

Abstract
BACKGROUND AND AIMS:
Neutrophil infiltration is a hallmark of nonalcoholic steatohepatitis (NASH), but how this occurs during the progression from steatosis to NASH remains obscure. Human NASH features hepatic neutrophil infiltration and upregulation of major neutrophil-recruiting chemokines (e.g., CXCL1 and IL-8). However, mice fed a high-fat diet (HFD) only develop fatty liver without significant neutrophil infiltration or elevation of chemokines. The aim of this study was to determine why mice are resistant to NASH development and the involvement of p38 mitogen-activated protein kinase (p38) activated by neutrophil-derived oxidative stress in the pathogenesis of NASH.
APPROACHES AND RESULTS:
Inflamed human hepatocytes attracted neutrophils more effectively than inflamed mouse hepatocytes due to the greater induction of CXCL1 and IL-8 in human hepatocytes. Hepatic overexpression of Cxcl1 and/or IL8 promoted steatosis-to-NASH progression in HFD-fed mice by inducing liver inflammation, injury, and p38 activation. Pharmacological inhibition of p38α/β or hepatocyte-specific deletion of p38a (a predominant form in the liver) attenuated liver injury and fibrosis in the HFD+Cxcl1-induced NASH model that is associated with strong hepatic p38α activation. In contrast, hepatocyte-specific deletion of p38a in HFD-induced fatty liver where p38α activation is relatively weak exacerbated steatosis and liver injury. Mechanistically, weak p38α activation in fatty liver upregulated the genes involved in fatty acid β-oxidation via PPARα phosphorylation, thereby reducing steatosis. Conversely, strong p38α activation in NASH promoted CASP3 cleavage, CHOP expression, and BCL2 phosphorylation, thereby exacerbating hepatocyte death.
CONCLUSIONS:
Genetic ablation of hepatic p38a increases simple steatosis but ameliorates oxidative stress-driven NASH, indicating that p38α plays distinct roles depending on the disease stages, which may set the stage for investigating p38α as a therapeutic target for the treatment of NASH.
Keywords: Steatosis, neutrophils, inflammation, CXCL1, IL-8
Introduction
Inflammation plays a key role in the progression of nonalcoholic fatty liver disease (NAFLD),(1–3) which has emerged as the leading cause of chronic liver disease(4,5) and includes a spectrum of disorders that range from steatosis to nonalcoholic steatohepatitis (NASH), fibrosis, and hepatocellular carcinoma. However, the underlying mechanisms that drive NAFLD progression still remain obscure.(6,7) Overnutrition-associated obesity is a major risk factor for hepatic steatosis which is characterized by excessive deposition of triglyceride in hepatocytes.(6) Simple steatosis may further progress to NASH in approximately 25% of patients. NASH is a more severe type of NAFLD, accompanied by hepatocellular injury and ballooning with lobular inflammation in addition to aberrant fat accumulation.(7) A hallmark of NASH is the marked hepatic infiltration of neutrophils in parallel with elevated expression of chemokines that attract neutrophils such as C-X-C motif chemokine ligand 1 (CXCL1) and interleukin (IL)-8, which is not evident in fatty liver of obese individuals.(2,8,9) Mice fed a high-fat diet (HFD) develop steatosis but rarely NASH. This is in line with the marginal elevation in hepatic neutrophil infiltration and expression of neutrophil-recruiting chemokines (e.g., CXCL1) upon HFD feeding.(10) To understand the potentially different susceptibility of human and mice to develop NASH, in the current study we compared mouse and human hepatocytes and found that mouse hepatocytes recruited neutrophils much less effectively than human hepatocytes under inflammatory conditions due to a lower expression of CXCL1 and lack of IL-8 (the Il8 gene is not present in the mouse genome). In addition, the number of circulating neutrophils in mice (∼1×109/L) is much lower than in humans (∼4×109/L).(11,12) Collectively, all of these factors suggest that mice may be more resistant to NASH development than humans due to a lower recruitment of neutrophils. Indeed, we have previously demonstrated that hepatic overexpression of Cxcl1 promotes steatosis-to-NASH development in HFD-fed mice through the stimulation of hepatic neutrophil infiltration, which in turn promotes liver injury via neutrophil-derived oxidative stress.(13) Herein, we further demonstrate that simultaneous hepatic overexpression of Cxcl1 and IL8 concomitantly promotes the simple steatosis-to-NASH progression in HFD-fed mice with greater oxidative stress, inflammation, and p38 mitogen-activated protein kinase (MAPK) (p38) activation in the liver. Because HFD feeding alone causes prominent fatty liver, whereas HFD feeding with Cxcl1 overexpression causes NASH, this HFD+Cxcl1 model can be used to study different stages of NAFLD including steatosis and NASH. By using this model, here we dissected the role of p38 in the pathogenesis of NAFLD and found that p38 plays beneficial and detrimental roles in the pathogenesis of steatosis and NASH, respectively.
The p38 family consists of four subtypes including p38α, p38β, p38γ, and p38δ. These subtypes can be activated by a variety of extracellular stimuli such as oxidative stress, inflammatory cytokines, and growth factors.(14) Among the four subtypes, p38α is the predominant form in the liver, playing an important role in glucose homeostasis and lipid metabolism.(15) With regard to lipid metabolism, a recent study demonstrated that liver-specific p38α knockout mice were more susceptible to HFD-induced obesity and steatosis accompanied by reduced fatty acid (FA) β-oxidation.(16) FA β-oxidation is mainly controlled by peroxisome proliferator-activated receptor alpha (PPARα), a ligand-activated transcription factor that is highly expressed in the liver where FA is actively oxidized.(17,18) PPARα activity is known to be controlled by many factors and signaling pathways including p38α, which has been shown to phosphorylate and activate PPARα in cardiac myocytes.(19) However, it remains elusive whether p38α controls lipid metabolism through PPARα phosphorylation in the liver. By performing reverse transcription quantitative PCR (RT-qPCR) analyses of various genes related to hepatic lipid metabolism, here we demonstrated that hepatic expression of PPARα and its target genes was downregulated in liver-specific p38a knockout mice compared to wild-type (WT) mice after HFD feeding, suggesting p38α protects against fatty liver in HFD-fed mice by activating PPARα, which in turn upregulates its target genes involved in FA β-oxidation.
Compared to HFD feeding alone that mainly induces steatosis with weak p38α activation, HFD+Cxcl1 challenge induces NASH with greater p38α activation.(13) Our data in the present study suggest that such strong p38α activation exacerbates NASH by regulating B-cell lymphoma 2 (BCL2) and C/EBP homologous protein (CHOP). BCL2 is a critical factor for cell survival and is modulated by p38 through phosphorylation in response to oxidative stress.(20,21) Phosphorylation of BCL2 decreases the anti-apoptotic effect of BCL2 and sensitizes cells to apoptosis.(22) CHOP is an ER stress-activated transcription factor that upregulates an array of apoptotic genes.(23) p38 directly phosphorylates and transactivates CHOP as well as elevates expression of CHOP, thereby promoting ER stress-associated apoptosis.(24,25) Despite the compelling evidence for the regulation of cell death mechanisms by p38α, there is a knowledge gap as to whether p38α activity contributes to NASH development by modulating CHOP or BCL2. The present study answered this question by providing in vitro and in vivo evidence suggesting that activation of p38α exacerbates hepatocyte death in CXCL1-induced NASH by regulating CHOP and BCL2.
Materials and Methods
Mice
C57BL/6J and Albumin Cre mice were obtained from the Jackson Laboratory (Bar Harbor, ME). p38a-floxed mice were kindly provided by Dr. Yibin Wang (University of California, Los Angeles) and were crossed with Albumin Cre mice via several steps to generate hepatocyte-specific p38a knockout (p38aHep−/−) mice. All animal experiments were approved by the NIAAA Institutional Animal Care and Use Committee.
HFD-induced fatty liver in mice
To induce steatosis in mice, p38aHep−/− mice and p38a-floxed littermate control mice (defined as WT mice) (male, 6-7 weeks of age) were fed an HFD (60 kcal% fat; D12492, Research Diets, New Brunswick, NJ) for 3 months and sacrificed for analyses.
HFD+Cxcl1-induced NASH in mice
To induce NASH, C57BL/6J mice, p38aHep−/−, and p38a-floxed littermate WT control mice (male, 6-7 weeks of age) were fed an HFD for 3 months followed by a tail vein injection with adenovirus expressing mouse CXCL1 (Ad-Cxcl1) or green fluorescence protein (Ad-Gfp) as a control. In some experiments, mice were also injected with Ad-Cxcl1 concomitantly with adenovirus expressing human IL-8 (Ad-IL8). Adenoviruses were purchased from Applied Biological Materials (Richmond, Canada). Mice were continued on an HFD for 2 or 4 weeks after the adenovirus infection, before sacrifice for analyses. To test the efficacy of p38α/β inhibitors (LY-2228820 and PH-797804)(Selleckchem, Houston, TX), mice were injected intraperitoneally with LY-2228820 (3 mg/kg) and PH-797804 (10 mg/kg) on days 7, 9, 11, and 13 post adenoviral infection.
Statistical analysis
Data are expressed as mean ± SEM and were analyzed using GraphPad Prism (v. 7.0a; GraphPad Software, La Jolla, CA). To compare values obtained from two groups, the Student’s t-test was performed. Data from multiple groups were compared with one-way ANOVA followed by Tukey’s post hoc test. P values of <0.05 were considered significant.
