

Abstract
Background:
Many different cellular systems and molecular processes become compromised in Alzheimer’s disease (AD) including proteostasis, autophagy, inflammatory responses, synapse and neuronal circuitry, and mitochondrial function. We focused in this study on mitochondrial dysfunction owing to the toxic neuronal environment produced by expression of Aβ42, and its relationship to other pathologies found in AD including increased neuronal apoptosis, plaque deposition, and memory impairment.
Methods:
Using super-resolution microscopy, we have assayed mitochondrial status in the three distinct neuronal compartments (somatic, dendritic, axonal) of mushroom body neurons of Drosophila expressing Aβ42. The mushroom body neurons comprise a major center for olfactory memory formation in insects. We employed calcium imaging to measure mitochondrial function, immunohistochemical and staining techniques to measure apoptosis and plaque formation, and olfactory classical conditioning to measure learning.
Results:
We found that mitochondria become fragmented at a very early age along with decreased function measured by mitochondrial calcium entry. Increased apoptosis and plaque deposition also occur early yet interestingly, a learning impairment was found only after a much longer period of time – 10 days, which is a large fraction of the fly’s lifespan. This is similar to the pronounced delay between cellular pathologies and the emergence of a memory dysfunction in humans.
Conclusion:
Our studies are consistent with the model that mitochondrial dysfunction and/or other cellular pathologies emerge at an early age and lead to much later learning impairments. The results obtained further develop this Drosophila model as a useful in vivo system for probing the mechanisms by which Aβ42 produces mitochondrial and other cellular toxicities that produce memory dysfunction.
Keywords: Mitochondrial fragmentation, Alzheimer’s disease, Aβ toxicity, learning
Background
Sporadic and genetic forms of Alzheimer’s disease (AD) are characterized by the failure of many different cellular and molecular systems of the brain and other tissues 1–4 For instance, the associated neuropathology reported for AD and AD animal models include the generation of toxic cleavage products of APP 5; impairments in the autophagy-lysosomal pathway leading to protein aggregation 6; chronic inflammation initiated by microglial activation 3,7; synapse failure and loss, reduced dendritic complexity, and impaired connectivity and brain circuit function 8; glutamate excitotoxicity and cell death 9; increased oxidative stress 10,11; impairments in mitochondrial dynamics and function 12,13 and others. Despite the broad effects of the disease, research into AD etiology has been driven largely by insights obtained from genetic forms of the disease. Thus, the last two decades have witnessed a concentrated effort to understand the metabolism of amyloid precursor protein (APP), the generation of Aβ oligomers, the deposition of amyloid plaque and the effects of genetic insults to this molecular system on other aspects of brain function. Although this focus is understandable in retrospect, many other facets of AD neuropathology have remained relatively sketchy. Moreover, extensive efforts made in developing therapeutics based on the APP/Aβ hypothesis have generally failed in clinical trials.
This history has prompted a sense of urgency to develop a better understanding of other aspects of AD neuropathology. Such an understanding will likely lead to two very significant gains. First, providing detailed knowledge of other system failures that occur in AD should lead to insights into their relationships, and help establish the hierarchy for the progression of sporadic AD. Second, combinatorial drug therapies may be developed using a mixture of drugs that protect the major system failures in the disease. Without clear knowledge of the initiating trigger(s) for sporadic AD, therapeutic advances will necessarily be combinatorial and require detailed knowledge of the different neuropathologies that define the disease.
Mitochondrial dysfunction represents a hallmark neuropathology for AD and other brain disorders. Indeed, the failure of the mitochondrial system has been found to be an early neuropathology and proposed to be a trigger 14,15. Each neuron contains thousands of mitochondria that are thought to be born in the soma and then trafficked to distal sites in axons and dendrites; each organelle offering its functions to remote regions of the neuron including synaptic specializations 16. They provide many different functions that help maintain neuronal health, the two most recognized being the generation of ATP through respiration and buffering of cytoplasmic calcium. In addition to trafficking to and from distal sites, mitochondria divide in the process of fission and join together in the process of fusion. These processes along with mitochondrial movement fall under the rubric of mitochondrial dynamics.
Drosophila metanogaster has been employed as a model for studying the neuropathology found in diseases that include Parkinson’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis 17–21. The organism offers the convenience of small size, rapid generation time, and the ability to generate hundreds of organisms at a relatively low expense; while providing the toxic, cellular disease environment related to genetic forms of the diseases using transgenic approaches analogous to those used for the mouse.
For AD-related neuropathology, many of the prior studies have employed the expression of Aβ42 or other toxic peptides in retinal cells to assay cell death 22–25. This approach offers a rapid way of screening cell death based on external morphology but prompts the concern that retinal cells are unique and may not offer the physiology of neurons found in the central brain. Other studies have employed pan-neuronal expression of the toxic peptides 26,27, but this approach fails to mimic the accepted progression of the disease in humans from the medial temporal lobe to other brain regions. The medial temporal lobe is intimately involved in memory formation, which accounts for the initial symptom displayed by AD patients – forgetfulness.
Here, we have expressed toxic peptide Aβ42 in the mushroom body neurons (MBn), neurons that constitute a major brain center for olfactory memory 28,29. We focused our efforts on understanding how impairments in mitochondrial dynamics and function emerge and evolve with the age of the fly and relating these to the temporal evolution of other AD pathologies including apoptosis, plaque deposition, and memory dysfunction. We found that mitochondria are more numerous and smaller in size at a very early age in the Aβ42 environment. Consistent with this, we found that mitochondrial calcium import was also impaired in young flies along with apoptosis and plaque deposition. Interestingly, learning became impaired much later than the mitochondrial deficits and other cellular pathologies.
Methods
Drosophila husbandry
Fly strains were raised on standard food medium at room temperature. Crosses were made at 70% relative humidity with a 12 hours’ light-dark cycle in a 25 °C incubator. Fly lines used in this study are listed in STAR ★ METHODS.
Immunostaining for structure illumination microscopy
For structured illumination microscopy, brains were isolated from female flies at various ages, processed as previously described and imaged with high-resolution microscope 49. Additional details are available in STAR ★ METHODS.
Mitochondrial calcium entry
Brains were isolated from female flies of various ages and continually perfused with saline solution (124 mM NaCl, 3 mM KCl, 20 mM MOPS, 1.5 mM CaCl2, 4 mM MgCl2, 5 mM NaHCO3, 1 mM NaH2PO4, 10 mM trehalose, 7 mM sucrose, 10 mM glucose; adjusted to pH 7.2 at 25 °C). Neurons were depolarized by adding 15% KCI to the perfusion solution. Additional details are available in STAR ★ METHODS.
Cell death assay
Brains isolated from female flies of various ages were processed as previously described for immunohistochemistry 49. The primary antibody against Dcp-1 was used to monitor cell death of MBn. Additional details are available in STAR ★ METHODS.
