

Abstract
Xeno-free and chemically-defined poly(ethylene glycol) (PEG)-based hydrogels are increasingly used for in vitro culture and differentiation of induced pluripotent stem cells (iPSCs). These synthetic matrices provide highly tunable gelation kinetics and adaptable material properties that are crucial for guiding stem cell fate processes. In this work, we integrated sequential norbornene-click chemistries for forming synthetic and dynamically tunable PEG-peptide hydrogels to promote human iPSCs culture and differentiation. Specifically, iPSCs were photo-encapsulated in xeno-free thiol-norbornene hydrogels crosslinked by multi-arm PEG-norbornene (PEG-NB) and protease-labile peptide crosslinkers. These matrices were used to evaluate growth of iPSCs under the influence of matrix degradability, adhesion ligands, cell density, and ROCK inhibition. We further employed tetrazine-norbornene (Tz-NB) inverse electron demand Diels-Alder (iEDDA) reaction to dynamically stiffen these cell-laden hydrogels. Fast reactive Tz and its more stable derivative methyltetrazine (mTz) were tethered to multi-arm PEG, yielding mono-functionalized PEG-Tz and PEG-mTz, as well as dual-functionalized PEG-Tz/mTz that readily reacted with PEG-NB to form additional crosslinks in the cell-laden hydrogels. The versatility of this stiffening approach was demonstrated with different Tz-modified macromers (e.g., hyaluronic acid-Tz and heparin-Tz) or by intermittent incubation of PEG-Tz for temporally regulated stiffening. Finally, the Tz-NB-mediated dynamic stiffening was explored for 4D culture and definitive endoderm differentiation of human iPSCs. Overall, this dynamic hydrogel platform affords exquisite controls of hydrogel crosslinking for serving as a xeno-free and dynamic stem cell niche.
Keywords: Dynamic hydrogels, pluripotent stem cells, click chemistry, endocrine differentiation
Graphical Abstract

1. Introduction
Three-dimensional (3D) cell-laden hydrogels are increasingly used for in vitro and ex vivo cell expansion, controlled stem cell differentiation, and regenerative medicine.[1] Hydrogels can be fabricated by derivatives of purely synthetic polymers (e.g., poly(ethylene glycol) or PEG) and/or naturally derived biomacromolecules (e.g., gelatin, hyaluronic acid, or heparin).[2] Tumor basement membrane derived Matrigel is routinely used as a convenient matrix for 3D cell culture owing to its inherent cell affinity provided by laminin, collagen IV, and residual growth factors. However, Matrigel contains ill-defined and batch-to batch variability of extracellular matrix (ECM) components that may present challenges for studying the contributions of individual matrix cues on cell fate processes. To this end, synthetic PEG-based hydrogels afford a blank slate in which a bottom-up approach can be used to impart precise biochemical and biophysical properties in the cell-laden network. Advances in material chemistry have also enabled the conjugation of a variety of ECM motifs to the otherwise inert PEG-based network, as well as for spatiotemporal manipulation of matrix properties to facilitate the study of cell fate processes within a highly defined and controllable microenvironment.
Chemically defined PEG-based hydrogels are particularly advantageous for studying iPSC behaviors in 3D.[3] For example, PEG hydrogels formed by Michael-type additions have proven effective in cell encapsulation owing to the reaction specificity between vinyl sulfone/maleimide (VS/MAL) and thiol (SH) moieties. Hubbell and colleagues utilized VS-SH reactions to determine the role of integrin activation on embryonic stem cell (ESC) self-renewal.[4] Lim et al. later use PEG-based VS-SH hydrogels for maintaining stemness of three human ESC lines.[5] Nonetheless, the reaction rates of Michael-type additions may be difficult to control and vary greatly from seconds to hours, depending on the type of Michael donor-acceptor pairs (e.g., VS-SH ~30–120 min and MAL-SH ~tens of seconds).[4a] Alternatively, enzymatic crosslinking provides excellent cytocompatibility and tunable gelation kinetics for in situ cell encapsulation. For instance, transglutaminase (TG), which ligates lysine- and glutamine-containing peptides, has been successfully employed to create PEG-based, PSCs-laden hydrogels.[6] Unfortunately, TG does not recognize specific substrates that may yield non-specific conjugation of extracellular and cell surface proteins. Chain-growth photopolymerized hydrogels have also been used for PSC culture.[7] However, many reports have highlighted the cytotoxicity of chain-growth photopolymerization, in large part due to the random propagation of carbonyl radical species.[8] In contrast, step-growth photopolymerization, particularly the orthogonal thiol-norbornene ‘photoclick’ chemistry, maintains the advantages of light-initiated polymerization, but generally requires lower radical concentration during initiation and yields a homogeneous distribution of crosslinks.[9] Using this gelation chemistry, Kloxin and colleagues[1b] determined the impact of key ECM properties, including protease sensitivity and cell adhesion ligands, on neuronal differentiation of human iPSCs (hiPSCs).
Tissue stiffness and the presence of growth factors and cytokines regulate PSC differentiation, migration, and morphogenesis.[10] To elucidate the influence of matrix properties on stem cell fate, Lutolf and colleagues developed a microfluidic-based droplet formation and an enzymatic crosslinking approach to generate a library of iPSC-laden microgels for combinatorial screening of biochemical niches.[6, 11] On the other hand, various synthetic strategies have been employed to induce stiffening of cell-laden matrices for understanding cancer cell growth,[12] MSC spreading and differentiation,[13] and fibroblast behaviors;[14] however, regulation of pluripotent stem cell fate by niche stiffening has not been extensively investigated. Generally, cells are encapsulated in hydrogels and cultured to a specified time point, in which matrix stiffening or softening is initiated. To this end, we have reported several thiol-norbornene hydrogels with enzymatic sensitivity for dynamic cell culture. In one example, mushroom tyrosinase (MT), which induces tyrosine dimerization, was utilized to induce temporal matrix stiffening for studying epithelial-mesenchymal transition (EMT) of pancreatic cancer cells.[12a] Other click chemistry has been developed to enable both matrix stiffening and modification of biochemical milieu. The inverse electron demand Diels-Alder (iEDDA) reaction, a chemistry widely used for site-specific protein labeling[15] and live cell imaging[16] was recently adapted for forming cell-laden hydrogels.[17] Jia and colleagues demonstrate the use of trans-cyclooctene (TCO)-tetrazine (Tz) reaction for temporally modifying the stiffness and bioactivity of cell-laden hydrogels. Hydrogels were first prepared using macromers decorated with methyltetrazine (mTz). Dynamic modification of the hydrogel was achieved by swelling in TCO modified hyaluronic acid (HA) and RGD, respectively.[17c] While significant changes in cell morphology were observed after tethering RGD to the network, TCO-Tz reaction did not lead to substantial increases in gel moduli (~300 Pa).
Matrix mechanics play a central role in stem cell fate processes. For example, previous work has shown that PEG-peptide hydrogels with a Young’s modulus (E) of ~3 kPa permitted aggregation and proliferation of iPSCs in 3D.[1b] Other studies show that higher moduli (E ~6–7 kPa) promote endoderm differentiation.[18] However, to the best of our knowledge, the effect of dynamic matrix stiffening on endodermal differentiation of iPSCs has not been reported. Here, we utilized thiol-norbornene photopolymerization in conjunction with Tz-NB crosslinking to encapsulate hiPSCs and enable investigation of dynamic ECM factors on cell survival and differentiation (Figure 1A). As demonstrated previously, thiol-ene gelation enables precise tuning of material properties to independently investigate each variable (i.e., matrix stiffness, adhesion ligands, chemical treatment, etc.). By fabricating hydrogels using thiol-norbornene photopolymerization with excess NB groups, further iEDDA reactions were permitted through temporal diffusion of multi-arm, Tz-functionalized macromers into the gels. Lastly, we showed that Tz-NB-mediated stiffening can be used to investigate the effect of temporal stiffening on definitive endoderm (DE) differentiation of hiPSCs.
Figure 1.