Results
Differential abilities of human and mouse hepatocytes to recruit neutrophils
To investigate the differential ability of human and mouse hepatocytes to stimulate neutrophil recruitment under inflammatory conditions, we examined the levels of CXCL1 and IL-8, two major chemokines for neutrophil recruitment, in the supernatant of cultured primary human and mouse hepatocytes treated with inflammatory cytokines such as TNF-α, IL-1β, and IL-6. As illustrated in Fig. 1A, TNF-α and IL-1β, but not IL-6, increased the levels of CXCL1 in the culture supernatants of primary human hepatocytes in a dose-dependent manner, which were approximately three times greater than their counterparts in mice. TNF-α and IL-1β also increased the supernatant levels of IL-8 in human hepatocytes but not in mouse hepatocytes due to absence of the Il8 gene in mouse genome. Next, the functional aspect of the differential release of these chemokines was tested with transwell migration assays. To simulate the fact that humans have approximately four times more circulating neutrophils than mice (~4×109/L vs. ~1×109/L),(11,12) we added 1×106 mouse neutrophils onto the insert, whereas 1×106 or 4×106 human neutrophils were added. Regardless of the number of neutrophils added, human hepatocytes treated with TNF-α or IL-1β attracted approximately two to three times higher number of neutrophils than mouse hepatocytes did (Fig. 1B). Taken together, inflamed human hepatocytes recruit neutrophils more effectively than mouse hepatocytes.
Fig. 1. Inflamed human hepatocytes recruit neutrophils more effectively than mouse hepatocytes via the greater production of CXCL1 and IL-8, promoting steatosis-to-NASH progression.
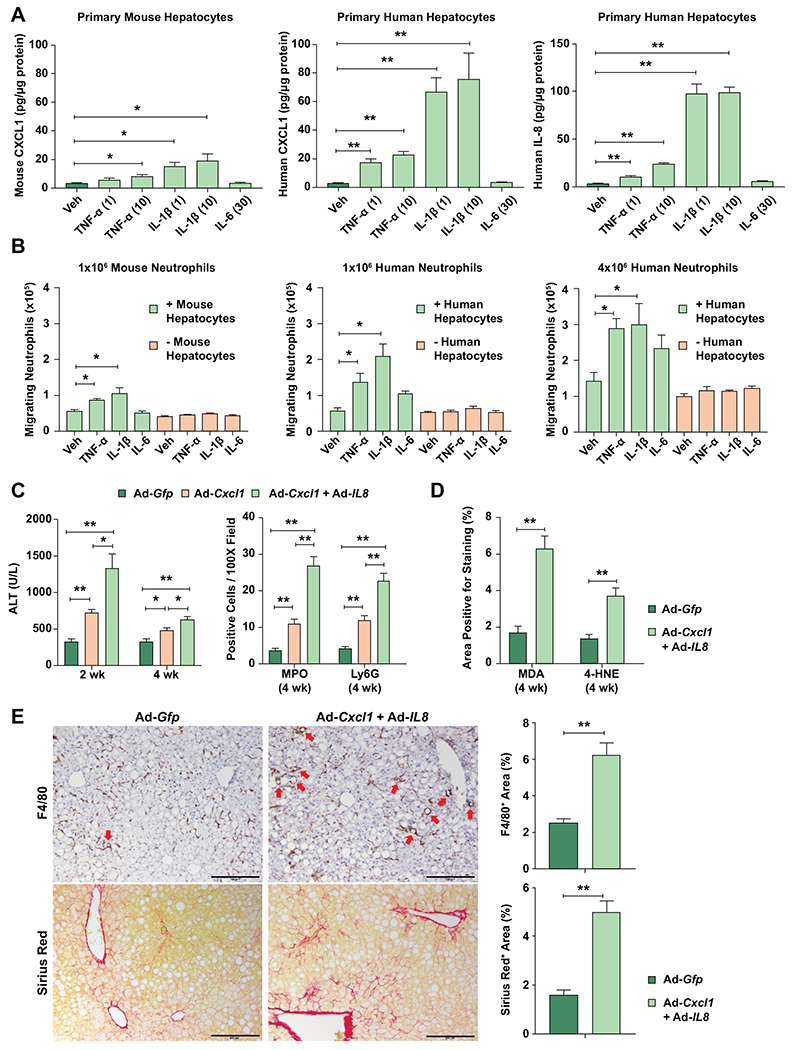
(A) Primary human or mouse hepatocytes were treated with TNF-α (1 or 10 ng/mL), IL-1β (1 or 10 ng/mL), or IL-6 (30 ng/mL) for 12 hr. The supernatant from cell culture was subjected to ELISA analyses of mouse CXCL1 and human CXCL1 and IL-8. The values were normalized to the protein concentration of hepatocytes. (B) Primary human or mouse hepatocytes cultured in the lower chamber of a transwell system, or absence of cells as control, were treated with TNF-α (10 ng/mL), IL-1β (10 ng/mL), or IL-6 (30 ng/mL) for 9 hr. Mouse or human neutrophils were added onto the insert of the transwell system. After incubation for 3 hr, the number of neutrophils that migrated to the lower chamber was counted. Values represent the mean from three independent experiments with triplicates in each experiment (panels A and B). (C) Three-month HFD-fed C57BL/6J mice were injected with Ad-Gfp, Ad-Cxcl1, or Ad-Cxcl1 plus Ad-IL8 for 4 weeks. Serum ALT levels at 2 and 4 weeks after adenoviral infection are shown (left). The number of cells positive for MPO or Ly6G staining per field in the liver sections (right). The representative images for each staining are included in Supporting Fig. S1C. (D-E) HFD-fed mice were injected with Ad-Gfp or Ad-Cxcl1 plus Ad-IL8 for 4 weeks. Quantification of the area positive for MDA or 4-HNE staining in the paraffin-embedded liver sections (panel D). The representative images for each staining are included in Supporting Fig. S2A. Liver sections were subjected to Sirius red and F4/80 staining (panel E, left), and the areas positive for each staining were quantified (panel E, right). Red arrows indicate macrophage aggregates surrounding lipid droplets (hepatic crown-like structures), and scale bars indicate 200 μm. Values represent mean ± SEM. Statistical evaluation was performed by Student’s t-test or one-way ANOVA with Tukey’s post hoc test for multiple comparisons (*p<0.05; **p<0.01). Veh, Vehicle.
To determine the contribution of CXCL1 and IL-8 to steatosis-to-NASH progression in vivo, male C57BL/6J mice were fed an HFD for 3 months followed by a 4-week infection with an adenoviral vector overexpressing Cxcl1 (Ad-Cxcl1), alone or in combination with an adenoviral vector overexpressing IL8 (Ad-Cxcl1+Ad-IL8), or Gfp (Ad-Gfp) as a control (Supporting Fig. S1A–B). Since we previously reported that Cxcl1 overexpression is sufficient to induce NASH in HFD-fed mice by enhancing neutrophil-driven oxidative stress,(13) we set out to determine whether concomitant overexpression of Cxcl1 and IL8 can further enhance the liver injury. Analysis of serum alanine aminotransferase (ALT) levels and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) revealed that overexpression of Cxcl1 and IL8 induced more severe hepatocyte death than Cxcl1 alone with a greater degree of neutrophil infiltration as evidenced by MPO and Ly6G staining (Fig. 1C, Supporting Fig. S1C), suggesting that Cxcl1 and IL8 additively enhance neutrophil infiltration and hepatocyte death. Concomitant overexpression of Cxcl1 and IL8 markedly elevated oxidative stress, inflammation, and fibrosis in the liver as evidenced by histological analyses of malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), F4/80, Sirius Red, and alpha-smooth muscle actin (α-SMA) and RT-qPCR analyses of genes involved in neutrophil oxidative burst and fibrogenesis (Fig. 1D–E, Supporting Fig. S2A–B).
Pharmacological inhibition of p38α/β attenuates HFD+Cxcl1-induced NASH
Since p38 pathway relays oxidative stress to cell death signaling following the activation of upstream mediators such as apoptosis signal-regulating kinase 1 (ASK1)(20) that is activated in our NASH model,(13) we tested whether overexpression of Cxcl1 alone or Cxcl1 and IL8 simultaneously activates p38. Cxcl1 overexpression led to 2-fold induction of p38 phosphorylation, which was further induced by additional overexpression of IL8 (Fig. 2A). Caspase-3 (CASP3) cleavage, an effector of apoptosis, was also induced by overexpression of Cxcl1 and/or IL8 (Fig. 2A). We then sought to determine the extent to which p38 contributes to the development of neutrophil-driven NASH. Because overexpression of Cxcl1 alone is sufficient to induce NASH similarly to the concomitant overexpression of both Cxcl1 and IL8, we used HFD with Ad-Cxcl1 injection alone (HFD+Cxcl1) in the rest of our study.
Fig. 2. Pharmacological inhibition of p38α/β ameliorates liver injury, oxidative stress, inflammation, and fibrosis in HFD+Cxcl1-treated mice.
