

Abstract
While gene and cell therapies have emerged as promising treatment strategies for various neurological conditions, heavy reliance on viral vectors could hamper widespread clinical implementation. Here, we explored the use of Tissue Nano-Transfection (TNT) as a platform nanotechnology to drive non-viral gene delivery to nerve tissue via nanochannels, in an effective, controlled, and benign manner. TNT facilitated plasmid DNA delivery to the sciatic nerve of mice in a voltage-dependent manner. Compared to standard bulk electroporation (BEP), TNT did not cause impairment in toe-spread and pinprick response, and had limited to no impact on electrophysiological parameters. BEP, however, induced significant nerve damage and increased macrophage immunoreactivity. TNT was subsequently used to deliver vasculogenic cell therapies to crushed nerves via delivery of reprogramming factor genes Etv2, Foxc2, and Fli1 (EFF). Our results indicate the TNT-based delivery of EFF in a sciatic nerve crush model led to increased vascularity, reduced macrophage infiltration, and improved recovery in electrophysiological parameters compared to crushed nerves that were TNT-treated with sham/empty plasmids. Altogether, our results indicate that TNT could be a powerful platform nanotechnology for localized non-viral gene delivery to nerve tissue, in vivo, and the deployment of reprogramming-based cell therapies for nerve repair/regeneration.
Keywords: Tissue nano-transfection, non-viral gene delivery, peripheral nerve
1. INTRODUCTION:
Peripheral nerve injuries (PNIs) are often multi-faceted and present various degrees of severity, potential ischemia, tissue deficits, etc.[1,2] In many cases, treatment of the nerve injury is delayed for several weeks to handle more immediate concerns of vascular and orthopedic stabilization, and the prevention of infections.[3,4] Delaying PNI treatment, however, can result in severe long-term consequences.[4] Therefore, there is a clear need for early intervention strategies that could either lead to a paradigm shift in PNI treatment, or significantly curve progressive degeneration until surgical management can be performed. Today’s treatments for PNI, however, have been greatly informed by direct experiences from old military conflicts.[5,6] Surgical management typically involves the use of cellular autografts, which possess key cues (e.g., cellular, molecular, structural) to support nerve tissue repair.[7–13] However, complete functional recovery is often hindered due to the extended distance requiring regeneration, and the body’s inability to drive endogenous repair processes for prolonged periods of time.[6] Additional concerns include donor site morbidity and donor tissue scarcity.[14] While acellular nerve allografts (ANAs) could potentially circumvent these issues, ANAs lack important cues for nerve tissue repair (e.g., Schwann and endothelial cells) and as such have poorer outcomes.[15–17] Alternative strategies focused on the use of scaffolds or other means typically fail to address distal effects from Wallerian degeneration, and/or fall short in being able to sustain growth over large defects.[6,18,19] Therefore, alternative approaches combining cellular/molecular and surgical innovation are needed to fully address PNI treatment.
Therapies solely based on the administration of neurotrophic/angiotrophic factors have had limited impact.[20–23] While gene therapies have shown promise in preclinical studies,[24,25] heavy reliance on viral vectors could hamper widespread clinical implementation due to safety concerns stemming from potentially adverse virus-host interactions/immunity, as well as capsid size constraints.[26–30] Cell therapy has emerged as a promising alternative strategy for the treatment of nerve injury.[20,22,31–34] Angiogenic cell therapies, in particular, have attracted a great deal of attention due the synergistic nature of angiogenesis and neurogenesis during nerve tissue repair/development,[35–38] with newly formed vasculature, for example, acting as a neurotrophic scaffold to support axonal growth.[38–40] Most cell therapies developed so far, however, rely on progenitor-like cells, which tend to be scarce and difficult to isolate, or pose major risks in terms of uncontrolled differentiation, tumorigenesis, immunogenicity, etc.[41–43] Recent advances in direct reprogramming could enable the development of autologous cell therapies that are based on more readily available cell sources (e.g., fibroblasts), and mitigate risks associated with progenitor cells.[44,45] However, similar to gene therapy, current approaches to reprogramming-based cell therapies are fraught with caveats, including the need for viral vectors and extensive ex-vivo pre-processing (e.g., isolation, expansion, viral transformation, etc.), precluding them as a solution compatible with early intervention strategies. Thus, novel non-viral tools for gene- and reprogramming-based cell therapies are needed to facilitate early intervention in PNI.
Recently we developed a Tissue Nano-Transfection (TNT) technology to deliver non-viral gene- and reprogramming-based cell therapies to skin tissue, in vivo, via nanochannels.[46] Compared to standard bulk electroporation (BEP), nanochannel-based poration minimizes electric field-driven damage to cells, and leads to enhanced cargo delivery.[45–52] TNT was successfully used to drive reprogramming-based vasculogenic cell therapies in the skin via co-delivery of Etv2, Foxc2, and Fli1 (EFF) genes, which can induce direct conversion of fibroblasts into induced endothelial cells (iECs).[46] Here we explored, for the first time, the use of TNT as a platform nanotechnology for the delivery of gene and reprogramming-based cell therapies to peripheral nerve tissue.
2. RESULTS:
To examine whether TNT can effectively deliver genetic cargo to peripheral nerves, we proceeded to conduct gene delivery experiments in the sciatic nerve of 8–10-week-old C57BL/6 mice using fluorescently labeled plasmid DNA (PCMV6, 4.9 kb, Origene) as model cargo (Figure 1). The sciatic nerves were surgically accessed through a longitudinal incision, posterior and parallel to the femur, and the TNT platform was placed against the exposed nerve surface prior to applying a pulsed electric field across electrodes (Figure 1 a). TNT conditions included 10 pulses of 200 Volts (V) and a duration of 10 millisecond (ms) per pulse with a 100 ms interval, for a total duration of 1 second per run. The sciatic nerve was collected within 5 minutes after gene delivery, and subsequently sectioned and processed for inspection via fluorescence microscopy. Imaging of TNT-treated nerve tissue revealed that under these conditions, the delivered plasmid DNA accumulated preferentially within the epineurium (Figure 1 d), the outermost layer of the nerve, which is made from dense irregular connective tissue (e.g., collagen, fibroblasts), and plays a key protective role (e.g., mechanical and diffusion barrier) to the underlying axons.[53–55]
Fig. 1.
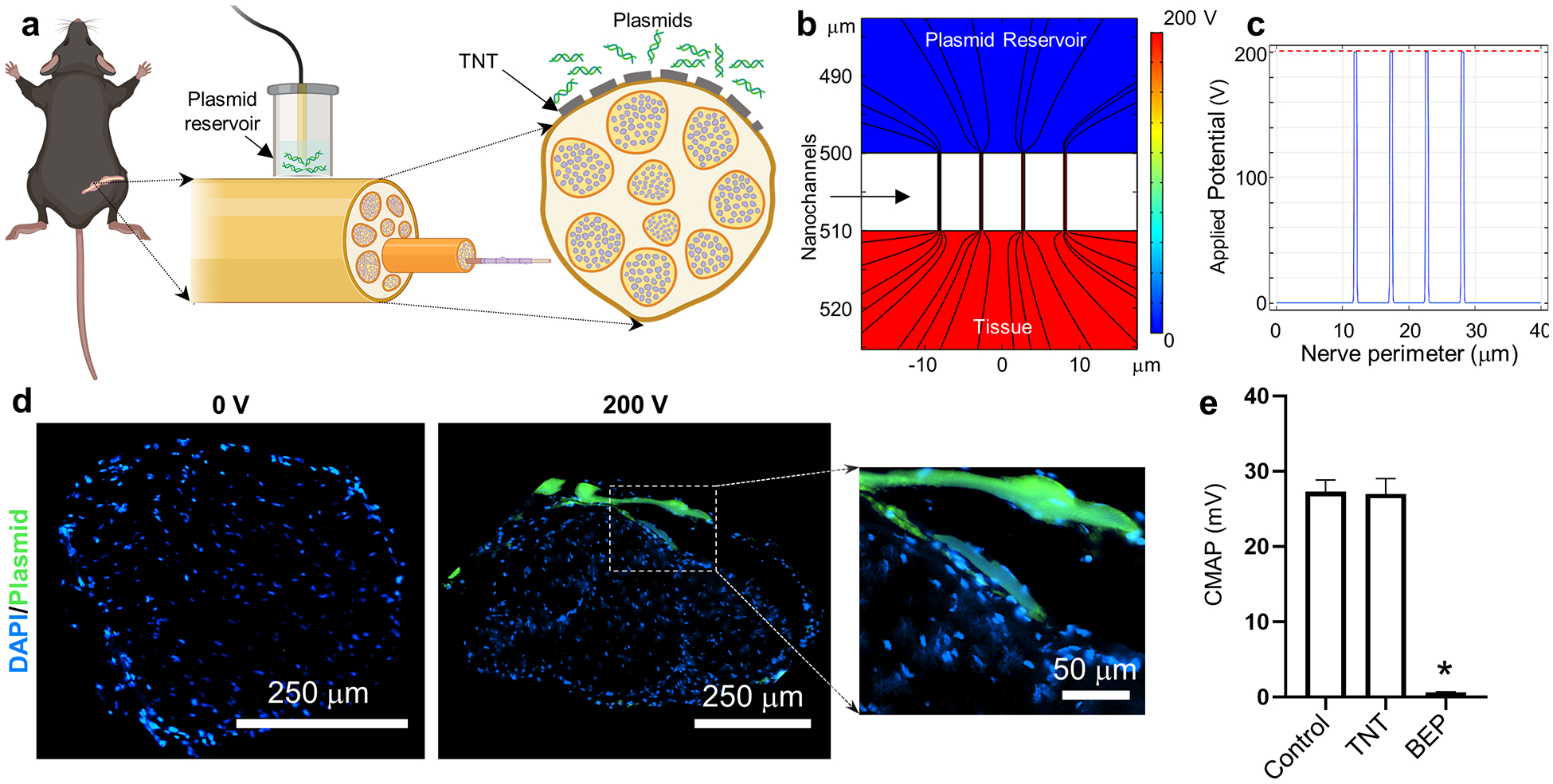
TNT can be used to deliver nucleic acids to genes in a safe and efficient manner. (a) Schematic diagram of the experimental procedure. The sciatic nerve is first surgically exposed, and the nanochanneled surface of the TNT platform is put in direct contact with the nerve. A negative electrode is immersed into the plasmid reservoir and a positive electrode is inserted into the muscle adjacent to the nerve. A pulsed electric field is then applied across electrodes to nanoporate the tissue surface and electrophoretically drive nucleic acids into the nerve. (b, c) Finite element modeling simulations of the electric field distribution when the poration is mediated by nanochannels. Dashed red line shows the voltage distribution for bulk electroporation. Nanochannel-mediated poration enables focused implementation of the electric field (solid blue line). (d) Fluorescence micrograph of the nerve cross-section following TNT with labeled plasmid DNA (green) at 0 vs. 200 volts. The plasmid DNA accumulates preferentially within the epineurium of the TNT-treated nerve surface. Inset to the right shows higher magnification image of the labeled plasmid within the epineurium. (e) Neuromuscular activity was evaluated via compound muscle action potential (CMAP) measurements in mice that underwent surgery to expose the sciatic nerve (control) vs. mice that underwent the surgery in addition to TNT- or BEP-based poration of the nerve (n = 3). While BEP led to a significant decrease in CMAP amplitude, TNT did not cause any significant changes. Mean ± sem, *p<0.001 vs. control (One Way Anova).
