

Abstract
Background:
Arginases (ARG-isoforms, ARG-1/ARG-2) are key regulatory enzymes of inflammation and tissue repair, however their role after neonatal brain hypoxia (H) and hypoxia-ischemia (HI) remains unknown.
Methods:
C57BL/6-mice subjected to the Vannucci procedure on postnatal day 9 were sacrificed at different timepoints. The degree of brain damage was assessed histologically. ARG spatiotemporal localization was determined via immunohistochemistry. ARG expression was measured by Western blot and activity spectrophotometrically.
Results:
ARG-isoforms expression increased during neurodevelopment (P9-P17) in the cortex and hippocampus. This was suppressed with H and HI only in the hippocampus. In the cortex, both isoforms increased with H alone and only ARG-2 increased with HI at 3 d. ARG activity during neurodevelopment remained unchanged but increased at 1 d with H and not HI. ARG-1 localized with microglia at the injury site as early as 4 h after injury, while ARG-2 localized with neurons.
Conclusions:
ARG-isoforms expression increases with age from P9-P17 but is suppressed by injury specifically in the hippocampus and not in the cortex. Both, levels and activity of ARG-isoforms increase with H while ARG-1 immunolabelling is upregulated in the HI cortex. Evidently, ARG-isoforms in brain differ in spatiotemporal localization, expression and activity during neurodevelopment and after injury.
Introduction
A critical determinant of neuronal survival after brain hypoxia-ischemia (HI) is neuroinflammation, resulting from mechanisms of oxidant stress, inflammation, and excitotoxicity. The neonatal brain has a low antioxidant reserve, therefore oxidative stress such as that from excess nitric oxide (NO) may lead to neuroinflammation, neuronal death and poor clinical outcomes (1).
The key regulatory enzymes of inflammation and tissue repair are arginases (ARG), which are expressed in two isoforms; ARG-1 predominantly found in the liver and lung, and the more recently discovered ARG-2, confined mainly to the kidney, brain, prostate and pancreas (2). Arginases are generally described as urea cycle enzymes and recent findings suggest that the complete urea cycle could also occur in brain (3) and play a role in disease (4–6). As ARG regulate NO synthesis via competition for the common substrate arginine (7) (NO pathway), they are capable of regulating the acute phase of inflammation through processes such as oxidative stress (8), apoptosis (9), or microglia polarization (10). Additionally, certain products of the ARG metabolic pathways (such as polyamines via a polyamine pathway) are essential for cell proliferation and collagen formation (11), with predicted roles in neuronal growth (12), axonal regeneration (13) and glial scar formation (14). Consequently, ARG clearly impact neurodevelopment as well as processes after brain injury (15). The ability to regulate NO and polyamine synthesis gives the ARG-pathway potential to serve in new therapies of brain injury and recent findings in this field have reflected this. The number of studies describing a new role for ARG in various conditions is growing, and a role for ARG has already been recognized in adult brain pathologies such as inflammation after traumatic brain injury (4) and autoimmune encephalitis (5). Recently, increased ARG-1 was reported in the blood of stroke patients with levels correlating with infarct size (6), suggesting the involvement of ARG in post-hypoxic-ischemic mechanisms. So far, many studies suggest ARG-1 as being mostly associated with the role of a marker of “pro-repair” microglia, and ARG-2 mostly in regulation of cerebral blood flow (15). However, the exact role of ARG remains unclear. While changes in ARG expression and activity have been described in embryonal and adult ages (16–18), to our knowledge there are very few studies focused on ARG expression and activity during the most robust period of brain development in the postnatal age (15–18, 20–24) and none after HI injury. Here, we describe for the first time the isoform specific ARG expression during the postnatal period as a factor in development as well as after HI.
Materials and methods
Animals
All animal research was approved by the University of California San Francisco Institutional Animal Care and Use Committee and was performed in accordance with the Guide for the Care and Use of Laboratory Animals. Wild-type mice of the C57BL/6 strain (The Jackson Laboratory, Sacramento, CA), of both sexes were used for all experiments.
Induction of Hypoxia-Ischemia
Neonatal HI was induced in P9 mice using the Vannucci procedure. Briefly, unilateral ischemic injury was induced by coagulation of the left common carotid artery (CCA) under isoflurane anesthesia (4 % isoflurane, balance oxygen). The pups were recovered for one hour with their dam. Global hypoxic injury was triggered by exposure to 50 minutes of hypoxia in a humidified chamber at 37 °C with 10 % oxygen/balance nitrogen. The mice were euthanized at 1 h, 4 h, 12 h, 1 d, 3 d, 5 d and 8 d after the procedure. Naive and sham mice (N) were used as controls. The contralateral hypoxic hemisphere (H) in injured animals was studied separately as it was exposed to global hypoxia during the Vannucci procedure and hypoxia alone has been shown to be a stimulus for ARG expression (16). The hypoxic-ischemic (HI) hemisphere was exposed to the initial ischemic insult and subsequent global hypoxia.
Histology and Immunohistochemistry
At each specific time point, mice were flushed with 8 mL of PBS and perfused with 10 mL of 4 % paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were post-fixed in 4 % paraformaldehyde overnight and transferred to 30 % sucrose till they sank (3 days). Brains were then flash-frozen in 2-methyl butane on dry ice and stored at −80 °C. Coronal sections were cut on a cryostat (12 μm- thick serial sections, 120 μm apart). Brain injury was determined in eight coronal sections stained with Cresyl violet using histopathological scoring in a blinded manner. Sections were assessed rostral to caudal. Regions scored included the following: anterior cortex; middle cortex; posterior cortex; anterior, middle and posterior caudate/striatum; thalamus; hippocampus CA1, CA2, and CA3; and the dentate gyrus. Scores were assigned for each region as follows: 0 = no injury, 1 = minimal cell loss manifested by scattered shrunken neurons and glia, 2 = moderate cell loss with infarction in a columnar distribution in the cortex with concomitant gliosis or the shrunken hippocampus with cell loss throughout the Sommer’s sector, and 3 = severe cell loss and gliosis with cystic infarction, for a total score of 0–33.