Amyloid plaque staining
Brains from female flies of various ages were dissected and processed as previously described 49. The isolated brains were immunostained with anti-nc82 and after immunostaining were perfused with 0.5% Thioflavin S for 24 h. Additional details are available in STAR ★ METHODS.
Behavioral assays
For behavior experiments, young flies were collected and maintained in bottles to various ages in bottles. The flies were transferred to vials 12 h prior to the experiments. We used the negatively reinforced, olfactory classical conditioning to assay learning 50. Additional details are available in STAR ★ METHODS.
Results
Neuronal mitochondrial are smaller and more numerous in the soma of flies expressing Aβ42
We used the Gal4/UAS system to co-express Aβ42 and a mitochondrial-targeted GFP (mito-GFP)30 to probe the effects of the toxic cellular environment produced by Aβ42 on mitochondrial morphology in neurons. The Gal4 driver used, R13F02-Gal4, is a mushroom body neuron (MBn) specific driver (Figure 1A). The MB are insect brain structures that regulate the acquisition and storage of olfactory memories 28,29. We employed structured illumination microscopy (SIM) which allowed us to characterize the form of individual mitochondria and segment them. We performed this analysis on the mitochondria in a 10 μm3 volume at a standardized location in the somatic, dendritic, and axonal regions of the MBn.
Figure 1. Mitochondrial morphology in the soma of MBn expressing Aβ42.
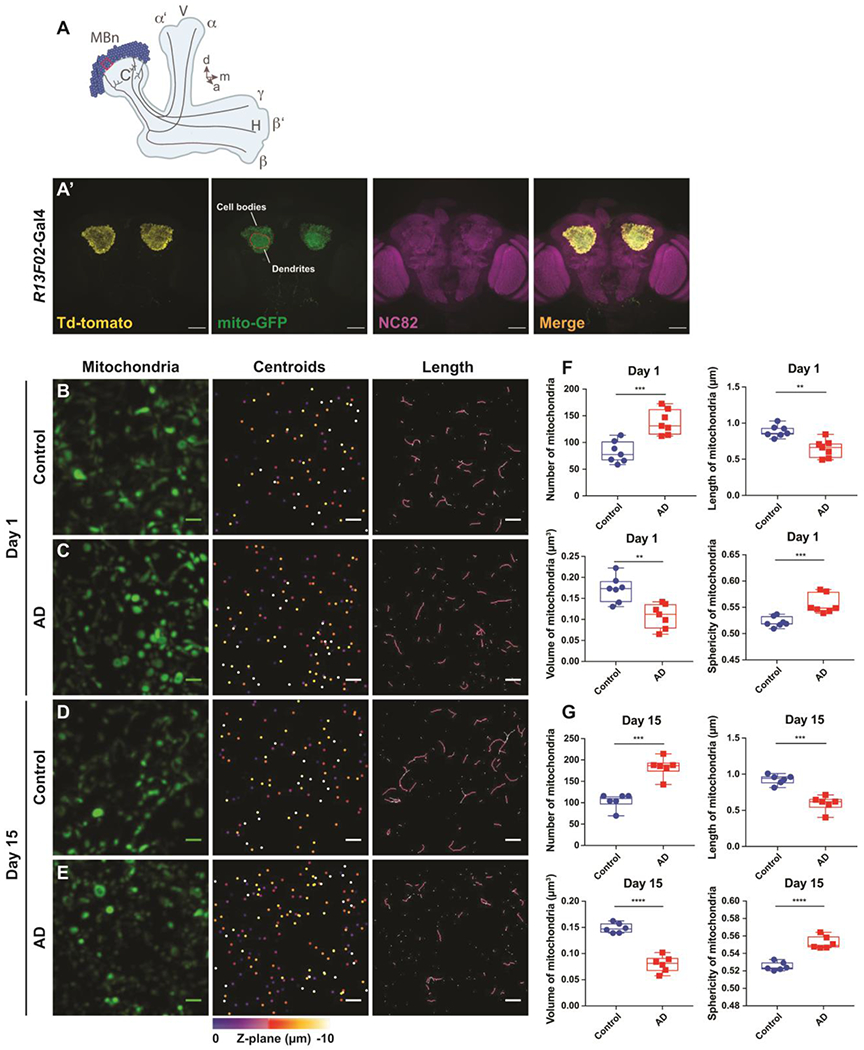
(A) Diagram of the MB in the fly’s right hemisphere. The somata of the MBn are colored dark blue with their associated neuropil areas colored light blue. The Calyx (C) contains the dendrites of the MBn and the Vertical (V) and Horizontal (H) lobes contain the axons of the MBn. The three major classes of MBn, α/β, α’/β’ and γ, classed according to the projection patterns of their axons, are illustrated along with the associated neuropil to which they project – the α/β lobes, α’/β’ lobes, and γ lobe, respectively. The ten μm3 volume ROI (red box), located at a one o’clock position viewed from a frontal perspective of the calyx in the fly’s left hemisphere (11 o’clock for the right hemisphere), was used to characterize the somatic mitochondria. d=dorsal, m=medial, a=anterior.
(A’) Low resolution maximum projection images of the soma and dendritic regions of the adult MB in both hemispheres. The R13F02-Gal4 expression was visualized using UAS-myr-Td-tomato and UAS-mito-GFP. Brain neuropil was stained with anti-nc82. Scale bar 50 μm.
(B) Maximum projection images of individual mitochondria, mitochondrial centroids and their lengths in a 10 μm3 ROI from the somatic region of a control brain at 1 day-of-age. Genotype: UAS-mito-GFP/+; R13F02-Gal4/+. Scale bar 1 μm.
(C) Same as panel A for an Aβ42-expressing brain at 1 day-of-age. Genotype: UAS-mito-GFP/UAS-Aβ42; R13F02-Gal4/UAS-Aβ42.
(D) Same as panel A for a control brain at 15 days-of-age.
(E) Same as panel C for an Aβ42-expressing brain at 15 days-of-age.
(F) Box plots of somatic mitochondrial parameters in control and Aβ42-expressing flies at 1 day-of-age. mitochondria are more numerous (***P< 0.001), shorter (**P< 0.01) and of reduced volume (**P< 0.01) with increased sphericity (***P< 0.001) in the Aβ42-expressing flies. Mann-Whitney U, n=7. Each box plot shows the median, interquartile range and the range.
(G) Same as panel F for control and Aβ42-expressing flies at 15 days-of-age. Mitochondria are more numerous (***P< 0.001), shorter (***P< 0.001) and of reduced volume (****P< 0.0001) with increased sphericity (****P< 0.0001) in the Aβ42-expressing flies. Mann-Whitney U, n=6.