To evaluate if TNT had a negative impact on functionality due to activation of an innate immune response or axonal damage, motor function was assessed 3 days post-TNT via compound muscle action potential (CMAP) measurements recorded from the sciatic innervated triceps surae muscle following sciatic nerve stimulation.[56,57] Our results indicate that TNT had no detrimental impact on functionality (Figure 1 e). To evaluate if standard BEP elicited similar responses, the plasmid solution was first injected into the sciatic nerve, and the nerve trunk was subsequently gently secured between two plate electrodes before applying a bias of 200 V (i.e., 10 pulses of 10 ms each) across electrodes (Supplementary Figure 1). Day-3 CMAP measurements for BEP-treated nerve, however, revealed that when the same conditions were applied under a BEP setup, there was a significant (i.e., ~97%) reduction in CMAP amplitude (Figure 1 e), clearly suggesting that BEP had a negative impact on neuromuscular function. Simulation studies indicate that nanochannel-based implementation of an electric field results in highly localized tissue poration (Figure 1 b, c) compared to BEP (Supplementary Figure 1), which presumably has a cytoprotective effect on highly sensitive electrically excitable/electrogenic tissue such as nerve tissue.[52] CMAP measurements in mice that had undergone sham surgeries (i.e., exposing the nerve but no TNT or BEP-driven electrical stimulation) revealed that the surgical procedure itself had no detrimental impact on neuromuscular function (Supplementary Figure 2), thus suggesting that the decrease in CMAP amplitude was primarily caused by the BEP procedure.
While the cells that reside in the epineurium (e.g., fibroblasts, immune cells) could represent a target of therapeutic interest for a number of conditions (e.g., demyelinating polyneuropathy, vasa nervorum disruption),[58,59] non-viral delivery of genetic cargo deeper into the nerve could potentially enable a wider range of therapeutic applications, as more cellular/molecular targets become available, including Schwann cells, endothelial cells, fibroblasts, immune cells, etc.[60,61] Therefore, we proceeded to develop a benign method to gently exfoliate the nerve to remove the epineurial barrier prior to TNT. We found that applying 0.25% trypsin for 5 minutes to the surgically exposed nerve surface in 8–10-week-old C57BL/6 mice successfully removed the outermost tissue layer of the sciatic nerve (Supplementary Figure 3). Functionality tests revealed that the exfoliation treatment had no negative impact on CMAP amplitude (Supplementary Figure 4). Moreover, histological analyses performed at days 3 and 7 post-exfoliation revealed that the epineurial barrier has the ability to regenerate (Supplementary Figure 3). Once we established that the trypsin treatment can be used to benignly and reversibly remove the epineurial barrier, we proceeded to run TNT experiments on exfoliated nerve tissue under different voltage conditions, and using fluorescently labeled plasmid DNA (PCMV6, 4.9 kb, Origene) as model cargo (Figure 2 a). The tissue was collected immediately after TNT and processed and analyzed via fluorescence microscopy. Imaging of the nerve tissue sections revealed that the delivery extent of plasmid DNA into the nerve can be modulated by the magnitude of the applied voltage and pulse length (Figure 2 b–e). Thus, these results confirm that trypsin-based exfoliation is a viable method to gain access to inner nerve tissue for TNT-driven gene delivery applications.
Fig. 2.
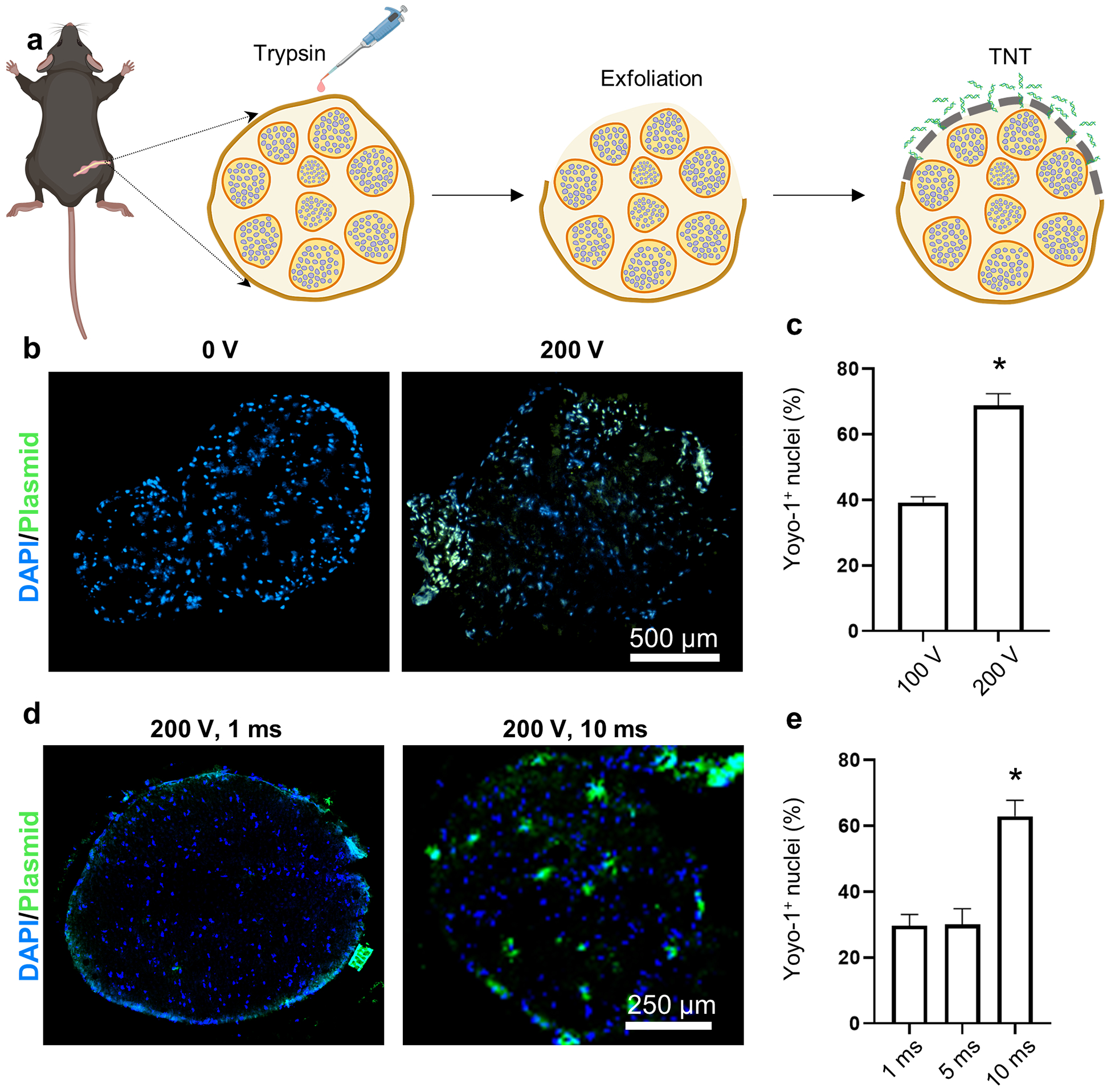
Trypsin treatment gently removes the epineurial membrane and enables nanochannel-mediated delivery of nucleic acids into the nerve core. (a) Schematic diagram of the experimental procedure. The exposed sciatic nerve surface is treated with 0.25% trypsin for 5 minutes to remove the epineurium prior to conducting TNT. The delivery efficiency (% of Yoyo-1+ nuclei) can be modulated via the (b, c) applied voltage or (n = 3–4) (d, e) pulse length (n = 3). Mean ± sem, *p<0.002.
To further contrast the performance of TNT vs. BEP, and to better understand the underlying principles driving impaired functionality in BEP, we proceeded to conduct gene delivery experiments with TNT and BEP at 200 V (i.e., 10 pulses, 10 ms/pulse) in exfoliated sciatic nerves of 8–10-week-old C57BL/6 mice, using plasmid DNA (PCMV6, 4.9 kb, Origene) as model cargo. Functional outcomes post-gene delivery were measured in terms of toe spread and pinprick response, and histological analyses of nerve damage and inflammatory markers were also conducted at day 3 post-gene delivery (Figure 3 a). While toe spread and pinprick assessments indicated that TNT and direct injection of the plasmid into the nerve prior to BEP yielded similar responses compared to untreated healthy control nerve tissue, histological analysis of nerve damage with Fluorojade-C (FJ-C) showed that plasmid injection, which is inherently needed for BEP-based gene delivery, can induce some degree of nerve damage (Figure 3 b). No FJ-C signal was detected for TNT-treated nerves. In addition, when plasmid injection was followed by BEP at 200V, histological damage was not only clearly visible by FJ-C staining, but abnormal toe spread and pinprick response were also recorded in all the mice subjected to BEP (Figure 3 a, b). Interestingly, analysis of the macrophage marker, F4/80, and the cellularity of the cross-section, revealed that only the nerves that underwent plasmid injection + BEP showed significantly increased immunoreactivity for F4/80, and a marked decrease in cellularity (i.e., DAPI signal) per cross-section (Figure 3 c, d), suggesting that the BEP procedure itself is cytotoxic to the nerve, and is conducive to more prolonged inflammation compared to the TNT procedure or plasmid injection alone, which could potentially contribute to the functional decline seen in the BEP group.[62–64]
Fig. 3.
