

Abstract
Direct interaction of cocaine with centrally located monoamine transporters is the primary mechanism underlying its reinforcing properties. It is also often assumed that this drug action is responsible for all the physiological and behavioral effects of this drug. The goal of this review is to challenge this basic mechanism and demonstrate the importance of peripheral actions of cocaine in inducing its initial, rapid neural effects. The use of high-resolution electrophysiological, neurochemical and physiological techniques revealed that the effects of intravenous cocaine at behaviorally relevant doses are exceptionally rapid and transient correlating with strong, quick, and transient increases in blood cocaine levels. Some of these effects are mimicked by cocaine-methiodide, a cocaine analog that cannot cross the blood-brain barrier and they are resistant to dopamine receptor blockade. Therefore, it appears that rapid neural effects of cocaine result from its direct interaction with receptive sites on afferents of sensory nerves densely innervating blood vessels. This interaction creates a rapid neural signal to the CNS that results in generalized neural activation and subsequent changes in different physiological parameters. This drug’s action appears to be independent from cocaine’s action on central neurons, which requires a definite time to occur and induce neural and physiological effects with longer latencies and durations. The co-existence in the same drug of two timely distinct actions with their subsequent interaction in the CNS could explain consistent changes in physiological and behavioral effects of cocaine following their repeated use, playing a role in the development of drug-seeking and drug-taking behavior.
Keywords: Neural activation, EEG desynchronization, Single-unit activity, Brain oxygen, Brain glucose, Metabolic activation, Arterial blood pressure, Dopamine release and uptake, Peripheral vasoconstriction, Rats
Introduction
Cocaine is a neuroactive drug with a strong potential for addiction. It is also a psychostimulant, which induces neural activation followed by increases in sympathetic and behavioral activity. According to the traditional viewpoint, monoamine transporters are the primary substrate of cocaine’s action in the brain; cocaine’s interaction with these transporters results in the inhibition of monoamine reuptake with subsequent enhancement of monoamine neurotransmission (Heikkila et al., 1975). While this action appears to be essential for the reinforcing properties of cocaine, i.e. its ability to serve as a drug reinforcer (Wise and Bozarth, 1987; Ritz et al., 1987; Kuhar at al., 1991), this action of cocaine in the CNS is often viewed as the cause of all physiological and behavioral effects of this drug. In this work, I will challenge this view and demonstrate that intravenous (iv) cocaine used in low, behaviorally-relevant doses in awake, freely moving animals induces exceptionally rapid neural effects, which correlate with rapid changes in blood cocaine levels.
These effects are often distorted or blocked in anesthetized animals and they are usually missed with slow, minute-scale time resolution analyses, but could be revealed using dynamic monitoring of neuronal activity, EEG, and high-resolution physiological and neurochemical techniques. This rapidity of neural effects is often viewed as a reflection of rapid drug entry in brain tissue, but this assumption is questioned by recent high-resolution assessments of brain cocaine levels, which increase with definite latencies and peak at much later time than blood levels after cocaine injection. The rapidity and transient nature of these effects are also inconsistent with the timing necessary for the drug to reach brain vessels, cross the blood-brain barrier (BBB) and diffuse within brain tissue to its receptive sites. Although collectively this evidence hints at the direct interaction of cocaine with peripherally located neural substrates, other critical proofs of this peripherally driven mechanism were obtained by using cocaine-methiodide, a cocaine’s analog that cannot cross the BBB and thus acts only in the periphery (Shriver and Long, 1971; Hemby et al., 1994; Wise et al., 2008). Despite the known ability of cocaine to inhibit dopamine (DA) uptake and increase DA neurotransmission, many important neural effects of this drug at behaviorally relevant doses are resistant to full DA receptor blockade, questioning the role of this mechanism in triggering and shaping of its neural effects.
Based on analysis of large body of experimental evidence, I consider the possible mechanisms underlying rapid affects of iv cocaine, specifically the role of its direct interaction with receptive substrates on afferents of visceral sensory nerves that densely innervate blood vessels. Under normal physiological conditions, these receptive substrates are activated by deviations of basic homeostatic parameters such as temperature, pH, osmolarity, pressure, and the levels of sodium, potassium, glucose, carbon dioxide, and oxygen, but they could be strongly but transiently activated by cocaine following robust increases in its concentration in blood vessels. While the exact nature of these peripheral receptive substrates still remains hypothetical, their activation creates a rapid neural signal to the CNS that results in generalized neural activation and subsequent changes in multiple physiological parameters that occur within seconds after drug injection. This peripherally triggered cocaine’s action appears to be independent from its direct actions on central neurons, which always have definite onset latencies, inducing weaker but more prolonged effects. The co-existence in the same drug of two timely distinct actions with their subsequent interaction in the CNS could explain the consistent changes in physiological and behavioral effects of cocaine following their repeated use, playing a role in the development of drug-seeking and drug-taking behavior.
Considering multiple evidence supporting this peripherally driven neural mechanism, I tried to be unbiased in analyses of available data obtained in different laboratories, stressing the existing contradictions in experimental findings and their interpretation. However, the main focus of this work was on our electrophysiological and neurochemical studies, which were designed to verify the role of peripheral actions of cocaine in triggering and mediating of its rapid neural effects. Some of my interpretations and conclusions may be viewed as speculative, requiring experimental verification and additional analytical support. The best reward for the author is attention to his work and substantive discussion and critique, which could result in clarification of existing knowledge, new ideas, new experiments to test these ideas, and an overall better understanding of the mechanisms underlying the unique addictive potential of cocaine.
Rapid neural effects of iv cocaine and their basic mechanisms
It is generally believed that numerous psycho-emotional, physiological and behavioral effects of cocaine are mediated via its action on centrally located neural substrates. The strength and duration of these effects are determined by the drug dose and the means of drug administration, with more rapid and stronger effects for iv than subcutaneous or intraperitoneal routes. Physiological and neural effects of cocaine as well as its addictive potential also depend on the speed of its iv delivery, being stronger with rapid injection speed and weaker with slow injection speed (Samaha et al., 2002; Minogianis et al., 2013; see however Crombag et al., 2008). The pattern of these effects also depends on temporal resolution of measurement, being accurate for dynamic parameters assessed with second-scale resolution and clearly distorted for slow-resolution measurements. Neural effects of cocaine also depend on the experimental paradigm used for their assessment. While the neuronal effects of cocaine and certain neurochemical evaluation are often conducted in anesthetized animals, general anesthetic drugs powerfully inhibit brain metabolism, affect numerous homeostatic functions, and significantly inhibit neuronal responses to sensory stimuli and cocaine (Michenfelder, 1988; Windel and Kiyatkin, 2006; Koulchitsky et al., 2012; Park et al., 2019).
The basic knowledge on cocaine’s effects on central neurons was obtained by using in vitro models. In contrast to in vivo preparations, when cocaine levels are highly dynamic after its systemic administration, cocaine in these experiments is typically applied to the recorded cells at high and constant concentrations. While this approach reveals the direct action of the drug on certain neurons, it is unclear whether these conclusions can be applicable for real conditions of the whole organism and awake brain, when the drug reaching brain receptive substrates and much lower and highly variable concentrations and induce multiple physiological and behavioral effects. These limitations are also applicable for some analytical experiments, when cocaine is administered in experimental animals at high doses that greatly exceed the typical range of human consumption and optimal doses for cocaine self-administration in rats that are within a 0.5–1.0 mg/kg or 1.5–3.0 μM/kg range (Pickens and Thompson, 1968; Gerber and Wise, 1989; Mandt et al., 2012). The issue of dose may be especially important for iv cocaine because, depending on dose, this drug will activate different neural substrates (monoamine transporters, various ionic channels, etc.). Furthermore, cocaine at doses exceeding 2–3 mg/kg can induce arrhythmias, robust changes in cardio-vascular parameters, and general toxicity. Microiontophoresis is an unique tool to study the direct action of neuroactive substances on single neurons, but its value for understanding the real effects of systemically administered cocaine also appears to be limited. While striatal neurons were uniformly inhibited by iontophoretic cocaine applications, these effects were insensitive to DA antagonists and mimicked by iontophoretic procaine, an anesthetic drug which shares with cocaine its ability to inhibit Na+ channels (Kiyatkin and Rebec, 2000). Therefore, it appears that cocaine-induced neuronal inhibitions result from the direct blockade of Na+ channels, the known anesthetic effect of cocaine, due to its high concentrations nearby the recorded neurons. Taking into account these technical and experimental limitations, when available my analysis of experimental data will be focused on the effects of iv cocaine at low, behaviorally relevant doses in awake freely moving rats.
Pharmacodynamics: Changes in cocaine levels in blood and brain after iv administration
The impact of the drug on brain functions critically depends on its temporal dynamics in the body and the brain. For iv cocaine delivery, accurate and highly temporally resolved concentration measurements both in the blood and brain’s extracellular space are necessary to understand the mechanisms underlying its neural effects. While the direct interaction of cocaine with central neurons is usually viewed as the primary target of its neural effects, blood vessels are densely innervated by afferents of sensory nerves (O’Regan and Majcherczyk, 1982; Lee et al., 2005; Michaelis et al., 1994) and interoceptors expressed on these afferents is the site of the initial organism’s interaction with cocaine. These afferents express various ionic channels (i.e., voltage-gated Na+ and K+ channels, Ca2+-activated channels, TRV channels, etc.; Zhang et al., 2002; Premkumar, 2005; Kobayashi et al., 2007; Chen et al., 2006; Lee et al., 2005; Wu et al., 2006) and are activated under normal physiological conditions by deviations of basic homeostatic parameters such as temperature, pH, osmolarity, and the levels of sodium, potassium, glucose, carbon dioxide, and oxygen. Changes in activity of visceral sensory nerves rapidly reach the spinal cord and the brain, where numerous adaptive responses are triggered to eliminate or minimize shifts in basic homeostatic parameters. In contrast to perception of external influences, stimulation of interoceptors under physiological conditions is usually not perceived and not specifically localized. Therefore, cocaine at high concentrations achieved by its iv administration may potentially interact with these peripherally located neural substrates, create a strong neural signal from the periphery to the CNS and induce neural activation with subsequent changes in physiological and ultimately behavioral parameters. To substantiate this hypothetical mechanism, it is critical to know cocaine’s precise dynamics both in peripheral blood and brain’s extracellular space.
Quantitative measurements of cocaine’s dynamics in venous blood of rats and rabbits revealed very rapid, strong, but transient increases (Parlaman et al., 2007; Tella and Goldberg et al., 1998). The levels of cocaine peaked at 30 s after the onset of a bolus 3 mg/kg iv injection and rapidly declined thereafter. The peak concentrations measured in both studies were similar (1.6 and 1.3 μg/ml or 5.3 and 4.3 μM). Even more rapid peaks in arterial blood concentrations (15 s) were found in humans iv injected with 16 and 32 mg of cocaine (Evans et al., 1996). Despite a lower dose in terms of body weight (0.23 and 0.46 mg/kg), peak concentration in arterial plasma transiently reached 3.8 and 7.6 μM for 16 and 32 mg doses, respectively. Based on these studies, it is possible to approximate that cocaine injected at optimal doses maintaining self-administration behavior in rats (1.0 mg/kg) will produce very rapid, strong (~1.5–1.75 μM) but transient rise in blood cocaine levels. While it is clear that the pattern of concentration changes and its quantitative parameters depend on time resolution, cocaine’s peak values in animal studies occurred at the first, either 15-s or 30-s, measurement points. While the samples of venous blood in animal studies were obtained far from the injection site after drug’s travel via pulmonary circulation, much larger concentration changes should occur within the route of cocaine’s travel from the injection site and following its distribution within the entire blood volume. As shown in human study (Evans et al., 1996), cocaine’s increases in arterial blood are substantially larger and more phasic than those in venous blood; their value for cocaine’s peak in arterial blood (16.5 μM for a 1 mg/kg dose) is much higher than those obtained in rats in venous blood. Therefore, due to low temporal resolution of sampling and essential limitations in obtaining clean and repeated blood samples in small animals, measurement data fail to provide accurate quantitative information on real-time dynamics of blood cocaine levels following iv drug injection. Moreover, this dynamic will be substantially different depending on the dose, duration of injection, injection volume, and the site of blood sampling.
Hypothetically, cocaine levels in brain’s extracellular space should be more delayed and weaker than in arterial blood reflecting drug travel from the injecting site, its crossing of three-cell layers of the BBB, and passive diffusion within brain tissue. While it is critical to know the dynamics of cocaine’s levels in brain’s extracellular space after its iv administration, this task is extremely complex and different approaches to solve this problem produced conflicting results. Three principal techniques, positron emission tomography (PET) with [C11] cocaine, in vivo microdialysis, and electrochemistry with cocaine-selective biosensors, were used to measure brain levels of cocaine. The earliest attempts to measure cocaine levels in brain tissue also employed post-mortem sample collection, when the animals were sacrificed at each time point after the injection; this approach is time- and animal-consuming, not very accurate due to sample contamination by blood, and greatly affected by drug instability in post-mortem environment.
