

Abstract
AIIt, a heterotetramer of S100A10 (P11) and Annexin A2, plays a key role in calcium dependent, membrane associations with a variety of proteins. We previously showed that AIIt interacts with the short cytoplasmic domain (12 amino acids) of CEACAM1 (CEACAM1-SF). Since the cytoplasmic domains of CEACAM1 help regulate the formation of cis- or trans-dimers at the cell membrane, we investigated the possible role of their association with AIIt in this process. Using NMR and molecular dynamics, we show that AIIt and its pseudoheterodimer interacts with two molecules of short cytoplasmic domain isoform peptides, and that interaction depends on the binding motif 454-Phe-Gly-Lys-Thr-457 where Phe-454 binds in a hydrophobic pocket of AIIt, the null mutation Phe454Ala reduces binding by 2.5 fold, and the pseudophosphorylation mutant Thr457Glu reduces binding by three fold. Since these two residues in CEACAM1-SF were also found to play a role in the binding of calmodulin and G-actin at the membrane, we hypothesize a sequential set of three interactions are responsible for regulation of cis- to trans-dimerization of CEACAM1. The hydrophobic binding pocket in AIIt corresponds to a previously identified binding pocket for a peptide found in SMARCA3 and AHNAK, suggesting a conserved functional motif in AIIt allowing multiple proteins to reversibly interact with integral membrane proteins in a calcium dependent manner.
Keywords: Annexin A2, AIIt, CEACAM1, P11, S100A10, NMR, Molecular Dynamics
Graphical Abstract
1. Introduction
CEACAM1 is a type 1 transmembrane protein expressed on many cell types with multiple splice forms that usually include 3–4 ectodomains and two cytoplasmic domains designated as long, CEACAM1-LF; or short, CEACAM1-SF [1]. The N-terminal ectodomain conveys cell-cell adhesive properties to cells, while the cytoplasmic domains, among other functions, exert inside-out signaling, affecting the formation of cis- and trans-dimers [1]. In this manner, the cis-dimer form of CEACAM1 is unable to convey cell-to cell signaling, while the trans-dimer forms intercellular contacts. Although the short cytoplasmic domain consists of only 12 amino acids, this domain has been shown to interact with multiple proteins, including G-actin [2, 3], AIIt [4], and calmodulin [3, 5], all of which are candidates for affecting cis-trans-dimer equilibrium. In the above studies, we have shown that the sequence 454-Phe-Gly-Lys-Thr-457 plays a key role in lumen formation [2] and the interactions with G-actin, Annexin A2, and calmodulin. While the interaction of CEACAM1-SF with calmodulin has been shown to stabilize the formation of trans-dimers [5, 6], the interactor that promotes or stabilizes cis-dimers has not been identified. As a first approach to this problem we considered AIIt as a candidate based on its membrane associated functions and binding to CEACAM1-SF.
AIIt, a heterotetramer of Annexin A2 and S100A10 (P11), is a key player in actin dynamics and epithelial cell polarization [7, 8]. These are also properties of CEACAM1-SF, a protein that plays a role in the polarization and lumen formation of epithelial breast cells, a process that involves extensive actin reorganization [2]. The symmetric heterotetramer structure of AIIt [9] suggests the possibility that peptide interactions could occur as pairs, which in the case of CEACAM1-SF would be compatible with its cis-dimer formation. Although this was not the case for the interaction of AIIt with the C-terminal AHNAK peptide (20 amino acids), derived from the large AHNAK protein [10, 11], but when a shorter AHNAK (15 amino acids) was studied, it was found that two peptides were bound to the two hydrophobic groves symmetrically on the heterotetramer AIIt [12]. The same binding mode was also true for an N-terminal peptide (14 amino acids) of SMARCA3 [12]. These studies demonstrated that short peptides (15 amino acids or less) can bind the AIIt heterotetramer with 1:1 molar ratio. Thus, we wanted to test the hypothesis that the short cytoplasmic domain peptide of CEACAM1-SF may bind to AIIt in a similar manner, stabilizing the formation of cis-dimers.