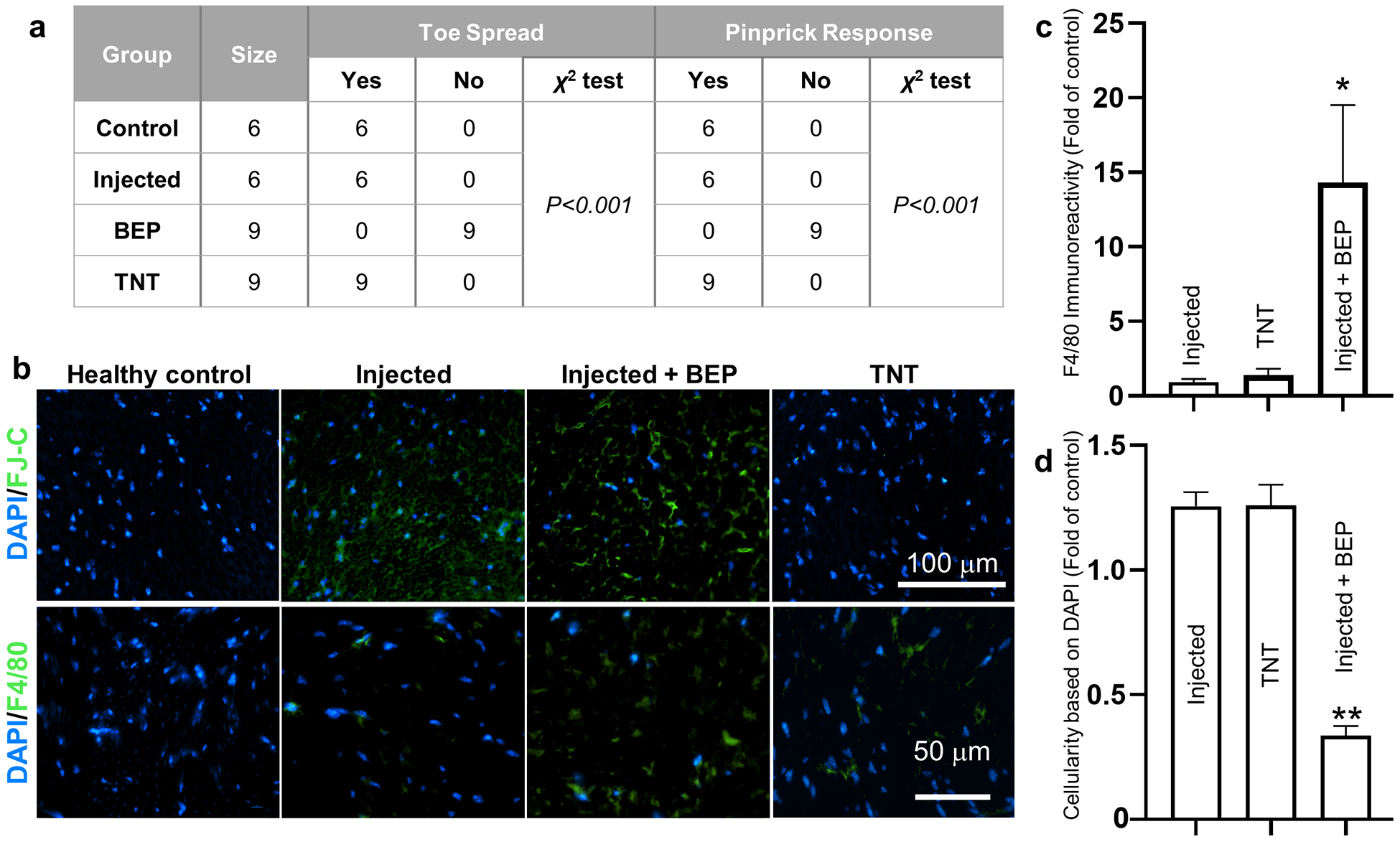
BEP leads to functional impairment, increased macrophage infiltration, and reduced cellularity. (a) Assessment of toe spread and pinprick response in mice subjected to TNT, plasmid injection, and plasmid followed by BEP (n = 6–9). Untreated mice served as control. (b) Fluorescence micrographs showing histological changes in response to TNT, plasmid injection, and plasmid injection + BEP. While plasmid injection with or without BEP appeared to lead to some degree of histological damage (t= 3 days post-injection/BEP), only the BEP group led to a significant increase in (c) macrophage marker immunoreactivity (F4/80) (n = 3–4), and a (d) marked decrease in cellularity (n = 3). Mean ± sem, *p<0.05 **p<0.001 vs. control/healthy nerve tissue (One Way Anova).
Once we established that TNT represented a safe and effective approach to deliver genetic cargo into nerve tissue, we proceeded to evaluate whether TNT can be used to drive reprogramming-based vasculogenic cell therapies in injured nerves (Figure 4 a). Vascular cell therapies have been shown to improve nerve tissue repair under neurodegenerative conditions.[35–40] To test this, we first performed an crush injury to the sciatic nerve of 8–10-week-old C57BL/6 mice using well-established procedures.[65–67] Briefly, the surgically exposed nerve was crushed with locked forceps, applying 2 mid-level crushes for 15 seconds (s), with a 15 s release time. This was immediately followed by trypsin-based exfoliation and TNT-based delivery of a vasculogenic reprogramming gene cocktail of Etv2, Foxc2, and Fli1 (EFF), which we had previously reported to drive vascular cell therapies in the skin.[46] TNT with sham/empty plasmids served as control. TNT conditions included 10 pulses of 200 V and a duration of 10 ms/pulse. Positive immunoreactivity for the Myc-DDK tag protein coupled with qRT-PCR analyses confirms successful expression of the delivered plasmids (Supplementary Figure 5). Histological analyses of the nerve at day 7 post-crush and TNT indicated that TNT-based delivery of EFF correlated with a significant increase in immunoreactivity for vascular markers, vWF and CD31, compared to crushed nerve tissue that was TNT-treated with sham/empty plasmids (Figure 4 b, c). No significant differences were noted between crushed/EFF-treated nerve and healthy/uncrushed controls (Figure 4 c). Notably, when neuromuscular function was evaluated via CMAP measurements, we found that while the crush injury had a clear negative impact on CMAP amplitude at t=7 days post-crush injury, the mice that were treated with EFF TNT showed improved and more pronounced recovery as early as t= 14 days post-injury compared to baseline measurements at day 7 (Figure 4 d). Mice treated with sham TNT, on the other hand, showed significantly reduced recovery in CMAP, suggesting that the TNT-driven vasculogenic intervention had a positive impact on the recovery rate. Analysis of macrophage marker, F4/80, showed that crushed nerve tissue TNT-treated with sham/empty plasmids exhibited more pronounced immunoreactivity compared to crushed nerve tissue TNT-treated with EFF and/or healthy/uncrushed nerve (Figure 5 a, b). Both sham- and EFF-treated nerves showed similarly elevated levels of immunoreactivity for S100B at day 7 post injury/intervention compared to healthy/uncrushed nerves (Figure 5 a, c), presumably reflective of increased Schwann cell and fibroblast activity in response to the crush injury.[68] Immunostaining for axonal marker, neurofilament heavy chain (NF), showed a significant decrease in immunoreactivity for crushed nerves treated with sham TNT compared to healthy control/uncrushed nerve tissue (Figure 5 a, d). No significant differences in NF immunoreactivity were noticed between healthy controls and crushed nerves treated with EFF TNT, suggesting that the vasculogenic intervention had a positive impact in the preservation of axonal processes following crush injury.
Fig. 4.