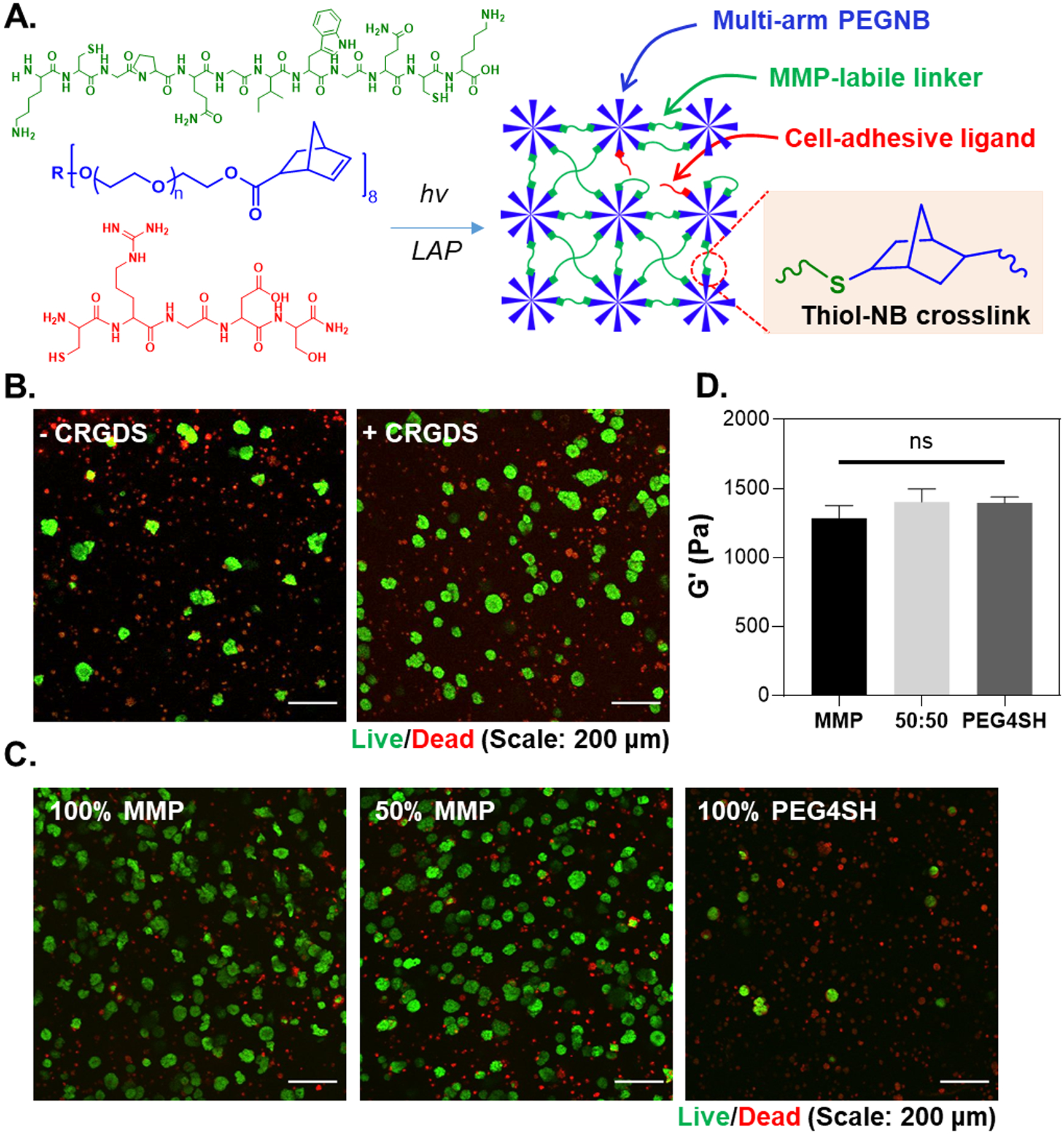
(A) Schematic of thiol-norbornene photopolymerization with 8-arm PEG-NB and bis-cysteine bearing, MMP-sensitive peptide KCGPQG↓IWGQCK and pendant CRGDS. R represents the tripentaerythritol core structure. (B) Effect of CRGDS (1 mM) and (C) network MMP-sensitivity on iPSC viability and aggregation. Hydrogel with 50% MMP sensitivity was crosslinked by an equal concentration of MMP peptide and a randomly selected, non-cleavable peptide (RSH:NB = 1). (D) Shear moduli of 2.5 wt% PEG-NB and peptide/PEG4SH hydrogels used for cell encapsulation. An ordinary one-way ANOVA was conducted to analyze significance (N = 3 gels per condition. ns: no significance).
2. Results
2.1. Encapsulation of iPSCs in thiol-norbornene photopolymerized hydrogels
Key parameters for iPSC survival in photopolymerized thiol-norbornene PEG-peptide hydrogels (Figure 1A) were first evaluated using the ChiPSC12 cells. Cells were encapsulated in PEG-NB hydrogels crosslinked by matrix metalloproteinase (MMP) labile peptide linker (KCGPQG↓IWGQCK), which was flanked with lysine residues to improve peptide solubility.[19] Integrin-binding ligand (i.e., 1 mM CRGDS) was also immobilized during gel crosslinking to support cell survival.[19–20] First, ROCK inhibition during cell encapsulation was deemed essential as very few cells survived in the absence of ROCK1 and ROCK2 inhibitor Y-27632 (Figure S1). Live/dead staining and imaging also showed that the inclusion of RGDS promoted the formation of iPSC clusters in 3D (Figure 1B). Higher degree of cell survival was apparent in hydrogels crosslinked by MMP labile peptides and very few cells survived in the gels crosslinked by non-degradable 4-arm PEG-thiol (PEG-SH) linker (Figure 1C). Of note, macromer concentrations were adjusted such that all gels were crosslinked to similar elastic moduli (G’ ~ 1.3 kPa. Figure 1D). Furthermore, while cell density during encapsulation did not alter iPSCs viability in the hydrogels (Figure S2A), the size of the aggregates were smaller at the highest cell density tested (i.e., 5 million cells/mL) (Figure S2B).
After identifying initial parameters for iPSC encapsulation in thiol-norbornene hydrogels, we sought to further investigate the effect of ROCK inhibition on iPSC aggregation and yes-associated protein 1 (YAP) nuclear accumulation. Compared with 3-day treatment of Y-27632 (Figure 2A), sustained presence of ROCK inhibition led to more ‘cystic’ aggregates with clear apico-basal polarity (Figure 2B). Aggregates with Y-27632 treatment for only 3 days appeared larger than those treated for 7 days (Figure 2C); however, no statistically significant difference was found between the two groups. Furthermore, pro-longed (7 days) Y-27632 treatment led to fewer nuclei stained positive for YAP.
Figure 2.
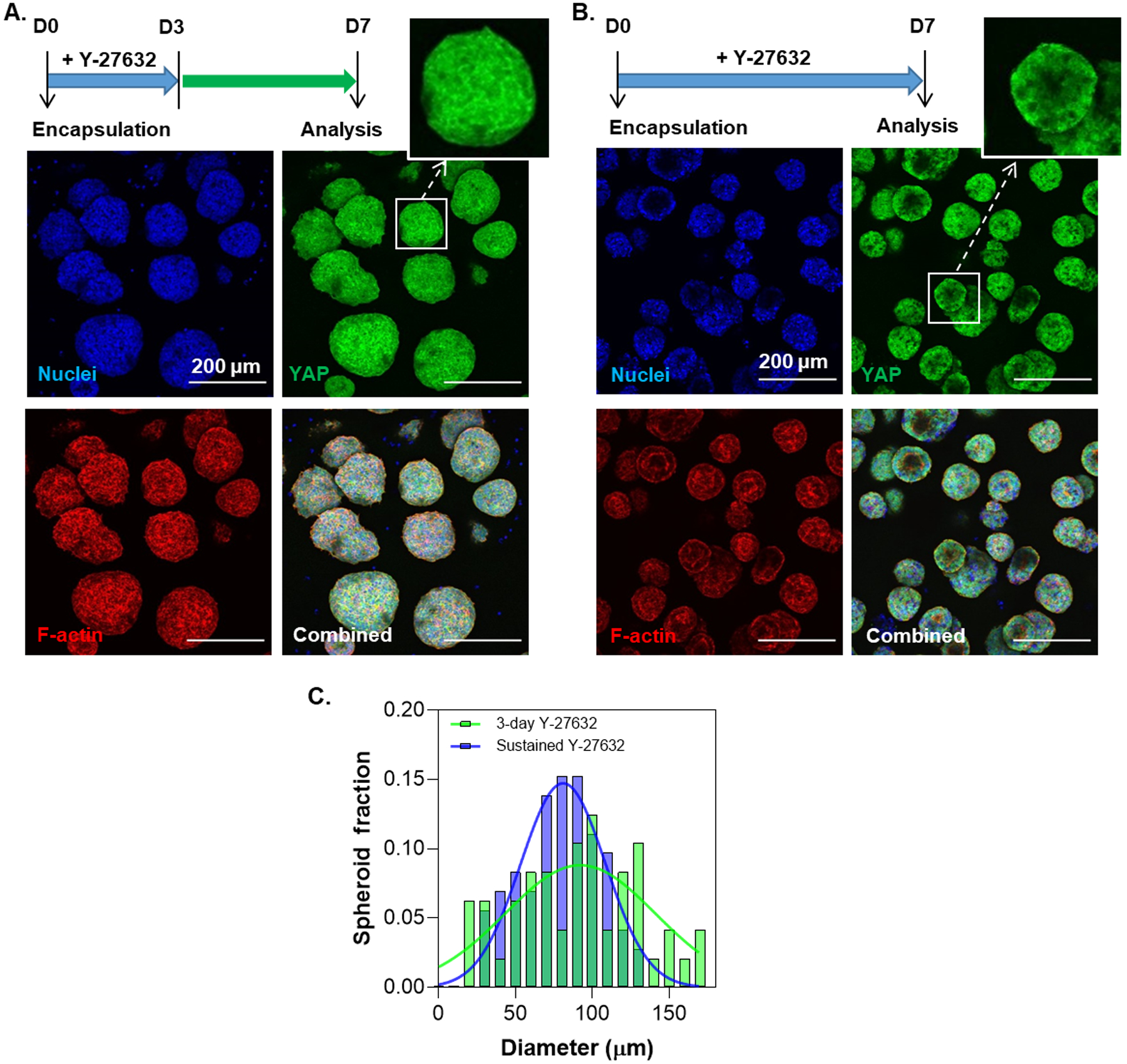
Effect of (A) 3-day or (B) sustained ROCK inhibition on YAP nuclear localization and iPSC aggregate morphology on day 7 post-encapsulation. (C) Distribution of aggregate diameters of the aggregates for 3-day and sustained Y-27632 treatment. The diameters of the aggregates for 3-day and sustained treatment conditions were 78.7 ± 3.0 μm (N = 72) and 90.3 ± 5.93 μm (N = 48) respectively. Aggregate diameters were quantified using live/dead staining images at day 7 post-encapsulation with ImageJ software. The diameters were analyzed using a Kolmogorov-Smirnov test of the cumulative frequency distributions (p = 0.67).
