

Abstract
Background:
The identification of genetic risk factors for chronic pancreatitis, such as PRSS1, CFTR and SPINK1, provides the opportunity to define key pathologic hallmarks and etiologic-specific changes. For example, pancreata from PRSS1 and CFTR patients exhibit progressive lipomatous atrophy without significant fibrosis. Considering the pathology of SPINK1-associated pancreatitis is ill-defined, we examined the pancreata of SPINK1 patients with chronic pancreatitis.
Methods:
Histologic sections after total pancreatectomy with islet autotransplantation and associated clinicopathologic data were collected from 28 patients with SPINK1 germline alterations. Clinical findings, germline data, anatomic anatomic anomalies and pathologic findings were descriptively evaluated.
Results:
Patients ranged in age from 5 to 48 years (median, 21.6 years) with abdominal pain between 2 and 25 years (median, 5.8 years). Most patients were SPINK1 heterozygous and 14 (50%) had co-occurring CFTR (n=12) and CTRC (n=2) mutations. Other pancreatitis risk factors included anatomic anomalies (n=9) and tobacco use (n=1). Overall, 24 (86%) patients had additional pancreatitis-associated germline alterations, SPINK1 homozygosity, anatomic anomalies or environmental factors. Examination of pancreata revealed a sequential pattern of exocrine parenchymal loss and replacement by prominent fibrosis, dependent on the duration of abdominal pain. No malignancies were identified, but low-grade pancreatic intraepithelial neoplasia was present for 2 cases.
Conclusions:
Within this descriptive study, SPINK1-associated pancreatitis is characterized by parenchymal fibrosis and suggests divergent pathophysiologic mechanisms from PRSS1 and CFTR-associated pancreatitis. Moreover, SPINK1 patients frequently had additional etiologic factors that did not impact the development of pancreatic fibrosis and may implicate SPINK1 as a disease modifier gene.
Keywords: alcohol, fibrosis, pancreatitis, genetics, pathology
Introduction
Chronic pancreatitis is a progressive condition that leads to irreversible damage to the pancreas and is frequently accompanied by chronic, disabling pain. However, rather than a single entity, chronic pancreatitis represents a heterogenous group of disorders with protean clinical and pathologic findings.1 The etiology of chronic pancreatitis is often multifactorial and known risk factors include chronic alcohol consumption, smoking, autoimmune conditions, pancreatic duct obstruction and anatomic anomalies. Genetics is also recognized as a risk factor and, in certain instances, influences disease penetrance and severity. Germline alterations strongly associated with chronic pancreatitis include those involving PRSS1, CFTR and SPINK1 and, to a lesser extent, CTRC and CASR.2 Importantly, the identification of germline carriers allows the opportunity to define the pathologic hallmarks and etiologic-specific changes of chronic pancreatitis. Further, the presence of distinctive histology could provide information on the pathogenesis of chronic pancreatitis associated with other etiologic factors.
Recently, the clinicopathologic features of PRSS1-associated chronic pancreatitis were reported. Germline alterations in PRSS1 are autosomal dominant with a 40% to 96% penetrance.3–6 Patients suffer from recurrent episodes of acute pancreatitis, which often progresses to chronic pancreatitis. PRSS1-associated chronic pancreatitis usually begins in early childhood, but onset can vary from infancy to the sixth decade of life. Histologically, the pancreata from PRSS1 carriers develop lipomatous atrophy that leads to pancreatic insufficiency.7 Similar observations have also been made with CFTR patients, and, thus, alterations in both genes may follow a common pathophysiologic mechanism.8, 9
However, among the genes strongly associated with chronic pancreatitis, the histopathologic features of pancreata from SPINK1 carriers have not been characterized. Therefore, we have collected pancreatic specimens from 28 SPINK1 patients of various ages with a reported history of chronic pancreatitis and who underwent total pancreatectomy with islet autotransplantation. We describe the patient demographics, clinical history, presence of anatomic anomalies, co-occurring germline alterations and the pancreatic pathology of this unique cohort.
Materials and Methods
Case Selection
Study approval was obtained from the institutional review boards at the University of Pittsburgh and University of Minnesota. A search for all patients undergoing total or complete pancreatectomy with islet autotransplantation at the University of Pittsburgh Medical Center and the University of Minnesota that harbored a SPINK1 gene mutation was performed between 2004 and 2018. All patients are participants in a prospective observational study collecting laboratory, medical records, radiographic/endoscopic imaging, intraoperative findings and pathology reports. Informed parental consent and assent or informed consent, as age appropriate, was obtained for all participants. Upon identification of 28 SPINK1 carriers, germline testing reports were reviewed for each patient to confirm the presence of SPINK1 alterations and the identification of other germline variants. Germline testing data included reports from Ambry Genetics (Aliso Viejo, CA), Associated Regional and University Pathologists (Salt Lake City, UT), Quest Diagnostics (Madison, NJ) and the Molecular and Genomic Pathology Laboratory at the University of Pittsburgh Medical Center (Pittsburgh, PA). For all testing platforms, extended testing of not only SPINK1 was performed, but also PRSS1 and CFTR. A germline analysis for CTRC was done for 19 (68%) patients and an evaluation of CASR was performed for 10 (36%) patients.