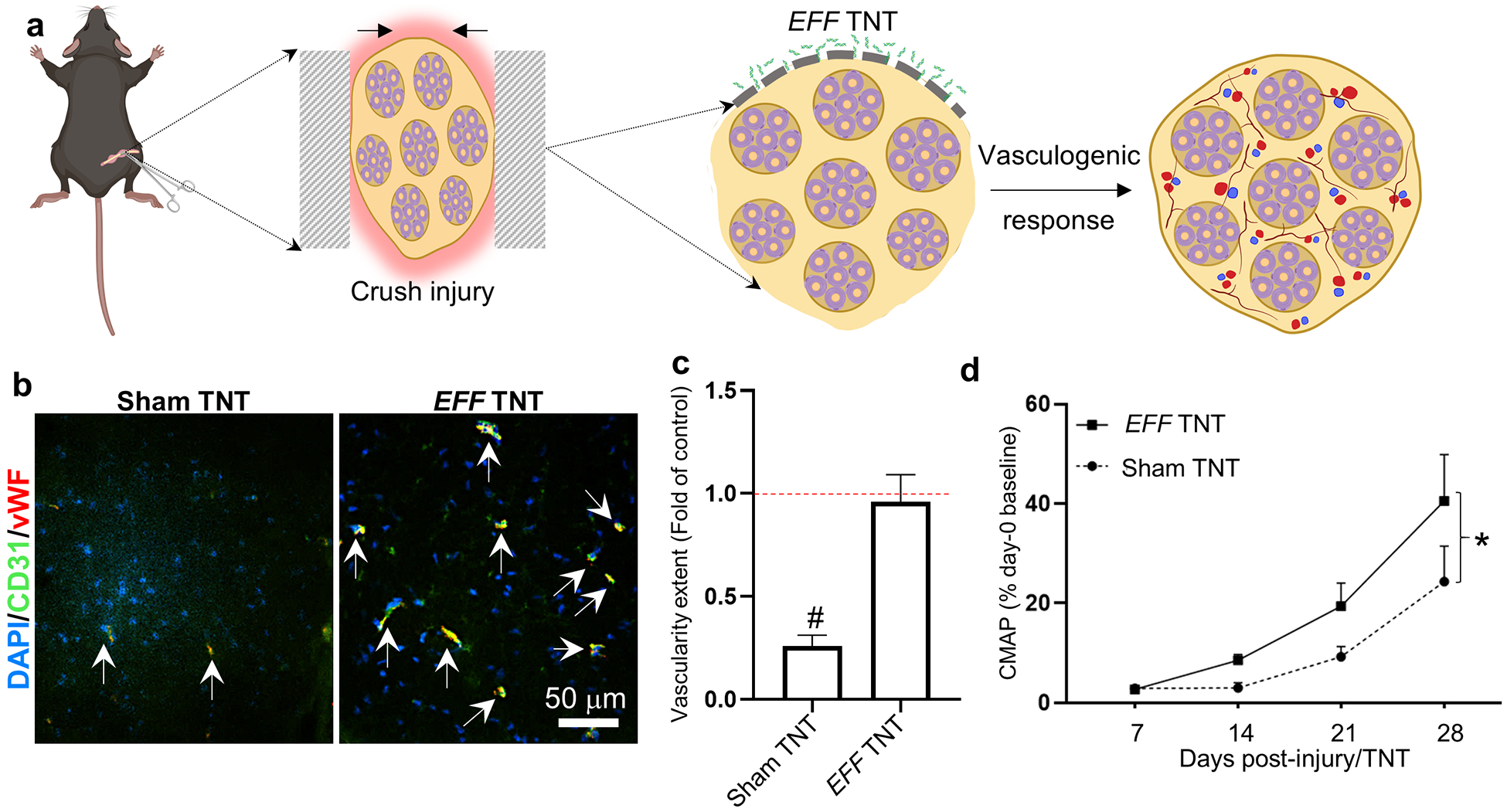
TNT-based delivery of EFF to crushed nerve tissue leads to an increase in vascularity and correlates with improved functional outcomes. (a) Schematic diagram of the experimental design. The sciatic nerve was crushed, exfoliated, and TNT-treated with EFF or sham/control plasmids. Histological changes were evaluated at day 7 post-injury/treatment, and changes in neuromuscular function were assessed via CMAP measurements at days 7–28 post-injury/treatment. (b) Histological analysis of vascularity confirmed (c) increased immunoreactivity for CD31 and vWF in EFF-treated nerves compared to sham (n = 3). (d) CMAP measurements show significant recovery for both groups over time (p=0.0003) and reveals that TNT-based delivery of EFF led to significantly improved recovery compared to baseline measurements (n = 5–6). Significant spontaneous recovery in sham-treated mice was not seen until 28 days post-injury. Mean ± sem, #p<0.005 vs. control/healthy nerve tissue (One Way Anova), * p<0.05 (repeat measure, mixed effect analysis).
Fig. 5.
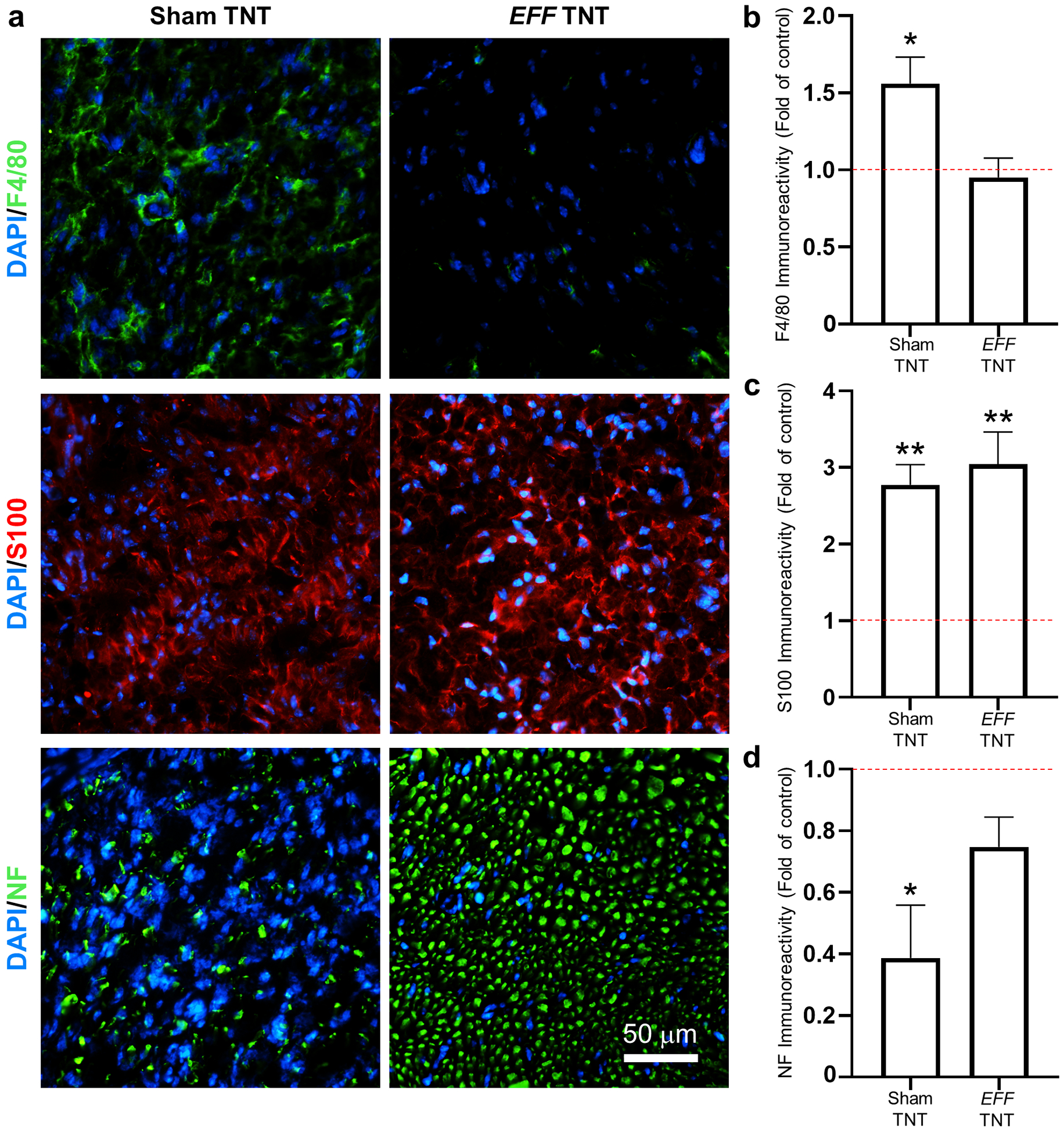
TNT-based delivery of EFF to crushed nerve tissue correlates with reduced macrophage infiltration and improved protection/regeneration of axonal processes. (a) Immunofluorescence staining and (b-d) quantification of F4/80, S100, and Neurofilament (NF) immunoreactivity in sham- vs. EFF-treated crushed nerve tissue (n = 3). Mean ± sem, *p<0.05 **p<0.006 vs. control/healthy nerve tissue (One Way Anova).
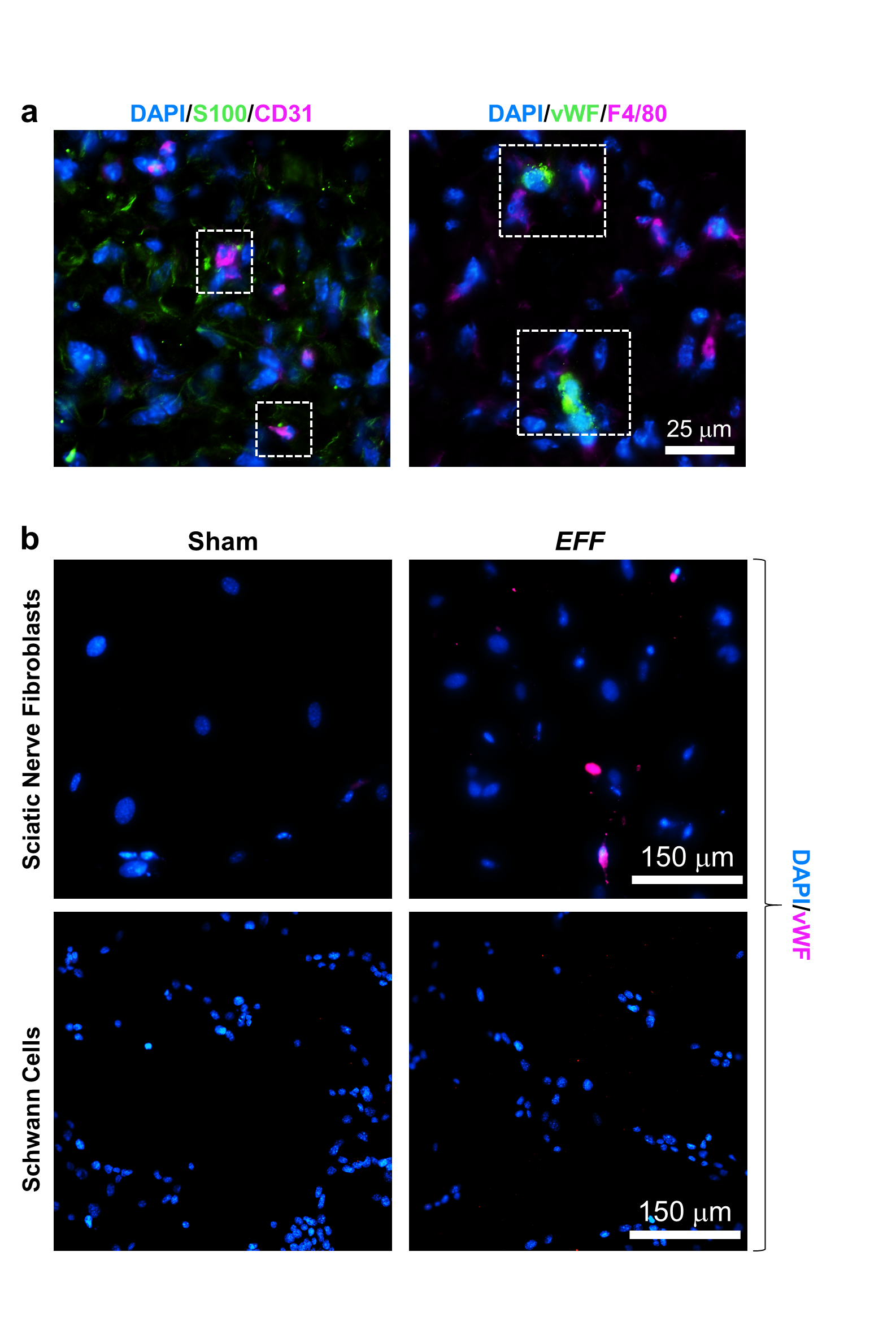
The increase in vascular tissue immunoreactivity in response to TNT-based delivery of EFF is likely due to the combined effect of paracrine angiogenesis from pre-existing blood vessels, and lineage conversions between somatic cells towards an induced endothelial cell (iEC) phenotype.[46] To investigate the extent to which specific subpopulations of nerve-resident cells were responsive to EFF-driven vasculogenic reprogramming, we proceeded to run ex vivo nanoelectroporation (NEP) experiments with EFF plasmids and select cell populations of the sciatic nerve.[45,46] High magnification co-immunofluorescence imaging of vascular markers (e.g., CD31, vWF) and S100B or F4/80 indicate that some of the vascular cells observed in the cross-sections of crushed nerves TNT-treated with EFF showed potential traces of S100B immunoreactivity (Supplementary Figure 6). No such traces were observed for F4/80, thus suggesting that some of the iECs may be originating from S100B+ cell populations. As such, we proceeded to evaluate the reprogrammability of Schwann cell cultures and sciatic nerve fibroblast cultures into iECs in response to NEP of EFF. NEP of sham/empty plasmids served as control. Immunofluorescence analysis of the Schwann cell and fibroblast cultures 7 days post-NEP revealed the presence of cells that stained positive for vascular markers in the fibroblast cultures that were NEP-treated with EFF (Supplementary Figure 6). No such cells were seen in fibroblast cultures that were NEP-treated with sham/empty plasmids, or Schwann cell cultures NEP-treated with EFF or sham plasmids (Supplementary Figure 6), thus suggesting that sciatic nerve fibroblasts are more prone to exhibiting vasculogenic plasticity compared to Schwann cells, which could be of relevance to reprogramming-based vasculogenic cell therapies for peripheral nerve tissue.