2.2. iEDDA-mediated stiffening of PEG-peptide hydrogels
Tz-NB iEDDA reaction (Figure 3A) was employed for post-gelation matrix stiffening. The reaction kinetics of NB- and Tz-modified macromers were first determined from UV/vis spectrometry (Figure S3A). Under a non-gelling condition, the reactions between PEG-NB and PEG-Tz were approximately ~13-fold faster than that of PEG-NB and PEG-mTz, as determined by the apparent second order rate constants (kobs, Figure S3B). Dynamic stiffening was conducted using PEG-NB hydrogels crosslinked with dithiothreitol (DTT) at RSH:NB of 0.75. The hydrogels were stiffened simply by swelling in buffer solution containing either PEG-Tz, PEG-Tz/mTz, or PEG-mTz. A remarkably wide range of stiffening was achieved (i.e., 10 to 20-fold increase, from G’0 ~300 Pa to G’t ~ 3–6 kPa) after 24 hours of incubation (Figure 3B). The rate of hydrogel stiffening was analyzed by taking the time derivative of G’ (Figure S4). During the first 24 hours, Tz-NB stiffening kinetic was approximately ~3 fold higher than that of mTz-NB reaction. After 24 hours, both reactions approached completion (i.e., dG’/dt =0) with minimal increases in modulus afterwards.
Figure 3.

(A) Schematic of tetrazine-norbornene click reaction showing conjugated tetrazine and norbornene reactants and cycloaddition product. (B) Influence of Tz derivatization on the magnitude rate of photopolymerized 2.5 wt% PEG-based hydrogel stiffening in the presence of 1 wt% PEG-Tz, PEG-mTz, or PEG-Tz/mTz. (C) Effect of Tz substitution on Hep-Tz (0.5 wt%) stiffening of 3 wt% PEG-NB and 1 wt% PEG-SH hydrogels. Low, mid, and high Hep-Tz correspond to 1.4, 2.6, and 4 mM/wt%, respectively. (D) Effect of thiol-ene ratio on HA-Tz (0.5 wt%) of 3 wt% PEG-NB and PEG-SH hydrogels. (E) Stepwise stiffening of PEG-based hydrogels with PEG-Tz (0.5 wt%) incubation. Shaded regions indicate time period of stiffening (4 hours).
Hydrogels could also be stiffened with Tz-modified heparin or HA (i.e., Hep-Tz or HA-Tz), and varying the degree of substitution (DS) on Hep yielded hydrogels with different magnitudes of stiffening (e.g., 1, 5, and 10 kPa for low, medium, and high DS of Hep-Tz, respectively (Figure 3C). Adjusting the stoichiometric ratio of thiol to norbornene (RSH:NB = 0.33 to 0.5, 0.67) at a fixed concentration of incubating HA-Tz also yielded up to 5-fold increases in the elastic modulus (Figure 3D). Not surprisingly, no stiffening was observed for hydrogels crosslinked at an equimolar ratio (i.e., RSH:NB = 1) as no excess NB group was available for the infiltrating HA-Tz macromer. Finally, Tz-NB click reaction also provides a mechanism for step-wise temporal stiffening of hydrogels. An approximate ~1 kPa increase in G’ was observed with each of the two 4-hour PEG-Tz incubation periods (0–4, 24–28 hr. Figure 3E), and there was minimal stiffening following the removal of PEG-Tz solution.
Non-dynamic soft and stiff hydrogels were prepared with 2.5 wt% or 3.5 wt% PEG-NB, respectively, whereas dynamic stiffening could be achieved by incubation the gels with different PEG-Tz concentrations. Optimal PEG-Tz concentration was identified to match initial non-dynamic soft gels (G’ ~ 1 kPa) and final non-dynamic stiff gels (G’ ~ 2.5 kPa) (Figure S5). All hydrogels were crosslinked by MMP-labile peptide (RSH:NB = 0.85) and were immobilized with integrin ligand CRGDS (1 mM). After determining that 0.2 wt% of PEG-Tz was appropriate for stiffening, we utilized this formulation for characterization of gel swelling, mesh size, and solute diffusivity (Figures S6). As expected, soft hydrogels swelled more than both non-dynamically stiff and dynamically stiffened hydrogels (Figure S6A). The swelling ratio data were used to calculate hydrogel mesh size, which affected diffusivity of proteins in the hydrogels. After accounting the changes in average molecular weight between crosslinks using multi-arm PEG-Tz macromer for stiffening (see method section), we determined that there was no statistical significant difference in the mesh size between the stiffened and stiff hydrogels (Figure S6B). Importantly, we determined the effect of the mesh size on diffusivity of select proteins (i.e., insulin, bovine serum albumin, BSA, and IgG). While there were significant differences in diffusion coefficients of these proteins between soft and stiff/stiffened hydrogels (Figure S6C), diffusion modeling results showed that there was only a slight difference in the time required to achieve equilibrium throughout the gel (Figure S6D). All proteins reached equilibrium in the hydrogels within 4 hours of incubation. We also characterized network degradability using exogenously added type 1 collagenase, which cleaves MMP-sensitive peptide crosslinkers (Figure S7). The soft hydrogels degraded within 1.5 hours of collagenase treatment, whereas both the stiff and stiffened gels degraded completely after 2.5 hours but with no noticeable difference in degradation rate.
2.3. Tz-NB click stiffening for xeno-free culture of iPSCs
Two human iPSC lines (i.e., ChiPSC12 and SOX2-iPSC) were encapsulated in PEG-peptide hydrogels with soft, stiff, or dynamically stiffened mechanics. For dynamic stiffening, soft hydrogels were treated with 0.2 wt% PEG-Tz on D3–D4 of the experiment (Figure 4A). On day 7 post-encapsulation, viable aggregates were observed across all conditions (Figure 4B); however, significant morphological differences were apparent. While the non-dynamic stiff hydrogels yielded larger and irregularly shaped aggregates, iPSCs formed smaller and more rounded spheroids in the soft and dynamically stiffened conditions (Figure 4B, 4D). Furthermore, F-actin and DAPI staining revealed that ChiPSC12s formed cyst-like cell clusters in soft and dynamically stiffened hydrogels, but fewer cystic aggregates appeared in the non-dynamic stiff gels (Figure 4C). Regardless of hydrogel stiffness, encapsulated iPSCs remained pluripotent, as demonstrated by high level of SOX2 expression (Figure 5A). This was substantiated by the similarity in the expression of an array of pluripotency markers, including LIN28, NANOG, POUF5, SOX2, TDGF1, and TERT (Figure 5B). Further analyses of expressions of germ layer commitment genes in the absence of differentiation cues revealed a slight but not statistically significant upregulation of mesoderm marker T in the stiff and dynamically stiffened hydrogels (Figure 5C).
Figure 4.
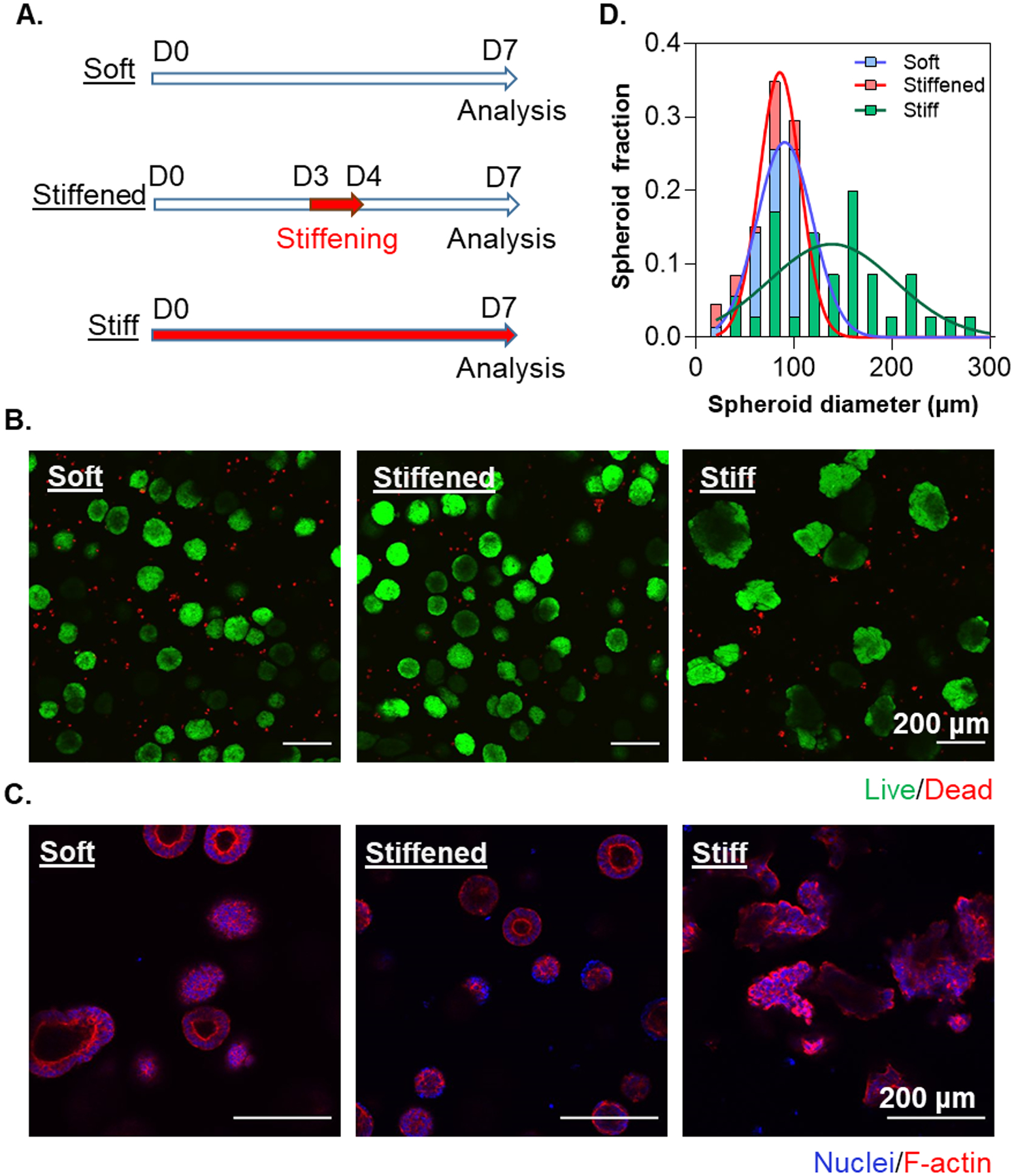
(A) Timeline of ChiPSC12 cells in statically soft, statically stiff, and dynamically stiffened hydrogels on day 7 post-encapsulation. For the dynamically stiffened condition, PEG-Tz (0.2 wt%) was added to the media for 24 hours on D3–D4. (B) Live/Dead staining and imaging of ChiPSC12 cell line on day 7 post-encapsulation. (C) Representative slice of F-actin and nuclear staining and imaging on day 7 post-encapsulation. (D) Distribution of aggregate diameters with or without Tz-NB stiffening. The diameters of the aggregates for non-dynamic soft, non-dynamic stiff and dynamically stiffened conditions were 95.8 ± 3.13 μm (N = 140), 144.1 ± 10.0 μm (N = 35), and 80.1 ± 1.9 μm (N = 152), respectively. Aggregate diameters were analyzed using a Kolmogorov-Smirnov test of the cumulative frequency distributions.. Significance between soft and stiff (p < 0.001),stiffened and stiff (p < 0.001), and soft and stiffened (p < 0.01) was determined.