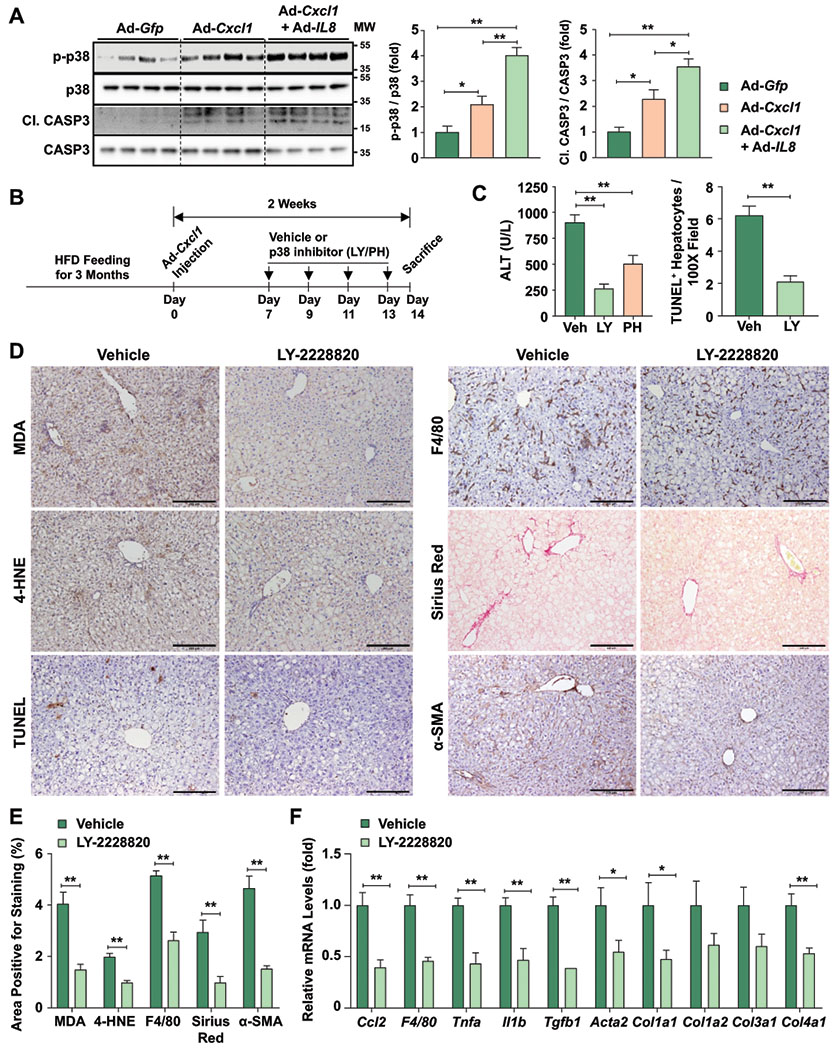
(A) Western blot analyses of liver tissues from HFD-fed mice injected with Ad-Gfp, Ad-Cxcl1, or Ad-Cxcl1 plus Ad-IL8 for 2 weeks (n=4/group). The blot images are shown on the left, and the blots were quantified (right). The p38 antibodies used here detect all four isoforms of p38. (B-F) HFD+Cxcl1-treated mice were injected with vehicle or p38α/β inhibitors intraperitoneally (n=5-6/group) as described in panel B. Serum ALT levels and the number of TUNEL+ hepatocytes/field in the liver sections are shown in panel C. The representative images of various types of staining (scale bars indicate 200 μm) and their quantifications are shown in panels D and E, respectively. Liver tissues were subjected to RT-qPCR analyses of the inflammatory or fibrogenic genes (panel F). Values represent mean ± SEM. Statistical evaluation was performed by Student’s t-test or one-way ANOVA with Tukey’s post hoc test for multiple comparisons (*p<0.05; **p<0.01). C1. CASP3, cleaved form of CASP3; Veh, Vehicle; LY, LY-2228820; PH, PH-797804; MW, molecular weight.
Mice fed an HFD for 3 months were infected with Ad-Cxcl1 and administered with selective p38α/β inhibitors (LY-2228820 or PH-797804)(26,27) or vehicle (Fig. 2B). p38α/β inhibitors significantly reduced serum ALT levels with a greater inhibitory efficacy from LY-2228820 than PH-797804 (Fig. 2C). Thus, we further focused on the LY-2228820 compound and found that treatment with this compound inhibited p38α/β activation in primary mouse hepatocytes and the liver of mice (LY-2228820 inhibits p38α/β to phosphorylate the downstream MK2 rather than directly inhibits p38α/β phosphorylation) (Supporting Fig. S3A–B). Moreover, LY-2228820 markedly attenuated hepatocyte death and reduced oxidative stress, neutrophil infiltration, inflammation, and fibrosis in the liver as evidenced by histological analyses and RT-qPCR analyses of inflammatory genes and fibrogenic genes (Fig. 2C–F, Supporting Fig. S3C–D). These findings support the notion that p38α/β activation is important for the development of HFD+Cxcl1-induced NASH.
Hepatocyte p38α accelerates HFD+Cxcl1-induced NASH
p38α is the most abundant in the liver among the four subtypes of p38.(28) Therefore, we investigated the role of the hepatic p38α subtype in HFD+Cxcl1-induced liver injury by using hepatocyte-specific p38α knockout mice (p38aHep−/−). After HFD+Cxcl1 challenge, p38aHep−/− and WT mice had comparable levels of body weight, liver weight, and liver triglyceride (Fig. 3A–B), as well as similar expression of several genes involved in hepatic lipid metabolism (Supporting Fig. S4A–B), and similar levels of circulating CXCL1 (Supporting Fig. S5A). However, Ad-Cxcl1 infection failed to elevate ALT levels in p38aHep−/− mice in contrast to WT mice whose ALT levels were doubled after 2 weeks of Cxcl1 overexpression (Fig. 3C–D). Resistance of p38aHep−/− mice to HFD+Cxcl1-induced hepatocyte death was further confirmed by TUNEL staining (Fig. 3D, Supporting Fig. S5B). It is noteworthy that Ad-Cxcl1-infected WT (p38a-floxed) mice showed lower ALT levels compared to Ad-Cxcl1-infected C57BL/6J mice (300 U/L vs. 700 U/L) presumably due to the difference in the background of mice (Fig. 1C, Fig. 3C–D). Likewise, the ability of Ad-Cxcl1 to induce hepatic neutrophil infiltration was lower in WT (p38a-floxed) mice than C57BL/6J mice (Supporting Fig. S5C).
Fig. 3. Hepatocyte-specific deletion of p38a ameliorates liver injury and oxidative stress in HFD+Cxcl1-treated mice.
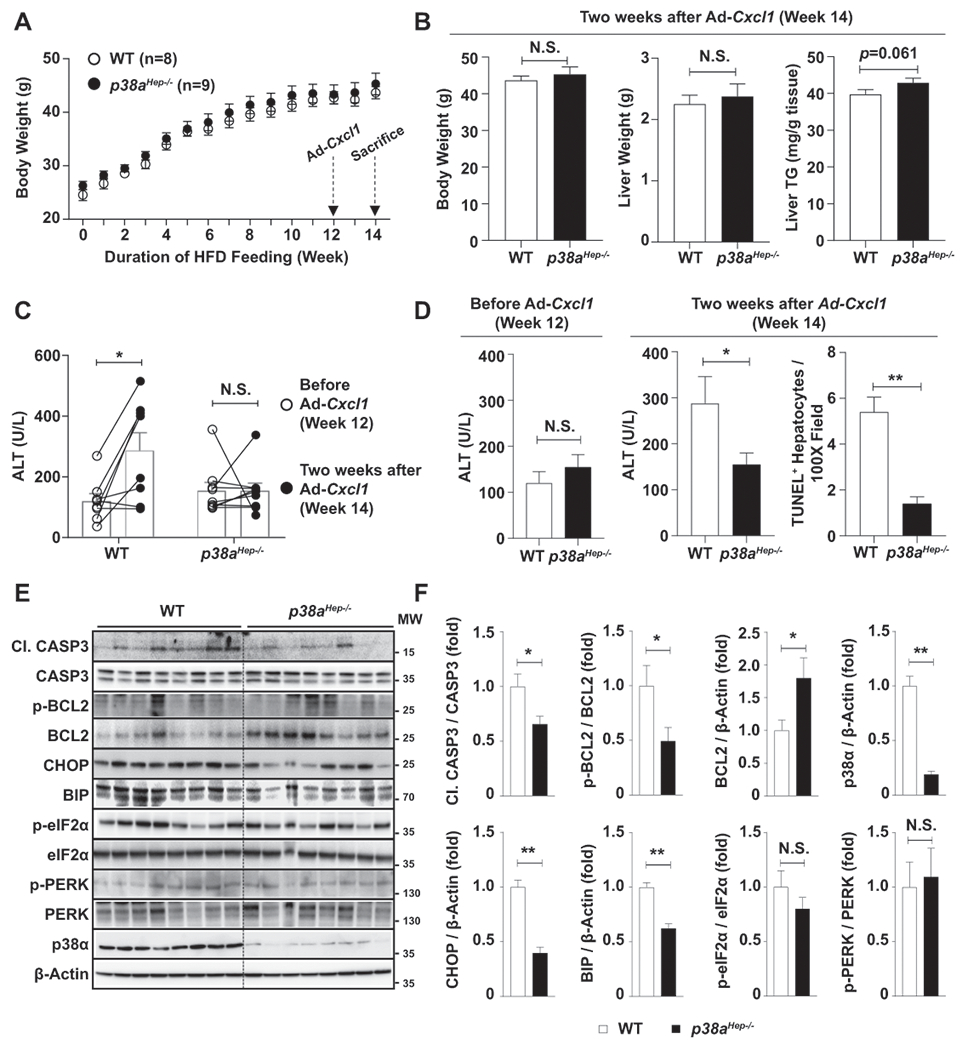
WT and p38aHep−/− mice were fed an HFD for 3 months and injected with Ad-Cxcl1 for 2 weeks (n=8-9/group). (A) Body weight of mice was measured. (B) Body weight, liver weight, and liver triglyceride content at sacrifice. (C, D) Serum ALT levels before Ad-Cxcl1 infection and 2 weeks after Ad-Cxcl1 infection. The number of TUNEL+ hepatocytes/field at sacrifice were statistically compared (panel D, right). The representative images for TUNEL staining are included in Supporting Fig. S5B. (E-F) Liver tissues were subjected to western blot analyses of factors involved in apoptosis and ER stress (panel E), and the blots were quantified (panel F). p38α western blot images were obtained with an antibody specific to p38α isoform. Values represent mean ± SEM. Statistical evaluation was performed by Student’s t-test or one-way ANOVA with Tukey’s post hoc test for multiple comparisons (*p<0.05; **p<0.01). N.S., not significant; TG: triglyceride; Cl. CASP3, cleaved form of CASP3; MW, molecular weight.
