

Abstract
Multiple sclerosis (MS) is an autoimmune demyelinating and neurodegenerative disease of the central nervous system, and the leading cause of non-traumatic neurological disability in young adults. Effective management requires a multifaceted approach to control acute attacks, manage progressive worsening, and remediate bothersome or disabling symptoms associated with this illness. Remarkable advances in treatment of all forms of MS, and especially for relapsing disease, have favorably changed the long-term outlook for many patients. There also has been a conceptual shift in understanding the immune pathology of MS, away from a purely T-cell-mediated model to recognition that B cells have a key role in pathogenesis. The emergence of higher-efficacy drugs requiring less frequent administration have made these preferred options in terms of tolerability and adherence. Many experts now recommend use of these as first-line treatment for many patients with early disease, before permanent disability is evident.
Keywords: Multiple Sclerosis, MS Therapy, B cell Therapy
INTRODUCTION
The autoimmune disease multiple sclerosis (MS) is the leading cause of non-traumatic neurological disability arising in young adults.1,2 MS is characterized by two pathological hallmarks: 1) inflammation with demyelination, and 2) astroglial proliferation (gliosis) and neurodegeneration. Tissue damage in MS is restricted to the central nervous system (CNS), sparing the peripheral nervous system. Clinically, MS can follow two paths: relapsing or progressive. Most commonly, onset is a relapsing form of MS (RMS), manifested as discrete episodes of neurological dysfunction followed by partial, complete, or no remission. Over time, relapses usually decrease in frequency but a gradual worsening often supervenes, resulting in uninterrupted progression (termed secondary progressive MS [SPMS]); (Figure 1).3 Less than 10% of patients with MS experience progression from onset, a category termed primary progressive MS (PPMS).3 Despite these distinctions, all clinical forms of MS appear to reflect the same underlying disease process. And although inflammation is typically associated with relapses, and neurodegeneration with progression, it is now recognized that both pathologies are present in essentially all patients across the entire disease continuum.
Figure 1. The New Natural History of MS.
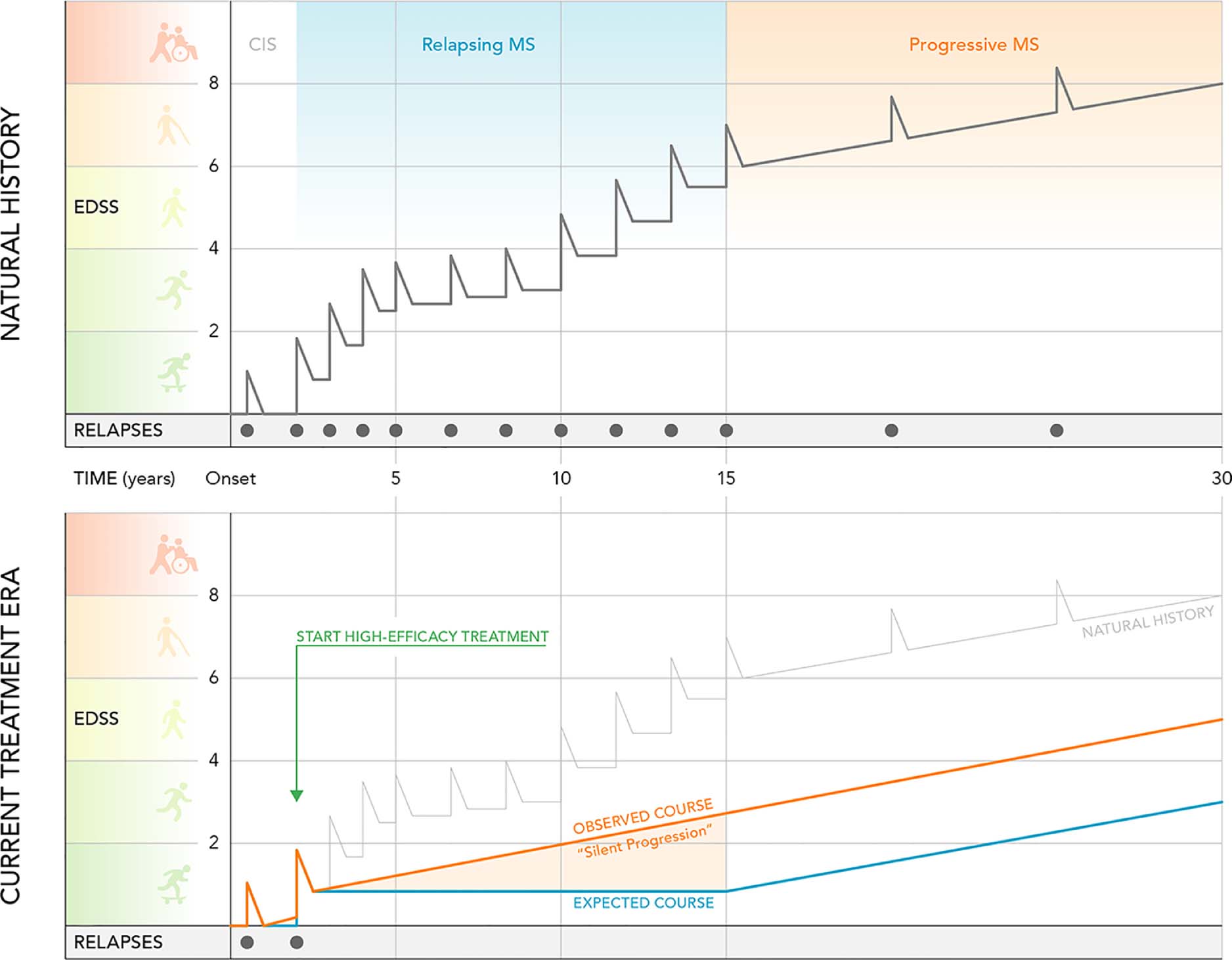
The top half of the figure illustrates the natural history of relapse-onset MS in the pretreatment era. During the relapsing phase, disability accumulation was thought to result from incomplete recovery from relapses, until relapse-independent disability, designated SPMS, supervened. The “new” natural history of MS in the current treatment era is shown in the bottom half. With use of highly effective therapies, attacks are abolished in most patients, but insidious progression independent of relapse activity, termed “silent progression”, is now evident during the relapsing phase.