We focused initially on mitochondria in the soma (Figure 1A) and found that, compared with the control group, the Aβ42-expressing animals contained increased numbers of mitochondria in the MB soma as early as 1 day after eclosion (Figure 1B, C, F). We also measured the length, surface area, volume, and sphericity of the somatic mitochondria. In Aβ42 animals, we found that mitochondrial length was shortened, the average size reduced – as reflected in surface area and volume – and the sphericity was increased at Day 1 (Figure 1B, C, F). In the process of analyzing the mitochondrial morphology, we searched for hemispheric differences in these parameters and found none for both the control and Aβ42 animals (Figure S1). Because of this, both hemispheres were imaged in each fly and we collapsed the morphological data from both hemispheres to obtain data on a per fly rather than a per hemisphere basis. The mitochondrial morphological differences observed between control and Aβ42 animals at 1 day-of-age were preserved at older ages. Aβ42-expressing flies at 5, 10 and 15 days of age also contained more numerous, shorter, smaller, and more spherical mitochondria compared to their age-matched controls (Figures S2, 1D, E, G). Collectively, these data indicate that expression of Aβ42 induces fragmentation of somatic mitochondria that is evident at 1 day-of-age and that this fragmentation persists across the first two weeks of adulthood.
Mitochondria in dendrites and axons are smaller and more numerous in flies expressing Aβ42 but detectable differences emerge at later ages
We then wondered whether the mitochondrial fragmentation observed soon after eclosion in the MBn soma of Aβ42-expressing flies was a neuron-wide effect or whether the other cellular compartments – the axons and dendrites – might exhibit differences. In the dendrites, we observed an increase in mitochondrial number at 1 day-of age, but no significant difference in length, volume or sphericity (Figure 2A, B). Other mitochondrial defects to include shortening, reduction in size and increased sphericity were observed at 5, 10 and 15 days of age (Figure 2C; S3, S4). In the MBn axons, we failed to observe any changes in these mitochondrial morphological parameters at 1, 5 or 10 days-of-age (Figure 2D, E; S5, S6). However, by 15 days-of-age, the mitochondria were more numerous, shortened, smaller and more spherical in the Aβ42-expressing flies (Figure 2F).
Figure 2. Mitochondrial morphology in the dendrites and axons of MBn expressing Aβ42.
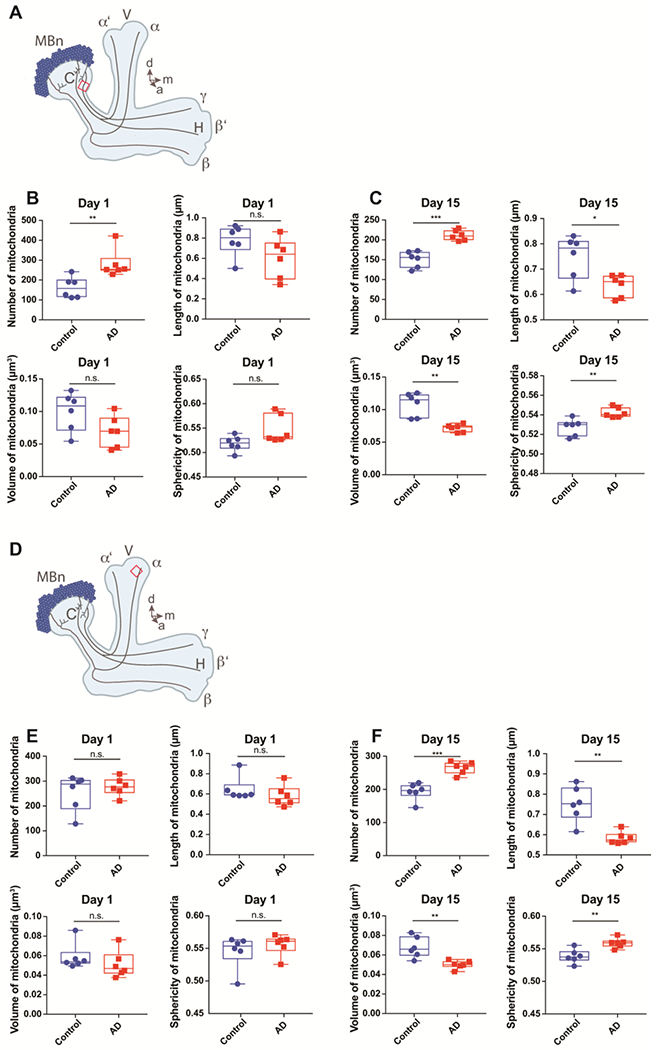
(A) Diagram of the MB in the fly’s right hemisphere. See the legend of Figure 1A for a complete description. The ten μm3 volume ROI (red box) located at the medial edge of the calyx was used to characterize the dendritic mitochondria. d=dorsal, m=medial, a=anterior.
(B) Box plots of dendritic mitochondrial parameters in control and Aβ42-expressing flies at 1 day-of-age. Mitochondria are more numerous (**P< 0.01) in the Aβ42-expressing flies, but no significant differences were observed in length, volume, or sphericity in the Aβ42-expressing flies. Mann-Whitney U, n=6. Each box plot shows the median, interquartile range and the range.
(C) Same as panel B for flies at 15 days-of-age. Mitochondria are more numerous (***P< 0.001), shorter (*P< 0.05) and of reduced volume (**P< 0.01) with increased sphericity (**P< 0.01) in the Aβ42-expressing flies. Mann-Whitney U, n=6.
(D) Same as panel A showing the ROI (red box) for characterizing axonal mitochondria located at a standardized and central location in the bulbous tip of the α lobe as viewed from a frontal perspective.
(E) Box plots of axonal mitochondria parameters in control and Aβ42-expressing flies at 1 day-of-age. No significant differences were observed in number, length, volume or sphericity in the Aβ42-expressing flies at 1 day-of-age. Mann-Whitney U, n=6. Each box plot shows the median, interquartile range and the range.
(F) Same as panel E for flies at 15 days-of-age. Mitochondria are more numerous (***P< 0.001), shorter (**P< 0.01) and of reduced volume (**P< 0.01) with increased sphericity (**P< 0.01) in the Aβ42-expressing flies. Mann-Whitney U, n=6.
In summary, detectable mitochondrial fragmentation occurs first in the soma of MBn expressing Aβ42, subsequently in the MBn dendrites and then in the distal axons. This is somewhat surprising given that current dogma posits that mitochondria are born in the soma from biogenesis followed by transport into the more distal regions of the neuron. It may be that some biogenesis occurs locally, with the toxicity of Aβ42 reduced in axons and dendrites compared to the soma, or the toxicity may be generally equivalent in all parts of the neuron with the mitochondria having differential sensitivity between compartments. Alternatively, all biogenesis could occur in the soma, but gating mechanisms present in the neurites may only allow healthier mitochondria to be transported.