Double immunofluorescence labeling was performed on brain sections that were defrosted and air dried at room temperature (RT) for 1 h. Following antigen retrieval in 10 mM citrate buffer (pH 6.0) for 3 min at 90.5 °C and a PBS wash, sections were incubated in blocking solution (10 % normal donkey serum, 0.3 % Triton X-100, and 10 mg/mL of bovine serum albumin in PBS) for 1 h at RT. Primary antibody incubation was done overnight at 4 °C with rabbit anti-arginase-1 (1:50, #93668, Cell Signaling Technology); rabbit anti-arginase-2 (1:50, #203071, Abcam); mouse anti-NeuN (NeuN) for neurons (1:500, MAB377, MilliporeSigma), goat anti-Iba1 (Iba1) for microglia (1:100, NB100-1028, Novus Biologicals), mouse anti-glial fibrillary acidic protein (GFAP) for astrocytes (1:300, MAB360, MilliporeSigma) and goat anti-oligodendrocyte transcription factor 2 (Olig2) for oligodendrocytes (1:50, AF2418, Novus Biologicals). After three 5 minute PBS washes, sections were incubated for 1 h at RT with appropriate secondary antibodies: donkey anti-goat Alexa Fluor 647 (1:100, A21447, Thermofisher), donkey anti-mouse Alexa Fluor 568 (1:100, A10037, Thermofisher) and donkey anti-rabbit Alexa Fluor 488 (1:100, A21206, Thermofisher). For nuclear staining, sections were stained with 4’,6-diamino-2-phenylindol (DAPI) for 5 minutes. Slides were then washed and coverslipped with ProLong Gold antifade (P36930, Invitrogen). Images were taken with a Leica TCS SP5 Spectral Confocal Microscope.
Western Blotting
Brain tissue was dissected on ice and separated into injured cortex and hippocampus from ipsilateral (HI) and contralateral (H) sides. Tissue extracts for Western blot analysis were prepared in RIPA lysis buffer with added protease and phosphatase inhibitors (Thermofisher Scientific, Waltham, MA). Protein concentration was determined using the BCA assay (Pierce). 30 ug of protein was separated by Bis-Tris SDS polyacrylamide gel electrophoresis using 12-well gels (Invitrogen, Carlsbad, CA) and blotted to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA) at 30 V overnight at 4 °C. After membrane incubation in blocking buffer (1× TBS, 5 % nonfat dry milk, and 0.1 % Tween-20) for 1 h at RT, the membranes were probed with following antibodies: rabbit polyclonal anti-Arginase-1 antibody and monoclonal anti-Arginase-2 antibody (#9819 ARG-1 polyclonal, #5503 ARG-2 monoclonal, Cell Signaling Technology, 1:100 in blocking buffer) and β-actin (A5316 Sigma Aldrich) overnight at 4 °C. Liver tissue extract was used as an arginase positive control. Mouse β-actin (Sigma, 1:2000) was used as a loading control. After 3 washes in TBST, membranes were incubated with anti-rabbit IgG-HRP conjugate (Cell Signaling Technology, 1:1000). Signal was visualized with the ECL chemiluminescence system (Amersham, Little Chalfont, United Kingdom). Optical densities (OD) were measured on scanned radiographs using Image Studio Lite (Li-Cor Biosciences, version 5.2.5) and all markers were normalized to β-actin.
Arginase Assay
Total arginase activity was determined spectrophotometrically (Quantichrom ™ Arginase Assay kit, BioAssay Systems) per manufacturer instructions. Briefly, hippocampal and cortical tissues were dissected on ice, weighed, and processed in Tris-HCl with protease and phosphatase inhibitor mixtures (Sigma-Aldrich) at 10 μL/mg of tissue. Tissue was homogenized with a Dounce tissue homogenizer and centrifuged at 14,000 rpm for 15 min at 4 °C. Supernatant fractions were incubated at 37 °C for 120 min with 10 mm MnCl2 in arginase buffer to activate the enzyme. The reaction was stopped by adding urea reagent and samples were further incubated for 60 min at RT. Samples were analyzed with a spectrophotometer at 430 nm and arginase activity in brain samples was calculated as U/L (1 unit of arginase converts 1 μmol of L-arginine to ornithine and urea per minute at pH 9.5 and 37 °C).
Microscopy Analysis
To define the anatomical localization of ARG expressing cells, we grossly evaluated all areas of brain from rostral to caudal at 5X magnification. For final analysis we chose the anatomical areas with ARG-positive cells and compared these to the contralateral side and N (for ARG-1 cortex and striatum, for ARG-2 hippocampus). Confocal-like Z-stacks (25X oil objective, 10 μm thick, 1 μm Z step) were acquired using a Zeiss microscope equipped with the confocal-like optigrid device and Volocity software (Improvision). Analysis was performed in anatomical areas corresponding to slide 66 of the Allen Mouse Brain Atlas (19). Matching regions of interest (ROIs) (500 × 400 μm) in N mice served as a control. Every brain had a control with no primary antibodies for staining. Image capturing (using Volocity software) and analysis (Fiji, ImageJ) was done in a blinded manner. The number of cells that express ARG-1 was quantified manually.
Statistical Analysis
Analyses were performed using Prism 7 (Graphpad Software, San Diego, CA). All data are shown as mean ± SEM. Grouped data were analyzed using 2-way ANOVA and subsequently subjected to Bonferroni t-tests for post-hoc analyses and differences were considered significant at p < 0.05. Comparisons were made between HI, H and N groups at different time points. For Western blot expression data, t-tests were used to compare expression at P9 vs PI7 only for the N animals to clarify the significant changes due to neurodevelopment.
Results
Evolution of HI brain injury over time
Brain HI injury evolves over time with corresponding histological changes at different timepoints ranging from 1 h (P9) to 5 d (P14) after the injury (Fig 1). The early changes that occur within 1 h after the injury are characterized by swelling of cortical neurons seen with cresyl violet stain (Fig 1 A). At later timepoints, the injury spreads from cortex (Fig 1 B, C) into deeper brain structures such as hippocampus, thalamus and striatum (Fig 1 D, E). This pattern corresponds to lower scores in the more immediate period after the injury compared to higher scores at the later timepoints (Fig 1 F).
