

Abstract
T cells engineered to express chimeric antigen receptors (CARs) targeting CD19 have produced impressive outcomes for the treatment of B cell malignancies, but different products vary in kinetics, persistence, and toxicity profiles based on the co-stimulatory domains included in the CAR. In this study, we performed transcriptional profiling of bulk CAR T cell populations and single cells to characterize the transcriptional states of human T cells transduced with CD3ζ, 4-1BB-CD3ζ (BBζ), or CD28-CD3ζ (28ζ) co-stimulatory domains at rest and after activation by triggering their CAR or their endogenous T cell receptor (TCR). We identified a transcriptional signature common across CARs with the CD3ζ signaling domain, as well as a distinct program associated with the 4-1BB co-stimulatory domain at rest and after activation. CAR T cells bearing BBζ had increased expression of human leukocyte antigen (HLA) class II genes, ENPP2, and interleukin (IL)-21 axis genes, and decreased PD1 compared to 28ζ CAR T cells. Similar to previous studies, we also found BBζ CAR CD8 T cells to be enriched in a central memory cell phenotype and fatty acid metabolism genes. Our data uncovered transcriptional signatures related to costimulatory domains and demonstrated that signaling domains included in CARs uniquely shape the transcriptional programs of T cells.
Keywords: adoptive cellular therapy, CAR T cells, single-cell RNA sequencing, transcriptional profiling, T cell activation
Graphical Abstract
Maus, Regev, and colleagues performed a deep transcriptional analysis of CAR T cells bearing different signaling domains, at rest and after activation. Findings include CAR-specific signatures and a distinct program activated in CARs with 4-1BB domains that was characterized by expression of central memory markers, MHC class II, and IL-21, among others.
Introduction
Chimeric antigen receptor (CAR) T cells targeting CD19 are an effective treatment option for relapsed or refractory B cell malignancies,1, 2, 3, 4, 5 including B cell leukemias6,7 and large-cell lymphomas.8,9 The incorporation of co-stimulatory domains is thought to be critical to the clinical efficacy of CAR T cells, since co-stimulation enhances cytokine production, proliferation, and persistence of the modified T cells.10 The two main co-stimulatory domains contained in US Food and Drug Administration (FDA)-approved CAR T cells in clinical use are CD28, a member of the B7 family, and 4-1BB, a member of the tumor necrosis factor (TNF) receptor superfamily.
In CARs directed to CD19, the clinical response rates of adults with relapsed or refractory large B cell lymphoma to CARs with the CD28 co-stimulation domain (axicabtagene ciloleucel) and the 4-1BB domain (tisagenlecleucel) are very similar.8,11 Likewise, in vitro co-culture experiments using 4-1BB- and CD28-containing CARs against the same antigen have strikingly similar cytotoxicity effects. However, clinical studies have found profound differences in the kinetics and phenotypes of these two CAR T cell products. CD28-based CARs undergo a more rapid expansion but have less persistence than do 4-1BB-based CARs, which can persist for more than 8 years.12,13 CD28-CD3ζ CAR T cells also have enhanced glycolytic metabolism in vitro and induce an effector memory phenotype, whereas 4-1BB-CD3ζ CAR T cells rely more heavily on fatty acid metabolism and typically have a central memory phenotype.14
The underlying molecular basis of these differences is not well understood. Given the complexity of a CAR T cell population, it is likely that the overall effects of the expression and activation of each CAR signaling domain (co-stimulatory domain and CD3ζ) are integrated into the overall function of each modified T cell, and those in turn integrate to a population-level response. Although multiple studies have assessed key markers and cytokines by flow cytometry and functional assays,12,14, 15, 16, 17 interpretations are complicated by selection of markers that reflect prior assumptions and the heterogeneity of the starting and analyzed T cell populations, which are composed of multiple phenotypes. Conversely, there is a paucity of deep, unbiased, transcriptional profiling studies in CAR T cells, at either the population or single-cell level.
In this study, we combined population (bulk) RNA sequencing (RNA-seq) and single-cell RNA-seq (scRNA-seq) of first- and second-generation human CAR T cells, which were designed to be identical in all aspects except their signaling domains. Through RNA-seq, we profiled 240 samples, spanning all combinations of CAR constructs, expressed in either CD4 or CD8 human T cells from three healthy donors. We further used scRNA-seq to uncover heterogeneity within each CAR T cell product from two additional donors. Based on gene expression co-variation patterns within each population at a given time point, we inferred regulatory programs active in subsets of CAR T cells. We identified transcriptional signatures common across CAR constructs, compared CAR activation to the T cell receptor (TCR) in CAR T cells, and identified a program unique to 4-1BB-bearing CARs that is present at rest and after antigen stimulation through the CAR. Notably, we found that the central memory phenotype, as defined by expression of CCR7, is driven by the 4-1BB program and is likely to be a key driver of the prolonged persistence of 4-1BB-based CAR T cells.
Results
CAR Designs, Transduction, and Preparation of CAR T Cells for Transcriptional Profiling
To identify transcriptional differences downstream of various CARs, we synthesized four CD19 CAR constructs bearing distinct signaling domains. These constructs closely resemble those in clinical use but with slight deviations to enable direct comparison of intracellular signaling domains (Figure 1A). The CARs consisted of a first-generation CAR with only a CD3ζ signaling domain (ζ), a second-generation CAR with a CD28 costimulatory domain (28ζ; using a different vector, promoter, and transmembrane domain than axicabtagene ciloleucel), a second-generation CAR with a 4-1BB co-stimulatory domain (BBζ; same as tisagenlecleucel), and a control CAR with a truncated, non-signaling CD3ζ chain (Δζ). All CARs had the same single-chain variable fragment (scFv) antigen binding domain against CD19 with identical CD8 hinge and transmembrane domains and were expressed behind an EF1α promoter using a third-generation self-inactivating lentiviral vector. To facilitate evaluation of CAR transduction, we included mCherry as a fluorescent reporter gene following a 2A ribosomal skip element. To cross-validate our findings on the signaling domains independently from the scFv, we also synthesized CARs with an anti-EGFR scFv domain (based on cetuximab) and kept all other components the same (Figure S1).
Figure 1.

Antigen Stimulation of CAR T Cells through Their CAR Yields a Weaker but Similar T Cell Activation Signal Compared to Stimulation via Anti-CD3-TCR
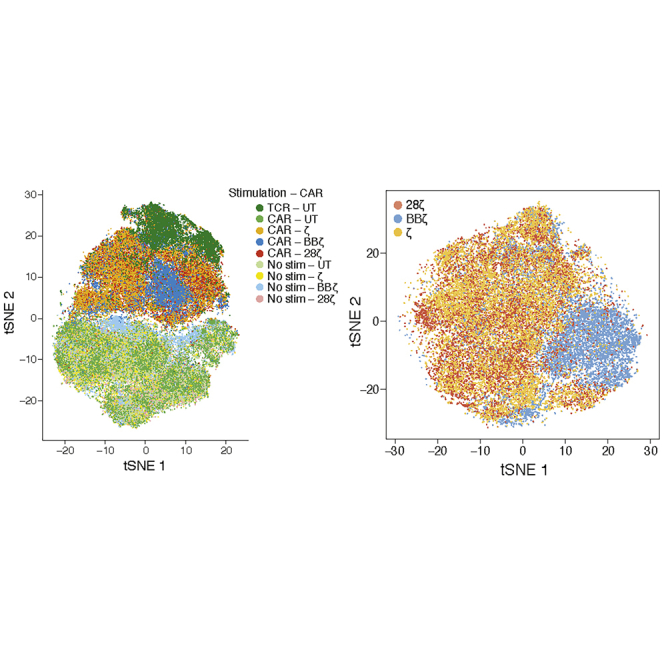
(A) Vector maps of CD19 CAR constructs (L, leader sequence; TM, hinge and transmembrane domain). (B) Experimental design of bulk RNA sequencing (RNA-seq). T cells were isolated from a leukopak from three normal donors, sorted on CD3+ and CD8+ or CD4+, mixed at a 1:1 CD4-to-CD8 ratio, activated with anti-CD3/CD28 T cell expansion beads, and transduced with one of the four CAR constructs or left untransduced (UT). Cells were expanded for 7 days before the beads were removed. T cells were then rested for an additional 7 days prior to stimulation with anti-CD3/CD28 beads or irradiated Nalm6 cells at a 1:1 effector-to-target cell ratio for 0, 4 or 24 h (see Figures S2A and S2B for transduction efficiencies). Samples were then sorted on mCherry+ CD4+ or CD8+ T cells and sequenced separately as bulk populations. Data were collected for T cells from three human donors with technical duplicates. (C) Experimental design of single-cell RNA-seq. T cells from two additional normal donors were isolated, activated, and transduced and expanded as in (B). UT T cells, BBζ, 28ζ, and ζ CAR T cells were either left unstimulated or stimulated with irradiated Nalm6 cells at a 1:1 effector-to-target cell ratio for 24 h. In addition, the UT T cells were stimulated through the TCR with irradiated K562 cells expressing an scFv against CD3 at a 1:1 effector-to-target cell ratio. Stimulated and unstimulated samples were then sorted as live mCherry+ cells and then sequenced using 10x Technology. More than 3,500 cells were sequenced per sample from two human donors. (D–F) Principal component analysis of the expression profiles from the bulk RNA-seq samples corrected for donor variation. Colored from left to right by (D) stimulation condition, (E) CD4+ versus CD8+, and (F) CAR construct. (G) t-Distributed Stochastic Neighbor Embedding (tSNE) of 83,123 single-cell expression profiles (dots) of all single cells sequenced from two donors after donor-specific batch correction (see Figures S2B and S2C). Cells are labeled by their CAR (color hue) and the type of stimulation (light tones for unstimulated). (H) tSNE expression profiles as in (G), colored by the degree of CD8A and CD4 expression. (I) Violin plots showing the distribution of T cell activation signature scores across each condition in CD4+ or CD8+ T cells. T cell activation gene signature were defined as the 15 most upregulated genes shared between CAR-activated CAR T cells and anti-CD3-activated UT cells compared to resting T cells (∗p < 2.2 × 10−16, Wilcoxon test; gene signatures are shown in Tables S1–S3).