In vivo microdialysis is widely used for assessing brain neurochemicals but measurements are typically done with relatively slow time-resolution (5–20 min). The first study using this approach (Pan et al., 1991) examined cocaine levels in the nucleus accumbens (NAc) with 5-min time resolution following iv injection of cocaine at 7.5 mg/kg dose in anesthetized rats. In this study, cocaine levels peaked (2.5 μM) at the first data point (0–5 min) and rapidly declined within the next 15–20 min. Taking into account that only a portion of cocaine will diffuse from extracellular space into dialysate (~5–10% recovery), estimated peak concentrations were ~30 μM, or 2–4 μM for behaviorally relevant doses (1 mg/kg). Similar results were obtained in another study using a lower cocaine dose (1 mg/kg) in anesthetized rats (Bradberry et al., 1993). In this case, measurements were conducted in the striatum with 4-min time resolution, revealing concentration peak of 3.5 μM at the second measurement point (4–8 min). More complex picture of cocaine’s dynamics in brain tissue was recently revealed, when measurements were conducted with 2-min time resolution in freely moving rats (Minogianis et al., 2019). In this study, cocaine was delivered at 2 mg/kg dose at different injection speeds. With ultra-fast (5-s) injection, cocaine slowly increased and peaked (0.22 μM) at 4–6 min post-injection. With a slower injection speed (45 s), cocaine increase occurred with ~3-min latency reaching a slightly lower peak (~0.19 nM) at ~6–8 min post-injection. Taking into account ~10% recovery, in vivo cocaine peaks after its iv delivery could be within 0.7–1.1 μM for 1 mg/kg dose.
Larger increases in brain cocaine levels were shown using PET with [C11]-cocaine in anesthetized baboons (Fowler et al., 1998). In this study, peak increases in radioactivity assessed in the striatum after 0.5 and 1.0 mg/kg injections occurred at 2–3 min at 9 and 26 μM, respectively. While this is a non-invasive technique, thus eliminating the problem of possible BBB damage typical to any other sampling technique, high values of cocaine and its rapid rise could result from contribution of much larger levels of cocaine in arterial blood. Concentrations of C11 in this study were determined in the striatum as a whole, but blood vessels account for ~50% of brain tissue volume and radioactive cocaine contained in these vessels can be summated at least partially with cocaine contained in brain’s extracellular space.
Electrochemical techniques have second-scale resolution, and advantages of this technology for detecting rapid fluctuations in brain chemicals are well known (see below). The development of aptamer cocaine biosensors, which have high substrate selectivity, allowed the assessment of fluctuations in brain cocaine levels with exceptional, 4-s time resolution (Taylor et al., 2017). When anesthetized rats were injected with cocaine at 2 mg/kg, cocaine levels in the dorsal striatum began to increase with ~2 min latencies, sharply peaked at 6.6 min at 14.8 μM concentrations (or 3.7 and 7.4 μM for 0.5 and 1.0 mg/kg doses), and slowly decreased for subsequent 40 min. In contrast to relatively large dialysis probes that produce damage in brain tissue, cocaine biosensors are much smaller, greatly reducing possible contributions from arterial blood. This factor as well as exceptional measurement resolution made it possible to reveal that extracellular cocaine levels began to increase after iv administration with a certain latency, peaking at a later time at values exceeded those detected that in microdialysis studies. While this approach provides a valuable alternative to microdialysis, it has a number of limitations, which will be addressed in future studies.
Rapid cardiovascular effects of iv cocaine
Important findings that suggest cocaine’s action on peripheral neural substrates were obtained by analyzing its cardiovascular effects. When injected intravenously at behaviorally relevant doses (0.3–2.0 mg/kg; Pickens and Thompson, 1968; Gerber and Wise, 1989), cocaine induced a rapid but relatively short-term increase in arterial blood pressure, which has second-scale latencies and peaks within 10–30 s from the injection start (Pitts et al., 1987; Poon and van den Buuse 1998). Although this hypertensive effect is centrally-mediated, resulting primarily from peripheral vasoconstriction (Knupfer and Branch, 1992), its rapidity is difficult to explain considering the time that is necessary for the drug to reach brain blood vessels, cross the BBB, passively diffuse in brain tissue, bind to centrally located receptive sites, and affect neuronal activity. Furthermore, this rapid hypertensive effect is resistant to DA receptor blockade (Kiritsy-Roy et al. 1990; Poon and van den Buuse 1998) and is mimicked by a BBB-impermeable cocaine-methiodide (Dickerson et al. 1999). While the initial, transient rise in arterial blood pressure was the most prominent effect of iv cocaine, it was followed by weaker and more prolonged pressure increase (Pitts et al., 1987), which was blocked by DA antagonists (Tella and Goldberg, 1998). Therefore, the effect of iv cocaine on arterial blood pressure appears to be biphasic, with the initial, peripherally triggered and subsequent centrally mediated components. While it is evident that ultra-fast hypertensive effect of iv cocaine results from neural mechanism and involves the CNS, it occurs before the measured changes in blood cocaine levels (see above), suggesting the importance of dynamic and highly time-resolved physiological measurements.
Rapid neural effects of iv cocaine and their possible mechanisms: electrophysiological evidence
To examine the pattern of neural activity induced by iv cocaine, we used cortical electroencephalography (EEG) with subsequent analysis of its power and frequency characteristics (Kiyatkin and Smirnov, 2010). In contrast to other parameters assessed with a slow time resolution, EEG activity can be monitored in freely moving rats dynamically and analyzed with second-scale resolution. To minimize any external sensory influences, cocaine injections (0.25–1.0 mg/kg) were conducted in quietly resting rats extensively habituated to the recording environment using a catheter extension from outside of the recording chamber. Monitoring of EEG activity was coupled with recording of neck electromyographic (EMG) activity, a centrally mediated physiological parameter that reflects tonic and phasic changes in motor output. In contrast to single-unit recording in freely moving rats, which are technically more complex and have a number of significant limitations (i.e., cell heterogeneity, silent/active cells, limited recording time, necessity of large data samples, possible movement artifacts), EEG and EMG recordings could be conducted for an extended period of time following repeated daily sessions in the same animals, thus providing an integral measure of event-related neural and motor responses. As a primary parameter to assess the changes in EEG activity, we used a total power of the EEG signal--a universal measure of electrical synchronization or desynchronization (Buzsaki, 2008; Steriade and McCarley, 2005). Additional information on changes in EEG activity was obtained by EEG spectral analysis, which reveals the dynamics of time-dependent alterations in specific activity bands.
These experiments revealed that iv cocaine administration induced rapid, powerful and prolonged changes in cortical EEG and neck EMG at each dose tested (Figure 1 and 2). EEG total power rapidly decreased to its minimum within the injection interval (10, 15, and 20 s for 0.25, 0.5 and 1.0 mg/kg injections, respectively) and remained at these low levels for at least five minutes post-injection. Averaged EMG had a similar but inverted pattern, rapidly and strongly increasing during the injection and remaining increased for the entire analysis interval. Our control tests with a short auditory stimulus and iv saline injections revealed that these mild sensory stimuli also induce phasic EEG desynchronization and EMG activation, but these effects were much weaker and more transient than those elicited by cocaine at each dose.
Figure 1. Phasic and tonic changes in cortical EEG (top) and neck EMG (bottom) total powers induced by iv cocaine administration at different doses within the self-administration range.
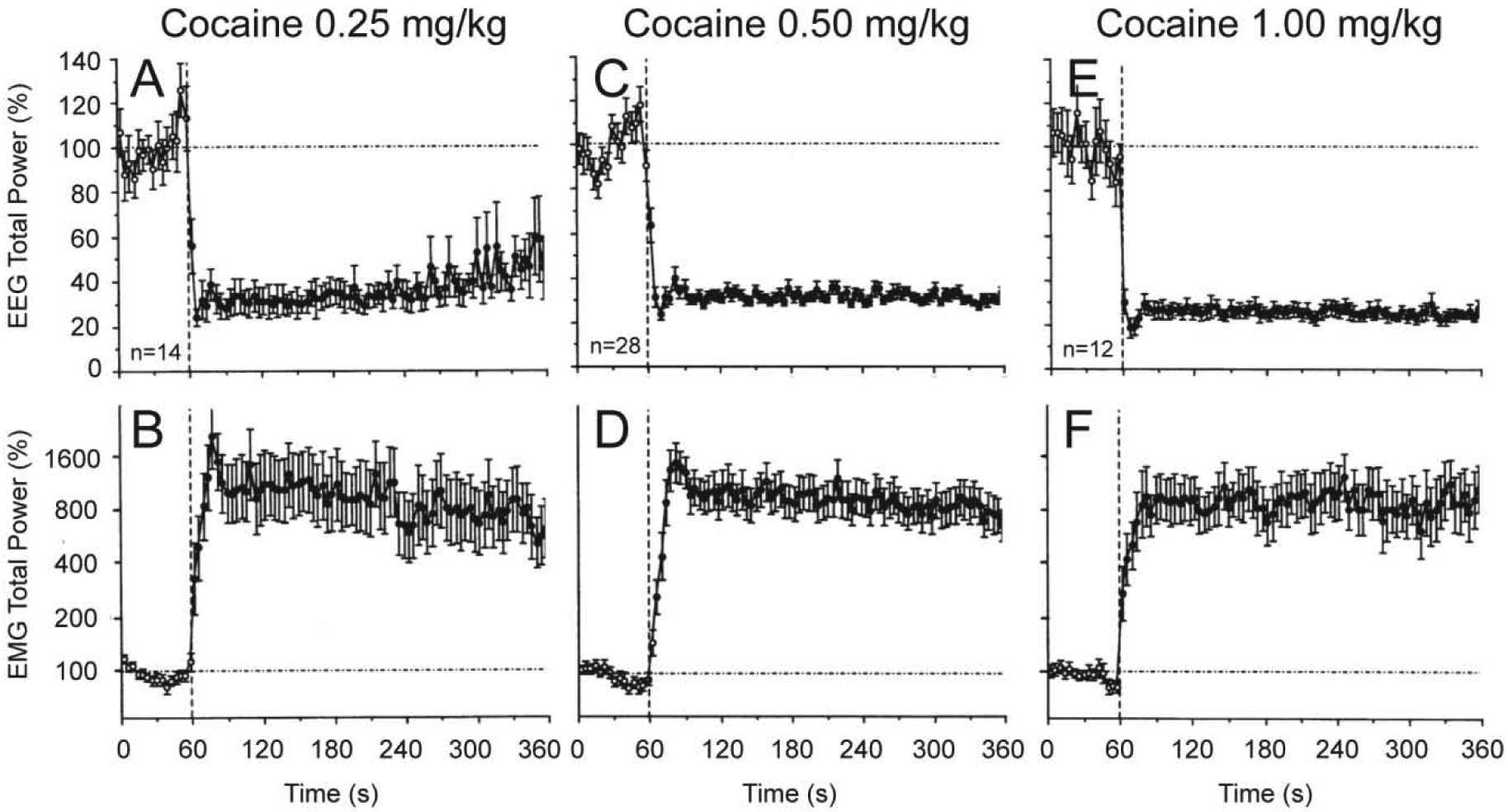
Mean (±SEM) values (for 4-s bins) are shown in percent vs. the pre-injection baseline. Vertical hatched lines (at 60 s) show the start of injections and horizontal hatched lines show basal values (100%). Filled symbols show values significantly different (Fisher F-test, p<0.05) from baseline. n=number of averaged tests. Original data were reported in Kiyatkin and Smirnov, 2010 (free PMC article).
Figure 2. Original records of changes in EEG and EMG activity (μV at final amplification) induced in freely moving rats by short auditory stimulus (clap; top), iv cocaine (middle) and iv saline (bottom) injections.