AIIt comprises two S100A10 molecules (2 × 12 kDa) and two Annexin A2 (AnxA2) molecules (2 × 39 kDa) to form a heterotetramer (102 kDa) through the interaction of the first 12 amino acids of AnxA2 with S100A10 [13]. This complex is a rather large species for an NMR based approach. However, a number of studies [14, 15] have shown that only the first 13–15 N-terminal residues of AnxA2 are required to generate a pseudocomplex with S100A10 that mimics many of the known interactions of AIIt. In this approach, full length S100A10 (92 amino acids) was expressed as a fusion protein with residues 1–15 of Anx A2 joined by an eight amino acid linker peptide (ENLYFQGD) that contains a TEV cleavage site [15]. By expressing the protein in defined media enriched in 15NH4Cl and 13C6-glucose Shaw and coworkers [15] determined NMR chemical shift assignments, and were able to show binding to the alpha1-subunit of the F-actin capping protein TRTK12 and the CapZ protein that caps the barbed ends of F-actin. Additional studies by Shaw and coworkers [16] led to the conclusion that most S100 proteins form homodimers with a symmetrical binding site that recognize two copies of the target protein. With this in mind we thought it likely that AIIt, and by extension, the pseudocomplex, would bind two copies of the cytoplasmic domain of CEACAM1-SF leading to cis-dimer formation.
S100A10 is unusual among the S100 proteins in that it forms an open conformation with Annexin A2 in the absence of Ca2+ [17]. In the Ca2+ free conformation, S100A10 is juxtaposed to the plasma membrane [18]. Based on cryo-EM studies on AIIt [18], the complex was shown to undergo a conformation change in the presence of calcium, moving S100A10 away from the membrane. To determine if these changes in the S100A10 conformation of AIIt apply to CEACAM1-SF, thereby affecting its cis-dimer status, it was first necessary to establish that the CEACAM1-SF interaction with AIIt was primarily through S100A10 and was compatible with dimer formation.
2. Materials and methods
2.1. Peptide Synthesis
Fifteen amino acid long synthetic peptides representing the cytoplasmic membrane proximal region of CEACAM1 long form (LW) and CEACAM1 short form (SW) and some peptides with mutations were synthesized by the City of Hope Peptide Synthesis Core Facility with N-terminal either capped with an acetyl or DOTA group. All the synthesized peptides were purified by HPLC and the molecular masses were confirmed by mass spectrometry. Both 15N-Fmoc-Phe and 15N-Fmoc-Gly (Cambridge Isotope Labs) were used to introduce 15N-labeleing for three residues in one of the peptides. A total of nine CEACAM1 peptides were synthesized in this work. The loading of Y3+ and Gd3+ to the DOTA ring of the peptides were carried out in 50 mM glycine buffer at 80°C following the published procedure [19]. The DOTA chelated Y3+ and Gd3+ peptides were purified using C18 Sep Pak cartridge (Waters), the purity and molecular weight were checked with mass spectrometry, and additionally with NMR for Y3+ loaded peptide. The synthesized peptide sequences and corresponding abbreviations used in this work were listed in (Table 1).
Table 1:
Peptides used in this study1
| Peptide Name | Peptide Sequence |
|---|---|
| 15N-SW-12 | 453-HFGKTGSSGPLQ-464 |
| Ac-15N-F454G458G461-SW | Ac-CFLHF*GKTG*SSG*PLQ |
| Ac-SW | Ac-CFLHFGKTGSSGPLQ |
| Ac-SW-F/A | Ac-CFLHAGKTGSSGPLQ |
| Ac-SW-T/E | Ac-CFLHFGKEGSSGPLQ |
| Ac-SW-S/E | Ac-CFLHFGKTGESGPLQ |
| Ac-SW-TS/EE | Ac-CFLHFGKEGESGPLQ |
| Ac-LW | Ac-CFLHFGKTGRASDQR |
| Y-DOTA-SW | Y3+-DOTA-GFLHFGKTGSSGPLQ |
| Gd-DOTA-SW | Gd3+-DOTA-GFLHFGKTGSSGPLQ |
15N-SW-12 was uniformly 15 N-labeled, and the residues are numbered according to the intact protein.
The remaining peptides were synthesized with a few 15N labeled residues indicated by *.