Mitochondria in Aβ42-expressing flies exhibit impaired calcium import
To evaluate the toxic consequences of Aβ42-expression on mitochondria, we measured mitochondrial calcium import in MBn. We used isolated brains (Figure 3) from control and Aβ42-expressing flies that carried a transgene expressing the calcium reporter, UAS-4mtGCaMP3 30, targeted to the mitochondrial matrix. These flies also expressed UAS-RFP to provide a normalization signal for the 4mtGCaMP3 responses. The MBn-specific GAL element R13F012-Gal4 was used to drive the transgenes, providing for specific expression in the soma, dendrites and axons of MBn (Figure 3A, S5). 15 mM KCl was perfused into the bath containing the isolated brains in order to stimulate calcium influx into the cytoplasm and mitochondria 30 For consistency, we measured the normalized 4mtGCaMP3 signal from the same somatic area used for experiments examining mitochondrial morphology.
Figure 3. Mitochondrial calcium import is impaired in the MBn of Aβ42-expressing flies.
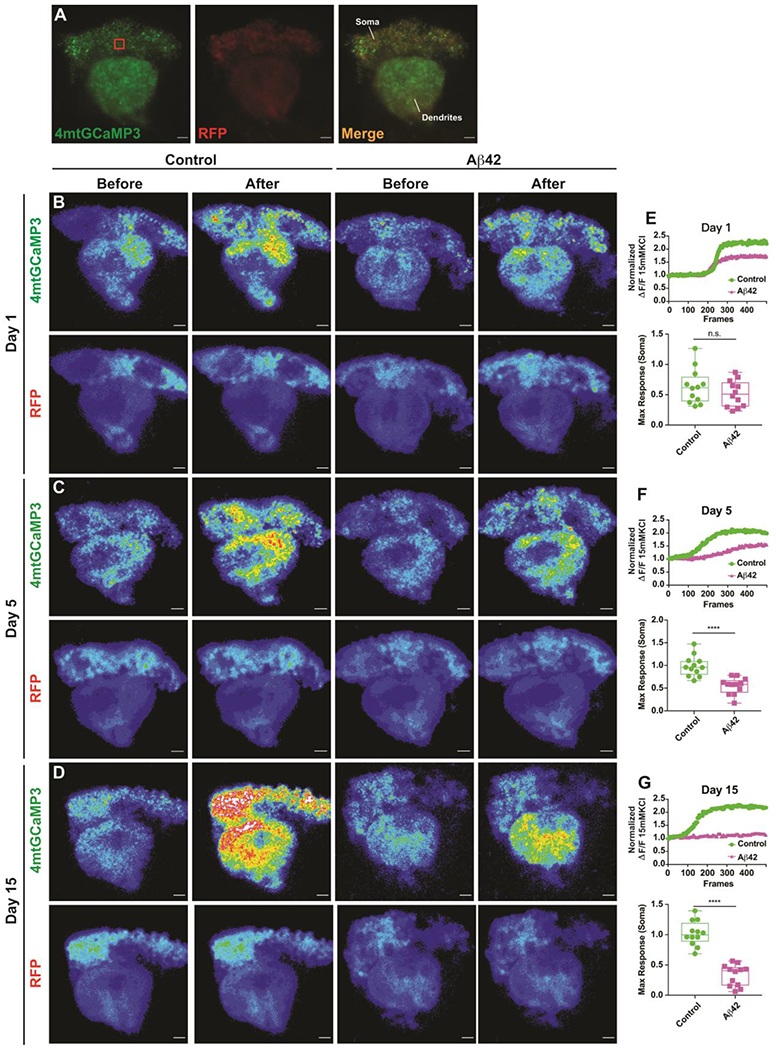
(A) Maximum projection images showing the expression of UAS-4mtGCaMP3 and UAS-RFP in the MBn ex vivo. The red box in the left panel represents the 10 μm3 volume used to quantify somatic responses. Genotype: UAS-4mtGCaMP3, UAS-RFP /+; R13F02-Gal4/+. Scale bar 50 μm.
(B) Representative pseudocolor images of the mito-GCaMP3 response in the soma and dendrites of MBn for control and Aβ42-expressing flies at 1 day-of-age, before and after stimulation with 15 mM KCl. The bottom row of images illustrates the RFP signal which was used to normalize the mito-GCaMP3 signal. Genotypes: UAS-mito-GCaMP3, UAS-RFP /+; R13F02-Gal4/+; and UAS-4mtGCaMP3, UAS-RFP / UAS-Aβ42; R13F02-Gal4/UAS-Aβ42.
(C) Same as panel B for flies at 5 days-of-age.
(D) Same as panel B for flies at 15 days-of-age.
(E) Quantification of the normalized 4mtGCaMP3 response in the soma of MBn (10 μm3 volume, red box in panel A) of control and Aβ42-expressing flies at 1 day-of-age. The upper panel illustrates a representative response to KCl in the two genotypes. Group data are shown in the lower panel as a box plot of the median, interquartile range, and the range. The maximum response of 4mtGCaMP3 in the Aβ42-expressing flies was depressed but not significantly different from the control. Mann-Whitney U. n=12.
(F) Same as panel E for flies at 5 days-of-age. ****P< 0.0001. n=12. Note also the depressed response in the dendrites.
(G) Same as panel E for flies at days-of-age. ****P< 0.0001. n=12. Note also the depressed response in the dendrites.
Figure 3B–D shows representative fluorescence images from the MB somata and dendrites in control and Aβ42-expressing flies at 1, 5, and 15 days-of-age. KCl perfusion generated a robust signal from these neurons in control and Aβ42-expressing flies at 1 day-of-age, with no significant difference in the peak magnitude of the response between the two genotypes (Figure 3E). In contrast, the mitochondrial calcium import in Aβ42-expressing MBn at 5, 10 (not shown) and 15 days-of-age was significantly depressed relative to the control, with essentially no import occurring in the Aβ42-expressing MBn at 15 days-of-age (Figure 3F, G). These data reveal a functional impairment in the somatic mitochondria from Aβ42-expressing MBn. However, the morphological impairment was significantly different from the control at 1 day-of-age but the functional impairment was only trending at this same age. This could indicate that the functional impairment follows the morphological impairment in time, or it could be due to differences in the sensitivity of the two assays. It is notable from the fluorescent assays that the functional impairment extends into the MBn dendrites.
Increased apoptosis in Aβ42-expressing MBn
Mitochondria play a fundamental role in regulating cell survival and cell death. Multiple types of stimuli such as activation of the proapoptotic Bcl-2 associated X protein (BAX), oxidants, and stress cause the mitochondria to release caspase-activating proteins like cytochrome c which leads to apoptosis 31–34. We wondered whether the expression of Aβ42 specifically in the MBn and the subsequent impairments in mitochondrial structure and function were associated with increased apoptosis. The Drosophila death caspase-1 (Dcp-1) – the homolog of mammalian Caspase-3 – is an effector caspase that triggers cell death after activation upon cleavage by initiator caspases. Immunohistochemistry with antibodies to the activated Dcp-1 has been used extensively to quantitate apoptosis 35–37 We stained brains from control and Aβ42-expressing flies of various ages with an anti-Dcp-1 (cleaved) antibody 38–40 to determine whether apoptosis was increased.