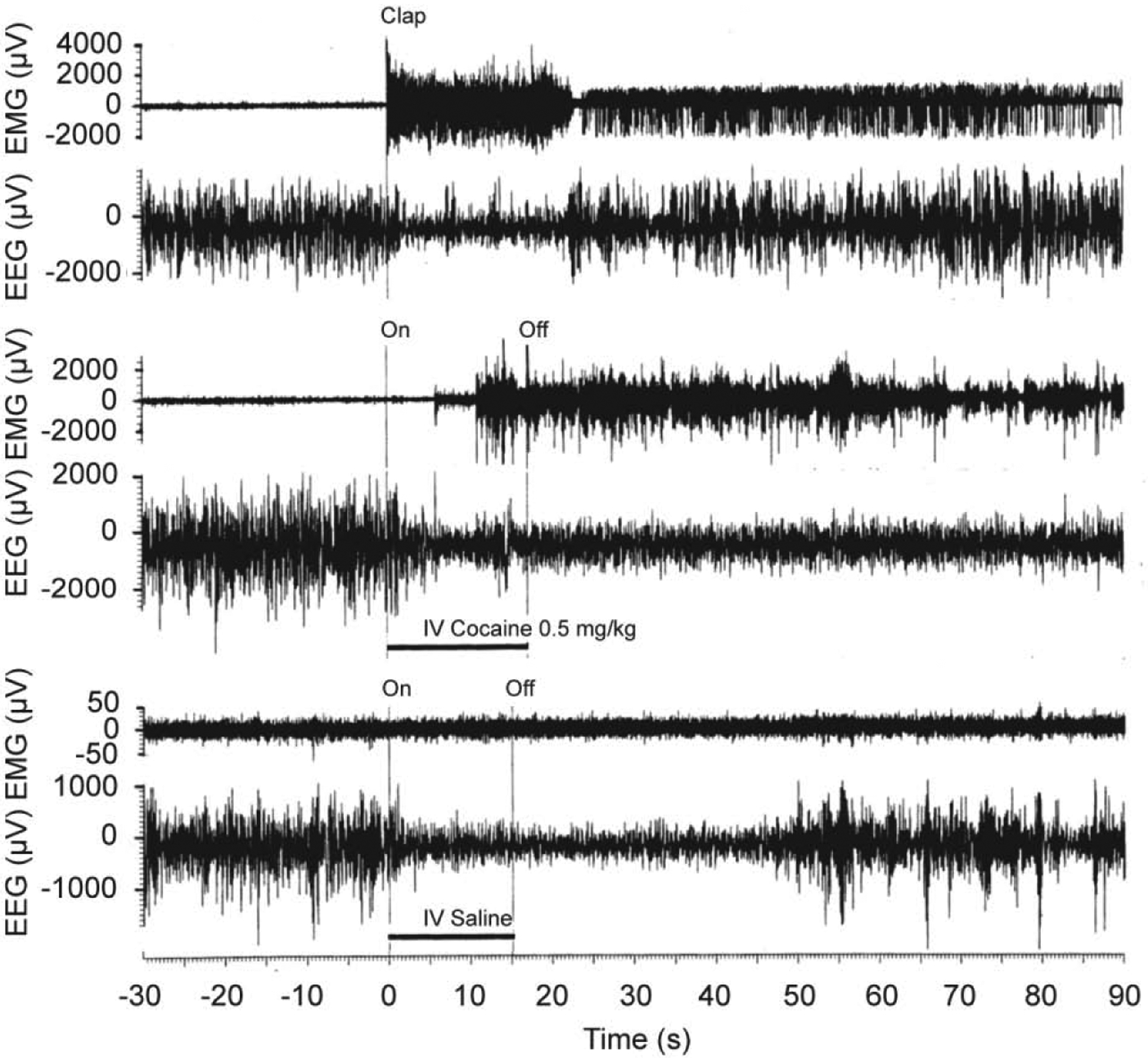
Original data were reported in Kiyatkin and Smirnov, 2010 (free PMC article).
Since it is often assumed that neural effects of cocaine are mediated via its action on the DA transporter and subsequent changes in DA transmission, we examined how changes in EEG and EMG activity, induced by iv cocaine are affected by DA receptor blockade. These tests revealed that full DA receptor blockade induced by a mixture of D1- and D2-like DA antagonists (SCH 23390 + eticlopride at 0.2 mg/kg, sc, each) had no significant effect on rapid changes in the EEG signal induced by iv cocaine at 0.5 mg/kg dose (Figure 3A). However, the effects of cocaine during DA receptor blockade had some small differences compared to the effects seen in control, untreated rats. The decrease in EEG total power in DA antagonist group was slightly slower than that in controls and had a tendency to recover within the observation period. While similarly rapid, the EMG response to cocaine during DA receptor blockade (B) was also slightly weaker and more transient than that in control tests. These between-group differences in the relative increase in EMG activity could be partially related to a higher basal EMG activity seen after the DA antagonist treatment. Since rats during DA receptor blockage were generally adynamic, a relatively large change in EMG total power obviously results from tonic increases in EMG activity.
Figure 3. Mean changes in EEG (A and C) and EMG (B and D) total powers induced by iv cocaine (0.5 mg/kg) in drug-free, freely moving conditions (control), during DA receptor blockade and urethane anesthesia (1.25 g/kg).
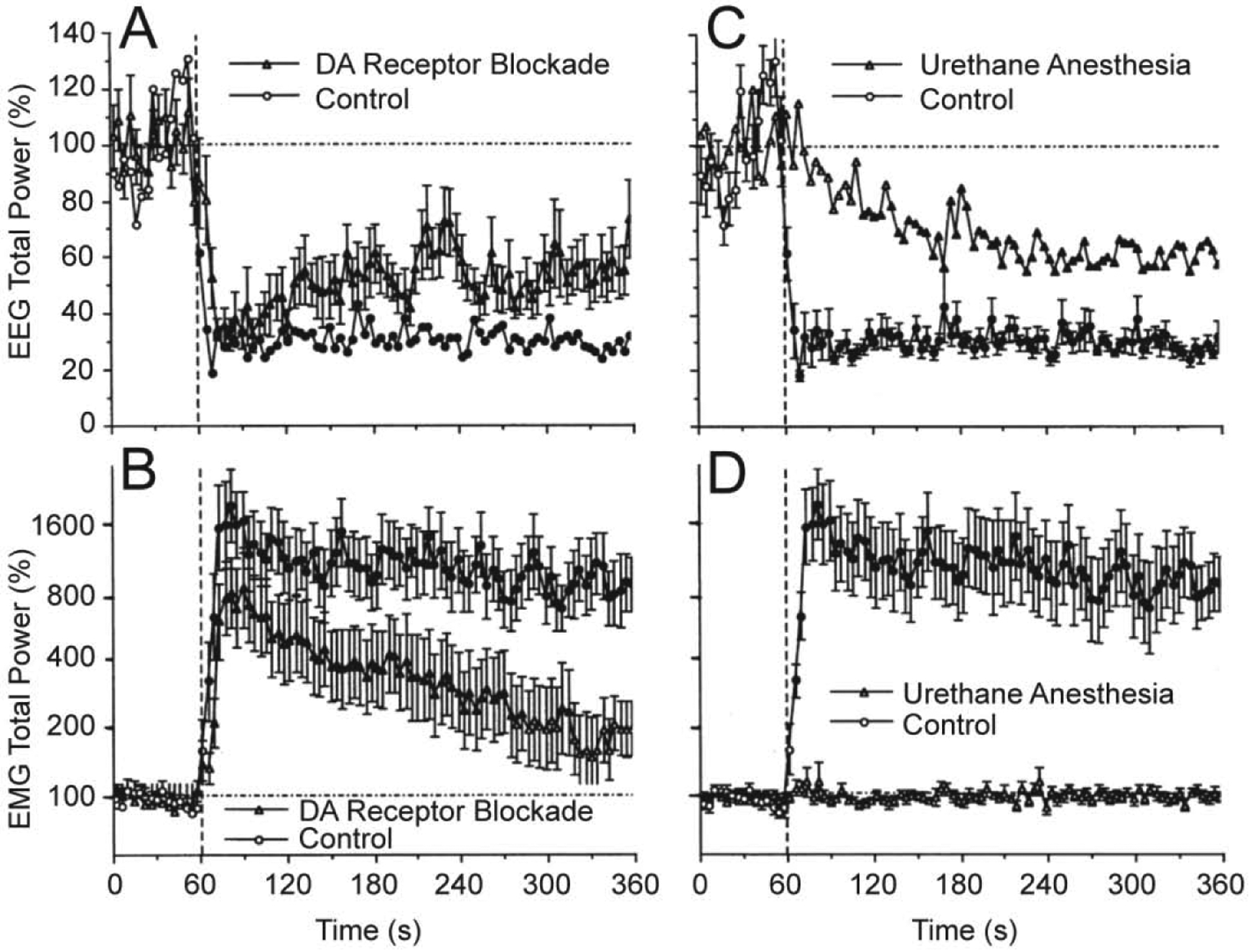
Filled symbols show values significantly different from baseline. Original data were reported in Kiyatkin and Smirnov, 2010 (free PMC article).
It is well known that general anesthesia inhibits or blocks neural responses to arousing stimuli. When the effects of cocaine were assessed during urethane anesthesia (1.25 mg/kg, ip), the patterns of EEG and EMG activity were dramatically changed (Figure 3C and D). Instead of a rapid decrease in EEG total power occurring during cocaine injection in controls, in anesthetized rats this parameter decreased much slower, being significantly different from the baseline from ~ 50 s after the injection and minimal at the end of the analysis interval. In contrast to EEG, EMG response was fully blocked in anesthetized animals (Figure 3D).
To validate the source of neural activation induced by cocaine, we examined electrophysiological effects of cocaine-methiodide--a peripherally acting cocaine’s analog. When used at an equimolar dose, cocaine-methiodide induced very similar changes in EEG and EMG activity to those induced by BBB-permeable cocaine-HCl (Figure 4). While both drugs induced very similar changes in EEG total power, the effects of cocaine-methiodide were less prolonged, with a significantly weaker decrease vs. regular cocaine at the last minute of analysis. Cocaine-methiodide also mimicked regular cocaine-HCl in its ability to increase EMG activity rapidly and strongly, but its effects were also somewhat shorter.
Figure 4. Mean changes in EEG (A) and EMG (B) total powers induced by iv administration of cocaine methiodide (0.67 mg/kg or 1.4 μM) shown together with those induced by regular cocaine-HCl at an equimolar dose (0.5 mg/kg).
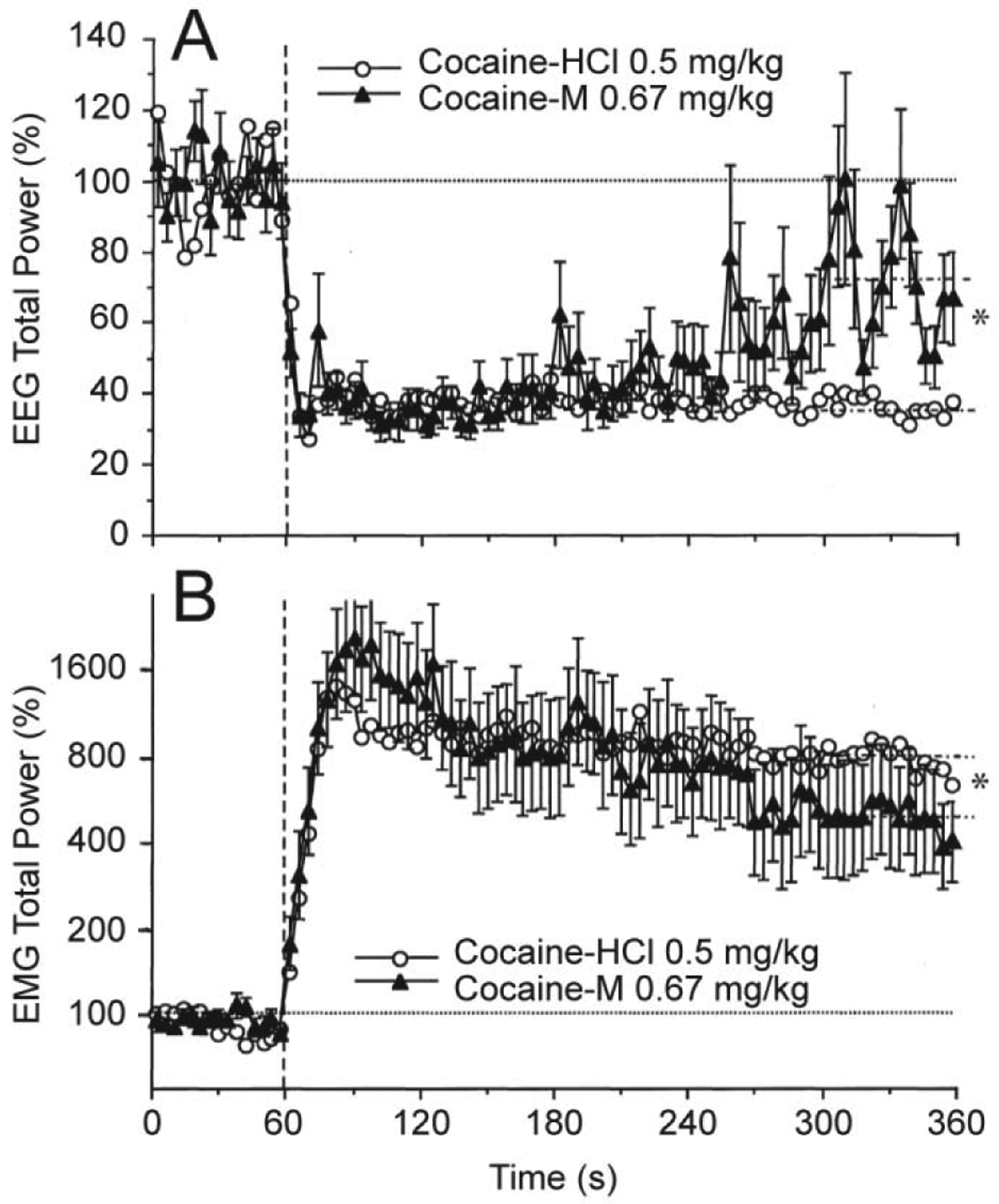
For clarity, standard errors are shown only for cocaine-methiodide. Vertical hatched lines (at 60 s) show the start of injections and horizontal hatched lines show basal values (=100%). Interrupted hatched lines at 300–360 s time interval in both graphs show mean values of EEG and EMG total powers for 5th min post-injection and asterisks denote significant between-group differences. Original data were reported in Kiyatkin and Smirnov, 2010 (free PMC article).