Abbreviations: EDSS, extended disability status score; CIS, clinically isolated syndrome; MS, multiple sclerosis; SPMS, secondary progressive multiple sclerosis.
MS is a global problem, and its prevalence is on the rise.4 The prevalence is highest in North America, Western Europe and Australasia (>100 cases per 100,000 population), and lowest in countries centered around the equator (<30 cases per 100,000 population).4 In the US, a recent study estimated that nearly 1 million individuals are affected. In RMS, women are affected nearly three times more often than men and the mean age of onset is ~30 years, whereas in PPMS the rates of men and women affected are similar and the mean age of onset is ~40 years.5–7
The development of increasingly effective therapies for RMS, and partially effective therapy for PPMS and SPMS, represents a profound success that has dramatically improved prospects for lives free from disability. For patients with RMS, the mean time to development of SPMS was historically estimated at approximately 19 years after onset but in the treatment era has been lengthened substantially. On highly effective therapy relapses are markedly reduced or eliminated. However, control of RMS has uncovered a relapse-independent “silent” progression that was previously obscured by attacks and remissions in RMS.8,9 This recognition has also led to an increasing reliance on highly effective therapies early in the course of MS in order to maximally control both relapses and progression. In this review we summarize recent advances in MS treatment and speculate on future directions.
DIAGNOSIS
Clinical Manifestations
MS symptoms vary according to location and severity of lesions occurring within the CNS. Clinical features of RMS may present acutely or subacutely over hours to days, sometimes followed by gradual spontaneous remission over weeks to months. Conversely, PPMS is characterized by slowly progressive symptoms from onset. Table 1 summarizes common clinical and laboratory features of MS. Symptoms may be severe at onset or begin insidiously, sometimes unnoticed for months or years. Once the patient seeks medical attention, and if MS is suspected, prompt referral to a specialist is indicated.
Table 1.
Approach to the Diagnosis of Multiple Sclerosis
| Symptoms | Magnetic Resonance Imaging (MRI) |
|---|---|
|
|
| Cerebrospinal Fluid | |
| |
| Evoked Potentials | |
| |
| Uncommon Symptoms (Red Flags) | |
| |
Investigation
Diagnosis requires objective evidence of inflammatory CNS injury and often additional details of dissemination of the disease process “in space and time”, i.e. affecting more than one CNS location with evolution over time (Table 2). Symptoms must last for >24 hours and occur as distinct episodes separated by at least 1 month. The main tests used to support diagnoses are magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis.
Table 2.
Diagnostic Criteria for Multiple Sclerosis65
| Criteria for diagnosis of multiple sclerosis in patients with an attack at onset |
|---|
| ≥2 attacks; objective clinical evidence of ≥2 lesions or objective clinical evidence of 1 lesion with reasonable historical evidence of a prior attack |
| ≥2 attacks; objective clinical evidence of 1 lesion Dissemination in space, demonstrated by:
|
| 1 attack; objective clinical evidence of ≥2 lesions Dissemination in time, demonstrated by:
|
| 1 attack; objective clinical evidence of 1 lesion (clinically isolated syndrome) Dissemination in space and time, demonstrated by: For dissemination in space
|
|
Criteria for diagnosis of multiple sclerosis in patients with a disease course characterized by progression from onset (primary progressive multiple sclerosis) Insidious neurologic progression suggestive of primary progressive multiple sclerosis 1 year of disease progression (retrospectively or prospectively determined) AND 2 out of the 3 following criteria:
|
CNS, central nervous system; MRI, magnetic resonance imaging; MS, multiple sclerosis.
In most patients an abnormal MRI is observed.10 Leakage of intravenous gadolinium is caused by breakdown in the blood-brain barrier that occurs early in the development of an MS lesion and is a marker of acute inflammation. Gadolinium enhancement typically persists for <1 month; however, the residual MS plaque remains visible indefinitely as a focal area of hyperintensity (a lesion) on fluid-attenuated inversion recovery or T2-weighted MRI scans. A periventricular (adjacent to the ventricles) distribution of lesions, indicative of perivenular inflammation, is characteristic. Lesions in juxtacortical (adjacent to the cerebral cortex) white matter, infratentorial white matter, and within the spinal cord are also suggestive of MS and contribute to “dissemination in space”. Although MS plaques can involve subcortical white matter, lesions in this location are considered non-diagnostic because other pathologies are associated with similar lesions.
Lumbar puncture is helpful, especially in uncertain cases and in all cases of suspected PPMS. CSF abnormalities include a mononuclear cell pleocytosis and an increased level of intrathecally synthesized IgG. Oligoclonal bands reflect the products of a highly focused immune response by activated B cells in the CNS. The abnormal intrathecal synthesis of gamma globulins, measured by an elevated IgG index or two or more discrete oligoclonal bands not present in a paired serum sample, is present in >90% of MS patients. Elevated intrathecal antibody production can also be used to fulfil “dissemination in time” criteria in patients presenting with their first clinical manifestation of MS. Although sensitive, elevated CSF antibody production is not specific for MS, and also occurs with CNS infections. More than 50 cells/mm3 are rare in MS, and polymorphonuclear leukocytes, eosinophils or a markedly elevated total protein level should call the diagnosis into question.
Other useful tests include evoked potentials to assess nerve conduction in CNS pathways, and retinal imaging by optical coherence tomography.
ADVANCES IN TREATMENT
Current management strategies are focused on treating acute attacks, ameliorating symptoms, and reducing biologic activity through disease-modifying therapies. The approach to treating acute attacks and symptom-based management is summarized in the supplement.
Disease-Modifying Therapies
Disease-modifying therapies modify the course of MS through suppression or modulation of immune function. They exert anti-inflammatory activity primarily in the relapsing phase of MS; they reduce the rate of relapses, reduce accumulation of MRI lesions and stabilize, delay, and in some cases modestly improve disability.