We observed a significantly increased number of Dcp-1 puncta in the MB somata of Aβ42-expressing flies at 1 day-of-age compared to their age matched controls. (Figure 4A, B, G). This measure of apoptosis was markedly increased with age in the experimental flies at 5, 10 (not shown) and 15 days-of-age (Figure 4C–F, H, I). Notably, the Dcp-1 puncta were observed only in the MB somata, and not in other regions of the brain consistent with the spatially limited expression of the driver/Aβ42. Collectively, these data reveal an increased level of apoptosis in the somata of Aβ42-expressing flies that parallels the onset and severity of mitochondrial morphological and functional deficits.
Figure 4. Increased Dcp-1 expression, a marker for apoptosis, in the MBn of Aβ42-expressing flies.
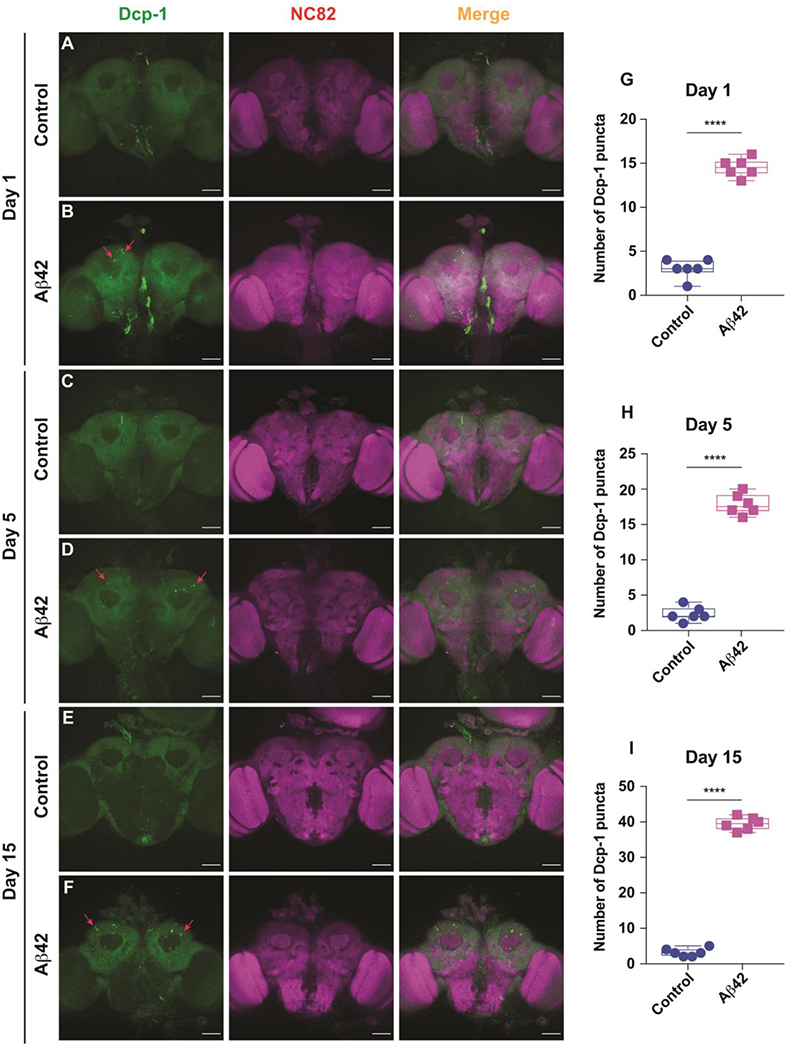
(A) Representative maximum projection images of a brain from a control fly at 1 day-of-age co-stained with anti-cleaved Dcp-1 and anti-NC82. Genotype: +/+; R13F02-Gal4/+. Scale bar 50 μm.
(B) Representative maximum projection images of a brain from an Aβ42-expressing fly at 1 day-of-age co-stained with anti- cleaved Dcp-1 and anti-NC82. Some of the Dcp-1 positive puncta are highlighted by red arrows. Genotype: +/ UAS-Aβ42; R13F02-Gal4/UAS-Aβ42. Scale bar 50 μm.
(C) Same as panel A for a control fly at 5 days-of-age.
(D) Same as panel B for an Aβ42-expressing fly at 5 day-of-age.
(E) Same as panel A for a control fly at 15 days-of-age.
(F) Same as panel B for an Aβ42-expressing fly at 15 day-of-age.
(G) Quantification of the number of Dcp-1 positive puncta in the MBn somata at 1 day-of age. A significant increase in the number of puncta were observed in the Aβ42-expressing brains compared to the control. Box plots show the median, interquartile range, and the range. Mann-Whitney U, ****P<0.0001. n=6.
(H) Same as panel G for 5 days-of age. A significant increase in the number of puncta were observed in the Aβ42-expressing brains compared to the control. ****P<0.0001. n=6.
(I) Same as panel G for 15 days-of age. A significant increase in the number of puncta were observed in the Aβ42-expressing brains compared to the control. ****P<0.0001. n=6.
Amyloid plaque develops near the Aβ42 expressing MBn
A main feature of AD is the formation of amyloid plaque in the hippocampus and other brain areas 41–43. To measure amyloid plaque and its co-occurrence with mitochondrial impairment and apoptosis, we stained Aβ42-expressing brains with Thioflavin S, a stain that binds to the amyloid fibrils 23,44,45. We observed a slight increase in Thioflavin S staining in MB somata of Aβ42-expressing brains at 1 day-of-age compared to the control (Figure 5A, B, G). The amyloid plaque burden increased at 5, 10 (not shown) and 15 days-of-age (Figure 5C–F, H, I). Thus, amyloid plaque burden generally follows the other cellular phenotypes observed – mitochondrial morphological and functional deficits and increase apoptosis.
Figure 5. Amyloid plaque formation in the MBn of Aβ42-expressing flies.
