

Abstract
Background:
KCHN2 encodes the KV11.1 potassium channel responsible for IKr, a major repolarization current during the cardiomyocyte action potential. Variants in KCNH2 that lead to decreased IKr have been associated with Type 2 Long QT syndrome (LQT2). The mechanism of LQT2 is most often induced loss of KV11.1 trafficking to the cell surface. Accurately discriminating between variants with normal and abnormal trafficking would aid in understanding the deleterious nature of these variants; however, the volume of reported nonsynonymous KCNH2 variants precludes the use of conventional methods for functional study.
Objective:
We report a high-throughput, multiplexed screening method for KCNH2 genetic variants capable of measuring the cell surface abundance of hundreds of missense variants in the resulting KV11.1 channel.
Methods:
We developed a method to quantitate KV11.1 variant trafficking on a pilot region of 11 residues in the S5 helix.
Results:
We generated trafficking scores for 220/231 missense variants in the pilot region. For 5/5 variants, high-throughput trafficking scores validated when tested in single variant flow cytometry and confocal microscopy experiments. We further explored these results with planar patch electrophysiology and found that loss-of-trafficking variants indeed do not produce IKr. Conversely, but expectedly, some variants that traffic normally were still functionally compromised.
Conclusions:
Here, we described a new method for detecting KV11.1 trafficking-deficient variants in a multiplexed assay. This new method accurately generated trafficking data for variants in KV11.1 and is extendable both to all residues in Kv11.1 and to other cell surface proteins.
Keywords: KCNH2, KV11.1, hERG, Deep Mutational Scanning, Membrane Trafficking
INTRODUCTION
KCNH2, also known as the human Ether-à-go-go-Related Gene (hERG), encodes the rapid component of the delayed inward-rectifying, voltage-gated, cardiac potassium channel KV11.1. This channel produces a major repolarizing current, IKr, in cardiomyocytes. KV11.1 loss-of-function variants are the second most common cause of congenital long QT syndrome (LQTS2; MIM: #613688, Figure 1A)1 whilst KV11.1 gain-of-function variants are the most common cause of short QT syndrome (SQTS; MIM: #609620).2 LQTS2 is characterized by a prolonged QT interval on the electrocardiogram, and SQTS, a rarer disease, is characterized by a short QT interval. Both LQTS2 and SQTS predispose individuals to syncope and sudden death.2, 3 To date, more than 1,000 unique non-synonymous KCNH2 variants have been discovered in patients presenting with arrhythmias and in public databases of mostly unaffected individuals.1, 4 With the onset of large population sequencing projects and increase in clinical genetic testing, the number of observed KCNH2 variants is rapidly growing, much faster than the detailed characterization of these variants.9 An important challenge for KCNH2 variant annotation is the identification and characterization of potentially disease-causing KCNH2 variants found in individuals with or without a clinical phenotype.
Figure 1: KV11.1 trafficking deficiency and Long QT Syndrome.
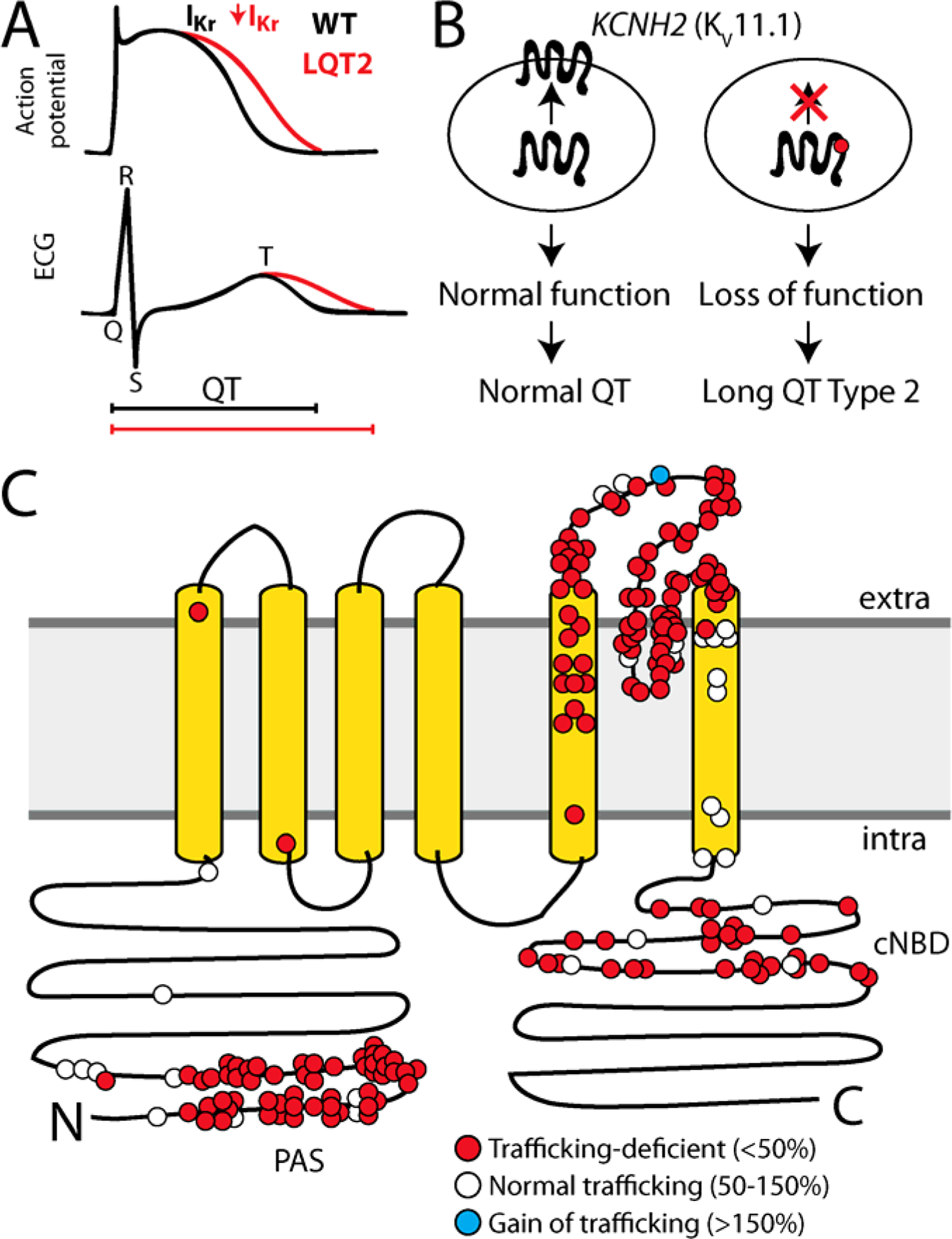
A) KCNH2 variants can cause Long QT Syndrome Type 2 (LQTS2) by reducing IKr inward potassium current. LQTS2 manifests as a prolonged action potential in cardiomyocytes and a prolonged QT interval on the electrocardiogram. B) Schematic of KV11.1 trafficking to the plasma membrane. A common mechanism of LQTS2 is reduced trafficking of KV11.1 to the cell surface. C) For missense variants, trafficking-deficiency is the dominant mechanism of LQTS2 (178/210 variants; 85%)8–10. LQTS2 variants that have been assessed for cell surface abundance are labeled in red (trafficking-deficient; <50% of wildtype) or white (trafficking-normal; 50–150% of wildtype). A Short QT Syndrome-associated variant, N588K, is labeled in blue; this variant has increased cell surface abundance (>150%). PAS indicates the approximate location of the PAS (Per-Arnt-Sim) domain thought to play a role in cell surface expression, cNBD indicates the Cyclic Nucleotide Binding Domain.