Figure 5.
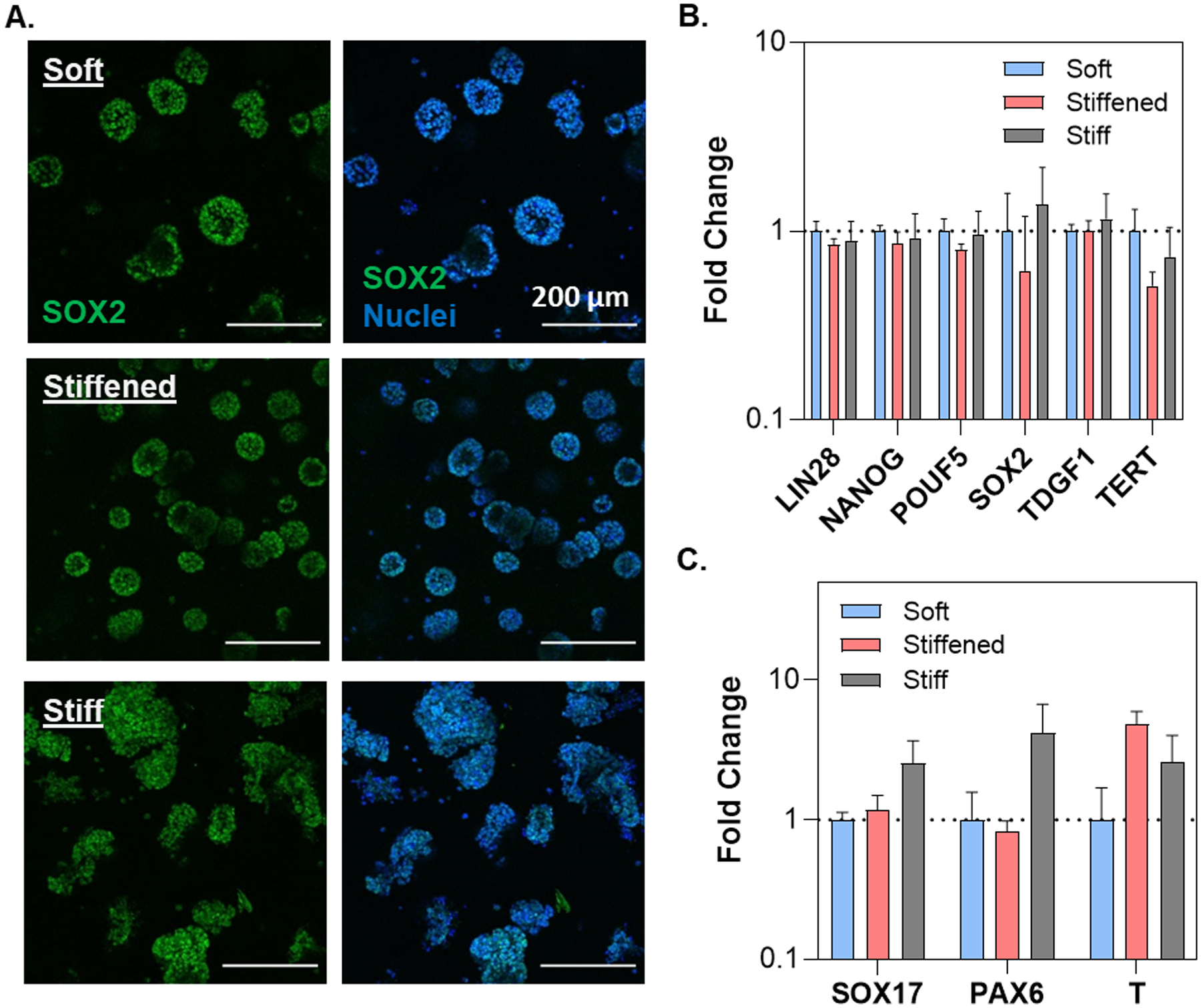
(A) Confocal images of SOX2-iPSC cell lines in soft, stiff and stiffened PEG-peptide hydrogels on day 7 post-encapsulation. PEG-Tz (0.2 wt%) was added to the media for 24 hours on D3–D4 to induce dynamic stiffening. RNA expression levels of (B) key pluripotency markers and (C) germ layer specific markers on day 7 post-encapsulation. A two-way ANOVA with a Tukey’s multiple comparison test was conducted on the dCt values prior to log transformation (N =3 replicates).
2.4. DE differentiation of encapsulated iPSCs.
With an interest in establishing a xeno-free dynamic hydrogel platform for pancreatic differentiation, endoderm lineage commitment was performed using a DE differentiation kit with SOX2-iPSCs encapsulated in the Tz-NB dynamic hydrogels (Figure 6A). Following the two-day DE differentiation in either non-dynamic soft, non-dynamic stiff, and dynamically stiffened hydrogels, SOX2 expression was substantially decreased and SOX17 positive cells became apparent in all conditions (Figure 6B). Semi-quantitation of the immunostaining images revealed similarly low SOX2 expression in all conditions. Significantly higher fraction of cells stained positive for SOX17 in the stiffened hydrogels when compared with both the non-dynamic soft and stiff conditions (Figure 6C).
Figure 6.
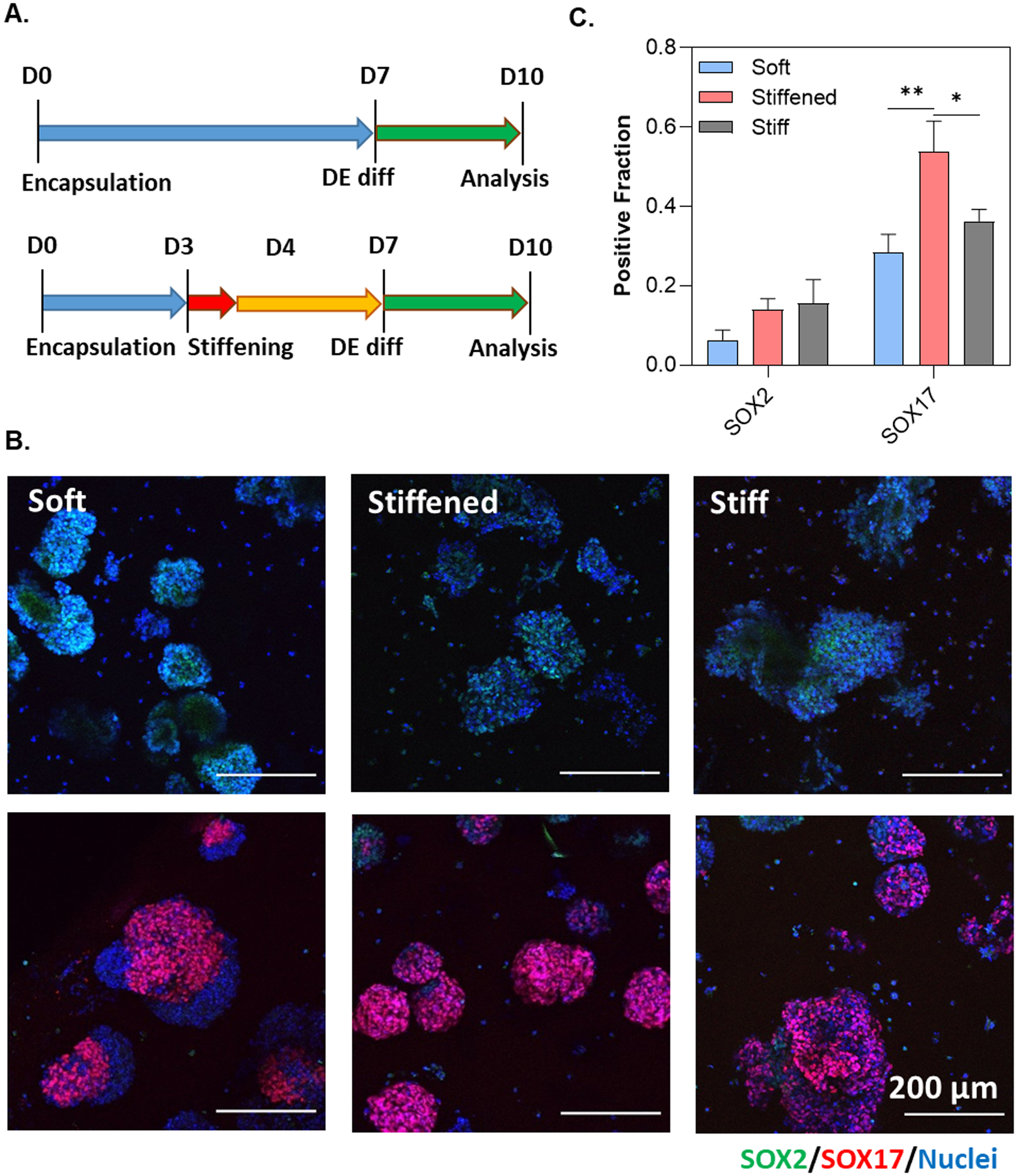
(A) Timeline of DE differentiation experiments. PEG-Tz (0.2 wt%) was added to the media for 24 hours on D3–D4. (B) Immunofluorescence staining and imaging of SOX2-iPSCs pre- and post-DE differentiation. (C) Semi-quantitation of SOX2-iPSCs post-DE differentiation. Positive fraction of either SOX2 or SOX17 was determined using ImageJ software. Statistical analysis was conducted using an ordinary one-way ANOVA (N = 9 images). **** and ** represents p < 0.0001 and p < 0.001, respectively.