Figure 1:
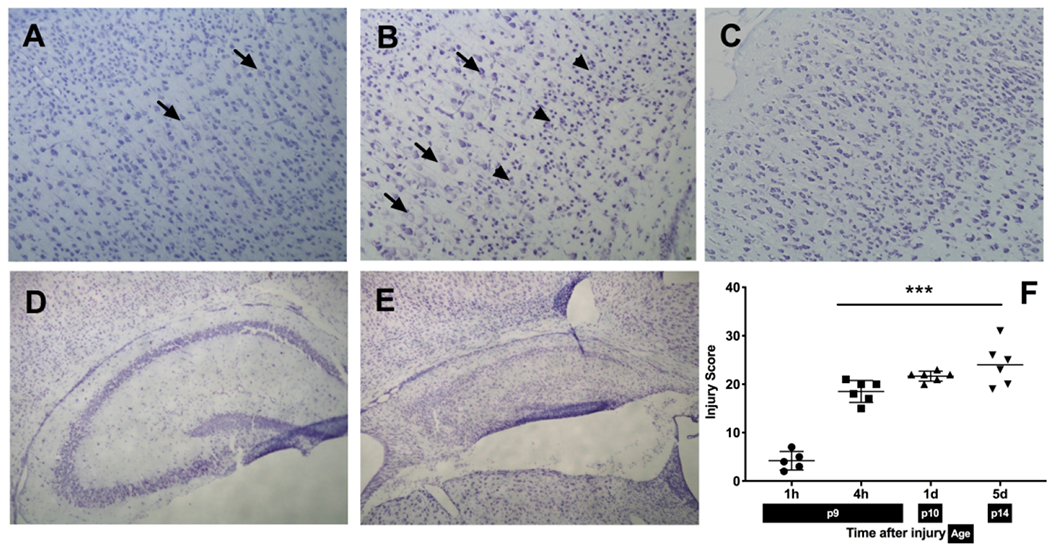
A-E: Histopathological analysis of hypoxic-ischemic brain injury at different timepoints: cortex at 1 h (A) showing swelling of a few neuronal cells, 4 h (B) with increased area of swollen neurons (arrows), pyknotic cells (arrowheads) and 5 d (C) after the injury, showing the disruption in cortical structure and cell loss (10X magnification); hippocampus after HI at 4 h with no apparent injury (D) and 5 d (E) showing injured area with accumulation of shrunken, pyknotic cells and cystic infarction (4X magnification) F: Injury scores after HI at different timepoints: Injury extends from cortex at earlier timepoints to involvement of deep brain nuclei and adjacent cortex at the later timepoints.
ARG expression changes during neurodevelopment and as a result of HI
To investigate if ARG expression changes during neurodevelopment and how brain H and HI affect their expression, we measured the expression of ARG isoforms ARG-1 and ARG-2 in cortex and hippocampus at different timepoints after the injury using Western blotting.
ARG-1 Expression
There was a developmental increase in ARG-1 expression from P9-P17 in both regions of N mice (Fig 2 A, B): cortex at P9 (n = 13) vs P17 (n = 17), (Fig 2 A, p < 0.001), hippocampus at P9 (n = 18) vs P17 (n = 14), (Fig 2 B, p < 0.0001). Following the Vannucci procedure, ARG-1 expression increased in the H cortex (n = 7) 5 d after the injury compared both to the HI cortex (n = 7) (Fig 2 A, p < 0.0001) and age-matched N mice (n = 10) as well as to the H cortex at all other time points (Fig 2 A, p < 0.0001). Hippocampal expression of ARG-1 was reduced at later timepoints after the injury compared to N age-matched animals (n = 10-14) in both H hemisphere (n = 7-11) (3 d, p = 0.0467; 5 d, p = 0.0028; 8 d, p < 0.0001) and HI hemisphere (8 d, p = 0.0003) (Fig 2 B).
Figure 2: Arginase expression in naive mice vs after brain HI at different timepoints:
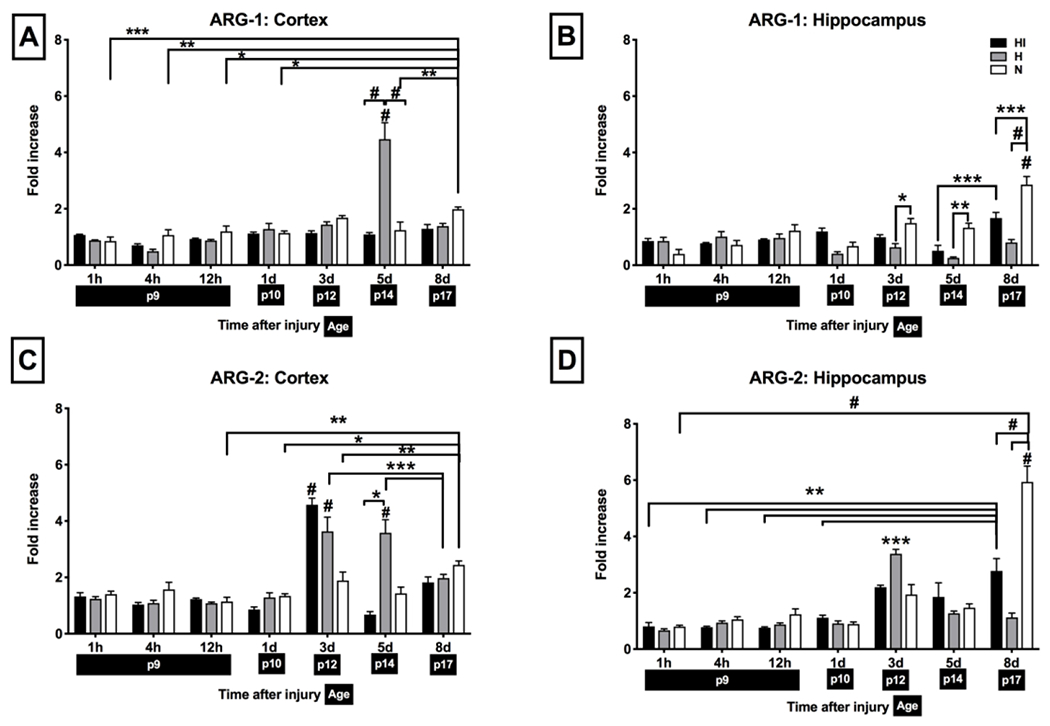
Protein expression was measured by Western blotting and presented as the OD ratio to (β-actin (for H and HI groups: n = 5 for timepoints 1 h, 4 h, 12 h, n = 9-11 for day 1, n = 6-11 for 3 d, n = 7 for 5 d, n = 6-11 for 8 d after the injury; for N group n = 5-17 per timepoint). Shown are HI (black), H (gray) and N (white) hemisphere for ARG-1 in cortex (A) and hippocampus (B) and ARG-2 in cortex (C) and hippocampus (D). Brackets show significances as follows: *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.0001. For ARG-1, expression at 5 d in H cortex and expression at 8 d in N hippocampus were both significant against all other time points in that group (shown by significance on top of the bar). For ARG-2, expression at 3 d in H and HI cortex, at 3 d in H hippocampus and 8 d in HI and N hippocampus were significant against all other time points in that group (shown by significance on top of the bar).