The most common mechanism by which pathogenic variants cause protein dysfunction is destabilization-induced misfolding.5–7 KV11.1 is a 1,159 residue protein with six membrane-spanning helices that forms a homotetrameric voltage-gated potassium channel at the plasma membrane surface in cardiomyocytes. For KV11.1, misfolding most commonly causes loss of trafficking to the plasma membrane, resulting in fewer functional channels at the cell surface (Figure 1B).8–10 178/211 (84%) KV11.1 variants associated with LQT2 whose cell surface abundance has been assayed have defective trafficking to the cell surface (Figure 1C).8, 11
Here, we develop an assay which combines the emerging technology of saturation mutagenesis with a high-throughput flow-cytometry-based trafficking assay. Our approach builds on previously developed methods of deep mutational scanning (DMS)12, 13 and cell surface abundance assays8, 11, 14, 15 to create a high-throughput cell surface trafficking assay. Although our method measures the same cell surface abundance of KV11.1 protein as previously established Western blot-based and flow cytometry methods,15 the major advancement here is the ability to readily scale to all 22,021 (1159 × 19) missense substitutions in KV11.1. We can now collect the necessary data to evaluate trafficking which would take years with traditional western blot assays. This provides complementary information to in silico predictions12, 16 at a scale currently inaccessible to other functional characterization techniques, such as automated patch clamp electrophysiology17 and CRISPR-generated, or naturally-occurring, variants in induced pluripotent stem cell-derived cardiomyocytes.18
METHODS
Methods for KCNH2 mutagenesis, generation of stable cell lines, sequencing of libraries, flow cytometry, confocal microscopy, patch clamping, and corresponding analyses are described in detail in the Supplemental Material.
RESULTS
We modified a flow cytometry-based cell-surface trafficking assay15 for compatibility with a DMS of KCNH2 (Figure S1). To accomplish this, we inserted an hemagglutinin (HA) tag19 into the first extracellular loop of KV11.1, overexpressed this plasmid in HEK293T cells, and isolated cells using flow cytometry.20 Control variants of KCNH2 were generated within the HA-KCNH2 construct, and trafficking characteristics were validated with flow cytometry and confocal microscopy (Figure 2A). Wildtype HA-KCNH2 yielded a high KV11.1 surface abundance, detected by both flow cytometry and confocal microscopy (Figure 2B). In contrast, G601S and A561V variants, which have previously been described to have mistrafficking-related loss-of-function defects,21, 22 had reduced cell surface abundance using both methods (Figure 2B). Additionally, the G601S variant, but not A561V, could be rescued by incubation with E-4031, as previously observed (Figure S2).10
Figure 2: KV11.1 cell-surface trafficking assay.