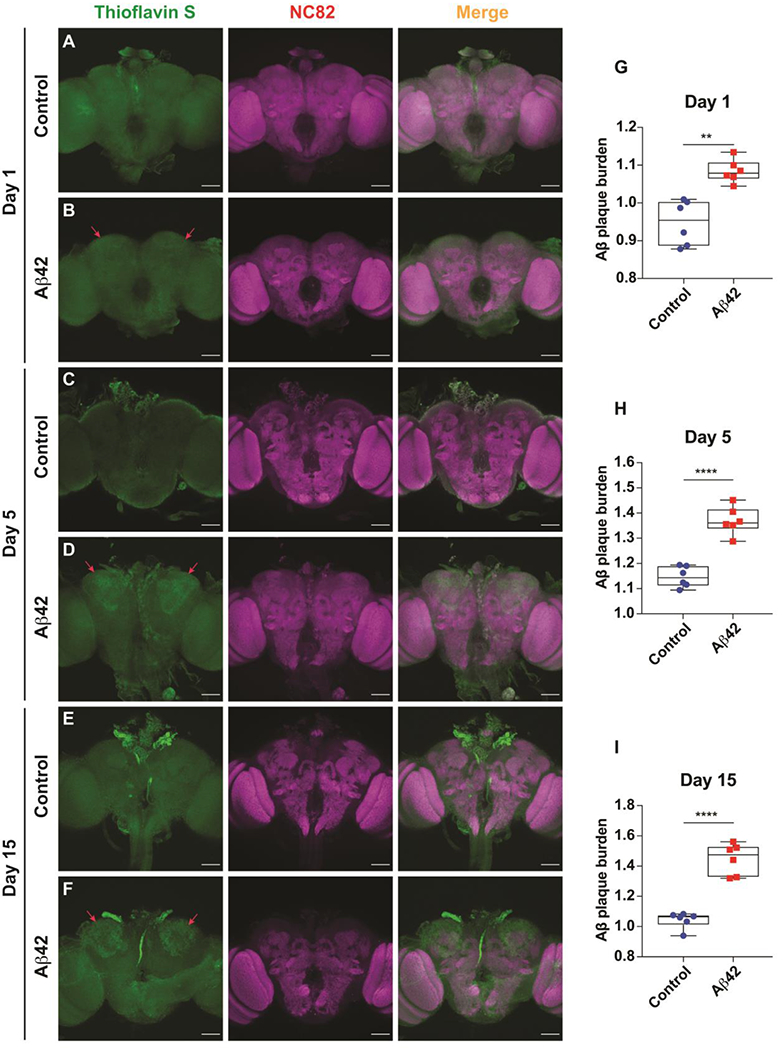
(A) Representative maximum projection images of a brain from a control fly at 1 day-of-age co-stained with Thioflavin S and anti-NC82. Genotype: +/+; R13F02-Gal4/+. Scale bar 50 μm.
(B) Representative maximum projection images of a brain from an Aβ42-expressing fly at 1 day-of-age co-stained with Thioflavin S and anti-NC82. Emerging amyloid plaque staining is highlighted by red arrows. Genotype: +/ UAS-Aβ42; R13F02-Gal4/ UAS-Aβ42. Scale bar 50 μm.
(C) Same as panel A for a brain from a control fly at 5 days-of-age.
(D) Same as panel B for a brain from an Aβ42-expressing fly at 5 days-of-age. Amyloid plaque staining is highlighted by red arrows and increasingly visible.
(E) Same as panel A for a brain from a control fly at 15 days-of-age.
(F) Same as panel B for a brain from an Aβ42-expressing fly at 15 days-of-age. Amyloid plaque staining is highlighted by red arrows and robust.
(G) Quantification of amyloid plaque burden in the MBn somata at 1 day-of age. The normalized Thioflavin S staining was significantly increased over the control. Box plots show the median, interquartile range, and the range. Mann-Whitney U, **P<0.01. n=6.
(H) Same as panel G for 5 days-of age. ****P<0.0001. n=6.
(I) Same as panel G for 15 days-of age. ****P<0.0001. n=6.
An impairment in olfactory learning occurs by 15 days-of-age for the Aβ42 expressing flies
A prominent behavioral consequence of Alzheimer’s Disease is the decline of learning and memory ability 45,46. However, the developmental relationship between the cellular pathologies such as mitochondrial impairment and the memory impairment have not been fully examined. We assayed olfactory memory tested at 3 min after training in the control and the Aβ42-expressing flies. Expression of Aβ42 in the MBn failed to impair 3 min memory performance at 1, 5, and 10 days-of-age (Figure 6). However, we detected a significant memory deficit in the experimental flies at 15 and 20 (not shown) days-of-age compared to the control genotypes. Interestingly, the 3 min behavioral impairment in the experimental flies fails to follow the early-developing mitochondrial, apoptotic, and amyloid pathology observed in the MB somata and dendrites. Rather, the age-dependent development of memory impairment follows most closely the mitochondrial impairment observed in the MBn axons (Figure 2D–F, S5, S6).
Figure 6. Impaired learning occurs later than mitochondrial defects, cell death and plaque formation.
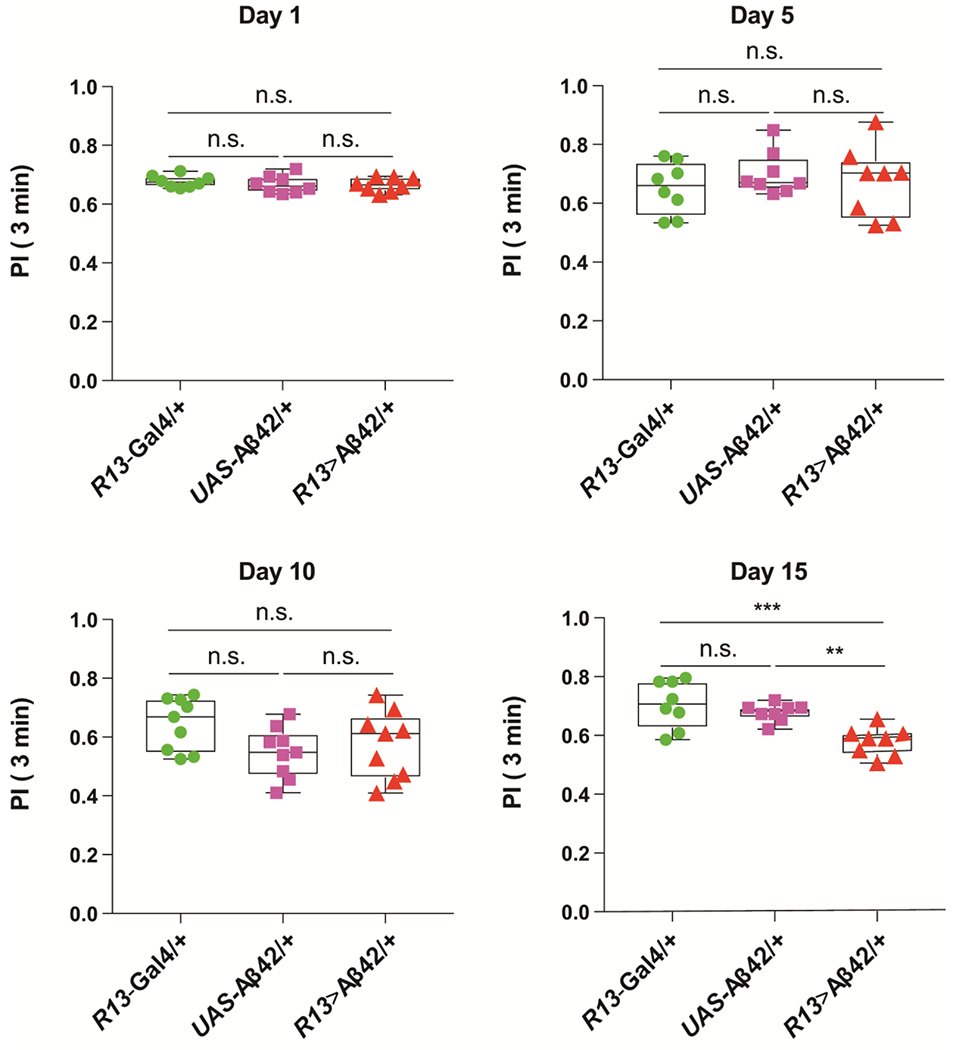
Performance Index (PI) when tested at 3 min after olfactory and negatively reinforced, classical conditioning. The PI ranges from zero to 1.0, where zero represents no memory observed and 1.0 represents complete memory. Flies carrying only the R13-Gal4 driver transgene (+/ +; R13F02-Gal4/ +) or the UAS-Aβ42 transgene (+/ UAS-Aβ42; +/ UAS-Aβ42) were used as controls for the experimental group carrying both transgenes (+/ UAS-Aβ42; R13F02-Gal4/UAS-Aβ42). Flies were trained and tested at 1, 5, 10, and 15 days-of-age. No significant differences between the controls and the experimental groups were observed at any age except at 15 days-of-age. One-way ANOVA followed by Tukey’s multiple comparison, **P<0.01, ***P<0.001. n=8. Box plots show the median, interquartile range, and the range.