Histopathologic analysis
For each surgical specimen, the relevant gross findings were documented to include color, texture, lobularity, concretions, cysts and mass lesions. Prior to islet cell isolation, representative sections of the pancreas were obtained and formalin-fixed and paraffin-embedded (FFPE). For each specimen, FFPE sections ranged between 1 to 9 (mean, 4 sections; median, 4 sections) with at least 1 section taken from the head of the pancreas. Hematoxylin-and-eosin stained slides for each case were independently reviewed by 2 surgical pathologists (T.E.J. and A.D.S.) that were blinded to clinical data. Particular attention was paid to document the number, distribution, and changes of pancreatic acini, ducts, and islets of Langerhans. Furthermore, the amount and location of both acute and chronic inflammation, presence of concretions, extent and distribution of parenchymal fibrosis and fat, and neoplasia were noted. Histopathologic findings were reviewed independently by both surgical pathologists and then were discussed and re-reviewed collectively with correlation to patient age, disease onset, abdominal pain duration, social history, germline genetics, and anatomic anomalies. As accepted guidelines for the interpretation of pancreata for chronic pancreatitis and associated changes are not available, the histopathologic findings were documented (Supplementary Table 1) as previously reported for patients with genomic alterations in PRSS1.7 The presence of fibrosis was semi-quantitatively graded as mild, moderate or severe with respect to intralobular, interlobular and perilobular parenchymal locations.10
Results
The study cohort consisted of 28 patients with a germline mutation in SPINK1. For each patient, the clinical features, pancreatobiliary/gastrointestinal tract anatomic anomalies and genomic findings are summarized in Table 1. At the time of total pancreatectomy, the patients ranged in age from 5 to 48 years (mean, 21.9 years; median, 20.5 years) and the majority were female (n = 17, 61%). All patients reported a history of intermittent abdominal pain with an age of onset ranging from infancy to 44 years and a duration of symptoms that ranged between 2 and 25 years (mean, 8.8 years; median, 5.7 years), which did not correlate with patient age. Other risk factors for pancreatitis, including alcohol consumption (>5 drinks per day), were absent; but, a history of tobacco smoking was identified for 1 patient. Of note, 9 (31%) patients were remarkable for pancreatobiliary or gastrointestinal tract anatomic anomalies that included pancreatic divisum (n = 6), pancreatobiliary maljunction (n = 1), ampullary stenosis (n = 1) and intestinal malrotation (n = 1). In addition, a history of exocrine insufficiency was documented 15 (54%) patients and more likely to be seen in patients with longstanding duration of symptoms.
Table 1.
Clinical features, anatomic anomalies and genomic findings of 28 patients with SPINK1-associated chronic pancreatitis.
| Case | Age (years) | Sex | Duration of Pain (years) | Tobacco Use & Alcohol History* | Exocrine Insufficiency (Age [years] of Diagnosis) | Pancreatobiliary/Gastrointestinal Tract Anatomic Anomalies | Genotype |
|---|---|---|---|---|---|---|---|
| 1 | 26 | F | 2.1 | None | No | Pancreatic divisum | SPINK1 N34S heterozygote; CFTR T1299A heterozygote |
| 2 | 22 | F | 2.5 | None | No | Pancreatobiliary maljunction | SPINK1 N34S heterozygote |
| 3 | 35 | F | 2.7 | None | No | Pancreatic divisum | SPINK1 N34S heterozygote; CFTR G576A and R668C heterozygote |
| 4 | 5 | M | 2.8 | None | Yes (5 y/o) | None | SPINK1 N34S heterozygote |
| 5 | 18 | F | 3.0 | None | No | None | SPINK1 N34S heterozygote; CFTR M470V heterozygote |
| 6 | 48 | F | 3.3 | 45 pack year smoking history | Yes (45 y/o) | None | SPINK1 N34S heterozygote |
| 7 | 12 | M | 3.3 | None | No | None | SPINK1 N34S heterozygote; CFTR deltaF508 heterozygote |
| 8 | 8 | M | 3.3 | None | No | Pancreatic divisum | SPINK1 N34S heterozygote |
| 9 | 11 | F | 3.4 | None | No | None | SPINK1 heterozygote**; CFTR heterozygote** |
| 10 | 6 | F | 3.9 | None | Yes (4 y/o) | None | SPINK1 R67H heterozygote; CTRC G103VfsX31 heterozygote |
| 11 | 26 | F | 4.2 | None | No | Ampullary stenosis | SPINK1 N34S heterozygote |
| 12 | 27 | M | 4.3 | None | Yes (Unknown) | None | SPINK1 N34S heterozygote |
| 13 | 45 | F | 4.6 | None | Yes (43 y/o) | None | SPINK1 N34S heterozygote; CFTR R117H heterozygote |
| 14 | 14 | F | 5.7 | None | No | Pancreatic divisum | SPINK1 N34S heterozygote |
| 15 | 25 | F | 5.8 | None | No | Intestinal malrotation | SPINK1 N34S heterozygote |
| 16 | 11 | F | 6.5 | None | No | None | SPINK1 N34S heterozygote; CFTR deltaF508 heterozygote |
| 17 | 25 | F | 7.9 | None | Yes (Unknown) | None | SPINK1 N34S heterozygote; CFTR IV8–5T heterozygote |
| 18 | 18 | M | 8.1 | None | Yes (Unknown) | None | SPINK1 N34S homozygote |
| 19 | 18 | M | 9.5 | None | Unknown | None | SPINK1 N34S homozygote |
| 20 | 27 | M | 10.0 | None | Yes (Unknown) | None | SPINK1 N34S homozygote |
| 21 | 15 | F | 12.1 | None | No | None | SPINK1 N34S heterozygote; CTRC V235I heterozygote |
| 22 | 20 | M | 14.8 | None | Yes (19 y/o) | Pancreatic divisum | SPINK1 N34S heterozygote; CFTR L997F heterozygote |
| 23 | 17 | M | 14.8 | None | Yes (12 y/o) | None | SPINK1 N34S heterozygote; CFTR 394delTT heterozygote |
| 24 | 20 | F | 15.0 | None | Yes (19 y/o) | None | SPINK1 N34S heterozygote; CFTR A1285V heterozygote |
| 25 | 21 | M | 21.0 | None | Yes (17 y/o) | None | SPINK1 heterozygote**; CFTR heterozygote** |
| 26 | 41 | F | 22.4 | None | Yes (41 y/o) | None | SPINK1 N34S heterozygote |
| 27 | 24 | M | 24.0 | None | Yes (23 y/o) | None | SPINK1 heterozygote**; CFTR heterozygote** |
| 28 | 29 | F | 24.8 | None | Yes (Unknown) | Pancreatic divisum | SPINK1 N34S heterozygote |
Abbreviations: F, female; M, male.
Heavy alcohol consumption was defined by more than 5 drinks per day.
Information regarding the exact genomic alteration identified was not available.
Germline testing for SPINK1, PRSS1 and CFTR was performed in all cases. In addition, 19 (68%) patients had germline testing for CTRC. A heterozygous mutation in SPINK1 was identified in 25 patients, while the remaining 3 patients had a SPINK1 homozygous mutation. Detailed genomic information was available for 25 patients and the most prevalent SPINK1 alteration was the p.N34S missense mutation (n = 24, 96%). The remaining patient was reported to harbor a heterozygous SPINK1 p.R67H missense mutation. Among SPINK1 heterozygous carriers, 14 (56%) patients also had a heterozygous mutation in either CFTR (n = 12) or CTRC (n = 2).