3. Discussion
In this study, thiol-norbornene photopolymerization was used for 3D culture of iPSCs to support proliferation, permit aggregation, and maintain pluripotency of the cells (Figure 1A). Consistent with previous finding,[1b] integrin binding and protease sensitivity were indispensable for iPSC survival in PEG-based hydrogels (Figures 1B and 1C). ChiPSC12 was used for its confirmed differentiation to endocrine cells, including hepatocytes and pancreatic β-cells. Since detachment of iPSCs from a cell-substrate or cell-cell interaction induce anoikis, Y-27632 (a ROCK1 and ROCK2 inhibitor) is routinely added during thawing or passaging of iPSCs. ROCK inhibition not only reduces dissociation-induced anoikis, but also leads to higher proliferation and clonal expansion.[21] Addition of Y-27632 also improved survival of iPSCs during encapsulation (Figure S1). Although ROCK inhibition has been shown to increase iPSC survival here and elsewhere,[1b] we sought to examine its impact on YAP expression, a signaling pathway central to many mechano-sensitive events.[22] In particular, stem cell differentiation and self-renewal are linked to the translocalization of YAP and PDZ-binding motif (TAZ).[23] YAP/TAZ nuclear translocation results in the activation of genes associated with pluripotency and proliferation of PSCs.[24] Importantly, Rho/ROCK signaling pathway has been demonstrated to affect YAP/TAZ signaling in PSCs.[25] We hypothesize that Rho/ROCK pathway inhibition turns ‘on’ the Hippo pathway which phosphorylates YAP leading to its cytoplasmic degradation. With Rho/ROCK signaling in 3D, nuclear YAP is increased, which in turn upregulates genes associated with iPSC proliferation. Indeed, prolonged (7-day) ROCK inhibition by Y-27632 led to retention of YAP in the cytoplasm (Figure 2B, inlet image), a phenomenon not found in cells with 3-day ROCK inhibition (Figure 2A). Removing Y-27632 from the media on day 3 likely restored Rho/ROCK signaling, which caused YAP nuclear translocation.
Another objective of the current study was to exploit the kinetics of Tz-NB reactions (Figure 3A) for tuning hydrogel crosslinking to facilitate xeno-free differentiation of human hiPSCs. To that end, we determined the rate constants for iEDDA reaction using Tz and mTz-conjugated 8-arm PEG (i.e., PEG-Tz, PEG-mTz) (Figure S3). Here, we used low concentrations of PEG-Tz or PEG-mTz to react with PEG-NB, as this allowed characterization of reaction kinetic constants without formation of a crosslinked network. The relative fold difference in reaction kinetics between Tz and mTz macromers was consistent with small molecule results reported in the literature.[26] Compared with Tz, mTz exhibits significantly higher stability and decreased reactivity due to the electron donating methyl group that increases the energy required to drive the mTz-NB reaction.[27] The apparent kinetic constant for PEG-Tz (0.22 M−1s−1) was approximately 8.6-fold lower than the value for soluble Tz and NB described in the literature (kTz-NB = 1.9 M−1s−1 [28]). This discrepancy might be due to the presence of long PEG chains that reduced the diffusivity of Tz/NB macromers or accessibility of the reacting species. After determining the kinetics of PEG-Tz or PEG-mTz and PEG-NB reaction, thiol-norbornene crosslinked hydrogels were stiffened through simple diffusion of either PEG-Tz, PEG-mTz, or PEG-Tz/mTz. As with the kinetics assay, the rate of stiffening was affected by the derivatization of the Tz moiety (Figure S3). However, only a 3-fold difference in rate was observed compared to a 13-fold decrease under non-gelling conditions. This may be due to the required diffusion of the Tz/mTz-modified macromers in the hydrogel; however, further investigation is required. Nevertheless, the magnitude and rate of stiffening could be tuned by adjusting the nature and concentration of the diene and dienophile. For example, Tz-TCO reaction exhibits second order rate constants on the order of 106 M−1s−1,[29] whereas Tz-NB reactions can reach 10−1 M−1s−1 magnitude.[30]
Given the similar substitutions of mTz and Tz-modified PEG, we originally hypothesized that the magnitudes of stiffening would be similar. However, hydrogels were stiffened to a higher degree with PEG-Tz than with PEG-mTz. It was recently reported that PEG-Tz and PEG-NB interact both covalently through Tz-NB iEDDA and non-covalently through physical association.[31] As a result, Tz-NB gels were significantly stiffer than their counterpart crosslinked by pure thiol-norbornene photopolymerization. We hypothesize that the non-covalent interactions are more prevalent in the PEG-Tz stiffened gels than in the PEG-mTz counterpart, leading to a lesser degree of stiffening when using PEG-mTz. Future work may explore the differences of non-covalent interactions between NB/Tz and NB/mTz. Tz-NB reaction can also be utilized to modify the physicochemical properties of hydrogels by using ligands as described elsewhere or macromolecules like PEG, heparin, or HA (Figures 3B–3D). Various degree of stiffening could be achieved by simply varying the substitution of Tz on heparin (Figure 3C). While we have achieved 10 to 20-fold of stiffening, the rate of Hep-Tz stiffening was significantly slower than data shown in Figure 3B, likely due to the bulky polysaccharide structure of heparin when compared to the well-defined 4-arm PEG. By adjusting RSH:NB, we also demonstrated that HA-Tz could be used to stiffen hydrogels for up to 5-fold increases in elastic modulus (Figure 3D). Interestingly, the high degree of stiffening achieved by HA-Tz was in contrast to the only ~300 Pa difference in a previous report where HA-TCO was used.[17c] We reasoned that the high degree of stiffening achieved in this report was due to the differences in macromer reactivity (Tz vs. mTz used by Jia and colleagues) and differences in substitution or molecular weight of HA. By adjusting the incubation time with PEG-Tz, a stepwise stiffening strategy was achieved (Figure 3E). This approach may be useful in creating hydrogels with dynamic and temporally tunable increases in crosslinking density, thus matching physiological timescales and magnitudes of tissue stiffening (e.g., embryological development and tumor metastatic progression).
Changing the stiffness of the hydrogels by increasing the crosslinking density can alter other properties of the hydrogels including transport of soluble factors and network degradation. To this end, we investigated the effect of Tz-NB-mediated hydrogel stiffening on mesh size, solute diffusivity, and collagenase degradability of the network. Indeed, matrix stiffening induced significant reduction in gel mesh size and estimated diffusion coefficients of proteins with different sizes (Figures S6). However, by modeling the diffusion of largest of the three model proteins, IgG, we found minimal differences in the distribution of proteins in hydrogels with different stiffness and all proteins achieved equilibrium in approximately 4 hours (Figure S6D). This is not surprising as the mesh sizes of the gels were not drastically altered (i.e., ~19.1, 16.5, 17.2 nm for soft, stiffened, and stiff gel, respectively) and were all much larger than the hydrodynamic radius of IgG (6.4 nm).[32] This result was corroborated by the enzymatic gel degradation study, where the dynamically stiffened and non-dynamic stiff hydrogels degraded almost at identical rate (Figure S7). Nonetheless, it may be possible that the increased crosslinking density with an inert PEG-Tz crosslinker might alter local protease-mediated degradation. This could in turn affected the capability of cells to remodel their local microenvironment. Future study may consider the effect of local matrix degradation on stem cell fate processes.
Matrix stiffness is critical for self-renewal and lineage-specific commitment of pluripotent stem cells. To this end, we studied the effect of dynamic hydrogel stiffening on the growth and DE differentiation of hiPSCs, which to our knowledge has not been reported before. We found minimal cell death and high proliferation of ChiPSC12 cells in all PEG-peptide hydrogels, as demonstrated by the formation of large and viable cellular aggregates after 7 days of 3D culture (Figure 4B). Interestingly, aggregates in the soft and stiffened hydrogels were mostly round and with a clear lumen, a morphology not apparent in the statically non-dynamic hydrogels (Figure 4C). Rather, the aggregates formed in non-dynamic stiff hydrogels adopted an irregular morphology. Cell spheroids formed in stiff hydrogels (G’ ~ 3 to 5 kPa) typically were smaller and more compact.[33] In the current study, while matrix stiffening did not significantly change the averaged sizes of iPSC aggregates (dia. ~90 μm), it did increase the fraction of smaller cell aggregates (dia. ~40–80 μm) (Figure 4D). On the other hand, cells grown in non-dynamic stiff hydrogels formed larger and irregular clusters (dia ~150 μm). These results suggest that temporal regulation of matrix stiffness could have a drastic impact in the iPSC growth. Matrix stiffening induced by a non-degradable PEG-Tz crosslinker might reduce local MMP sensitivity that could in turn alter cell-mediated matrix remodeling. However, it is worth noting that the concentration of additional PEG-Tz macromer in the stiffened hydrogel only accounted for 15% of the maximum crosslinking density (i.e., the gels were initially formed with RSH:NB = 0.85). We reason that the difference in local MMP-degradability would not alter cell fate process as we showed that there was no significant difference on iPSC growth in hydrogels with 100% or 50% MMP-degradability (Figure 1C). Nonetheless, collagenase-mediated degradation of the hydrogels revealed an almost identical degradation profile for non-dynamically stiff and dynamically stiffened hydrogels (Figure S7). Future work may explore dynamic stiffening using bi-functional Tz-MMP-Tz peptide linker, which will result in stiffening while still permitting 100% cell-mediated matrix degradation
While some morphological variations were observed in iPSCs grown in different gels, expressions of SOX2 and other pluripotency markers were similar in all three conditions (Figure 5A, 5B). Further evaluation of the germ layer specific markers - SOX17, PAX6, and T - also showed no statisticalyl significant changes (Figure 5C). In the stiff and stiffened gels, however, mesoderm marker T was slightly higher compared with the non-dynamic soft condition. This could indicate a potential bias for mesoderm development using dynamically stiffened hydrogels. Future study will focus on tri-lineage differentiation of iPSCs encapsulated in chemically defined hydrogels with spatiotemporal stiffening. Additional research is also required to understand the mechanisms by which the formation of ‘cystic’ aggregates occurs in the soft and stiffened conditions but not in the non-dynamic stiff hydrogels.