To generate CAR T cells, we isolated CD4+ and CD8+ primary T cells from the peripheral blood of healthy donors and mixed these two populations in equal proportions prior to activation with anti-CD3/CD28 beads (Figure 1B). The following day, we transduced bulk T cells with lentiviral vectors to express one of the four CAR constructs (ζ, 28ζ, BBζ, and Δζ) or left them untransduced (UT). We expanded CAR T cells for 1 week, then removed beads and continued culture for another week, approximating a resting cell state prior to experimental analysis. All T cell cultures had comparable transduction and transgene expression as determined by the mCherry surrogate marker (Figures S2A and S2B). Furthermore, CAR T cells had equivalent cytotoxicity against Nalm6 (Figure S2C).
Because T cells are composed of a heterogeneous population of cells, we hypothesized that co-stimulation domains would have a differential effect on CD4 and CD8 T cell sub-populations. Therefore, we generated a high-quality, bulk RNA-seq atlas of CD19-targeting CAR T cell profiles from three normal donors by simulating their CAR T cells for 4 or 24 h through their CAR (using irradiated Nalm6 leukemia cells, which express CD19 endogenously) or through their TCR (using beads), and then sorting mCherry+CD4+ and mCherry+CD8+ T cell populations for each construct analyzed (Figure 1B). Using two additional normal donors, we also generated a scRNA-seq atlas of 83,123 profiles from ζ, 28ζ, and BBζ CAR T cells at rest and at 24 h post-stimulation through their CAR (Figure 1C). UT T cells at rest or post-stimulation through their TCR were used as controls. We used K562 cells expressing a surface scFv against human CD3 (K562-αCD3) to stimulate the TCR, avoiding the agonistic anti-CD28 antibody coated on beads. Quality was consistent across samples from the same donor, but differed by donor and stimulation type, as expected (Figure S2D). We aligned the datasets and corrected for donor-specific batch effects with canonical correlation analysis18 (Figure S2E).
Stimulation of CAR T Cells through Their CAR Yields a Weaker but Similar T Cell Activation Signal Compared to Stimulation via Their TCR
To investigate the main drivers of variation across different CAR T cell populations, we first performed principal-component analysis (PCA) of the bulk RNA-seq profiles, combined with linear modeling to account for donor variation. We observed a difference in transcriptional response following T cell activation according to the time and type of stimulation the T cell received (Figures 1D and S2F). TCR-activated CAR T cells grouped with TCR-activated UT cells and further away from the unstimulated cells on PC1 compared to CAR-activated CAR T cells. This is consistent with findings from mouse CAR T cells, where stimulation through the CAR versus TCR induces distinct gene expression signatures.19 The distinction between CAR T cells stimulated for 4 and 24 h through their CAR was less clear when looking at the CAR-activated groups, but when PCA was performed for each donor, there was a clear separation between the 4 and 24 h CAR-activated groups (Figure S2F). While samples primarily grouped according to the type of stimulation, samples within the stimulation groups separated secondarily by CD4+ versus CD8+ cell types (Figure 1E) but not by the type of CAR construct expressed (Figure 1F). The only exception was for Δζ CAR T cells stimulated with CD19-expressing targets, which grouped with unstimulated cells. This indicated that lentiviral transduction with Δζ CARs has little to no discernible effect on the T cell transcriptome and, therefore, serves as an appropriate negative control for CAR-mediated signaling.
To determine whether these distinctions stemmed from differences in the percentage of cells being stimulated as opposed to the degree or kind of stimulation within each cell, we turned to the scRNA-seq profiles. Overall, the bulk differences with T cell stimulation were also reflected in the single-cell profiles, suggesting an overall shift across the entire population rather than a change in the proportion of responding cells (Figure 1G). Furthermore, unstimulated UT and CAR T cells grouped together, indicating that the separation with activation is a result of the stimulation type rather than the CAR transduction. Similar to the bulk sequencing, T cells in the scRNA-seq analysis also separated by CD4+ and CD8+ cell types within the stimulation groups (Figure 1H). When pseudobulk CD4+ and CD8+ profiles were generated by averaging the respective scRNA-seq data and included with the true bulk RNA-seq samples in the PCA with correction for donor variation, the cells again separated on PC1 according to the type of receptor stimulated (CAR versus TCR; Figure S2F). Thus, TCR stimulation is distinct from CAR stimulation, and this held true whether the TCR was engaged by K562-αCD3 cells or αCD3/CD28 beads.
Next, we compared the genes expressed in TCR- and CAR-stimulated T cells versus unstimulated T cells. Comparing the top genes expressed across cells with the three stimuli, the genes specific to TCR-stimulated cells were known T cell activation genes (e.g., IFNG, IL3, and CCL4; Table S1). Genes specific to resting CAR and UT T cells were known resting T cell genes (e.g., IL7R and KLF2). However, CAR-stimulated CAR T cell genes were less clearly associated with T cell activation (with the exception of IL2RA). Nevertheless, 75% of the top 20 genes upregulated in CAR-activated CAR T cells were among the top 20 genes induced in TCR-stimulated cells (Table S2). We used the 75% of these genes that overlapped to define a T cell activation signature (Table S3). TCR-stimulated cells expressed the activation signature at an increased level compared to CAR-activated CAR T cells (Figure 1I; p value <10−16, Wilcoxon test), suggesting that CAR-activated CAR T cells have a weaker but similar response to activation as TCR-stimulated cells.
Resting CAR T Cells Have a Tonic CD3ζ Signaling Signature
Any receptor expressed on the surface of a cell has the potential to induce low levels of signaling that may lead to detectable transcriptional changes in the absence of triggers, particularly CARs that are constitutively driven by strong promoters. This ligand-independent signaling, otherwise known as “tonic signaling,” can be evident in the CAR T cells at rest, or before antigen stimulation through their CAR. We examined the difference among the resting CAR T cells to look for evidence of tonic transcriptional activity from the various CARs. Based on bulk sequencing profiles, we identified a set of genes that was significantly induced in all three T cell subsets with functional CARs compared to Δζ (Wald test, Benjamini-Hochberg false discovery rate [FDR] <0.1), whereas no genes were significantly differentially expressed between Δζ CAR and UT T cells. This gene set was identified by first compiling three lists of differentially expressed genes between ζ and Δζ, 28ζ and Δζ, and BBζ and Δζ in CD8 T cell samples. We then calculated the overlap of genes that existed in all three gene sets as the tonic CD3ζ signaling signature (Figure 2A). Many of the genes in this set followed the same expression trends in the CD4 T cell samples (although in some cases the genes did not have an FDR <0.1 in all three comparisons).
Figure 2.

Transcriptional Signatures of Resting CAR T Cells Indicate Tonic Signaling through Both CD3ζ and the Co-stimulatory Domain
(A) Heatmap of normalized expression from bulk RNA-seq of both the upregulated and downregulated genes that were differentially expressed (DE). FDR < 0.1 in all three functional CD19 CARs: 28ζ, BBζ, and ζ at rest in each case compared to Δζ at rest in either CD8 or CD4 T cells. Each column represents a different donor, and columns of heatmaps are always ordered as donors 1 to 3. (B) tSNE of scRNA-seq from all CAR T cells at rest, colored by the cluster assigned using a graph-based clustering approach. Assigned clusters were then defined based on the gene expression of several key genes (see also Figures S3H and S3I). (C) The same tSNE plot as in (B), depicting degree of CD8A and CD4 expression. (D) Violin plots of cells showing the degree of expression of SELL (CD62L) and CCR7 used to describe the clusters shown in (B). (E) Violin plots of cells as clustered in (B), showing degree of S phase and G2M phase signatures. (F) Of all the cells in a defined cluster, the percent contribution of each CAR to these cells is shown. (G) Graphical display showing the correlation matrix of the chi-square test residual values (difference between observed and expected values; see also Table S4).
Therefore, CARs bearing a functional CD3ζ chain had a specific, stimulation-independent, transcriptional signature at rest. This signature was enriched in genes involved in the response to cytokine stimuli (gene set enrichment analysis [GSEA]; FDR = 1.51 × 10−6), including CCL3 and CCL4, which are involved in monocyte recruitment, and GZMB (encoding granzyme B), a key cytotoxicity gene. Although tonic signaling of CARs has been reported before, it has mainly been attributed to differences in the binding characteristics of the extracellular portions of the CARs rather than signaling independently through the costimulatory domains.20,21 To determine whether this signature was dependent on CD3ζ expression rather than the scFv of the CAR, we validated this signature in EGFR-specific CAR T cells. Using digital droplet PCR of selected upregulated and downregulated signature genes, we found that EGFR-specific CAR T cells had a similar stimulation-independent transcriptional signature as the CD19-targeting cells (Figure S3A–S3G). Thus, the expression of a CAR in T cells modulates their transcriptional expression profiles even at rest, prior to CAR-antigen engagement, and this profile is independent of the antigen to which the CAR is specific. It also demonstrated that the 4-1BB-specific effects are present across different antigen/CAR combinations.