The first approved therapies, the interferons and glatiramer acetate, were fortuitous discoveries. These well-tolerated drugs modestly reduce the frequency of MS relapses and soon became commonly prescribed.11 Preclinical studies in experimental autoimmune encephalomyelitis, an animal model of MS, advanced our understanding of crucial steps in the pathogenesis of CNS autoimmune diseases, specifically early peripheral expansion of autoreactive immune cells in secondary lymphoid organs, subsequent infiltration of activated cells into the CNS, and generation of inflammatory lesions in white matter.12,13 These studies showed that T cells play a critical role in experimental autoimmune encephalomyelitis;12 however clinical trials with purely T-cell-based treatment approaches were ineffective in RMS.14,15 In contrast, strategies that inhibited lymphocyte access to the CNS by blocking adhesion (natalizumab) or sequestering lymphocytes in primary lymphoid organs (the sphingosine-1-phosphate [S1P] receptor modulators fingolimod, siponimod, and ozanimod) were found to be effective in both MS and experimental autoimmune encephalomyelitis. A major advance was the development of disease models that more closely replicated the pattern of tissue damage in MS, leading to a new appreciation of the importance of humoral immunity in MS pathogenesis.16,17 This paved the way for clinical trials of B-cell-depleting therapies, initially with the anti-CD20 monoclonal antibody rituximab and followed by the more successful development of ocrelizumab and ofatumumab. Anti-CD20-mediated B-cell depletion demonstrated a high level of success in limiting new relapses and silent progression in RMS,18–20 and in reducing disability progression in PPMS.21 Thus, the past decade has seen a shift in the conceptual framework of MS pathophysiology, from the earlier model that MS is a T-cell-mediated autoimmune disorder, to the understanding of a central role for B cells.22 Although dramatic progress has been made against RMS, the development of highly effective therapies against progressive MS remains an unmet need (Figure 2).
Figure 2. Five Distinctive Pathologies Likely Contribute to Progressive MS.
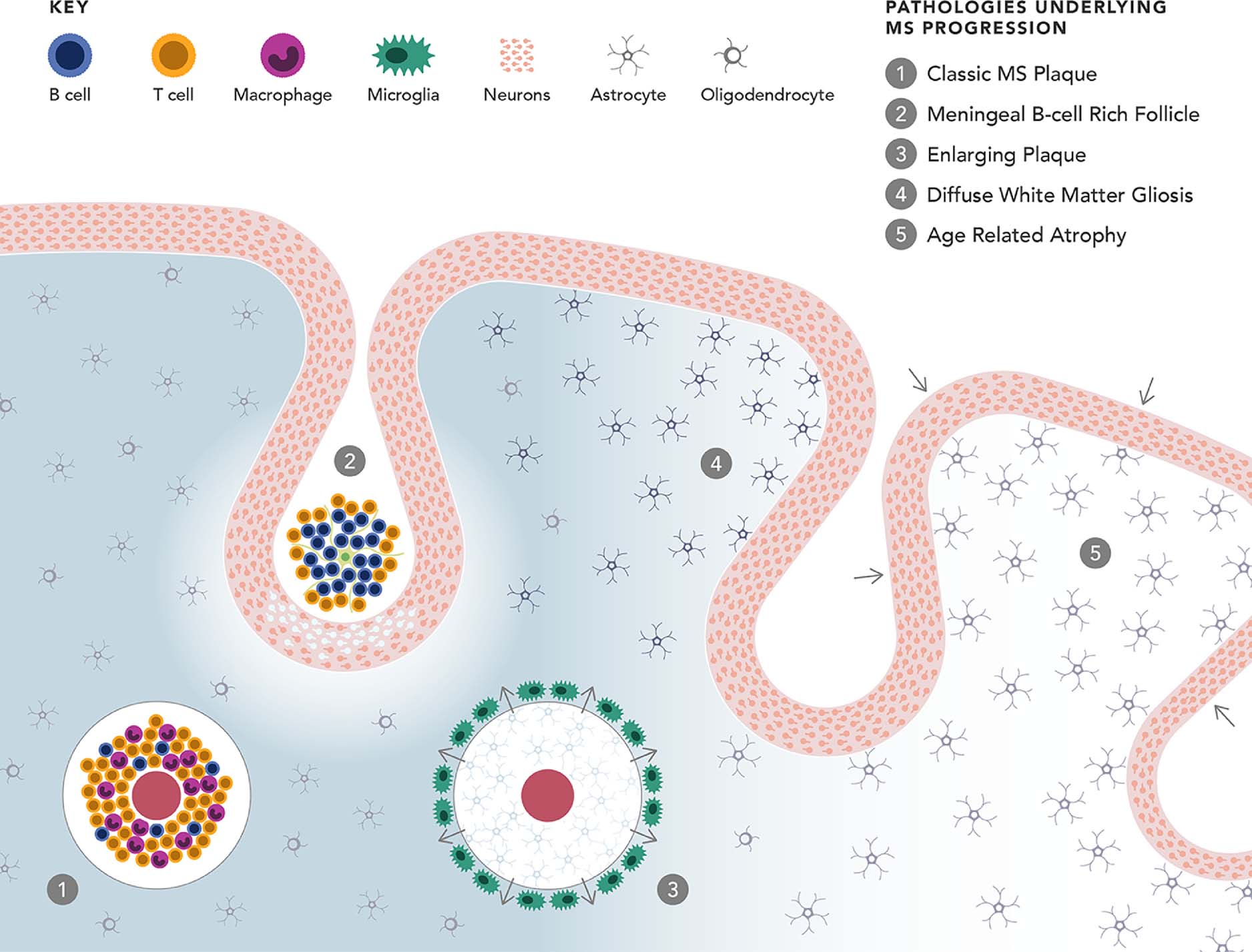
(1) Classic inflammatory white matter plaques are typically associated with relapses but also occur in patients who progress gradually without acute attacks. Hallmarks are perivenous inflammation dominated by lymphocytes and macrophages (detected by MRI as gadolinium enhancement indicating blood brain barrier disruption); demyelination associated with activated microglia; sharply demarcated plaques with glial scarring; and axonal loss with secondary retrograde and anterograde degeneration. (2) B cell rich lymphoid aggregates in the meninges, often in deep sulci, with underlying cortical demyelination and neuronal loss. (3) Slowly enlarging lesions due to gradual concentric expansion of chronic plaques, characterized by a rim of activated microglia at the leading edge, astrocytosis, stressed oligodendrocytes and progressive axonal injury. (4) Widespread diffuse microglial inflammation and astrogliosis throughout the CNS white matter, associated with decreased myelin density and ongoing axonal damage. (5) Age-related neurodegeneration. For a more detailed discussion, readers may consult references66–68
Abbreviations: CNS, central nervous system; MRI, magnetic resonance imaging; MS, multiple sclerosis.
