

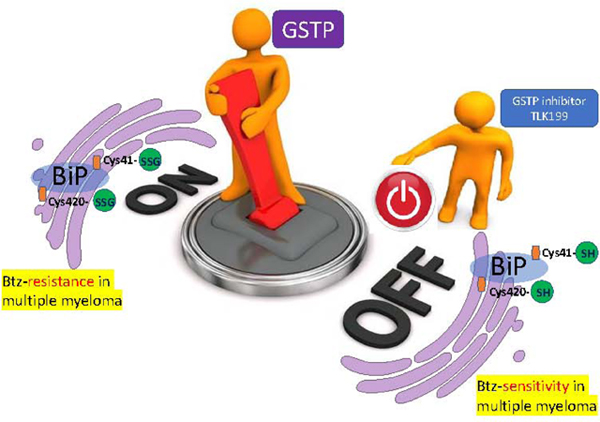
Abstract
Multiple myeloma (MM) cells have high rates of secretion of proteins rich in disulfide bonds and depend upon compartmentalized redox balance for accurate protein folding. The proteasome inhibitor bortezomib (Btz) is a successful frontline treatment for the disease, but its long-term efficacy is restricted by the acquisition of resistance. We found that MM cell lines resistant to Btz maintain high levels of oxidative stress and are cross resistant to endoplasmic reticulum (ER) stress-inducing agents thapsigargin (ThG), and tunicamycin (TuM). Moreover, cells expressing high/wild type levels of glutathione S-transferase P (GSTP) are more resistant than Gstp1/p2 knockout cells. In agreement, basal levels of S-glutathionylated proteins and redox regulation enzymes, including GSTP are elevated at mRNA and protein levels in resistant cells. GSTP mediated S-glutathionylation (SSG) regulates the activities of a number of redox active ER proteins. Here we demonstrated that the post-translational modification determines the balance between foldase and ATPase activities of the binding immunoglobulin protein (BiP), with Cys41-SSG important for ATPase, and Cys420-SSG for foldase. BiP expression and S-glutathionylation are increased in clinical specimens of bone marrow from MM patients compared to non-cancerous samples. Preventing S-glutathionylation in MM cells with a GSTP specific inhibitor restored BiP activities and reversed resistance to Btz. Therefore, S-glutathionylation of BiP confers pro-survival advantages and represents a novel mechanism of drug resistance in MM cells. We conclude that altered GSTP expression leads to S-glutathionylation of BiP, and contributes to acquired resistance to Btz in MM.
Keywords: BiP, bortezomib, endoplasmic reticulum, glutathione, glutathione S-transferase, S-glutathionylation, multiple myeloma, reactive oxygen species, redox, unfolded protein response
Graphical Abstract
Introduction
As a malignancy of monoclonal plasma cells, multiple myeloma (MM) comprises ~10% of hematologic malignant tumors and therapeutic treatments usually include some combination of alkylating agents, immunomodulators, microtubule-targeting agents and proteasome inhibitors. MM cells support a uniquely high rate of secretion of proteins with disulfide bonds1 and as a consequence are dependent upon redox balance to maintain accurate folding.2 Distinct from MM, normal hematopoietic progenitor cells require regulated and reduced rates of protein synthesis. Based on these differences, proteasome inhibitors (PIs) such as bortezomib (Btz; Velcade®) have been established as a frontline therapy for the disease. Patients are initially responsive to PIs, but invariably relapse as a result of resistance. Second-generation PIs are also subject to cross-resistance, both clinically and pre-clinically3,4. Btz resistance has been linked with reduced drug binding avidity through altered proteasome structure,5,6 but links between proteasome functions and efficiency of protein folding in the endoplasmic reticulum (ER) suggest that other resistance mechanisms exist. For example, the unfolded protein response (UPR) pathway regulates a cascade of transcriptional/translational events to preserve protein folding fidelity. When accumulation of unfolded proteins is not resolved, the UPR is insufficient to regain homeostasis and normal cells die.7 Mechanisms to circumvent cell death due to accumulation of unfolded proteins may contribute to drug resistance. In this regard, the binding immunoglobulin protein (BiP), which in the ER lumen mediates accurate folding or degradation of misfolded proteins via interaction with components of the proteasome, is of a particular interest.8
Glutathione transferase P (GSTP) known for phase II detoxification activities, can also function as a thiolase in S-glutathionylation (SSG) of proteins9 and as a chaperone-like regulator of kinase activities.10,11 Primarily cytosolic, GSTP also localizes to ER, and is subject to glycosylation.12 Its presence in ER correlates with SSG of ER resident proteins.13 During protein folding, levels of reactive oxygen species (ROS) in the ER can fluctuate, with redox potential of the organelle generally shifted towards an oxidative state.14,15 S-glutathionylation of ER resident proteins such as BiP, calnexin, calreticulin, endoplasmin and ER Ca2+ ATPase (SERCA), control organelle functions that codify protein folding and communicate inaccuracies through proteasome degradation. The absence of GSTP has been shown to reduce S-glutathionylation and Ca2+ signaling, affecting the UPR.13 This post-translational modification may supersede in its regulatory importance other mechanisms that regulate protein abundance and activity, such as protein turnover. Increased GSTP expression has frequently been linked to resistance to a range of anticancer drugs where their pharmacology precludes the requirement for detoxification as a means of resistance.16,17 Our current data show a direct relationship between GSTP expression and development of resistance to Btz, accompanied by cross-resistance to other drugs that cause cytotoxicity by affecting protein folding and ER function. We interrogated the hypothesis that differences in redox parameters impact BiP function contributing to the acquired resistant phenotype and tested whether manipulation of GSTP could reverse resistance.
Results
GSTP expression and cross-resistance to Btz and ER-targeting drugs.