2.2. Protein and peptides expression
Synthetic genes of S100A10, S100A10-AnxA2Nter fusion peptide [(S100A10(1–92)-ENLYFQGD linker-AnxA2 peptide (1–13)] [15] and 12 amino acid ceacam1 short form peptide (HFGKTGSSGPLQ) were produced using E. coli codon-optimized overlapping oligo DNA primers, and cloned into pET32b. The pET32b was modified with insertion of SMT3 to create an Ulp1 cleavage sites. The recombinant proteins were expressed in E.cloni or E. coli C41(DE3) as a thioredoxin (Trx)-SMT3 fusion protein with an N-terminal His-tag. The expressed proteins from 2 to 4 liters culture were first purified using a Ni-NTA resin, and further purified by cleaving the peptides off the N-terminal Trx-SMT3 portion by incubation with hexa-histidine tagged Ulp1. Expression of the recombinant proteins and cleaved peptides were confirmed by SDS-PAGE. The proteins were concentrated using Centricon YM10 centrifugal filters (Fisher Scientific) and purified by FPLC on a Superdex 75G column (GE Healthcare Life Science, Pittsburgh, PA) using 50 mM Tris buffer with 150 mM NaCl and 10% glycerol at pH 8. The purified proteins were stored at −80°C freezer for future buffer exchange and NMR studies. For quality control and identity of proteins and peptides, all fractions and gel bands were analyzed for their primary sequence by MS and MS/MS on an Agilent 1200 Mass spectrometry system. The data were analyzed using GPM X! Tandem search engine (The Global Proteome Machine Organization) with human protein database and Scaffold (Proteome Software). The human Anx A2 gene with a thrombin cleavage site and His tag attached at C-terminal were from GeneArt (Thermo), and it was cloned into pET28, and expressed in E. coli C41(DE3). The 15N-labeled proteins were expressed using M9 minimal media supplemented with 15NH4Cl as sole nitrogen source. All the purified procedures are similar to that described above except for the 15N-CEACAM1 short isoform, which was purified by HPLC after Ulp1 cleavage.
2.3. NMR sample preparation
For the 15N-labeled CEACAM1-SF synthetic peptide (Ac-15N-F454G458G461-SW) and uniformed 15N-labeled CEACAM1-SF 12 amino acid peptide study, 50 mM NaAc-d3 buffer (5% D2O) with pH 5.5 and 2mM DTT was used. The S100A10, S100A10-AnxA2Nter fusion protein and AnxA2 protein were buffer exchanged to the same buffer as described above. For quality control, the heterotetramer formation between S100A10 and AnxA2 was confirmed using HPLC. The concentration of 15N-labeled peptide stock solution is 1 mM determined by NMR spectroscopy with known concentration of TSP-d4 as an internal reference. Protein sample concentrations are between 0.3–0.4 mM determined by a ND-1000 spectrophotometer (NanoDrop). When the 15N-labeled S100A10-AnxA2Nter fusion protein was used to characterize the interaction with unlabeled CEACAM1 peptides, the protein was exchanged to 50 mM phosphate buffer containing 40 mM NaCl and 2 mM DTT at pH 7.0 in 5% D2O. After exchange and concentration, the protein was aliquoted, and fast frozen with liquid nitrogen and stored at −80°C for NMR experiments. The peptide solution was prepared from purified dry powder with the same buffer solution described above, and the pH was adjusted to 7.0 as needed. The stock solution of peptides was 2.5 mM except for Gd-DOTA-SW, which was 5 mM.
2.4. NMR Experiments and data analysis
The assignment of 15N-labeled SW peptide (12 amino acid, 15N-SW-12, see Table 1) was achieved by analysis spectra of 1H-15N-HSQC, 3D 15N-TOCSY-HSQC, 3D 15N-NOESY-HSQC, 1H-13C-HSQC and 1H-13C-HSQC-TOCSY. All experiments were carried out at 15°C on a 700 MHz Bruker Ascend with a TXI-triple resonance cryoprobe. For the titration of S100A10-AnxA2Nter fusion protein and AnxA2 protein to 15N-SW-12 peptide, the peptide concentration was 60 μM, and the 1H-15N HSQC Bruker sequence “hsqcetf3gpsi” was used at 25°C. For the titration of S100A10, S100A10-AnxA2Nter fusion protein and AnxA2 protein to Ac-15N-F454G458G461-SW, the peptide concentration was 25 μM, and the sofast HMQC [20] “sfhmqcf3gpph” was performed at 25°C. For the titration of CEACAM1 peptides to 15N-S100A10-AnxA2Nter fusion protein, 54 μM protein was used. The Bruker sequence “sfhmqcf3gpph” was used to monitor the chemical shift changes of 15N-S100A10-AnxA2Nter fusion protein at 35°C. The 1H-15N backbone assignment of 15N-S100A10-AnxA2Nter fusion protein at 35°C was mapped from reported data [15], and a private communication with Dr. Gary Shaw. All NMR data were processed and analyzed using NMRPipe [21], NMRView [22] or NMRFAM-SPARKY [23] and Bruker Topspin. The dissociation constant KD of the complex between CEACAM1 peptides with either S100A10-AnxA2Nter fusion protein or S100A10-AnxA2 tetramer was derived from global fitting of chemical shift perturbations of selected residues either on 15N-labeled CEACAM1 peptide or 15N-labeled S100A10-AnxA2Nter fusion protein using equation 6 described in the review [24]. The fitting was carried out using Prism software.
