

Abstract
N6-methyladenosine (m6A) is the most abundant internal modification in mRNA and this methylation constitutes an important regulatory mechanism for the stability and translational efficiency of mRNA. In this study, we found that the protein levels of adenylate kinase 4 (AK4) and m6A writer METTL3 are significantly higher in tamoxifen-resistant (TamR) MCF-7 cells than in parental cells. The TamR MCF-7 cells also exhibit increased methylation at multiple m6A consensus motif sites in the 5′ untranslated region (5′ UTR) of AK4 mRNA, and genetic depletion of METTL3 in TamR MCF-7 cells led to a diminished AK4 protein level and attenuated resistance to tamoxifen. In addition, we observed augmented levels of reactive oxygen species (ROS) and p38 activity in TamR MCF-7 cells, and both are diminished upon genetic depletion of AK4. Reciprocally, overexpression of AK4 in MCF-7 cells stimulates ROS and p38 phosphorylation levels, and it suppresses mitochondrial apoptosis. Moreover, scavenging of intracellular ROS leads to reduced p38 activity and re-sensitizes TamR MCF-7 cells to tamoxifen. Thus, our results uncover a novel m6A-mediated epitranscriptomic mechanism for the regulation of AK4, illustrate the cellular pathways through which increased AK4 expression contributes to tamoxifen resistance, and reveal AK4 as a potential therapeutic target for overcoming tamoxifen resistance.
Graphical Abstract
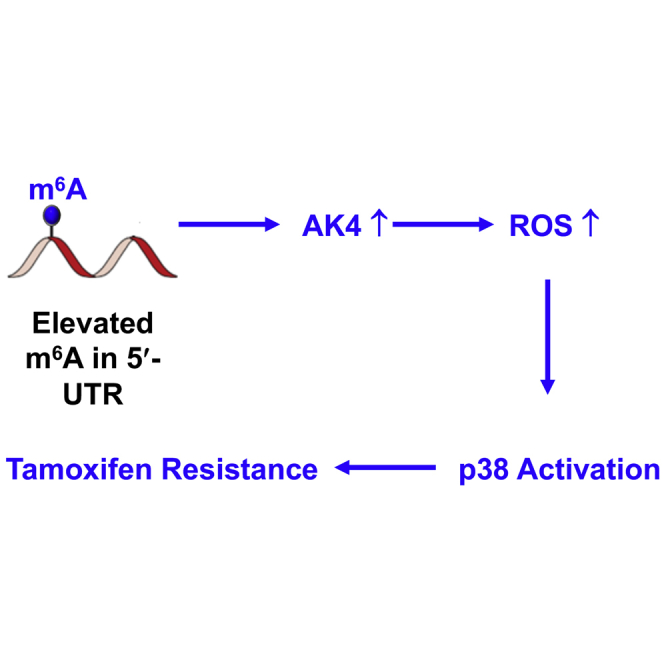
Liu et al. reported that elevated expression of adenylate kinase 4 (AK4) protein contributes to tamoxifen resistance in breast cancer cells, which involves augmented levels of reactive oxygen species, activation of p38 kinase, and suppression of mitochondrial apoptosis. Additionally, increased expression of AK4 arises from an N6-methyladenosine-based epitranscriptomic mechanism.
Introduction
Tamoxifen has been used for treating estrogen receptor (ER)-positive breast cancer for 40 years, and drug resistance has been the most challenging issue for this therapy.1, 2, 3, 4 Aberrant kinase protein expression has been found to be a major mechanism through which cancer cells acquire resistance toward chemotherapy.5 Adenylate kinases are abundant enzymes modulating cellular energy metabolism and homeostasis of cellular adenine nucleotides in different subcellular compartments.6 They catalyze the reversible transfer of a phosphate group from ATP to AMP, generating two molecules of ADP.6 Adenylate kinases differ in molecular weight, tissue distribution, subcellular localization, substrate and phosphate donor specificity, and kinetic properties.6 Among them, adenylate kinase (AK)3 and AK4 are localized in the mitochondrial matrix.7 AK4 may indirectly modulate the mitochondrial membrane permeability via its interaction with ADP/ATP translocase, thereby maintaining cellular energy homeostasis.6 In addition, AK4 was found to promote the progression of bladder cancer8 and confer the resistance of cancer cells toward radiation therapy and multiple chemotherapeutic agents.9,10
N6-methyladenosine (m6A) is the most prevalent internal modification in mRNA, and this methylation has emerged as a crucial and dynamic regulatory mechanism that controls gene expression in many physiological and pathological processes.11 In addition, the m6A-mediated epitranscriptomic pathway has been shown to contribute to chemoresistance and radioresistance. For instance, FTO, an m6A eraser,12 regulates therapeutic resistance of cervical squamous cell carcinoma by targeting β-catenin through demethylation of its mRNA.13,14 Likewise, METTL3, the catalytic subunit of the major m6A writer complex,15 promotes therapeutic resistance of pancreatic cancer cells,16 and m6A in FZD10 mRNA confers the resistance of BRCA-mutated epithelial ovarian cancers toward a poly(ADP-ribose) polymerase (PARP) inhibitor.17
In this study, we found that tamoxifen-resistant (TamR) MCF-7 breast cancer cells exhibit elevated levels of methylation at multiple m6A consensus motif sites in the 5′ untranslated region (5′ UTR) of AK4 mRNA, and the expression level of AK4 protein is influenced by m6A writer (METTL3) and eraser (ALKBH5) proteins. We also identify AK4 as a driver for tamoxifen resistance, which involves diminished mitochondrial apoptosis, increased ROS production, and augmented p38 activation.
Results
TamR MCF-7 Cells Exhibit Elevated Expression of AK4 Protein, Which Is Correlated with Poorer Survival of Breast Cancer Patients
A previous study showed that aberrant expression of AK4 is associated with increased malignancy in lung cancers.18 In this study, we examined whether differential expression of AK4 confers tamoxifen resistance in breast cancer cells by assessing first its expression levels in TamR MCF-7 and parental MCF-7 breast cancer cells. We found that the AK4 mRNA level exhibits a 2-fold increase, whereas its protein level displays a much more pronounced elevation in TamR MCF-7 relative to parental MCF-7 cells (Figures 1A–1C. We also examined the relative levels of expression of other members of the adenylate kinase family, and our results showed that AK2 also exhibits augmented expression in TamR MCF-7 over MCF-7 cells; the expression levels of AK1 and AK3 proteins, nevertheless, are very similar in these two cell lines (Figure 1A).