While changes in EEG activity are non-specific with respect to individual neurons and brain structures, we also used single-unit recordings to assess how neuronal activity of dorsal and ventral striatal neurons is affected by iv cocaine (Kiyatkin and Brown, 2007). The striatum, especially its ventral part or NAc is an important brain structure involved in sensorimotor integration and mechanisms of natural and drug reinforcement (Wise and Bozath, 1987; Mogenson et al., 1980; Di Chiara, 2002). Since robust changes in motor activity induced by iv cocaine greatly complicate single-unit recordings, rats in these experiments were pretreated with a mixture of DA-1 like and D-2 like DA antagonists (SCH 23390 and eticlopride at 1.0 mg/kg each), providing a full blockade of DA neurotransmission. This procedure drastically attenuated cocaine-induced motor responses and eliminated a possible involvement of DA in observed neuronal responses. As shown in Figure 5, cocaine at low, self-administering doses induced rapid, transient excitations of striatal neurons, with the response starting from ~ 7–8 s, peaking at ~20 s, and disappearing at ~32 s from the onset of 10-s cocaine injection. This effect was absent with control saline injections, fully blocked during urethane anesthesia and mimicked by cocaine-methiodide that induced equally rapid and even stronger excitations of striatal neurons. These drug-induced excitatory responses greatly resemble neuronal excitations induced in striatal neurons in response to mild sensory stimuli (Kiyatkin and Rebec, 1995, 1999). Rapid neuronal responses to iv cocaine were also found on ventral tegmental area (VTA) neurons in awake rats similarly treated with DA antagonists (Brown and Kiyatkin, 2008). In contrast to striatal cells that showed uniform excitations, cocaine had differential responses in long-spike, presumed DA neurons and short-spike non-DA neurons, but changes in activity of both cellular subgroups occurred with 5–10-s latencies, within the injection duration.
Figure 5. Original records (A) and mean values (B) of changes in impulse activity of striatal neurons induced by iv cocaine-HCl (0.5 mg/kg), cocaine-methiodide (0.67 mg/kg), and saline in awake, freely moving and urethane-anesthetized (U) rats.
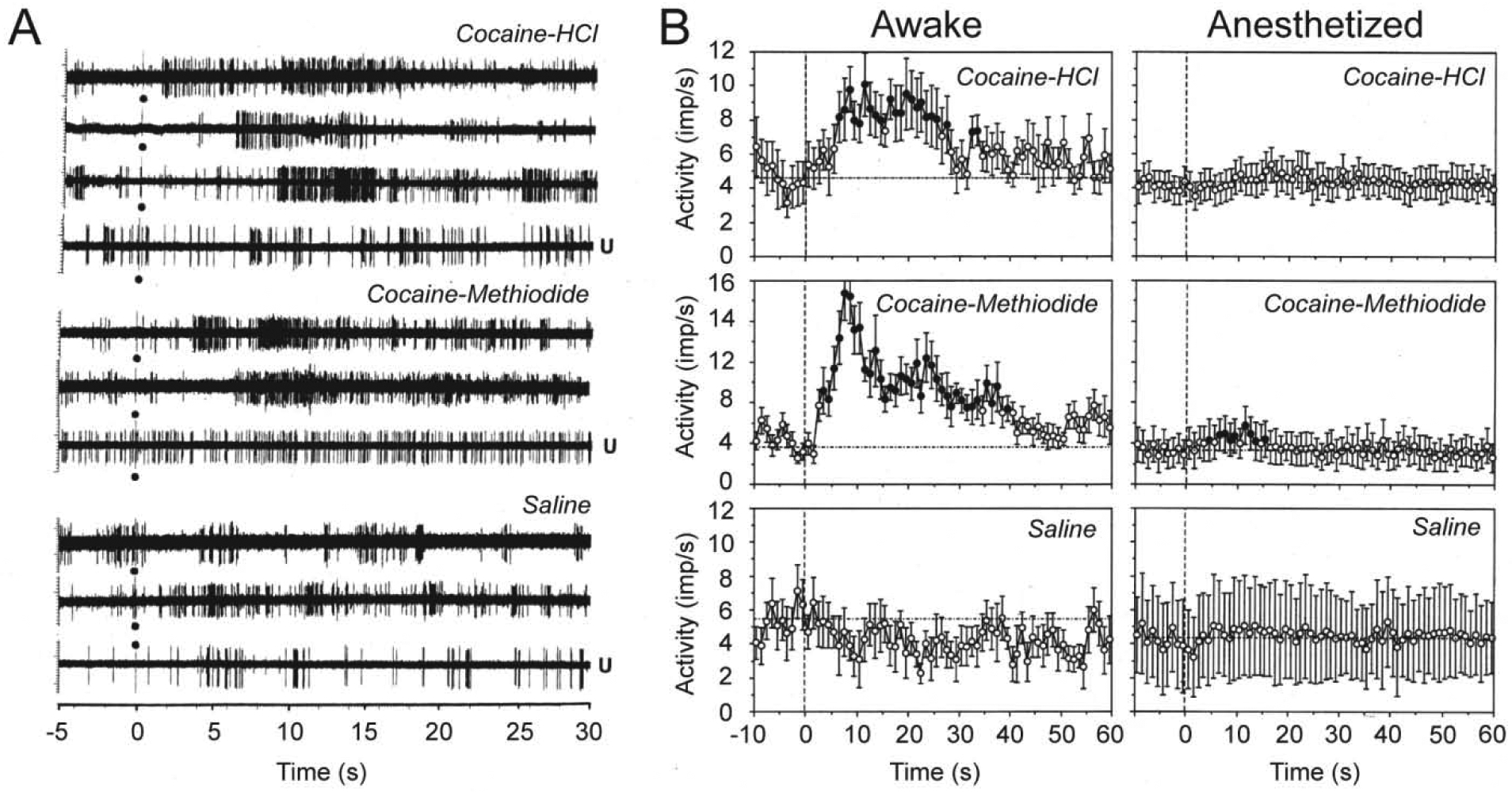
Black circle at 0 s shows the injection onset. Filled symbols show values significantly different (Fisher F-test, p<0.05) from baseline. Mean values are shown with 1-s time resolution. Original data for this picture were published in Kiyatkin and Brown, 2007.
Thus, our electrophysiological data suggest that iv cocaine at low, self-administering doses induces generalized neural activation that begins very rapidly, suggesting its neural triggering from the periphery. The initial components of cocaine-induced neural activation are similar to those induced by sensory stimuli and they are attenuated during general anesthesia. Despite the known effects of cocaine on DA system, full DA receptor blockade fails to affect this rapid neural activation. Finally, cocaine-methiodide that cannot cross the BBB, greatly mimics the immediate neural effects of cocaine, providing a strong evidence on its peripheral triggering.
Rapid changes in brain oxygen and glucose induced by iv cocaine
The brain’s metabolic activity essentially depends on proper delivery of oxygen and glucose from arterial blood, where their concentrations are higher than in the brain’s extracellular space. Both these substances are intensively consumed during metabolic activity and their utilization increased during functional neural activation (Siesjo, 1978; Sokoloff, 1999; Mergenthaler et al., 2013). Brain levels of oxygen and glucose directly depend on fluctuations in their levels in blood, and their blood and brain levels remain relatively stable under quiet resting conditions. Along with this basic gradient-dependent mechanism, brain entry of oxygen and glucose is modulated by neural activity via changes in the tone of cerebral vessels and fluctuations in local cerebral blood flow (Fox and Raichle, 1986; Fellows and Boutelle, 1993; Attwell et al., 2010; Lecrux and Hamel, 2011). Therefore, extracellular levels of oxygen and glucose reflect a balance between highly dynamic and opposing influences between their entry from arterial blood and their consumption in brain tissue.
Based on our previous electrophysiological studies that revealed that cocaine induces rapid neural activation, we assumed that that iv cocaine administration will also increase brain levels of oxygen and glucose. This was the case as shown by using enzyme-based glucose biosensors and oxygen sensors coupled with high-speed amperometry in awake, freely moving rats (Wakabayashi and Kiyatkin, 2015; Kiyatkin and Lenoir, 2012; Solis et al., 2018). These technologies allow second-scale measurement resolution that is critical for accurate assessment of rapid fluctuations of these substances in brain tissue. As shown in Figure 6, Iv cocaine delivered at a low, self-administering dose (1 mg/kg) significantly increased oxygen and glucose levels in the NAc, showing a bimodal response. Initially, both oxygen and glucose rapidly increased during the injection duration, but then they both showed a second elevation, which was stronger and more prolonged (A). Using high-speed resolution analysis, we observed a rapid and almost identical increase in glucose and oxygen that peaked ~30 s from the injection onset (B). Then both parameters slightly decreased but began to accelerate upwards again from ~60–90 s after the injection onset. As shown by time-dependent correlative analysis (C), the oxygen and glucose increases significantly correlated for the initial 20–25 s post-injection (r=0.454, p<0.05), suggesting a common trigger driving this synchronous response.
Figure 6. Changes in NAc oxygen and glucose induced by iv cocaine (1 mg/kg) delivered passively to awake, freely moving rats.
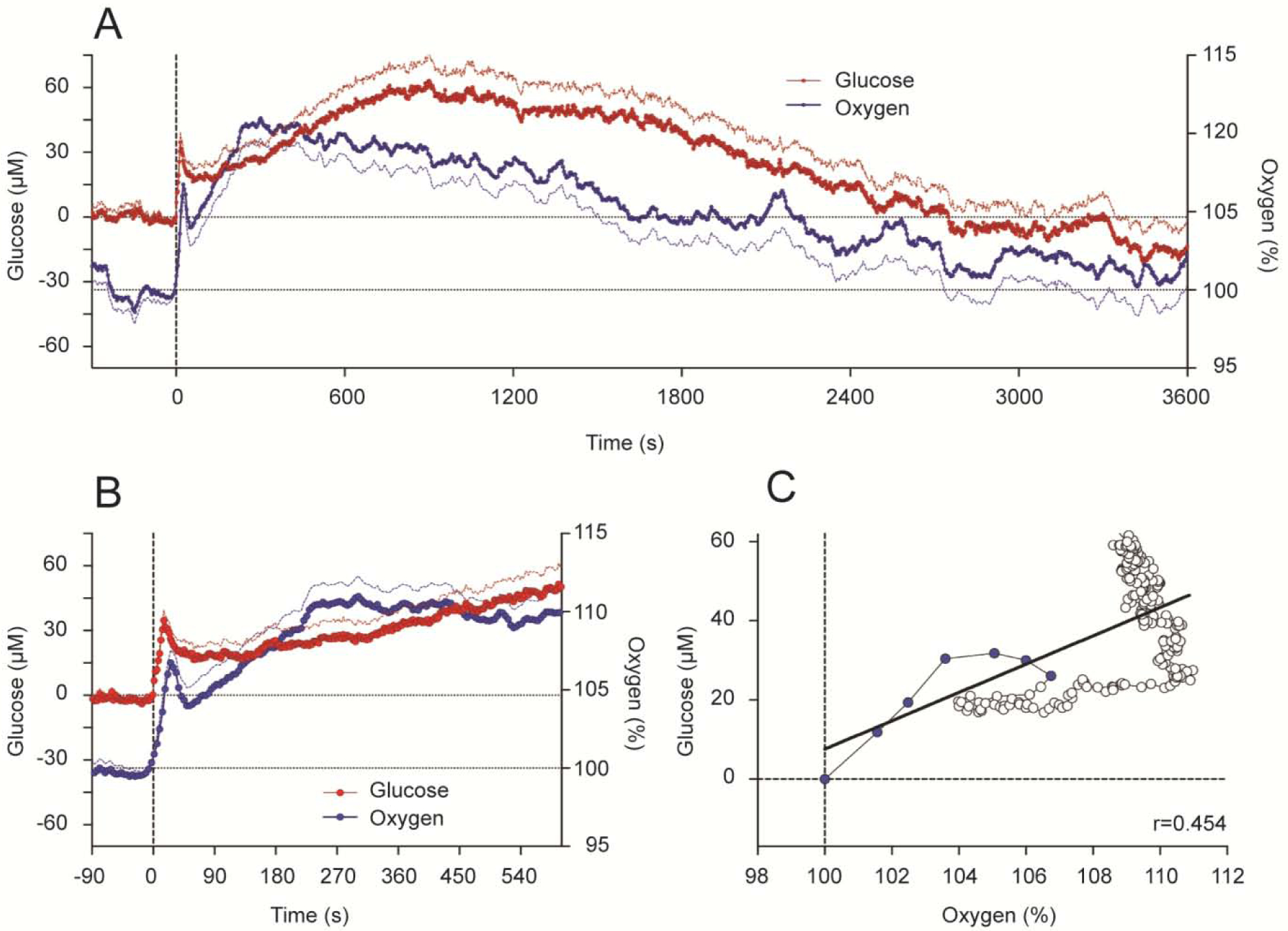
A shows mean (±SEM) changes for one hour after the injection. B shows changes for the initial time interval following the injection. C shows time-dependent correlative relationships between cocaine-induced changes in oxygen and glucose. Values to oxygen peak are shown as blue symbols.