ARG-2 Expression
ARG-2 also exhibited a developmental increase in expression from P9-P17 in N mice (Fig 2 C, D): cortex at P9 (n = 16) vs P17 (n = 17), (Fig 2 C, p < 0.001), hippocampus at P9 (n = 18) vs P17 (n = 15) (Fig 2 D, p < 0.0001). Following the Vannucci procedure, there was a significant increase in ARG-2 expression in the H cortex 3 d (n = 6; p < 0.0001) and 5 d (n = 7; p < 0.0001) after the injury compared to N age-matched animals (n = 10-16). In HI cortex, ARG-2 expression peaked at 3 d after the injury (n = 6; p < 0.0001) and was suppressed by 5 d (n = 7; p < 0.0001). Hippocampal expression of ARG-2 peaked transiently in H hemisphere 3 d after the injury (n = 6; p <0.001 vs all other time points in that group) and decreased 5 d (n=7, p < 0.001) and 8 d after the injury (n = 11; p < 0.001). In the HI hemisphere, there was a significant increase after HI by 8 d compared to earlier timepoints from that group (n = 6; p < 0.001 vs 1 h, 4 h, 12 h and 1 d). However, ARG-2 expression was significantly decreased at 8 d in HI (n=6; p < 0.0001), as well as in H (n=11; p < 0.0001) hemisphere compared to N age-matched animals (n = 14-15, P < 0.0001).
Arginase activity is suppressed by HI
As expression of the enzyme may not necessarily translate to its function, we measured ARG activity in N mice and after the Vannucci procedure using a spectrophotometric enzymatic activity measurement kit. As this kit measures the combined activity of ARG and does not separate the activity of ARG-1 and 2, our measures reflected the total ARG activity in cortex and hippocampus.
Total ARG activity in cortex and hippocampus was constant postnatally from P9-P14 and did not differ between cortex and hippocampus (P9: n = 11 per cortex and hippocampus group, p = 0.6063, P14: n = 8 per cortex and hippocampus group; p = 0.2786) (Fig 3). There was no significant change in ARG activity in cortex after the Vannucci procedure (n = 7-8 per HI and H groups, n = 6-11 per N group; p > 0.05 Fig 3 A). In the hippocampus however, there was a significant increase in ARG activity 1 d after the injury only in H hemisphere (n = 9 per H group and n = 11 per N group; p = 0.0452, Fig 3 B).
Figure 3: Arginase activity in cortex (A) and hippocampus (B) in N mice (white) and in H (gray) and HI hemisphere (black).
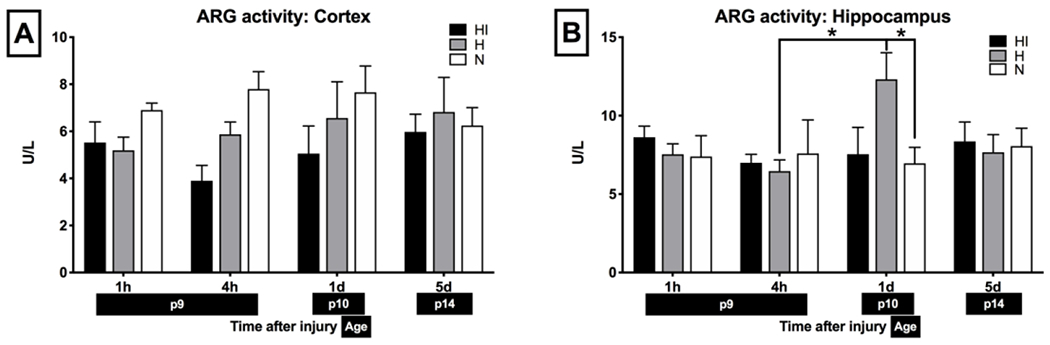
Unit definition: 1 unit of ARG converts 1 μmole of L-arginine to ornithine and urea per minute at pH 9.5 and 37 °C. Brackets show significance with *p < 0.05. n = 7-8 per HI and H group, n = 6-11 per N group.
Arginase isoforms differ in spatiotemporal expression patterns
Cellular Arginase-1 expression
Given the changes in ARG expression and activity noted, we wanted to clarify the cell types associated with these changes. Immunofluorescent double-labeling was performed with ARG-1 and either the neuronal marker NeuN, microglial marker Iba1, astroglial marker GFAP or oligodendrocyte marker Olig2 (Fig 4 A–D). ARG-1 expression was clearly co-localized only with Iba1+ cells (Fig 4 B).