APPROACH TO TREATING PATIENTS WITH MS
Table 3 provides an overview of the characteristics and pivotal data associated with approved agents, stratified according to frequency of use and perceived level of efficacy. Given this large array of available therapies, it may be prudent for clinicians to become conversant with a few agents. As such, the following section is focused on detailing MS disease-modifying therapies that are widely used in clinical practice.
Table 3.
Summary of Approved Disease-Modifying Therapies for Multiple Sclerosis
| Drug name | Mechanism of action | Indicatio n | Route of and frequency of administratio n | Pivotal efficacy data | Common adverse events |
|---|---|---|---|---|---|
| Highly effective | |||||
| Ocrelizumab18,21 | Anti-CD20 mAb | RMS and PPMS (1st line) | IV infusion, every 6 months | RMS: Relative reduction in ARR compared with IFN β- la:47% PPMS: Relative reduction in 12-week CDP compared with placebo: 24% |
RMS: Infusion-related reaction, nasopharyngitis, upper respiratory tract infection, headache, and urinary tract infection PPMS: Infusion-related reaction, upper respiratory tract infection, and oral herpes infection |
| Ofatumumab27 | Anti-CD20 mAb | RMS (1st line) | SC injection, every 4 weeks | Relative reduction in ARR compared with teriflunomid e: 54% | Injection-related reaction, nasopharyngitis, headache, upper respiratory tract infection, and urinary tract infection |
| Natalizumab30 | α4β1 integrin inhibitor | RRMS (2nd line) | IV infusion, every 4 weeks | Relative reduction in ARR compared with placebo: 68% Relative reduction in sustained disease progression compared with placebo: 42% | Fatigue and allergic reaction |
| Alemtuzumab59–61 | Anti-CD52 mAb | RMS (1st line) | IV infusion, once daily | Relative reduction in ARR compared with placebo: 49–69% | Headache, rash, nausea, and pyrexia |
| Mitoxantrone58 | DNA intercalator | RMS, SPMS (2nd or 3rd line) | IV infusion, every month or 3 months | Relative reduction in relapses compared with placebo: 61% | Dose-related cardiomyopathy, promyelocytic leukemia |
| Moderately effective | |||||
| FingolimodM34,36 | Sphingosine- 1- phosphate inhibitor | RMS (2nd line) | Oral, once daily | Relative reduction in ARR compared with placebo: 48–60% | Bradycardia, atrioventricular conduction block, macular edema, elevated liver-enzyme levels, and mild hypertension |
| Siponimod64 | Sphingosine 1- phosphate receptor modulator | CIS, RMS, active SPMS (1st Line) | Oral, once daily | Relative reduction in 12-week CDP compared with placebo: 21% | Headache, nasopharyngitis, urinary tract infection, and falls |
| Ozanimod39,40 | Sphingosine 1- phosphate receptor modulator | CIS, RMS, active SPMS | Oral, once daily | Relative reduction in ARR compared with placebo: 48% | Headache and elevated liver aminotransferas e |
| Dimethyl fumarate and diroximel Fumarate41,42 | Nuclear factor (erythroid- derived 2)- like 2 pathway inhibitor | RMS (1st line) | Oral, twice daily | Relative reduction in ARR compared with placebo: 48–53% | Flushing, diarrhea, nausea, upper abdominal pain, decreased lymphocyte counts, and elevated liver aminotransferas e |
| Cladribine62 | Not fully known | RMS (2nd or 3rd line) | Oral, 4–5 days over 2-week treatment courses | Relative reduction in ARR compared with placebo: 55–58% | Headache, lymphocytopeni a, nasopharyngitis, upper respiratory tract infection, and nausea |
| Modestly effective | |||||
| Teriflunomide47 | Dihydroorotat e dehydrogenas e inhibitor | RMS (1st line) | Oral, once daily | Relative reduction in ARR compared with placebo: 32–36% | Nasopharyngitis, headache, diarrhea, and alanine aminotransferas e increase |
| Glatiramer Acetate35 | Not fully known | RMS (1st line) | SC injection, once daily or 3 times weekly | Relative reduction in ARR compared with placebo: 29% | Injection-site reactions |
| IFN β-la (Rebif)69 | Not fully known | CIS and RMS (1st line) | SC injection, 3 times weekly | Relative reduction in ARR compared with placebo: 33% | Injection-site inflammation, flu-like symptoms, rhinitis, and headache |
| IFN β-la (Avonex)51 | Not fully known | CIS and RMS (1st line) | IM injection, once weekly | Relative reduction in 24-week CDP compared with placebo: 37% | Flu-like symptoms, muscle aches, asthenia, chills, and fever |
| PeglFN β-la (Plegridy)70 | Not fully known | CIS and RMS (1st line) | SC injection, every 2 weeks | Relative reduction in ARR compared with placebo: 39% | Injection-site erythema, influenza-like illness, pyrexia, and headache |
| IFN β-lb (Betaseron)50 | Not fully known | CIS and RMS (1st line) | SC injection, every other day | Relative reduction in ARR compared with placebo: 31% | Lymphopenia, flu-like symptoms, and injection-site reactions |
ARR, annualized relapse rate; CDP, confirmed disability progression; CIS, clinically isolated syndrome; IFN β−1a, interferon beta 1a; IM, intramuscular; IV, intravenous; mAb, monoclonal antibody; PPMS, primary progressive multiple sclerosis; RMS, relapsing forms of multiple sclerosis; SC, subcutaneous; SPMS, secondary progressive multiple sclerosis.