To determine the molecular players that mediate CXCL1-induced, p38α-dependent liver injury, we examined apoptosis-associated factors such as CASP3 and BCL2 in the liver of WT and p38aHep−/− mice. As illustrated in Fig. 3E–F, p38aHep−/− mice exhibited less cleavage of CASP3 which is an effector of apoptosis. Also, the expression of an anti-apoptotic factor BCL2 was higher in p38aHep−/− mice, whereas the percentage of phosphorylated BCL2, an inactive form of BCL2, was lower in p38aHep−/− mice compared to WT mice. Furthermore, p38aHep−/− mice exhibited less hepatic expression of ER stress markers such as CHOP and binding immunoglobulin protein (BIP), whose transcription is activated by p38.(24) Notably, phosphorylation of PERK or eIF2α, which is also closely associated with ER stress but not controlled by p38α, was not affected by p38a deletion. Altogether, these results indicate that hepatocyte-specific deletion of p38a protects against HFD+Cxcl1-induced liver injury by modulating factors related to apoptosis (e.g., CASP3 and BCL2) and ER stress-induced cell death (e.g., CHOP and BIP).
We further sought to determine whether p38α activation is involved in the development of inflammation and fibrosis in HFD+Cxcl1-induced NASH. Compared to WT mice, p38aHep−/− mice exhibited lower levels of F4/80+ area and macrophage aggregates surrounding lipid-laden hepatocytes (hepatic crown-like structures) which are indicative of NASH-associated inflammation (Fig. 4A).(29) In addition, p38aHep−/− mice also had a lower number of hepatic neutrophils and lower oxidative stress than WT mice after HFD+Cxcl1 challenge as shown by myeloperoxidase (MPO), MDA, and 4-HNE staining (Supporting Fig. S5C, Fig. 4B). Furthermore, deletion of p38a significantly reduced the activation of myofibroblasts as evidenced by a reduction in α-SMA+ area; however, collagen deposition showed a tendency of decrease without statistical significance (Fig. 4C). Improvement in inflammation was further confirmed by a downregulation of Ccl2 and Tnfa in the liver, whereas fibrogenic genes did not demonstrate a significant reduction by p38a deletion (Fig. 4D). Overall, p38α activation in hepatocytes plays a crucial role in CXCL1-induced tissue injury, oxidative stress, and inflammation in the liver, whereas p38α plays a redundant role in CXCL1-induced fibrogenesis which could involve multiple mechanisms that are independent of p38α.
Fig. 4. Hepatocyte-specific deletion of p38a ameliorates liver inflammation and to a lesser extent liver fibrosis in HFD+Cxcl1-treated mice.
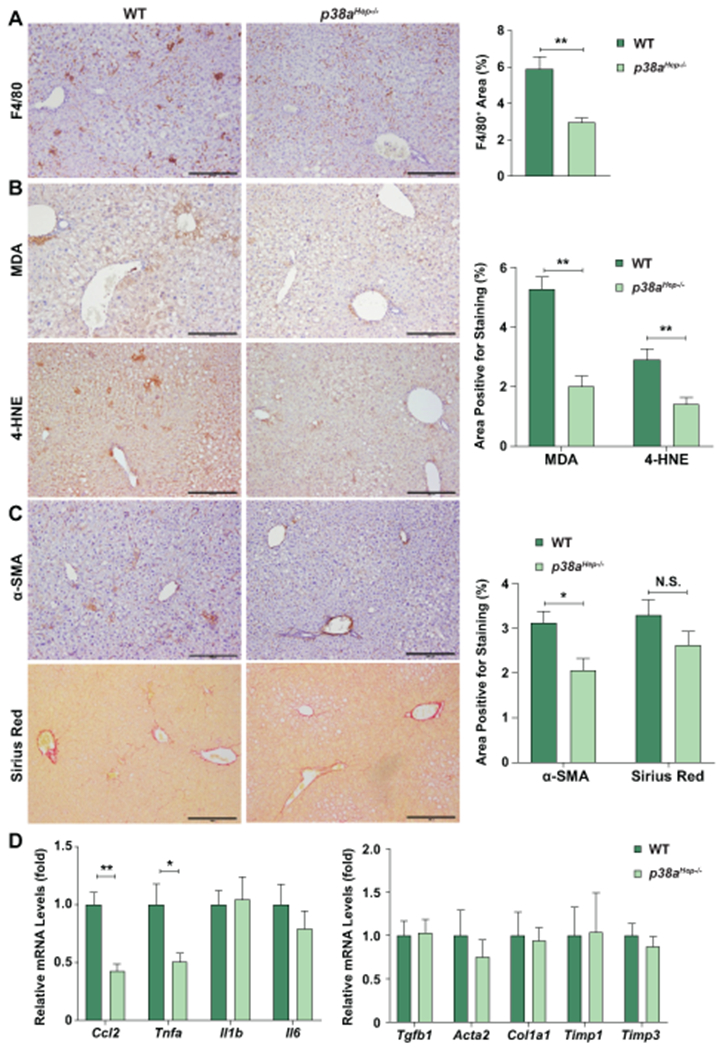
WT and p38aHep−/− mice were fed an HFD for 3 months and injected with Ad-Cxcl1 for 2 weeks (n=8-9/group). (A-C) Liver sections were subjected to F4/80 staining (panel A), MDA and 4-HNE staining (panel B), and α-SMA and Sirius red staining (panel C). Quantifications of the positive staining areas are shown on the right side. Scale bars indicate 200 μm. (D) Liver tissues were subjected to RT-qPCR analyses of inflammatory mediators (left) and fibrogenic genes (right). Values represent mean ± SEM. Statistical evaluation was performed by Student’s t-test (*p<0.05; **p<0.01). N.S., not significant.
Hepatocyte p38α protects mice from HFD-induced steatosis
Our finding that p38α mediates oxidative stress promotion of steatosis-to-NASH progression prompted us to further investigate whether p38α also plays a pathogenic role at an earlier stage of NAFLD progression in which fat accumulation is severe, whereas oxidative stress is less prominent. Therefore, we analyzed mice at steatosis stage after 3 months of HFD feeding alone. The data in Fig. 3D revealed a trend toward higher serum ALT levels in p38aHep−/− mice compared to WT mice after 3-month HFD feeding, but it did not reach statistical difference. To further clarify the role of p38α in steatosis development, we increased the sample size by performing three independent experiments. Notably, p38aHep−/− mice had greater steatosis as demonstrated by H&E staining and higher levels of hepatic triglyceride content than WT mice after HFD feeding (Fig. 5A–B). Deletion of p38a and the subsequent increase in fat accumulation promoted hepatocyte death as evidenced by TUNEL staining and elevation of serum ALT levels (Fig. 5C–D). In addition, p38aHep−/− mice exhibited higher level of oxidative stress and inflammation than WT mice (Fig. 5C–D, Supporting Fig. S6A), albeit to a lesser extent compared to those shown in NASH induced by Cxcl1 overexpression. However, p38aHep−/− mice did not exhibit an increase in fibrosis (Supporting Fig. S6A). There were no significant changes in apoptotic factors by p38a deletion in mice fed an HFD alone (Supporting Fig. S6B).
Fig. 5. Hepatocyte-specific deletion of p38a enhances steatosis, liver injury, and inflammation in HFD-fed mice.