ER resident GSTP plays a role in the control of organelle redox and the UPR. Drug naïve bone marrow derived dendritic cells (BMDDC) and mouse embryonic fibroblast (MEF) cells from wild type (WT) mice have been demonstrated to be more resistant to ThG and TuM than those from Gstp1/p2 knockout mice.13 Figures 1A and B show that BMDDC and MEF WT cells are also intrinsically resistant to Btz. Neither of these cell lines had previously been exposed to those drugs. The absence of GSTP in those drug naïve BMDDC and MEF cells resulted in 8- and 2- fold decrease in Btz IC50 values, respectively (Table 1). Reciprocally, we found that MM cell lines that had been selected for Btz-resistance by 6-month dose escalation were cross-resistant to ThG and TuM (Figure 1C-E (MM1.S), and Figure 1 F-H (U266)) with increased IC50 values from 2 to 6-fold (Table 1). Therefore, differences in GSTP expression can be linked to acquisition of resistance to Btz and ER-targeting drugs, and the results provided a rational platform for studying redox-based mechanisms as causative factors in drug response.
Figure 1.

BMDDC and MEF cells expressing high/wild type (WT) GSTP are more resistant to thapsigargin (ThG), tunicamycin (TuM), and bortezomic (Btz) (A and B) than those from Gstp1/p2 knockout cells. Moreover, multiple myeloma cell lines resistant to Btz have a cross resistance to ThG, and TuM (C-E for MM1.S cells, and F-H for U266 cells, respectively). Cells were treated with the drugs at the concentrations indicated for 48 h. Viabilities were determined by MTT assay with data normalized as percentage of untreated controls.
Table 1.
IC50 values for ThG, TuM and Btz in BMDDC, MEF, MM1.S and U266 cells.
| BMDDC* | MEF* | MM1.S | U266 | |||||
|---|---|---|---|---|---|---|---|---|
| WT | Gstp1/p2−/− | WT | Gstp1/p2−/− | Resistant | Sensitive | Resistant | Sensitive | |
| Btz (nM) | 23.6 ± 1.4 | 3.0 ± 0.9 | 49.0 ± 3.6 | 29.3 ± 1.6 | 26.5 ±3.2 | 5.3 ±2.4 | 27.9 ±1.7 | 5.4 ±0.8 |
| (fold change) | (~8) | (~2) | (~5) | (~5) | ||||
| ThG (nM) | 122 ± 28 | 17 ±1 | 5550 ±260 | 904 ±43 | 17.4 ± 2.7 | 9.7 ±2.4 | 28.3 ±3.4 | 5.0 ±2.1 |
| (fold change) | (~7) | (~6) | (~2) | (~6) | ||||
| TuM (ng/ml) | 209±6 | 92 ±11 | 5870 ±320 | 2333 ±112 | 43.7 ±2.9 | 10.3 ±2.1 | 48.0 ± | 15.8 ±1.9 |
| (fold change) | (~2) | (~3) | (~4) | 3.5 | ||||
| (~3) | ||||||||
The IC50 values of ThG and TuM in BMDDC and MEF cells were from our previous published paper.
Btz-resistant cells have higher basal levels of oxidative and ER stress.
To gain insight into the mechanism of the cross-resistance, cellular ROS and thiol levels were measured. Btz-resistant MM cells (both MM1.S and U266 cells) had lower total thiol, protein thiol and glutathione (GSH) levels, GSH/GSSG ratios, and higher GSSG and ROS levels (Figure 2A-2F), indicating a higher basal oxidative state associated with drug resistance. RNAseq of MM1.S sensitive and resistant cells identified altered expression of a number of oxidative stress associated genes in MM1.S resistant cells. GPX1 and 4, PRDX2 and 5, SRXN1, and GRX1 were decreased, GSR, SOD2, GSTP1, PRDX6, TRX1 and 2 were increased, and UPR associated ERN1 and HSP90B1 were also decreased (Figure 2G).
Figure 2.

Btz-resistant MM cells (MM1.SR and U266R) are under higher basal levels of oxidative stress compared to the sensitive cells (MM1.SS and U266S). Resistant cells possess significantly reduced levels of total thiols (A); protein thiols (B); GSH (C); GSH/GSSG ratios (D); increased levels of GSSG (E); and ROS (F); mRNA sequencing data revealed differential expression of 2227 genes in MM1.SR cells compared to MM1.SS. Among these, GPX1 and 4, PRDX2 and 5, SRXN1, GRX1, GSR, SOD2, GSTP1, PRDX6, TRX1 and 2 are genes encoding redox regulation enzymes, and ERN1 and HSP90B1 are genes encoding UPR proteins (G).
ER functions and ROS levels are inter-connected since high levels of ROS may disrupt protein folding, induce ER stress, which in turn may further increase ROS production.18 Therefore, expression of the UPR sensor inositol-requiring enzyme 1 (IRE1), ER chaperones and redox regulatory enzymes was compared between MM1.S and U266 sensitive and resistant cells using real-time PCR (Figure 3A-C) and immunoblots (Figure 3D-G). Resistance to Btz in MM cells demonstrated significant decreases in the expression of proteins associated with ER stress management, e.g. IRE1, glucose-regulated protein 94 (GRP94) and protein disulfide isomerase (PDI) (encoded by ERN1, HSP90B1, and P4HB genes, respectively), as well as changes in antioxidant enzymes, e.g. glutathione peroxidase 1 and 4 (GPx1 and 4), peroxiredoxin 2, 5, and 6 (Prx2, 5 and 6), sulfiredoxin (Srx), glutaredoxin 1 (Grx1), glutathione disulfide reductase (GR), superoxide dismutase 2 (SOD2), GSTP, thioredoxin 1 and 2 (Trx1 and 2). Down-regulation of GPx1 and 4, Prx2 and 5, Srx1, and Grx1 (encoded by GPX1 and 4, PRDX2 and 5, SRXN1, andGRX1 genes, respectively) may contribute to the higher basal levels of oxidation prone thiols/proteins observed in Btz-resistant MM cells. The up-regulation of GR, SOD2, GSTP, Prx6, Trx1 and 2 (encoded by GSR, SOD2, GSTP1, PRDX6, TRX1 and 2 genes, respectively) could be interpreted as adaptive responses, to protect those cells from ROS damage. Therefore, resistance is characterized by re-establishing redox homeostasis towards a more oxidative state.
Figure 3.

UPR proteins and redox-regulated enzymes are differentially expressed in Btz resistant and sensitive MM cells. (A) Heat map for UPR and anti-oxidant enzyme gene expression patterns shows significant changes in expression patterns in both resistant cell lines (MM1.S and U266) when compared to sensitive cells. (B and C) Real time PCR quantification of UPR protein and redox enzyme mRNA expression in MM1.S and U266 sensitive and resistant cells. (D-G) Immunoblot and quantification of UPR protein and anti-oxidant enzyme expression in MM1.S and U266 sensitive and resistant cells.