Evidence of Tonic Signaling through the 4-1BB Co-stimulation Domain
An additional contributing factor to understanding the effect of co-stimulation on CAR T cells is that there are multiple sub-populations of T cells present at homeostasis, including CD4+ and CD8+ T cells and their respective naive, effector, and memory subsets.22,23 The effects of co-stimulation could be different in the different populations or shift the make-up of these populations. To explore this factor, we leveraged the single-cell profiles to estimate the heterogeneity of UT and CAR T cells prior to antigen stimulation through their CAR. Since CAR T cells were resting, we could identify T cell sub-populations based on known surface markers, whereas once the T cells are activated, these markers are no longer able to differentiate sub-populations of T cells.24 Using a graph-based clustering approach, we clustered the resting T cells into nine groups (Figure 2B), annotated them by the expression of known T cell sub-population marker genes (CD4, CD8A, CCR7, and SELL [CD62L]; Figures 2C and 2D), and scored them for cell cycle gene signatures to rule out changes due to cell division (Figures 2E, S3H, and S3I). Among CD8 cells, we distinguished effector memory-like (cluster 2, CD8A+, CCR7−, SELL−) and central memory-like (cluster 3, CD8A+, CCR7+, SELL+) T cells. Within CD4 cells, we distinguished central memory-like (cluster 1, CD4+, CCR7+, SELL+), effector memory-like (cluster 0, CD4+, CCR7−, SELL−), and an intermediate population, which expressed CD62L but not CCR7 (cluster 7, CD4+, CCR7−, SELL+). When evaluating where the different CARs fell within the T cell sub-population clusters, only BBζ CAR T cells had a distribution across the clusters that was distinct from all other cells (p value < 2.2 × 10−16, χ2 test). Although cluster 4 was highly enriched in BBζ CAR T cells, cells in this cluster were actively dividing, with a high S phase gene score, making it hard to interpret their phenotype beyond dividing cells. Additionally, this cluster was only found in donor 4, whereas all other clusters contained cells from both donors. Therefore, this cell cluster was not further analyzed. In non-dividing cell clusters, the BBζ CAR T cells were enriched in CD8 central memory-like cells (cluster 3) and depleted among CD8 effector memory-like cells (cluster 2) and CD4 central memory-like cells (cluster 1; Figures 2F and 2G; Table S4). Previous studies showed that BBζ CAR T cells are enriched for CD8 central memory over effector memory after CAR stimulation.14 In this study, we found this enrichment present prior to stimulation. Thus, the 4-1BB co-stimulation domain may also have tonic signaling effects that are distinct from CD3ζ tonic signaling.
To further support the hypothesis of constitutive signaling from the 4-1BB co-stimulatory domain, we analyzed differentially expressed genes between BBζ and 28ζ bulk RNA-seq profiles in resting CAR T cells (Figure S4A). Recently, in vitro studies have demonstrated that several days after CAR activation, 28ζ CAR T cells have enhanced glycolytic metabolism, whereas BBζ CAR T cells rely more on fatty acid metabolism.14 We also found that fatty acid oxidation genes were enriched in BBζ versus 28ζ CAR T cells at rest (Figure S5). This further illustrates that signaling from the co-stimulatory domain, particularly 4-1BB, can modulate T cell programs independent of antigen stimulation.
Antigen Stimulation in BBζ CAR T Cells Results in Persistent Upregulation of an Activation Program Associated with Expression of MHC Class II Genes, but Not PD1
Given our finding of constitutive transcriptional changes occurring in CAR T cells, we next asked what transcriptional changes occur following antigen stimulation of CAR T cells and if transcriptional differences remain between the co-stimulatory domains (Figures 3A and S4A; Table S5). In the bulk RNA-seq, we found that many of the genes differentially expressed in BBζ versus 28ζ CAR T cells at rest were also differentially expressed following activation but were more pronounced. For example, activated BBζ CAR T cells were still enriched for fatty acid oxidation (Figures 3A and S5). Likewise, major histocompatibility complex class II (MHC class II) genes were upregulated in BBζ CAR T cells prior to stimulation, and this difference increased with activation. MHC class II genes and their regulators were among the most differentially expressed genes with activation when comparing BBζ to 28ζ CAR T cells (Figure 3B). We validated this finding at the protein level by flow cytometry of human leukocyte antigen (HLA)-DR expression in anti-CD19 CAR T cells at rest and after activation. HLA-DR was uniquely upregulated in BBζ CAR T cells compared to all other CAR T cells at rest and after activation (Figures 3C and 3D). This was not only true for the CD19-directed CAR T cells, but also for anti-EGFR T cells following activation (Figure 3E), suggesting that this differential expression in BBζ CAR T cells was independent of the antigen-binding domain of the CAR.
Figure 3.

CAR T Cells with the 4-1BB Co-stimulation Domain Have Persistent Upregulation of Markers Associated with Activation, Particularly MHC Class II Genes but Not PD1
(A) Volcano plots of DE genes between BBζ (positive x axis) and 28ζ CARs at 24 h post-CAR activation in CD4+ (left) and CD8+ (right) T cells. Genes with FDR <0.05 are colored red (see also Figure S4 for 0- and 4-h time points; see Table S5 for gene list). (B) Heatmap showing normalized HLA class II gene expression in CD4+ T cells from all three donors of 28ζ and BBζ CAR T cells 24 h post-CAR stimulation. (C–E) Mean fluorescence intensity (MFI) of HLA-DR surface expression measured by flow cytometry on CD19-CAR T cells (C) at rest or (D) 24 h after activation of CD19-specific CARs with Nalm6 cells at a 1:1 ratio, or (E) U87-mediated activation of EGFR-specific CAR T cells at a 2:1 effector-to-target cell ratio. (F) IL21R expression in bulk RNA-seq BBζ and 28ζ CAR T samples with CAR stimulation. Individual TPM values shown with mean and SEM; adj-p values were calculated by DESeq2 using a Holm-Bonferroni correction. (G) IL-21 cytokine levels measured in the supernatants of bulk CD4+/CD8+ CD19-CAR T cells stimulated for 24 h with Nalm6 cells at a 1:1 ratio or (H) EGFR-CAR T cells stimulated with U87 cells at a 2:1 effector-to-target cell ratio. (I) MFI of PD1 expression in CD19-CAR T cells with CAR stimulation (via Nalm6 cells, 1:1 effector-to-target cell ratio). (C–I) N = 3 normal donors that were not used for RNA-seq studies; mean and SEM plotted. (C–F) When required, p values were adjusted for multiple comparisons using a Holm-Bonferroni method adjustment (∗adj-p < 0.05, ∗∗adj-p < 0.01) or (G–I) p values were determined using a paired Student’s t test between BBζ and 28ζ (∗p < 0.05, ∗∗p < 0.01). See also Table S6.
To determine whether these genes were specific to the expression of 4-1BB, rather than the lack of CD28 in BBζ CAR T cells, we further compared the differentially expressed genes between BBζ and ζ CAR T cells. We found that many of the same genes upregulated in the BBζ versus 28ζ analysis were also upregulated in this analysis, suggesting that the presence of 4-1BB was inducing the genes’ expression (Figure S6). Additionally, this analysis revealed that immunosuppressive genes (interleukin [IL]-10) and pro-apoptotic genes (BCL2L11) were upregulated in the first-generation CAR (ζ) compared to BBζ, which highlights the role of costimulation in inhibiting apoptotic pathways.25
We also used these gene expression profiles to analyze the signaling pathways that were differentially regulated between activated BBζ and 28ζ CAR T cells that were not present in our tonic signaling analysis. We hypothesize that genes identified in this analysis would represent 4-1BB-specific genes that require a threshold of activation in order to be produced (i.e., baseline activation through tonic expression is not enough for them to be induced). In CAR-activated cells, there was upregulation of multiple cytokine and immune signaling pathways in BBζ CAR T cells compared to 28ζ CAR T cells (Figure S4B). The pathway containing the most differentially expressed genes was the “response to cytokine.” Among the specific cytokine pathways that appeared in the analysis were TNF-α and interferon (IFN)-γ signaling pathways. Genes in these pathways were upregulated after stimulation in BBζ CARs versus 28ζ (Table S6).
Another set of cytokine genes that were differentially expressed in activated BBζ and 28ζ CAR T cells were part of the IL-21 axis. This axis is particularly important for the formation of long-term memory CD8 T cell responses,26,27 so we examined differences in genes involved with this axis between CAR T cells. The cytokines and cytokine receptors IL21, IL21R, IL12RB2, and IL23R were upregulated in BBζ CARs versus 28ζ CARs, in both CD4+ and CD8+ T cells, by bulk RNA-seq (Figures 3F, S7A, and S7B). We confirmed these transcriptional findings at the protein level by measuring the amount of IL-21 produced following antigen stimulation. BBζ CAR T cells indeed produced more soluble IL-21 than 28ζ and ζ CARs in both anti-CD19 CARs (Figure 3G) and in anti-EGFR CAR T cells (Figure 3H).