In a previous study, Banerjee and colleagues showed that DE differentiation of pluripotent stem cells was strongly promoted in alginate-based capsule with an optimized stiffness (i.e., E ~ 9 to 20 kPa, or G’ ~ 3 to 7 kPa).[18] In our study, we found more SOX17 positive cells in dynamically stiffened hydrogels than in soft and non-dynamic stiff hydrogels. This may be attributed to stiffness-mediated activation of signaling pathways key to DE differentiation.[34] Mechanistically, Banerjee et al. demonstrated that phosphorylated SMAD (pSMAD) and phosphorylated AKT (pAKT) levels were higher in cells within stiff hydrogels which increased the level of TGFβ signaling leading to higher degrees of DE differentiation.[18] Nonetheless, the advantages of dynamically stiffening hydrogels are evident as the growth of iPSCs can be promoted in hydrogels with a lower modulus (~1 kPa) until aggregates were formed. Subsequently, gel stiffness could be dynamically increased to promote stem cell differentiation. In theory, Tz-NB stiffening can be combined with other bioorthogonal chemistries such as SrtA-mediated transpeptidation to achieve on-demand stiffening and softening of hydrogels or dynamic labeling of bioactive ligands.[35] This could enable stage-specific ECM modulation to enhance differentiation efficiency and pancreatic progenitor quality.
4. Conclusion
A chemically-defined, dynamic PEG-peptide hydrogel system was designed for xeno-free culture and differentiation of iPSCs. Parameters including protease sensitivity, cell adhesion, and cell density were identified for encapsulation of iPSCs. In addition, YAP signaling and spheroid size were found to be directly affected by inhibition of the ROCK signaling pathway. The use of iEDDA reaction facilitated the tuning of cell-laden hydrogel properties. Tz-NB crosslinking was advantageous for investigation of dynamic stiffening on iPSC behaviors. Tz-mediated stiffening was cytocompatible and did not alter expression of iPSC pluripotency markers, but it did improve DE differentiation of the cells. This approach affords a simple, synthetically accessible strategy for 4D stem cell culture for regenerative medicine and basic stem cell biology. In addition to eventual pancreatic progenitor differentiation, future work may also focus on investigating the effect of dynamic stiffening or changes of ECM composition on other lineage-specific differentiation.
5. Experimental
5.1. Materials
8-arm PEG-OH (20 kDa) and 4-arm PEG-amido succinic acid (PEG-ASA, 10 kDa) were purchased from JenKem Technology and Laysan Bio, respectively. 5-norbornene-2-carboxylic acid, N,N’-dicyclohexylcarbodiimide (DCC), 4-(dimethylamino)pyridine (DMAP), N,N-dimethylformamide (DMF), and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) were purchased from Sigma-Aldrich. Tetrazine-amine (Tz-NH2) and methyltetrazine amine (mTz-NH2) were obtained from Click Chemistry Tools. N,N’-diisopropylethylamine (DIEA) and 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) were purchased from TCI and Anaspec, respectively. Heparin sodium salt was purchased from Celsus Laboratories and hyaluronic acid was obtained from Lifecore Biomedical. 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Acros and TCI, respectively. All other chemicals were purchased from Fisher Scientific unless noted otherwise. All peptides (>90% purity) were purchased from GenScript.
5.2. Macromer synthesis
Macromer 8-arm PEG-norbornene (PEG-NB, 20 kDa) was synthesized as described previously.[33b] Conjugation efficiencies for PEG-NB was confirmed by 1H NMR (CDCl3, 500 MHz, Bruker Advance 500) and fluoraldehyde assay, respectively.
Tz, mTz, and Tz/mTz-modified PEG conjugates (i.e., PEG-Tz, PEGm-Tz, PEG-Tz/mTz) were synthesized through carbodiimide coupling chemistry. To a reaction vessel containing PEG-ASA dissolved in DMF, HATU and DIEA were added in 4-fold excess to the carboxylic acid groups on PEG. Next, Tz-NH2, mTz-NH2, or an equal concentration of Tz-NH2 and mTz-NH2 were dissolved in DMSO (5% v/v relative to total reaction volume) and added at an equimolar ratio to the carboxylic acid functional groups. The reactions were protected from light and proceeded for 16 hours at room temperature. The conjugates were purified by dialysis against ddH2O for three days prior to lyophilization and storage at −20 °C.
Hep-Tz was synthesized as described elsewhere[36] with modification to adjust the substitution. To a round bottom flask, heparin sodium salt (500 mg, 16.7 kDa) was dissolved in 15 mL ddH2O. 2.5-fold, 5-fold, and 10-fold excess of EDC and NHS to the carboxyl groups on heparin was added to the flask for low, mid, and high substitution, respectively. The reaction proceeded for 30 minutes prior to adding 2.5-fold, 5-fold, and 10-fold Tz-NH2 to the flask for low, mid, and high substitution, respectively. The reaction proceeded for 16 hours in the dark. For synthesis of HA-Tz, EDC/NHS was again used in 5-fold excess along with 5-fold excess of Tz-NH2. The conjugates were purified by dialysis against ddH2O for three days in the dark prior to lyophilization and storage at −20 °C. The degree of substitution for all Tz-modified macromers was determined by measuring the absorbance of the product at 523 nm and comparing against a Tz-NH2 or mTz-NH2 standard curve. The degree of substitution for PEG-Tz, PEG-Tz/mTz, and PEG-mTz were 94%, 95%, and 96%, respectively. The substitution of low, mid, and high Hep-Tz was 1.4, 2.6, and 4 mM/wt% respectively. For HA-Tz, the substitution was 2.6 mM/wt%. All PEG materials were also characterized using 1H NMR (Bruker Advanced 500) (Figures S8 and S9).
5.3. iPSC culture
Cellartis iPSC-12s (ChiPSC12, Takara) and mono-allelic mEGFP-tagged SOX2 WTC iPSCs (AICS-0074–026, genetically engineered by the Allen Institute for Cell Science and distributed by the Coriell Institute) were cultured on tissue culture dishes coated with vitronectin (Gibco) following manufacturer’s protocol. Essential 8™ (E8) Medium (Gibco) was used to maintain both iPS cell lines and was changed daily. Passaging was conducted every 3–4 days using TrypLE Select reagent (Gibco) treatment for 5 minutes. For the first 24 hours after passaging, media was supplemented with ROCK inhibitor Y-27632 (10 μM) to prevent cell death.
5.4. Encapsulation of iPSCs in thiol-norbornene photopolymerized hydrogels.
Immediately following trypsinization and counting, either ChiPSC12s or SOX2-iPSCs were encapsulated in PEG-peptide hydrogels via thiol-norbornene photopolymerization. The cells were mixed with pre-polymer solution consisting of PEG-NB, adhesion ligand CRGDS, and either MMP-sensitive crosslinker KCGPQG↓IWGQCK or PEG-SH, and photoinitiator LAP (1 mM) to achieve a final thiol-norbornene ratio (RSH:NB) of 1. After homogenizing, the mixture was dispensed into a cylindrical mold and exposed to 365 nm light (5 mW/cm2) for 2 minutes to achieve gelation. Concentrations of PEG-NB, crosslinkers, adhesion peptide were modified to evaluate individual effects on iPSC behaviors. Cell-laden gels were maintained in E8 media supplemented with Y-27632 ROCK inhibitor (10 μM). Y-27632 addition was either sustained daily throughout the duration of the experiment or for 3 days post-encapsulation as specified. Media were refreshed daily for the duration of the experiment. A live/dead staining kit (Life Technologies) was implemented to ascertain the viability of encapsulated cells. Cell-laden gels were incubated in PBS with calcein AM and an ethidium homodimer for 1 hour, following washes with fresh PBS. For experiments varying the duration Y-27632 incubation, hydrogels were stained with an anti-YAP antibody to visualize YAP localization in hydrogels. Live/dead images and all immunofluorescence images were captured with a confocal microscope (Olympus Fluoview FV100 laser scanning microscope). For all confocal imaging, at least three images were taken per sample.
5.5. Tz-NB iEDDA-mediated stiffening of hydrogels
For stiffening studies, hydrogels were fabricated using thiol-norbornene photopolymerization. Briefly, PEG-NB and either crosslinker DTT (RSH:NB =0.75) or PEG-SH (RSH:NB =0.33) were mixed with photoinitiator LAP (1 mM) and injected between two glass slides separated by 1 mm Teflon spacers. Gelation was conducted by UV-light treatment (365 nm, 5 mW/cm2) for 2 minutes. Gels were placed in wells containing PBS for overnight swelling at 37 °C. Hydrogels were stiffened by reacting excess NB groups with Tz or mTz through incubating the gels in wells containing either PEG-Tz, PEG-mTz, PEG-Tz/mTz, HA-Tz, or Hep-Tz for predetermined lengths of time. Moduli were obtained using oscillatory rheometry in strain-sweep mode prior to and at specified time points throughout the duration of stiffening.