S-glutathionylation is increased in Btz-resistant cells.
Btz-resistant MM cells have higher GSTP, but lower levels of Grx1 (Figure 3). Since GSTP catalyzes the forward and Grx1 the reverse reactions of S-glutathionylation, such data predict that S-glutathionylation levels should be higher in resistant cells. The human GSTP locus comprises four different alleles: GSTP1*A (WT, Ile105; Ala114), GSTP1*B (Val105; Ala114), GSTP1*C (Val105; Val114), and GSTP1*D (Ile105; Val114).19 mRNA sequencing demonstrated that only the GSTP1*A allele was expressed in Btz-sensitive and resistant MM1.S and U266 cells (Figure 4A). To investigate GSTP mediated S-glutathionylation, we used MCF7 cells, which have limited endogenous GSTP, whereby its function can be studied by introducing recombinant forms of each GSTP allelic variant into the cells. Figure 4B shows the S-glutathionylation levels in MCF7 cells with or without recombinant GSTP after PABA/NO treatment.20 All the GSTP allelic variants promoted S-glutathionylation, with higher levels found for GSTP1*A. To enhance the sensitivity of detection of S-glutathionylation, we employed a modified protocol using deglutathionylation catalyzed by Grx for quantitative comparison of basal S-glutathionylation in MM cells. Elevated S-glutathionylation levels were observed in both Btz-resistant MM1.S and U266 cells compared to their Btz-sensitive counterparts (Figure 4C and D), in agreement with the immunoblot data (higher GSTP and lower Grx1). Inhibition of GSTP by TLK199 reduced S-glutathionylation levels, especially in MM1.S Btz-resistant cells (Figure 4C and D).
Figure 4.

Btz resistant MM cells have higher global S-glutathionylation levels, which are reduced after TLK199 treatment. (A) The mRNA sequencing results showed that GSTP1*A is the only allele present in all MM1.S and U266 cells (sensitive and resistant). (B and C) GSTP1*A has the highest catalytic activities for protein S-glutathionylation compared to other GSTP polymorphic variants. MCF7 cell lysates, which have undetectable endogenous GSTP1 levels, were freshly prepared, treated with PABA/NO in the absence or presence of different E. Coli-expressed and purified GSTP1 allelic variants (A, B, C or D), and immediately subjected to SDS-PAGE under non-reducing conditions. (D and E) S-glutathionylation levels in MM1.S and U266 cells (sensitive and resistant) with or without TLK199 treatment were quantified after deglutathionylation by Grx. Free thiols were blocked by adding 40 mM N-ethylmaleimide for 30 min, and the S-glutathionylated thiols were then deglutathionylated by adding 1 mM GSH, 1 mM NADPH, 35 μg/ml GSSG reductase, and 13.5 μg/ml Grx at 37°C for 30 min. The regenerated free thiols were quantified by western blotting using the streptavidin secondary antibodies after labeling with Maleimide-PEG11-Biotin.
GSTP inhibition rescues Btz cytotoxicity.
Since upregulated S-glutathionylation levels were found in MM resistant cells, we then investigated whether the GSTP specific inhibitor TLK199 could restore Btz-sensitivity in MM cells. The cytotoxic effects of TLK199 in combination with Btz, ThG and TuM in MM cells were assessed by MTT assays. Inhibition of GSTP by TLK199 enhanced Btz, ThG and TuM cytotoxicity in both sensitive (Figure 5A-C for MM1. Ss and Figure 5G-I for U266S) and resistant MM cells (Figure 5D-F for MM1.SR and Figure 5J-L for U266R). The effect was most prominent in Btz-resistant cells (Figure 5D, E, F, J, K and L). In the presence of TLK199, the IC50 values of Btz decreased from 5.3 to 4.1 nM (~1.3 fold), and 5.4 to 3.2 nM (~1.7 fold) in Btz-sensitive MM1.S and U266 cells respectively; in Btz-resistant MM1.S and U266 cells, the IC50 values of Btz decreased from 26.5 to 9.8 nM (~2.7 fold), and 27.9 to 10.1 μM (~2.8 fold) respectively (Table 2). Similar synergistic effects of TLK199 were also observed on the IC50 values of ThG (~1.9-, 1.4- fold change versus 2.0-, 2.1- fold change) and TuM (~1.6-, 1.2- fold change versus 2.8-, 1.6- fold change) in MM1.S and U266 sensitive versus resistant cells (Table 2). Therefore, inhibition of GSTP decreases the doses of Btz, ThG and TuM required to reach maximal cytotoxic effects.
Figure 5.

Pharmacological inhibition of GSTP by TLK199 reverses Btz resistance in MM cells. Cytotoxicity of Btz (A, D, G, J), ThG (B, E, H, K), and TuM (C, F, I, L) was determined in both sensitive and resistant MM1.S and U266 cells with or without the GSTP inhibitor TLK199. Cells were treated with varying concentrations of drugs as indicated for 48 h in each cell line, and the cell viabilities were evaluated using the MTT assay. Data were normalized to percentage of control cells.
Table 2.
IC50 values for Btz, ThG and TuM in MM1.S and U266 cells in combination with GSTP inhibitor TLK199.
| MM1.SS | MM1.SR | U266S | U266R | |||||
|---|---|---|---|---|---|---|---|---|
| Control | TLK199 | Control | TLK199 | Control | TLK199 | Control | TLK199 | |
| Btz (nM) | 5.3 ±2.4 | 4.1 ±2.6 | 26.5 ±3.2 | 9.8 ±1.7 | 5.4 ±0.8 | 3.2 ±0.8 | 27.9 ± 1.7 | 10.1 ±1.6 |
| (fold change) | (~1.3) | (~2.7) | (~1.7) | (~2.8) | ||||
| ThG (nM) | 9.7 ±2.4 | 5.1 ±2.6 | 17.4 ±2.7 | 8.9 ±1.9 | 5.0 ± 2.1 | 3.7 ±2.1 | 28.3 ±3.4 | 13.7 ±2.4 |
| (fold change) | (~1.9) | (~2.0) | (~1.4) | (~2.1) | ||||
| TuM (ng/ml) | 10.3 ±2.1 | 6.6 ±2.0 | 43.7 ±2.9 | 15.4 | 15.8 | 12.7 ±2.0 | 48.0 ±3.5 | 29.4 ±3.3 |
| (fold change) | (~1.6) | (~2.8) | ±2.6 | ±1.9 | (~1.6) | |||
| (~1.2) | ||||||||
S-glutathionylation of BiP on Cys41 and Cys420 residues regulates the balance of its foldase and ATPase activities by altering protein structure.