Figure 1.

TamR MCF-7 Cells Exhibit Elevated Expression of AK4 Protein Relative to Parental MCF-7 Cells, and a Higher Level of AK4 Expression Contributes to 4-OHT Resistance in TamR MCF-7 Cells
(A) Western blot analysis revealed the increased expression of AK4 protein in TamR MCF-7 cells. (B) The changes in AK4 protein expression were quantified based on its band intensities using ImageJ, normalized against that of the loading control (i.e., GAPDH), and displayed relative to the level observed in parental MCF-7 cells. (C) Relative mRNA levels of AK4 in MCF-7 and TamR MCF-7 cells. (D–F) Parental MCF-7 cells with stable overexpression of AK4 (D) and TamR MCF-7 cells with stable knockdown of AK4 (E) or AK2 (F) were treated with serially diluted 4-OHT for 72 h, and cell viability was measured using CCK8. The data were normalized against the control groups (ethanol). The data in (B)-(F) represent mean ± SD (n = 3). The p values were calculated based on an unpaired, two-tailed Student’s t test. #p ≥ 0.05, ∗0.01 ≤ p < 0.05, ∗∗0.001 ≤ p < 0.01, ∗∗∗p < 0.001.
We next assessed whether AK4 expression confers prognostic values in breast cancer patients. To this end, we performed Kaplan-Meier (KM) survival analysis by querying the Gene Expression Omnibus (GEO) database for breast cancer patients (n = 1,764). We found that higher levels of mRNA expression of the AK4 gene are significantly associated with worse survival of breast cancer patients (hazard ratio [HR] = 1.568; 95% confidence interval [CI] = 1.299−1.893; log rank p = 4.2e−06) (Figure S1A). There is, however, no apparent correlation between the mRNA expression level of the AK2 gene and breast cancer patient survival (HR = 1.0472; 95% CI = 0.931−1.164; log rank p = 0.7886) (Figure S1B).
Higher Level of AK4 Confers (Z)-4-Hydroxytamoxifen (4-OHT) Resistance
We next examined whether a higher level of AK4 expression contributes to tamoxifen resistance in breast cancer cells. To this end, we generated MCF-7 cells with stable overexpression of the AK4 gene and TamR MCF-7 cells with stable knockdown of this gene using two separate short hairpin RNAs (shRNAs) (Figures S2A and S2B). Results from a proliferation assay showed that overexpression of AK4 elicited increased resistance of MCF-7 cells to 4-OHT, and reciprocally depletion of AK4 re-sensitized TamR MCF-7 cells to 4-OHT (Figures 1D and 1E). Moreover, clonogenic survival assay results revealed that genetic depletion of AK4 in TamR MCF-7 cells led to increased sensitivity to 4-OHT (Figure S2D).
Because AK2 protein also showed a significant increase in TamR MCF-7 cells, we asked whether augmented expression of AK2 also promotes tamoxifen resistance. Thus, we generated TamR MCF-7 cells with stable knockdown of AK2 using two different sequences of shRNAs (Figure S2C). However, stable knockdown of AK2 in TamR MCF-7 cells did not elicit any alteration in their sensitivity toward 4-OHT (Figure 1F). These results demonstrate that elevated expression of AK4, but not AK2, renders MCF-7 cells resistant to tamoxifen.
TamR MCF-7 Cells Exhibit Augmented METTL3 Expression, Which Stimulates AK4 Protein Expression
The above results revealed a much more pronounced increase in AK4 protein over its mRNA in TamR MCF-7 cells than in the parental MCF-7 cells. Considering that m6A in mammalian mRNA can modulate the stability and translation efficiency of mRNA,19 we next asked whether increased expression of AK4 protein in TamR MCF-7 cells arises, in part, from this m6A-mediated epitranscriptomic mechanism. We first analyzed the publicly available m6A sequencing data by interrogating the Met-DB v2.0 database.20,21 The result shows that most m6A peaks are distributed in the 5′ and 3′ UTR of AK4 mRNA (Figure 2A). Our liquid chromatography-multi-stage mass spectrometry (LC-MS/MS/MS) quantification results also unveiled that the global m6A level was significantly higher in mRNA isolated from TamR than parental MCF-7 cells (Figure S3A).
Figure 2.

Different Levels of m6A Modification in the 5′ UTR of AK4 mRNA and Differential Expression of m6A Reader, Writer, and Eraser Proteins in MCF-7 and the Isogenic TamR Cells
(A) Genome browser track showing the distribution of m6A peaks in AK4 mRNA. The data were retrieved from MeT-DB v2.0 (http://180.208.58.19/metdb_v2/), where the 5′ UTR, coding sequence (CDS), and 3′ UTR are represented by blue, red, and green bars, respectively, and intron regions are indicated by a thin yellow line. (B) Putative m6A sites in the 5′ UTR of AK4 mRNA. (C) Relative template abundances of AK4 5′ UTR measured by the SELECT method (n ≥ 3). A total of nine sites were chosen, seven from the m6A motif site (G(A)C/A(A)C) and two from the C(A)G or G(A)G site (as negative controls [Neg]), where the 'A' highlighted in bold designates the adenosine sites that were interrogated by the SELECT method. The p values were calculated versus the mean value of the two negative controls. (D) Parental and TamR MCF-7 cells were treated with 1 μM 4-OHT for 48 h and cell lysates were used to monitor the levels of YTHDF1, YTHDF2, YTHDF3, FTO, ALKBH5, and METTL3 proteins by western blot analyses. (E) Relative expression levels of YTHDF1, YTHDF2, YTHDF3, FTO, ALKBH5, and METTL3 proteins in MCF-7 and TamR MCF-7 cells. The protein levels were quantified from gel band intensities using ImageJ. The values were normalized against the band intensities of GAPDH. The data are presented as the mean ± SD (n = 3), where the p values were calculated using an unpaired, two-tailed Student’s t test. #p ≥ 0.05, ∗0.01 ≤ p < 0.05, ∗∗0.001 ≤ p < 0.01, ∗∗∗p < 0.001.