Upon microscopic examination of representative sections from patients’ pancreata, a sequential pattern of histologic changes became apparent based on the duration of reported abdominal pain and could be subdivided into four pathologic stages of progressive chronic pancreatitis (Supplementary Table 1). The pancreata of SPINK1 carriers with a history of abdominal pain of < 4 years (n = 10) exhibited mild atrophy that was characterized by decreasing size and variable shape of the pancreatic lobules (Figure 1A and 1B). These histologic findings were often associated with multifocal, cytoplasmic vacuolization of the acinar parenchyma (n = 6) (Figure 1C) and the presence of mild intralobular fibrosis at the periphery of the pancreas (n = 7) (Figure 1D). Intralobular (n = 3) and extralobular (n = 4) lipomatous infiltration into the pancreatic parenchyma was also seen but focal in distribution. The presence of lipomatous infiltration was distinctly seen among CFTR carriers.
Figure 1.
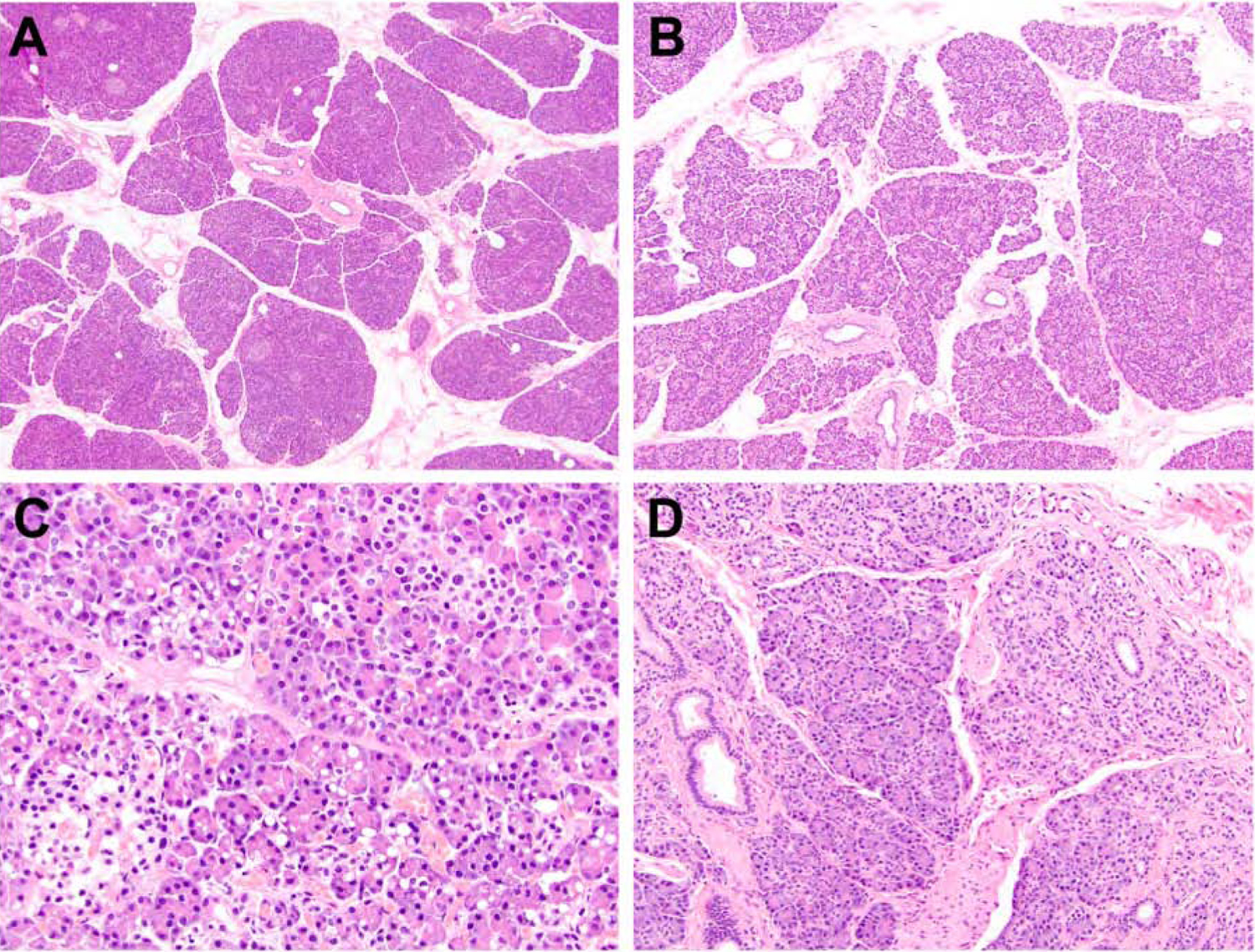
The pancreata of SPINK1 carriers with a history of abdominal pain of < 4 years. An examination of pancreatic specimens identified mild atrophy that was characterized by decreasing size and variable shape of the pancreatic lobules (A and B). For a subset of cases, multifocal, cytoplasmic vacuolization of the acinar parenchyma (C) and focal intralobular fibrosis at the periphery of the pancreas (D) were seen.
Among SPINK1 patients with a history of abdominal pain ranging from 4 to < 7 years (n = 6), their pancreata showed a progressive loss of acinar parenchyma. Further, mild interlobular (n = 6), mild intralobular (n = 5) and mild perilobular (n = 5) fibrosis was frequently identified and composed of thick bundles of collagen, fibroblasts and scattered islands of neutrophilic inflammation (Figure 2A and 2B). In fact, neutrophilic inflammation was seen in the majority of cases (n = 4) and present within and in between the pancreatic lobules (Figure 2C and 2D). In addition, fat necrosis and saponification were found within the pancreatic periphery for 3 cases. Extralobular lipomatous infiltration was seen in 2 patient that were also heterozygous for CFTR.
Figure 2.
