al (1999) Homocysteine‐induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood, 94, 959–967. [PubMed] [Google Scholar]
- Papa, F.R. (2012) Endoplasmic reticulum stress, pancreatic β‐cell degeneration, and diabetes. Cold Spring Harbor Perspectives in Medicine, 2, a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, K.H. and Park, W.J. (2015) Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. Journal of Korean Medical Science, 30, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, J.D. (1999) Endothelial cell function and thrombosis. Best Practice & Research Clinical Haematology, 12, 329–341. [DOI] [PubMed] [Google Scholar]
- Pearson, P.J. and Vanhoutte, P.M. (1993) Vasodilator and vasoconstrictor substances produced by the endothelium. Reviews of Physiology Biochemistry and Pharmacology, 122, 1–67. [DOI] [PubMed] [Google Scholar]
- Rafieian‐Kopaei, M. , Setorki, M. , Doudi, M. , Baradaran, A. and Nasri, H. (2014). Atherosclerosis: Process, indicators, risk factors and new hopes. International Journal of Preventive Medicine, 5, 927–946. [PMC free article] [PubMed] [Google Scholar]
- Raposo, G. , van Santen, H.M. , Leijendekker, R. , Geuze, H.J. and Ploegh, H.l. (1995) Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER‐Golgi intermediate compartment. The Journal of Cell Biology, 131, 1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remondelli, P. and Renna, M. (2017) The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Frontiers in Molecular Neuroscience, 10, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, E.S. (1963) The use of lead citrate at high pH as an electron‐opaque stain in electron microscopy. The Journal of Cell Biology, 17, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.G. , Fox, K.M. , Bullano, M.F. and Grandy, S. (2009) Atherosclerosis profile and incidence of cardiovascular events: a population‐based survey. BMC Cardiovascular Disorders, 9, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanoski, C.E. , Lee, S. , Kim, M.J. , Ingram‐Drake, L. , Plaisier, C.L. , Yordanova, R. et al (2010) Systems genetics analysis of gene‐by‐environment interactions in human cells. The American Journal of Human Genetics, 86, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubanyi, G.M. (1993) The role of endothelium in cardiovascular homeostasis and diseases. Journal of Cardiovascular Pharmacology, 4, S1–S14. [DOI] [PubMed] [Google Scholar]
- Sandoo, A. , van Zanten, J.J. , Metsios, G.S. , Carroll, D. and Kitas, G.D. (2010) The endothelium and its role in regulating vascular tone. The Open Cardiovascular Medicine Journal, 4, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazonova, M.A. , Sinyov, V.V. , Ryzhkova, A.I. , Galitsyna, E.V. , Melnichenko, A.A. , Demakova, N.A. et al (2017) Ultrastructural analysis of mitochondrial cristae in leukocytes of patients with atherosclerosis. Atherosclerosis, 263, e280–e281. [Google Scholar]
- Schönthal, A.H. (2012) Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica (Cairo), 2012, 857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, M. and Kaufman, R.J. (2005) ER stress and the unfolded protein response. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 569, 29–63. [DOI] [PubMed] [Google Scholar]
- Singhal, P. , Singh, N.N. , Sreedhar, G. , Banerjee, S. , Batra, M. and Garg, A. (2016) Evaluation of histomorphometric changes in tissue architecture in relation to alteration in fixation protocol ‐. an invitro study. Journal of Clinical and Diagnostic Research, 10, 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenkle, N.T. , Sims, S.G. , Sánchez, C.L. and Meares, G.P. (2017) Endoplasmic reticulum stress and inflammation in the central nervous system. Molecular Neurodegeneration, 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacchiotti, A. , Gia Favero, G. and Rezzani, R. (2013) Endoplasmic reticulum stress in the endothelium: A contribution to athero‐susceptibility In: Rezzani R. (Ed.), Current Trends in Atherogenesis. Houston, TX: InTech. [Google Scholar]
- Stary, H.C. , Chandler, A.B. , Dinsmore, R.E. , Fuster, V. , Glagov, S. , Insull, W. et al (1995) A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis. American Heart Association. Circulation, 92, 1355–1374. [DOI] [PubMed] [Google Scholar]
- Sun, H.J. , Wu, Z.Y. , Nie, X.W. and Bian, J.S. (2019) Role of endothelial dysfunction in cardiovascular diseases: The link between inflammation and hydrogen sulfide. Frontiers in Pharmacology, 10, 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I. , Seimon, T. , Timmins, J. , Li, G. and Lim, W. (2009) Macrophage apoptosis in advanced atherosclerosis. Annals of the New York Academy of Sciences, 1173, E40–E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I. (2010) The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation Research, 107, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend, N. , Wilson, L. , Bhatnagar, P. , Wickramasinghe, K. , Rayner, M. and Nichols, M. (2016) Cardiovascular disease in Europe: Epidemiological update 2016. European Heart Journal, 37, 3232–3245. [DOI] [PubMed] [Google Scholar]
- van Hinsbergh, V.W.M. (2012) Endothelium ‐ role in regulation of coagulation and inflammation. Seminars in Immunopathology, 34, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welihinda, A.A. , Tirasophon, W. and Kaufman, R.J. (1999) The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expression, 7, 293–300. [PMC free article] [PubMed] [Google Scholar]
- Widmer, R.J. and Lerman, A. (2014) Endothelial dysfunction and cardiovascular disease. Global Cardiology Science and Practise, 2014(3), 291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav, R.K. , Chae, S.W. , Kim, H.R. and Chae, H.J. (2014) Endoplasmic reticulum stress and cancer. Journal of Cancer Prevention, 19, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, H. (2007) ER stress and diseases. FEBS Journal, 274, 630–658. [DOI] [PubMed] [Google Scholar]
- Zeng, L. , Zampetaki, A. , Margariti, A. , Pepe, A.E , Alam, S. , Martin, D. et al (2009) Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proceedings of the National Academy of Sciences, 106, 8326–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Cai, Y. , Adachi, M.T. , Oshiro, S. , Aso, T. , Kaufman, R.J et al (2001) Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. Journal of Biological Chemistry, 276, 35867–35874. [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Syed, T.W. , Liu, R. and Yu, J. (2017) Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Frontiers in Cardiovascular Medicine, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Werstuck, G.H. , Lhotak, S. , de Koning, A.B.L. , Sood, S.K. , Hossain, G.S. et al (2004) Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E‐deficient mice. Circulation, 110, 207–213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1