To evaluate the mesh size and solute diffusivity in hydrogels used for the cell studies, we first determined the volumetric swelling ratio of the gels. Hydrogels were fabricated using either 2.5 wt% (for non-dynamic soft and dynamic stiffened conditions) or 3.5 wt% (for non-dynamic stiff) PEG-NB with KCGPQG↓IWGQCK (R = 0.85), and CRGDS (1 mM) with LAP (1 mM) and UV-light exposure (2 min., 5 mW/cm2). After 24 -hour swelling in PBS at 37 °C, gels in the dynamically stiffened condition were incubated with 0.2 wt% PEG-Tz at 37 °C for an additional 24 hours. The gels were washed with additional PBS for 3 hours to remove unreacted PEG-Tz solution. The gels were dried in vacuo for 24 hours and weighed to obtain mdried. The hydrogels were then swollen for 24 hours in PBS and weighed to obtain mswollen. The following equations were utilized to determine the mass swelling ratio (q), volumetric swelling ratio (Q), and mesh size (ξ) of the hydrogels.
| (1) |
After obtaining q, we use this parameter as well as the densities (ρ) of water (ρH2O = 0.994 g/cm3) and PEG (ρPEG = 1.087 g/cm3) to obtain the volumetric swelling ratio (Q):
| (2) |
After obtaining Q, the mesh size (ξ) of the gel can be determined using the following equation:
| (3) |
Where l is the average bond length (1.47 Angstroms) in the backbone of an ethylene glycol subunit, 3 is the total number of bonds in a PEG repeat subunit, Cn is the Flory characteristic ratio (4 for PEG-based hydrogel) and Mn is the number for the average molecular weight of PEG8NB. Mc is defined as the following for equation for non-dynamic soft and stiff hydrogels:
| (4) |
Mc in the dynamically stiffened hydrogel was defined as the following:
| (5) |
Where MWA, MWB, and MWc correspond to the molecular weight of PEG-NB (20 kDa), MMP peptide (1303.56 Da), and PEG-Tz (10 kDa), respectively. The parameters fA, fB, and fC correspond to the functionality of PEG-NB (8), MMP peptide (2), and PEG-Tz (4), respectively.
Given the mesh size values we next sough to determine the effect of stiffening on the diffusivity of components of both the E8 and DE differentiation media by employing the Lustig-Peppas relationship[37]:
| (6) |
Where Dssol is the diffusivity of a solute in solution, Rs is the hydrodynamic radius of the solute, and Y is the critical volume required for translational movement of a substrate with respect to the average free volume of a water molecule (1 for PEG-based hydrogels). We calculated protein diffusivity in gel for insulin and bovine serum albumin using literature values of the diffusion coefficients in solution, 216 and 89 μm2/s, and the hydrodynamic radii of 1.47 and 3.56 nm, respectively.[38] The solution diffusion coefficient of IgG (51.28 μm2/s) was first estimated using the Stokes-Einstein equation below and a literature value of the hydrodynamic radius of 6.4 nm.[32]
| (7) |
Here, k is Boltzman’s constant; T is temperature (37 °C = 310 K); η is the viscosity of the solvent, water, at 37 °C (6.915*10−4 N*m/s2)[39] and Rs is the radius of the solute. With the diffusion coefficient of IgG in solution and the hydrodynamic radius, we then calculated the protein diffusivity in gel using equation (6) as descrbed previously.
Diffusion of the largest protein, IgG, in hydrogel was modeled using Fick’s second law of diffusion and MATLAB software:
| (8) |
with initial conditions of C(x,0) = 0 and boundary conditions of C(0,t) = C(L,t) = 1. Here, Dg is the calculated diffusion coefficient in hydrogels.
For collagenase degradation studies, hydrogels were fabricated using either 2.5 wt% (for non-dynamic soft and dynamic stiffened conditions) or 3.5 wt% (for non-dynamic stiff) PEG-NB with KCGPQG↓IWGQCK (R = 0.85), and CRGDS (1 mM) with LAP (1 mM) and UV-light exposure (2 min., 5 mW/cm2). After 24-hour swelling in PBS at 37 °C, gels in the dynamically stiff condition were stiffened with 0.2 wt% PEG-Tz at 37 °C for an additional 24 hours. The hydrogels were weighed to obtain m0 and placed in wells containing type 1 collagenase (30 U/mL, Worthington) hydrogels were weighed periodically. Data is presented using the following equation:
| (9) |
5.6. Tz-NB-mediated stiffening of iPSC-laden hydrogels and DE differentiation
For cell-laden hydrogel stiffening studies, encapsulation was conducted as described in section 5.4. Briefly, the cells (2*106 cells/mL) were mixed with pre-polymer solution consisting of PEG-NB, adhesion ligand CRGDS (1 mM), MMP sensitive crosslinker KCGPQG↓IWGQCK, and photoinitiator LAP (1 mM) to achieve a final thiol-norbornene ratio (RSH:NB) of 0.85. After homogenizing, the mixture was dispensed into a cylindrical mold and exposed to 365 nm light (5 mW/cm2) for 2 minutes to achieve gelation. Non-dynamic soft, dynamically stiffened, and non-dynamic stiff hydrogels consisted of 2.5 wt%, 2.5 wt%, and 3.5 wt% PEG-NB, respectively. Cell laden hydrogels were stiffened on day 3 post-encapsulation by adding PEG-Tz (0.2 wt%) to E8 media and incubating for 24 hours. As before, live/dead staining and imaging was conducted to ascertain the viability of the cells. Pluripotency was assessed by confocal imaging of SOX2-iPSCs with cell nuclei counterstained with DAPI on day 7 post-encapsulation. Additional analysis of pluripotency was conducted using a TaqMan® array pluripotency panel as described below. DE differentiation of soft, stiffened, and stiff was initiated using the STEMdiff™ Definitive Endoderm Kit (STEMCELL Technologies) per manufacturer’s protocol. Following DE differentiation, SOX2-iPSCs were stained SOX17 antibody and counterstained with DAPI. Non-differentiated controls were also stained with anti-SOX17 antibody and counter-stained nuclei with DAPI. Imaging was conducted on a confocal microscope as described previously.
5.7. Immunofluorescence staining and imaging of iPSC-laden hydrogels
iPSC-laden hydrogels were washed twice with Dulbecco’s phosphate buffered saline (DPBS) for 5 minutes and fixed with 4% paraformaldehyde for 45 minutes. Hydrogels were rinsed once with PBS and twice with DPBS containing 1% bovine serum albumin (BSA) and 0.3% Trition-X-100 for 5 minutes each. Blocking and permeabilization was conducted with 1% BSA and 0.3% Triton X-100 in DPBS at 4 °C overnight. Samples containing non-differentiated ChiPSC12s or differentiated SOX2-iPSCs were incubated with rabbit anti-YAP (Cell Signaling Technologies, 1:100) or rabbit anti-SOX17 antibody (Cell Signaling Technologies, 1:100), respectively, with 1% BSA, 0.3% Trition-X-100 at 4 °C overnight. After 3 45-minute washes with PBS, the gels were incubated with goat anti-rabbit IgG-Texas Red antibody (Santa Cruz Biotechnology, 1:200), 1% BSA, and 0.3% Triton-X-100 overnight at 4 °C. After three 30-minute DPBS washes, ChiPSC12-laden hydrogels were incubated for an additional day with rhodamine-phalloidin (Cytoskeleton, Inc, 1:200). The samples were rinsed thrice with DPBS for 10 minutes each and counterstained with DAPI for 1 hour. The cells were rinsed and imaged using confocal microscopy.
ImageJ was used to quantify the fraction of SOX2-positive and SOX17-positive cells. The mean of background areas was multiplied by the integrated density to obtain the background contribution. The determined background contribution was subtracted from the total pixel area of the entire image to ascertain the contribution of each fluorochrome. The fluorescent contribution of SOX17 or SOX2 was subtracted from the fluorescent contribution of DAPI to determine the positive fraction of the image. Three randomly selected areas of one sample in each condition were imaged and analyzed. A total of three samples per condition were analyzed.
5.8. RNA extraction, reverse transcription PCR, and real-time PCR of encapsulated iPSCs
Three cell-laden hydrogels were transferred to DNase/RNase free microtubes, flash frozen in liquid nitrogen, and stored at −80 °C until ready for use. RNA was extracted from the encapsulated cells using a combination of bead disruption and a NucleoSpin RNA II kit (Clontech) following manufacturer’s protocol. A NanoDrop 2000 Spectrophotometer (Thermo Scientific) was used to obtain the concentration and purity of the RNA. The isolated RNA was converted into single-stranded cDNA using a PrimeScript RT reagent kit (Clontech). A 96-well TaqMan® array (Thermo Fisher Scientific) was utilized to assess the pluripotency of the stem cells. cDNA samples were diluted to 100 ng/mL and added to an equal volume of TaqMan® fast universal master mix. After transferring 10 μL of the mixture to each well, the plate was sealed, and vortexed. Data was collected using an Applied Biosystems 7500 Fast Real-Time PCR machine. For each condition, a total of 3 biological replicates were used. The relative gene expression was analyzed by the 2−ΔΔCT method with GUSB as the housekeeping gene, and the expression level of the stiffened condition was presented relative to the control group (i.e., soft hydrogel condition).
5.9. Statistical Analysis
Either an ordinary one-way or two-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc test were conducted for statistical analysis. Statistical significance was considered at a p value < 0.05. P value < 0.05, 0.01, 0.001, 0.0001 were represented by single, double, triple, and quadruple asterisks, respectively. At least three independent repeats were conducted for each study and mean ± SEM was used to represent the quantitative results.
Supplementary Material
Acknowledgement
This work was supported in part by the National Institutes of Health via R01CA227737 (to CL) and R01DC017461 (to KRK) and the National Science Foundation via a Graduate Research Fellowship Program (GRFP) to MRA.