Treating RMS
Frequently used disease-modifying therapies for RMS
Ocrelizumab, a humanized monoclonal antibody against the CD20 molecule on the surface of mature B cells,23 has been widely used since its approval in 2017. Ocrelizumab is highly effective against relapses and silent progression in RMS patients, and has dramatic benefits in halting development of new white matter lesions detected by MRI. Ocrelizumab selectively depletes CD20-expressing B cells, preserving pre-existing humoral immunity and the capacity for B-cell reconstitution. B-cell depletion is associated with potent interruption in B-cell trafficking from the periphery to the CNS, reduced B cell antigen presentation to T cells, modulation of proinflammatory cytokine secretion by B cells, and reduced activation and differentiation to immunoglobulin secreting plasmablasts.24,25 Ocrelizumab is administered as an intravenous infusion every 24 weeks. Initial findings from the phase 3 trials indicated a possible low risk of increased malignancies including breast cancer, although longer follow-up revealed cancer rates identical to epidemiologic expectations. Post-marketing studies are generally consistent with the clinical trials although serious herpes virus infections have become a recognized complication.26 Rituximab is a chimeric anti-CD20 monoclonal antibody that appears to be similarly effective against RMS and PPMS in preliminary trials and real world experience and is widely used despite never receiving approval from any regulatory body. Ofatumumab27 is a fully human anti-CD20 monoclonal antibody administered by monthly subcutaneous injection at home and has an efficacy profile similar to ocrelizumab with a high degree of safety in the phase 3 trials for RMS.
Natalizumab, a humanized monoclonal antibody, is an inhibitor of the α4β1 integrin, an adhesion molecule expressed on the surface of lymphocytes and involved in transmigration across endothelia into the CNS.28 Natalizumab is administered as an intravenous infusion once every 4 weeks. Natalizumab is highly effective in reducing relapses and slowing disease progression in RMS patients compared with placebo or IFN β−1a,29,30 benefits sustained over longer-term in real-world studies.31 Natalizumab is generally well tolerated; however, long-term treatment carries risk of progressive multifocal leukoencephalopathy (an opportunistic brain infection caused by the John Cunningham virus), occurring in ~0.4% of natalizumab-treated patients.32 Progressive multifocal leukoencephalopathy incidence increases with natalizumab exposure and this risk can be stratified according to the levels of John Cunningham virus antibodies in serum. In antibody-negative patients, progressive multifocal leukoencephalopathy risk is minimal, <1:10,000. Conversely, in antibody-positive patients, risk may be as high as ≥1.1% annually.32 Thus, natalizumab is generally recommended only for antibody-negative patients. Extending the dosing interval from 4 to 6 weeks appears to reduce the risk of progressive multifocal leukoencephalopathy by more than 90% in seropositive patients, providing a useful strategy in reducing risk.33 A disadvantage of natalizumab is that discontinuation can trigger ‘rebound’ disease activity, a problem that may be encountered in patients who are noncompliant, or John Cunningham virus seroconvert, or who discontinue prior to attempting to conceive.
Fingolimod was the first oral therapy approved for RMS. It is a S1P inhibitor that prevents egress of lymphocytes from secondary lymphoid organs, blocking infiltration of autoreactive lymphocytes into the CNS. Fingolimod is well tolerated, although adverse events include mild abnormalities on routine laboratory evaluation (e.g. elevated liver function tests or lymphopenia).34–37 Heart block and bradycardia can also occur when therapy is initiated, thus a 6-hour observation period (including electrocardiogram monitoring) is recommended for all patients receiving their first dose.38 Other side effects include macular edema and, rarely, disseminated varicella-zoster virus, cryptococcal infections, and progressive multifocal leukoencephalopathy; prior to initiating therapy, an ophthalmic examination and varicella-zoster virus vaccination for seronegative individuals are indicated. Ozanimod, a recently approved selective S1P receptor modulator, was also shown to be effective in RMS, with favorable safety and tolerability.39,40
Dimethyl fumarate41,42 exerts anti-inflammatory and cytoprotective effects through activation of the nuclear factor (erythroid-derived 2)–like 2 (Nrf2) pathway and Nrf2-independent pathways.43,44 Dimethyl fumarate is generally well tolerated, but treatment has been linked with some progressive multifocal leukoencephalopathy risk.45 Most of these cases were lymphopenic, thus monitoring for lymphopenia every 6–12 months is recommended. Diroximel fumarate was recently approved and, like dimethyl fumarate, is metabolized to mono-methyl fumarate.46
Teriflunomide47 is the active metabolite of leflunomide, an immune suppressant medication used in rheumatoid arthritis. Teriflunomide inhibits dihydroorotate dehydrogenase, an enzyme involved in pyrimidine synthesis. Teriflunomide inhibits proliferation of activated lymphocytes presumed to be autoreactive. Boxed warnings include risk of hepatotoxicity and teratogenesis. Common adverse events include headache, diarrhea, nausea, alopecia, and increase in hepatic alanine transferase. When necessary, teriflunomide can be rapidly eliminated by ingestion of cholestyramine.
Interferon beta (IFN-β) is a class I interferon whose mechanism of action likely involves immunomodulation through downregulating expression of MHC molecules on antigen-presenting cells, decreasing proinflammatory and increasing anti-inflammatory cytokines, inhibiting T-cell proliferation, and blocking trafficking of inflammatory cells to the CNS.48 IFN-β modestly reduces the relapse rate and MRI disease measures and slows accumulation of disability.49–53 Common adverse events include flu-like symptoms and mild abnormalities on routine laboratory evaluation, as well as injection-site reactions with subcutaneous administration.50,53 Side effects can usually be managed with nonsteroidal anti-inflammatory medications.
Glatiramer acetate is the acetate salt of a mixture of random polypeptides composed of four amino acids. Its mechanism of action may involve favorably altering the balance between proinflammatory and regulatory cytokines.54–56 It modestly reduces relapse rates and some disease severity measures, and can be considered as an equally effective alternative to IFN-β in RMS.54,55,57 Common adverse events include injection-site reactions, flushing, chest tightness, dyspnea, palpitations, anxiety after injection, and less commonly, lipoatrophy that can rarely be disfiguring and require treatment cessation.54,55
Infrequently used disease-modifying therapies for RMS
These include mitoxantrone,58 alemtuzumab,59–61 and cladribine.62 All are of proven benefit in RMS; however, serious treatment emergent adverse events limit their use. Finally, a growing body of evidence indicates that autologous hematopoietic stem cell transplantation can induce a prolonged remission in many RMS patients. Randomized trials comparing this procedure with other highly effective therapies are now underway. For the time being, autologous hematopoietic stem cell transplantation is recommended only for RMS patients who have exhausted all other reasonable treatment options, and is not recommended for patients with progressive MS because available data suggest that progression often continues despite this treatment.63
Treating Progressive MS
SPMS
Siponimod64 is a selective S1P modulator that is approved for relapsing forms of MS including active SPMS, meaning patients with SPMS who have had recent clinical relapses and/or evidence of new or enlarging MRI lesions. Ocrelizumab, cladribine, and diroximel fumarate can also be used for patients with active SPMS.