Recent studies showed that, in response to heat shock stress, cells overexpress HSP70 and DNAJB4 proteins through a mechanism involving cap-independent translation enabled by m6A in the 5′ UTRs of the mRNAs of HSP70 and DNAJB4.21, 22, 23, 24 Thus, we examined whether a similar mechanism is at play for the elevated expression of AK4 in TamR MCF-7 cells. Results from the single-base elongation- and ligation-based qPCR amplification (SELECT) assay25 showed that seven adenosines at the m6A motif sites in the 5′ UTR of AK4 mRNA from TamR MCF-7 cells were with increased m6A levels relative to parental MCF-7 cells (Figures 2B, 2C, and S3B).
We also examined the relative levels of expression of m6A reader, writer, and eraser proteins in this pair of cell lines. We found that that the m6A reader proteins YTHDF1 and YTHDF3 exhibit no pronounced differences in the two cell lines, whereas the YTHDF2 protein level was lower in TamR MCF-7 cells than in parental cells (Figures 2D and 2E). FTO, a demethylase for m6A and m6Am,12 displays a markedly higher level of expression in TamR MCF-7 cells than in MCF-7 cells (Figures 2D and 2E). Similarly, METTL3, the catalytic subunit of the m6A methyltransferase complex,15 exhibits an augmented level of expression in TamR MCF-7 cells than in MCF-7 cells (Figure 2E).
Next, we investigated whether these m6A reader, writer, and eraser proteins affect AK4 protein expression in MCF-7 cells. We found that overexpression of FTO in TamR MCF-7 cells does not lead to any apparent alteration in AK4 protein expression (Figure 3A), whereas genetic depletion of FTO in TamR MCF-7 cells results in an increased AK4 protein level (Figure 3B), suggesting that the elevated expression of AK4 in TamR MCF-7 cells is not attributed to increased FTO expression. Alternatively, small interfering RNA (siRNA)-mediated knockdown of ALKBH5 elicits increased AK4 protein expression, and overexpression of ALKBH5 confers diminished level of AK4 protein (Figures 3C and 3D). These results support that, although the expression level of ALKBH5 is similar in these two cell lines, AK4 protein expression could be subjected to regulation by this m6A eraser protein. Moreover, depletion of METTL3 in TamR MCF-7 cells leads to significantly decreased AK4 protein level; reciprocally, the AK4 protein level increases slightly in MCF-7 cells upon overexpression of METTL3 (Figures 4A, 4B, S4A, and S4B), substantiating that the AK4 expression level is regulated by the METTL3-containing m6A writer complex.
Figure 3.

The Expression Level of AK4 Protein Is Modulated by the m6A Eraser Proteins
(A–D) Western blot for assessing the expression levels of AK4 protein after overexpression (A and C) or knockdown (B and D) of FTO (A and B) and ALKBH5 (C and D) in TamR MCF-7 cells. (E–H) AK4 protein changes were quantified from band intensities detected in (A)-(D), respectively, using ImageJ and are displayed relative to the level detected for control cells. The data are presented as the mean ± SD (n = 3), where the p values were calculated using an unpaired, two-tailed Student’s t test. ∗0.01 ≤ p < 0.05, ∗∗0.001 ≤ p < 0.01, ∗∗∗p < 0.001.
Figure 4.

The Expression Level of AK4 Protein Is Modulated by the m6A Writer Protein METTL3
(A and B) Images from western blot for monitoring the changes in AK4 protein after overexpression of METTL3 in MCF-7 cells (A), or knockdown of METTL3 in TamR MCF-7 cells (B). (C) Western blot for examining the changes in expression of AK4 protein after overexpression of YTHDF2 or YTHDF2 W432A in TamR MCF-7 cells. (D–F) Quantification of AK4 protein change based on results obtained from (A) (A)–(C), respectively. The values in the bar graphs represent the values that were normalized against the band intensities of GAPDH. (G and H) Parental MCF-7 cells with stable overexpression of METTL3 (G) and TamR MCF-7 cells with stable knockdown of METTL3 (H) were treated with serially diluted 4-OHT for 72 h and the cell viability was measured using CCK8. The data were normalized against the control groups (ethanol). The data in (D)–(H) represent mean ± SD (n = 3). The p values were calculated by using the unpaired, two-tailed Student’s t test. #p ≥ 0.05, ∗0.01 ≤ p < 0.05, ∗∗0.001 ≤ p < 0.01, ∗∗∗p < 0.001.
We also measured global levels of m6A in mRNA samples by using LC-MS/MS/MS. Our results showed that genetic depletions of ALKBH5 and METTL3 in TamR MCF-7 cells gave rise to increased and decreased global levels m6A in cellular mRNA samples, respectively (Figures S4C and S4D). This is in agreement with the established functions of ALKBH5 and METTL3 in serving as the eraser and writer proteins of m6A.15,26
Since YTHDF2 exhibits a lower level of expression in TamR MCF-7 cells than in MCF-7 cells, we investigated whether AK4 is also regulated by this m6A reader protein. The results show that overexpression of YTHDF2 or YTHDF2 W432A, which does not bind to m6A,27 did not alter AK4 protein expression (Figure 4C) in TamR MCF-7 cells, suggesting that the stability or translation efficiency of AK4 mRNA in these cells is not modulated by YTHDF2.
On the grounds that AK4 protein expression is positively regulated by METTL3 and elevated expression of AK4 confers resistance of MCF-7 cells to tamoxifen, we also examined whether resistance to tamoxifen is modulated by the expression level of METTL3. To this end, we produced MCF-7 cells with stable overexpression of METTL3 and TamR MCF-7 cells with stable knockdown of METTL3 using two independent sequences of shRNAs. Results from a cell proliferation assay showed that overexpression of METTL3 elicits slightly increased resistance of MCF-7 cells to tamoxifen (Figure 4G), whereas genetic depletion of METTL3 re-sensitizes TamR MCF-7 cells to tamoxifen (Figure 4H).