Discussion
The overriding challenge in finding causes and developing therapies for Alzheimer’s disease (AD) is that the disease causes the failure of many different tissue, cellular and molecular systems, as noted earlier. The dizzying array of neuropathological consequences are undoubtedly linked but the hierarchy of disease etiology and progression for sporadic AD remains unclear. Yet, emerging evidence indicates that mitochondrial dysfunction may be one of the more upstream failures, as captured in the mitochondrial cascade hypothesis originally championed by Swerdlow and colleagues14,15
We offer with this study a relatively deep, in vivo investigation of mitochondrial dysfunction due to the toxicity associated with Aβ42 expression. We employed super-resolution microscopy to gain insights into the structural changes that occur in mitochondria due to this toxicity, including quantifying the number, length, surface area, volumes and sphericity of mitochondria in the soma of MBn and their dendrites and axons. We found that the mitochondria were shorter and smaller in size, consistent with increased mitochondrial fragmentation that has been reported by others 1. It has frequently been reported that mitochondrial number or content is reduced in AD, in contrast to our results, but this may be related to disease progression. Increased fragmentation would lead to increased mitochondrial numbers early in the disease with mitochondrial loss observed at later stages due to other contributing factors including perhaps a greater effect of decreased mitochondrial biogenesis. Other researchers have also reported increased mitochondrial number in AD models 47,48. We found functional defects in mitochondria due to Aβ42 toxicity in the form of impaired import of calcium. And similar to changes in mitochondrial morphology, mitochondrial function declines with age of the fly. In addition, we provide evidence for increased apoptosis and amyloid plaque deposition in the vicinity of the MBn engineered to express Aβ42. Moreover, a learning impairment develops in flies that express Aβ42 in the MBn. Our studies offer a facile, in vivo model to query the status of mitochondria in the presence of the toxic peptide involved in AD. For instance, the various pathologies develop over a period of 2 weeks, compared to 4-6 months for the most vigorous mouse model for AD. Cost differences for similar experiments using flies versus the mouse would vary by orders of magnitude. The flies and techniques described here could be used to probe the mechanism by which Aβ42 expression leads to structural and functional impairments of neuronal mitochondria and how a dysfunction of the mitochondrial system and/or others, leads to a late-developing impairment in memory. In addition, it offers an expedient model to test the protective potential of mitochondrial therapeutics.
We made two observations that are of high interest. First, the abnormal morphology of mitochondria due to Aβ42 expression was observed to occur at different ages in the three compartments of MBn. Mitochondrial fragmentation occurs by 1 day after eclosion in the soma of the MBn, but not until day 5 in the dendrites and day 15 in axons. In addition, we observed an increased number of mitochondria both in the somata and dendrites at day 1 but not in the axons until day 15. There are two broad explanations for these compartment differences. It may be that the mitochondria in the various compartments are differentially sensitive to the toxicity due to inherent differences in the mitochondria themselves, including, for instance, differences in protein composition. Alternatively, the compartments themselves may present differing levels of toxicity due to differential expression or localization of the toxic peptide or other compartment-specific factors that influence the magnitude of the toxicity. Additional research is required to understand this issue.
Second, the learning impairments occur relatively late, observable at day 15, whereas the mitochondrial structural and functional impairments occur at day 1 along with increased apoptosis and plaque deposition. This observation is consistent with the interpretation that the learning impairment stems from deficits in mitochondrial function and/or the other intervening pathologies. We favor this interpretation but a counterargument that cannot be dismissed is that each assay has its own inherent sensitivity. It is formally possible that learning impairments also exist at day 1 but that the behavioral assay lacks the sensitivity to extract the difference. This seems unlikely because of the large difference in time-of-onset: the Aβ42-expressing flies show no learning impairment at 10 days-of-age yet the mitochondrial impairments are pronounced at day 1. The long delay between the detection of cellular pathologies and the learning impairment, which is essentially 15-20% of the fly’s lifespan, parallels the decades-long progression of the disease in humans.
Conclusions
Collectively, our data show that mitochondrial dysfunction occurs early in hierarchy of cellular behavioral pathologies associated with Alzheimer’s disease and that this function occurs at different times in the somatic, dendritic and axonal compartments of brain neurons.
Supplementary Material
Acknowledgements:
Funding: This research was supported by NIH grant 5R01AG049037.
Abbreviations
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- MBn
mushroom body neurons
- mito-GFP
mitochondrial-targeted GFP
- SIM
structured illumination microscopy
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Ethical Approval and Consent to participate: Not applicable.
Consent for publication: Not applicable.
Availability of supporting data: All data generated and/or analyzed during the current study are included in this published article and supplementary files.
Competing interests: The authors declare no competing interests.
Authors’ information: Not applicable.