PPMS
Ocrelizumab is the only approved disease-modifying therapy for the treatment of PPMS. Dosing is the same as for RMS. Ocrelizumab reduces progression of clinical disability by approximately one-quarter, and improves other clinical and MRI markers of inflammatory and degenerative disease activity in this population.21
Decision Making for Treatment Of MS
Given the increasingly complex landscape of therapeutics for MS, the treatment algorithm in Figure 3 may aid in decision making. In general, therapy should be initiated in patients with a first attack who are at high risk for MS or in patients diagnosed with RMS.65
Figure 3. A Therapeutic Algorithm for Disease-Modifying Therapy Use in MS.
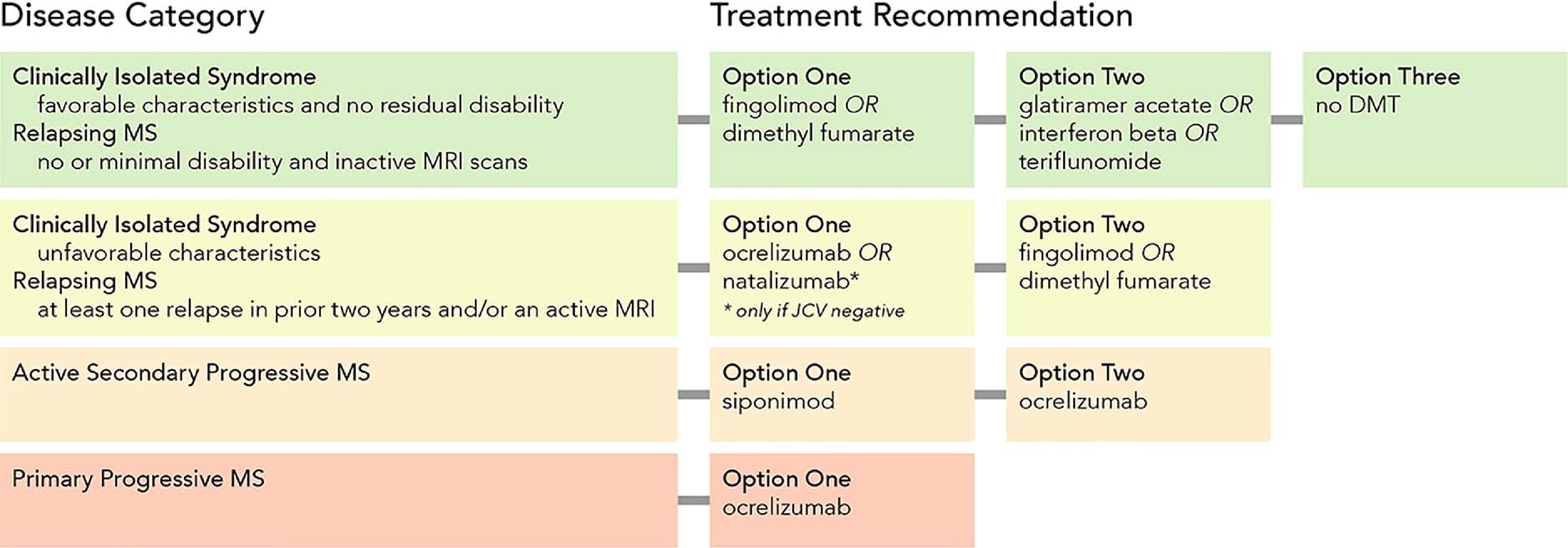
A simplified MS treatment algorithm based on the authors’ practice pattern. Several drugs with proven effectiveness for MS are not mentioned, including: diroximel fumarate (presumably comparable to dimethyl fumerate); cladribine (reserved for MS refractory to at least two other treatments); alemtuzumab and mitoxantrone (not generally recommended due to severe toxicities); hematopoietic stem cell transplantation (reserved for severe treatment refractory disease unresponsive to high efficacy treatments); ozanimod (approved but not yet commercially available).
Abbreviations: DMT, disease-modifying therapy; JCV, John Cunningham virus; MRI, magnetic resonance imaging; MS, multiple sclerosis.
Although historically high-dose IFN β−1a and glatiramer acetate were the preferred first-line options, we now recommend the use of highly effective disease-modifying therapies as first-line options for the majority of patients with active MS. This approach supervenes the traditional “treat to target” method in which a treatment of modest or moderate effectiveness is first used, and therapy advanced to a more effective agent when breakthrough disease (evident clinically or by MRI) occurs. Observational studies suggest that early use of high efficacy therapy improves long-term outcomes. We favor initiating therapy for many patients with ocrelizumab or another anti-CD20 drug, or with natalizumab in John Cunningham virus negative patients. Anti-CD20 therapies represent an attractive option given their high level of efficacy, infrequent infusions or injections, favorable safety profile, and absence of rebound following discontinuation. Patients with PPMS with new or enlarging MRI lesions should also be considered for ocrelizumab treatment. Switching therapies may be required in the following situations: suboptimal response, experiencing more than one relapse with active MRI scans in the prior year while on treatment, and safety issues including development of persistent high-titer neutralizing antibodies in patients receiving IFN-β. Discontinuation of disease-modifying therapies is required in cases of serious adverse events that may be drug-related and for many disease-modifying therapies in women who become pregnant while on treatment. Exceptions include glatiramer acetate that can be continued during pregnancy, and in some cases prior use of ocrelizumab, alemtuzumab and cladribine that have prolonged pharmacodynamic effects which persist after the drug has been eliminated.