Elevated AK4 Induces Increased Production of Reactive Oxygen Species (ROS) and Activation of p38 Kinase
Exposure of human cancer cells to chemotherapeutic agents or ionizing radiation is known to upregulate ROS.28,29 Chronic exposure of cancer cells to high levels of ROS may trigger therapeutic resistance in cancer cells.28 It was previously reported that overexpression of AK4 shifts metabolism toward aerobic glycolysis and elevates intracellular ROS.30 Therefore, we also assessed whether higher level of AK4 elicits increased generation of ROS. Results from fluorescence measurement of cells treated with 2′,7′-dichlorofluorescin diacetate (DCF-DA) showed that ROS production was elevated in TamR MCF-7 cells compared with parental MCF-7 cells (Figure 5A). Additionally, overexpression of AK4 in parental MCF-7 cells results in a pronounced increase in ROS generation, whereas knockdown of AK4 in TamR MCF-7 cells markedly diminishes the ROS level (Figures 5B and 5C).
Figure 5.

Elevated Expression of AK4 Gene Led to Increased ROS and p38 Phosphorylation
(A–C) Differences in the levels of ROS in MCF-7 and TamR MCF-7 cells (A), MCF-7 cells transfected with empty vector or a vector for the stable overexpression of AK4 (B), or TamR MCF-7 cells with stable expression of control shRNA or shRNA for knockdown of AK4 gene (C). Cells were pre-incubated with or without 1 μM 4-OHT for 24 h followed by 10 μM DCF-DA for 30 min. The cells were then washed with PBS twice, and DCF-DA fluorescence was recorded at 525 nm using an excitation wavelength of 488 nm on a BioTek reader. The data were normalized against the control group (DMSO). (D) Images from western blot for monitoring the changes in levels of phosphorylated p38 (p-p38) and p38 in parental and TamR MCF-7 cells. (E and F) Alterations in p-p38 and p38 protein levels after ectopic overexpression of AK4 in MCF-7 cells (E) or knockdown of AK4 in TamR MCF-7 cells (F). (G–I) Quantification of p-p38 and p38 protein changes based on Western blot images shown in (D)–(F), respectively. The band intensities were quantified using ImageJ and are displayed relative to the level in parental MCF-7, MCF-7 vector, and TamR cells with stable expression of control shRNA (shCtrl), respectively. The values were normalized against the band intensities of p38. The data in (A)-(C) and (G)-(I) are presented as the mean ± SD (n = 3). The p values were calculated using an unpaired, two-tailed Student’s t test. #p ≥ 0.05, ∗0.01 ≤ p < 0.05, ∗∗0.001 ≤ p < 0.01, ∗∗∗p < 0.001.
Mitochondrial ROS is known to initiate phosphorylation and subsequent activation of p38 mitogen-activated protein (MAP) kinase (MAPK) during hypoxia in cardiomyocytes.31 Next, we assessed whether p38 activity is elevated in cellular stress environment arising from long-term exposure to tamoxifen. Our results showed that p38 activity was augmented in TamR MCF-7 cells relative to parental MCF-7 cells (Figure 5D). Additionally, overexpression of AK4 gave rise to elevated phosphorylation of p38 in MCF-7 cells (Figure 5E), whereas stable knockdown of AK4 in TamR MCF-7 cells led to diminished activity of p38 (Figure 5F). These results demonstrate that elevated AK4 expression elicits augmented production of ROS, which results in the activation of p38 kinase.
We also monitored the activities of two other mitogen-activated protein kinases, i.e., extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), and our results showed that ERK activity displayed the same trend as p38 upon overexpression or knockdown of AK4, whereas JNK activity was not affected by upregulation or downregulation of AK4 gene expression (Figure S5).
High Level of ROS Activates p38 and Confers 4-OHT Resistance
Modulation of ROS in cancer cells, for example, through targeted production of ROS via ATP-driven transmembrane efflux pumps in mitochondria,32 was advocated as a viable strategy to overcome drug resistance in cancers.33 Hence, we also asked whether diminution in ROS levels by treating cells with N-acetyl-l-cysteine (NAC) could overcome tamoxifen resistance. Our results from a proliferation assay showed that scavenging ROS with NAC could re-sensitize TamR MCF-7 cells and AK4-overexpressing MCF-7 cells to 4-OHT (Figures 6A and 6B). In addition, treatment with NAC diminishes p38 phosphorylation level in both TamR MCF-7 cells and AK4-overexpressing MCF-7 cells (Figures 6C and 6D).
Figure 6.

High Level of ROS Activates p38 and Confers 4-OHT Resistance
(A and B) Cells were treated with 5 mM NAC for 2 h, washed with PBS twice, and treated with serially diluted 4-OHT for 72 h. Shown in (A) are comparisons for MCF-7 cells transfected with an empty vector and without NAC treatment, or AK4-overexpressing MCF-7 cells with or without NAC treatment. Displayed in (B) are the comparisons for MCF-7 cells without NAC treatment, or TamR MCF-7 cells with or without NAC treatment. Cell viability was measured using CCK8, and luminescence was recorded with a BioTek reader. The data were normalized to control groups (DMSO) and are represented by the mean ± SD (n = 3). (C and D) Cells were treated with 5 mM NAC for 1 h, and the lysates were used to monitor the levels of AK4, p-p38, and p38 by western blot analyses. Shown in (C) are the results for MCF7 cells without NAC treatment, and TamR MCF-7 cells with or without NAC treatment; displayed in (D) are the results for MCF-7 cells transfected with an empty vector and without NAC treatment, or AK4-overexpressing MCF7 cells with or without NAC treatment.
Next, we investigated whether active p38 confers resistance of MCF-7 cells to 4-OHT, and proliferation results showed that treatment of TamR cells and AK4-overexpressing MCF-7 cells with a small-molecule inhibitor of p38, i.e., SB202190, re-sensitized these cells toward 4-OHT (Figure S6). These results substantiate the notion that elevated AK4 confers increased ROS generation, which results in p38 activation, thereby rendering MCF-7 cells resistant to tamoxifen.