2.5. Prediction of CEACAM1-SF binding to S100A10-AnxA2Nter fusion protein
The crystal structure of S100A10-AnxA2Nter fusion protein was obtained from the PDB databank (pdb ID: 4HRG) [12]. The CEACAM1-SF peptide was created as an extended chain in Maestro [25], and two of the peptides were placed approximately 30Å above the binding pocket of the S100A10 dimer. This configuration was exported as a pdb file and further prepared using tleap of the AmberTools package [26], when hydrogen atoms were added and neutral capping groups acetyl and N-methyl amide were added to the N and the C termini of S100A10-AnxA2Nter. The N and the C termini of CEACAM1-SF peptides were capped using acetyl and charged carboxylic groups respectively, in line with the experimental construct. In order to dock the CEACAM1-SF peptides into the binding pockets of the S100A10-AnxA2Nter dimer, molecular dynamics simulations were performed using the Generalized Born implicit solvation model (igb=8) [27] of the AMBER 16 simulation package [26] in conjunction with the FF14SBonlysc force-field [28, 29], as recommended in the AMBER manual. During simulations, the default PB radii set was set to mbondi3, corresponding to the igb8 implicit solvent model. Applying hydrogen mass repartitioning allowed a timestep of 4 fs to be used in both implicit and explicit solvent simulations [30].
The structure was minimized using 7000 steps of conjugate gradient minimization, followed by an annealing procedure, where the system was initially heated to 450K over 20 ns and simulated at 450K for another 100 ns, before slowly cooling down to 290K over 200 ns. During this and the successive steps, the S100A10-AnxA2Nter heavy atoms were restrained with weak harmonic positional restraints with a force constant of 1 kcal/mol. The annealing step ensured that the docking results were independent of the memory effect of the initial peptide configurations. The system was then simulated at 290K for 2.5μs with weak distance restraints (force constant of 0.5 kcal/mol) applied between the residue L78 of both S100A10-AnxA2Nter monomers and the F454 of the two CEACAM1-SF peptides, to guide the docking. In total, five independent docking simulations were performed.
2.6. Explicit solvent simulations of S100A10-AnxA2Nter/CEACAM1-SF complex
The docked CEACAM1-SF conformations were clustered using the Cα coordinates with a cutoff of 2Å root mean square deviation, and the representative conformation from the largest cluster was chosen to perform the explicit solvent simulations. The complex structure was solvated in a water box of dimensions 77Å x 86Å x 93Å and sodium ions were added to neutralize the charges. The system was parameterized using the FF14SB force-field for protein [28] and TIP3P for water [31]. The system was equilibrated by first minimizing over 2000 steps of conjugate gradient minimization, then heating to 310K over 30 ns at constant volume, followed by simulation at a constant pressure of 1 atmosphere. During these steps, the protein heavy atoms were restrained to their original positions with a harmonic force constant of 5 kcal/mol. The position restraints were then gradually relaxed over 50 ns at constant pressure and temperature, followed by 50 ns of completely unrestrained equilibration. The temperature was controlled using the Langevin thermostat [32] with a collision frequency of 1 ps−1. The pressure was controlled by isotropic position scaling using the Berendsen barostat [33] with a relaxation time of 2 ps. The production runs consisted of six independent simulations of 200 ns each, with different starting velocities.