Contributor Information
Matthew R. Arkenberg, Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907, USA
Karl R. Koehler, Departments of Otolaryngology and Plastic and Oral Surgery, F.M. Kirby Neurobiology Center, Boston Children’s Hospital/Harvard Medical School, Boston, MA 02115, USA
Chien-Chi Lin, Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907, USA; Department of Biomedical Engineering, Purdue School of Engineering & Technology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA.
References
- [1].a) Marquardt LM, Heilshorn SC, Current stem cell reports 2016, 2, 207; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ovadia EM, Colby DW, Kloxin AM, Biomaterials science 2018, 6, 1358; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bai T, Li J, Sinclair A, Imren S, Merriam F, Sun F, O’Kelly MB, Nourigat C, Jain P, Delrow JJ, Nature medicine 2019, 25, 1566; [DOI] [PubMed] [Google Scholar]; d) Lei Y, Schaffer DV, Proceedings of the National Academy of Sciences 2013, 110, E5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Zieris A, Prokoph S, Levental KR, Welzel PB, Grimmer M, Freudenberg U, Werner C, Biomaterials 2010, 31, 7985; [DOI] [PubMed] [Google Scholar]; b) Burdick JA, Prestwich GD, Advanced materials 2011, 23, H41; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tabata Y, Ikada Y, Biomaterials 1999, 20, 2169; [DOI] [PubMed] [Google Scholar]; d) Lin C-C, Anseth KS, Pharmaceutical research 2009, 26, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677; [DOI] [PubMed] [Google Scholar]; b) Zhu Y, Li X, Janairo RRR, Kwong G, Tsou AD, Chu JS, Wang A, Yu J, Wang D, Li S, J Cell Physiol 2019, 234, 7569; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sun M, Chi G, Li P, Lv S, Xu J, Xu Z, Xia Y, Tan Y, Xu J, Li L, Li Y, Int J Med Sci 2018, 15, 257; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Smith LR, Cho S, Discher DE, Physiology (Bethesda) 2018, 33, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Lee ST, Yun JI, Jo YS, Mochizuki M, van der Vlies AJ, Kontos S, Ihm JE, Lim JM, Hubbell JA, Biomaterials 2010, 31, 1219; [DOI] [PubMed] [Google Scholar]; b) Jang M, Lee ST, Kim JW, Yang JH, Yoon JK, Park JC, Ryoo HM, van der Vlies AJ, Ahn JY, Hubbell JA, Song YS, Lee G, Lim JM, Biomaterials 2013, 34, 3571. [DOI] [PubMed] [Google Scholar]
- [5].Jang M, Lee ST, Kim JW, Yang JH, Yoon JK, Park J-C, Ryoo H-M, Van Der Vlies AJ, Ahn JY, Hubbell JA, Biomaterials 2013, 34, 3571. [DOI] [PubMed] [Google Scholar]
- [6].Ranga A, Gobaa S, Okawa Y, Mosiewicz K, Negro A, Lutolf MP, Nat Commun 2014, 5, 4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Kerscher P, Turnbull IC, Hodge AJ, Kim J, Seliktar D, Easley CJ, Costa KD, Lipke EA, Biomaterials 2016, 83, 383; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hwang NS, Varghese S, Zhang Z, Elisseeff J, Tissue Eng 2006, 12, 2695; [DOI] [PubMed] [Google Scholar]; c) Schukur L, Zorlutuna P, Cha JM, Bae H, Khademhosseini A, Adv Healthc Mater 2013, 2, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS, Adv Mater 2010, 22, 3484; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin-Gibson S, Jones RL, Washburn NR, Horkay F, Macromolecules 2005, 38, 2897. [Google Scholar]
- [9].Lin CC, Ki CS, Shih H, J Appl Polym Sci 2015, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Handorf AM, Zhou Y, Halanski MA, Li WJ, Organogenesis 2015, 11, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barriga EH, Franze K, Charras G, Mayor R, Nature 2018, 554, 523; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Thompson AJ, Pillai EK, Dimov IB, Foster SK, Holt CE, Franze K, Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kolb L, Allazetta S, Karlsson M, Girgin M, Weber W, Lutolf MP, Biomater Sci 2019, 7, 3471. [DOI] [PubMed] [Google Scholar]
- [12].a) Liu HY, Greene T, Lin TY, Dawes CS, Korc M, Lin CC, Acta Biomater 2017, 48, 258; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arkenberg MR, Lin CC, Biomater Sci 2017, 5, 2231; [DOI] [PubMed] [Google Scholar]; c) Arkenberg MR, Moore DM, Lin CC, Acta Biomater 2019, 83, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guvendiren M, Burdick JA, Nat Commun 2012, 3, 792. [DOI] [PubMed] [Google Scholar]
- [14].Stowers RS, Allen SC, Suggs LJ, Proc Natl Acad Sci U S A 2015, 112, 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Mayer SV, Murnauer A, von Wrisberg M-K, Jokisch M-L, Lang K, Angewandte Chemie 2019; [DOI] [PMC free article] [PubMed]; b) Umlauf BJ, Mix KA, Grosskopf VA, Raines RT, Shusta EV, Bioconjug Chem 2018, 29, 1605; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blizzard RJ, Gibson TE, Mehl RA, Methods Mol Biol 2018, 1728, 201. [DOI] [PubMed] [Google Scholar]
- [16].a) Werther P, Yserentant K, Braun F, Kaltwasser N, Popp C, Baalmann M, Herten DP, Wombacher R, Angewandte Chemie International Edition 2020, 59, 804; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wieczorek A, Werther P, Euchner J, Wombacher R, Chemical science 2017, 8, 1506; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Baalmann M, Ziegler MJ, Werther P, Wilhelm J, Wombacher R, Bioconjugate chemistry 2019, 30, 1405. [DOI] [PubMed] [Google Scholar]
- [17].a) Alge DL, Azagarsamy MA, Donohue DF, Anseth KS, Biomacromolecules 2013, 14, 949; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS, Biomaterials 2015, 50, 30; [DOI] [PubMed] [Google Scholar]; c) Hao Y, Song J, Ravikrishnan A, Dicker KT, Fowler EW, Zerdoum AB, Li Y, Zhang H, Rajasekaran AK, Fox JM, Jia X, ACS Appl Mater Interfaces 2018, 10, 26016; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hu Y, Mao AS, Desai RM, Wang H, Weitz DA, Mooney DJ, Lab on a Chip 2017, 17, 2481; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jivan F, Yegappan R, Pearce H, Carrow JK, McShane M, Gaharwar AK, Alge DL, Biomacromolecules 2016, 17, 3516. [DOI] [PubMed] [Google Scholar]
- [18].Richardson T, Barner S, Candiello J, Kumta PN, Banerjee I, Acta biomaterialia 2016, 35, 153. [DOI] [PubMed] [Google Scholar]
- [19].McKinnon DD, Kloxin AM, Anseth KS, Biomaterials Science 2013, 1, 460. [DOI] [PubMed] [Google Scholar]
- [20].Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS, Advanced Materials 2009, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, Sasai Y, Nat Biotechnol 2007, 25, 681; [DOI] [PubMed] [Google Scholar]; b) Claassen DA, Desler MM, Rizzino A, Mol Reprod Dev 2009, 76, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Varelas X, Development 2014, 141, 1614. [DOI] [PubMed] [Google Scholar]
- [23].Liu H, Jiang D, Chi F, Zhao B, Protein & cell 2012, 3, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Qin H, Hejna M, Liu Y, Percharde M, Wossidlo M, Blouin L, Durruthy-Durruthy J, Wong P, Qi Z, Yu J, Qi LS, Sebastiano V, Song JS, Ramalho-Santos M, Cell Rep 2016, 14, 2301; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, Ding S, Guan KL, Genes Dev 2010, 24, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ohgushi M, Minaguchi M, Sasai Y, Cell stem cell 2015, 17, 448. [DOI] [PubMed] [Google Scholar]
- [26].Karver MR, Weissleder R, Hilderbrand SA, Bioconjugate chemistry 2011, 22, 2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oliveira B, Guo Z, Bernardes G, Chemical Society Reviews 2017, 46, 4895. [DOI] [PubMed] [Google Scholar]
- [28].Devaraj NK, Weissleder R, Hilderbrand SA, Bioconjug Chem 2008, 19, 2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Selvaraj R, Fox JM, Current opinion in chemical biology 2013, 17, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Knall A-C, Hollauf M, Slugovc C, Tetrahedron letters 2014, 55, 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Holt SE, Rakoski A, Jivan F, Pérez LM, Alge DL, Macromolecular Rapid Communications 2020, 2000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Karlsson D, Zacchi G, Axelsson A, Biotechnology progress 2002, 18, 1423. [DOI] [PubMed] [Google Scholar]
- [33].a) Liu H-Y, Korc M, Lin C-C, Biomaterials 2018, 160, 24; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arkenberg MR, Lin C-C, Biomaterials science 2017, 5, 2231. [DOI] [PubMed] [Google Scholar]
- [34].Pucéat M, Cardiovascular research 2007, 74, 256. [DOI] [PubMed] [Google Scholar]
- [35].a) Cambria E, Renggli K, Ahrens CC, Cook CD, Kroll C, Krueger AT, Imperiali B, Griffith LG, Biomacromolecules 2015, 16, 2316; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arkenberg MR, Moore DM, Lin C-C, Acta biomaterialia 2019, 83, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Peuler K, Dimmitt N, Lin C-C, Carbohydrate Polymers 2020, 234, 115901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lustig SR, Peppas NA, Journal of Applied Polymer Science 1988, 36, 735. [Google Scholar]
- [38].Weber LM, Lopez CG, Anseth KS, Journal of Biomedical Materials Research Part A: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2009, 90, 720. [Google Scholar]
- [39].Geankoplis CJ, Transport processes and separation process principles:(includes unit operations), Prentice Hall Professional Technical Reference, 2003.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.