Augmented AK4 Leads to Decreased Mitochondrial Apoptosis, Which May Confer Resistance of MCF-7 Cells to 4-OHT
Inhibition of mitochondrial apoptosis is known to contribute to drug resistance.34 Furthermore, AK4 is localized in mitochondrial matrix and modulates mitochondrial activity.35 Hence, we examined whether a higher level of AK4 affects mitochondrial apoptosis. To this end, we monitored the differential expression of pro-apoptotic proteins Bim, Bad, and anti-apoptotic protein Bcl-236 in MCF-7 and TamR MCF-7 cells. The results show that pro-apoptotic protein Bim is downregulated, whereas the anti-apoptotic protein Bcl-2 is elevated in TamR cells relative to parental MCF-7 cells (Figures 7A and 7B). Moreover, overexpression of AK4 in MCF-7 cells decreases Bim and Bad levels while concomitantly upregulates the Bcl-2 level (Figures 7C and 7D). Conversely, genetic depletion of AK4 in TamR MCF-7 cells restores the protein levels of Bim and Bad and diminishes that of Bcl-2 (Figures 7E and 7F). These results indicated that high levels of AK4 may confer tamoxifen resistance through attenuating mitochondrial apoptosis.
Figure 7.

Augmented AK4 Leads to Decreased Mitochondrial Apoptosis, Which May Confer Resistance of MCF-7 Cells to 4-OHT
(A) Western blot analyses of Bim, Bad, and Bcl-2 protein levels in parental and TamR MCF-7 cells. (B) Quantification of changes in protein levels of Bim, Bad, and Bcl-2. (C) Images from western blot for monitoring the levels of Bim, Bad, and Bcl-2 proteins after overexpressing AK4 in MCF-7 cells. (D) Quantification of Bim, Bad, and Bcl-2 protein changes based on results shown in (C). (E) The relative levels of Bim, Bad, and Bcl-2 proteins after knockdown of AK4 in TamR MCF-7 cells. (F) Quantification of Bim, Bad, and Bcl-2 protein changes based on results shown in (E). All of the band intensities were quantified using ImageJ and were displayed relative to the level in parental MCF-7, MCF-7 vector, and TamR shCtrl cells, respectively. The values were normalized against the band intensities of GAPDH, and the data are presented as the mean ± SD (n = 3). The p values were calculated using an unpaired, two-tailed Student’s t test. #p ≥ 0.05 ∗0.01 ≤ p < 0.05, ∗∗0.001 ≤ p < 0.01, ∗∗∗p < 0.001.
Discussion
Tamoxifen has been approved for treating ER-positive breast cancer for more than four decades, and drug resistance constitutes the major hurdle for this therapy.4 Understanding the molecular mechanisms of resistance to tamoxifen may provide new venues for overcoming therapeutic resistance. In this study, we uncover, for the first time, that elevated expression of AK4, a mitochondrial nucleotide kinase, constitutes a novel mechanism contributing to tamoxifen resistance in MCF-7 breast cancer cells, and AK4’s role in modulating drug resistance involves the elevated production of ROS, augmented activation of p38 MAPK, and suppression of mitochondrial apoptosis. Our findings are in keeping with the prior observations that overexpression of AK4 shifts metabolism toward aerobic glycolysis and elevates intracellular ROS.30 Our results are also consistent with the previous findings that mitochondrial ROS stimulate phosphorylation of p38 MAPK during hypoxia in cardiomyocytes31 and suppression of mitochondrial apoptosis modulates drug resistance.34
We also reveal that mechanistically the elevated expression of AK4 in tamoxifen-resistant cells arises from increased expression of METTL3 and the ensuing elevated levels of m6A in the 5′ UTR of AK4 mRNA. Although m6A-mediated epitranscriptomic mechanism is known to modulate therapeutic resistance,14,16,37 our work unveils a novel molecular target for this mechanism (i.e., AK4 mRNA) and reveals the epitranscriptomic modulator for this target (i.e., METTL3). Furthermore, elevated level of METTL3 and its function in tamoxifen resistance are in line with recent observations that METTL3 assumes an oncogenic role in breast and ovarian cancer progression38,39 and promotes chemoresistance and radioresistance in pancreatic cancer.16 Future studies are needed to understand further how chronic exposure to tamoxifen elicits increased expression of METTL3.
A limitation of the present study resides in that most cellular experiments were conducted with a single pair of cell lines, i.e., MCF-7 and its paired tamoxifen-resistant line. Nevertheless, bioinformatics analyses of publicly available data revealed a strong correlation between a higher level of AK4 expression and poorer survival of breast cancer patients. Thus, our result suggests that this mechanism contributes to tamoxifen resistance in a large fraction of breast cancer patients.
Collectively, our study demonstrates that chronic exposure to tamoxifen induces increased m6A in the 5′ UTR of AK4 mRNA, which promotes its translation, and a higher level of AK4 suppresses mitochondrial apoptosis and promotes ROS production, thereby activating p38 and ultimately resulting in elevated resistance of MCF-7 cells to tamoxifen. Thus, our results uncovered AK4 as a potential therapeutic target for overcoming tamoxifen resistance.
Materials and Methods
Materials
4-OHT, DCF-DA, and NAC were purchased from Sigma-Aldrich. SB202190 was purchased from MedChemExpress.
Cell Culture
Parental and TamR MCF-740 human breast cancer cells were generous gifts from Dr. David Eastmond at the University of California, Riverside and Dr. Guangdi Wang at Xavier University, respectively. The cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and antibiotics. In addition, 1 μM 4-OHT was included in the culture medium to maintain the tamoxifen-resistant subline. The cells were cultured at 37°C in an incubator containing 5% CO2.
The initial passage numbers for breast cancer cells used were MCF-7 (p4) and TamR (p5). All of the relevant experiments were conducted within 20 passages from revival of the initial frozen seeds. A LookOut mycoplasma PCR detection kit (MP0035, Sigma-Aldrich, St. Louis, MO, USA) for detection of 19 mycoplasma species was used following the manufacturer’s protocol. PCRs were performed using Taq DNA polymerase. Results were visualized on a 2% agarose gel, where mycoplasma-positive samples would show a band at 259 bp, and mycoplasma-negative internal control DNA has a band at 481 bp. MCF-7 and TamR breast cancer cell lines were tested on November 22, 2019, and results indicated no mycoplasma contamination. In addition, these two breast cancer cell lines were authenticated by ATCC on July 11, 2018 using short tandem repeat (STR) analysis as described in 2012 in the American National Standards Institute (ANSI) standard (ASN-0002) for authentication of human cell lines.