2.7. Modeling of the CEACAM1-SF transmembrane helical dimer and S100A10-AnxA2Nter complex
The structure of the CEACAM1-SF helical transmembrane (TM) dimer was predicted using the PREDDIMER web server [34, 35]. The details of the method can be obtained from the associated citations. In brief, the TM helical monomers of the CEACAM1-SF were oriented next to each other and a potential map was constructed based on hydrophobicity and surface complementarity. The best structures corresponding to the lowest potential were further filtered according to the interaction between the GxxxG motifs of the TM monomers [36]. The final structure was selected based on compatibility with the distance between the N terminal cysteines of the bound CEACAM1-SF peptides in S100A10 (8.5Å). Both the CEACAM1-SF TM and the peptides bound S100A10-AnxA2Nter structures were loaded in Maestro and oriented such that the C terminal ends of the TM helices were proximal to the N terminal ends of the bound peptides in S100A10-AnxA2Nter. Then peptide bonds were created between the TM helices and the bound peptides and the whole structure was minimized using Prime [25]. The CECAM1-SF/S100A10-AnxA2Nter complex was embedded in the lipid bilayer using the Desmond module of Maestro. No MD simulations were performed on the lipid bilayer bound complex of CEACAM1-SF/S100A10-AnxA2Nter.
2.8. Modeling of DOTA-peptides and calculation of positional density distribution
The CEACAM1-SF conformations were obtained from the explicit solvent MD simulations. The structure of the Gd-DOTA was obtained from the PDB Databank (pdb ID: 1NC4). In each CEACAM1-SF conformation, the DOTA groups were fused to the N termini of the CEACAM1-SF peptides in-silico and the center of the gadolinium atoms were used to calculate the positional density distribution. In total, 3000 CEACAM1-SF conformations were used for the density distribution calculation.
3. Results
3.1. NMR Studies of the interaction between 15N-labeled CEACAM1-SF peptide and S100A10-AnxA2Nter fusion protein, AnxA2, S100A10 and AIIt
Previously, we showed that CEACAM1-SF interacted with AIIt by GST-pull down and SPR binding studies [3]. To directly observe the interaction between CEACAM1-SF cytoplasmic domain peptide SW with either AnxA2 or S100A10-AnxA2Nter fusion by NMR, the assignments of the 15N-labeled 12 amino acid SW (see Table 1 for sequence) were obtained by the analysis of 2D and 3D NMR experiments (supplementary Figure S1). The cross peak of Phe-454 was not seen, likely due to its close proximity to the N-terminus. In addition to major cross peaks, minor cross peaks for residues close to Pro are indicated, likely due to both cis- and trans-configurations of Pro-462. When either AnxA2 or the S100A10-AnxA2Nter fusion protein were gradually titrated to 15N-SW-12 up to molar ratio 1:1, no chemical shift perturbation was observed. The possible reason for no interactions with AnxA2 or S100A10-AnxA2Nter fusion protein with this peptide may be due to the fact that Phe-454 was too close to the N-terminus, and the positive charge of the free N-terminus at pH 5.5 would interfere with binding.
To overcome the potential interference of the positive charge of the N-terminal residue, we synthesized a second peptide of 15 residues with its N-terminus acetylated and with three 15N-labeled residues, Ac-15N-F454G458G461-SW (see Table 1). Four potential binding partners, S100A10, AnxA2, S100A10-AnxA2Nter tetramer and S100A10-AnxA2Nter fusion proteins were titrated to the Ac-15N-F454G458G461-SW peptide. The overlaid cross peaks of three 15N-labeled residues from free peptide (in blue color) and from the final titration point with different potential binding partners (in red color) are presented in Figure 1. The molar ratio between the protein binding partners to Ac-15N-F454G458G461-SW was around 4:1. There were no chemical shift perturbations observed for Ac-15N-F454G458G461-SW when it was mixed with AnxA2 (Figure 1A and 1B) or with S100A10 (Figure 1C and 1D). On the other hand, chemical shift perturbations were observed for Ac-15N-F454G458G461-SW when it was mixed with AIIt, the S100A10-AnxA2 tetramer (Figure 1E and 1F) or with S100A10-AnxA2Nter fusion protein (Figure 1G and 1H). The chemical shift perturbation patterns of three 15N labeled residues in Ac-15N-F454G458G461-SW were very similar when S100A10-AnxA2 tetramer or S100A10-AnxA2Nter fusion were added to Ac-15N-F454G458G461-SW with Phe-454 showing the greatest perturbation. Thus, this peptide interacted with either AIIt or the S100A10-AnxA2Nter fusion protein in a similar pattern, but did not interact with AnxA2 or S100A10 alone.