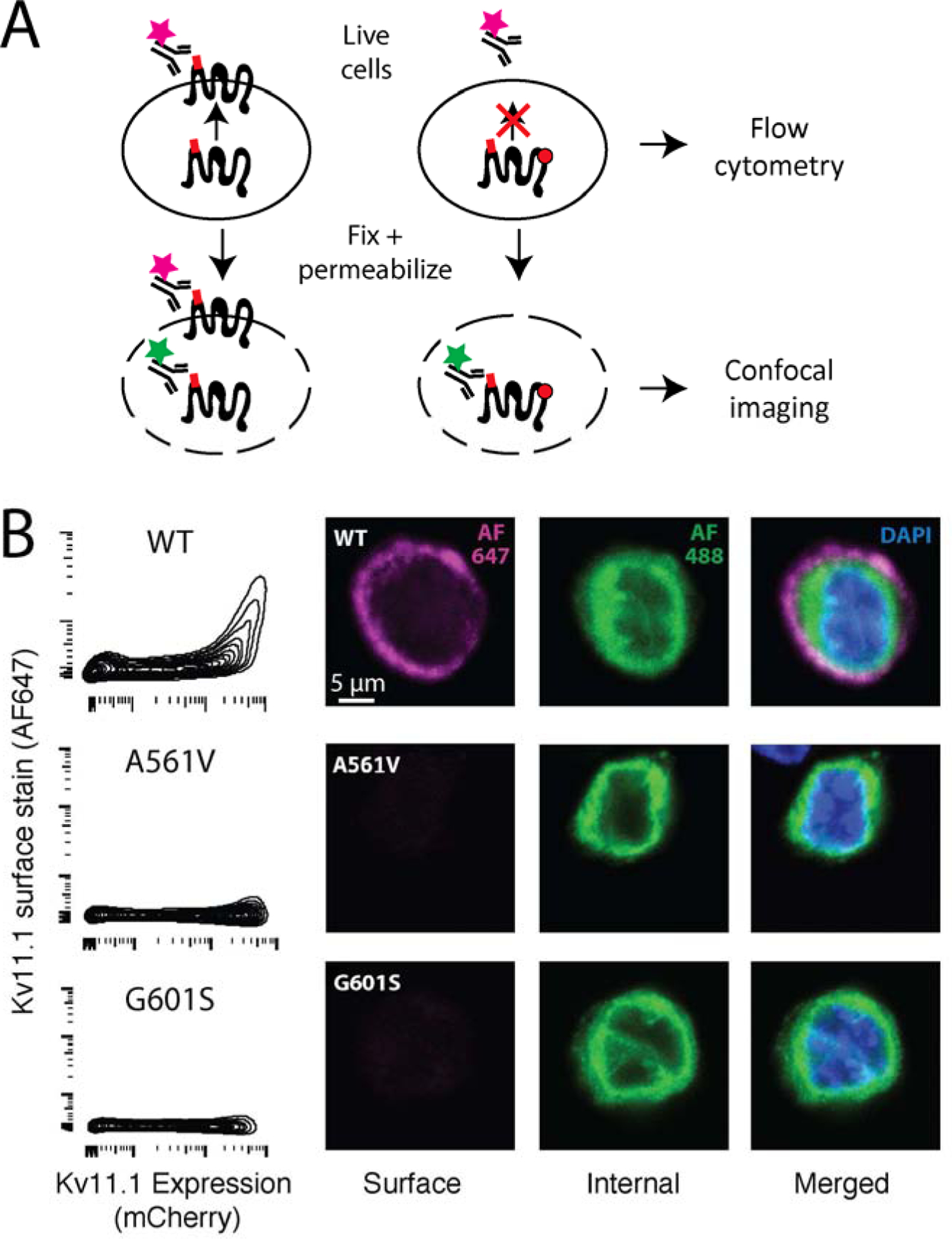
A) Diagram of antibody labeling of surface or both surface and internal KV11.1. Live cells are stained with Alexa 647-labeled anti-HA antibody, which only labels surface KV11.1. Cells are assayed by flow cytometry. For confocal imaging, cells are fixed, permeabilized, and stained with an Alexa 488-labeled anti-HA antibody, which labels internal KV11.1. B) Flow cytometry assay of HEK293T cells expressing wildtype KV11.1, G601S (partial loss-of-function), or A561V (near total loss-of-function). The x-axis indicates mCherry level, which is a marker of protein expression. C) Confocal imaging of HEK293T cells expressing wildtype, G601S, or A561V KV11.1.
Deep mutational scan of an 11-amino-acid region of KV11.1
We next generated a barcoded plasmid library of nearly every possible variant in an 11-amino-acid region (residues 545–555) of KCNH2 using PCR with degenerate primers23 in a cDNA construct broken into tiles (Table S1, Figure S3). The 11-amino-acid region covers a segment stretching from outside the membrane on the S4-S5 linker to within the membrane on the S5 helix. This segment was chosen for its location, which changes from adjacent to within the membrane, and structural diversity, which varies between flexible and helical. After subassembly24, 14,466 unique barcodes were detected, which included 220/231 possible missense variants (Figure S4). The 11-amino-acid experiment successfully distinguished between two control classes of variants: nonsense variants that were predicted to be undetectable at the cell surface and synonymous variants that were predicted to have wildtype channel abundance at the cell surface (Figure 3A). Initial trafficking scores were calculated from a weighted average of each variant’s abundance in the four sorted pools (see Supplemental Materials for details). These scores were averaged across two independently transfected and sorted replicate experiments. Nonsense variants had an average score of 5 (0 was the lowest possible score) and synonymous variants had an average score of 101 (100 is the score for wildtype; Figure 3). The missense variant scores ranged from loss-of-function to wildtype-like; the mean score for missense variants was 55 (Figure 3A and B). Trafficking scores were highly consistent across the two replicate experiments (Spearman’s ρ = 0.77). Seven variants were unable to be analyzed due to lack of barcoding during the generation of the KCNH2 plasmid library (grey boxes in Figure 3B). Of note, methionine is encoded by only one codon and therefore lack synonymous variants.
Figure 3: Deep mutational scan identifies trafficking-defective variants.