shRNA and Plasmids
The sequences for shRNA and primers are shown in Table S1. Scrambled shRNA with a hairpin sequence of 5′-CCT AAG GTT AAG TCG CCC TCG CTC TAG CGA GGG CGA CTT AAC CTT AGG-3′ (Addgene, Cambridge, MA, USA) was used as a negative control, as described previously.41 All primers and oligodeoxynucleotides used for the construction of shRNA plasmids were purchased from Integrated DNA Technologies. All shRNAs were cloned into the AgeI/EcoRI site of the pLKO.1 vector (Addgene, plasmid #10878). Coding sequences of AK4 and METTL3 plasmids were cloned into the NheI and AgeI restriction sites of the pLJM1-EGFP vector (Addgene, plasmid #10878). Expression plasmids for YTHDF2 and YTHDF2 W432A were previously described.27 All constructs were confirmed by Sanger sequencing.
siRNA and Transfection
The sequences for siMETTL3-1 and siMETTL3-2 were 5′-CUG CAA GUA UGU UCA CUA UGA-3′ and 5′-AGG AGC CAG CCA AGA AAU CAA-3′, respectively.15 The sequences for siALKBH5-1 and siALKBH5-2 were 5′-ACA AGU ACU UCU UCG GCG A-3′ and 5′-GCG CCG UCA UCA ACG ACU A-3′, respectively.42 siRNA was transfected using RNAiMAX (Invitrogen) following the manufacturer’s protocol, where non-targeting siRNA (Dharmacon, D-001210-02-20) was used as control. The pcDNA3.1-DYK-ALKBH5 and pcDNA3.1-DYK-FTO were kindly provided by Prof. Chuan He.14,37,43
Lentivirus Production and Stable Cell Line Generation
HEK293T cells were transfected with pLKO.1/puro-shRNAs and pLJM1-EGFP-AK4/METTL3 plasmids together with pLTR-G (plasmid #17532) envelope plasmid and pCMV-dR8.2 dvpr (plasmid #8455) package plasmid using PolyFect transfection reagent (QIAGEN). Viral particles were collected 48 h later and filtered through a 0.45-μm sterile filter. Tamoxifen-resistant MCF-7 cells were infected with a 5:1 mixture of viral particles and DMEM containing 1 μM 4-OHT for 48 h. The cells were selected with 2 μg/mL puromycin for a week and cultured in complete DMEM supplemented with 1 μM 4-OHT and 1 μg/mL puromycin.
Western Blot
MCF7 and tamoxifen-resistant MCF7 cells were cultured in a six-well plate and the cells, when their confluency levels reached 40%–50%, were treated for 24 h with 4-OHT at the indicated concentrations. The cells were lysed with CelLytic M cell lysis reagent (Sigma-Aldrich), and the supernatant was used for western blot analysis. Antibodies recognizing human AK1 (Santa Cruz Biotechnology, sc-365316), AK2 (Santa Cruz Biotechnology, sc-374095), AK3 (Santa Cruz Biotechnology, sc-398571), AK4 (Santa Cruz Biotechnology, sc-271161), phosphorylated (p-)p38 (Santa Cruz Biotechnology, sc-166182), p38α (Santa Cruz Biotechnology, sc-271120), Bcl-2 (Santa Cruz Biotechnology, sc-7382), Bim (Santa Cruz Biotechnology, sc-374358), Bad (Santa Cruz Biotechnology, sc-8044), YTHDF1 (Proteintech, 17479-1-AP), YTHDF2 (EMD Millipore, Q2490423), YTHDF3 (Santa Cruz Biotechnology, sc-377119), FTO (Santa Cruz Biotechnology, sc-271713), METTL3 (Proteintech, 15073-1-AP), ALKBH5 (Proteintech, 16837-1-AP), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cell Signaling Technology, 4370T), ERK1/2 (Santa Cruz Biotechnology, sc-514302), p-JNK (Santa Cruz Biotechnology, sc-6254), and JNK (Santa Cruz Biotechnology, sc-7345) were used as primary antibodies for western blot analysis. Anti-rabbit immunoglobulin G (IgG) (whole molecule)-peroxidase antibody produced in goats (Sigma, #A0545) and mouse (m-)IgGκ binding protein (BP)-horseradish peroxidase (HRP) (Santa Cruz Biotechnology, sc-516102) were used as secondary antibodies. Membranes were also probed with anti-GAPDH antibody (Santa Cruz Biotechnology, sc-32233) to confirm equal protein loading.
Proliferation Assay
Cells were grown in 96-well plates (6,000 cells/well). Compounds of various concentrations were added into the plates after the cells were cultured for 12 h. Cell proliferation was determined after treatment with compounds for 72 h. Cell viability was measured using the Cell Counting Kit-8 (CCK8) (Dojindo Molecular Technologies) following the manufacturer’s instructions, and luminescence was recorded in a multi-label reader (BioTek, Synergy H1). The data were normalized to control groups (ethanol or DMSO) and represented by the mean and SD of results from three independent measurements using Prism 7.0 (GraphPad).
Clonogenic Survival Assay
Cells were cultured in six-well plates at a density of 300 cells/well. Compounds of various concentrations were added into the wells after the cells were cultured for 12 h, and cells were grown in the presence of 4-OHT for 14 days. The cells were subsequently washed once with phosphate-buffered saline (PBS), fixed with 5% glutaraldehyde for 30 min, stained with 0.5% crystal violet for 1 h, and subsequently washed with PBS five times. Single colonies, which contained a minimum of 50 cells per colony, were subsequently counted; the data were normalized to the control groups and represented as mean ± SD of results from three independent measurements.
Measurement of Intracellular ROS
Cells were cultured in 96-well plates. After pre-incubation with or without 1 μM 4-OHT for 24 h, the cells were treated with 10 μM DCF-DA (Sigma) for 30 min. The cells were subsequently washed twice with PBS. DCF-DA fluorescence was recorded at 525 nm using an excitation wavelength of 488 nm in a multi-label reader (BioTek, Synergy H1). The data were normalized to control groups (DMSO) and represented as mean ± SD of results from three independent measurements.