Finally, we looked for the genes that were the most differentially expressed between BBζ and 28ζ CAR T cells across all time points. ENPP2 (encoding autotaxin) was the most significant upregulated gene between the two types of CAR T cells, induced in all BBζ samples at rest and following stimulation (Figure S7C). ENPP2 encodes a phosphodiesterase, which hydrolyzes lysophospholipids to produce lysophosphatidic acid and is involved in chemotaxis and proliferation of various cell types.28 Conversely, LGMN (encoding asparaginyl endopeptidase) and PDCD1 (encoding the PD1) were the most strongly suppressed genes in stimulated BBζ versus 28ζ CD4+ CAR T cells (Figure 3A). Although there were no genes suppressed in BBζ CD8+ CAR T cells that had an adjusted-p (adj-p) value <0.1 at 24 h, PDCD1 had one of the higher expression differences between 28ζ and BBζ CAR T cells (Figure S7D). PD1 is both an inhibitory receptor and a known marker of T cell activation that also suppresses CAR T cell function. We validated this finding using flow cytometry, confirming that surface expression of PD1 was lower in stimulated BBζ CAR T cells compared to 28ζ CAR T cells manufactured from multiple healthy human donors (Figure 3I). These data suggest that 4-1BB signaling prevents the induction of PD1 since PDCD1 was up on both ζ and 28ζ compared to BBζ (Figures 3A, 3I, and S6).
Antigen Stimulation of CARs Bearing the 4-1BB versus CD28 Co-stimulation Domains Induces a Th1 versus Th2 Polarization Program in CD4 T Cells
CD4 T cells can polarize to T helper (Th)1 or Th2, which causes them to produce different cytokines in response to stimulation. To determine whether CAR co-stimulatory domains differentially polarize CD4 T cells, we analyzed expression of Th1 and Th2 polarizing genes in CD4+ BBζ or 28ζ CAR T cells. We found that Th1 polarizing genes were enhanced in CD4+ BBζ CAR T cells after CAR stimulation and at rest (Figures 4A), whereas activated 28ζ cells were enriched for Th2 early polarizing genes29 (differentially expressed genes up in 28ζ; FDR < 0.1, GSEA: p value = 9.12e−9, FDR = 4.44e−5; Figure S7E). In particular, IL12RB2, which encodes a subunit of the IL-12 receptor and is upregulated in Th1 polarized cells,5,30 was upregulated in both 28ζ and BBζ cells at 4 h post-stimulation, but only BBζ cells maintained this higher expression at 24 h (Figure S7B). Other genes induced in CD4+ BBζ CAR T cells included the Th1 transcription factor (TF) genes EGR1 and TBX21 and the Th17 TF gene RORC (Figure 4B). When confirming these results at the protein level, CAR-activated 28ζ CAR T cells (both CD19-CAR and EGFR-CAR) consistently secreted higher levels of the Th2 cytokines IL-4 and IL-5 compared to BBζ CAR T cells (Figures 4C, 4D, and S7F). Thus, the type of CAR co-stimulatory domain plays a strong role in the cytokine polarization profile of CAR T cells, particularly after stimulation of the CAR.
Figure 4.

Antigen Stimulation of CARs Bearing the 4-1BB Co-stimulation Domain Results in Marked Th1 Polarization
(A) Gene set enrichment analysis of an early polarizing Th1 signature. Position of signature genes depicted in a rank fold-change list of the DE genes between RNA-seq BBζ and 28ζ profiles 24 h post-CAR activation with Nalm6 cells (see also Figure S7E). (B) Heatmap of known Th1 cell polarizing genes in CD4+ T cells from three donors in 28ζ versus BBζ CARs 24 h post-Nalm6 stimulation (see also Figure S7B). (C) IL-4 soluble cytokine detected in the supernatants of CD19-CAR T cells after 24 h of Nalm6 stimulation at a 1:1 ratio and in (D) EGFR-CAR T cells stimulated for 24 h with U87 cells at a 2:1 effector-to-target ratio measured by Luminex (see Figure S7F for IL-5). (C and D) N = 3 normal donors that were not used for RNA-seq studies. Mean and SEM plotted. p values were determined using a paired Student’s t test between BBζ and 28ζ. ∗p < 0.05.
Stimulation Activates a Range of Cellular Programs in Different CAR T Cells
To examine the spectrum of activated CAR T cell populations that underlie the global distinctions between BBζ and 28ζ CAR T cells that we observed via bulk sequencing, we turned to the scRNA-seq profiles. In this study, we observed separation of BBζ CAR T cells from the other activated CAR T cells, whereas profiles of 28ζ- and ζ-containing CAR T cells were similarly distributed (Figure 5A). We identified gene programs underlying these cellular distributions with a latent Dirichlet allocation (LDA), or “topic modeling,” approach. This unsupervised approach was recently used to analyze profiles of single immune cells,31 capturing the continuous nature of their variation and showing that different T cell subsets can activate similar programs. For instance, a CD4 T cell can have an effector memory program, a Th1 program, and a Th17 program during a particular antigen response.32 We followed the program analysis with a network analysis33 to associate certain programs with putative TF regulators with the aim of identifying mechanisms driving differences in CAR T cell behavior in all three activated CAR T cell profiles.
Figure 5.

Antigen-Specific Activation of 4-1BB CAR T Cells Induces a Distanced Program with Additional Genes Networks Than 4-1BB Ligand-Mediated Triggering of 4-1BB
Single-cell expression profiles of CAR T cells after 24 h of stimulation with Nalm6 cells were normalized and aligned across two donors. (A) tSNE plot of single-cell expression profiles (dots) colored by CAR T cell construct. (B) Results of latent Dirichlet allocation on T cells with 16 topics and a tolerance parameter of 0.1 (see Materials and Methods). For each topic shown, there is a bar plot of top-scoring genes (y axis), ranked by a uniqueness score. The genes in topic 11, which were also found as DE and upregulated (adj-p < 0.05) in BBζ versus 28ζ CAR T cells at any time point from bulk RNA-seq data, are in red. Each cell (dot) of the tSNE is colored by the weight of the topic (see also Figure S8 and Table S7). (C) Degree of CD4 and CD8A expression in cells plotted in the tSNE as in (A). (D) Gene regulators discovered using network analysis of the top 100 genes in each topic. Transcription factors with a statistical association (FDR < 0.1) were plotted by −log(pval) for each topic of interest (see also Table S8). (E) T cell activation signature expression level in cells plotted in the tSNE as in (A) (see Table S3 for activation signature). (F) Box-and-whisker plot and cumulative distribution plot of the topic 11 weights per cell across the different CAR T cell groups. (G) Network of predicted transcription factors identified and the genes they regulate in the 4-1BB program (topic 11). (H) UT T cells were stimulated for 24 h with irradiated K562 expressing αCD3 with or without 4-1BBL at a 1:1 effector-to-target cell ratio. Heatmap of 4-1BB CAR signature genes in UT T cells stimulated with and without 4-1BBL (normalized by row) gene signatures. N = 3 normal donors.
The programs we identified (referred to as “topics”) reflected the main facets of known T cell biology and varied among the different stimulated CAR T cells (Figures 5B and S8). A CD8 memory-specific program (topic 9) was clearly expressed in the CD8A-expressing cells (Figure 5C) and was highly enriched in 4-1BB CAR T cells (Figures S9 and S10). Genes encoding granzyme B and CD27 were two of the most significant contributors to this pathway. This was similar to what we observed in the bulk sequencing, where BBζ CAR T cells were enriched in the CD8 central memory phenotype (Figures 2F and 2G). A Th2 program (topic 8) scored highly in a small subset of ζ CAR T cells, and to a lesser extent 28ζ and BBζ CAR T cells, suggesting only a small subset of cells accounts for the Th2 response observed in 28ζ CAR T cells during bulk sequencing (p value <10−16 and 3.5 × 10−5 respectively, Kolmogorov-Smirnov [KS] test). When identifying TF regulators by network analysis, the Th2 program was associated with ETS1, a known regulator of Th2 cytokines34 (Figure 5D). A polyfunctional program of multiple cytokines and chemokines, including IFN-γ, IL-3, and IL-9 (topic 14), was expressed in cells with high activation signatures (from Figure 1I) and was enriched in ζ and 28ζ cells (Figures 5E, S9, and S10; Table S3; p value <10−16 and 1.21 × 10−10 respectively, KS test). This polyfunctional program was associated with nuclear factor κB (NF-κB) and nuclear factor of activated T cells (NFAT) TFs (Figure 5D). A cell proliferation program (topic 12) was associated with many known regulators of proliferation (Table S8) and contained cells from all three CAR T cell types. Cells scoring high for proliferation scored low for topics that included cytokine production, suggesting that proliferation and cytokine production are mutually exclusive. Topic 16 was identified to be a Th17-like program including IL17RB, IL17F, and IL22 as well as IL10 and associated with STAT1, STAT3, and SMAD1 as its regulators. Topic 16 was slightly enriched in ζ and 28ζ cells compared to BBζ cells (Figures S9 and S10).