CONCLUSION
Spectacular progress has been made in the treatment of MS as a result of advances in understanding of the pathogenesis and course of this disease. The development of highly effective therapies has produced near-complete control of relapsing disease and focal brain inflammation. However, effective treatment of progression remains an unmet need because current therapies confer only partial protection against the neurodegenerative component of MS. Although natural history studies suggest that the long-term course of disease has been dramatically improved in the treatment era, further clinical and real-world assessments are needed to gather long-term efficacy and safety evidence for these therapies. Additional studies of the value of highly effective agents for early treatment and determination of patients who benefit most will also be essential as we seek to achieve evidence-based and personalized approaches to MS treatment and management.
Supplementary Material
CLINICAL SIGNIFICANCE.
Multiple sclerosis, a common autoimmune demyelinating disease of the central nervous system, has two major components: inflammation causing episodic attacks and neurodegeneration responsible for progressive worsening.
Recently developed MS therapies are highly effective in preventing attacks and can partially protect against progression.
Many experts now recommend the use of these therapies early in the disease course to protect against late disability.
ACKNOWLEDGMENTS
Supported is by grants from the National Institute of Neurological Disorders and Stroke (R35NS111644), the National Multiple Sclerosis Society (RR 2005-A-13), and the Valhalla Foundation. The authors thank Natalie Nwkor for invaluable editorial assistance, and Andrew Barnecut for superb work creating figures and finalizing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure
Outside of the submitted work, Dr. Cree reports personal fees for consulting from Akili, Alexion, Atara, Biogen, EMD Serono, Novartis, Sanofi, and TG Therapeutics. Outside of the submitted work, Dr. Hauser reports stock options received for serving on the Board of Directors for Neurona; Scientific Advisory Board for Alector, Annexon, Bionure, and Molecular Stethoscope. He has also received non-financial support (travel reimbursement and writing support for anti-CD20 related meetings) from F. Hoffmann-La Roche Ltd and Novartis AG.
REFERENCES
- 1.Cree BAC, Hauser SL. Multiple Sclerosis In: Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J, eds. Harrison's Principles of Internal Medicine, 20e New York, NY: McGraw-Hill Education; 2018. [Google Scholar]
- 2.Browne P, Chandraratna D, Angood C, et al. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology. 2014;83(11):1022–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet neurology. 2019;18(3):269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamm CP, Uitdehaag BM, Polman CH. Multiple sclerosis: current knowledge and future outlook. European neurology. 2014;72(3–4):132–141. [DOI] [PubMed] [Google Scholar]
- 6.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain : a journal of neurology. 2006;129(Pt 3):606–616. [DOI] [PubMed] [Google Scholar]
- 7.Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology. 2019;92(10):e1029–e1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Annals of neurology. 2019;85(5):653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kappos L, Butzkueven H, Wiendl H, et al. Greater sensitivity to multiple sclerosis disability worsening and progression events using a roving versus a fixed reference value in a prospective cohort study. Multiple sclerosis (Houndmills, Basingstoke, England). 2018;24(7):963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippi M, Rocca MA, Ciccarelli O, et al. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet neurology. 2016;15(3):292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galetta SL, Markowitz C, Lee AG. Immunomodulatory agents for the treatment of relapsing multiple sclerosis: a systematic review. Arch Intern Med. 2002;162(19):2161–2169. [DOI] [PubMed] [Google Scholar]
- 12.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Annals of neurology. 2006;60(1):12–21. [DOI] [PubMed] [Google Scholar]
- 13.Martin R Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis and their application for new therapeutic strategies. J Neural Transm Suppl. 1997;49:53–67. [DOI] [PubMed] [Google Scholar]
- 14.van Oosten BW, Lai M, Hodgkinson S, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology. 1997;49(2):351–357. [DOI] [PubMed] [Google Scholar]
- 15.Weinshenker BG, Bass B, Karlik S, Ebers GC, Rice GP. An open trial of OKT3 in patients with multiple sclerosis. Neurology. 1991;41(7):1047–1052. [DOI] [PubMed] [Google Scholar]
- 16.Massacesi L, Genain CP, Lee-Parritz D, Letvin NL, Canfield D, Hauser SL. Active and passively induced experimental autoimmune encephalomyelitis in common marmosets: a new model for multiple sclerosis. Annals of neurology. 1995;37(4):519–530. [DOI] [PubMed] [Google Scholar]
- 17.Hauser SL. The Charcot Lecture | beating MS: a story of B cells, with twists and turns. Multiple sclerosis (Houndmills, Basingstoke, England). 2015;21(1):8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. The New England journal of medicine. 2017;376(3):221–234. [DOI] [PubMed] [Google Scholar]
- 19.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing remitting multiple sclerosis. The New England journal of medicine. 2008;358(7):676–688. [DOI] [PubMed] [Google Scholar]
- 20.Kappos L, Wolinsky JS, Giovannoni G, et al. Progression Independent of Relapse Activity in Patients With Typical Relapsing Multiple Sclerosis: Pooled Analysis of 2 Randomized Clinical Trials. JAMA neurology. 2020. [In Press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. The New England journal of medicine. 2017;376(3):209–220. [DOI] [PubMed] [Google Scholar]
- 22.Greenfield AL, Hauser SL. B-cell Therapy for Multiple Sclerosis: Entering an era. Annals of neurology. 2018;83(1):13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorensen PS, Blinkenberg M. The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Therapeutic advances in neurological disorders. 2016;9(1):44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nature immunology. 2018;19(7):696–707. [DOI] [PubMed] [Google Scholar]
- 25.Sabatino JJ Jr, Probstel AK, Zamvil SS B cells in autoimmune and neurodegenerative central nervous system diseases. Nature reviews Neuroscience. 2019;20(12):728–745. [DOI] [PubMed] [Google Scholar]
- 26.Hauser SL, Kappos L, Montalban X, et al. Safety of ocrelizumab in multiple sclerosis: updated analysis in patients with relapsing and primary progressive multiple sclerosis. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2019;26(Supplement 1):239. [Google Scholar]
- 27.Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–557 [DOI] [PubMed] [Google Scholar]