SELECT Assay
A SELECT assay was performed as previously reported.25 Briefly, total RNA and up and down primers were mixed and annealed. A 3-μL solution containing Bst 2.0 DNA polymerase, ATP, and SplintR was subsequently added to the mixture to make the final volume 20 μL. The final reaction mixture was incubated at 40°C for 20 min, denatured at 80°C for 20 min, and subsequently cooled to 4°C. Real-time quantitative PCR was subsequently performed using an iQ SYBR Green supermix kit (Bio-Rad) on a Bio-Rad iCycler system (Bio-Rad). The comparative cycle threshold (Ct) method (ΔΔCt) was used for the relative quantification of template abundance.44 The sequences for up and down primers and qPCR primers are shown in Table S2.
LC-MS/MS/MS Measurements of the Levels of m6A in mRNA
mRNA was isolated from cells and digested using previously published procedures.45 Briefly, 100 ng of mRNA was digested, at 37°C for 2 h, with 1 U of nuclease P1 in a 25-μL buffer containing 25 mM NaCl and 2.5 mM ZnCl2. Alkaline phosphatase (0.25 U) and 3 μL of 1.0 M NH4HCO3 buffer were subsequently added to the mixture. After incubation at 37°C for another 2 h, the digestion mixture was dried by using a SpeedVac and reconstituted in 100 μL of double distilled (dd)H2O.
LC-tandem MS (LC-MS/MS) and LC-MS/MS/MS were used for the quantification of adenosine (rA) and m6A, respectively, in mRNA samples isolated from cells, as described previously.46 In short, to 2 ng of digested mRNA were added 1.5 pmol of [13C5]-rA and 8.5 fmol of [D3]-m6A. The enzymes in the digestion mixture were removed by chloroform extraction, and the ensuing aqueous layer was dried, reconstituted in water, and subjected to LC-MS/MS and LC-MS/MS/MS measurements.
Selected reaction monitoring (SRM) experiments were performed on an LTQ-XL linear ion trap mass spectrometer coupled with an EASY-nLC II (Thermo Fisher Scientific, San Jose, CA, USA). The mass spectrometer was operated in the positive-ion mode with the electrospray, capillary, and tube lens voltages being 2.0 kV, 12 V, and 100 V, respectively. Samples were loaded onto a 5-cm in-house packed pre-column containing Magic C18-AQ (5 μm in particle size, 100 Å in pore size, Michrom BioResources) at a flow rate of 2.5 μL/min. Analytes were resolved on a 20-cm Magic C18-AQ analytical column (75-μm inner diameter [i.d.]) at a flow rate of 300 nL/min. The gradient used was 0%–15% B in 40 min, 55%–90% B in 1 min, and 90% B in 10 min, where 0.1% (v/v) formic acid in water and 0.1% (v/v) formic acid in acetonitrile were used as mobile phases A and B, respectively.
Bioinformatics Analysis of Publicly Available Datasets
The prognostic values of AK4 and AK2 genes in breast cancer patients were assessed by using the KM Plotter database (http://kmplot.com/analysis/) derived from the GEO repository.47 KM survival analysis was conducted with patient data stratified by median AK2 and AK4 mRNA expression levels, and the HR, 95% CIs, and log-rank p values were calculated and displayed. The distribution of m6A in AK4 mRNA data was retrieved from the MeT-DB v2.0 website.20,21
Author Contributions
X.L., G.G., X.D., W.M., J.Y., M.H., D.B., L.L., and Y.S. conducted the experiments, and X.L. and Y.W. designed the experiments and wrote the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors would like to thank Dr. David Eastmond (UC Riverside) and Dr. Guangdi Wang (Xavier University) for providing the cell lines used in this study. This work was supported by the National Institutes of Health, US (R01 CA210072 to Y.W.), and M.H. was supported in part by an NRSA Institutional Training Grant (T32 ES018827).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.09.007.
Supplemental Information
References
- 1.Ali S., Rasool M., Chaoudhry H., N Pushparaj P., Jha P., Hafiz A., Mahfooz M., Abdus Sami G., Azhar Kamal M., Bashir S. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation. 2016;12:135–139. doi: 10.6026/97320630012135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee S., Saxena N., Sengupta K., Banerjee S.K. 17α-estradiol-induced VEGF-A expression in rat pituitary tumor cells is mediated through ER independent but PI3K-Akt dependent signaling pathway. Biochem. Biophys. Res. Commun. 2003;300:209–215. doi: 10.1016/s0006-291x(02)02830-9. [DOI] [PubMed] [Google Scholar]
- 4.Chang M. Tamoxifen resistance in breast cancer. Biomol. Ther. (Seoul) 2012;20:256–267. doi: 10.4062/biomolther.2012.20.3.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duncan J.S., Whittle M.C., Nakamura K., Abell A.N., Midland A.A., Zawistowski J.S., Johnson N.L., Granger D.A., Jordan N.V., Darr D.B. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panayiotou C., Solaroli N., Karlsson A. The many isoforms of human adenylate kinases. Int. J. Biochem. Cell Biol. 2014;49:75–83. doi: 10.1016/j.biocel.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Noma T., Fujisawa K., Yamashiro Y., Shinohara M., Nakazawa A., Gondo T., Ishihara T., Yoshinobu K. Structure and expression of human mitochondrial adenylate kinase targeted to the mitochondrial matrix. Biochem. J. 2001;358:225–232. doi: 10.1042/0264-6021:3580225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xin F., Yao D.W., Fan L., Liu J.H., Liu X.D. Adenylate kinase 4 promotes bladder cancer cell proliferation and invasion. Clin. Exp. Med. 2019;19:525–534. doi: 10.1007/s10238-019-00576-5. [DOI] [PubMed] [Google Scholar]
- 9.Zang C., Zhao F., Hua L., Pu Y. The miR-199a-3p regulates the radioresistance of esophageal cancer cells via targeting the AK4 gene. Cancer Cell Int. 2018;18:186. doi: 10.1186/s12935-018-0689-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei W., Yan C., Ya J., Yong D., Yujun B., Kai L. miR-199a-3p affects the multi-chemoresistance of osteosarcoma through targeting AK4. BMC Cancer. 2018;18:631. doi: 10.1186/s12885-018-4460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaccara S., Ries R.J., Jaffrey S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 12.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.G., He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou S., Bai Z.L., Xia D., Zhao Z.J., Zhao R., Wang Y.Y., Zhe H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol. Carcinog. 2018;57:590–597. doi: 10.1002/mc.22782. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bögler O. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taketo K., Konno M., Asai A., Koseki J., Toratani M., Satoh T., Doki Y., Mori M., Ishii H., Ogawa K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018;52:621–629. doi: 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- 17.Fukumoto T., Zhu H., Nacarelli T., Karakashev S., Fatkhutdinov N., Wu S., Liu P., Kossenkov A.V., Showe L.C., Jean S. N6-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019;79:2812–2820. doi: 10.1158/0008-5472.CAN-18-3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jan Y.H., Tsai H.Y., Yang C.J., Huang M.S., Yang Y.F., Lai T.C., Lee C.H., Jeng Y.M., Huang C.Y., Su J.L. Adenylate kinase-4 is a marker of poor clinical outcomes that promotes metastasis of lung cancer by downregulating the transcription factor ATF3. Cancer Res. 2012;72:5119–5129. doi: 10.1158/0008-5472.CAN-12-1842. [DOI] [PubMed] [Google Scholar]