Antigen-Specific Activation of 4-1BB CAR T Cells Induces a Distinct Program Shared in CD4 and CD8 Cells
In the program analysis, topic 11 was highly specific to BBζ CAR T cells (Figure 5F; p value <10−16, KS test). Many of its top-scoring genes corresponded to our previous observations of differentially expressed genes between BBζ and 28ζ activated CAR T cells (Figure 5B; Table S7), including HLA class II genes, CCR7, ENPP2, the TF BATF3, and the TNF receptor-associated signaling molecule TRAF1 (Table S7), which were also identified during the bulk sequencing analysis (Figures 3A and S4). HLA class II genes (HLA-DR) were also previously confirmed by flow cytometry (Figure 3C). The physiological significance of this upregulation is not certain at this time. Similar to our results, CCR7 has been well described to be induced by 4-1BB signaling.14 CCR7 has an impact on the formation of memory T cells.35,36 TRAF1 is thought to directly bind to the 4-1BB intracellular domain during signaling, resulting in a positive feedback loop.37 A network analysis of topic 11 predicted NF-κB TFs and TNFAIP3 (A20)38,39 as the regulators of this program (Figure 5G). IL-21 was also present in the top 200 genes distinguishing topic 11 (Table S7), which we previously showed was also upregulated at the protein level (Figure 3G), and is known to modulate memory and effector T cell phenotypes.27,40
Finally, to determine whether the genes induced by 4-1BB-containing CARs were also induced after endogenous 4-1BB signaling,41 we used the top genes defining topic 11 that were also differentially expressed in at least one time point in BBζ versus 28ζ CAR T cells to define a 4-1BB gene signature. We then scored this signature in UT cells activated via their TCR and the 4-1BB ligand (4-1BBL) compared to their TCR alone. We generated a K562 cell line expressing a membrane-bound anti-CD3 scFv with or without 4-1BBL expression. As before, we expanded UT T cells with beads for 7 days, rested them for 7 days, activated them for 24 h with irradiated K562-anti-CD3 or K562-anti-CD3/4-1BBL, and measured the 4-1BB signature expression with nCounter. Although many of the genes in our 4-1BB signature were induced by endogenous 4-1BB signaling, a subset was unique to stimulated CAR T cells, including all the MHC class II genes and ENPP2 (Figure 5H). We hypothesize that 4-1BB signaling from the synthetic CAR molecule is strong enough to induce programs that would otherwise not be induced in T cells, such as the MHC class II molecules known to be upregulated after dendritic cell (DC) maturation using 4-1BB/4-1BBL stimulation.42,43 This may also be in part due to constitutive expression of the 4-1BB intracellular signaling domain when it is part of the CAR, in contrast to the activation-dependent expression of the natural 4-1BB molecule.
Discussion
In this study, we generated a transcriptional atlas of human CAR T cells at rest and following activation to understand tonic and antigen-dependent signaling mediated by the intracellular domains of CARs. We found that CAR T cells have ligand-independent signaling, prior to stimulation through their CAR, that varies based on the co-stimulatory domain expressed. It is possible that this constitutive signaling present with baseline CAR expression, along with signaling changes that occur following CAR activation, could impact CAR T cell phenotype, cell fate, and persistence depending on the co-stimulatory domain that is used. The differences observed in this study will help to develop hypotheses behind the clinical differences observed in 4-1BB- and CD28-containing CARs, as well provide guidance on how CAR T cells might be endowed or genetically modified to enhance desirable properties in the future.
This work used two methods of sequencing with unbiased analyses to determine the phenotypes of CAR T cells associated with different CAR intracellular signaling domains. In both cases, we used genes strongly expressed in cell clusters (Figure 2) or topics (Figure 5) to assign phenotypes or properties to the cells based on the genes’ known functions. By analyzing CAR T cells at rest, we were able to identify memory T cell populations along with tonic signaling events specific to the CAR’s intracellular signaling domain (Figure 2). These distinctions were made even clearer when analyzing activated CAR T cells (Figures 3, 4, and 5). Many of the genes identified as differentially expressed between BBζ and 28ζ in the bulk RNA-seq and confirmed at the protein level (Figure 3) overlapped with genes in the scRNA-seq topic that was unique to BBζ cells (Figure 5, topic 11, genes in red). Additionally, the Th cell polarization identified by bulk RNA-seq (Figure 4) was similarly discerned in the topic analysis (Figure 5, topic 8).
Previous studies that identified tonic signaling in CAR T cells used CARs with different extracellular moieties that induced dramatic functional changes in the T cell. These studies found that c-Met and GD2-directed CAR T cells have ligand-independent signaling during CAR T cell expansion, which results in changes in proliferation, cytokine production, and induced T cell exhaustion.20,21 In these studies, the CARs were undergoing clustering due to interactions between their extracellular domains, which was inducing downstream signaling and measurable functional changes that did not occur in CD19 CAR T cells. Therefore, it was hypothesized that scFvs with different specificities can induce tonic signaling, but that this does not occur in CD19 CAR T cells.
Similar to these studies, we did not find profound functional changes in CD19 CAR T cells that are not activated.44 To our knowledge, previous studies have not assessed the transcriptional effects of constitutive CAR expression in resting human T cells. However, we hypothesized that every CAR induces some baseline signaling changes simply from being expressed in a cell, even if it does not result in dramatic functional changes prior to activation. To test this hypothesis, we chose to examine transcriptional changes with CAR expression in human T cells expressing functional and non-functional CD3ζ domains prior to CAR-induced activation. This stringent control allowed us to identify the presence of ligand-independent activity at the transcriptional level from baseline CAR expression, and we observed that the profile was present in all CAR T cells transduced with a functional CD3ζ domain compared to the truncated Δζ domain. Although these changes were weaker than the strong functional changes observed in c-Met and GD2 CARs, they became more pronounced with CAR activation, suggesting the differences observed with ligand-independent signaling can prime the cell for the changes that occur after activation. This baseline signaling could have meaningful implications as to how specific CAR constructs will behave in patients, prior to the CAR T cell interacting with its antigen on the tumor and triggering activation.
We also identified a ligand-independent signaling signature that was specific to 4-1BB-expressing CARs both at the population and the single-cell level. At rest, 41BB cells had increased markers associated with CD8 central memory T cells and favored fatty acid metabolism, similar to what has been shown previously in 4-1BB CAR T cells following activation.14 These two characteristics have also been attributed to the increased proliferation and persistence of BBζ CAR T cells compared to CD28ζ.14 Since we found these differences in resting CAR T cells, it suggests that the degree of 4-1BB tonic signaling in CAR T cells could have profound effects on the heterogeneity and functional state of the cells even before they are administered to a patient.
Similarly, genes that differentially changed with CAR T cell activation could be responsible for differences in 4-1BB CAR T cell persistence. Following activation, we recovered a unique gene program induced in 4-1BB CAR T cells, including MHC class II genes ENPP2, CXCL10, and CCR7 and the TFs BATF3 and JUNB. ENPP2 promotes proliferation in other cell types28 and may have a similar effect in CAR T cells, although this hypothesis remains to be explored. The predicted regulators of the 4-1BB signature were NF-κB family members and A20. Tonic signaling from NF-κB has been found to be critical for both T and B cell longevity;45,46 therefore, tonic signaling from the 4-1BB co-stimulation domain in CAR T cells may similarly promote the increased persistence seen in BBζ CAR T cells. This extends on recent findings showing the NF-κB is necessary for 4-1BB co-stimulatory enhancement of CAR T cell function and that non-canonical NF-κB activation by 4-1BB promotes CAR T cell survival.47,48
The brisk upregulation of MHC class II on T cells with 4-1BB-containing CARs was much higher than after activation of CARs that did not contain 4-1BB (i.e., 28ζ and ζ). The physiologic relevance of this finding is unclear and is unlikely to have an impact on patients receiving 4-1BB-based CAR T cell products, since CAR T cells would be a miniscule population of the total antigen-presenting compartment. Alternatively, we speculate that if CAR T cells were to be combined with tumor vaccines, 4-1BB-based CARs may enhance epitope spreading via increased antigen presentation. Furthermore, investigators and sponsors generating allogeneic “off-the-shelf” CAR T cells may want to consider whether MHC class II upregulation could result in increased rejection of CAR T cells.
Other aspects of these CAR T cells that could affect their phenotypes in patients include differences in cytokines and cytokine receptors. For example, IL21 and IL21R were upregulated in BBζ CAR T cells. IL-21 secretion from CD4+ cells is known to support the formation of memory CD8+ T cells, which could be important in the production of a lasting anti-tumor CD8+ immune response49 and may further explain the increased persistence of BBζ CARs. Additionally, IL12RB2, which encodes a subunit of the IL-12 receptor, was significantly upregulated in BBζ CARs. IL-12 is a pro-inflammatory cytokine that increases the anti-tumor potency of CAR T cells.50 IL-12-secreting CARs with CD28 co-stimulatory domains are being developed by various groups,50 but these data suggest that BBζ CARs may be more sensitive to the additional IL-12.
Following stimulation, we also identified Th1 polarizing genes in BBζ CAR T cells by bulk sequencing, reflecting a shift across the entire cell population. In contrast, 28ζ CAR T cells induced Th2 polarizing genes by bulk sequencing that was reflective of a minor subset of cells at the single-cell level, as identified by topic 8 in the topic analysis. Additionally, one of the key genes distinguishing the BBζ-specific topic (topic 11) was TRAF1, a known inhibitor of Th2 differentiation.51 This is similar to observations made in CARs directed toward the solid tumor antigen GPC3, where BBζ was associated with Th1 cytokines and CD28ζ was associated with Th2.52 This difference is important because polarization affects the types of cytokines released from T cells, which can mediate toxicities that occur with CAR T cell treatment, such as cytokine release syndrome (CRS). Therefore, the difference in cytokine profiles based on the CAR’s co-stimulatory domain may help refine the treatment of CRS based on the CAR product causing the syndrome. Furthermore, Th1 CD4+ T cells are known to be important for CD8+ T cell activity and are required for their anti-tumor activity;50,53 therefore, a skew toward the Th1 phenotype could support CAR T cell efficacy.