Figure 4: Cellular expression of ARG-1:
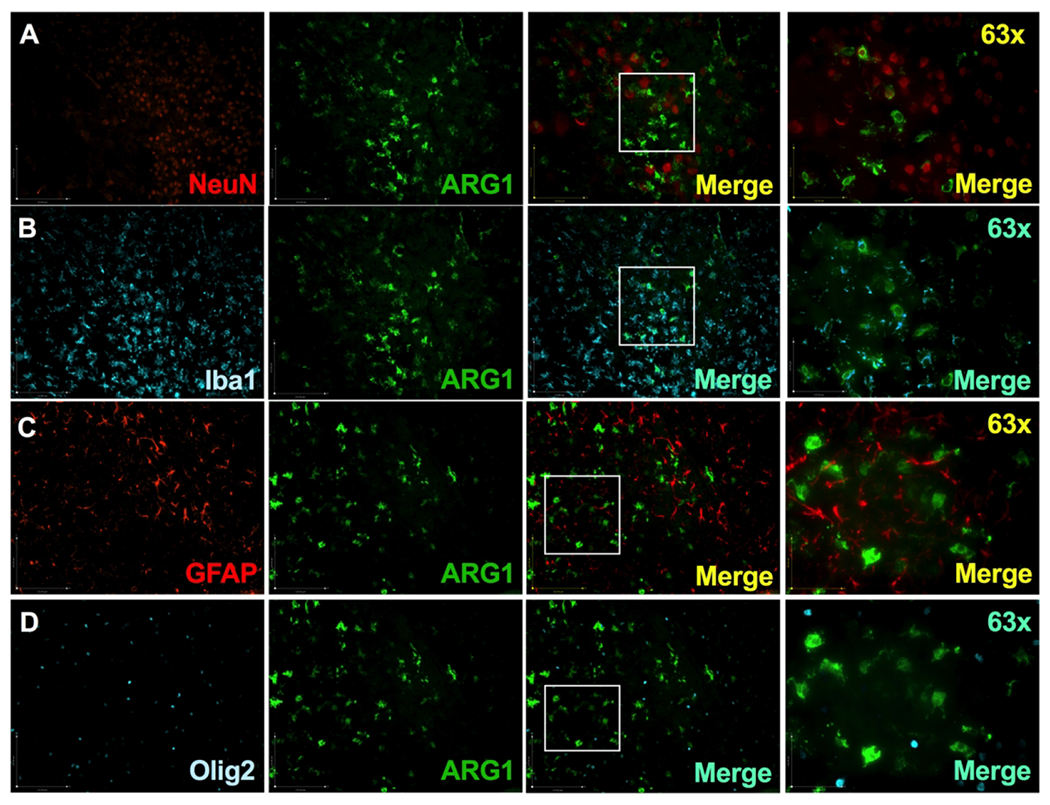
Images of double immunofluorescent staining with ARG-1 antibody (green, middle panels) paired with an antibody specific for neuron (A: NeuN, red panel), microglia (B: Iba1, cyan panel), astrocyte (C: GFAP, red panel) and oligodendrocyte (D: Olig2, cyan panel). 63X image captures region denoted by a box in the 3rd image of that row. Pictured is the injury site of a P10 mouse exposed to the Vannucci HI model on P9. ARG-1 colocalized with Iba1+cells (B).
Anatomically, ARG-1 was colocalized with Iba1+ microglial cells in the areas of olfactory tubercle, pyriform cortex, pallidum, striatum, external capsule of corpus callosum, anterior commissure-temporal limb, amygdala, hypothalamus and caudate (Fig 5 A). Notably, under normal circumstances in N mice, ARG-1+/Iba1+ cells were absent from the thalamus, hippocampus, internal capsule and other parts of fiber tracts or cortex at all timepoints studied (P9, P10, P14). After HI, ARG-1+ microglia morphology was clearly transformed to the ameboid phenotype at the injury site (Fig 5 B, C, D) when compared to the ramified phenotype evident in the N mice (Fig 5 E). The changes in ARG-1+ microglial morphology and accumulation at the injury occurred as early as 4 h after the injury (Fig 5 F, G, H). At later timepoints, ARG-1+/Iba1+ cell numbers remain elevated both at the HI core in striatum (24 h: n = 6 per HI and H group, n = 8 per N group; 5 d: n = 6 per HI and H group, n = 14 per N group; 24 h: p < 0.0001 for HI vs H, p < 0.0001 for HI vs N; 5d: p < 0.0001 for HI vs H and p < 0.0001 for HI vs N) and the HI hemisphere (24 h: p = 0.0025 for HI vs H; p = 0.0002 for HI vs N; 5 d: p < 0.0001 for HI vs H; p < 0.0001 for HI vs N) (Fig 5 I, J).
Figure 5: Changes in ARG-1+microglia after brain HI:
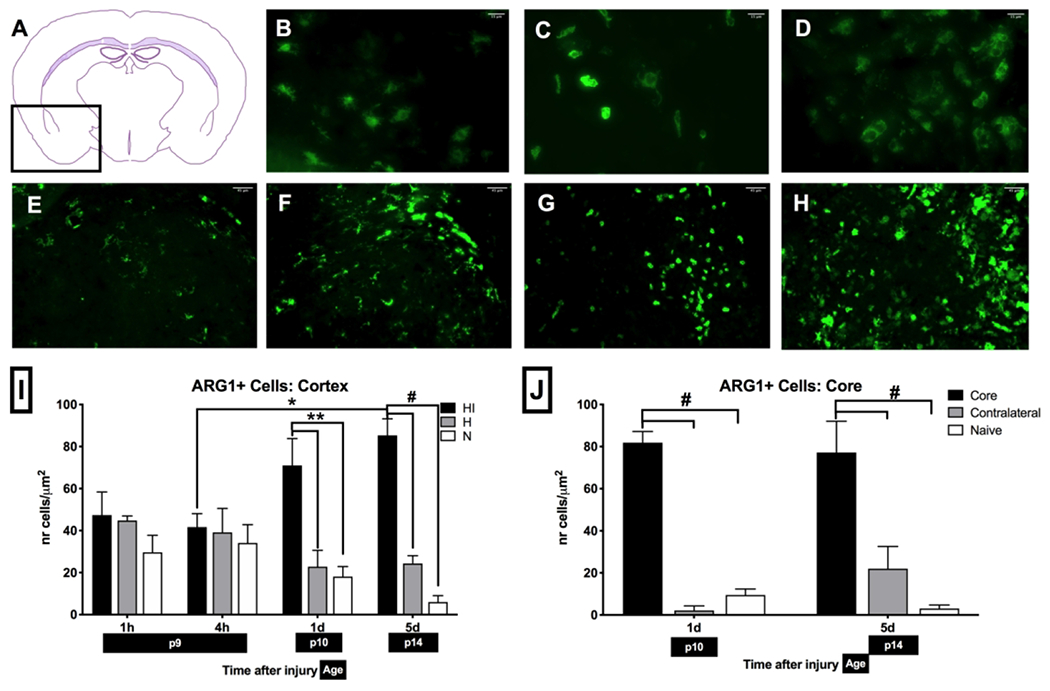
(A) Anatomical expression of ARG-1 in mice localizes to pyriform cortex, olfactory tubercle, pallidum, striatum, external capsule of corpus callosum, anterior commissure-temporal limb, amygdala, hypothalamus, caudoputamen. Change in ARG-1+ microglia morphology as a result of HI from bushy (B) into ameboid (C) and round phagocyting (D) phenotype. Changes in ARG-1+ microglia accumulation in striatum at different timepoints: N mice (E), and 4 h (F), 1 d (G) and 5 d (H) after injury. ARG-1+ microglia count increases in HI cortex (I) and HI core in striatum (J) at 1 d after injury and remains elevated for a prolonged time (n = 5-6 per HI and H group; n = 8 per N group, *p < 0.05, **p < 0.01, #p < 0.0001).