Cocaine-induced neural activation is a possible trigger that drives these rapid synchronous changes in NAc glucose and oxygen. To verify this link, we compared cocaine-induced changes in NAc glucose and oxygen with changes in EEG gamma-activity, the high-frequency component of EEG spectrum (29–50 Hz) that is phasically increased by sensory stimuli and iv cocaine (Kiyatkin and Smirnov, 2010). As shown in Figure 7, both parameters phasically increased following iv cocaine administration, but changes in gamma activity that reflect neural activation clearly preceded changes in both glucose (A) and oxygen (C).
Figure 7. Relationships between cocaine-induced changes in NAc glucose and (A) EEG activation as assessed by changes in gamma power and (B) NAc glutamate levels.
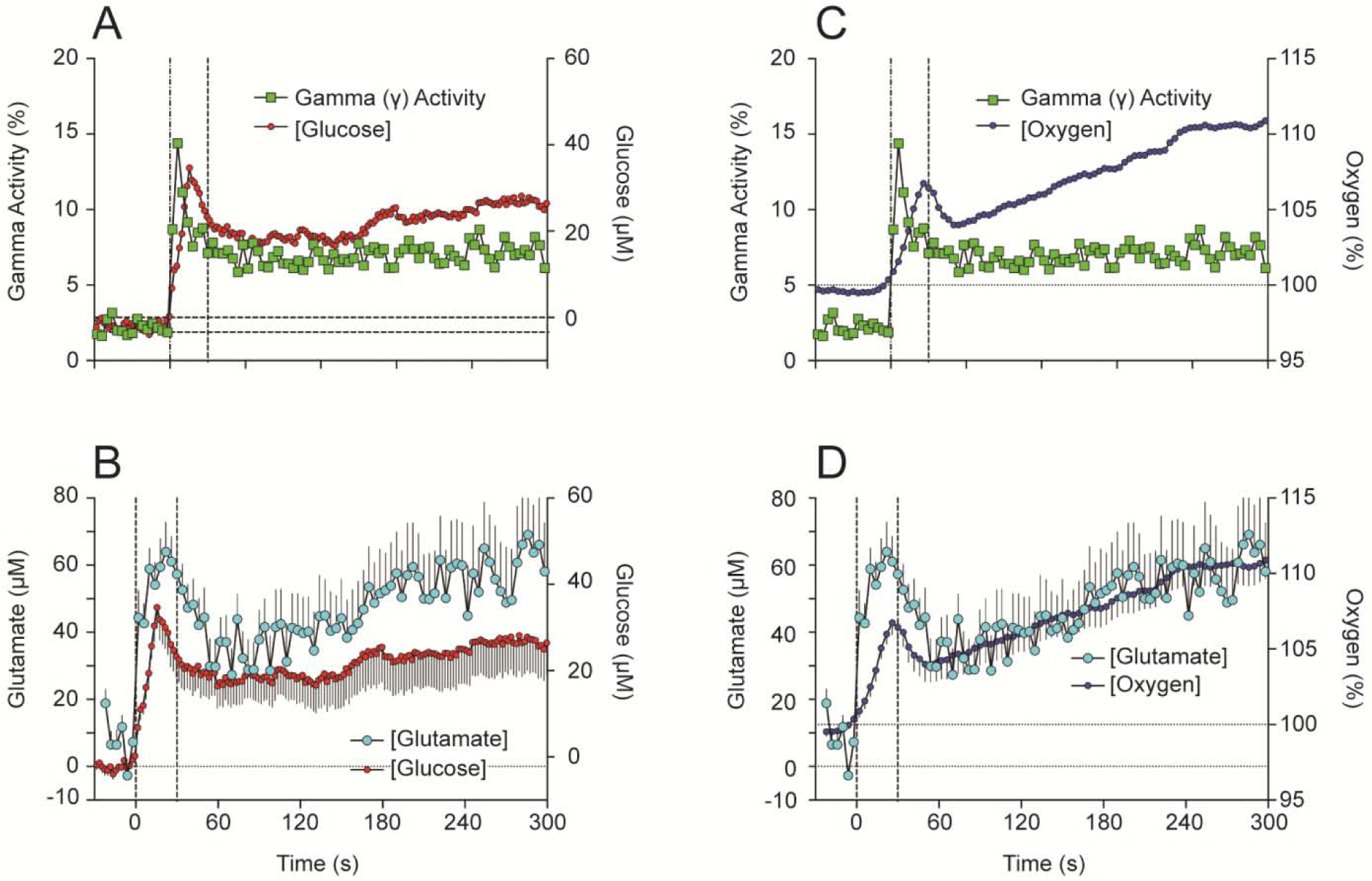
Relationships between changes in NAc oxygen and (C) EEG activation as assessed by changes in gamma power and (D) NAc glutamate levels. Drug-induced changes in glutamate levels were assessed by using Pinnacle glutamate-oxidase biosensors; these data were described in detail in Wakabayashi et al., 2015 and Wakabayashi and Kiyatkin, 2015.
Since accumbal neurons receive dense glutamate inputs and are very sensitive to iontophoretic applications of glutamate (Kiyatkin and Rebec, 1999), both neuronal activation and subsequent changes in NAc oxygen and glucose can be caused by cocaine-induced release of this neurotransmitter. This assumption was verified by comparing changes in NAc oxygen and glucose with changes in glutamate as assessed in this brain structure by enzyme-based glutamate biosensors (Wakabayashi and Kiyatkin, 2012, 2014, 2015). Changes in NAc glutamate also showed very rapid increases, with a strong phasic change within the injection duration followed by a more prolonged tonic increase (Figure 7B and D). Importantly, increases in glutamate levels occurred more rapidly than increases in both glucose and oxygen. Therefore, similar to excitations of accumbal neurons elicited by natural arousing stimuli, cocaine-induced excitation of these cells triggers the enhanced entry of oxygen and glucose into brain tissue due to local cerebral vasodilation.
A critical role of peripheral actions of cocaine in mediating its acute effects on brain glucose and oxygen was demonstrated by using cocaine-methiodide (Kiyatkin and Lenoir, 2012; Wakabayashi and Kiyatkin 2015; Solis et al., 2018). As shown in Figure 8A, NAc glucose changes induced by cocaine-HCl and cocaine-methiodide drastically differed when data were analyzed with slow (1-min) time resolution. In contrast to the prolonged biphasic effect of regular cocaine, its methiodide analog induced only a weaker and more transient response. However, the immediate changes in brain glucose were virtually identical, when the data were analyzed with rapid (4-s) time resolution (Figure 8B). Therefore, it appears that the immediate increases in brain glucose induced by iv cocaine are triggered via its action in the periphery, but the drug’s action in the CNS appears to be responsible for subsequent, larger and more prolonged glucose increases.
Figure 8. Slow (A) and rapid (B) changes in NAc glucose induced by iv cocaine-HCl and cocaine-methiodide in freely moving rats.
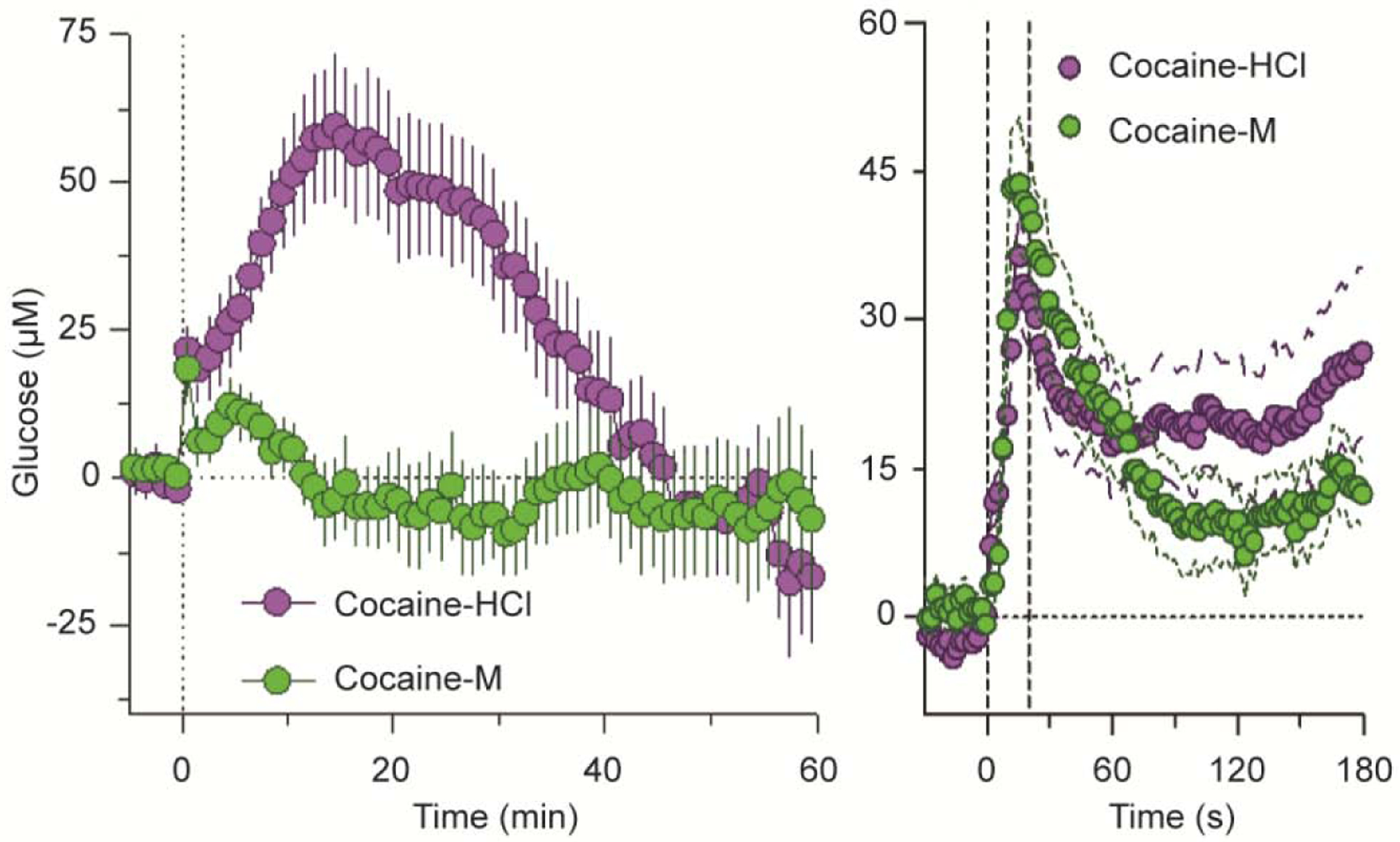
n=number of averaged responses. Original data were reported in Wakabayashi and Kiyatkin, 2015 (open access article).