Many, but not all, of the genes in our CAR 4-1BB program were also upregulated following endogenous 4-1BB signaling in UT T cells; a subset was only induced when 4-1BB is activated in the context of a CAR co-stimulatory domain. This highlights the importance of studying these costimulatory programs in the context of a CAR construct, which may induce additional signaling pathways that provide additional functions to BBζ CAR T cells, such as antigen presentation through MHC class II. The role that MHC class II genes have in CAR T cell function remains to be explored but is intriguing since it was identified in both the bulk and single-cell sequencing and these genes among the most significantly differentially expressed between BBζ and 28ζ CAR T cells at multiple time points.
Profiling of stimulated CAR T cells showed that while the same genes are upregulated following TCR and CAR activation, TCR signaling induces far stronger induction for the whole population and at the cell-intrinsic level. This is consistent with other studies that have demonstrated that CARs have poorer organization of the supramolecular activation cluster (SMAC) and fewer immunoreceptor tyrosine-based activation motifs (ITAMs) of the CD3ζ chain compared to a full TCR complex.54 However, one caveat in the interpretation of these data is that we used anti-CD3, a highly potent activator of the TCR, which binds with higher affinity than typical MHC-peptide binding.
One limitation to our study is that we focused on costimulation domains of CAR constructs that bear resemblance to those in clinical use, but do not represent commercial therapeutic products. For example, our 28ζ CAR used a different vector and promoter than axicabtagene ciloleucel, and the transmembrane domain was derived from CD8a instead of CD28. Our BBζ construct design closely resembled the tisagenlecleucel transgene. We performed the analyses on CAR T cells that were manufactured using one process, on a small scale in plates, and in a research laboratory, not using a large-scale manufacturing process in good manufacturing practice (GMP) facilities. To better understand the specific role of the costimulation domains CD28 versus 4-1BB, we kept all of the other components of our CAR T cells identical, in contrast to commercial products, which use different manufacturing processes and have some variations in the transgene design and vectors used.
Taken together, our data expand our understanding of how CAR signaling domains affect the gene expression profile, functional state, and ultimate fate of engineered human T cells. They also highlight unexpected differences between endogenous TCR and 4-1BB signaling compared to 4-1BB signaling through a CAR. This information will enhance CAR therapies by providing greater insight into the selection and engineering of costimulatory domains that lead to changes in gene expression. Knowing that these differences can be detected even prior to the stimulation through the CAR provides a rationale for understanding a specific CAR’s signaling phenotype at baseline, prior to administration to the patient. Designing CARs with specific signaling domains to achieve a desired function could make CAR T cells more effective in specific cancers where a particular profile would be beneficial.
Materials and Methods
Generation of CAR Lentiviral Vectors and Isolation of Human T Cells
CD19- and EGFR-specific CARs were synthesized and cloned into a third-generation lentiviral plasmid backbone under the regulation of a human EF-1α promoter (GenScript, Piscataway, NJ, USA). Replication-defective lentiviral vectors were produced by four plasmids co-transfected into human embryonic kidney cell line 293 (HEK293T) cells using TransIT-2020 transfection reagent (Mirus Bio, Madison, WI, USA). Supernatants were collected 24 and 48 h after transfection and filtered. Virus was concentrated by ultracentrifugation. Vector was harvested and stored at −80°C. Healthy donor leukopaks were obtained from the Blood Transfusion Services at Massachusetts General Hospital under an institutional review board (IRB)-approved protocol to obtain discarded tissues. CD4+ and CD8+ T cells were negatively selected using RosetteSep kits with a Ficoll gradient (STEMCELL Technologies, Vancouver, BC, Canada). For the bulk RNA-seq experiments, enriched T cells were further purified by CD3+CD4+ or CD3+CD8+ using fluorescence-activated cell sorting (FACS) with a BD FACSAria III (BD Biosciences, San Jose, CA, USA). CD4+ and CD8+ T cells were mixed at a 1:1 ratio prior to expansion. For single-cell experiments, T cells were used at the donor-specific CD4/CD8 ratio.
Target Cell Lines
The HEK392T and Nalm6 cell lines were purchased from American Tissue Culture Collection (ATCC, Manassas, VA, USA). Both cell lines were expanded in RPMI 1640 with 1× l-GlutaMAX and 25 mM HEPES (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific). The U87 cell line was from ATCC and expanded in Eagle’s minimal essential medium (EMEM) with 10% FBS. U87 cells were passaged with 0.05% trypsin (Gibco, Thermo Fisher Scientific). Target cells were irradiated with 10,000 rad and frozen in FBS with 10% DMSO to be thawed prior to stimulation of CAR T cells. K562 cells were transduced to express anti-CD3 scFv and 4-1BBL as described previously.55
T Cell In Vitro Expansion and Transduction for RNA-Seq
Bulk RNA-seq samples were collected from T cells of three donors stimulated under the different conditions. T cells were plated in a 24-well plate at 1 million cells/mL in RPMI 1640 with 1× l-GlutaMAX and 25 mM HEPES (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS and IL-2 (20 IU/mL; PeproTech, Rocky Hill, NJ, USA). Anti-CD3/CD28 beads (Dynabeads; Invitrogen, Carlsbad, CA, USA) were added with a 3:1 bead-to-cell ratio. T cells were cultured for 1 day and then transduced with one of the four lentiviral constructs at an MOI of 5. Cells were counted and maintained at 5e5/mL with IL-2 replaced every 2 days during the bead expansion and resting periods. Beads were removed on day 7 using a magnet, and cells were rested for a further 7 days. T cells were checked for mCherry expression and purity by flow cytometry analysis on day 13. On day 14, each CAR T cell population was divided into five wells at 5e5 CAR T cells/mL in a six-well plate and left unstimulated or stimulated with Nalm6 (at a 1:1 effector-to-target ratio) or anti-CD3/CD28 beads (1:1 T cell-to-target/bead ratio) for 4 or 24 h prior to staining and sorting. Cells were stained with CD3-FITC (UCHT1; BioLegend, San Diego, CA, USA), CD4-Brilliant Violet 786 (BV786) (SK3; BD Biosciences), CD8-allophycocyanin (APC)-H7 (SK1; BD Biosciences), and CD69-APC (FN50; BioLegend). DAPI was added before FACS. Technical duplicates of 5,000 cells were sorted on CD3+, CD4+ or CD8+, and mCherry+ with a MoFlo Astrios EQ cell sorter (Beckman Coulter, Indianapolis, IN, USA) and resuspended in lysis buffer to be bulk sequenced at the Broad Institute.
For single-cell RNA-seq, samples were generated from the same three donors as the bulk RNA plus two new donors (donor 4 and donor 5). The same CAR T cell subsets were generated except that the Δζ construct was not included in the experiment, and cells were either left unstimulated or stimulated for 24 h with irradiated Nalm6 cells prior to sorting. In addition to Nalm6 cell stimulation, the UTD sample was also stimulated for 24 h with K562 cells expressing anti-CD3 scFv. Both types of stimulation were at a 1:1 effector T cell/target cell ratio. Following stimulation, T cells were sorted on a CD4+ or CD8+ gate and mCherry+. All functional validation studies were performed on CAR T cells generated from additional normal donors, as described in the figure legends.
Flow Cytometry
All antibodies were purchased from BioLegend unless otherwise stated. The following antibodies were used: HLA-DR-Pacific Blue (PacBlue) (clone L243), CD4-FITC (clone OKT4), CD8-APC-H7 (clone SK1, BD Pharmingen), and PD1-Brilliant Violet (BV711) (clone EH122H7). Cells were stained for 30 min in the dark at 4°C and washed twice in PBS with 2% FBS. DAPI was added for cell viability directly before running flow. We collected events on a LSRFortessa X-20 (BD Biosciences) and analyzed the data with FlowJo software (Tree Star, Ashland, OR, USA).
Bulk RNA-Seq
CAR T cells were collected using a FACS machine, resuspended at 200 cell/μL in lysis buffer comprised of TCL buffer (1031576; QIAGEN, Hilden, Germany) plus 1% 2-mercaptoethanol (63689; Sigma-Aldrich, St. Louis, MO, USA) and immediately frozen at −80°C.
For preparation of RNA-Seq libraries, cells were thawed (1,000 cells per sample) and purified with 2.2× RNAClean SPRI beads (Beckman Coulter) without a final elution.56 RNA capture beads were air-dried and processed immediately for cDNA synthesis. A SMART-Seq2 protocol was carried out as previously described57 with minor modifications in the reverse transcription step.58 Each PCR was performed in a 25-μL reaction with 15 cycles for cDNA amplification. We used 0.25 ng of cDNA of each sample and one-fourth of the standard Nextera XT reaction volume in both the fragmentation and final PCR amplification steps. Up to 30 libraries were pooled per one NextSeq run (∼500 million reads) and sequenced 50 × 25 paired-end reads using a single kit on a NextSeq 500 instrument (Illumina, San Diego, CA, USA).
Bulk RNA-Seq Initial Read Alignment and QC
BAM files from bulk RNA-seq were converted to merged, demultiplexed FASTQ files. Reads were mapped to the UCSC hg19 human transcriptome using Bowtie.59 and transcripts-per-million (TPM) values were calculated with RSEM v1.2.8 in paired-end mode.60
Samples passed quality control (QC) if the number of aligned reads was greater than 107, the percent of reads mapped was above 30%, and the percent of rRNA was in the range of 10%–30%. We calculated the correlation between technical duplicates and further interrogated any duplicates with an r2 <0.9. Duplicates were then averaged for downstream analysis.