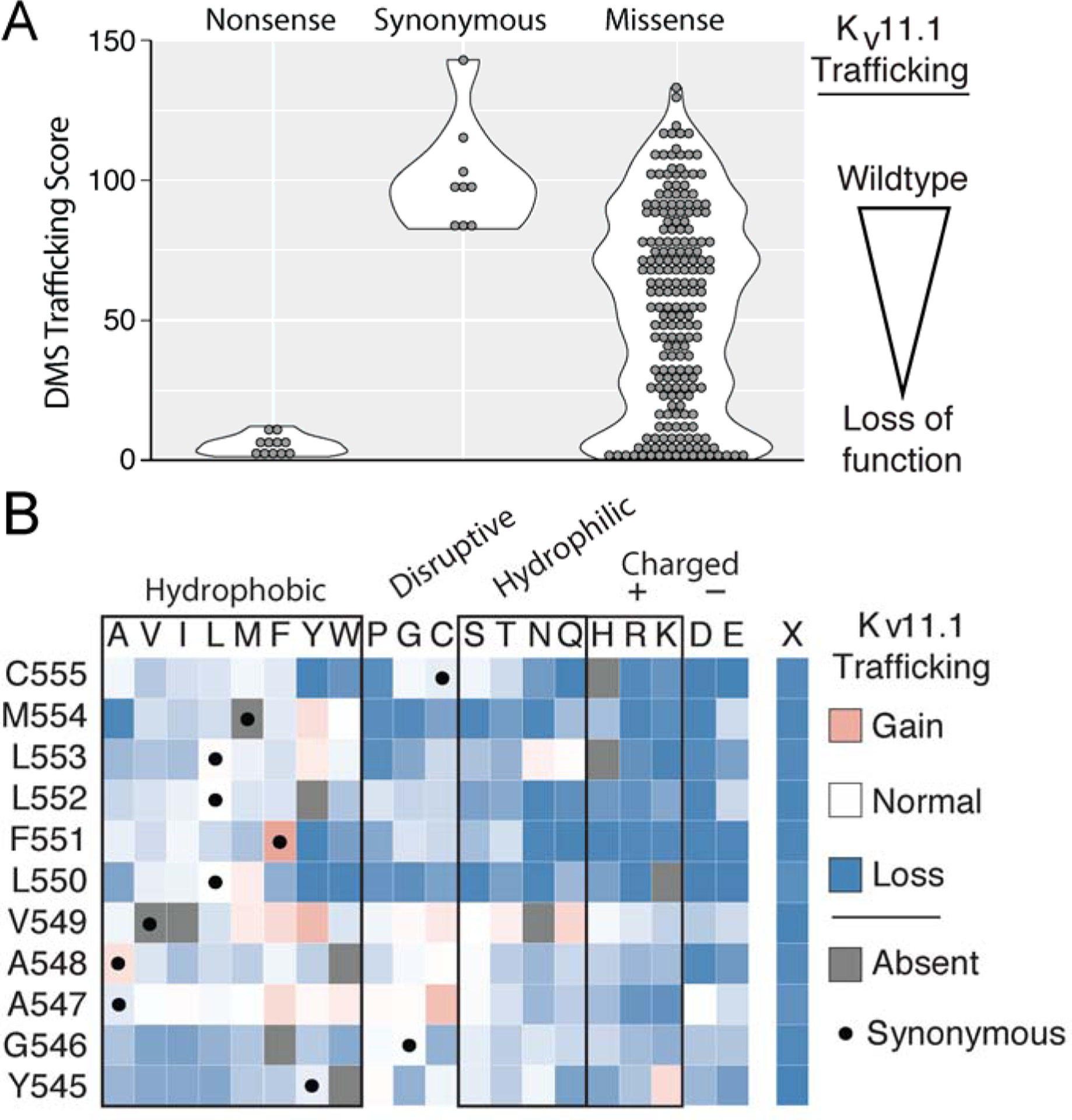
A) Trafficking scores for nonsense, synonymous, and missense KV11.1 variants in the pilot region from Y545 to C555. Synonymous and nonsense variants were well-separated, with a mean score of 5 and 101, respectively. Missense variants had a range of scores from near-wildtype to loss-of-function, with a mean score of 55. B) A heatmap of the trafficking scores from the pilot region also displayed in A). KV11.1 residue number and identity are indicated in the leftmost column and amino-acid substitution is indicated on the top row. The black dot indicates synonymous variants. Color of the box indicates trafficking score compared to wildtype with blue as low expression to the plasma membrane and red as increased expression at the plasma membrane. Gray boxes indicate variants were absent in either the barcode libraries or trafficking results and therefore a trafficking score could not be determined. Out of the 220 variants assayed, 86 were WT-like (score > 75), 57 were mild loss-of-trafficking (75 > score > 50), 48 were loss-of-trafficking (50 > score > 25), and 73 were severe loss-of-trafficking (score < 25). Note the low trafficking scores of charged or polar amino-acids closer to the middle of the membrane (residues 550–555) (see Figure 4).
Trafficking scores and KV11.1 topology
We explored the structural basis of the trafficking scores with a three-dimensional structure of KV11.1 (Figure 4, PDB: 5VA1).25 Residues Y545-V549 tolerated substitutions more than residues L550-C555, especially secondary structure-disruptive substitutions (glycine and proline) and hydrophilic substitutions (Figure 3B). These observations are consistent with the topology and position within the membrane of the pilot region: residues 545–549 join the S4 and S5 helices on the intracellular membrane leaflet, a segment transitioning from random coil to α-helical secondary structure (Figure 4). Missense substitutions at residues 545–549 were less likely on average to induce mistrafficking compared to residues 550–555, a mean trafficking score of 70.2 versus 42.2, respectively. In contrast, residues 550–555, closer to the membrane interior, were less tolerant to any substitution, especially polar and charged residues. The trafficking scores correlated modestly with computational predictions, with a Spearman’s ρ of 0.31, 0.21, and 0.42 with PROVEAN,26 PolyPhen-2,27 and REVEL,28 respectively (Table S3).
Figure 4. Polar and charged residues close to the middle of the membrane cause trafficking-deficiency.