Similar results were obtained with NAc oxygen when comparing the effects of the two cocaine analogs (Solis et al., 2018). Both drugs induced rapid increases in NAc oxygen (Figure 9A and B). Despite being larger in magnitude and longer in duration, these effects were similar to those induced by a short auditory stimulus that induced very rapid but transient oxygen increase (C). The initial, most rapid component of the oxygen increase was virtually identical for both drugs, but the oxygen increase elicited by cocaine-methiodide was monophasic while the increase elicited by cocaine-HCl was biphasic, showing a second, much slower acceleration from ~ 45–50 s following injection onset (D). Importantly, this difference was evident only with rapid-time course analyses. As shown by time-dependent correlative analysis (E), the initial effects of both cocaine analogs on NAc oxygen were virtually identical, showing high correlation (r=0.988, p<0.0001). However, oxygen concentration curves diverged from ~35 s after the injection onset, possibly reflecting the timing when cocaine reaches centrally located neural substrates and begins to directly affect neural activity.
Figure 9. Mean (±SEM) changes in NAc oxygen induced by cocaine-HCl (A), cocaine-methiodide (B) and auditory stimulus (C) in awake, freely moving rats.
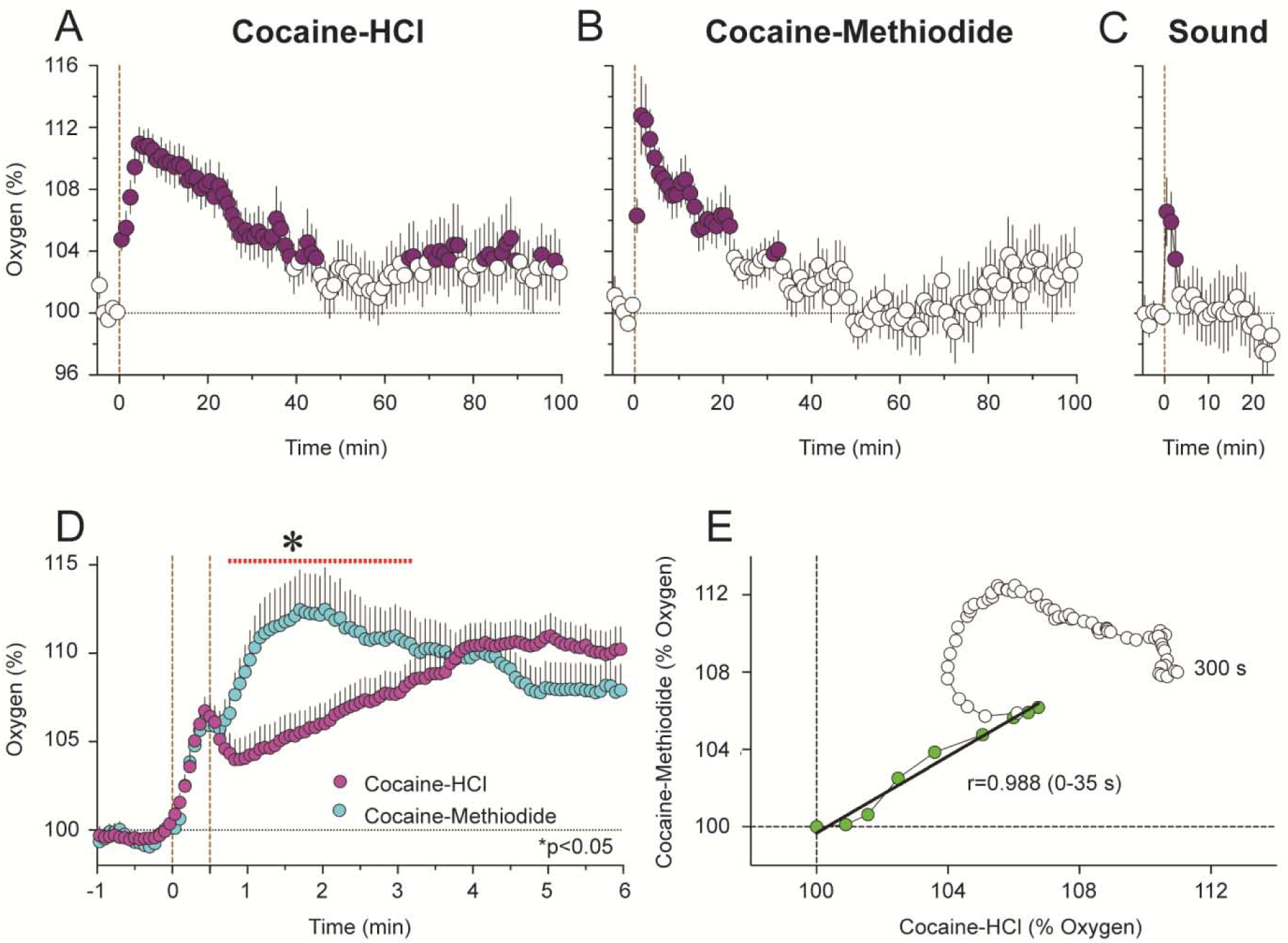
Filled symbols show values significantly different from pre-injection baseline. D shows rapid time-course analysis of immediate effects of two cocaine analogs. The red line with an asterisk shows between-group differences. E shows time-dependent correlation between NAc oxygen changes induced by two cocaine analogs.
While the immediate effects of both cocaine analogs on NAc oxygen and glucose were surprisingly similar, there were important differences in the delayed effects of these drugs and in their behavioral outcomes. In contrast to cocaine-HCl, cocaine-methiodide does not induce notable locomotor activation (Brown and Kiyatkin, 2006) and its effects on NAc glutamate are much weaker, showing within-session tolerance (Wakabayashi and Kiyatkin 2012). These differences obviously arise from the direct action of cocaine in the brain, whereas such action appears to be absent with cocaine-methiodide.
Thus, our electrochemical data suggest that iv cocaine at low, self-administering doses rapidly increases intra-brain entry of oxygen and glucose and these effects are tightly related to neural activation induced by this drug. While rapid, these neurochemical changes followed increases in neuronal activity and glutamate release. The rapidity of these increases and their similarity to changes induced by BBB-impermeable cocaine-methiodide strengthen the evidence of the critical role of peripheral neural substrates in mediating rapid neural effects of cocaine.
Rapid and slow changes in DA induced by iv cocaine: interplay of DA release and DA uptake
Classic data obtained in vitro revealed that cocaine fails to affect DA release but potently inhibits DA uptake (Heikkila et al., 1975; Lacey et al., 1990). Therefore, cocaine-induced increases in DA revealed in limbic and cortical projection sites of DA neurons using in vivo microdialysis were explained as the consequence of inhibiting action of cocaine on DA reuptake. When tested in freely moving rats, iv cocaine at low, behaviorally relevant doses (0.5–1.0 mg/kg) modestly increased NAc DA levels (Pontieri et al., 1995). Although the maximal increase was detected in the first post-injection dialysate sample, suggesting a rapid response, the low temporal resolution (10 min) and significant damage of brain tissue by large-size dialysis probes made it impossible to know the precise DA dynamics following drug injection. More detailed picture of the initial DA response to iv cocaine was obtained by using smaller dialysis probes and higher, 2-min time resolution in awake, freely moving rats (Minogianis et al., 2019). In this case, changes in striatal DA levels induced by iv cocaine at 2 mg/kg were dependent on the rate of cocaine infusion, increasing at the second and third minutes and peaking at the third and six minutes following ultra-fast (5-s) and slow (45-s) cocaine injections.
The use of fast-cyclic voltammetry, an electrochemical technique that employed very miniature carbon fiber sensors and allowed extra-fast (100 ms) temporal resolution, revealed that changes in extrasynaptic DA occur much more rapidly, questioning the involvement of DA uptake in their mediation. DA levels assessed in the NAc shell in freely moving rats began to increase at ~12 s and peaked at 25 s after the start of 6-s cocaine 3 mg/kg injections (Aragona et al., 2008). Similarly short, ~25-s latency of NAc DA increase was found in another study using freely moving rat and a similar, 3 mg/kg cocaine injection (Cheer et al., 2007). Consistent with rapid increases in other neural effects of iv cocaine, DA increases detected by high-resolution electrochemical techniques rapidly peaked and then slowly decreased toward baseline, suggesting impulse-dependent DA release but not DA uptake as their underlying cause. Although fast-cyclic voltammetry provides excellent measurement sensitivity and selectivity, and it has exceptional time resolution, reliable quantifications of drug-induced DA responses could be conducted for a limited time interval (~90 s; Heien et al., 2005). This restricts our knowledge of cocaine-induced DA dynamics during prolonged, behaviorally relevant time intervals.
It appears that electrochemical data, which suggest rapid DA release due to DA cell activation, contradict to single-unit data obtained on presumed DA ventral tegmental area (VTA) in anesthetized rats, most of which were rapidly (10–20 s latency) but modestly (60–80% of baseline) inhibited by iv cocaine at behaviorally relevant doses (Einhorn et al., 1988; Pitts and Marwah, 1987, 1988). These inhibitions were blocked by D2-like DA receptor antagonists, suggesting inhibition of DA cell firing due to stimulation of DA autoreceptors resulting from cocaine-induced increases in DA levels. However, these partial inhibitions were seen only in a portion of long-spike VTA neurons and were absent in presumed DA neurons in substantia nigra, pars compacts (Pitts and Marwah, 1987). In contrast, data obtained in long-spike presumed DA VTA neurons in awake rats revealed that more than half of these cells (10/16) showed short-latency excitations, which also have relatively small magnitude and duration (Brown and Kiyatkin, 2008; Figure 10). Although a portion of these cells (6/16) did not show evident increases in discharge rate, some of these “insensitive” cells showed bursting, resulting in increased variability of discharge rate in entire subgroup of tested cells (standard deviation or SD in Figure 10B). However, in contrast to previous studies, these neuronal recordings were conducted in rats during full DA receptor blockade (SCH 23390 + eticlopride at 0.2 mg/kg each) and cocaine was used at a relatively low dose (0.25 mg/kg) at the lower border of self-administration range. Moreover, neuronal activity in this study was assessed for a couple minutes post-injection, making it impossible to observe tonic changes in activity of these cells in intact animals. It is not fully clear why this response pattern differs from that observed in anesthetized animals. Possibly, the initial excitatory effect of cocaine is blocked or strongly attenuated during deep chloral hydrate or urethane anesthesia used in previous studies. However, neuronal excitations induced by cocaine were also revealed in histochemically identified VTA DA neurons in urethane-anesthetized rats (Mejias-Aponte et al., 2015). These responses were also relatively small in amplitude, rapid but more delayed than in awake rats, and they were seen only in a portion of these cells.
Figure 10. Changes in impulse activity of long-spike, presumed dopamine VTA neurons induced by iv cocaine at low reinforcing dose (0.25 mg/kg, 5–15 s) in freely moving rats.
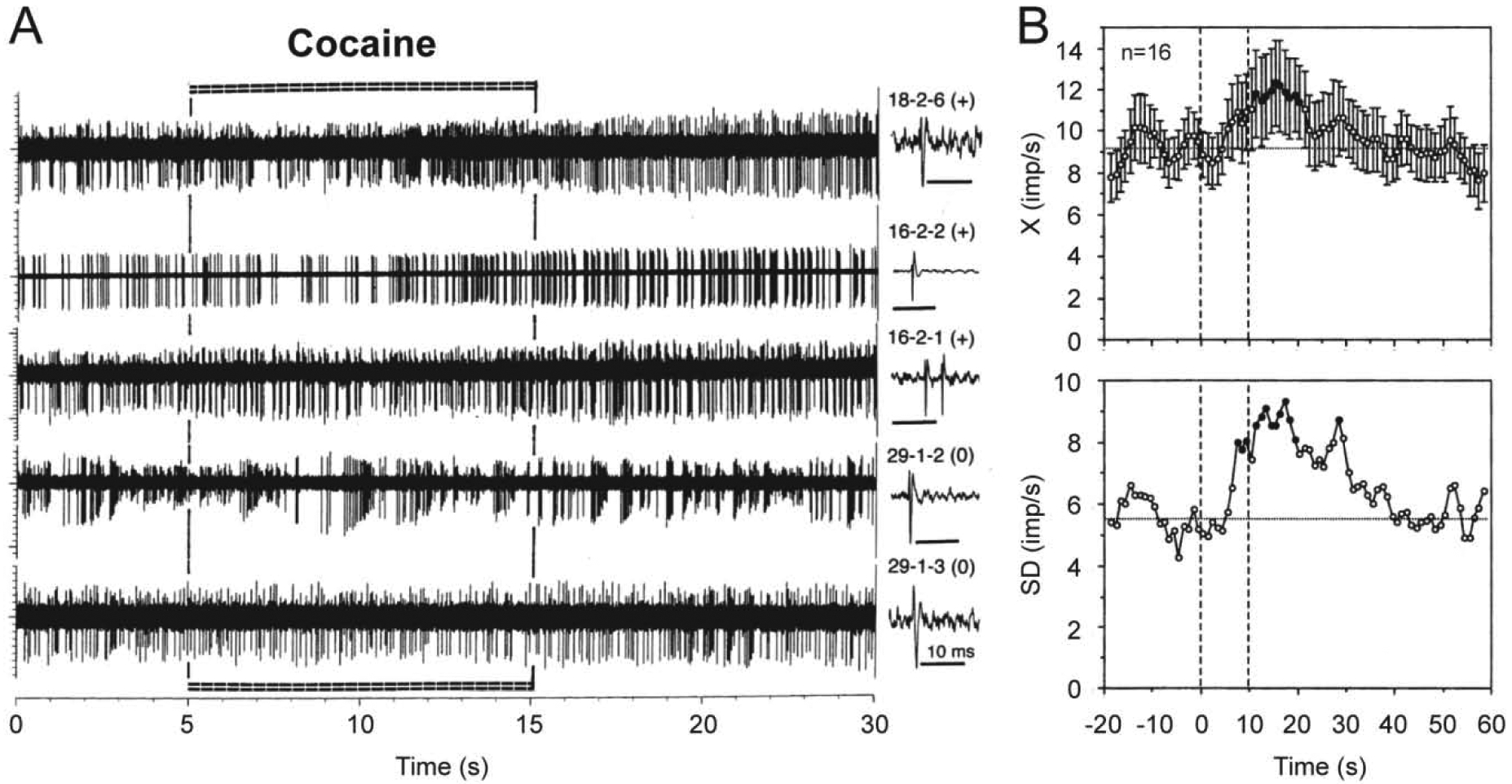
A shows original records of neuronal responses for 20 s following cocaine injection and B shows mean (±SEM) changes in discharge rate (top; X, imp/s) and standard deviation of rate (bottom; SD, imp/s) for 60 s post-injection. Each original record was obtained from a different cell, whose numbers and single spikes are shown on the right panel of A. Note that three top traces show neuronal excitations (+) and the two lower traces show no changes in rate (0) but clear bursting. Mean data are shown for the entire group of tested cells, but 10 of 16 cells show excitations and 6 rats show no changes in rate of impulse activity. Filled symbols show values significantly different from baseline. Two vertical hatched lines on B at 0 and 10 s show the duration of cocaine injection. Original data were reported in Brown and Kiyatkin, 2008.