3. CONCLUSION
In conclusion, we demonstrated that TNT can be used to safely and effectively deliver genetic cargo to peripheral nerve tissue, non-virally, which could potentially be used to drive a myriad of gene and/or cell therapies of relevance to neuropathic and neurodegenerative conditions. Benchmarking studies with BEP indicated that TNT-based delivery of genetic cargo led to little to no damage to the nerve cytoarchitecture, and had negligible impact on inflammatory cell infiltration and neuromuscular function. BEP, on the other hand, caused visible damage to nerve tissue, promoted marked inflammatory cell infiltration, and had a detrimental impact on neuromuscular function, thus suggesting that nanoscale confinement of electroporation in TNT is better suited for electric field-based delivery of genetic cargo to nerve tissue. Subsequent studies in a mouse model of crush nerve injury indicate that TNT-based delivery of a vasculogenic gene cocktail of Etv2, Foxc2, and Fli1 (EFF), which we had previously reported to mediate reprogramming of dermal fibroblasts into induced endothelial cells,[46] leads to improved injury outcomes compared to TNT-based intervention with sham/empty plasmids, including increased vascularization, reduced inflammation, improved preservation of axonal processes, and speedier recovery of neuromuscular function. Altogether, these studies suggest that TNT is a powerful platform nanotechnology for non-viral gene delivery to nerve tissue, and the deployment of reprogramming-based cell therapies of potential relevance to a wide variety of conditions.
4. METHODS
Plasmid preparation
All plasmids were purchased from Origene (Table 1). All plasmids were expanded via bacterial inoculation and purified as directed by the protocol for plasmid purification (ZymoPURE II Plasmid Midiprep Kit, cat. no. D4201). Concentrations of isolated plasmids were obtained using a Nanodrop 2000c Spectrophotometer (ThermoFisher Scientific). The plasmids were labeled for some experiments using Yoyo-1 Iodide (cat. no. Y3601, ThermoFisher Scientific) following the instructions provided by the manufacturer.
Table 1.
Plasmid information.
Animal husbandry
All animal procedures were approved by the Animal Care and Use Committee of The Ohio State University (2016A00000074-R1). C57BL/6J mice were purchased from Jackson Laboratory. Mice were 8–10 weeks at the time of experimentation. Both male and female sexed mice were included in the studies. All animals were anesthetized via isoflurane inhalation before experimental manipulations.
Sciatic nerve exposure, crush injury, and gene delivery
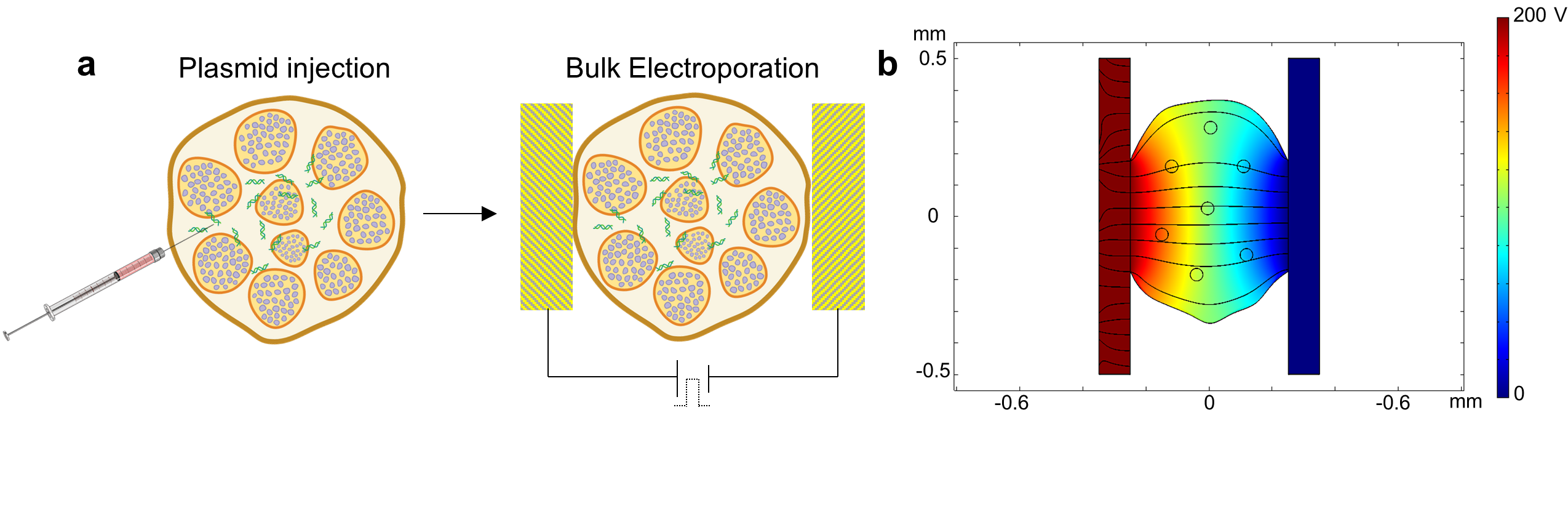
A skin and muscle incision of ~1cm was made in the medial aspect of the limb to expose the sciatic nerve. Sciatic nerves were separated from the surrounding tissue and fascia using Vannas spring scissors. For the crush injury model, the sciatic nerve was crushed using 1-mm-wide hemostatic forceps (3 clicks) 2 times for 15 seconds. Prior to the second crush, the forceps were dipped in carbon powder to mark the site of injury. For TNT experiments, a track-etched Polyethylene terephthalate (PET) membrane with a pore size of ~400 nm and a density of ~108 pores/cm2 was adapted into a TNT platform. The basal surface of the membrane was gently pressed against the sciatic nerve, with the apical compartment/membrane containing the plasmid solution (at a 0.05 μg/μl per plasmid concentration) and the negative electrode. A positive needle electrode was then inserted into the biceps femoris, and a pulsed electric field (0–200V, 10 ms pulses, 10 pulses) was applied across electrodes to drive plasmid DNA across the nanochannels and into the nerve. For BEP experiments, the plasmid solution (0.05 μg/μl) was pre-injected into the nerve using a 1cc U-100 insulin syringe (BD 329424). The nerve was subsequently gently clamped between two plate electrodes, and a pulsed electric field (200V, 10 ms pulses, 10 pulses) was applied across electrodes to facilitate electroporation-based plasmid uptake. Exfoliation (i.e., epineurium removal) of the sciatic nerve was conducted for some experiments by applying 0.25% trypsin (50–100μl) to the exposed nerve surface for a total of 5 minutes. The trypsin was then removed via cotton tip applicator prior to DNA delivery experiments. COMSOL Multiphysics version 5.4 was used to study and compare the electric field distribution for both BEP and TNT. Static electric field physics was used to analyze the physical model set up. For BEP, a nerve cross-section of 500 μm was sandwiched between two copper plates and voltage of 200 V was applied. A conductivity of 0.2 S/m was used for the nerve tissue. For TNT we used 400 nm diameter and 10 mm-long nanochannels in direct contact with the nerve surface. Positive electrode was modeled underneath the nerve to replicate the experimental set up. An electric potential of 200V was applied between the top surface and the positive electrode under the nerve. The conductivity for plasmid, nerve, and membrane were 0.8 S/m, 0.2 S/m and 5×10−7 S/m, respectively.[52]
Nerve functionality measurements
Outcomes of toe-spread and pinprick were documented and recorded after visual observations prior to treatment and at the time of tissue collection[69]. These measurements were reported in a binary manner. Compound muscle action potentials were recorded from the triceps surae muscle using a Sierra Summit EMG System (Cadwell, Kennewick, WA), as previously described. [70–72] Briefly, stimulation was applied to the sciatic nerve supramaximally. CMAP amplitudes were recorded using two ring electrodes (Catalog # 9013S0312, Natus Neurology, Middleton, Wisconsin, USA); the active electrode was positioned over triceps surae muscles, whereas the reference electrode was positioned on the foot in the mid-metatarsal region. A ground electrode was placed on the animal’s tail. Amplitudes were measured peak-to-peak.