- 28.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356(6364):63–66. [DOI] [PubMed] [Google Scholar]
- 29.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. The New England journal of medicine. 2006;354(9):911–923. [DOI] [PubMed] [Google Scholar]
- 30.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. The New England journal of medicine. 2006;354(9):899–910. [DOI] [PubMed] [Google Scholar]
- 31.Myhr K-M, Schüler S, Alstadhaug KB, et al. Long-term, real-world effectiveness of natalizumab treatment in relapsing-remitting multiple sclerosis (RRMS): Data from ≥6 years in the TYSABRI® Observational Program (TOP) Nordic and global cohorts. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2019;26(Supplement 1):680. [Google Scholar]
- 32.Ho PR, Koendgen H, Campbell N, Haddock B, Richman S, Chang I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet neurology. 2017;16(11):925–933. [DOI] [PubMed] [Google Scholar]
- 33.Ryerson LZ, Foley J, Chang I, et al. Risk of natalizumab-associated PML in patients with MS is reduced with extended interval dosing. Neurology. 2019;93(15):e1452–e1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet neurology. 2014;13(6):545–556. [DOI] [PubMed] [Google Scholar]
- 35.Kappos L, Cohen J, Collins W, et al. Fingolimod in relapsing multiple sclerosis: An integrated analysis of safety findings. Multiple sclerosis and related disorders. 2014;3(4):494–504. [DOI] [PubMed] [Google Scholar]
- 36.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. The New England journal of medicine. 2010;362(5):387–401. [DOI] [PubMed] [Google Scholar]
- 37.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. The New England journal of medicine. 2010;362(5):402–415. [DOI] [PubMed] [Google Scholar]
- 38.Laroni A, Brogi D, Morra VB, et al. Safety of the first dose of fingolimod for multiple sclerosis: results of an open-label clinical trial. BMC neurology. 2014;14:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Comi G, Kappos L, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet neurology. 2019;18(11):1009–1020. [DOI] [PubMed] [Google Scholar]
- 40.Cohen JA, Comi G, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet neurology. 2019;18(11):1021–1033. [DOI] [PubMed] [Google Scholar]
- 41.Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. The New England journal of medicine. 2012;367(12):1087–1097. [DOI] [PubMed] [Google Scholar]
- 42.Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. The New England journal of medicine. 2012;367(12):1098–1107. [DOI] [PubMed] [Google Scholar]
- 43.Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. Pharmacol Exp Ther. 2012;341(1):274–284. [DOI] [PubMed] [Google Scholar]
- 44.Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain : a journal of neurology. 2011;134(Pt 3):678–692. [DOI] [PubMed] [Google Scholar]
- 45.Mills EA, Mao-Draayer Y. Aging and lymphocyte changes by immunomodulatory therapies impact PML risk in multiple sclerosis patients. Multiple sclerosis (Houndmills, Basingstoke, England). 2018;24(8):1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naismith RT, Wundes A, Ziemssen T, et al. Diroximel Fumarate Demonstrates an Improved Gastrointestinal Tolerability Profile Compared with Dimethyl Fumarate in Patients with Relapsing-Remitting Multiple Sclerosis: Results from the Randomized, Double-Blind, Phase III EVOLVE-MS-2 Study. CNS Drugs. 2020;34(2):185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. The New England journal of medicine. 2011;365(14):1293–1303. [DOI] [PubMed] [Google Scholar]
- 48.Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 2011;25(6):491–502. [DOI] [PubMed] [Google Scholar]
- 49.Kappos L, Kuhle J, Multanen J, et al. Factors influencing long-term outcomes in relapsing-remitting multiple sclerosis: PRISMS-15. Journal of neurology, neurosurgery, and psychiatry. 2015;86(11):1202–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):655–661. [DOI] [PubMed] [Google Scholar]
- 51.Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Annals of neurology. 1996;39(3):285–294. [DOI] [PubMed] [Google Scholar]
- 52.Rudick RA, Goodkin DE, Jacobs LD, et al. Impact of interferon beta-1a on neurologic disability in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Neurology. 1997;49(2):358–363. [DOI] [PubMed] [Google Scholar]
- 53.PRISMS-4: Long-term efficacy of interferon-beta-1a in relapsing MS. Neurology. 2001;56(12):1628–1636. [DOI] [PubMed] [Google Scholar]
- 54.Johnson KP, Brooks BR, Cohen JA, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1998;50(3):701–708. [DOI] [PubMed] [Google Scholar]
- 55.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45(7):1268–1276. [DOI] [PubMed] [Google Scholar]
- 56.Lalive PH, Neuhaus O, Benkhoucha M, et al. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs. 2011;25(5):401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cadavid D, Wolansky LJ, Skurnick J, et al. Efficacy of treatment of MS with IFNbeta-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology. 2009;72(23):1976–1983. [DOI] [PubMed] [Google Scholar]
- 58.Goodin DS, Arnason BG, Coyle PK, Frohman EM, Paty DW. The use of mitoxantrone (Novantrone) for the treatment of multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2003;61(10):1332–1338. [DOI] [PubMed] [Google Scholar]
- 59.Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. The New England journal of medicine. 2008;359(17):1786–1801. [DOI] [PubMed] [Google Scholar]
- 60.Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–1828. [DOI] [PubMed] [Google Scholar]
- 61.Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–1839. [DOI] [PubMed] [Google Scholar]
- 62.Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. The New England journal of medicine. 2010;362(5):416–426. [DOI] [PubMed] [Google Scholar]
- 63.Cohen JA, Baldassari LE, Atkins HL, et al. Autologous Hematopoietic Cell Transplantation for Treatment-Refractory Relapsing Multiple Sclerosis: Position Statement from the American Society for Blood and Marrow Transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2019;25(5):845–854. [DOI] [PubMed] [Google Scholar]
- 64.Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–1273. [DOI] [PubMed] [Google Scholar]
- 65.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet neurology. 2018;17(2):162–173. [DOI] [PubMed] [Google Scholar]
- 66.Lassmann H Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front Immunol. 2018;9:3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain : a journal of neurology. 2009;132(Pt 5):1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haider L, Zrzavy T, Hametner S, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain : a journal of neurology. 2016;139(Pt 3):807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352(9139):1498–1504. [PubMed] [Google Scholar]
- 70.Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon beta-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet neurology. 2014;13(7):657–665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.