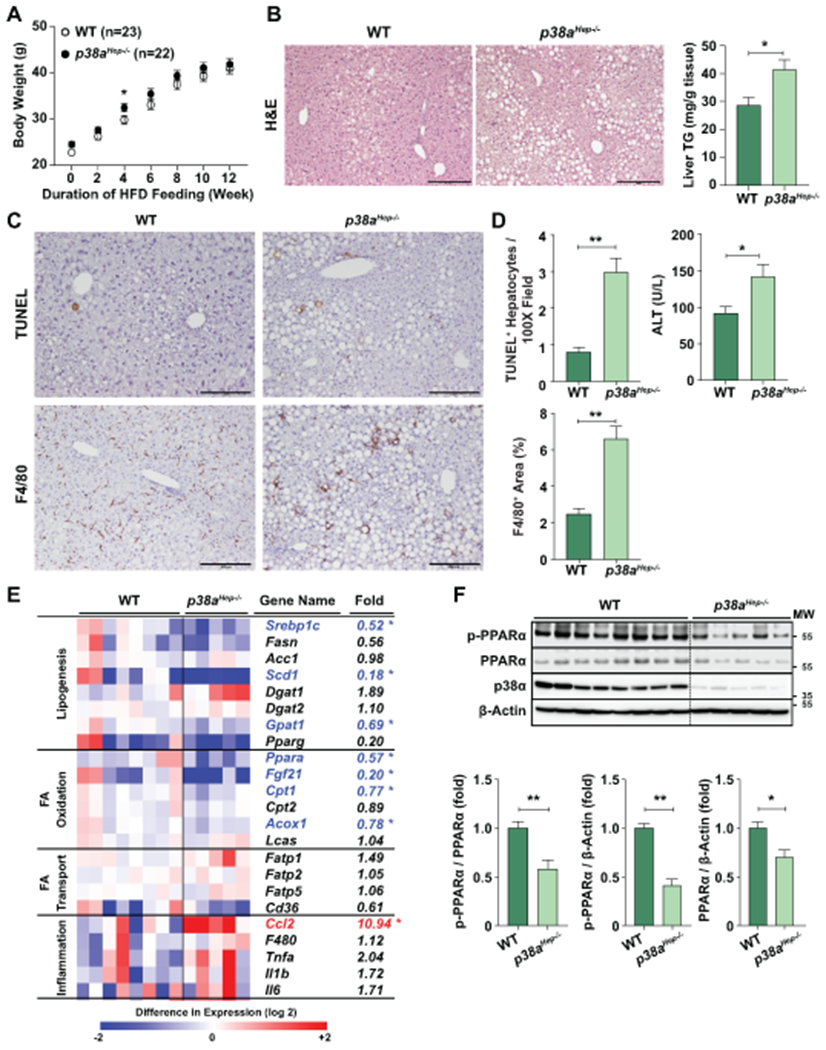
WT and p38aHep−/− mice were fed an HFD for 3 months without Ad-Cxcl1 injection. (A) Body weight of mice was measured (n=22-23/group, pooling of three independent experiments). (B) Liver sections were subjected to H&E staining (left), and liver tissues were subjected to the measurement of triglyceride levels (right) (n=5-8/group). (C) Liver sections were subjected to TUNEL and F4/80 staining. Scale bars indicate 200 μm. (D) The number of TUNEL+ hepatocytes and F4/80+ area was quantified (n=5-8/group), and serum ALT levels were measured (n=22-23/group, pooling of three independent experiments). (E) RT-qPCR analyses of hepatic genes related to lipid metabolism from WT and p38aHep−/− mice (n=5-8/group). Heat map illustration is shown in the left, and the fold induction is shown in the right. * indicates statistical difference (p<0.05). (F) Liver tissues were subjected to western blot analyses of phosphorylated PPARα and total PPARα (top) (n=5-8/group), and the blots were quantified (bottom). p38α western blot images were obtained with an antibody specific to p38α isoform. Values represent mean ± SEM. Statistical evaluation was performed by Student’s t-test (*p<0.05; **p<0.01). TG: triglyceride; MW, molecular weight.
To understand why p38aHep−/− mice are more susceptible to HFD-induced steatosis, we performed RT-qPCR analyses of various genes involved in lipogenesis, FA oxidation, FA transport, and inflammation all of which contribute to homeostatic control of hepatic fat content (Fig. 5E). Notably, hepatic expression of the genes involved in FA oxidation such as Fgf21, Cpt1, and Acox1 was significantly lower in p38aHep−/− mice than in WT mice. Interestingly, hepatic expression of Ppara, a gene encoding PPARα that is an upstream regulator of these FA oxidation genes,(30) was also markedly lower in p38aHep−/− mice versus WT mice. Surprisingly, despite more steatosis, HFD-fed p38aHep−/− mice had lower hepatic expression of several lipogenic genes than HFD-fed WT mice, which may be due to the compensatory inhibition by an excessive hepatic fat accumulation in p38aHep−/− mice. Also, mRNA levels of Pparg that transactivates lipogenic genes (e.g., Cidea and Cidec) showed a trend of decrease by p38a ablation in our study although it did not reach statistical difference. This is consistent with a recent report that inhibition of p38 slightly reduced PPARγ and its target genes in HFD-fed C57BL/6J (WT) mice although it markedly inhibited these genes in HFD-fed MAPK phosphatase 5 KO mice that were associated with severe steatosis.(31) Collectively, p38aHep−/− mice are more susceptible to HFD-induced steatosis due to the reduced hepatic expression of PPARα, not lipogenic genes, and subsequent reduced FA oxidation. Therefore, we further examined PPARα phosphorylation, which stimulates its ligand-induced transactivation, and its own expression.(32) As illustrated in Fig. 5F, western blot analyses revealed that hepatocyte-specific deletion of p38a markedly reduced the phosphorylation of PPARα, along with a mild reduction in total PPARα expression.
p38α attenuates fat accumulation in hepatocytes
The above data suggest that p38α reduces fat accumulation in hepatocytes in HFD-fed mice in vivo. To determine whether p38α directly inhibits fat accumulation in hepatocytes and does not require the interaction with other nonparenchymal and immune cells, we performed in vitro cell culture experiments. As illustrated in Fig. 6A–B, p38a-deleted hepatocytes from p38aHep−/− mice had higher basal levels of fat content than WT hepatocytes as illustrated by oil red O staining. Palmitic acid (PA) treatment markedly increased fat content in hepatocytes, which was further elevated by p38a deletion. Furthermore, pharmacological inhibition of p38α/β by LY-2228820 or PH-797804 similarly enhanced fat accumulation in mouse AML12 hepatocytes (Fig. 6C, Supporting Fig. S7A). Next, we compared PPARα phosphorylation and expression of its target genes involved in FA β-oxidation between WT and p38a-deleted primary hepatocytes. Our data in Fig. 6D revealed that deletion of p38a attenuated PPARα phosphorylation and mildly reduced PPARα expression in both vehicle- and PA-treated cells. As a result, mRNA levels of PPARα target genes involved in FA β-oxidation (e.g., Cpt1, Cpt2, and Acox1) were significantly reduced in p38a-deficient hepatocytes (Fig. 6E). These findings suggest that p38α inhibits hepatocyte fat accumulation through phosphorylation of PPARα and transactivation of its target genes which activate β-oxidation of FA that is derived from exogenous supply (with PA treatment) as well as endogenous synthesis (without PA treatment).
Fig. 6. Inhibition of p38α downregulates PPARα target genes and enhances fat accumulation in mouse hepatocytes.
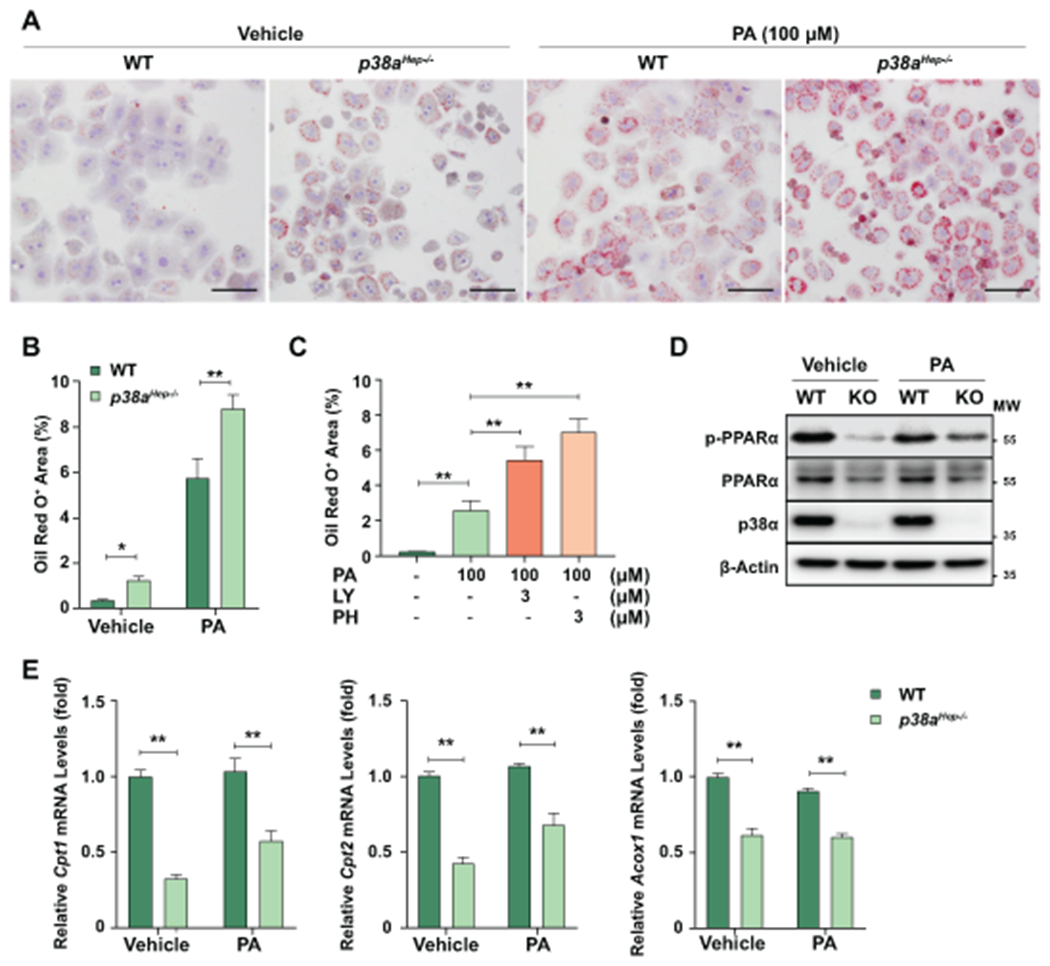
(A-B) WT and p38aHep−/− mouse hepatocytes were treated with vehicle or PA (100 μM) for 24 hr. (A) Representative images of oil red O staining from three independent experiments. Scale bars indicate 100 μm. (B) Quantification of the area positive for oil red O staining. (C) AML12 cells were treated with LY-2228820 or PH-797804 for 1 hr followed by 24-hr treatment with PA. Quantification of the area positive for oil red O staining. Representative staining images are included in Supporting Fig. S7A. (D-E) WT and p38aHep−/− mouse hepatocytes were treated with vehicle or PA (100 μM) for 24 hr. Total cell lysates were subjected to western blot analyses of phosphorylated PPARα and total PPARα (panel D). p38α western blot images were obtained with an antibody specific to p38α isoform. RNA extracted from hepatocytes were subjected to RT-qPCR analyses of PPARα target genes (panel E). Values represent mean ± SEM. Statistical evaluation was performed by one-way ANOVA with Tukey’s post hoc test for multiple comparisons (*p<0.05; **p<0.01). MW, molecular weight.