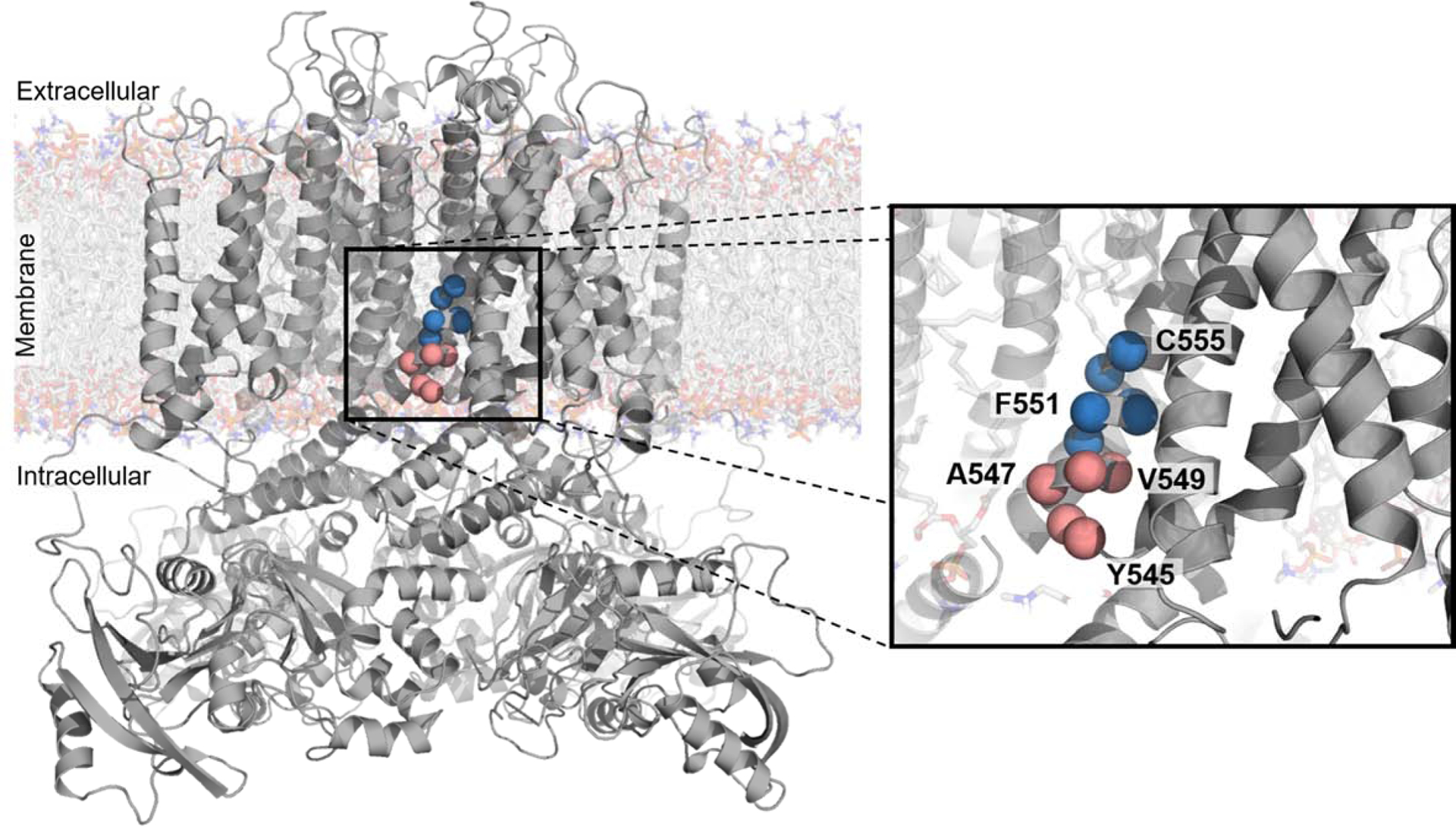
CryoEM structure of KV11.1 (PDB: 5VA1)41 with residues mutagenized in a pilot study of a high-throughput trafficking assay highlighted as red or blue spheres. These residues are on the intracellular half of the S5 transmembrane helix. The red to blue transition from V549 to L550 reflects the transition from residues tolerant of hydrophilic substitution to residues intolerant of hydrophilic substitution, respectively, as observed in the high-throughput trafficking assay (Figure 3B).
Validation of trafficking assay scores
To validate our trafficking scores, we selected five variants for further characterization. These variants were selected based on their unintuitive DMS trafficking results: L553N and A547D are in regions intolerant of hydrophilic substitution yet had a trafficking score like wildtype (scores of 108 and 99, respectively); M554A is in a region tolerant of hydrophobic substitution yet does not traffic (score of 4) yet M554W is a disruptive substitution and traffics like wildtype (score of 99); F551Y is a relatively conservative substitution yet is not tolerated (score of 2). In single variant validation experiments, these five variants had confocal and flow cytometry cell surface abundance concordant with their DMS scores (Figure 5 and Table 1).
Figure 5: Validation of trafficking scores.
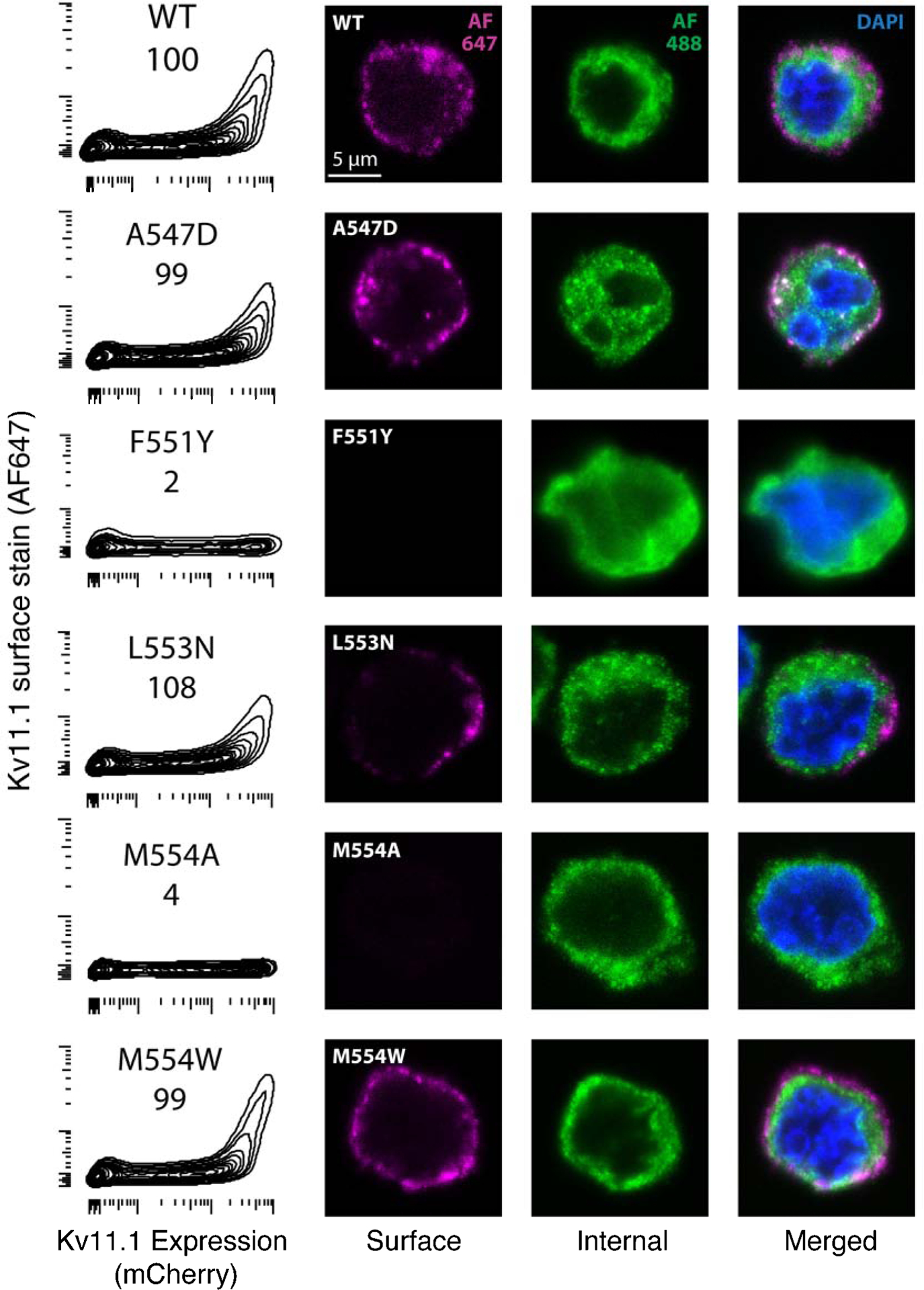
Validation of selected variants by analytical flow cytometry (left) and confocal microscopy (right). In the leftmost column, DMS score is indicated directly below variant name. X-axis is mCherry fluorescence (indicating KV11.1 expression), the y-axis is surface stained KV11.1. The remaining columns are extracellularly stained KV11.1 (AF647, purple) intracellularly stained KV11.1 (AF488, green), and both merged with DAPI stained nucleus (blue).