To understand how S-glutathionylation contributes to resistance, we focused on its effect on the activities of BiP. BiP was chosen for analysis because it has a critical role in protein folding and UPR activation, and it has two cysteine residues that are predicted to be susceptible to redox regulation, but their actual modification states in human cells haven’t been tested. To compare S-glutathionylation of BiP in Btz-sensitive and resistant MM cells (MM1.SS, MM1.SR, U266S, and U266R), BiP was immunoprecipitated with anti-BiP antibodies from the lysates prepared from both resistant and sensitive cells and subjected to immunoblots with anti-PSSG antibodies. For each cell line, higher S-glutathionylated BiP levels were found in the resistant compared to sensitive (Figure 6A and B).
Figure 6.

Btz resistant MM cells have higher BiP S-glutathionylation levels. Proteomic analysis of recombinant BiP revealed that Cys41 and Cys420 were subject to S-glutathionylation, and this post-translational modification altered both BiP structure and catalytic activities. (A and B) Cell lysates from MM1.S and U266 sensitive and resistant cells were incubated with rabbit anti-BiP antibody or rabbit IgG control overnight at 4°C. Antibody-antigen complexes were then IP by incubating with protein A/G-agarose beads overnight at 4°C. Immuno-precipitates were solubilized in the SDS-loading buffer and subjected to SDS-PAGE under non-reducing conditions, probing of the immunoblots with mouse anti-PSSG antibody. The western blots were quantified by Li-cor image studio 4.0. (C) Recombinant BiP proteins, native and S-glutathionylated (10 μM disulfiram and 5 mM GSH at 37°C for 30 min), were separated on a non-reducing gel. One part of the gel was transferred to a PVDF membrane and blotted with rabbit anti-BiP (green) and mouse anti-PSSG antibodies (red) to confirm the S-glutathionylation, and the other part stained by colloidal Coomassie stain. Bands were excised, destained, enzymatically digested, and subjected to LC-MS/MS identification. (D and E) Proteomic identification of sites of S-glutathionylation on BiP. (F) circular dichroism spectra were measured at 25°C in 40mM sodium acetate buffer, pH 4. (G) Tryptophan emission spectra for native BiP (solid line) and S-glutathionylated BiP (dashed line) were measured with fluorescence excitation at 295 nm. Each spectrum is representative of three independent experiments. (H) The ATPase activities of native and S-glutathionylated BiP were determined by the Malachite Green phosphate assay measuring phosphate release. (I) The foldase activities of native and S-glutathionylated BiP were determined by the rhodanese aggregation assay. comparative ATPase (J) and foldase (K) activities of native and mutated BiP (C41A and C420A).
Two cysteine residues are located at positions 41 and 420 of BiP. We used purified human full-length BiP to identify whether they could be S-glutathionylated. Disulfiram was used to induce ROS-mediated S-glutathionylation, which was confirmed by immunoblotting (Figure 6C). S-glutathionylation was found in two distinct peptides (residues 34–46 and 418–435), confirming that both Cys41 and Cys420 of BiP were susceptible residues (Figure 6D and E).
Next, we investigated whether S-glutathionylation of these cysteines altered BiP structure and function. Circular dichroism (CD) spectroscopy was used to show that the secondary structure of BiP-SSG was different from that of native BiP-SH. Differences included decreased alpha helix and increased beta sheets (Figure 6F). In addition, the intrinsic tryptophan fluorescence of BiP was increased following S-glutathionylation (Figure 6G). Changes in secondary structure were obvious, therefore we tested their precise effect on the BiP catalytic activities. The ATPase activity of S-glutathionylated BiP was significantly decreased (Figure 6H), while the corresponding foldase activity was significantly enhanced compared to native BiP-SH (Figure 6I). To attain a clearer understanding of the contribution of S-glutahionylation for each individual cysteine, recombinant BiP protein was obtained with mutations (Cys to Ala) at either Cys41 or Cys420. ATPase and foldase activity assays showed that Cys41 is important for ATPase activity while Cys420 is important for foldase activity. As shown in Figure 6J, BiP with the Cys41 to Ala41 (C41A) had decreased ATPase activity, whereas Cys420 to Ala420 (C420A) increased foldase activity (Figure 6K). In conclusion, S-glutathionylated BiP has enhanced foldase activity, which can maintain more accurate protein folding during oxidized conditions contributing to a reduced requirement for proteasome activity and thus the Btz resistant phenotype.
BiP expression and S-glutathionylation levels are upregulated in MM tissues.
To confirm clinical and translational relevance of our findings, MM tissue arrays were analyzed by immunohistochemical (IHC) studies for BiP expression and S-glutathionylation levels in normal bone marrow and bone marrow tissues with MM. The representative IHC stained photomicrographs are shown in Figure 7A for BiP and Figure 7B for S-glutathionylation. Higher levels of both BiP and S-glutathionylation levels (Figure 7A and B) were found in bone marrow tissues from patients with MM. These data suggest that acquisition of higher levels of BiP S-glutathionylation may be a general mechanism for Btz-resistance.
Figure 7.