p38α mediates oxidative stress-induced hepatocyte death
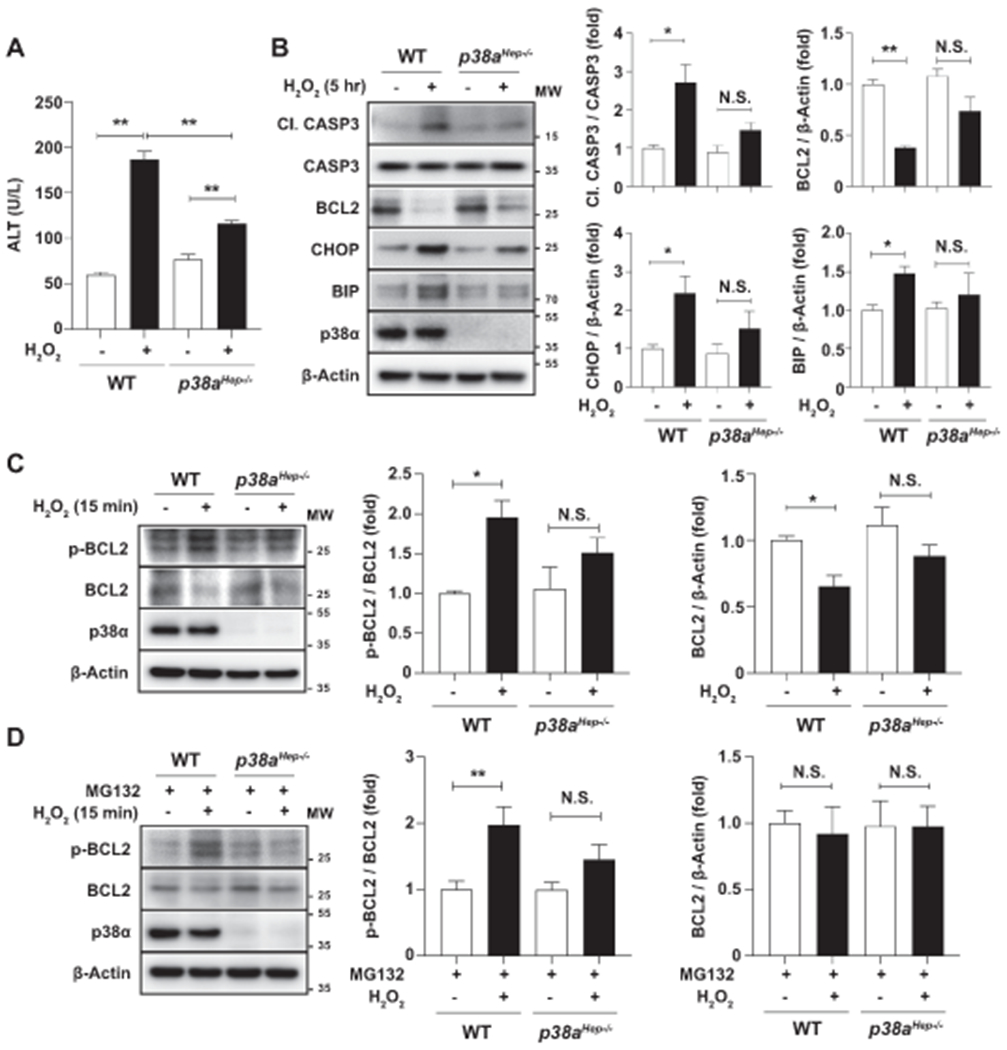
We next sought to determine the molecular mechanisms by which p38α activation mediates oxidative stress-stimulated hepatocyte death in HFD+Cxcl1-induced NASH. In above experiments, a low concentration of PA (100 μM) failed to induce cell death (Supporting Fig. S7B–C), which is in agreement with previous studies reported by others.(33) Next, we used H2O2, which is a strong inducer of oxidative stress, to treat primary mouse hepatocytes isolated from WT and p38aHep−/− mice. As illustrated in Fig. 7A, H2O2 treatment induced hepatocyte death within 5 hr of treatment, which was lowered in p38a-deficient hepatocytes as demonstrated by measuring ALT levels in the supernatant. Moreover, Fig. 7B shows that H2O2 treatment increased the cleavage of CASP3 and downregulated the expression of an anti-apoptotic factor BCL2 in WT hepatocytes, which were reversed in p38a-deficient hepatocytes. Furthermore, H2O2 treatment increased the expression of CHOP and BIP in WT hepatocytes, but such induction was diminished in p38a-deficient hepatocytes.
Fig. 7. Deletion of p38a attenuates H2O2-induced death of primary mouse hepatocytes.
(A-B) WT and p38aHep−/− mouse hepatocytes were treated with vehicle or H2O2 (500 μM) for 5 hr. ALT levels were determined from the supernatant of culture media to examine the death of hepatocytes (panel A). Total cell lysates were subjected to western blot analyses of the factors involved in apoptosis and ER stress (panel B, left), and the blots were quantified (panel B, right). (C) WT and p38aHep−/− mouse hepatocytes were treated with vehicle or H2O2 (500 μM) for 15 min. Total cell lysates were subjected to western blot analyses of phosphorylated and total BCL2 (left), and the blots were quantified (right). (D) WT and p38aHep−/− mouse hepatocytes were first treated with MG132 (20 μM) for 3 hr and then with H2O2 (500 μM) or vehicle for 15 min. Total cell lysates were subjected to western blot analyses of phosphorylated and total BCL2 (left), and the blots were quantified (right). p38α western blot images were obtained with an antibody specific to p38α isoform. Values represent mean ± SEM from three independent experiments. Statistical evaluation was performed by one-way ANOVA with Tukey’s post hoc test for multiple comparisons (*p<0.05; **p<0.01). N.S., not significant; MW, molecular weight.
Since p38 phosphorylates BCL2 at serine 87 and suppresses its anti-apoptotic function,(22) we examined the change in the phosphorylation status of BCL2 in hepatocytes treated with H2O2 for a shorter period of time (15 min). H2O2 reduced the expression of BCL2 as early as 15 min, and the ratio of phosphorylated BCL2 (serine 87) to total BCL2 was increased in WT hepatocytes, which was reversed in p38a-deleted hepatocytes (Fig. 7C). As it is unlikely that transcriptional regulation occurs within 15 min of H2O2 treatment, we postulated that reduction of BCL2 protein levels could be due to the post-transcriptional or post-translational regulation. Thus, we continued to study the degradative regulation of BCL2 as it is known that BCL2 is targeted to proteasomal degradation when phosphorylated at serine residues.(34) When proteasomal degradation was blocked by MG132 (Fig. 7D), H2O2 treatment failed to reduce BCL2 protein levels. Under this condition where BCL2 is stabilized, we could better observe H2O2 stimulation of BCL2 phosphorylation, which was reversed by deletion of p38a. Similarly, H2O2-induced death of AML12 cells was attenuated by pharmacological inhibition of p38α/β which modulated the factors involved in apoptosis and ER stress (Supporting Fig. S8A–D). The above results indicate that p38α mediates oxidative stress-induced hepatocyte death through mechanisms involving CASP3 cleavage, the phosphorylation and degradation of BCL2, and CHOP/BIP-associated ER stress.