Cellular ARG-2 expression
The pattern of ARG-2 expression was different compared to that of ARG-1. ARG-2 expression was noted to colocalize largely to NeuN+ cells rather than Iba1+, GFAP+ or Olig2+ cells (Fig 6 A–D). Anatomically, ARG-2 expression was localized in areas of indusium griseum, fasciola cinerea, and CA1 and CA2 of hippocampus, dentate gyrus and M1 area of the neocortex (Fig 6 E–J). Following HI, ARG-2 expressing cells appeared to have an altered morphology consistent with being injured, in that they appeared pyknotic and shrunken (Fig 6 F, G).
Figure 6: Cellular and anatomical expression of ARG-2:
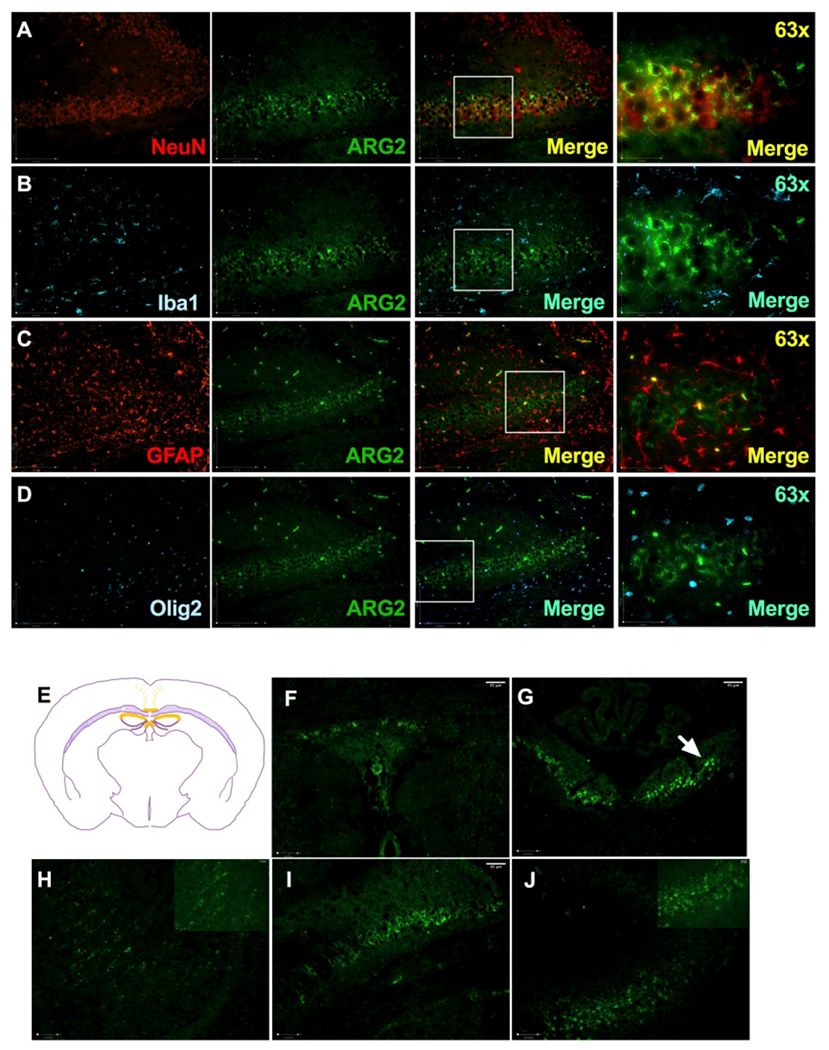
Images of double immunofluorescent staining with ARG-2 antibody (green, middle panels) paired with an antibody specific for neuron (A: NeuN, red panel), microglia (B: Iba1, cyan panel), astrocyte (C: GFAP, red panel) and oligodendrocyte (D: Olig2, cyan panel). 63X image captures region denoted by a box in the 3rd image of that row. ARG-2 colocalized with NeuN+ cells (A). Anatomical localization of ARG-2 (E): ARG-2 was found in indusium griseum (F), fasciola cinerea (G), neurons of the neocortex (H), hippocampus CA1 (I), CA2 (J). Note the pyknotic cells in the HI hemisphere (arrow); n = 5-6 per H and HI group, n = 8 per N group.
Discussion
In this study, we elucidate, for the first time, alterations that occur in ARG expression and activity during development from P9 to P14-P17 as well as after H and HI injury. We found that the expression of ARG-1 and 2 increases significantly with age in both the cortex and hippocampus. The expression of ARG-1 and 2 after injury appear to be intrinsically modulated by H alone or HI. Total ARG activity is only modulated by H alone. On the cellular level, both ARG-isoforms have very specific expression patterns with ARG-1 expression predominantly microglial and ARG-2 largely neuronal, both before and after HI. The highly region- and cell-specific expression suggests the possibility of specific functions of ARG-isoforms in brain under physiological and pathological conditions.
While ARG expression has been described in embryonic and very early postnatal (E13-P1)(16–17), young (4-months) and aged (24-months) brains in rat (18), to the best of our knowledge this study is the first to describe isoform-specific ARG expression patterns during the early postnatal period P9-P17 in mice. We show that ARG-1 expression increases during normal neurodevelopment from neonatal (P9) to infantile age (P17) and is predominantly localized to microglia. Our findings are consistent with Crain et al (20) who studied ARG-1 expression exclusively in microglia and detected high mRNA expression for the ARG-1 gene in the early postnatal period (P3) that diminished by 70 % at P21 and by 90 % at 12 months. Similar to ARG-1, we detected increase in ARG-2 expression from P9 to P17 in neurons. There is no previous data on ARG-2 expression during P9-P17 in both the hippocampus and the cortex and while previous studies did not detect ARG-2 increase E13-P1 (16), they did so in adulthood (21).
Currently, it is not possible correlate isoform specific expression to activity as available kits do not differentiate between ARG-1 and ARG-2. This may be why we failed to detect significant changes in ARG activity despite an obvious increase in microglial ARG-1 expression in the cortex. Alternatively, it is possible that ARG-1+ microglia represent only a small fraction of the expressed ARG-1. Hippocampal activity however is most likely related to ARG-2 expression as this is the predominant isoform expressed in the hippocampus. In our study, ARG activity did not change significantly during neurodevelopment in either region at the ages studied (P9, P10, P14), consistent with previous findings (22). The regional variation in ARG activity across different cortical regions and subregions of the hippocampus has also been described previously (23, 24). These age-, isoform- and region-specific alterations in ARG expression and activity highlight the tightly regulated functions of this enzyme during neurodevelopment.