Table 1:
Trafficking score validation
| Variant | DMS score | Flow/Confocal validation | Patch clamp |
|---|---|---|---|
| A547D | 99 | WT-like | LOF |
| L553N | 108 | WT-like | Fast deactivation |
| M554A | 4 | No trafficking | LOF |
| M554W | 99 | WT-like | LOF |
| F551Y | 2 | No trafficking | -- |
In addition, we examined four variants in the studied region that were previously observed in literature reports or in gnomAD.4 L552S was previously reported as trafficking deficient variant8 and has been observed in 90 cases of Long QT syndrome in the literature, 28 unaffected individuals, and 20 alleles in the gnomAD database. Our DMS score for L552S was 25, consistent with the strong association with LQTS2 and loss of trafficking phenotype as previously reported.8 Three other variants, A547T, V549M, and F551L, were not observed in any literature cases of Long QT Syndrome and have been observed in 1–2 cases in gnomAD, too few to assess relevance from patient data alone. All of these variants had normal trafficking scores (Table S2). Thus, the DMS trafficking scores are consistent with the available data for variants in this region.
To further assess the trafficking results, we collected planar patch clamp data on four of the five validation variants. M554A also had a total loss of IKr, consistent with its loss of trafficking DMS score (score of 4) and its loss of trafficking in validation experiments. Surprisingly, the other three validation variants, which had detectable trafficking by DMS and in validation experiments, also had loss-of-function defects by patch clamp (Figure 6). Despite being present at the cell surface (Figure 5), M554W and A547D did not produce measurable IKr, while L553N had detectable IKr, but with lowered peak tail current and faster deactivation rate, essentially producing a loss-of-function phenotype (Figure 6). Therefore, the high-throughput trafficking scores were successful at identifying loss-of-function variants that acted through a trafficking mechanism, but not by other mechanisms, unlike PROVEAN, PolyPhen-2, and REVEL, which predicted all variants in the validation set to be deleterious.
Figure 6: Functional characterization of L553N.
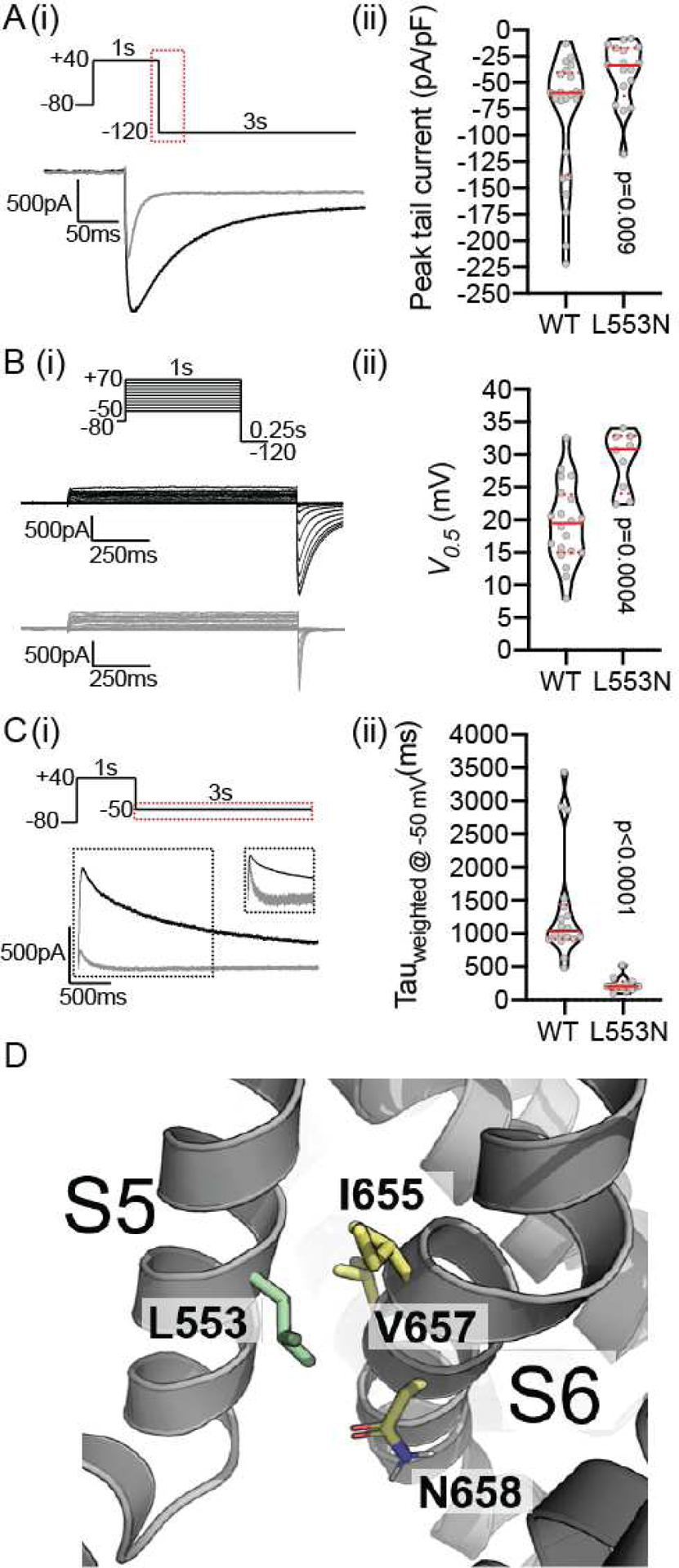
Ai) All channels were fully activated at +40mV for 1s before stepping to −120mV for 3s to allow for recovery from inactivation and measure current density. The peak tail current traces for WT (black) and L553N (grey) are shown for the region highlighted in the voltage protocol. Aii) Violin plot of peak tail current amplitudes normalized to cell capacitance. Bi) Typical families of current traces recorded during Isochronal activation protocols for WT (black) and L553N (grey). Bii) Violin plot of V0.5 values derived by fitting Boltzmann function to the tail current amplitudes (see Supplementary Methods for details). Ci) The current traces for WT (black) and L553N (grey) were shown for the region highlighted in the voltage protocol. Cii) Violin plot of the weighted time constant (Tweighted) for channel deactivation at −50mV, (see methods for detail of calculation of Tweighted). In all violin plots, median and quartiles are shown as solid and dotted lines, respectively. The reported two-tailed P-values were derived from Mann-Whitney tests. D) Structural context of L553 (on helix S5), which points between residues V657 and N658 on the S6 helix within the same subunit.
DISCUSSION
A high-throughput assay for identifying KCNH2 variant trafficking
We developed a scalable, multiplexed flow cytometry and high-throughput sequencing assay to measure the cell surface abundance of hundreds (and potentially thousands) of variants in KCNH2. We generated trafficking scores for 220/231 possible missense variants in an 11-amino-acid region of KCNH2. We validated these scores in several ways. First, the scores correlated very well (Spearman ρ = 0.77) across the two replicate experiments. Second, the scores distinguished nonsense from synonymous variants (Figure 3). Finally, 5/5 missense variants with a range of surprising, unintuitive trafficking scores were validated by flow cytometry and confocal immunocytochemistry on single variants (Figure 5). Therefore, we believe these scores accurately capture cell surface abundance, which is the mechanism for ~80% of all long QT variants studied to date.8–10
Improved understanding of KCNH2 trafficking deficiency