Immunohistochemistry
Harvested nerve tissue was embedded in optimal cutting temperature (OCT) solution and frozen for cryosectioning. Immunostaining was performed using specific antibodies and standard procedures. Briefly, OCT-embedded tissues were cryosectioned at 10 μm thickness, fixed in cold methanol, and blocked for non-specific binding with either 10% normal goat serum or 10% BSA. Tissue samples were incubated with specific antibodies diluted in the blocking solutions overnight at 4°C (Table 2). The signal was visualized by subsequent incubation with appropriate fluorescence-tagged secondary antibodies Alexa 488-tagged α-chicken, 1:200; (Alexa 488-tagged α-mouse, 1:200; Alexa 488-tagged α-rabbit, 1:200; Alexa 568-tagged α-rabbit, 1:200, Alexa 647-tagged α-rat, 1:200) before being counter-stained with DAPI. Images were captured on an inverted fluorescence microscope (Nikon Ti-2e).
Table 2.
Antibody information.
| Target | Company | Catalog Number | Species | Dilution |
|---|---|---|---|---|
| CD31/PECAM-1 | BD Pharmigen | 550274 | Rat | 1:50 |
| DDK (FLAG) | Origene | TA50011 | Mouse | 1:500 |
| F4/80 | ThermoFisher | 14-4801-85 | Rat | 1:50 |
| Neurofilament heavy polypeptide (NF-H) | Abcam | ab72996 | Chicken | 1:400 |
| S100B | Abcam | ab52642 | Rabbit | 1:500 |
| Von Willebrand Factor (vWF) | Abcam | ab6994 | Rabbit | 1:500 |
Histological analysis
Quantitative image analysis was performed using ImageJ (National Institutes of Health). Cellularity was calculated as total number of cells/mm2. Briefly, identical sized regions of interest were obtained for each sample and the total number of DAPI+ cells counted. The cell count was normalized to the region of interest size and converted from microns to millimeters. Nuclear delivery was calculated as the ratio of localized Yoyo-1+ and DAPI+ nuclei to the total number of DAPI+ cells/nuclei. Briefly, ImageJ was used to manually count DAPI+ cells/nuclei and DAPI+/Yoyo-1+ nuclei. The total for each population was recorded and a ratio/percentage for Yoyo-1+ nuclei was presented. Immunoreactivity analysis was calculated as the area of immunostained nerve tissue. Briefly, images were split by channel and converted to binary images. A threshold algorithm was applied and the sum of positive particles calculated as a percent coverage of the nerve bundle. To calculate vascular coverage, the CD31+ and vWF+ regions were manually traced. The areas of the co-stained regions were summed and converted to a ratio of coverage of the nerve bundle. All analyses were compared to healthy controls and represented as a ratio/fold-change of the normal state. Each analysis was performed based on the average results of 3–4 tissue sections/images from 3–4 biological replicates per group.
In vitro reprogramming assays
To evaluate whether specific cell populations from the nerve were susceptible to reprogramming, we conducted in vitro reprogramming experiments Schwann cells (CRL-2766, ATCC) and primary sciatic nerve fibroblasts.[73] The EFF (Etv2, Foxc2, Fli1) or sham/empty PCMV6 plasmids were delivered into the cells via nanochannel-based or standard electroporation approaches, as described elsewhere.[45,46] The cells were maintained in culture for an additional 7 days post-gene delivery, and then processed for immunostaining against vascular markers, as described above with few modifications. Briefly, the cells were fixed in a 10% formalin solution and permeabilized via 0.1% Triton X-100. Cells were then washed and immunostained.
Gene Expression
Sections of sciatic nerves were used to evaluate transcription factor expression after TNT. Samples were placed TRIzol reagent (Thermofisher Scientific, ref. no. 15596026) and total RNA was extracted according to the manufacturer’s protocol. Subsequently, cDNA was generated through a reverse-transcriptase reaction using SuperScript™ IV VILO™ Master Mix (Thermofisher Scientific, ref. no. 11756500). Targets were detected using FAM labeled TaqMan probes (Table 3; Thermofisher Scientific) and the QuantStudio™ 3 Real-Time PCR System (Thermofisher Scientific). Relative transcript levels were reported as ΔΔCT values, where ΔΔCT is ΔCTTreated – ΔCTControl/Untreated).
Table 3.
Primer information.
| Gene Symbol | Gene Name | Gene Aliases | Species | Company | Ref. no. |
|---|---|---|---|---|---|
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | Gapd | Mouse | Thermofisher Scientific | Mm99999915_g1 |
| Etv2 | ets variant 2 | Etsrp71 | Mouse | Thermofisher Scientific | Mm00468389_m1 |
| Fli1 | Friend leukemia integration 1 | EWSR2, Fli-1, SIC-1, Sic1 | Mouse | Thermofisher Scientific | Mm00484410_m1 |
| Foxc2 | forkhead box C2 | Fkh14, Hfhbf3, MFH-1, Mfh1 | Mouse | Thermofisher Scientific | Mm00546194_s1 |
Statistical Analysis
When possible, coding was used for samples, and blinding introduced in data collection. Reported data is represented as the mean ± standard error of 3–9 biological replicates. In the case of unsuccessful gene delivery or misfires (potentially the result of poor connection between the nanochannels and nerve, or clogging of nanochannels), results were excluded from the analysis. Experiments were replicated at least twice to confirm reproducibility. Comparisons between groups were made by analysis of variance (ANOVA). Statistical differences were determined using parametric/non-parametric tests, as appropriate, with SigmaPlot version 14.0. Toe spread and pinprick data were then evaluated using a chi-square analysis.
Supplementary Material
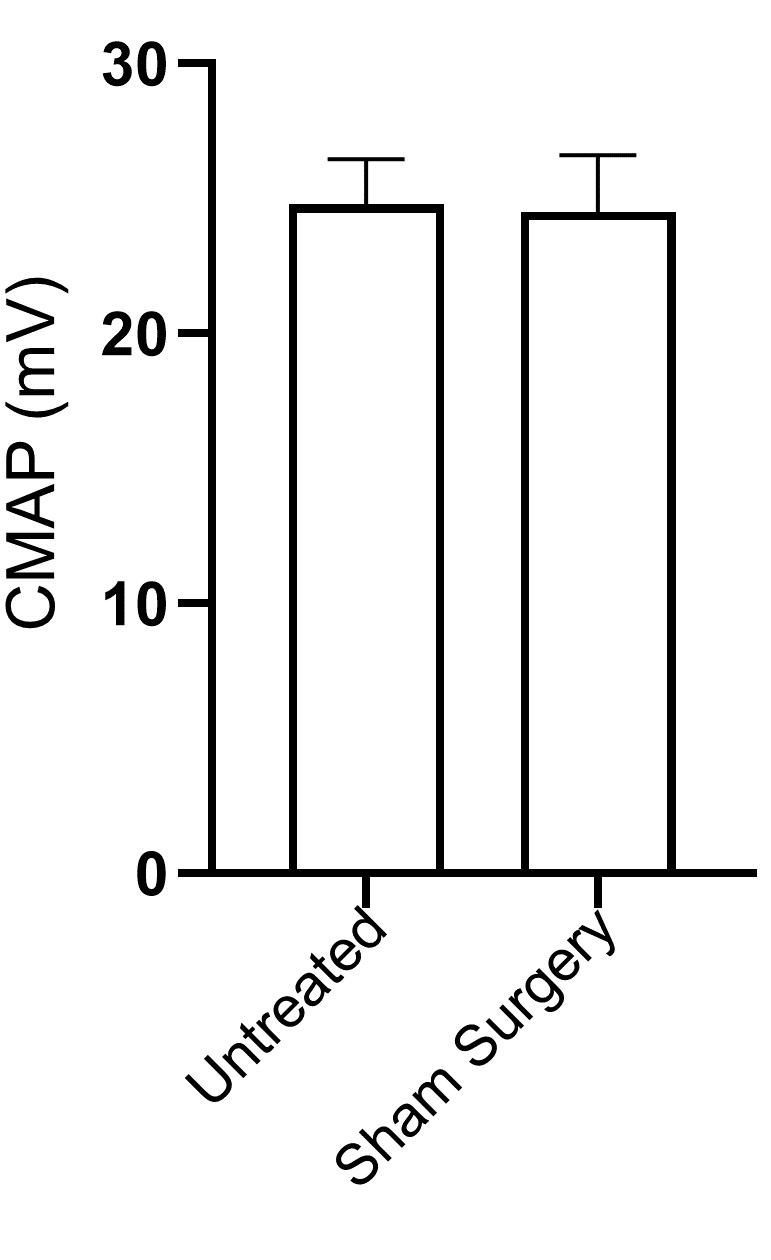
Fig. S2. CMAP measurements in untreated vs. operated mice show that the surgical procedure followed to expose the sciatic nerve does not have a significant impact in neuromuscular function (n = 6).
{kind=link}
Fig. S1. (a) Schematic diagram of the bulk electroporation (BEP) procedure. The plasmid solution is injected into the nerve tissue prior to applying a pulsed electric field between a pair of plate electrodes that are gently clamping the nerve. (b) Finite element modeling simulation showing widespread electric field distribution under BEP.