Discussion
The pathogenic mechanisms of NAFLD are complex. A subset of patients with steatosis progresses to NASH which possesses more aggressive form of pathology such as liver injury and inflammation, whereas others remain in less severe form of simple steatosis.(7) Despite the effort to address the crucial players in the steatosis-to-NASH progression, it is still obscure what determines the severity of the different stages of NAFLD due to the lack of mouse models that recapitulate the full spectrum of human NAFLD.(35) One of the reasons for difficulty in development of a mouse NASH model is probably the resistance of mouse liver to recruit neutrophils due to the low number of circulating neutrophils in mice and the low expression of neutrophil-recruiting chemokines in mouse hepatocytes as demonstrated in the current study. Indeed, overexpression of these chemokines promoted steatosis-to-NASH progression in HFD-fed mice via the induction of reactive oxygen species (ROS) production and p38 activation. Interestingly, we further demonstrate that activation of p38 plays distinct/complex roles in the pathogenesis of fatty liver and NASH, which are summarized in a scheme in Fig. 8 and discussed below.
Fig. 8. A schematic overview of the distinct roles of p38α depending on the disease stages during NAFLD progression.
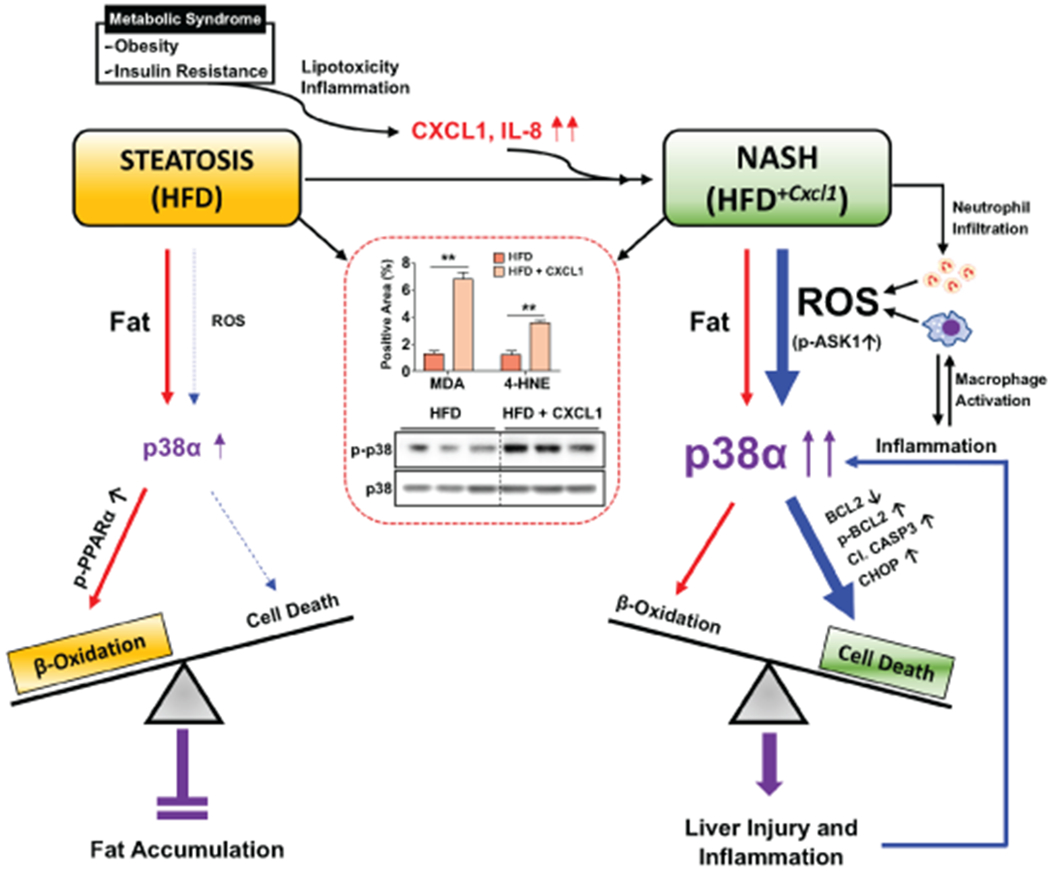
Steatosis is a hepatic manifestation of metabolic syndrome. Lipotoxicity and inflammation induce hepatic expression of CXCL1 and IL-8, which promotes NASH development. Fatty liver induced by 3-month HFD feeding has prominent fat accumulation with relatively low levels of ROS and oxidative stress. Under this condition, activation of p38α is mild, which preferentially activates the signaling pathways that lead to FA β-oxidation via PPARα phosphorylation than those activating hepatocyte death. NASH induced by HFD+Cxcl1 challenge has significant hepatic neutrophil infiltration and subsequent ROS production, which causes a greater degree of oxidative stress and p38 phosphorylation than HFD feeding alone via upstream kinases (e.g., ASK1) as reported by our previous work (illustrated in the red square).(13) This condition further stimulates the signaling pathway leading to hepatocyte apoptosis via BCL2 phosphorylation, CASP3 cleavage, and CHOP upregulation. Therefore, in simple steatosis induced by HFD, p38α plays an adaptive role to protect against fat accumulation, whereas in NASH induced by HFD+Cxcl1, robust p38α activation promotes hepatocyte death and liver inflammation, thereby exacerbating NASH.
To understand why mice are resistant to NASH development, we compared the susceptibility to steatosis-to-NASH progression between mouse and human hepatocytes by examining their neutrophil-recruiting abilities. Our data clearly revealed that mouse hepatocytes had much lower ability to recruit neutrophils than human hepatocytes under inflammatory conditions due to two mechanisms. First, compared to human hepatocytes, mouse hepatocytes not only lacked IL-8 expression but also induced less CXCL1 in response to inflammatory cytokines such as TNF-α and IL-1β. Second, mice have fewer circulating neutrophils than humans.(11,12) These two factors likely at least in part explain why mice are less susceptible to NASH development than humans, although there are various factors responsible for the development of NASH other than neutrophil infiltration. Furthermore, our data demonstrated that TNF-α and IL-1β, but not IL-6, stimulated CXCL1 production by human and mouse hepatocytes, suggesting that CXCL1 induction in hepatocytes is likely to be dependent on NF-kB signaling pathway, whose upstream regulators include TNF-α and IL-1β, but not IL-6.(36) It would be interesting to explore why TNF-α and IL-1β induce greater production of CXCL1 in human hepatocytes than in mouse hepatocytes. Further studies are required to explore the regulatory mechanisms of CXCL1 such as comparative analysis of NF-κB response elements in the promoter region of the CXCL1 gene between species, which will help understand the reason for resistance of mice to NASH development.
Zhang et al. recently reported that deletion of hepatocyte p38a exacerbated steatosis in HFD-fed mice.(16) Surprisingly, in their study, WT and p38aHep−/− mice gained 7 to 11 g body weight after 12-week HFD feeding, which is much lower than the typical body weight gain (∼20 g) after 12-week HFD feeding as observed in our study. Moreover, Zhang et al. reported that p38aHep−/− mice gained much more body weight than WT mice (11 g vs. 7 g) after 12-week HFD feeding. This greater body weight gain might contribute to stronger steatosis phenotypes observed in p38aHep−/− mice than in WT mice by Zhang et al. In contrast, in our study, we observed similar body weight gain (20 g) between WT and p38aHep−/− mice after 3-month HFD feeding. The discrepancy between our study and theirs may be due to the differences in animal facility, the source of HFD, and background of mice. Interestingly, despite similar body weight gain after HFD feeding in our study, p38aHep−/− mice had greater steatosis than WT mice, indicating that p38α indeed protects against HFD-induced fatty liver development. Mechanistically, we provided in vivo and in vitro evidence suggesting that p38α can directly phosphorylate PPARα and subsequently activates transcription of its target genes involved in FA β-oxidation, thereby ameliorating steatosis. We found that hepatic PPARα activation and expression of its downstream FA β-oxidation-related genes were markedly attenuated in HFD-fed p38aHep−/− mice compared to HFD-fed WT mice. Furthermore, hepatic expression of several FA synthesis genes such as Srebp1c, Scd1, and Gpat1 was not enhanced, but rather decreased in mice with hepatic p38a deletion, suggesting that p38α may promote FA synthesis. Collectively, the beneficial role of p38α in the control of steatosis is likely mediated via the promotion of FA β-oxidation, which may conceal its promotion of FA synthesis.
In contrast to HFD feeding alone, HFD+Cxcl1 challenge strongly induced neutrophil infiltration and ROS production, which robustly activated p38α in the liver. Pharmacological inhibition of p38α/β or genetic deletion of p38a in hepatocytes ameliorated HFD+Cxcl1-induced liver injury, inflammation, and fibrosis with reductions in BCL2 phosphorylation, CASP3 cleavage, and CHOP expression, suggesting that p38α promotes NASH development via the regulation of cell survival and ER stress. In addition to ROS, several inflammatory cytokines such as IL-1β and TNF-α,(37) can also activate p38α. Thus, activation of p38α exacerbates liver injury and inflammation which subsequently enhance p38α activation, forming a positive feedback loop for p38α acceleration of NASH.
Deletion of hepatocyte p38a exacerbated steatosis in HFD-fed mice as discussed above but surprisingly, did not increase liver triglyceride levels or reduce expression of FA β-oxidation genes (such as Ppara and its target genes) in HFD+Cxcl1-induced NASH model. After 3-month HFD feeding, liver triglyceride levels in WT and p38aHep−/− mice were approximately 30 and 40 mg/g liver tissue, respectively; however, an additional overexpression of Cxcl1 increased liver triglyceride levels up to 40 mg/g liver tissue in both WT and p38aHep−/− mice. Among various possibilities, it is probable that CXCL1-induced oxidative stress and inflammation, which are known to stimulate hepatic triglyceride accumulation,(38,39) could maximize triglyceride-accumulating signal in hepatocytes such that p38a deletion fails to further increase liver triglyceride levels and enhance subsequent fat-driven detrimental effects, making it possible to study NASH pathogenesis without interference from processes affected by a difference in hepatic fat content.