Figure 1. 1H-15N HSQC spectra of Ac-15N-F454G458G461-SW in the presence and absence of binding partners.

A and B: Cross peaks of F454, G458 and G461 residues in the absence of AnxA2 (Blue) and in the presence of AnxA2 (Red) with molar ratio of AnxA2: Ac-15N-F454G458G461-SW = 4:1. C and D: Cross peaks of F454, G468 and G461 residues in the absence of S100A10 (Blue) and in the presence of S100A10 (Red) with molar ratio of S100A10: Ac-15N-F454G458G461-SW = 4:1. E and F: Cross peaks F454, G458 and G461 residues in the absence of AIIt (Blue) and in the presence of AIIt (Red) with molar ratio of AIIt: Ac-15N-F454G458G461-SW = 4:1. G and H: Cross peaks of F454, G458 and G461 residues in the absence of S100A10-AnxA2Nter fusion (Blue) and in the presence of S100A10-AnxA2Nter fusion (Red) with molar ratio of S100A10-AnxA2Nter fusion: Ac-15N-F454G458G461-SW = 4.3:1.
Based on the chemical shift perturbation data versus the molar ratio of protein sample titrated to the Ac-15N-F454G458G461-SW, the dissociation constants were derived from global curve fitting. The chemical shift perturbations (CSP) of the Phe-454 cross peak in 1H-15N-HSQC spectra titrated by S100A10-AnxA2Nter fusion protein is shown in Figure 2A. The curve fitting of CSPs versus the concentration of S100A10-AnxA2Nter fusion protein for residues Phe-454 and Gly-458 are presented in Figure 2B. The dissociation constant, KD, was 0.46 ± 0.16 mM. Similarly, the KD between Ac-15N-F454G458G461-SW and AIIt complex was 0.30 ± 0.12 mM. The KD values are comparable considering the associated errors in the measurements. These results showed the S100A10-AnxA2Nter fusion protein can be used as a model system for the intact heterotetramer (AIIt) formed by dimers of S100A10 and AnxA2 as reported before for other complexes such as AIIt with AHANK peptide [10] and with SMARCA3 peptide [12].
Figure 2. S100A10-AnxA2Nter titration to Ac-15N-F454G458G461-SW.

A: Chemical shift changes of F454 cross peak versus the titration of S100A10-AnxA2Nter fusion protein. The molar ratio between S100A10-AnxA2Nter fusion protein and Ac-15N-F454G458G461-SW was color coded as follows: Red 0:1; Black 1:1; Green 1.5:1; Blue 2:1; Yellow 2.75:1; Magenta 3.5:1 and Cyan 4.3:1. B: The curve fitting of chemical shift perturbation for residues F454 and G458 of Ac-15N-F454G458G461-SW titrated by S100A10-AnxA2Nter fusion protein. The CSP values in unit of HZ was calculated using the following equation. . The ΔωN and ΔωH are the nitrogen and proton chemical shift difference between free Ac-15N-F454G458G461-SW and that in the mixture with addition of S100A10-AnxA2Nter.
3.2. 15N-S100A10-AnxA2Nter fusion protein interactions with CEACAM1-SF wild type and mutant peptides
Since the S100A10-AnxA2Nter fusion protein was shown to interact with the Ac-SW peptide in a similar way to AIIt, a 15N-labeled S100A10-AnxA2Nter fusion protein was generated to further investigate the interaction with various Ac-SW mutants (see Table 1 for the sequences). The assigned chemical shifts of 15N-labeled S100A10-AnxA2Nter fusion protein from an earlier study [15] were mapped to our spectra. When the Ac-SW peptides were titrated to 15N-labeled S100A10-AnxA2Nter fusion protein, the KD values were determined from CSPs of seven representative well-resolved residues. The global curve fitting of chemical shift perturbation of 15N-labeled S100A10-AnxA2Nter fusion protein with Ac-SW titration is shown in Figure 3. The KD of Ac-SW peptides as well as a truncated version of the long isoform CEACAM1-LF peptide are listed in Table 2. The primary sequence of the long isoform is identical to the short isoform over the first nine residues, but diverges after that (Table 1) due to the effect of alternative mRNA splicing. Thus, it was important to determine if the equivalent length long isoform peptide would bind to the S100A10-AnxA2Nter fusion peptide. Since their KDs to S100A10-AnxA2Nter are similar (Table 2), we conclude that the interaction mainly depends upon on the first nine N-terminal amino acids of the peptides.