Both, hypoxia and ischemia-reperfusion injury have been described as a potent stimulus for ARG-1 and ARG-2 expression (25) and consistent with this, we detected a peak in ARG-1 and ARG-2 expression in the H cortex 5 d after the injury, similar to that reported for ARG-1 by Hamzei et al (26). Current data on the extent of HI vs H stimulus on ARG-isoforms expression is limited and we speculate that H could be a more potent activator of ARG-isoforms than HI, especially in uninjured regions. However, 8 d after the injury we see suppression of ARG-1 and ARG-2 expression in hippocampus. A similar transient upregulation followed by suppression of ARG-1 was reported by Quirie et al. (27) in an adult rat photothrombotic stroke model at 15 d after the injury. It is possible that the variation in timeline of expression between our studies is related to the age, model and species studied and suppression of ARG-isoforms may be related to the presence of endogenous inhibitors (27).
Cellular localization of ARG was isoform-specific, confined to certain anatomical areas and changed during development and with injury. Notably, we found ARG-1+ microglia in specific regions of the ventral forebrain and midbrain. A unique population of ARG-1+ microglia has been shown to represent only 0.5 % of microglia formed at E14.5/P4/5 with higher specificity for females (28). It is possible that ARG-1+ microglia play role in neurodevelopment and/or sex-specific response to injury (29). ARG-1+ microglia have been widely reported after HI injury (30) and are thought to mediate neuroprotective effects. While we detected similarly high numbers of ARG-1+ microglia at the injury site, not all microglia at the injury site were ARG-1+. This dimorphic expression could relate to the origin of the microglia or to the reported microglial “switch” that can happen at the injury site (31). Interestingly, despite significant evident injury in the hippocampus, ARG-1+Iba1+ cells were only minimally present in this area compared to the injured cortex where ARG-1+Iba1+ were detectable as early as 4 h after the injury and persisted for 5 d. This finding demonstrates the heterogeneity of regional responses to injury processes and confirms that injury, protection and recovery mechanisms after injury are modulated in a spatiotemporal fashion. Despite evidence for ARG-1 expression in astrocytes along with neurons and microglia after injury (25, 26), we failed to detect colocalization with GFAP. It is possible that differences in ARG expression and activity during development are based on species, timing after the injury and injury type (25, 26).
ARG-2 belongs to a group of less studied enzymes in the brain and there are currently few reports on its expression, during neurodevelopment and particularly after injury, in the developing brain. Our findings suggest this isoform may have roles that are distinct to those of ARG-1, as is evident from the altered cellular and anatomical expression in the brain. Our study is the first to describe ARG-2 expression in structures such as indusium griseum (IG), fasciola cinerea (FC) or hippocampus. These structures create a continuum referred to as hippocampal rudiments (HR) (32, 33). A few neurons expressing ARG-2 were found also in the M1 part of the neocortex. Although the exact function of HR structures is not well known, they are believed to play an important role in axonal guidance across the midline during development of brain commissures and interhemispheric connections (34). The damage of these structures during HI may lead to a spectrum of neurodevelopmental conditions (35). Expression of ARG-2 in HR suggests important roles for ARG-2 in mediating at least some of these functions. The presence of ARG-2+ NeuN+ cells in the CA1, CA2 of the hippocampus also associates this enzyme with processes of learning and memory formation (36), emotion, neural cell birth (37), among others. While recent reports indicate that acetylcholine, dopamine, noradrenaline, 5-hydroxytryptamine and GABA neurons innervate the IG (38), we speculate ARG-2 to primarily localize to GABA-ergic neurons considering it plays important roles in glutamine synthesis (39). After HI, we see considerable damage of ARG-2+ cells in almost all of these areas, potentiating a role for ARG-2 in mediating at least some of the learning, memory and motor deficits seen after HI (40) and consequently may present an attractive therapeutic target that needs to be validated carefully.
In summary, expression and activity of ARG enzymes change during neurodevelopment and in response to hypoxic and hypoxic-ischemic brain injury. As ARG-1 localizes mostly to microglia at the injury site, it is possible that its primary role is mediating neuroinflammatory processes. ARG-2 expression on the other hand is largely neuronal predominantly in neurodevelopmental structures, suggesting involvement in developmental processes. Further studies are needed to elucidate the precise roles of ARG isoforms in neonatal brain hypoxic and hypoxic-ischemic injury.
Impact:
What is the key message of your article?
Arginase isoforms change during neurodevelopment and after neonatal brain hypoxia-ischemia.
What does it add to the existing literature?
This is the first study investigating the key enzymes of inflammation and tissue repair called arginases following murine neonatal brain hypoxia-ischemia.
What is the impact?
The highly region- and cell-specific expression suggests the possibility of specific functions of arginases.
Arginase-1 in microglia at the injury site may regulate neuroinflammation, while arginase-2 in neurons of developmental structures may impact neurodevelopment.
While further studies are needed to describe the exact role of arginases after neonatal brain hypoxia-ischemia, our study adds valuable data on anatomical localization and expression of arginases in brain during development and after stroke.
Acknowledgements
We would like to thank to Sandrijn Vanschaik, Jeff Fineman and Division of Pediatric Critical Care for their help and support.
Statement of Financial Support: This work was supported by supported by NIH National Institute of Neurological Disorders and Stroke R35-5R35NS097299 to DMF, UCSF Pediatric Critical Care Division and the Thrasher Research Fund P0530684.
Footnotes
Competing interests: Material is original, has not been previously published, and has not been submitted for publication elsewhere while under consideration.
Disclosure statement: The authors have no conflicts of interest and no financial relationships relevant to this article to disclose.