From our trafficking data, we could observe two segments in the 11-residue pilot with distinct substitution tolerances. In the segment comprised of residues 545–549, hydrophilic and secondary structure-disrupting substitutions were tolerated. However, in the segment comprised of residues 550–555, hydrophilic substitutions were more likely to lead to compromised trafficking. The tolerance of hydrophobic or hydrophilic substitution reflect the observed topology of the KV11.1 S5 helix41 (Figures 3B and 4). Our trafficking data have a Spearman’s ρ of 0.31, 0.21, and 0.42 with PROVEAN, PolyPhen-2, and REVEL respectively, indicating our DMS scores provide distinct information to that available in in silico these predictive models.
In addition, we unexpectedly found L553N trafficked like wildtype and produced measurable peak tail current. However, this variant had a dramatically faster deactivation time (the time it takes the channel to stop conducting after the cell is repolarized; Figure 6). L553 points in between residues N658 and V659 (both known to accelerate deactivation) in the S6 helix within the same subunit.29 Given our result, we speculate the asparagine (N) substitution at residue 553 stabilizes an interaction between N658 and L553N resulting in a more stable deactivated state which allows this variant to escape trafficking quality control but does not allow the channel to function normally (Figure 6D).
Approaches to studying KCNH2 variants
This work represents a new tool for studying KCNH2 variants in high-throughput. Several studies have previously quantified trafficking of KV11.1, typically using assays that rely on Western blotting or confocal imaging, which are not scalable to the thousands of possible missense variants within the protein.10, 15, 30 Recent advances in automated patch clamp electrophysiology have enabled the rapid characterization of dozens of voltage-gated potassium channel variants,17, 31 however, the throughput of these methods does not allow for the rapid screening of the thousands of possible variants in KCNH2. In contrast, computational predictions like Polyphen2, PROVEAN, or REVEL can be easily generated for every variant in KCNH2. However, while these computational predictions can partially predict disease risk, they are imperfect and often cannot capture the variant-induced defect (Table S3). Lastly, this method may also be used to investigate variant-specific and therapy-specific trafficking rescue for a multitude of variants per experiment (Figure S3).
Improving the prediction of KCNH2 variant pathogenicity
Our assay has the potential to sample all possible amino acid substitutions in KCNH2 in an unbiased manner. This will provide a database for patients and clinicians to identify the effect a previously uncharacterized mutation may have on patient disease propensity. Future work will expand this method to much larger regions of KCNH2, with a focus on regions enriched for known long QT variants (Figure 1). However, our electrophysiological results highlight the need to approach trafficking data with caution. A547D and M554W trafficked normally in the high-throughput assay and in single variant validation experiments, yet did not produce IKr currents in patch clamp experiments. This result indicates all variants with normal trafficking scores need to be interpreted with caution, as they may still cause loss of function through other mechanisms.
Limitations
Here, we described a method to investigate whether a missense variant in KCNH2 affects the steady-state concentration of mutant KV11.1 channels at the plasma membrane. The trafficking score derived from this method will indicate if the variant channel traffics normally; however, we cannot assess whether a low-trafficking variant will have a dominant negative effect or whether wildtype-trafficking variants will produce wildtype IKr. The assay described was developed for the dominant isoform in HEK293T cells and therefore does not account for the expression of additional KCNH2 splice variants, common variants (e.g. T897 and L1047), and other subunits known to contribute to physiologically relevant IKr in cardiomyocytes.32–34 To better understand the implications of the DMS trafficking scores in the clinical context, future work is needed to investigate KV11.1 variant trafficking in these contexts.