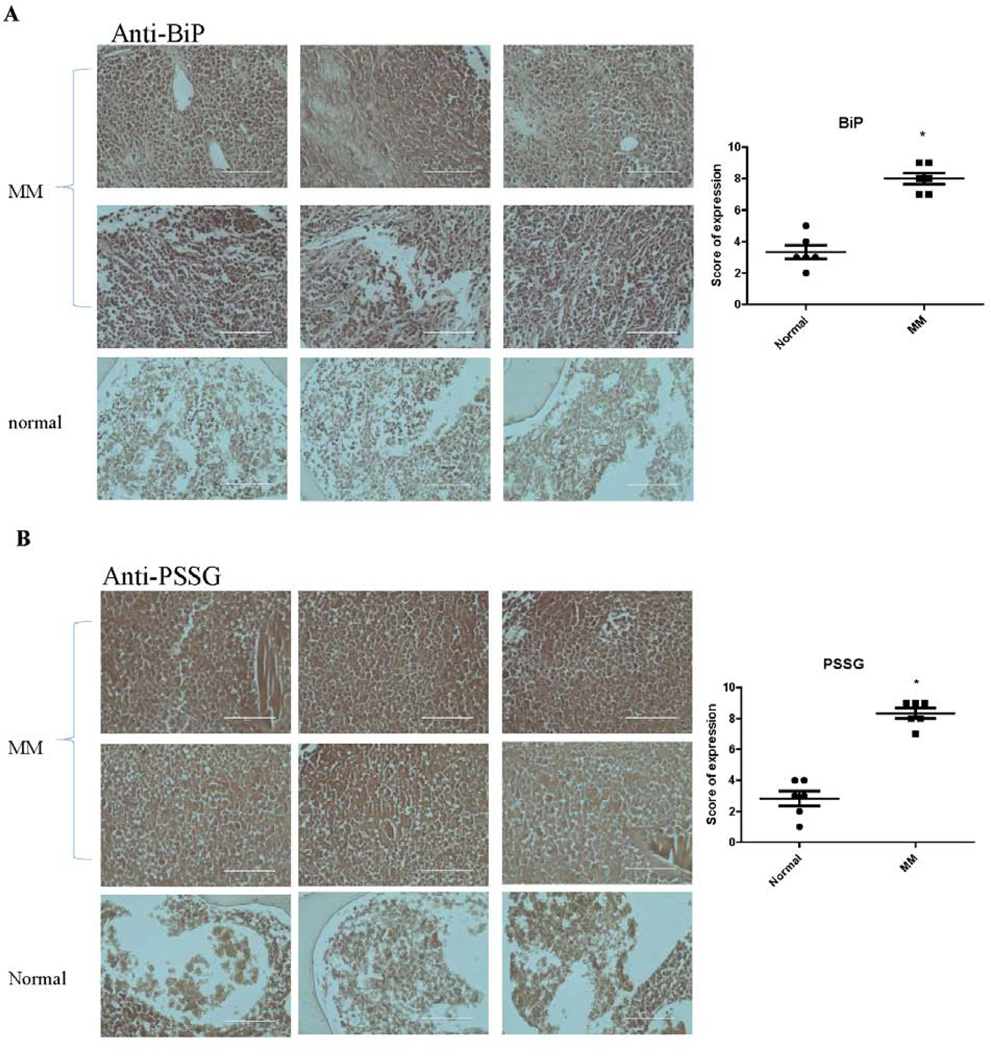
BiP and S-glutathionylation levels are upregulate in clinical samples of MM compared to normal BM tissues. MM tissue arrays was blotted with rabbit anti-BiP (A) and mouse anti-PSSG (B) antibodies, then treated with appropriate HRP conjugated secondary antibodies followed by DAB visualization. Sections were photographed with the EVOS® FL Cell Imaging System. Immunoreactivity was scored using a semi-quantitative system combining intensity of staining (0–3) and percentage of positive cells (0–3) and shown in the scatter plots adjacent to each panel. The higher levels in MM tissues are shown by the plots using 12 cases of MM tissue and 6 cases of normal tissue.
Discussion
Since FDA approval of Btz in 2003, the ubiquitin-proteasome system has been considered an effective target for cancer therapy.21,22 Btz targets the 20S proteasome, but its therapeutic value is limited by development of acquired resistance, a process that to date has been linked with cellular adaptations that include: altered efflux transporter levels, over-expression of a subunit of the 26S proteasome, increased expression of heat shock proteins such as Hsp27, or altered tolerance for aggregated misfolded proteins (aggresome).5,23 We showed that ablation of GSTP increased oxidative conditions in the ER, induced ER stress and sensitized cells to ThG or TuM13, drugs that exert toxic effects through interfering with ER functions by mechanisms distinct from Btz - inhibition of SERCA (ThG), interference with glycosylation (TuM). We therefore investigated whether GSTP-mediated changes in ER redox homeostasis are involved in the acquisition of resistance to Btz.
MM cells are aberrantly developed professional secretory cells, producing antibodies and necessarily have a built-in capacity to fold proteins and deal with oscillations in ROS levels. This capacity can come from redox mediated signaling events, which occur via modification of susceptible cysteine residues within certain proteins. The variable valence and nucleophilic nature of sulfur confers flexibility, where thiol groups provide a platform for various post-translational modifications,24 among which S-glutathionylation can alter both the structure and function of modified proteins.9 We used multiple MM cell lines that expressed WT GSTP1*A implying no intrinsic differences in their capacities to catalyze S-glutathionylation. RNAseq showed a dysregulation of expression of a number of transcripts in Btz-resistant MM cells, with redox pathway participants well represented. We demonstrated that those redox changes have regulatory effects on the activity of the ER chaperone BiP. Reconstitution of oxidative stress demonstrated that each cysteine (Cys41 and Cys420) in BiP is subject to S-glutathionylation. Primary amino acid sequence in the vicinity of those cysteines showed the presence of basic residues, implying that the Cys pKa values are low and predisposed to oxidation. BiP-SSG changed its tertiary and quaternary structure and led to blunted ATPase activity and increased foldase activity. This result is consistent with a similar observation for the single cysteine in yeast.25,26 Insight into the importance of redox signaling mediated by S-glutathionylation of BiP is provided by mammalian NPGPx, a stress sensor that transmits oxidative signals by forming disulfide bonds between Cys57 and Cys86. The oxidized form of NPGPx binds to BiP and forms covalent bonded intermediates between Cys86 of NPGPx and Cys41/Cys420 of BiP. Subsequently, the formation of the disulfide bond between Cys41 and Cys420 of BiP enhances its chaperone activity. It appears that NPGPx is essential for releasing excessive ER stress by enhancing BiP chaperone activity to maintain functional homeostasis.27
Given the non-overlapping mechanisms of action for Btz, ThG and TuM, the observed cross-resistance patterns are intriguing. None of these drugs produce electrophilic intermediates that require GSH based detoxification. We have shown that drug resistance accompanied by elevated levels of GSTP occurs with a variety of drugs that are not bona fide substrates for thiolation.28 Also, bone marrow niches and the hematopoietic progenitor cells therein, are susceptible to changes in localized tissue redox gradients29,30 and progenitor cell proliferation is accelerated in Gstp1/p2 knockout mice.11 Thiol-active drugs (even as simple as N-acetyl cysteine) have been used in treatment of various hematopoietic and immune disorders.31,32 Nevertheless, without target identification and the understanding of how S-glutathionylation changes the function of each specific target, our ability to improve and further develop these drug is limited.