- 19.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H., Flores M.A., Meng J., Zhang L., Zhao X., Rao M.K., Chen Y., Huang Y. MeT-DB: a database of transcriptome methylation in mammalian cells. Nucleic Acids Res. 2015;43:D197–D203. doi: 10.1093/nar/gku1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J., Wan J., Shu X.E., Mao Y., Liu X.M., Yuan X., Zhang X., Hess M.E., Brüning J.C., Qian S.-B. N6-methyladenosine guides mRNA alternative translation during integrated stress response. Mol. Cell. 2018;69:636–647.e7. doi: 10.1016/j.molcel.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miao W., Li L., Zhao Y., Dai X., Chen X., Wang Y. HSP90 inhibitors stimulate DNAJB4 protein expression through a mechanism involving N6-methyladenosine. Nat. Commun. 2019;10:3613. doi: 10.1038/s41467-019-11552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.B., Jaffrey S.R. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J., Wan J., Gao X., Zhang X., Jaffrey S.R., Qian S.B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Y., Wang Y., Tang Q., Wei L., Zhang X., Jia G. An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N6-methyladenosine modification. Angew. Chem. Int. Ed. Engl. 2018;57:15995–16000. doi: 10.1002/anie.201807942. [DOI] [PubMed] [Google Scholar]
- 26.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vågbø C.B., Shi Y., Wang W.L., Song S.H. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai X., Wang T., Gonzalez G., Wang Y. Identification of YTH domain-containing proteins as the readers for N1-methyladenosine in RNA. Anal. Chem. 2018;90:6380–6384. doi: 10.1021/acs.analchem.8b01703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahalingaiah P.K.S., Singh K.P. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS ONE. 2014;9:e87371. doi: 10.1371/journal.pone.0087371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stojković S., Podolski-Renić A., Dinić J., Stanković T., Banković J., Hadžić S., Paunović V., Isaković A., Tanić N., Pešić M. Development of resistance to antiglioma agents in rat C6 cells caused collateral sensitivity to doxorubicin. Exp. Cell Res. 2015;335:248–257. doi: 10.1016/j.yexcr.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Jan Y.H., Lai T.C., Yang C.J., Lin Y.F., Huang M.S., Hsiao M. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1α to drive lung adenocarcinoma metastasis. J. Hematol. Oncol. 2019;12:12. doi: 10.1186/s13045-019-0698-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kulisz A., Chen N., Chandel N.S., Shao Z., Schumacker P.T. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L1324–L1329. doi: 10.1152/ajplung.00326.2001. [DOI] [PubMed] [Google Scholar]
- 32.Wang H., Gao Z., Liu X., Agarwal P., Zhao S., Conroy D.W., Ji G., Yu J., Jaroniec C.P., Liu Z. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat. Commun. 2018;9:562. doi: 10.1038/s41467-018-02915-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okon I.S., Zou M.H. Mitochondrial ROS and cancer drug resistance: implications for therapy. Pharmacol. Res. 2015;100:170–174. doi: 10.1016/j.phrs.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giménez-Bonafé P., Tortosa A., Pérez-Tomás R. Overcoming drug resistance by enhancing apoptosis of tumor cells. Curr. Cancer Drug Targets. 2009;9:320–340. doi: 10.2174/156800909788166600. [DOI] [PubMed] [Google Scholar]
- 35.Liu R., Ström A.L., Zhai J., Gal J., Bao S., Gong W., Zhu H. Enzymatically inactive adenylate kinase 4 interacts with mitochondrial ADP/ATP translocase. Int. J. Biochem. Cell Biol. 2009;41:1371–1380. doi: 10.1016/j.biocel.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chao D.T., Korsmeyer S.J. BCL-2 family: regulators of cell death. Annu. Rev. Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z., Chen Y., Sulman E.P., Xie K., Bögler O. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang S., Guan H., Lin X., Li N., Geng F., Li J. METTL3 serves an oncogenic role in human ovarian cancer cells partially via the AKT signaling pathway. Oncol. Lett. 2020;19:3197–3204. doi: 10.3892/ol.2020.11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H., Xu B., Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076. doi: 10.1016/j.gene.2019.144076. [DOI] [PubMed] [Google Scholar]
- 40.Zhou C., Zhong Q., Rhodes L.V., Townley I., Bratton M.R., Zhang Q., Martin E.C., Elliott S., Collins-Burow B.M., Burow M.E., Wang G. Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells reveals expression signatures associated with enhanced migration. Breast Cancer Res. 2012;14:R45. doi: 10.1186/bcr3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 42.Cheong A., Low J.J.A., Lim A., Yen P.M., Woon E.C.Y. A fluorescent methylation-switchable probe for highly sensitive analysis of FTO N6-methyladenosine demethylase activity in cells. Chem. Sci. (Camb.) 2018;9:7174–7185. doi: 10.1039/c8sc02163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y., Deng X., Wang Y., Weng X., Hu C. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172:90–105.e23. doi: 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu L., Amato N.J., Wang P., McGowan S.J., Niedernhofer L.J., Wang Y. Simultaneous quantification of methylated cytidine and adenosine in cellular and tissue RNA by nano-flow liquid chromatography-tandem mass spectrometry coupled with the stable isotope-dilution method. Anal. Chem. 2015;87:7653–7659. doi: 10.1021/acs.analchem.5b00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Györffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J., Li Q., Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.