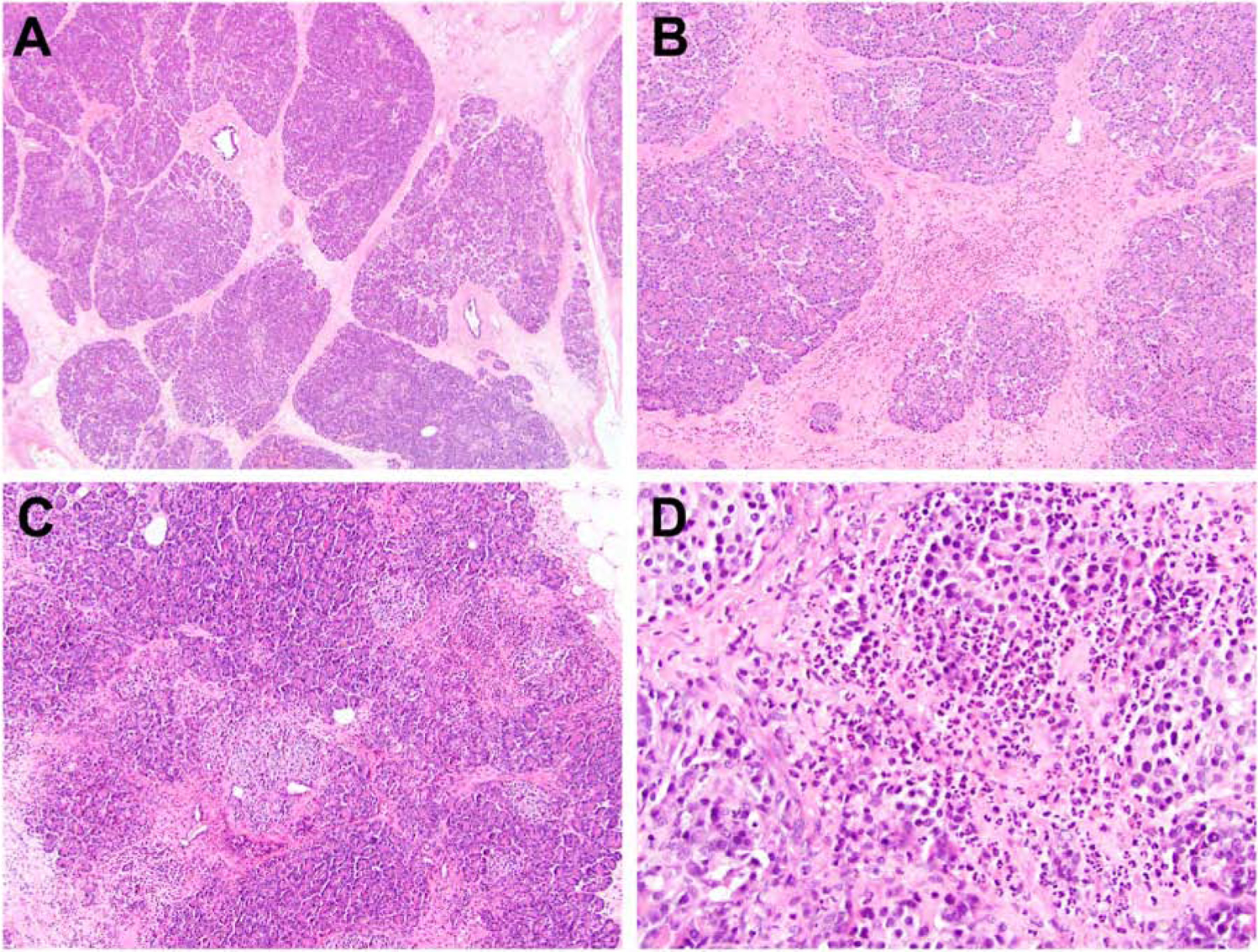
SPINK1 patients with a history of abdominal pain ranging from 4 to < 7 years. The pancreata exhibited a progressive loss of acinar parenchyma with coinciding mild interlobular and mild perilobular fibrosis. Fibrosis was typically composed of thick bundles of collagen, fibroblasts and scattered islands of neutrophilic inflammation (A and B). A brisk acute inflammatory infiltrate was also seen present within the pancreatic lobules (C and D).
Pancreatic specimens from patients with a 7-to-15-year history of abdominal pain (n = 8) displayed increasing loss of not only acinar cell epithelium, but also intralobular ducts. These findings coincided with a moderate amount of intralobular, interlobular and perilobular fibrosis (Figure 3A and 3B). Main duct and interlobular ductal alterations were identified (n = 7) and included ductal ectasia (n = 6), squamous metaplasia (n = 5) and intraductal concretions (n = 5) (Figure 3C). The periphery of the pancreas was remarkable for complete loss of acinar and ductal epithelium, but preservation of islets of Langerhans and surrounding nerves (Figure 3D) and, for 5 cases, the presence of fat necrosis and saponification. Of note, 3 patients within this group were SPINK1 homozygous and did not show appreciable differences from the remaining 6 SPINK1 heterozygous patients (Figure 3E and 3F). Lipomatous infiltration within an extralobular distribution was focally present in 2 patients with a germline alteration in CFTR.
Figure 3.