To identify possible translational relevance, we interrogated the hypothesis that inhibition of S-glutathionylation, and thiol-redox signaling, could provide a therapeutic option for treatment of resistant cells. We used TLK199 (Telintra®), a nanomolar inhibitor of GSTP, which has therapeutic activity in Phase II clinical trials in myelodysplastic syndrome patients33,34. TLK199 possesses some tropism for hematologic tissues and the pathologies thereof.11 In the present study, TLK199 reversed resistance to Btz, TuM and ThG, indicating some commonality in redox-sensitive pathways, despite their different mechanisms of action. Moreover, we analyzed a series of pathology sections of samples from patients with MM and compared them to normal bone marrow, verifying earlier reports35 that BiP levels are increased in malignant tissues, but also demonstrating increased protein S-glutathionylation in MM samples, thus causally linking BiP S-glutathionylation to the disease.
BiP S-glutathionylation can have profound effects on protein folding under oxidized conditions in the ER. Post-translational S-glutathionylation of BiP on two cysteine residues alters its structure, blunts its ATPase, while enhancing its foldase activities. Therefore S-glutathionylation makes BiP more efficient in protein folding under oxidative conditions, with less unfolded proteins for proteasomal degradation and less UPR sensors freed to convey the stress signaling. Within the Btz resistant cell population, conditions of step-wise, selective drug exposure have resulted in higher levels of GSTP expression, causal to this enhanced S-glutathionylation and augmenting the capacity of these cells to tolerate levels of Btz (proteasomal malfunction) that are lethal to WT cells. Cross resistance to ThG and TuM and subsequent reversal by GSTP inhibition implies resistance that is upstream and distinct from individual drug targets and may indicate a novel approach to enhance therapeutic utility of proteasome inhibitors such as Btz.
Materials and methods
Tissue culture
MM cell lines were provided by Dr. Dolloff (Medical University of South Carolina) and maintained in RPMI-1640 medium (HyClone, Logan, UT), supplemented with 15% FBS (Atlanta biologicals, Flowery Branch, GA), 1x sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin (Corning, Manassas, VA), 55 μM 2-mercaptoethanol, and 1x GlutaMax (Gibco, Grand Island, NY). BMDDC and MEF cells were generated and maintained as reported.13
RNA isolation and quantitative real-time PCR
Total RNA was extracted from MM cells using the Isolate II RNA mini kit (Bioline, Taunton, MA), and cDNA was then generated with the iScript™ cDNA synthesis kit (Bio-Rad) according to the manufacturers’ protocols. Subsequently, quantification of gene expression was performed by real-time PCR.13 The primers used are listed in Supplementary Table 1. Relative gene expression quantification was based on the comparative threshold cycle (CT) method (2−ΔΔCt) with normalization of the raw data to the included housekeeping gene (GAPDH).
RNA sequencing and data analysis
Total RNA was extracted and subjected to commercial RNAseq analysis (Novogene, China). The abundance of transcript reflects gene expression level directly. FPKM (the expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced) was used to estimate the gene expression level.36 Volcano diagram were plotted according to the different expression of genes from sensitive and resistant cells, and the threshold of differential expression genes is: |log2(FoldChange)| > 1 and q-value < 0.005.
Cytotoxicity assay
The cytotoxic effect of Btz (0.01–200 nM), ThG (0.01–800 nM), and TuM (0.01–800 ng/ml) was evaluated by MTT assay13 after 48 h treatment.
Measurement of intracellular reduced thiol, GSH and GSSG levels
Cell lysates were subjected to the measurement as reported.37 Fluorescent intensities were detected at 400Ex/465Em by CLARIOstar Microplate Reader (BMG LABTECH, Cary, NC).
ROS detection
Reduced 2’,7’dichlorodihydrofluorescein diacetate (H2DCFDA, Thermo Fisher Scientific, Waltham, MA) is oxidized and converted into fluorescent 2’, 7’-dichlorofluorescein (DCF) by intracellular ROS, and fluorescent signals were detected by CytoFLEX S flow cytometer (Beckman coulter, Indianapolis, IN). 10,000 cells were analyzed per sample.38
Immunoblotting and detection of S-glutathionylation
Immunoblot assays were performed as described30 and the antibodies used are listed in Supplementary Table 2.
Comparative analysis of protein S-glutahionylase activities of different GSTP1 allelic variants was enacted following treatment with PABA/NO.20 MCF7 cell lysates, which have undetectable endogenous GSTP1 levels, were freshly prepared, treated with PABA/NO in the absence or presence of different E. Coli-expressed and purified GSTP1 allelic variants (A, B, C or D),19 and immediately subjected to SDS-PAGE under non-reducing conditions.
In MM cells, global S-glutathionylation was quantified after deglutathionylation by Grx, and BiP S-glutathionylation was quantified by immunoprecipitation using rabbit anti-BiP antibody, and immunoblotting using mouse anti-PSSG antibody under non-reducing conditions.13
BiP and S-glutathionylation levels in clinical samples were evaluated by immunohistochemistry on MM tissue microarray slides (US Biomax, Derwood, MD).38 Immunoreactivity was scored using a semi-quantitative system combining intensity of staining (0–3) and percentage of positive cells (0–3).