Electrochemical monitoring of DA responses induced by electrical stimulation of VTA DA cells was used to examine the time-course of DA uptake inhibition induced by cocaine (Mateo et al., 2004; Espana et al., 2008). These studies were conducted in urethane-anesthetized rats and changes in the descending curve of electrically induced DA increase (apparent Km) were used as a presumed measure of DA uptake. Based on rapid increases in this measure (5-s latency and 60-s peak after an ultra-fast 2-s injection at 0.75–3.0 mg/kg), it was suggested that iv cocaine induces ultra-fast inhibition of DA uptake. Similarly rapid changes in electrically stimulated DA release were found in anesthetized mice, but these studies revealed that reinforcing doses of cocaine produce only very modest occupancies/blockade at DA transporter (2% of maximum), suggesting dissociation between DA transporter occupancy and changes in DA uptake (Brodnik et al., 2017).
While cocaine-HCl had clear effects on electrically evoked DA increases at each dose tested (0.75–3.0 mg/kg), no changes in this parameter were found following administration of BBB-impermeable cocaine-methiodide (Espana et al., 2008). However, in freely moving rats, both cocaine analogs induced similar increases in DA transients in the NAc, suggesting increased DA release (Wang et al., 2013). These latter data were obtained in rats previously trained to self-administer cocaine-HCl and no control data on the effects of cocaine-methiodide in drug-intact freely moving rats were provided in this study.
It is not fully clear why cocaine begins to affect the descending part of DA overflow with so short latencies. Maybe it reflects the change not induced by cocaine itself, but a combination of stimulation-induced and cocaine-induced DA release; the latter change (see above) was not recognized at that time and not considered in calculations of changes in DA dynamics. To assess the time-course of cocaine-induced DA uptake inhibition, we developed another approach that employed fast-scan cyclic voltammetry to evaluate DA clearance after its microiontophoretic delivery in brain tissue near the DA detecting electrode (Kiyatkin et al., 2000). To accomplish this task, we prepared combined multi-channel glass electrodes, which allowed DA detection (30 μm carbon fiber), precise current-regulated DA delivery, and its retention to counteract spontaneous DA diffusion into brain tissue. The recording procedure had high selectivity, second-scale time resolution, and it allowed to assess how cocaine at a low, behaviorally relevant dose affects uptake of iontophoretically delivered DA in awake, freely moving rats. Using this approach, we found that iv cocaine at a 1 mg/kg dose did not affect the amplitude and time to peak of the DA response, but significantly increases its decay time, suggesting inhibition of DA uptake. However, this change occurred at 2 min, peaked at 6–7 min and disappeared at 16–18 min, suggesting that cocaine-induced inhibition of DA uptake is much slower than cocaine-induced increases in DA release. This effect at behaviorally relevant doses of cocaine was also relatively weak (30–40% above baseline), consistent with weak inhibiting effects of cocaine on DA uptake shown in analytical studies (Brodnik et al., 2017). Relatively slow dynamics of cocaine-induced DA uptake was later demonstrated by analyzing changes in impulse activity of striatal neurons induced by iontophoretic DA (Wakazono and Kiyatkin, 2008). In this case, inhibiting action on DA reuptake was assessed by changes in the strength and aftereffect of DA-induced neuronal inhibition. Despite large neuronal sample, changes in these parameters following 1.0 mg/kg cocaine injection were weak and they occurred within several minutes and peaked at 6–10 min post-injection, suggesting that this direct central effect of cocaine is much slower and weaker than its effect on DA release.
Hence, despite the existing controversies in data and essential limitations of each technique, it appears that changes in DA levels induced by cocaine in awake animals results from the interaction of two independent drug actions. The first action is triggered from the periphery and it may be responsible for rapid DA release, which that appears within seconds following drug injection. The second action depends on direct interaction of cocaine with neuronal DA transporters, which appears with certain latencies and is more prolonged, being dependent on slower and more prolonged changes in cocaine levels in brain tissue. While relatively weak at behaviorally relevant doses of cocaine, this action could be responsible for potentiation of DA response due to inhibition of reuptake of previously release DA.
Concluding Remarks
The use of high-resolution electrophysiological, neurochemical and physiological techniques revealed that iv cocaine at low, behaviorally relevant doses induces rapid neural effects that appear within seconds after the onset of drug injection. While it is quite difficult and possibly impossible to measure real-time fluctuations in cocaine concentrations in blood and brain’s extracellular space, the time-course of these neural effects better correlates with rapid and robust increases in blood cocaine levels preceding more slowly appearing and weaker changes in brain cocaine levels. This rapidity of neural effects appears to be inconsistent with the time that is necessary for cocaine to reach cerebral vessels, cross the BBB, passively diffuse to brain receptor site and interact with them. Moreover, the patterns of rapid neural effects of iv cocaine share basic similarities with those induced by arousing stimuli, suggesting a possible common underlying mechanism involving activation of peripherally located neural substrates and rapid neural transmission. Many of these rapid neural effects of iv cocaine are also mimicked by BBB-impermeable cocaine-methiodide, providing additional support for the critical role of cocaine’s interaction with peripherally located neural substrates. However, the exact nature of these substrates and pathways transmitting this cocaine-induced neural signal from the periphery to the CNS remain unclear. Since blood vessels are densely innervated by afferents of sensory nerves, cocaine travelling via these vessels at high concentrations can stimulate multiple ionic channels (i.e., voltage-gated Na+, K+, Ca2+, TRP channels) densely expressed on these afferents. Therefore, these receptive sites (interoceptors), in addition to their physiological role in detecting deviations of basic homeostatic parameters, can be over-activated by cocaine, playing a critical role in triggering ascending excitatory drive to the CNS using neural pathways that normally transmit the effects of visceral somatosensory stimuli. While analytical research is necessary to determine the nature of peripheral neural substrates activated by cocaine, it is a difficult challenge to undertake because of the inability to pharmacologically block multiple neural targets and the complexity of neural pathways, which transmit visceral signals to the CNS.
However, after rapid, strong and transient rise in blood levels following iv administration, cocaine’s levels in brain tissue begin to increase and interact with centrally located neural substrates, thus modulating the activity of central neurons and contributing to physiological and behavioral effects of this drug. Considering cocaine’s dynamics in brain tissue, these direct effects should occur with definite onset latencies and they are relatively weak but more prolonged. Therefore, the observed effects of cocaine reflect a combination of two independent mechanisms: a rapid, transient action of peripheral neural substrates followed by more prolonged direct action on centrally located neural substrates. The co-existence in the same drug of two independent and timely distinct actions with their subsequent interaction in the CNS could explain consistent changes in physiological and behavioral effects of cocaine following their repeated use, playing a role in the development of drug-seeking and drug-taking behavior.
Highlights:
Iv cocaine induces rapid neural effects revealed by high-resolution physiological techniques in freely moving animals;
The rapidity of these neural effects cannot be explained by cocaine’s action on brain receptive substrates;
Rapid effects of cocaine are mimicked by peripherally acting cocaine-methiodide and resistant to dopamine receptor blockade;
The initial sites of cocaine action are receptive sites on afferents of visceral sensory nerves innervating blood vessels;
This action creates neural signal to CNS, resulting in neural activation and rapid changes in physiological parameters.
Acknowledgements
The study was supported by the Intramural Research Program of the NIH, NIDA. I want to thank all post-doc and post-bac fellows (Anum Afzal; P. Leon Brown, PhD; Magalie Lenoir, PhD; Michael Smirnov, PhD; Ernesto Solis Jr., PhD; Ken T. Wakabayashi, PhD), who were involved in experiments described in this review paper and in discussions on the functional significance of the data obtained in these experiments. I also want to specially thank David Perekopskiy and Shruthi Thomas for editing the final text and providing valuable suggestions on its improvement. Special thanks to Carlos Curay for final editing of figures.