Single-Cell RNA-Seq
CAR T cells were sorted into PBS with 0.04% BSA, and live cells were processed directly for droplet-based 3′ end parallel scRNA-seq. We used the 10x Genomics chromium single cell 3′ library & gel bead kit v2 according to the manufacturer’s protocol (10x Genomics, Pleasanton, CA, USA). An input of 10,000 T cells was added to each 10× channel with a median recovery of 4,051 cells. scRNA-seq libraries were sequenced on an Illumina HiSeq (132 bp reads) at the Broad Institute Genomics Platform.
scRNA-Seq Initial Data Processing and QC
Gene counts were obtained by aligning reads to the human genome GRCh38 using CellRanger software (v2.2) (10x Genomics, single cell 3′ v2)61. The scCloud pipeline62 was used for an overview analysis and to create a Seurat_h5ad file that could be loaded into R as a Seurat object for further QC and analysis. Seurat pipeline version 2.3.418 was used to remove poor quality cells (200 < number of unique molecular identifiers [nUMI] < 6,000 and ribosomal RNA <10% of the reads). Reads were normalized using Seurat’s normalization function based on log normalization with a scale factor of 10,000. For each analysis (all samples, resting samples, or CAR-stimulated samples) samples were selected and batch corrected for donor variation. Seurat’s FindVariableGenes() function was used to determine the variable genes. Data were scaled using the ScaleData() function in Seurat using the identified variable genes.
Batch Correction, Dimensionality Reduction, Clustering, and Visualization
The union of the top 1,000 most variable genes from the samples in each donor was used to run canonical correlation analysis (CCA) using the RunCCA() function in Seurat. The donors’ CCA subspaces were aligned using the cca.aligned() dimensional reduction function in Seurat, which was then used to run tSNE and for clustering purposes with the following parameter settings: reduction.use = “cca.aligned,” dims.use = 1:20, do.fast = T. Clustering was performed using Seurat function FindClusters() with parameters: reduction.type = “cca.aligned,” resolution = 0.6, dims.use = 1:20. Subsequent analysis confirmed that our results were not sensitive to the exact number of dimensions chosen. Violin plots were calculated using Seurat’s VlnPlot() function on normalized gene expression values or signature scores.
G2M and S phase gene signatures were calculated using Seurat’s CellCycleScoring() function, which calculates gene scores using a set of defined S phase and G2M phase genes.63 Other scores from gene signatures defined were calculated by the methods described previously.64 Individual gene expression differences were calculated as average log fold change, and p values were calculated using a Wilcoxon rank sum test.
In Figure 2D, to bin the level of CD62L (SELL) expression x of a cluster, we used the average gene expression log2 fold change (logFC) with respect to the cluster with the highest CD62L expression y (cluster 3). CD62Lhigh (log2(x/y) < 0.25), CD62Lmid (0.25 < (log2(x/y) < 0.5 and adj-p < 1e−50, Wilcoxon test), and CD62Llow (log2(x/y) > 0.5, adj-p < 1e−50, Wilcoxon test)
Topic Modeling
We fit an LDA topic model on the sparse matrix from the CAR-stimulated samples that included all genes as described.31 We calculated topics for set K number of topics ranging from 4 to 20 with the tolerance parameter (Tol) set to 0.1. We chose K to be 16 by calculating the Akaike and Bayesian information criteria for each K tested, and we set K as the value where the Akaike information criterion (AIC) curve changed from a steep gradient to a more gradual one. The “top n genes” for a topic were generated by the CountClust function ExtractTopFeatures(), which selects genes critical for separating one topic from the others with the following parameters: top_features = n, method = “poisson,” options = “min,” shared = TRUE. Empirical cumulative distribution function of the topic weights was calculated as previously described.65
Network Analysis
TF binding targets were downloaded from the RegNetwork database.33 TFs were associated with each topic by testing for the hypergeometric enrichment of the TF-binding targets in the top 100 genes contributing to that topic (identified by the function ExtractTopFeatures() described under Topic Modeling). We report TFs showing statistically significant association (FDR < 0.1, Benjamini-Hochberg correction).
4-1BBL-Endogenous 4-1BB Stimulation Experiment
Pan T cells were expanded UT as described above with anti-CD3/CD28 beads and IL-2. Cells were frozen on day 14 and thawed 1 day before re-stimulation into in R10 supplemented with IL-2 (20 IU/mL). UT cells were co-cultured with irradiated K562-GFP-OKT3 or K562-GFP-OKT3-4-1BBL at a 1:1 3e6 cells/6 mL in a six-well plate in R10. Co-cultures were performed with three normal donors in technical duplicates during 24 h, after which they were washed and resuspended in sorted buffer with DAPI. Samples were sorted with 100,000–200,000 cells per sample from live, GFP− cells. RNA from sorted T cells was isolated using the QIAGEN RNeasy Plus micro kit and loaded on the NanoString nCounter SPRINT instrument according to the manufacturer’s instructions with the CAR T panel plus additional 4-1BB signature probes (NanoString, Seattle, WA, USA). Data were analyzed using nCounter software (NanoString). The 4-1BB signature gene set was defined as those genes that were both in the top 100 feature genes of topic 11 and differentially expressed by RNA-seq during at least one time point between BBζ and 28ζ CAR T cells.
Statistical Analysis
Data are presented as means ± SEM as stated in the figure legends. Unless specifically indicated, comparison between different groups was conducted with a two-tailed, paired Student’s t tests. Unless otherwise stated, p values below 0.05 were considered significant. If adjustments for multiple comparisons were needed, they were performed using the Holm-Bonferroni method, with adj-p <0.05 considered significant. Statistical analysis was performed with Prism (GraphPad, San Diego, CA, USA), and Holm-Bonferroni adj-p and chi-square distribution were calculated with R software package (Thermo Fisher Scientific). A Wilcoxon rank sum test was used to calculate p values for single-cell gene expression data with the R software package.
Author Contributions
A.C.B., N.D.M., R.C.L., A.R., O.R.-R., and M.V.M. designed the study. A.C.B., N.D.M., R.C.L., A.P.C., L.S.R., and S.J.L. performed experiments. A.C.B., N.D.M., K.G., C.B.M.P., O.A., G.S.-R., J.G., S.J.R., B.D.C., R.M., and L.J. analyzed data. A.C.B., T.R.B., A.R., and M.V.M. wrote the manuscript. All authors edited and approved the manuscript.
Conflicts of Interest
A.R. is a member of the scientific advisory board of Cambridge EpiGenetix. M.V.M. is a co-founder of Century Therapeutics and holds equity in the company; M.V.M. also has equity in TCR2 and is a consultant for multiple CAR T cell companies. The remaining authors declare no competing interests.
Acknowledgments
We would like to thank Leslie Gaffney and Jennifer Rood for help in manuscript preparation, as well as the Broad Flow Cytometry Facility and the Flow Cytometry Core. M.V.M. was supported by NIH CA166039, the Gabrielle's Angel Foundation, and V Foundation. A.C.B. was supported by NIH 5T32AI118692-02. We thank the Klarman Cell Observatory for sequencing support.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.07.023.
Contributor Information
Aviv Regev, Email: aregev@broadinstitute.org.
Marcela V. Maus, Email: mvmaus@mgh.harvard.edu.