Proteomic analysis: Identification of S-glutathionylated cysteines
Recombinant human BiP protein (MyBioSource, San Diego, CA) was buffer exchanged into 50 mM Tris-HCl buffer pH 7.4 via Bio-Spin 6 columns, purified by Amicon Ultra-0.5 Centrifugal Filter Unit (50 kDa cutoff), S-glutathionylated with 5 mM GSH and 10 μM disulfiram at 37°C for 30 min, and separated on a non-reducing Bis-Tris gel. Following colloidal Coomassie staining, gel bands were digested with trypsin (Sigma) and Asp-N (Thermo) and extracted peptides were desalted and analyzed on an Orbitrap Elite mass spectrometer (Thermo) equipped with an Ultimate3000 nanoHPLC (Dionex) with trap column (300 μm i.d. x 5 mm C18 PepMap 100, 5 μm, 100 Å) (Thermo) and analytical column (75 μm i.d. x 15 cm C18 YMC ODS-AQ 120Å S5) (Waters Corporation, Milford, MA) using a 180-minute gradient from 3%−50% of solvent B (98% ACN, 0.2% FA) at 200 ul/min. A top10 data-dependent acquisition experiment was performed with FT-MS scan at 60,000 resolution followed by alternating collision induced dissociation (35% NCE) and electron transfer dissociation (100 AT) in the ion trap. Dynamic exclusion was enabled with a repeat count of 3, duration of 30 sec, exclusion list size of 100, and exclusion duration of 100 sec.
The raw files were searched with Proteome Discoverer 1.4 against BiP with a precursor mass tolerance of 20 ppm, fragment mass tolerance of 0.8 Da, 2 missed cleavages, and variable modifications of glutathione or mono-tri oxidation of cysteine and methionine oxidation. Spectra were manually confirmed.
Plasmid construction
To construct the expression plasmids of BiP, the cDNA fragment that encodes human BiP was prepared. The PCR primers were 5’-CGCGCGGCAGC CATATGAAGCTCTCCCTGGTGG-3’ (the Ndel cloning site is underlined) and 5’-GTCATGCTAGCCATACTACTACAACTCATCTTTTTCTG-3’ (the Ndel cloning site is underlined). The cDNA was subcloned into the Ndel site of pET28a by In-Fusion® HD Cloning Kit (Takara Bio USA, Mountain View, CA). Individual cysteine-to-alanine mutations were generated by primers 5’-GACTGCGGAGTAGGTGGTC-3’ and 5’-GACCACCTACTCCGCAGTC-3’ for cysteine 41 and 5’-GGGTGCTACATCAAGCAGTAC-3’ and 5’-GTACTGCTTGATGTAGCACCC-3’ for cysteine 420. The constructed plasmids were designated as pET28a-BiP, pET28a-BiPC41A and pET28a-BiPC420A and confirmed by conventional PCR and sequence analysis. To express and purify the WT and mutated BiP proteins, the pET28a recombinant plasmids were transformed into a competent E. coli BL21(DE3) strain and purified with Ni-column ion-exchanged chromatography.19
ATPase activity assay
The ATPase activity of BiP was determined by measuring phosphate release using the Malachite Green phosphate assay (Sigma, St. Louis, MO) with modifications. 1 μg BiP protein was incubated in 160 μl of 20 mM Tris-HCl pH 7.4 – 50 mM KCl - 5 mM MgCl2 - 250 μM ATP at 37°C for 45 min. 40 μl of working solution (2.36 mM malachite green solution in 2.36 M sulfuric acid, 2.95% (w/v) ammonium molybdate, and 0.17% (v/v) Tween 20), and 20 μl of 30% (w/v) sodium citrate were added and incubated at 37°C for 10 min. The absorbance at 630 nm was recorded.
Foldase activity assay
The foldase activity of BiP was determined by its ability to prevent rhodanese aggregation as reported39 with modifications. Denaturation of rhodanese (Sigma) was accomplished by diluting samples four-fold into denaturation solution (100 mM HEPES pH7.4 - 6 M guanidine hydrochloride - 100 mM NaCl− 5 mM DTT) and incubating at room temperature for 1 h. Aggregation assays were performed in 1 μM denatured rhodanese, 1 mM ATP, and 10 μM BiP in assay buffer (100 mM HEPES pH7.4 - 50 mM KCl - 5 mM MgCl2). The absorbance at 320 nm was recorded until the signal plateaued.
Spectroscopic analysis of BiP
CD spectra were recorded in 50 mM sodium acetate buffer pH4.0 using AVIV 202 CD Spectrometer (Lakewood, NJ). BiP samples (50 μM) were maintained at 22°C using a Pelletier element. Spectra were recorded while scanning in the far-UV region (190–260 nm). The output of the CD spectrometer was recalculated according to the protein concentration, amino acid content, and cuvette thickness into molar ellipticity units (degrees×cm2/dmol). Protein tryptophan fluorescence was recorded on a F-2500 spectrofluorometer (Hitachi). The excitation wavelength was 295 nm to minimize an effect of protein tyrosines and phenylalanines. Background spectra were subtracted from final emission of the protein.
Statistical analysis
Student’s t tests were used to analyze significant differences. P values < 0.01 were regarded as statistically significant. Data were expressed as means ± SD. GraphPad Prism 6 (GraphPad Prism, La Jolla, CA) for statistical analysis of data and one-way ANOVA analyses were used.
Supplementary Material
Acknowledgement
This work was supported by grants from the National Institutes of Health (CA085660, CA117259, S10OD010731, NCRR C06RR015455, and NCRR P20RR024485 - COBRE in Oxidants, Redox Balance and Stress Signaling) and by the American Cancer Society Institutional Research Grant #IRG-19-137-20, from the American Cancer Society.
Abbreviations:
- ACN
acetonitrile
- BiP
binding immunoglobulin protein
- BMDDC
bone marrow derived dendritic cells
- BMDM
bone marrow derived malignancies
- Btz
bortezomib
- CD
circular dichroism
- ER
endoplasmic reticulum
- FA
formic acid
- GM-CSF
mouse granulocyte macrophage colony-stimulating factor
- GPx
glutathione peroxidase
- GR
glutathione disulfide reductase
- GRP78
the 78-kDa glucose-regulated protein
- GRP94
glucose-regulated protein 94
- Grx1
glutaredoxin 1
- GSH
glutathione
- GSSG
glutathione disulfide
- GSTP
glutathione S-transferase P
- H2DCFDA
2’,7’dichlorodihydrofluorescein diacetate
- IRE1
inositol-requiring enzyme 1
- MEF
mouse embryonic fibroblast
- MM
multiple myeloma
- NCE
normalized collision energy
- PDI
protein disulfide isomerase
- Prx
peroxiredoxin
- ROS
reactive oxygen species
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- SOD2
superoxide dismutase 2
- Srx
sulfiredoxin
- ThG
thapsigargin
- TuM
tunicamycin
- Trx
thioredoxin
- UPR
unfolded protein response
- WT
wild type
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References.