Figure 3. Curve fitting of 15N-S100A10-AnxA2Nter titrated by Ac-SW.

Curve fitting of some well resolved cross peaks of 15N-S100A10-AnxA2Nter titrated by Ac-SW peptide. The selected residue numbers of S100A10-AnxA2Nter are shown at the right side of the figure. The KD for the complex derived from this global curve fitting is 0.88 ± 0.1 mM. The CSP values in unit of HZ was calculated using the following equation. . The ΔωΝ and ΔωΗ are the nitrogen and proton chemical shift difference between free 15N-S100A10-AnxA2Nter and that in the mixture with addition of Ac-SW.
Table 2:
Dissociation constants of CEACAM1-SF peptide complexes with their binding partners.
| Peptide Name | KD (mM) | Conditions |
|---|---|---|
| Ac-15N-F454G458G461-SW | 0.30 ± 0.12 | Titrated by AlIt at 25°C, pH 5.5 |
| Ac-15N-F454G458G461-SW | 0.46 ± 0.16 | Titrated by S100A10 AnxA2Nter fusion at 25°C, pH 5.5 |
| Ac-SW | 0.88 ± 0.10 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
| Ac-SW-F/A | 2.10 ± 0.81 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
| Ac-SW-T/E | 2.33 ± 0.39 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
| Ac-SW-S/E | 1.58 ± 0.20 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
| Ac-SW-TS/EE | 3.53 ± 1.20 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
| Ac-LW | 1.22 ± 0.08 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
| Y-DOTA-SW | 1.07 ± 0.12 | Titrated to 15N-S100A10-AnxA2Nter fusion at 35°C, pH 7.0 |
When Phe-454 was mutated to Ala, the KD increased by more than two fold, indicating that Phe-454 is important for Ac-SW peptide binding to the S100A10-AnxA2Nter fusion protein. When Thr-457 or Ser-459 or both were mutated to Glu to mimic the phosphorylation of Thr and Ser, the KD values were increased compared to the wild type peptide, and the changes were additive. These results imply that the phosphorylation of these two residues may inhibit the interaction between Ac-SW peptide with the AIIt complex, or vice versa.
The interaction patterns between the S100A10-AnxA2Nter fusion protein and wild type CEACAM1 peptide or the mutant peptides were investigated by comparison of the S100A10-AnxA2Nter fusion protein CSPs from complexes with different peptides (Figure 4). There are no data for a few residues due to unassigned peaks or severe spectral overlaps. The residues with CSP values larger than two times the average RMSD (the horizontal line in each figure panel) are labeled. These residues are mainly from the second EF hands of S100A10, i.e., helix 3, helix 4, the loop between helix 3 and 4 and some residues at the C-terminus (the N-terminal fragment of AnxA2 fused to the C-terminal of S100A10 protein). This explains why CEACAM1 peptides only interact with the heterotetramer of S100A10 and AnxA2, but not with the individual S100A10 or AnxA2 proteins. The CSPs of S100A10-AnxA2Nter from all six peptide complexes presented similar patterns, suggesting that the mutations or the LF peptide did not change the basic interaction pattern between SW and S100A10-AnxA2Nter fusion protein.
Figure 4. Chemical shift perturbation of 15N-S100A10-AnxA2Nter in complexes with CEACAM1-SF peptides.

The chemical shift changes between free 15N-S100A10-AnxA2Nter and in complex with six CEACAM1-SF peptides. The molar ratio between peptides and 15N-S100A10-AnxA2Nter for all complexes is 10:1 except for the complex with Ac-SW, which is 8.7:1. The thin line in each panel is the two times RMSD of the chemical shift changes between free 15N-S100A10-AnxA2Nter and 15N-S100A10-AnxA2Nter in complex with peptides. The peptide in complex is Ac-SW for A, Ac-SW-F/A for B, Ac-SW-T/E for C, Ac-SW-S/E for D, Ac-SW-TS/EE for E and Ac-LF for F. The sequences for each peptide are listed in Table 1. The CSP values in unit of HZ was calculated using the following equation. . The ΔωΝ and ΔωΗ are the nitrogen and proton chemical shift difference between free 15N-S100A10-AnxA2Nter and that in the mixture with molar ratio between peptides and 15N-S100A10-AnxA2Nter listed above.