Conclusion
We have developed a high-throughput trafficking assay to characterize cell surface expression of KCNH2 variants in a massively parallel fashion. The vast majority of KCNH2 variants only have limited available patient data and most are currently annotated as variants of uncertain significance. The method we put forward here enables the collection of in vitro experimental variant defect data at a scale commensurate with the scale of new KCNH2 variants being discovered. Lastly, this method can be expanded to other ion channels and transmembrane proteins where altered cell surface abundance is a major mechanism of disease pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
Vanderbilt Flow Cytometry Shared Resource, the Vanderbilt Cell Imaging Shared Resource, the Vanderbilt VANTAGE genomics core and the Victor Chang Cardiac Research Institute Innovation Centre, funded by the NSW Government, Australia.
FUNDING
Funding: This work was supported by the National Institutes of Health [grant numbers R00HL135442 (BMK), F32HL137385 (AMG), and 1R01GM109110 (DMF)]; and the Leducq Transatlantic Network of Excellence Program [grant number 18CVD05 (BCK, BMK, and CLE)].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None.
The authors have no conflicts to disclose
Literature Cited
- 1.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AAM and Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION (R) long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjerregaard P. Diagnosis and management of short QT syndrome. Heart Rhythm. 2018;15:1261–1267. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G and Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63. [DOI] [PubMed] [Google Scholar]
- 4.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG and Exome Aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlebach JP and Sanders CR. The safety dance: biophysics of membrane protein folding and misfolding in a cellular context. Q Rev Biophys. 2015;48:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yue P, Li Z and Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–73. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z and Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17:263–70. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP and January CT. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nature communications. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith JL, Anderson CL, Burgess DE, Elayi CS, January CT and Delisle BP. Molecular pathogenesis of long QT syndrome type 2. J Arrhythm. 2016;32:373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong QM, Zhou ZF, Ackerman MJ and January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. [DOI] [PubMed] [Google Scholar]
- 11.Huo J, Zhang Y, Huang N, Liu P, Huang C, Guo X, Jiang W, Zhou N, Grace A, Huang CL and Ma A. The G604S-hERG mutation alters the biophysical properties and exerts a dominant-negative effect on expression of hERG channels in HEK293 cells. Pflugers Archiv : European journal of physiology. 2008;456:917–28. [DOI] [PubMed] [Google Scholar]
- 12.Fowler DM and Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glazer AM, Kroncke BM, Matreyek KA, Yang T, Wada Y, Shields T, Salem JE, Fowler DM and Roden DM. Deep Mutational Scan of an SCN5A Voltage Sensor. Circ Genom Precis Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin EC, Holzem KM, Anson BD, Moungey BM, Balijepalli SY, Tester DJ, Ackerman MJ, Delisle BP, Balijepalli RC and January CT. Properties of WT and mutant hERG K(+) channels expressed in neonatal mouse cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;298:H1842–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanner SA, Jain A and Colecraft HM. Development of a High-Throughput Flow Cytometry Assay to Monitor Defective Trafficking and Rescue of Long QT2 Mutant hERG Channels. Front Physiol. 2018;9:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J and Fowler DM. Variant Interpretation: Functional Assays to the Rescue. Am J Hum Genet. 2017;101:315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng CA, Perry MD, Liang W, Smith NJ, Foo B, Shrier A, Lukacs GL, Hill AP and Vandenberg JI. High-throughput phenotyping of heteromeric human ether-a-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Heart Rhythm. 2019. [DOI] [PubMed] [Google Scholar]
- 18.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M and Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. [DOI] [PubMed] [Google Scholar]
- 19.Ficker E, Dennis AT, Wang L and Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. 2003;92:e87–100. [DOI] [PubMed] [Google Scholar]
- 20.Matreyek KA, Stephany JJ and Fowler DM. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 2017;45:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furutani M, Trudeau MC, Hagiwara N, Seki A, Gong Q, Zhou Z, Imamura S, Nagashima H, Kasanuki H, Takao A, Momma K, January CT, Robertson GA and Matsuoka R. Novel mechanism associated with an inherited cardiac arrhythmia: defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation. 1999;99:2290–4. [DOI] [PubMed] [Google Scholar]
- 22.Sanguinetti MC, Curran ME, Spector PS and Keating MT. Spectrum of HERG K+ −channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci U S A. 1996;93:2208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain PC and Varadarajan R. A rapid, efficient, and economical inverse polymerase chain reaction-based method for generating a site saturation mutant library. Anal Biochem. 2014;449:90–98. [DOI] [PubMed] [Google Scholar]
- 24.Hiatt JB, Patwardhan RP, Turner EH, Lee C and Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W and MacKinnon R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell. 2017;169:422–430 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi Y, Sims GE, Murphy S, Miller JR and Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS one. 2012;7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS and Sieh W. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wynia-Smith SL, Gillian-Daniel AL, Satyshur KA and Robertson GA. hERG gating microdomains defined by S6 mutagenesis and molecular modeling. J Gen Physiol. 2008;132:507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou ZF, Gong QM, Epstein ML and January CT. HERG channel dysfunction in human long QT syndrome - Intracellular transport and functional defects. Journal of Biological Chemistry. 1998;273:21061–21066. [DOI] [PubMed] [Google Scholar]
- 31.Vanoye CG, Desai RR, Fabre KL, Gallagher SL, Potet F, DeKeyser JM, Macaya D, Meiler J, Sanders CR and George AL Jr. High-Throughput Functional Evaluation of KCNQ1 Decrypts Variants of Unknown Significance. Circ Genom Precis Med. 2018;11:e002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC and Robertson GA. hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci U S A. 2014;111:18073–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT and Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. [DOI] [PubMed] [Google Scholar]
- 34.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL Jr. and Schwartz PJ. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.