{kind=link}
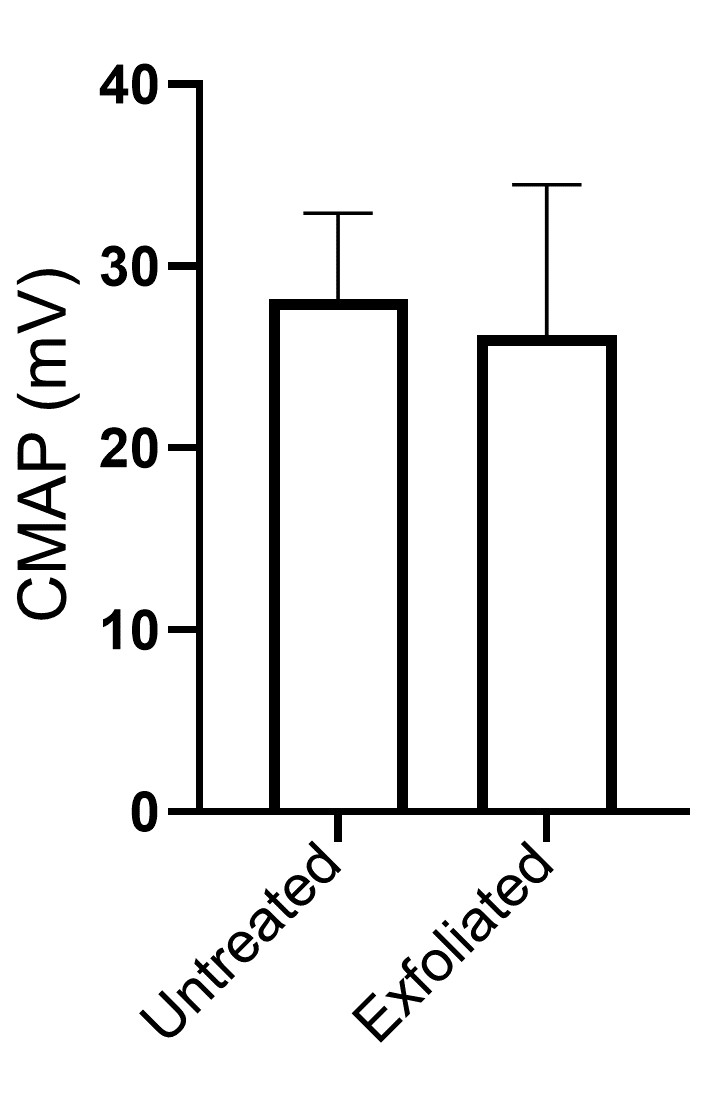
Fig. S4. Trypsin treatment does not have a significant impact in neuromuscular function as assessed by CMAP measurements (n = 7).
{kind=link}
Fig. S5. Successful plasmid expression was (a) confirmed via immunostaining of the Myc-DDK tag protein associated with sham and EFF expression plasmids. Furthermore, qRT-PCR analysis (b) revealed an upregulation of the delivered transcription factors Etv2, Fli1, and Foxc2, in EFF TNT vs. Sham TNT vs. control/untreated nerve (n=3–8). Mean ± sem, *p<0.05.
{kind=link}
Fig. S6. (a) High magnification fluorescence microscopy micrographs show that some CD31+ vascular cells had traces of S100 immunoreactivity (7 days post-crush), potentially suggesting that EFF-driven vascular reprogramming was impacting S100+ cells (e.g., Schwann cells, sciatic nerve fibroblasts). (b) In vitro reprogramming studies with primary sciatic nerve fibroblasts and a Schwann cell (SW10) show that EFF-treated fibroblast cultures showed positive immunoreactivity for vascular cell marker vWF, which were absent in EFF-treated Schwann cell cultures (7 days post-NEP).
{kind=link}
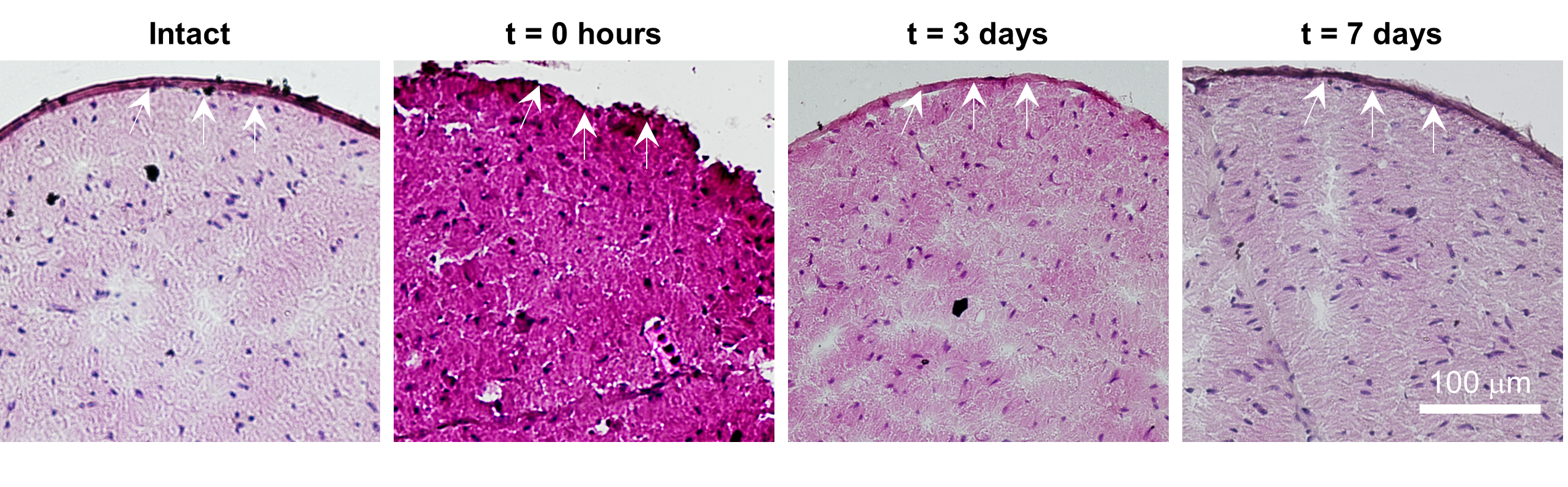
Fig. S3. Effects of trypsin treatment on the sciatic nerve as a function of time. Trypsin exposure leads to removal of the epineurium (t = 0 h), which enables TNT-based delivery of genetic cargo to the core of the nerve. However, the epineurium can regenerate/recover as a function of time (t = days 3 – 7).
{kind=link}
ACKNOWLEDGEMENTS
Funding for this work was provided by NIH/NIBIB via a New Innovator Award to D.G-P. (DP2EB028110), and NIH/NINDS (R21NS099869).
Footnotes
Supporting information
Schematic diagram and simulation results for bulk electroporation, CMAP measurements before and after nerve exposure surgeries; histological (hematoxylin/eosin) analysis and CMAP measurements of the impact of trypsin-based exfoliation on the epineurial layer; immunofluorescence imaging of reprogramming cell populations from the nerve in vitro and in vivo; methods. Supporting Information is available from the Wiley Online Library or from the author.
The authors declare no competing financial interests.
REFERENCES
- 1.Secer HI, Solmaz I, Anik I, Izci Y, Duz B, Daneyemez MK, Gonul E, J Brachial Plex Peripher Nerve Inj 2009, 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burtt K, Badash I, Wu B, Clinical Trials in Orthopedic Disorders 2017, 2, 123. [Google Scholar]
- 3.Ecklund JM, Ling GS, Neurosurg Clin N Am 2009, 20, 107. [DOI] [PubMed] [Google Scholar]
- 4.Smith JK, Miller ME, Carroll CG, Faillace WJ, Nesti LJ, Cawley CM, Landau ME, Muscle Nerve 2016, 54, 1139. [DOI] [PubMed] [Google Scholar]
- 5.Naff NJ, Ecklund JM, Neurosurgery Clinics 2001, 12, 197. [PubMed] [Google Scholar]
- 6.Gaudet AD, Popovich PG, Ramer MS, Journal of Neuroinflammation 2011, 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber RV, Mackinnon SE, Clin Plast Surg 2005, 32, 605. [DOI] [PubMed] [Google Scholar]
- 8.Bunge RP, Curr Opin Neurobiol 1993, 3, 805. [DOI] [PubMed] [Google Scholar]
- 9.Levi AD, Guenard V, Aebischer P, Bunge RP, J Neurosci 1994, 14, 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levi AD, Sonntag VK, Dickman C, Mather J, Li RH, Cordoba SC, Bichard B, Berens M, Exp Neurol 1997, 143, 25. [DOI] [PubMed] [Google Scholar]
- 11.Bixby JL, Lilien J, Reichardt LF, J Cell Biol 1988, 107, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitlock EL, Tuffaha SH, Luciano JP, Yan Y, Hunter DA, Magill CK, Moore AM, Tong AY, Mackinnon SE, Borschel GH, Muscle Nerve 2009, 39, 787. [DOI] [PubMed] [Google Scholar]
- 13.Jesuraj NJ, Santosa KB, Macewan MR, Moore AM, Kasukurthi R, Ray WZ, Flagg ER, Hunter DA, Borschel GH, Johnson PJ, Mackinnon SE, Sakiyama-Elbert SE, Muscle Nerve 2014, 49, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun X, Wang Y, Guo Z, Xiao B, Sun Z, Yin H, Meng H, Sui X, Zhao Q, Guo Q, Wang A, Xu W, Liu S, Li Y, Lu S, Peng J, Adv Healthc Mater 2018, 7, e1800276. [DOI] [PubMed] [Google Scholar]
- 15.Saheb-Al-Zamani M, Yan Y, Farber SJ, Hunter DA, Newton P, Wood MD, Stewart SA, Johnson PJ, Mackinnon SE, Exp Neurol 2013, 247, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Best TJ, Mackinnon SE, Evans PJ, Hunter D, Midha R, J Reconstr Microsurg 1999, 15, 183. [DOI] [PubMed] [Google Scholar]
- 17.Rovak JM, Mungara AK, Aydin MA, Cederna PS, J Reconstr Microsurg 2004, 20, 53. [DOI] [PubMed] [Google Scholar]
- 18.Faroni A, Mobasseri SA, Kingham PJ, Reid AJ, Adv Drug Deliv Rev 2015, 82–83, 160. [DOI] [PubMed] [Google Scholar]
- 19.Muheremu A, Ao Q, Biomed Res Int 2015, 2015, 237507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong JO, Kim MO, Kim H, Lee MY, Kim SW, Ii M, Lee JU, Lee J, Choi YJ, Cho HJ, Lee N, Silver M, Wecker A, Kim DW, Yoon YS, Circulation 2009, 119, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pittenger G, Vinik A, Exp Diabesity Res 2003, 4, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han JW, Sin MY, Yoon Y.-s., Diabetes Metab J 2013, 37, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terenghi G, The Journal of Anatomy 1999, 194, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Li W-Y, Sun P, Jin Z.-s., Liu G.-b., Deng L-X, Guan L-X, Neurological research 2016, 38, 242. [DOI] [PubMed] [Google Scholar]
- 25.Zacchigna S, Giacca M, International Review of Neurobiology 2009, 87, 381. [DOI] [PubMed] [Google Scholar]
- 26.Daya S, Berns KI, Clinical Microbiology Reviews 2008, 21, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sudres M, Ciré S, Vasseur V, Brault L, Da Rocha S, Boisgérault F, Le Bec C, Gross DA, Blouin V, Ryffel B, Molecular Therapy 2012, 20, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrand M, Da Rocha S, Corre G, Galy A, Boisgerault F, Molecular Therapy 2015, 23, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mays LE, Wilson JM, Molecular Therapy 2011, 19, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenberg B, Butler J, Felker G, Ponikowski P, Voors A, Pogoda J, Provost R, Guerrero J, Hajjar R, Zsebo K, Gene therapy 2016, 23, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Zhang Z, Kim HS, Han S, Kim SW, Mol Cell Neurosci 2014, 62, 60. [DOI] [PubMed] [Google Scholar]
- 32.Walsh S, Midha R, Neurosurgery 2009, 65, A80. [DOI] [PubMed] [Google Scholar]
- 33.Kitada M, Murakami T, Wakao S, Li G, Dezawa M, Glia 2019, 67, 950. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Z, Wang Y, Peng J, Ren Z, Zhan S, Liu Y, Zhao B, Zhao Q, Zhang L, Guo Q, Microsurgery 2011, 31, 388. [DOI] [PubMed] [Google Scholar]
- 35.Arai K, Lo EH, J Neurosci 2009, 29, 4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohab JJ, Fleming S, Blesch A, Carmichael ST, J Neurosci 2006, 26, 13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi A, Soma T, Tanaka H, Kanda T, Nishimura H, Yoshikawa H, Tsukamoto Y, Iso H, Fujimori Y, Stern DM, Naritomi H, Matsuyama T, J Clin Invest 2004, 114, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thored P, Wood J, Arvidsson A, Cammenga J, Kokaia Z, Lindvall O, Stroke 2007, 38, 3032. [DOI] [PubMed] [Google Scholar]