Abbreviations:
- BBB
blood-brain barrier
- EEG
electroencephalography, electroencephalogram
- EMG
electromyography, electromyogram
- Iv
intravenous
- NAc
nucleus accumbens
- SD
standard deviation
- VTA
ventral tegmental area of midbrain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The Author reports no conflict of interest
References
- Aragona BJ, Cleaveland NA, Stuber GD, Day JJ, Carelli RM, and Wightman M (2008). Preferential enhancement of dopamine transmission within the nucleus accumbens shell by cocaine is attributed to a direct increase in phasic dopamine release events. J. Neurosci 28, 8821–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, and Newman EA (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradberry CW, Nobiletti JB, Elsworth JD, Murphy B, Jatlow P, and Roth RH (1993). Cocaine and cocaethylene: Microdialysis comparison of brain drug levels and effects on dopamine and serotonic. J. Neurochem 60, 1429–1435. [DOI] [PubMed] [Google Scholar]
- Brodnik ZD, Ferris MJ, Jones SR, and Espana RA (2017). Reinforcing doses of intravenous cocaine produce only modest dopamine uptake inhibition. ACS Chem. Neurosci 8, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PL, and Kiyatkin EA (2006). The role of peripheral Na(+) channels in triggering the central excitatory effects of intravenous cocaine. Eur. J. Neurosci 24, 1182–1192. [DOI] [PubMed] [Google Scholar]
- Brown PL, and Kiyatkin EA (2008). Sensory effects of intravenous cocaine on dopamine and non-dopamine ventral tegmental area neurons. Brain Res. 1218, 230–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G (2006). Rhythms of the brain. Oxford: Oxford University Press. [Google Scholar]
- Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, Phillips PE, and Wightman RM (2007). Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J. Neurosci 27, 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Lin CH, Lin PL, and Tsai MC (2006). Cocaine elicits action potential burst in a central snail neuron: the role of delayed rectifying K+ channels. Neuroscience 138, 257–280. [DOI] [PubMed] [Google Scholar]
- Crombag HS, Ferrario CR, and Robinson TE (2008). The rate of intravenous cocaine or amphetamine delivery does not influence drug-taking and drug-seeking behavior in rats. Pharmacol. Biochem. Behav 90, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson LW, Rodak DJ, Kuhn FE, Wahlstrom SK, Tessel RE, Visner MS, Schaer GL, and Gillis RA (1999). Cocaine-induced cardiovascular effects: lack of evidence for a central nervous system site of action based on hemodynamic studies with cocaine methiodide. J. Cardiovasc. Pharmacol 33, 36–42. [DOI] [PubMed] [Google Scholar]
- Di Chiara G (2002). Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav. Brain Res 137, 75–114. [DOI] [PubMed] [Google Scholar]
- Einhorn LC, Johansen PA, and White FJ (1988). Electrophysiological effects of cocaine in the mesoaccumbrns dopamine system: Studies in the verntral tegmental area. J. Neurosci 8, 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espana RA, Roberts DC, and Jones SR (2008). Short-acting cocaine and long-acting GBR-12909 both elicit rapid dopamine uptake inhibition following intravenous delivery. Neuroscience 155, 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SM, Cone EJ, and Henningfiled JE (1996). Arterial and venous cocaine plasma concentrations in hmans: relaionships to the route of administration, cardio-vascular effects and subjective effects. J. Pharmacol. Exp. Ther 279, 1335–1356. [PubMed] [Google Scholar]
- Fellows LK, and Boutelle MG (1993). Rapid changes in extracellular glucose levels and blood flow in the striatum of the freely moving rat. Brain Res. 604, 225–231. [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Logan J, Gatley SJ, Pappas N, King P, Ding Y-S, and Wang G-J (1998). Measuring dopamine transporter occupancy by cocaine in vitro: radiotracer considerations. Synapse 28, 111–116. [DOI] [PubMed] [Google Scholar]
- Fox PT, and Raichle ME (1986). Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc. Natl. Acad. Sci. USA 83, 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber GJ, and Wise RA (1989). Pharmacological regulation of intravenous cocaine and heroin self-administration in rats: A variable dose paradigm. Pharmacol. Biochem. Behav 32, 527–531. [DOI] [PubMed] [Google Scholar]
- Heien MLA, Khan AS, Ariansen JL, Cheer JF, Phillips PEM, Wassum KM, and Wightman RM (2004). Real-time measurements of dopamine fluctuations after cocaine in the brain of behaving rats. Proc. Natl. Acad. Sci. USA 102, 10023–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila RE, Orlanski H, and Cohen G (1975). Studies on the distinction between uptake inhibition and release of [3H]-dopamine in rat brain tissue slices. Biochem. Pharmacol 24, 847–852. [DOI] [PubMed] [Google Scholar]
- Hemby SE, Jones GH, Hubert GW, Neill DB, and Justice JB (1994). Assessment of the relative contribution of peripheral and central components in cocaine place conditioning. Pharmacol. Biochem. Behav 47, 973–979. [DOI] [PubMed] [Google Scholar]
- Kiritsy-Roy JA, Halter JB, Gordon SM, Smith MJ, and Terry LC (1990). Role of the central nervous system in hemodynamic and sympathoadrenal responses to cocaine in rats. J. Pharmacol. Exp. Ther 255, 154–160. [PubMed] [Google Scholar]
- Kiyatkin EA, and Brown PL (2007). Iv cocaine induces rapid, transient excitation of striatal neurons via itys action on peripheral neural elements: single-unit, iontophoretic study in awake and anesthetized rats. Neuroscience 148, 978–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Kiyatkin DE, and Rebec GV (2000). Phasic inhibition of dopamine uptake in nucleus accumberns induced by intravenous cocaine in freely behaving rats. Neuroscience 98, 729–741. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, and Lenoir M (2012). Rapid fluctuations in extracellular brain glucose levels induced by natural arousing stimuli and intravenous cocaine: fueling the brain during neural activation. J. Neurophysiol 108, 1669–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, and Rebec GV (1996). Dopaminergic modulation of glutamate-induced excitations of neurons in the neostriatum and nucleus accumbens of awake, unrestrained rats. J. Neurophysiol 75, 142–153. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, and Rebec GV (1999). Modulation of striatal neuronal activity by glutamate and GABA: iontophoresis in awake, unrestrained rats. Brain Res. 822, 88–106. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, and Rebec GV (2000). Dopamine-independent action of cocaine on striatal and accumbal neurons. Eur. J. Neurosci 12, 1789–1800. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, and Smirnov MS (2010). Rapid EEG desynchronization and EMG activation induced by intravenous cocaine in freely moving rats: a peripheral, nondopamine neural triggering. Am. J. Physiol. Regul. Integr. Comp. Physiol 298, R285–R300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuepfer MM, and Branch CA (1992). Cardiovascular responses to cocaine are initially mediated by the central nervous system in rats. J. Pharmacol. Exp. Ther, 263, 734–741. [PubMed] [Google Scholar]
- Kobayashi T, Nishizawa D, Iwamura T, and Ikeda K (2007). Inhibition by cocaine of G protein-activated inwardly rectifying K+ channels expressed in Xenopus oocytes. Toxicol. In Vitro 21, 656–664. [DOI] [PubMed] [Google Scholar]
- Koulchisky S, De Backer B, Quertemont E, Charlier C, and Seutin V (2012). Differential effects of cocaine on dopamine neuron firing in awake and anesthetized rats. Neuropgychopharmacology 37, 1559–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhar MJ, Ritz MC, and Boja JM (1991). The dopamine hypothesis of the reinforcing properties of cocaine. Trends Neurosci. 14, 299–302. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, and North RA (1990). Actions of cocaine on rat dopaminergic neurons. Br. J. Pharmacol 99, 731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrux C, and Hamel E (2011). The neurovascular unit in brain function and disease. Acta Physiol. (Oxf) 203, 47–59. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee CH, and Oh U (2005). Painful channels in sensory neurons. Mol. Cells 20, 315–324. [PubMed] [Google Scholar]
- Mandt BH, Johnston NL, Zahniser NR, Allen RM (2012). Acquisition of cocaine self-administration in male Sprague-Dawley rats: effects of cocaine dose but not initial locomotor response to cocaine. Psychopharmacology 219, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo Y, Budygin EA, Morgan D, Roberts DCS, and Jones SR (2004). Fast onset of dopamine uptake inhibition induced by intravenous cocaine. Eur. J. Neurosci 20, 2838–2842. [DOI] [PubMed] [Google Scholar]
- Mejias-Aponte CA, Ye C, Bonci A, Kiyatkin EA, and Morales M (2015). A subpopulation of histochemically-identified ventral tegmental area dopamine neurons is excited by intravenous cocaine. J. Neurosci 35, 1965–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis M, Goder R, Habler HJ, and Janig W (1994). Properties of afferent nerve fibers supplying the saphenous vein in the rat. J. Physiol 474, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michenfelder J (1988). Anesthesia and the Brain: Clinical, Functional and Vascular Correlates. New York: Churchill Livingstone. [Google Scholar]
- Minogianis EA, Levesque D, and Samaha AN (2013). The speed of cocaine delivery determines the subsequent motivation to self-administer the drug. Neuropsychopharmacology 38, 2644–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogianis EA, Shams WM, Mabrouk OS, Wong JT, Brake WG, Kennedy RT, du Soulch P, and Samaha AN (2019). Varying the rate of intravenous cocaine infusion influences the temporal dynamics of both drug and dopamine concentrations in the striatum. Eur. J. Neurosci 50, 2054–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel G, and Meisel A (2013). Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, and Yim CY (1980). From motivation to action: functional interface between the limbic system and the motor system. Prog. Neurobiol 14, 69–97. [DOI] [PubMed] [Google Scholar]
- O’Regan RG, and Majcherczyk S (1982). Role of peripheral chemoreceptors and central chemosensitivity in the regulation of respiration and circulation. J. Exp. Biol 100, 23–40. [DOI] [PubMed] [Google Scholar]
- Pan HT, Menacherry S, and Hustice JB (1991). Differences in the pharmacokinetics of cocaine in naive and cocaine-experienced rats. J. Neurochem 56, 1299–1306. [DOI] [PubMed] [Google Scholar]
- Park K, Chen W, Volkow ND, Allen CP, Pan Y, and Du C (2019). Hemodynamic and neuronal responses to cocaine differ in awake versus anesthetized animals: Optical brain imaging study. Neuroimage 188, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlaman JP, Thompson BL, Levitt P, and Stanwood GD (2007). Pharmacokinetic profile of cocaine following intravenous administration in the female rabbit. Eur. J. Pharmacol 563, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickens R, and Thompson T (1968). Cocaine-reinforced behavior in rats: effects of reinforcement magnitude and fixed-ratio size. J. Pharmacol. Exp. Ther 161, 122–129. [PubMed] [Google Scholar]
- Pitts DK, Udom CE, and Marwah J (1987). Cardiovascular effects of cocaine in anesthetized and conscious rats. Life Sci., 40, 1099–1111. [DOI] [PubMed] [Google Scholar]
- Pitts DK, and Marwah J (1987). Cocaine modulation of central monoaminergic neurotransmission. Pharmacol. Biochem. Behav 26, 453–461. [DOI] [PubMed] [Google Scholar]
- Pitts DK, and Marwah J (1988). Cocaine and central monoaminergic neurotransmission: a review of electrophysiological studies and comparison to amphetamine and antidepressants. Life Sci. 42, 949–468. [DOI] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, and Di Chiara G (1995). Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc. Natl. Acad. Sci. USA 92, 12304–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon J, and van den Buuse M (1998). Autonomic mechanisms in the acute cardiovascular effects of cocaine in conscious rats. Eur. J. Pharmacol 363, 147–152. [DOI] [PubMed] [Google Scholar]
- Premkumar LS (2005). Block of a Ca(2+)-activated potassium channel by cocaine. J. Membr. Biol 204, 129–136. [DOI] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, and Kuhar MJ (1987). Cocaine receptors of dopamine transporters are related to self-administration of cocaine. Science 237, 1219–1223. [DOI] [PubMed] [Google Scholar]
- Samaha AN, Li Y, and Robinson TE (2002). The rate of intravenous cocaine administration determines susceptibility to sensitization. J. Neurosci 22, 3244–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo B (1978). Brain Energy Metabolism. New York: Wiley. [Google Scholar]
- Shriver DA, and Long JP (1971). A pharmacological comparison of some quaternary derivatives of cocaine. Arch. Int. Pharmacodyn. Ther 189, 198–208. [PubMed] [Google Scholar]
- Sokoloff L (1999). Energetics of functional activation in neural tissues. Neurochem. Res 24, 321–329. [DOI] [PubMed] [Google Scholar]
- Solis E, Afzal A, and Kiyatkin EA (2018). Intravenous cocaine increases oxygen entry into brain tissue: critical role of peripheral drug actions. ACS Chem. Neurosci 8, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCarley RW (2005). Brain control of wakefulness and sleep. New York: Springer. [Google Scholar]
- Taylor IM, Du Z, Bigelow ET, Eles JR., Horner AR, Catt KA, Weber SG, Jamieson BG, Cui XT (2017). Aptamer-functionalized neural recording electrodes for the direct measurement of cocaine in vivo. J. Mater. Chem. B 5, 2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tella SR (1996). Possible novel pharmacodynamic action of cocaine: cardiovascular and behavioral evidence. Pharmacol. Biochem. Behav 54, 343–354. [DOI] [PubMed] [Google Scholar]
- Tella SR, and Goldberg SR (1998). Monoamine transporter and sodium channel mechanisms in the rapid pressor response to cocaine. Pharmacol. Biochem. Behav 59, 305–312. [DOI] [PubMed] [Google Scholar]
- Wakabayashi KT, and Kiyatkin EA (2012). Rapid changes in extracellular glutamate induced by natural arousing stimuli and intravenous cocaine in the nucleus accumbens shell and core. J. Neurophysiol 108, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi KT, and Kiyatkin EA (2014). Critical role of peripheral drug actions in experience-dependent changes in nucleus accumbens glutamate release induced by intravenous cocaine. J Neurochem. 128, 672–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi KT, and Kiyatkin EA (2015). Central and peripheral contributions to dynamic changes in nucleus accumbens glucose induced by intravenous cocaine. Front. Neurosci 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakazono Y, and Kiyatkin EA (2008). Electrophysiological evaluation of the time-course of dopamine uptake inhibition induced by intravenous cocaine at a reinforcing dose. Neuroscience 151, 824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, You ZB, Oleson EB, Cheer JF, Myal S, and Wise RA (2013). Conditioned contribution of peripheral cocaine actions to cocaine reward and cocaine-seeking. Neuropsychopharmacology 38, 1763–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windels F, and Kiyatkin EA (2006). General anesthesia as a factor affecting impulse activity and neuronal responses to putative neurotransmitters. Brain Res. 1086, 104–116. [DOI] [PubMed] [Google Scholar]
- Wise RA, and Bozarth MA (1987). A psychomotor stimulant theory of addiction. Psychol. Rev 94, 469–492. [PubMed] [Google Scholar]
- Wise RA, Wang B, and You ZB (2008). Cocaine serves as a peripheral interoceptive conditioned stimulus for central glutamate and dopamine release. PLoS One 3(8):e2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SN, Chang HD, and Sung RJ (2006). Cocaine-induced inhibition of ATP-sensitive K+ channels in rat ventricular myocytes and in heart-derived H9c2 cells. Basic Clin. Pharmacol. Toxicol 98, 510–517. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Cooper DC, and White FJ (2002). Repeated cocaine treatment decreases whole-cell calcium current in the rat nucleus accumbens neurons. J. Pharmacol. Exp. Ther 301, 1119–1125. [DOI] [PubMed] [Google Scholar]