Supplemental Information
References
- 1.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer J.N., Dudley M.E., Kassim S.H., Somerville R.P., Carpenter R.O., Stetler-Stevenson M., Yang J.C., Phan G.Q., Hughes M.S., Sherry R.M. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamza T., Barnett J.B., Li B. Interleukin 12 a key immunoregulatory cytokine in infection applications. Int. J. Mol. Sci. 2010;11:789–806. doi: 10.3390/ijms11030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K., Chung S.S., Stefanski J., Borquez-Ojeda O., Olszewska M. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak Ö., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., JULIET Investigators Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 12.van der Stegen S.J., Hamieh M., Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Guedan S., Posey A.D., Jr., Shaw C., Wing A., Da T., Patel P.R., McGettigan S.E., Casado-Medrano V., Kawalekar O.U., Uribe-Herranz M. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018;3:e96976. doi: 10.1172/jci.insight.96976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paulos C.M., Carpenito C., Plesa G., Suhoski M.M., Varela-Rohena A., Golovina T.N., Carroll R.G., Riley J.L., June C.H. The inducible costimulator (ICOS) is critical for the development of human TH17 cells. Sci. Transl. Med. 2010;2:55ra78. doi: 10.1126/scitranslmed.3000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong X.S., Matsushita M., Plotkin J., Riviere I., Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y., Kohler M.E., Chien C.D., Sauter C.T., Jacoby E., Yan C., Hu Y., Wanhainen K., Qin H., Fry T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017;9:eaag1209. doi: 10.1126/scitranslmed.aag1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frigault M.J., Lee J., Basil M.C., Carpenito C., Motohashi S., Scholler J., Kawalekar O.U., Guedan S., McGettigan S.E., Posey A.D., Jr. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taipale K., Liikanen I., Juhila J., Karioja-Kallio A., Oksanen M., Turkki R., Linder N., Lundin J., Ristimäki A., Kanerva A. T-cell subsets in peripheral blood and tumors of patients treated with oncolytic adenoviruses. Mol. Ther. 2015;23:964–973. doi: 10.1038/mt.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jameson S.C., Masopust D. Understanding subset diversity in T cell memory. Immunity. 2018;48:214–226. doi: 10.1016/j.immuni.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farber D.L., Yudanin N.A., Restifo N.P. Human memory T cells: generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2014;14:24–35. doi: 10.1038/nri3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 26.Xin G., Schauder D.M., Lainez B., Weinstein J.S., Dai Z., Chen Y., Esplugues E., Wen R., Wang D., Parish I.A. A critical role of IL-21-induced BATF in sustaining CD8-T-cell-mediated chronic viral control. Cell Rep. 2015;13:1118–1124. doi: 10.1016/j.celrep.2015.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loschinski R., Böttcher M., Stoll A., Bruns H., Mackensen A., Mougiakakos D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget. 2018;9:13125–13138. doi: 10.18632/oncotarget.24442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cholia R.P., Nayyar H., Kumar R., Mantha A.K. Understanding the multifaceted role of ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) and its altered behaviour in human diseases. Curr. Mol. Med. 2015;15:932–943. doi: 10.2174/1566524015666150921104804. [DOI] [PubMed] [Google Scholar]
- 29.Lund R., Aittokallio T., Nevalainen O., Lahesmaa R. Identification of novel genes regulated by IL-12, IL-4, or TGF-β during the early polarization of CD4+ lymphocytes. J. Immunol. 2003;171:5328–5336. doi: 10.4049/jimmunol.171.10.5328. [DOI] [PubMed] [Google Scholar]
- 30.Athie-Morales V., Smits H.H., Cantrell D.A., Hilkens C.M. Sustained IL-12 signaling is required for Th1 development. J. Immunol. 2004;172:61–69. doi: 10.4049/jimmunol.172.1.61. [DOI] [PubMed] [Google Scholar]
- 31.Bielecki P., Riesenfeld S.J., Kowalczyk M.S., Amezcua Vesely M.C., Kroehling L., Yaghoubi P. Skin inflammation driven by differentiation of quiescent tissue-resident ILCs into a spectrum of pathogenic effectors. bioRxiv. 2018 doi: 10.1101/461228. [DOI] [Google Scholar]
- 32.Geginat J., Paroni M., Maglie S., Alfen J.S., Kastirr I., Gruarin P., De Simone M., Pagani M., Abrignani S. Plasticity of human CD4 T cell subsets. Front. Immunol. 2014;5:630. doi: 10.3389/fimmu.2014.00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z.-P., Wu C., Miao H., Wu H. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database (Oxford) 2015;2015:bav095. doi: 10.1093/database/bav095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strempel J.M., Grenningloh R., Ho I.C., Vercelli D. Phylogenetic and functional analysis identifies Ets-1 as a novel regulator of the Th2 cytokine gene locus. J. Immunol. 2010;184:1309–1316. doi: 10.4049/jimmunol.0804162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moschovakis G.L., Förster R. Multifaceted activities of CCR7 regulate T-cell homeostasis in health and disease. Eur. J. Immunol. 2012;42:1949–1955. doi: 10.1002/eji.201242614. [DOI] [PubMed] [Google Scholar]
- 36.Jung Y.W., Kim H.G., Perry C.J., Kaech S.M. CCR7 expression alters memory CD8 T-cell homeostasis by regulating occupancy in IL-7- and IL-15-dependent niches. Proc. Natl. Acad. Sci. USA. 2016;113:8278–8283. doi: 10.1073/pnas.1602899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zapata J.M., Perez-Chacon G., Carr-Baena P., Martinez-Forero I., Azpilikueta A., Otano I., Melero I. CD137 (4-1BB) signalosome: complexity is a matter of TRAFs. Front. Immunol. 2018;9:2618. doi: 10.3389/fimmu.2018.02618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L., Soetandyo N., Wang Q., Ye Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim. Biophys. Acta. 2009;1793:346–353. doi: 10.1016/j.bbamcr.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shembade N., Ma A., Harhaj E.W. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J., Hasan F., Frey A.C., Li H.S., Park J., Pan K., Haymaker C., Bernatchez C., Lee D.A., Watowich S.S., Yee C. Histone deacetylase inhibitors and IL21 cooperate to reprogram human effector CD8+ T cells to memory T cells. Cancer Immunol. Res. 2020;8:794–805. doi: 10.1158/2326-6066.CIR-19-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maus M.V., Thomas A.K., Leonard D.G.B., Allman D., Addya K., Schlienger K., Riley J.L., June C.H. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat. Biotechnol. 2002;20:143–148. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 42.Kim Y.J., Li G., Broxmeyer H.E. 4-1BB ligand stimulation enhances myeloid dendritic cell maturation from human umbilical cord blood CD34+ progenitor cells. J. Hematother. Stem Cell Res. 2002;11:895–903. doi: 10.1089/152581602321080556. [DOI] [PubMed] [Google Scholar]
- 43.Macdonald D.C., Hotblack A., Akbar S., Britton G., Collins M.K., Rosenberg W.C. 4-1BB ligand activates bystander dendritic cells to enhance immunization in trans. J. Immunol. 2014;193:5056–5064. doi: 10.4049/jimmunol.1301723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scarfò I., Ormhøj M., Frigault M.J., Castano A.P., Lorrey S., Bouffard A.A., van Scoyk A., Rodig S.J., Shay A.J., Aster J.C. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood. 2018;132:1495–1506. doi: 10.1182/blood-2018-04-842708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pasparakis M., Schmidt-Supprian M., Rajewsky K. IκB kinase signaling is essential for maintenance of mature B cells. J. Exp. Med. 2002;196:743–752. doi: 10.1084/jem.20020907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knudson K.M., Pritzl C.J., Saxena V., Altman A., Daniels M.A., Teixeiro E. NFκB-Pim-1-Eomesodermin axis is critical for maintaining CD8 T-cell memory quality. Proc. Natl. Acad. Sci. USA. 2017;114:E1659–E1667. doi: 10.1073/pnas.1608448114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Philipson B.I., O’Connor R.S., May M.J., June C.H., Albelda S.M., Milone M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020;13:eaay8248. doi: 10.1126/scisignal.aay8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li G., Boucher J.C., Kotani H., Park K., Zhang Y., Shrestha B., Wang X., Guan L., Beatty N., Abate-Daga D., Davila M.L. 4-1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight. 2018;3:e121322. doi: 10.1172/jci.insight.121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian Y., Cox M.A., Kahan S.M., Ingram J.T., Bakshi R.K., Zajac A.J. A context-dependent role for IL-21 in modulating the differentiation, distribution, and abundance of effector and memory CD8 T cell subsets. J. Immunol. 2016;196:2153–2166. doi: 10.4049/jimmunol.1401236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pegram H.J., Lee J.C., Hayman E.G., Imperato G.H., Tedder T.F., Sadelain M., Brentjens R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bryce P.J., Oyoshi M.K., Kawamoto S., Oettgen H.C., Tsitsikov E.N. TRAF1 regulates Th2 differentiation, allergic inflammation and nuclear localization of the Th2 transcription factor, NIP45. Int. Immunol. 2006;18:101–111. doi: 10.1093/intimm/dxh354. [DOI] [PubMed] [Google Scholar]
- 52.Li W., Guo L., Rathi P., Marinova E., Gao X., Wu M.F., Liu H., Dotti G., Gottschalk S., Metelitsa L.S., Heczey A. Redirecting T cells to glypican-3 with 4-1BB zeta chimeric antigen receptors results in Th1 polarization and potent antitumor activity. Hum. Gene Ther. 2017;28:437–448. doi: 10.1089/hum.2016.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terhune J., Berk E., Czerniecki B.J. Dendritic cell-induced Th1 and Th17 cell differentiation for cancer therapy. Vaccines (Basel) 2013;1:527–549. doi: 10.3390/vaccines1040527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davenport A.J., Cross R.S., Watson K.A., Liao Y., Shi W., Prince H.M., Beavis P.A., Trapani J.A., Kershaw M.H., Ritchie D.S. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA. 2018;115:E2068–E2076. doi: 10.1073/pnas.1716266115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boroughs A.C., Larson R.C., Choi B.D., Bouffard A.A., Riley L.S., Schiferle E., Kulkarni A.S., Cetrulo C.L., Ting D., Blazar B.R. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight. 2019;5:e126194. doi: 10.1172/jci.insight.126194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shalek A.K., Satija R., Adiconis X., Gertner R.S., Gaublomme J.T., Raychowdhury R., Schwartz S., Yosef N., Malboeuf C., Lu D. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 58.Ding J., Adiconis X., Simmons S.K., Kowalczyk M.S., Hession C.C., Marjanovic N.D., Hughes T.K., Wadsworth M.H., Burks T., Nguyen L.T. Systematic comparative analysis of single cell RNA-sequencing methods. bioRxiv. 2019 doi: 10.1101/632216. [DOI] [Google Scholar]
- 59.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.10x Genomics. Cell Ranger. https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger.
- 62.Li B., Sarkizova S., Gould J., Tabaka M., Ashenberg O. 2020. Single cell Cloud tools (scCloud)https://sccloud.readthedocs.io/en/stable/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jerby-Arnon L., Shah P., Cuoco M.S., Rodman C., Su M.J., Melms J.C., Leeson R., Kanodia A., Mei S., Lin J.-R. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984–997.e24. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu H., Ding J., Porter C.B.M., Wallrapp A., Tabaka M., Ma S., Fu S., Guo X., Riesenfeld S.J., Su C. Transcriptional atlas of intestinal immune cells reveals that neuropeptide α-CGRP modulates group 2 innate lymphoid cell responses. Immunity. 2019;51:696–708.e9. doi: 10.1016/j.immuni.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.