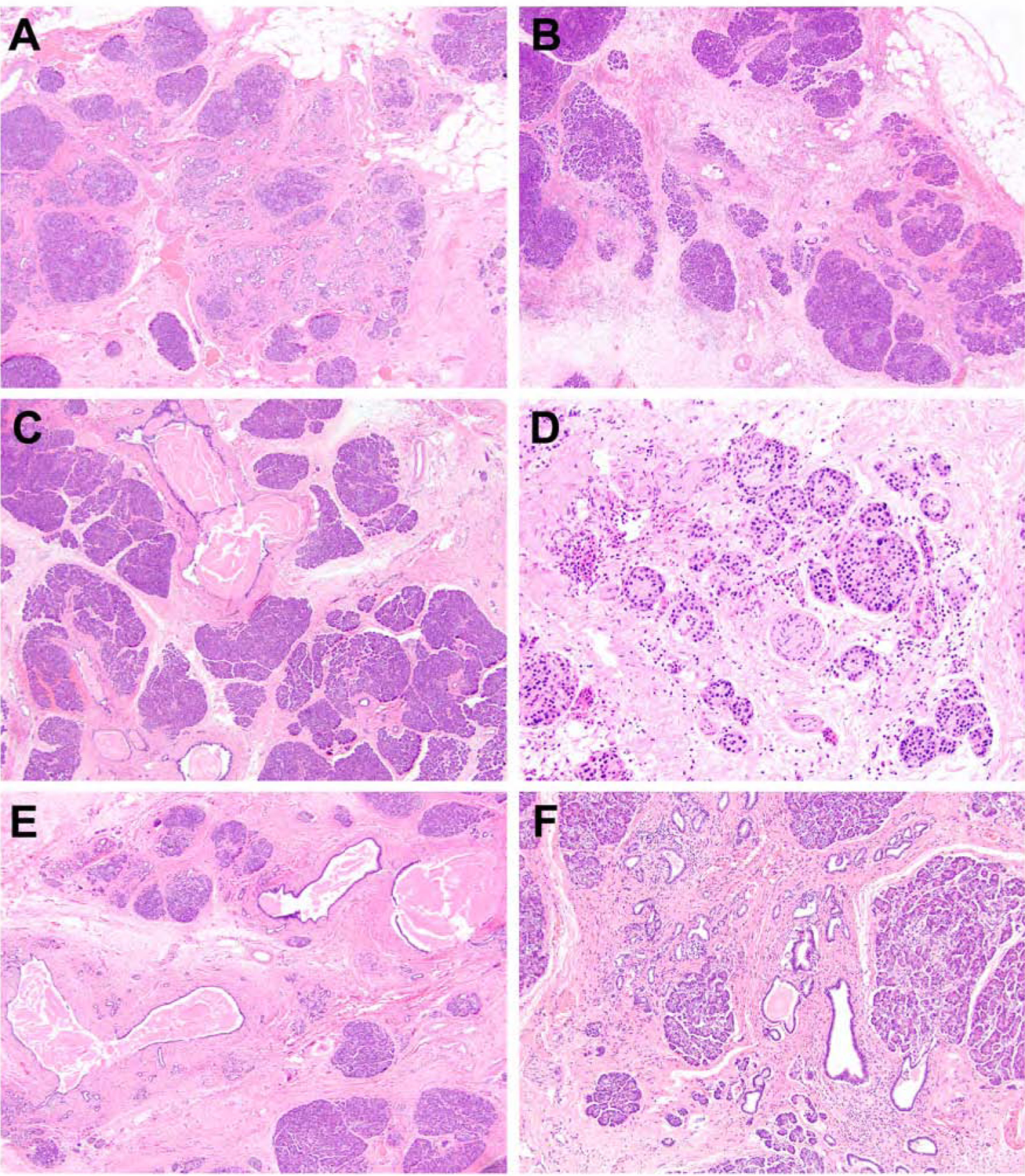
Pancreatic specimens from SPINK1 patients with a 7-to-15-year history of abdominal pain. The pancreata demonstrated increasing loss of acinar cell epithelium and intralobular ducts, which coincided with intralobular, interlobular and perilobular fibrosis (A and B). Ductal ectasia, squamous metaplasia and intraductal concretions were also present (C). The periphery of the pancreas was remarkable for complete loss of acinar and ductal epithelium, but preservation of islets of Langerhans and surrounding nerves (D). In addition, while the majority of patients were heterozygous for SPINK1, 3 patients were SPINK1 homozygotes with reported abdominal pain between 8.1 to 10 years and their pancreata exhibited similar histopathologic features as their SPINK1 heterozygous counterparts with ductal ectasia and intraductal concretions (E) and both intralobular and interlobular fibrosis (F).
Finally, pancreata from patients with a history of abdominal pain that ranged between 21 and 25 years (n = 4) exhibited near complete to complete loss of acinar and ductal epithelium, and extensive or severe parenchymal fibrosis (Figure 4A and 4B). The remnant exocrine pancreas consisted primarily of a dilated main pancreatic duct and associated interlobular ducts (Figure 4C). Three cases also had intraductal concretions/calcifications and, for 2 cases, scattered foci of reactive and hyperplastic intralobular ducts were also identified (Figure 4D). Pancreatic intraepithelial neoplasia (PanIN) of the highest grade PanIN-2 was also seen for 2 cases. The borders of the pancreata were ill-defined and the only clue were the presence of residual islets of Langerhans embedded in fibrosis (Figure 4E and 4F). Fat necrosis and saponification were absent. With regards to the presence of anatomic anomalies or germline status in not only SPINK1, but CFTR, CTRC and PRSS1, no significant pathologic differences that deviate from those associated with the duration of abdominal pain were identified. Of note, the presence of lipomatous infiltration was distinctly absent, even among the 2 CFTR patients within this group.
Figure 4.