- 1.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature reviews Cancer. 2014;14(9):581–597. [DOI] [PubMed] [Google Scholar]
- 2.Masciarelli S, Sitia R. Building and operating an antibody factory: redox control during B to plasma cell terminal differentiation. Biochimica et biophysica acta. 2008;1783(4):578–588. [DOI] [PubMed] [Google Scholar]
- 3.Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171–003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120(14):2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stessman HA, Baughn LB, Sarver A, et al. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Molecular cancer therapeutics. 2013;12(6):1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Wilt LH, Jansen G, Assaraf YG, et al. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem Pharmacol. 2012;83(2):207–217. [DOI] [PubMed] [Google Scholar]
- 6.Franke NE, Niewerth D, Assaraf YG, et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26(4):757–768. [DOI] [PubMed] [Google Scholar]
- 7.Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Current opinion in cell biology. 2004;16(6):653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oka OB, Pringle MA, Schopp IM, Braakman I, Bulleid NJ. ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol Cell. 2013;50(6):793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284(1):436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adler V, Yin Z, Fuchs SY, et al. Regulation of JNK signaling by GSTp. EMBO J. 1999;18(5):1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gate L, Majumdar RS, Lunk A, Tew KD. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J Biol Chem. 2004;279(10):8608–8616. [DOI] [PubMed] [Google Scholar]
- 12.Kuzmich S, Vanderveer LA, Tew KD. Evidence for a glycoconjugate form of glutathione S-transferase pI. International journal of peptide and protein research. 1991;37(6):565–571. [DOI] [PubMed] [Google Scholar]
- 13.Ye ZW, Zhang J, Ancrum T, Manevich Y, Townsend DM, Tew KD. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid Redox Signal. 2017;26(6):247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Anken E, Braakman I. Versatility of the endoplasmic reticulum protein folding factory. Critical reviews in biochemistry and molecular biology. 2005;40(4):191–228. [DOI] [PubMed] [Google Scholar]
- 15.Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nature reviews Molecular cell biology. 2002;3(11):836–847. [DOI] [PubMed] [Google Scholar]
- 16.Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54(16):4313–4320. [PubMed] [Google Scholar]
- 17.Tew KD. Glutathione-Associated Enzymes In Anticancer Drug Resistance. Cancer Res. 2016;76(1):7–9. [DOI] [PubMed] [Google Scholar]
- 18.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21(3):396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manevich Y, Hutchens S, Tew KD, Townsend DM. Allelic variants of glutathione S-transferase P1–1 differentially mediate the peroxidase function of peroxiredoxin VI and alter membrane lipid peroxidation. Free Radic Biol Med. 2013;54:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Townsend DM, Findlay VJ, Fazilev F, et al. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69(2):501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y. Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol Ther. 2003;2(6):623–629. [PubMed] [Google Scholar]
- 22.Montagut C, Rovira A, Mellado B, Gascon P, Ross JS, Albanell J. Preclinical and clinical development of the proteasome inhibitor bortezomib in cancer treatment. Drugs Today (Barc). 2005;41(5):299–315. [DOI] [PubMed] [Google Scholar]
- 23.Arkwright R, Pham TM, Zonder JA, Dou QP. The preclinical discovery and development of bortezomib for the treatment of mantle cell lymphoma. Expert Opin Drug Discov. 2017;12(2):225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. The Journal of biological chemistry. 2013;288(37):26497–26504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Pareja KA, Kaiser CA, Sevier CS. Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. Elife. 2014;3:e03496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Sevier CS. Formation and Reversibility of BiP Protein Cysteine Oxidation Facilitate Cell Survival during and post Oxidative Stress. J Biol Chem. 2016;291(14):7541–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei PC, Hsieh YH, Su MI, et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol Cell. 2012;48(5):747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Ye ZW, Singh S, Townsend DM, Tew KD. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic Biol Med. 2018;120:204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grek CL, Townsend DM, Tew KD. The impact of redox and thiol status on the bone marrow: Pharmacological intervention strategies. Pharmacology & therapeutics. 2011;129(2):172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Ye ZW, Gao P, et al. Glutathione S-transferase P influences redox and migration pathways in bone marrow. PLoS One. 2014;9(9):e107478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murata Y, Shimamura T, Hamuro J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int Immunol. 2002;14(2):201–212. [DOI] [PubMed] [Google Scholar]
- 32.Hadzic T, Li L, Cheng N, Walsh SA, Spitz DR, Knudson CM. The role of low molecular weight thiols in T lymphocyte proliferation and IL-2 secretion. J Immunol. 2005;175(12):7965–7972. [DOI] [PubMed] [Google Scholar]
- 33.Raza A, Galili N, Callander N, et al. Phase 1–2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J Hematol Oncol. 2009;2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyons RM, Wilks ST, Young S, Brown GL. Oral ezatiostat HCl (Telintra(R), TLK199) and idiopathic chronic neutropenia (ICN): a case report of complete response of a patient with G-CSF resistant ICN following treatment with ezatiostat, a glutathione S-transferase P1–1 (GSTP1–1) inhibitor. J Hematol Oncol. 2011;4:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdel Malek MA, Jagannathan S, Malek E, et al. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget. 2015;6(5):3098–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Ye ZW, Chen W, et al. S-Glutathionylation of estrogen receptor alpha affects dendritic cell function. J Biol Chem. 2018;293(12):4366–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Zhang J, Ye Z, et al. Isoflavone ME-344 Disrupts Redox Homeostasis and Mitochondrial Function by Targeting Heme Oxygenase 1. Cancer Res. 2019;79(16):4072–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber F, Hayer-Hartl M. Prevention of rhodanese aggregation by the chaperonin GroEL. Methods Mol Biol. 2000;140:111–115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.