References
- 1.Cenini G & Voos W Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front Pharmacol 10, 902, doi: 10.3389/fphar.2019.00902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holtzman DM, Morris JC & Goate AM Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3, 77sr71, doi: 10.1126/scitranslmed.3002369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henstridge CM, Hyman BT & Spires-Jones TL Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat Rev Neurosci 20, 94–108, doi: 10.1038/s41583-018-0113-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gan L, Cookson MR, Petrucelli L & La Spada AR Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci 21, 1300–1309, doi: 10.1038/s41593-018-0237-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardy J & Selkoe DJ The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356, doi: 10.1126/science.1072994 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Martini-Stoica H, Xu Y, Ballabio A & Zheng H The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci 39, 221–234, doi: 10.1016/j.tins.2016.02.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217, doi: 10.1016/j.cell.2017.05.018 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Iaccarino HF et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235, doi: 10.1038/nature20587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang R & Reddy PH Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J Alzheimers Dis 57, 1041–1048, doi: 10.3233/JAD-160763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo T et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487–496, doi: 10.1016/j.stem.2013.01.009 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Butterfield DA & Halliwell B Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20, 148–160, doi: 10.1038/s41583-019-0132-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burte F, Carelli V, Chinnery PF & Yu-Wai-Man P Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11, 11–24, doi: 10.1038/nrneurol.2014.228 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Cai Q & Tammineni P Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front Cell Neurosci 10, 24, doi: 10.3389/fncel.2016.00024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez Ortiz JM & Swerdlow RH Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol 176, 3489–3507, doi: 10.1111/bph.14585 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swerdlow RH & Khan SMA “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 63, 8–20, doi: 10.1016/j.mehy.2003.12.045 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Misgeld T & Schwarz TL Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96, 651–666, doi: 10.1016/j.neuron.2017.09.055 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai Y PINK1-Parkin signaling in Parkinson’s disease: Lessons from Drosophila. Neurosci Res, doi: 10.1016/j.neures.2020.01.016 (2020). [DOI] [Google Scholar]
- 18.Cha SJ et al. Parkin expression reverses mitochondrial dysfunction in fused in sarcoma-induced amyotrophic lateral sclerosis. Insect Mol Biol 29, 56–65, doi: 10.1111/imb.12608 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Garrido-Maraver J, Loh SHY & Martins LM Forcing contacts between mitochondria and the endoplasmic reticulum extends lifespan in a Drosophila model of Alzheimer’s disease. Biol Open 9, doi: 10.1242/bio.047530 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X et al. Establishment of a Drosophila AD model. J Biol Methods 3, e43, doi: 10.14440/jbm.2016.61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X et al. FoxO mediates APP-induced AICD-dependent cell death. Cell Death Dis 5, e1233, doi: 10.1038/cddis.2014.196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cutler T et al. Drosophila Eye Model to Study Neuroprotective Role of CREB Binding Protein (CBP) in Alzheimer’s Disease. PLoS One 10, e0137691, doi: 10.1371/journal.pone.0137691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iijima K & Iijima-Ando K Drosophila models of Alzheimer’s amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-beta 42. J Alzheimers Dis 15, 523–540, doi: 10.3233/jad-2008-15402 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Povellato G, Tuxworth RI, Hanger DP & Tear G Modification of the Drosophila model of in vivo Tau toxicity reveals protective phosphorylation by GSK3beta. Biol Open 3, 1–11, doi: 10.1242/bio.20136692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dourlen P Identification of Tau Toxicity Modifiers in the Drosophila Eye. Methods Mol Biol 1523, 375–389, doi: 10.1007/978-1-4939-6598-4_26 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Iijima K et al. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One 3, e1703, doi: 10.1371/journal.pone.0001703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S et al. The calcineurin inhibitor Sarah (Nebula) exacerbates Abeta42 phenotypes in a Drosophila model of Alzheimer’s disease. Dis Model Mech 9, 295–306, doi: 10.1242/dmm.018069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busto GU, Cervantes-Sandoval I & Davis RL Olfactory learning in Drosophila. Physiology (Bethesda) 25, 338–346, doi: 10.1152/physiol.00026.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis RL SnapShot: Olfactory Classical Conditioning of Drosophila. Cell 163, 524–524 e521, doi: 10.1016/j.cell.2015.09.043 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Drago I & Davis RL Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Rep 16, 2763–2776, doi: 10.1016/j.celrep.2016.08.017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chauhan D et al. BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1beta Maturation in Macrophages. Cell Rep 25, 2354–2368 e2355, doi: 10.1016/j.celrep.2018.10.087 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Bock FJ & Tait SWG Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21, 85–100, doi: 10.1038/s41580-019-0173-8 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Green DR & Reed JC Mitochondria and apoptosis. Science 281, 1309–1312, doi: 10.1126/science.281.5381.1309 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Wang C & Youle RJ The role of mitochondria in apoptosis*. Annu Rev Genet 43, 95–118, doi: 10.1146/annurev-genet-102108-134850 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song Z, McCall K & Steller H DCP-1, a Drosophila cell death protease essential for development. Science 275, 536–540, doi: 10.1126/science.275.5299.536 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Akagawa H et al. The role of the effector caspases drICE and dcp-1 for cell death and corpse clearance in the developing optic lobe in Drosophila. Dev Biol 404, 61–75, doi: 10.1016/j.ydbio.2015.05.013 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Ryoo HD & Bergmann A The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol 4, a008797, doi: 10.1101/cshperspect.a008797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Florentin A & Arama E Caspase levels and execution efficiencies determine the apoptotic potential of the cell. J Cell Biol 196, 513–527, doi: 10.1083/jcb.201107133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarkissian T, Timmons A, Arya R, Abdelwahid E & White K Detecting apoptosis in Drosophila tissues and cells. Methods 68, 89–96, doi: 10.1016/j.ymeth.2014.02.033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loewen C, Boekhoff-Falk G, Ganetzky B & Chtarbanova S A Novel Mutation in Brain Tumor Causes Both Neural Over-Proliferation and Neurodegeneration in Adult Drosophila. G3 (Bethesda) 8, 3331–3346, doi: 10.1534/g3.118.200627 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardy J & Allsop D Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12, 383–388, doi: 10.1016/0165-6147(91)90609-v (1991). [DOI] [PubMed] [Google Scholar]
- 42.Bloom GS Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508, doi: 10.1001/jamaneurol.2013.5847 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberson ED et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754, doi: 10.1126/science.1141736 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Greeve I et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci 24, 3899–3906, doi: 10.1523/JNEUROSCI.0283-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiang HC, Wang L, Xie Z, Yau A & Zhong Y PI3 kinase signaling is involved in Abeta-induced memory loss in Drosophila. Proc Natl Acad Sci U S A 107, 7060–7065, doi: 10.1073/pnas.0909314107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lazarov O, Lee M, Peterson DA & Sisodia SS Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 22, 9785–9793 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Onyango IG, Dennis J & Khan SM Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis 7, 201–214, doi: 10.14336/AD.2015.1007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calkins MJ, Manczak M, Mao P, Shirendeb U & Reddy PH Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20, 4515–4529, doi: 10.1093/hmg/ddr381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cervantes-Sandoval I, Phan A, Chakraborty M & Davis RL Reciprocal synapses between mushroom body and dopamine neurons form a positive feedback loop required for learning. Elife 6, doi: 10.7554/eLife.23789 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phan A et al. Stromalin Constrains Memory Acquisition by Developmentally Limiting Synaptic Vesicle Pool Size. Neuron 101, 103–118 e105, doi: 10.1016/j.neuron.2018.11.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.