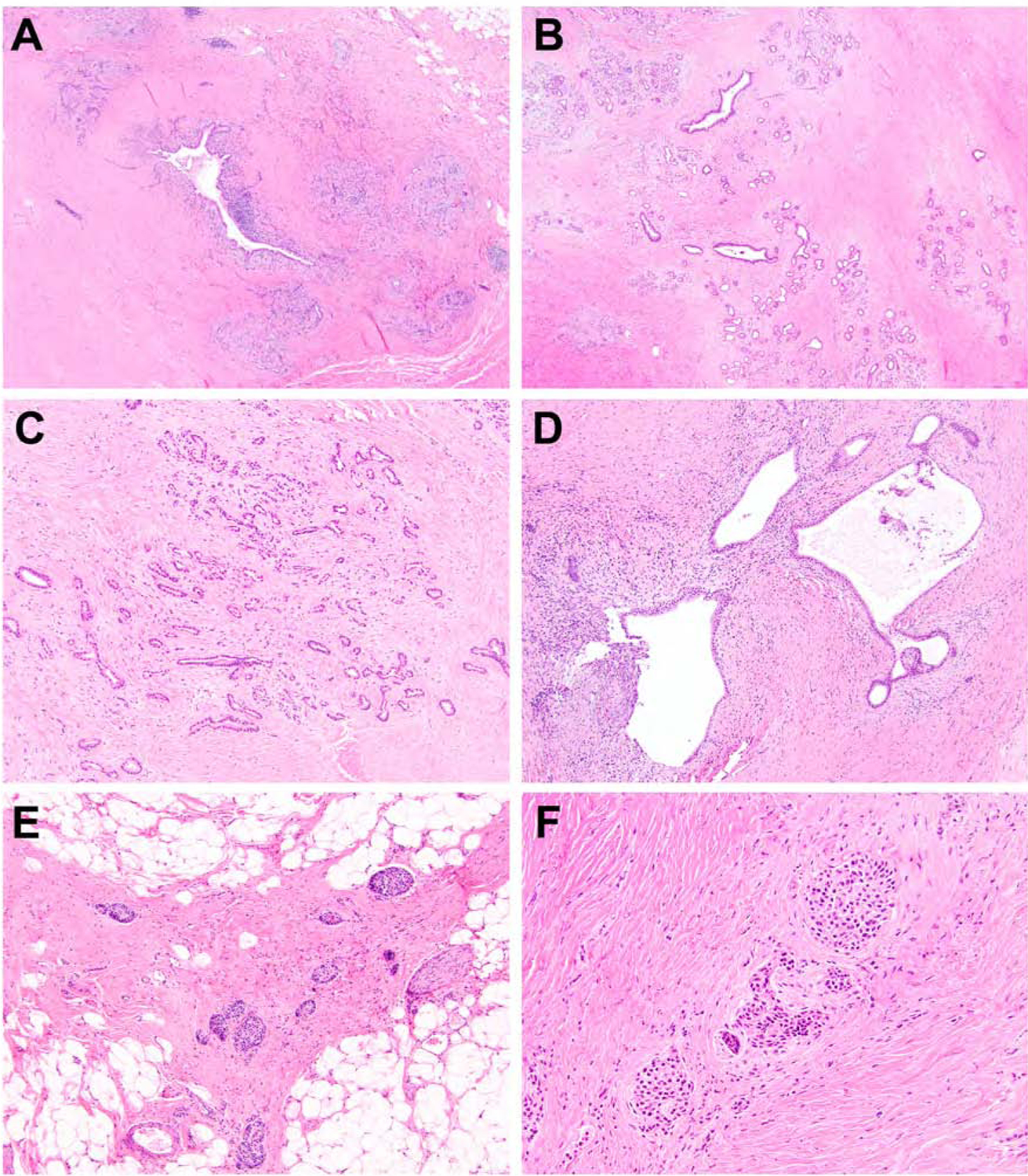
SPINK1 carriers with a history of abdominal pain that ranged between 21 and 25 years. Pancreatic specimens exhibited near complete to complete loss of acinar and ductal epithelium, and extensive parenchymal fibrosis (A and B). The remnant exocrine pancreas consisted primarily of a dilated main pancreatic duct and associated interlobular ducts (C). Scattered foci of reactive and hyperplastic intralobular ducts were also identified (D). At higher magnification, residual islets of Langerhans were found scattered throughout the pancreas but embedded in dense fibrosis (E and F).
Discussion
The serine protease inhibitor Kazal type 1 (SPINK1) protein is an antiprotease secreted by pancreatic acinar cells and is hypothesized to function by delaying the autoactivation cascade of converting trypsinogen to trypsin.11 This delay is important as it allows for the digestive proteases, such as trypsin, to transit from the pancreas to the duodenum in an inactive form and, thus, preventing autodigestion of the gland.12 Although the underlying molecular mechanisms between SPINK1 and pancreatitis are unclear, mutations in SPINK1 may lead to diminished inhibition of trypsin activity.13 Several mutations in SPINK1 have been documented, but the most common is the p.N34S haplotype and may increase the risk of chronic pancreatitis by up to 23%.14–17 However, this mutation is also a common polymorphism in the general population at a frequency of approximately 2%, which suggests that it may play a role as a disease modifier rather than a causative agent.18–20 In fact, when inherited in a heterozygous form, as is the case for the majority of patients with chronic pancreatitis, other inherited germline alterations, environmental factors and/or anatomic anomalies have been reported.21–26
Herein, we collected pancreatic specimens from patients with pathogenic SPINK1 variants and chronic pancreatitis. Among the reported SPINK1 variants, the p.N34S mutation was the most prevalent within our study cohort and were predominantly heterozygous. Furthermore, consistent with its potential status as a disease modifier, the majority of SPINK1 heterozygote patients (23 of 25, 92%) had either co-occurring alterations in other genes associated with chronic pancreatitis, anatomic anomalies and/or a history of tobacco use. Additional etiologic factors were not identified among patients with SPINK1 homozygous mutations. Examination of pancreata from SPINK1 carriers of various ages revealed a progressive loss of primarily exocrine parenchyma and replacement by prominent fibrosis. These findings do not seem dependent on patient age, but the duration of reported abdominal pain or clinical chronic pancreatitis.
The presence of progressive parenchymal fibrosis within the pancreas is intriguing and contrasts histopathologic findings reported for other germline variants associated with chronic pancreatitis, such as PRSS1 and CFTR. PRSS1 encodes for cationic trypsinogen and gain-of-function mutations lead to premature trypsinogen activation.27 As a result, PRSS1 patients exhibit chronic pancreatitis at a young age and develop progressive exocrine insufficiency.28 Previously, we published the histopathology of PRSS1-associated chronic pancreatitis and found these patients develop pancreatic lipomatous atrophy and not parenchymal fibrosis.7 Similarly, dysfunctional germline alterations in CFTR, which leads to retention of trypsinogen within the pancreas and, consequently, pancreatitis, also results in fatty replacement of the pancreas.8, 9 Pancreatic parenchymal fibrosis has not been described in CFTR patients. SPINK1 carriers with co-occurring mutations in CFTR exhibited prominent fibrosis of the pancreas and only focal intralobular and/or extralobular lipomatous infiltration into the pancreatic parenchyma. In addition, the presence of lipomatous atrophy was not seen in longstanding cases of SPINK1-associated chronic pancreatitis. Our observations would suggest that the pathophysiologic mechanisms of SPINK1-associated pancreatic parenchymal fibrosis are not only distinct from those described in PRSS1 and CFTR patients, but overcome those associated with lipomatous atrophy.