- 39.Pereira Lopes FR, Lisboa BC, Frattini F, Almeida FM, Tomaz MA, Matsumoto PK, Langone F, Lora S, Melo PA, Borojevic R, Han SW, Martinez AM, Neuropathol Appl Neurobiol 2011, 37, 600. [DOI] [PubMed] [Google Scholar]
- 40.Cattin A-L, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJ, Calavia NG, Guo Y, McLaughlin M, Rosenberg LH, Quereda V, Cell 2015, 162, 1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okano H, Nakamura M, Yoshida K, Okada Y, Tsuji O, Nori S, Ikeda E, Yamanaka S, Miura K, Circ Res 2013, 112, 523. [DOI] [PubMed] [Google Scholar]
- 42.Ben-David U, Benvenisty N, Nat Rev Cancer 2011, 11, 268. [DOI] [PubMed] [Google Scholar]
- 43.Bongso A, Fong CY, Gauthaman K, J Cell Biochem 2008, 105, 1352. [DOI] [PubMed] [Google Scholar]
- 44.Vierbuchen T, Wernig M, Nat Biotechnol 2011, 29, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallego-Perez D, Otero JJ, Czeisler C, Ma J, Ortiz C, Gygli P, Catacutan FP, Gokozan HN, Cowgill A, Sherwood T, Ghatak S, Malkoc V, Zhao X, Liao WC, Gnyawali S, Wang X, Adler AF, Leong K, Wulff B, Wilgus TA, Askwith C, Khanna S, Rink C, Sen CK, Lee LJ, Nanomedicine 2016, 12, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gallego-Perez D, Pal D, Ghatak S, Malkoc V, Higuita-Castro N, Gnyawali S, Chang L, Liao W-C, Shi J, Sinha M, Nature nanotechnology 2017, 12, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boukany PE, Morss A, Liao WC, Henslee B, Jung H, Zhang X, Yu B, Wang X, Wu Y, Li L, Gao K, Hu X, Zhao X, Hemminger O, Lu W, Lafyatis GP, Lee LJ, Nat Nanotechnol 2011, 6, 747. [DOI] [PubMed] [Google Scholar]
- 48.Chang L, Bertani P, Gallego-Perez D, Yang Z, Chen F, Chiang C, Malkoc V, Kuang T, Gao K, Lee LJ, Lu W, Nanoscale 2015. [DOI] [PubMed] [Google Scholar]
- 49.Chang L, Gallego-Perez D, Zhao X, Bertani P, Yang Z, Chiang CL, Malkoc V, Shi J, Sen CK, Odonnell L, Yu J, Lu W, Lee LJ, Lab Chip 2015, 15, 3147. [DOI] [PubMed] [Google Scholar]
- 50.Zhao X, Wu Y, Gallego-Perez D, Kwak KJ, Gupta C, Ouyang X, Lee LJ, Analytical Chemistry 2015, 87, 3208. [DOI] [PubMed] [Google Scholar]
- 51.Zhao X, Huang X, Wang X, Wu Y, Eisfeld A-K, Schwind S, Gallego-Perez D, Boukany PE, Marcucci GI, Lee LJ, Advanced Science 2015, 2, n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang L, Gallego-Perez D, Chiang CL, Bertani P, Kuang T, Sheng Y, Chen F, Chen Z, Shi J, Yang H, Huang X, Malkoc V, Lu W, Lee LJ, Small 2016, 12, 5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Q, Wang X, Yi S, Frontiers in neuroscience 2018, 12, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peltonen S, Alanne M, Peltonen J, Tissue barriers 2013, 1, e24956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Topp KS, Boyd BS, Physical Therapy 2006, 86, 92. [DOI] [PubMed] [Google Scholar]
- 56.Menorca RM, Fussell TS, Elfar JC, Hand Clin 2013, 29, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ydens E, Cauwels A, Asselbergh B, Goethals S, Peeraer L, Lornet G, Almeida-Souza L, Van Ginderachter JA, Timmerman V, Janssens S, Journal of Neuroinflammation 2012, 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mausberg AK, Szepanowski F, Odoardi F, Flügel A, Kleinschnitz C, Stettner M, Kieseier BC, Journal of Neuroinflammation 2018, 15, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mizisin A, Weerasuriya A, Acta neuropathologica 2011, 121, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zochodne DW. Neurobiology of peripheral nerve regeneration. (Cambridge University Press; Cambridge, UK, 2008). [Google Scholar]
- 61.Jurecka W, Ammerer H, Lassmann H, Acta neuropathologica 1975, 32, 299. [DOI] [PubMed] [Google Scholar]
- 62.Pongratz G, Straub RH, Nature Reviews Rheumatology 2013, 9, 117. [DOI] [PubMed] [Google Scholar]
- 63.Schomberg D, Ahmed M, Miranpuri G, Olson J, Resnick DK, Annals of neurosciences 2012, 19, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreau N, Mauborgne A, Bourgoin S, Couraud P-O, Romero IA, Weksler BB, Villanueva L, Pohl M, Boucher Y, PAIN 2016, 157, 827. [DOI] [PubMed] [Google Scholar]
- 65.Gao Y, Weng C, Wang X, Neural Regen Res 2013, 8, 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jacobs JM, Ro LS, J Neurol Sci 1994, 127, 143. [DOI] [PubMed] [Google Scholar]
- 67.Ju MS, Lin CC, Fan JL, Chen RJ, J Biomech 2006, 39, 97. [DOI] [PubMed] [Google Scholar]
- 68.Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh MC, White IJ, Woodhoo A, Neuron 2012, 73, 729. [DOI] [PubMed] [Google Scholar]
- 69.Wilder-Kofie TD, Lúquez C, Adler M, Dykes JK, Coleman JD, Maslanka SE, Comparative medicine 2011, 61, 235. [PMC free article] [PubMed] [Google Scholar]
- 70.Li J, Geisbush TR, Arnold WD, Rosen GD, Zaworski PG, Rutkove SB, PLoS One 2014, 9, e111428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnold WD, Sheth KA, Wier CG, Kissel JT, Burghes AH, Kolb SJ, J Vis Exp 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnold WD, Porensky PN, McGovern VL, Iyer CC, Duque S, Li X, Meyer K, Schmelzer L, Kaspar BK, Kolb SJ, Kissel JT, Burghes AH, Ann Clin Transl Neurol 2014, 1, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Li D, Wang G, Chen L, Chen J, Liu Z, Zhang Z, Shen H, Jin Y, Shen Z, Int J Biol Sci 2017, 13, 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S2. CMAP measurements in untreated vs. operated mice show that the surgical procedure followed to expose the sciatic nerve does not have a significant impact in neuromuscular function (n = 6).
Fig. S1. (a) Schematic diagram of the bulk electroporation (BEP) procedure. The plasmid solution is injected into the nerve tissue prior to applying a pulsed electric field between a pair of plate electrodes that are gently clamping the nerve. (b) Finite element modeling simulation showing widespread electric field distribution under BEP.
Fig. S4. Trypsin treatment does not have a significant impact in neuromuscular function as assessed by CMAP measurements (n = 7).
Fig. S5. Successful plasmid expression was (a) confirmed via immunostaining of the Myc-DDK tag protein associated with sham and EFF expression plasmids. Furthermore, qRT-PCR analysis (b) revealed an upregulation of the delivered transcription factors Etv2, Fli1, and Foxc2, in EFF TNT vs. Sham TNT vs. control/untreated nerve (n=3–8). Mean ± sem, *p<0.05.
Fig. S6. (a) High magnification fluorescence microscopy micrographs show that some CD31+ vascular cells had traces of S100 immunoreactivity (7 days post-crush), potentially suggesting that EFF-driven vascular reprogramming was impacting S100+ cells (e.g., Schwann cells, sciatic nerve fibroblasts). (b) In vitro reprogramming studies with primary sciatic nerve fibroblasts and a Schwann cell (SW10) show that EFF-treated fibroblast cultures showed positive immunoreactivity for vascular cell marker vWF, which were absent in EFF-treated Schwann cell cultures (7 days post-NEP).
Fig. S3. Effects of trypsin treatment on the sciatic nerve as a function of time. Trypsin exposure leads to removal of the epineurium (t = 0 h), which enables TNT-based delivery of genetic cargo to the core of the nerve. However, the epineurium can regenerate/recover as a function of time (t = days 3 – 7).