One interesting question raised from the current study is how certain p38α downstream targets are selected to be activated by particular factors under different conditions.(14) Several types of stimuli are known to induce p38α activation such as FA, ROS, and inflammatory cytokines with different strength depending on the stages of NAFLD progression. We observed that HFD-induced steatosis caused a mild p38α activation and promoted PPARα signaling without strong activation of cell death-related signaling, while HFD+Cxcl1-induced NASH exhibited a more robust activation of p38α which amplified cell death-related signaling such as BCL2 phosphorylation, CASP3 cleavage, and CHOP upregulation. Efficiency of p38α signaling is known to be determined by the interaction of p38α with multiple factors including receptors, upstream kinases (e.g., ASK1, TAK1, MKK3/6, and MKK4), and substrates.(40,41) It is probable that various stimuli activate p38α to different extents via diverse upstream kinases, and the outcome of this differential degree of activation is a selective stimulation of downstream processes, possibly determined by the efficiency to phosphorylate each substrate or by the mediators involved in phosphorylation reaction.(40,41) Moreover, CASP3 cleavage and CHOP expression which are indirectly controlled by p38α could involve more complex mechanisms for regulation. Further studies are warranted to elucidate these processes involved in the pathogenesis of steatosis and NASH.
Recent clinical trials reported that ASK1 inhibitors failed to improve NASH. As p38α is a downstream target of ASK1, the next question is whether p38 inhibitors, which have been actively investigated for the treatment of various diseases,(42) will have therapeutic potential for the treatment of NASH. Here, we tested the role of p38α in the development of NAFLD by taking both pharmacological and genetic approaches. Interestingly, pharmacological inhibition of p38α/β using LY-2228820 markedly attenuated liver injury and fibrosis in the HFD+Cxcl1-induced NASH model, whereas hepatocyte-specific deletion of p38a also ameliorated them, but to a lesser extent. A recent study reported that myeloid-specific deletion of p38a protected mice from development of steatohepatitis by repressing inflammatory cytokine secretion.(16) Thus, it is possible that LY-2228820 inhibits p38α/β in multiple types of cells including hepatocytes and myeloid cells, thereby attenuating HFD+Cxcl1-induced NASH more effectively than specific deletion of hepatocyte p38a. As NASH patients have higher levels of phosphorylated p38α in the liver,(16) inhibition of p38α may have therapeutic benefits for the treatment of NASH by ameliorating hepatocyte death, inflammation, and fibrosis. However, we also found that p38a deletion exacerbated hepatic fat accumulation and subsequently caused liver injury in HFD-induced fatty liver model, in opposition to the above notions that support the p38α inhibition therapy. Hence, caution is required when considering the p38α inhibition therapy for NASH treatment as p38α plays both protective and detrimental roles in different stages of NAFLD progression.
Supplementary Material
Acknowledgments
Financial support:
This work was supported by the intramural program of NIAAA, NIH (B.G.)
Abbreviations:
- ALT
alanine aminotransferase
- ASK1
apoptosis signal-regulating kinase 1
- BCL2
B-cell lymphoma 2
- BIP
binding immunoglobulin protein
- CASP3
caspase-3
- CHOP
C/EBP homologous protein
- CXCL1
C-X-C motif chemokine ligand 1
- FA
fatty acid
- HFD
high-fat diet
- 4-HNE
4-hydroxynonenal
- IL
interleukin
- MAPK
mitogen-activated protein kinase
- MDA
malondialdehyde
- MPO
myeloperoxidase
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PA
palmitic acid
- PPARα
peroxisome proliferator-activated receptor alpha
- ROS
reactive oxygen species
- RT-qPCR
reverse transcription quantitative PCR
- α-SMA
alpha-smooth muscle actin
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
wild-type
References
Author names in bold designate shared co-first authorship.
- 1.Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349–364. [DOI] [PubMed] [Google Scholar]
- 2.Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology 2016;150:1704–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018;67:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019;69:2672–2682. [DOI] [PubMed] [Google Scholar]
- 5.Zhou F, Zhou J, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Unexpected Rapid Increase in the Burden of NAFLD in China From 2008 to 2018: A Systematic Review and Meta-Analysis. Hepatology 2019;70:1119–1133. [DOI] [PubMed] [Google Scholar]
- 6.Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2019;16:377–386. [DOI] [PubMed] [Google Scholar]
- 7.Rinella ME, Tacke F, Sanyal AJ, Anstee QM, participants of the AEW. Report on the AASLD/EASL Joint Workshop on Clinical Trial Endpoints in NAFLD. Hepatology 2019;70:1424–1436. [DOI] [PubMed] [Google Scholar]
- 8.Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One 2010;5:e13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 10.Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology 2015;62:1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Vietinghoff S, Ley K. Homeostatic regulation of blood neutrophil counts. J Immunol 2008;181:5183–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47(phox)-oxidative stress pathway in neutrophils. Gut 2017;66:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005;15:11–18. [DOI] [PubMed] [Google Scholar]
- 15.Lawan A, Bennett AM. Mitogen-Activated Protein Kinase Regulation in Hepatic Metabolism. Trends Endocrinol Metab 2017;28:868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Fan L, Wu J, Xu H, Leung WY, Fu K, et al. Macrophage p38alpha promotes nutritional steatohepatitis through M1 polarization. J Hepatol 2019;71:163–174. [DOI] [PubMed] [Google Scholar]
- 17.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015;62:720–733. [DOI] [PubMed] [Google Scholar]
- 18.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest 2006;116:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem 2001;276:44495–44501. [DOI] [PubMed] [Google Scholar]
- 20.Tormos AM, Talens-Visconti R, Nebreda AR, Sastre J. p38 MAPK: a dual role in hepatocyte proliferation through reactive oxygen species. Free Radic Res 2013;47:905–916. [DOI] [PubMed] [Google Scholar]
- 21.Markou T, Dowling AA, Kelly T, Lazou A. Regulation of Bcl-2 phosphorylation in response to oxidative stress in cardiac myocytes. Free Radic Res 2009;43:809–816. [DOI] [PubMed] [Google Scholar]
- 22.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, et al. Bcl-2 Phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem 2006;281:21353–21361. [DOI] [PubMed] [Google Scholar]
- 23.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004;11:381–389. [DOI] [PubMed] [Google Scholar]
- 24.Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta 2014;1843:2150–2163. [DOI] [PubMed] [Google Scholar]
- 25.Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 1996;272:1347–1349. [DOI] [PubMed] [Google Scholar]
- 26.Campbell RM, Anderson BD, Brooks NA, Brooks HB, Chan EM, De Dios A, et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol Cancer Ther 2014;13:364–374. [DOI] [PubMed] [Google Scholar]
- 27.Selness SR, Devraj RV, Devadas B, Walker JK, Boehm TL, Durley RC, et al. Discovery of PH-797804, a highly selective and potent inhibitor of p38 MAP kinase. Bioorg Med Chem Lett 2011;21:4066–4071. [DOI] [PubMed] [Google Scholar]
- 28.Wang XS, Diener K, Manthey CL, Wang S, Rosenzweig B, Bray J, et al. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J Biol Chem 1997;272:23668–23674. [DOI] [PubMed] [Google Scholar]
- 29.Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, et al. Hepatic crown-like structure: a unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS One 2013;8:e82163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr Rev 2018;39:760–802. [DOI] [PubMed] [Google Scholar]
- 31.Tang P, Low HB, Png CW, Torta F, Kumar JK, Lim HY, et al. Protective Function of Mitogen-Activated Protein Kinase Phosphatase 5 in Aging- and Diet-Induced Hepatic Steatosis and Steatohepatitis. Hepatol Commun 2019;3:748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 2007;1771:952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007;46:823–830. [DOI] [PubMed] [Google Scholar]
- 34.Basu A, Haldar S. Signal-induced site specific phosphorylation targets Bcl2 to the proteasome pathway. Int J Oncol 2002;21:597–601. [PubMed] [Google Scholar]
- 35.Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, et al. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019;69:2241–2257. [DOI] [PubMed] [Google Scholar]
- 36.Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 2013;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, et al. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm 2014;2014:352–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grunfeld C, Soued M, Adi S, Moser AH, Dinarello CA, Feingold KR. Evidence for two classes of cytokines that stimulate hepatic lipogenesis: relationships among tumor necrosis factor, interleukin-1 and interferon-alpha. Endocrinology 1990;127:46–54. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med 2020;152:116–141. [DOI] [PubMed] [Google Scholar]
- 40.Yang SH, Sharrocks AD, Whitmarsh AJ. Transcriptional regulation by the MAP kinase signaling cascades. Gene 2003;320:3–21. [DOI] [PubMed] [Google Scholar]
- 41.Mayor F Jr., Jurado-Pueyo M, Campos PM, Murga C. Interfering with MAP kinase docking interactions: implications and perspective for the p38 route. Cell Cycle 2007;6:528–533. [DOI] [PubMed] [Google Scholar]
- 42.Buhler S, Laufer SA. p38 MAPK inhibitors: a patent review (2012 – 2013). Expert Opin Ther Pat 2014;24:535–554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.