While the histopathologic findings of SPINK1-associated chronic pancreatitis may seem unique, they are reminiscent of those seen among chronic pancreatitis patients with documented heavy alcohol consumption. Within a large autopsy series, Suda et al. found pancreata from patients with chronic alcohol abuse exhibited a combination of intralobular and perilobular fibrosis.29 Intralobular fibrosis was characterized by a uniform distribution of fibrosis within the pancreas that imparts a micronodular arrangement around atrophic lobules. In comparison, perilobular fibrosis is often dense with ductal ectasia and intraductal calcifications. In advanced cases, the pancreatic parenchyma may be completely replaced by extensive fibrosis. These histopathologic findings are analogous to pancreata from SPINK1 patients and, similarly, may represent a sequence of changes that correlates with duration of heavy alcohol consumption and clinical chronic pancreatitis. In fact, the reported prevalence of SPINK1 mutations in alcohol-induced chronic pancreatitis is higher than the general population at 5.6% to 6%.19, 24, 30, 31 It is interesting to speculate that there may be a common pathophysiologic mechanism shared between SPINK1-associated and alcohol-induced chronic pancreatitis.
In addition to defining the histopathologic features of chronic pancreatitis among SPINK1 carriers, our observations are also significant as they can be used to determine the validity of current and future animal models of SPINK1-associated chronic pancreatitis. Targeted disruption of Spink3 (the murine homologue of human SPINK1) results in is severe pancreatic damage and death within two weeks after birth.32 The histopathologic changes are restricted to pancreatic acinar cells, which are filled with numerous autophagic vacuoles with enhanced trypsin activity. These vacuoles closely resemble the vacuolated acinar parenchyma of SPINK1 patients with a history of chronic pancreatitis of < 4 years. Mosaic expression of human SPINK1 in Spink3 knockout mice rescued the perinatal lethality, but these mice gradually developed chronic pancreatitis, manifested by loss of acinar cells, intralobular fibrosis and dilatation of intralobular ducts, which bears resemblance to human SPINK1-associated chronic pancreatitis.33 However, in humans, homozygous SPINK1 mutations do not result in embryonic lethality, and heterozygous mutations are frequently present in the general population with development of chronic pancreatitis typically associated with the co-occurrence of other genetic alterations, anatomic anomalies and environmental factors. Hence, while current mouse models emulate several aspects of SPINK1-associated chronic pancreatitis, they likely do not recapitulate human disease.
It is worth noting that there are several limitations to our study. Although it is the first to assess the histopathologic features of SPINK1-associated chronic pancreatitis, the number of cases within our cohort is relatively small and a control cohort was not available for comparative histopathologic review. In addition, most of our patients were heterozygous for SPINK1, which as discussed previously is common within the general population, and had co-occurring genomic, anatomic and/or environmental risk factors. Thus, one could infer that the histopathologic findings described herein may be unrelated to SPINK1 status. However, our study cohort included 3 patients that were homozygous for SPINK1 and had no additional risk factors. The pancreata from these 3 patients exhibited similar histopathologic findings as SPINK1 heterozygotes within the reported timeframe of abdominal pain. Another limitation of this study is that we did not use a histopathologic scoring system for chronic pancreatitis, but rather graded the presence of fibrosis semi-quantitatively.10 While clinical, imaging and functional scoring systems have been developed for chronic pancreatitis, an accepted and reproducible method of evaluating the microscopic findings of chronic pancreatitis does not exist.34–36 In fact, within a recent joint publication by the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society and the European Pancreatic Club, there was strong agreement that there currently exists no reproducible or universally accepted histological grading system to assess the severity of chronic pancreatitis.37 Moreover, there was strong agreement that a three-tiered semi-quantitative scoring system is typically used to evaluate the pancreas.37 Therefore, we chose to report the clinical, genomic and histopathologic findings of SPINK1-associated chronic pancreatitis within a descriptive manner. As additional cases of SPINK1-associated chronic pancreatitis are evaluated, it is likely that the histopathologic features described herein may prove to represent a continuum of findings rather than discrete stages of chronic pancreatitis. Further, the duration of abdominal pain and subsequent time periods were determined retrospectively at the time of total pancreatectomy with islet autotransplantation and, thus, was dependent upon patient recall. Our study also suffers from selection bias because only patients undergoing total pancreatectomy with islet autotransplantation were evaluated. Finally, the isolation of islets during this procedure precludes complete histologic analysis of the pancreas. Therefore, only representative sections of the pancreas were reviewed.
In summary, SPINK1-associated chronic pancreatitis is characterized by progressive parenchymal fibrosis. These findings contrast the reported findings of lipomatous atrophy observed is PRSS1 and CFTR patients, suggesting divergent pathophysiologic mechanisms. Further, patients with SPINK1-associated chronic pancreatitis frequently have additional etiologic factors for pancreatitis, such as alterations in CFTR, that did not impact the development of pancreatic fibrosis and may indicate a role for SPINK1 as a disease modifier gene. While this study enhances our current understanding of chronic pancreatitis among SPINK1 carriers, certainly additional questions emerge regarding the pathogenesis of SPINK1 mutations and its interaction with other etiologic factors. However, as germline testing for chronic pancreatitis becomes more widespread, future studies should provide greater insight into this disease.