Category of the study: Basic science study
References
- 1.LaFemina MJ, Sheldon RA, Ferriero DM. Acute hypoxia-ischemia results in hydrogen peroxide accumulation in neonatal but not adult mouse brain. Pediatr Res. 2006;59(5):680–683. [DOI] [PubMed] [Google Scholar]
- 2.Choi S, Park C, Ahn M, Lee JH, Shin T. Immunohistochemical study of arginase 1 and 2 in various tissues of rats. Acta Histochem. 2012;114:487–494. [DOI] [PubMed] [Google Scholar]
- 3.Handley RR, et al. Brain urea increase is an early Huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc Natl Acad Sci USA. 2017; 114(52):E11293–E11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villalba N, et al. Traumatic Brain Injury Causes Endothelial Dysfunction in the Systemic Microcirculation through Arginase-1-Dependent Uncoupling of Endothelial Nitric Oxide Synthase. J Neurotrauma. 2017;34(1):192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn M, et al. Immunohistochemical study of arginase-1 in the spinal cords of Lewis rats with experimental autoimmune encephalomyelitis. Brain Res. 2012; 1453:77–86. [DOI] [PubMed] [Google Scholar]
- 6.Petrone AB, et al. The Role of Arginase 1 in Post-Stroke Immunosuppression and Ischemic Stroke Severity. Transl Stroke Res. 2016;7(2):103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris S, Wu G. Arginase: nitric oxide and beyond. Biochem. J. 1998;336, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen MC, et al. Arginase Inhibition Restores Peroxynitrite-Induced Endothelial Dysfunction via L-Arginine-Dependent Endothelial Nitric Oxide Synthase Phosphorylation. Yonsei Med J. 2016;57(6):1329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estévez AG, et al. Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neurons. J Neurosci. 2006;26:8512–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonathan D, Cherry JD, Olschowka JA, O’Banion KM. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. Journal of Neuroinflammation. 2014,11:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai D, et al. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35(4):711–9. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, Higginson DS, Hester L, Park MH, Snyder SH. Neuronal growth and survival mediated by eIF5A, a polyamine-modified translation initiation factor. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(10), 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng K, et al. Increased synthesis of spermidine as a result of upregulation of arginase I promotes axonal regeneration in culture and in vivo. J Neurosci. 2009;29(30):9545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alizadeh A, et al. Neuregulin-1 positively modulates glial response and improves neurological recovery following traumatic spinal cord injury. Glia. 2017;65(7):1152–1175. [DOI] [PubMed] [Google Scholar]
- 15.Krystofova J, Pathipati P, Russ J, Sheldon A, Ferriero D. The Arginase Pathway in Neonatal Brain Hypoxia-Ischemia. Dev Neurosci. 2019; 17:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H, et al. Arginase expression in mouse embryonic development. Mech Dev. 2002;115:151–5. [DOI] [PubMed] [Google Scholar]
- 17.Grillo MA, Fossa T, Dianzani U. Arginase, ornithine decarboxylase and Sadenosylmethionine decarboxylase in chicken brain and retina. Int J Biochem 1983;15:1081–4. [DOI] [PubMed] [Google Scholar]
- 18.Liu P, Smith P, Appleton I, Darlington C, Bilkey D. Age-related changes in nitric oxide synthase and arginase in the rat prefrontal cortex. Neurobiol Aging. 2004;25:547–552. [DOI] [PubMed] [Google Scholar]
- 19.© 2004. Allen Institute for Brain Science. Allen Mouse Brain Atlas. Available from: https://mouse.brain-map.org.
- 20.Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J Neurosci Res. 2013;91:1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H, et al. Expression of arginase isozymes in mouse brain. J Neurosci Res. 2001;66:406–422. [DOI] [PubMed] [Google Scholar]
- 22.Konarska L, Tomaszewski L. Studies on L-Arginase in Developing Rat Small Instestine, Brain, and Kidney I. Ontogenic Evolution of Arginase lsoenzymes. Biochem Med Metab Biol. 1986;35(2):156–69. [DOI] [PubMed] [Google Scholar]
- 23.Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Regional variations and age-related changes in nitric oxide synthase and arginase in the sub-regions of the hippocampus. Neuroscience 2003;119:679–87. [DOI] [PubMed] [Google Scholar]
- 24.Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Nitric oxide synthase and arginase in the rat hippocampus and the entorhinal, perirhinal, postrhinal, and temporal cortices: Regional variations and age-related changes. Hippocampus. 2003;13:859–867. [DOI] [PubMed] [Google Scholar]
- 25.Shosha E, et al. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis. 2016;7:e2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials. 2016;91:151–165. [DOI] [PubMed] [Google Scholar]
- 27.Quirié A, et al. Effect of stroke on arginase expression and localization in the rat brain. Eur J Neurosci. 2013;37:1193–1202. [DOI] [PubMed] [Google Scholar]
- 28.Hammond TR, et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 2019;50(1):253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, et al. Abstract TP507: Sex-Related Differences in Neonatal Stroke: International Maternal Newborn Stroke Registry. Stroke. 2019; 50(1): ATP507–ATP507. [Google Scholar]
- 30.Hellström Erkenstam N, et al. Temporal Characterization of Microglia/Macrophage Phenotypes in a Mouse Model of Neonatal Hypoxic-Ischemic Brain Injury. Front Cell Neurosci. 2016;10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni MG. The ischemic environment drives microglia and macrophage function. Front Neurol. 2015; 6(81): 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carpenter MB. Human Neuroanatomy, 7th edn. Williams and Wilkins Co., Baltimore, 1976; 532 pp. [Google Scholar]
- 33.Adamek GD, Shipley MT, Sanders MS. The indusium griseum in the mouse: architecture, Timm’s histochemistry and some afferent connections. Brain Res Bull. 1984; 12(6): 657–68. [DOI] [PubMed] [Google Scholar]
- 34.Morcom LR, Edwards TJ, Richards LJ. Chapter 14 - Cortical Architecture, Midline Guidance, and Tractography of 3D White Matter Tracts. Axons and Brain Architecture. Academic Press. 2016; 289–313. [Google Scholar]
- 35.Paul LK, et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8(4):287–99. [DOI] [PubMed] [Google Scholar]
- 36.Bendel O, et al. Reappearance of hippocampal CA1 neurons after ischemia is associated with recovery of learning and memory. Journal of Cerebral Blood Flow and Metabolism, 2005; 25(12): 1586–1595. [DOI] [PubMed] [Google Scholar]
- 37.Gulyaaeva N Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem Res. 2019; 44(6):1306–1322. [DOI] [PubMed] [Google Scholar]
- 38.Di Ieva A, Fathalla H, Cusimano MD, Tschabitscher M. The indusium griseum and the longitudinal strie of corpus callosum. Cortex. 2015; 62:34–40. [DOI] [PubMed] [Google Scholar]
- 39.Li H, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Metab. 2001;280:E75–E82. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Biarge M, Ferriero D, Cowan FM. Perinatal arterial ischemic stroke. Handb Clin Neurol. 2019; 162:239–266. [DOI] [PubMed] [Google Scholar]