Supplementary Material
Acknowledgments
Funding support: None
Footnotes
Disclosure/conflict of interest: The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kleeff J, Whitcomb DC, Shimosegawa T, et al. Chronic pancreatitis. Nat Rev Dis Primers 2017;3:17060. [DOI] [PubMed] [Google Scholar]
- 2.Zhan W, Shelton CA, Greer PJ, et al. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas 2018;47:924–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sossenheimer MJ, Aston CE, Preston RA, et al. Clinical characteristics of hereditary pancreatitis in a large family, based on high-risk haplotype. The Midwest Multicenter Pancreatic Study Group (MMPSG). Am J Gastroenterol 1997;92:1113–6. [PubMed] [Google Scholar]
- 4.Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004;2:252–61. [DOI] [PubMed] [Google Scholar]
- 5.de las Heras-Castano G, Castro-Senosiain B, Fontalba A, et al. Hereditary pancreatitis: clinical features and inheritance characteristics of the R122C mutation in the cationic trypsinogen gene (PRSS1) in six Spanish families. JOP 2009;10:249–55. [PubMed] [Google Scholar]
- 6.Rebours V, Boutron-Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut 2009;58:97–103. [DOI] [PubMed] [Google Scholar]
- 7.Singhi AD, Pai RK, Kant JA, et al. The histopathology of PRSS1 hereditary pancreatitis. Am J Surg Pathol 2014;38:346–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tham RT, Heyerman HG, Falke TH, et al. Cystic fibrosis: MR imaging of the pancreas. Radiology 1991;179:183–6. [DOI] [PubMed] [Google Scholar]
- 9.Soyer P, Spelle L, Pelage JP, et al. Cystic fibrosis in adolescents and adults: fatty replacement of the pancreas--CT evaluation and functional correlation. Radiology 1999;210:611–5. [DOI] [PubMed] [Google Scholar]
- 10.Kloppel G, Maillet B. Pseudocysts in chronic pancreatitis: a morphological analysis of 57 resection specimens and 9 autopsy pancreata. Pancreas 1991;6:266–74. [PubMed] [Google Scholar]
- 11.Kazal LA, Spicer DS, Brahinsky RA. Isolation of a crystalline trypsin inhibitor-anticoagulant protein from pancreas. J Am Chem Soc 1948;70:3034–40. [DOI] [PubMed] [Google Scholar]
- 12.Pubols MH, Bartelt DC, Greene LJ. Trypsin inhibitor from human pancreas and pancreatic juice. J Biol Chem 1974;249:2235–42. [PubMed] [Google Scholar]
- 13.Kume K, Masamune A, Ariga H, et al. Do genetic variants in the SPINK1 gene affect the level of serum PSTI? J Gastroenterol 2012;47:1267–74. [DOI] [PubMed] [Google Scholar]
- 14.Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000;25:213–6. [DOI] [PubMed] [Google Scholar]
- 15.Teich N, Bauer N, Mossner J, et al. Mutational screening of patients with nonalcoholic chronic pancreatitis: identification of further trypsinogen variants. Am J Gastroenterol 2002;97:341–6. [DOI] [PubMed] [Google Scholar]
- 16.Schneider A, Barmada MM, Slivka A, et al. Clinical characterization of patients with idiopathic chronic pancreatitis and SPINK1 Mutations. Scand J Gastroenterol 2004;39:903–4. [DOI] [PubMed] [Google Scholar]
- 17.Truninger K, Kock J, Wirth HP, et al. Trypsinogen gene mutations in patients with chronic or recurrent acute pancreatitis. Pancreas 2001;22:18–23. [DOI] [PubMed] [Google Scholar]
- 18.Pfutzer RH, Barmada MM, Brunskill AP, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000;119:615–23. [DOI] [PubMed] [Google Scholar]
- 19.Threadgold J, Greenhalf W, Ellis I, et al. The N34S mutation of SPINK1 (PSTI) is associated with a familial pattern of idiopathic chronic pancreatitis but does not cause the disease. Gut 2002;50:675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandak GR, Idris MM, Reddy DN, et al. Absence of PRSS1 mutations and association of SPINK1 trypsin inhibitor mutations in hereditary and non-hereditary chronic pancreatitis. Gut 2004;53:723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider A, Larusch J, Sun X, et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011;140:162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witt H Chronic pancreatitis and cystic fibrosis. Gut 2003;52 Suppl 2:ii31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitcomb DC. Value of genetic testing in the management of pancreatitis. Gut 2004;53:1710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoun E, Chang CC, Greer JB, et al. Pathways to injury in chronic pancreatitis: decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS One 2008;3:e2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimosegawa T, Kume K, Masamune A. SPINK1 gene mutations and pancreatitis in Japan. J Gastroenterol Hepatol 2006;21 Suppl 3:S47–51. [DOI] [PubMed] [Google Scholar]
- 26.Reddy DN, Prasad SS. Genetic basis of chronic pancreatitis in Asia Pacific region. J Gastroenterol Hepatol 2011;26 Suppl 2:2–5. [DOI] [PubMed] [Google Scholar]
- 27.Sahin-Toth M, Toth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 2000;278:286–9. [DOI] [PubMed] [Google Scholar]
- 28.Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141–5. [DOI] [PubMed] [Google Scholar]
- 29.Suda K, Takase M, Takei K, et al. Histopathologic study of coexistent pathologic states in pancreatic fibrosis in patients with chronic alcohol abuse: two distinct pathologic fibrosis entities with different mechanisms. Pancreas 1996;12:369–72. [DOI] [PubMed] [Google Scholar]
- 30.Drenth JP, te Morsche R, Jansen JB. Mutations in serine protease inhibitor Kazal type 1 are strongly associated with chronic pancreatitis. Gut 2002;50:687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witt H, Luck W, Becker M, et al. Mutation in the SPINK1 trypsin inhibitor gene, alcohol use, and chronic pancreatitis. JAMA 2001;285:2716–7. [DOI] [PubMed] [Google Scholar]
- 32.Ohmuraya M, Hirota M, Araki M, et al. Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 2005;129:696–705. [DOI] [PubMed] [Google Scholar]
- 33.Sakata K, Araki K, Nakano H, et al. Novel method to rescue a lethal phenotype through integration of target gene onto the X-chromosome. Sci Rep 2016;6:37200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider A, Lohr JM, Singer MV. The M-ANNHEIM classification of chronic pancreatitis: introduction of a unifying classification system based on a review of previous classifications of the disease. J Gastroenterol 2007;42:101–19. [DOI] [PubMed] [Google Scholar]
- 35.Conwell DL, Lee LS, Yadav D, et al. American Pancreatic Association Practice Guidelines in Chronic Pancreatitis: evidence-based report on diagnostic guidelines. Pancreas 2014;43:1143–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tirkes T, Shah ZK, Takahashi N, et al. Reporting Standards for Chronic Pancreatitis by Using CT, MRI, and MR Cholangiopancreatography: The Consortium for the Study of Chronic Pancreatitis, Diabetes, and Pancreatic Cancer. Radiology 2019;290:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esposito I, Hruban RH, Verbeke C, et al. Guidelines on the histopathology of chronic pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with the International Association of Pancreatology, the American Pancreatic Association, the Japan Pancreas Society, and the European Pancreatic Club. Pancreatology 2020;20:586–593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.