3.3. The role of Phe-454 in the interaction with S100A10-AnxA2Nter fusion protein
A critical Phe residue in both AHANK and SMARCA3 peptides were shown to play important roles in binding to the S100A10-AnxA2Nter fusion protein by sitting in a hydrophobic pocket of the S100A10-AnxA2Nter fusion protein [10, 12]. The key residues forming the hydrophobic pocket in the fusion protein are Phe-41, I-54, L-58, L-74, L-78, A-81, L-107, L-110 and L-112 with the last three residues from the N-terminal peptide of AnxA2 fused to the C-terminus of S100A10. Since our earlier studies identified a critical Phe (labeled as Phe-454 here) in CEACAM1-SF played a key role in binding to Actin [2, 37] and calmodulin [5], we investigated the binding of the null mutant (Phe-454-Ala) to the fusion protein. Representative 1H-15N HSQC spectra overlays between free S100A10-AnxA2Nter (in blue color) and in complex with molar ratio of 8.7:1 of S100A10-AnxA2Nter:Ac-SW (in red color) or with molar ratio of 10:1 of S100A10-AnxA2Nter:Ac-SW-F454A (in red color) are shown in Figure 5A and 5B, respectively. In complex with Ac-SW, the chemical shift of residues L-42, I-54, L-58, L-74, I-75, L-78, L-110 showed significant changes compared to free S100A10-AnxA2Nter. However, all of these changes are significantly reduced in complex with Phe-454-Ala mutant peptide. This suggests that Phe-454 sits in the hydrophobic pocket of S100A10-AnxA2Nter similar to the critical Phe in the AHANK or SMARCA3 peptides. Thus, Phe-454 of Ac-SW plays an important role in binding to S100A10-AnxA2Nter. It is also worth noting that Phe-451 of Ac-SW did not replace Phe-454 in binding to the hydrophobic pocket of S100A10 when Phe-454 was mutated to Ala. This finding suggests that the position of Phe in the cytoplasmic domain of CEACAM1-SF is also critical to the interaction with S100A10-AnxA2Nter.
Figure 5. Selected region of 1H-15N HSQC spectra of 15N-S100A10-AnxA2Nter in complexes with Ac-SW and Ac-SW-F/A.

The cross peaks in blue in both A and B are from free 15N-S100A10-AnxA2Nter fusion protein. A: the peaks in red are from the complex in which the molar ratio between Ac-SW and 15N-S100A10-AnxA2Nter is 8.7:1. B: the peaks in red are from the complex in which the molar ratio between Ac-SW-F/A and 15N-S100A10-AnxA2Nter is 10:1. The residues of 15N-S100A10-AnxA2Nter with significant chemical shift perturbation from free to complex with Ac-SW are labeled in both A and B.
3.4. Predicted docked structure of CEACAM-1 peptides bound to S100A10-AnxA2Nter
Based on the above NMR studies and the X-ray structures of the complexes between the S100A10-AnxA2Nter fusion protein with AHANK and SMARAC3 peptides, docking simulations were carried out in which Phe-454 of Ac-SW was placed in the hydrophobic pocket composed of I54, L74, L78, A81 and L110 of the S100A10-AnxA2Nter fusion protein.
The predicted docked structure was subjected to multiple independent simulations in explicit water and ions, in order to sample the bound peptide conformations. The ensemble of peptide conformations from the explicit water simulations is presented in Figure 6A. The N-termini of the Ac-SW peptide cluster are well defined where Phe-454 sits in the hydrophobic pocket of the S100A10-AnxA2Nter fusion protein. The C-termini of the peptide conformations are more dynamic and water exposed. This is in agreement with the NMR observation in which the KD of Ac-SW and Ac-LW are very similar in which their N-terminal are identical but their sequences differ at their C-termini. This suggests that the N-terminal residues play more important roles in defining the interaction with S100A10-AnxA2Nter fusion protein than the C-terminal residues. To analyze the orientation preference of the docked peptide conformations, we calculated the population distribution of the docked conformations as a function of the N-terminus to N- terminus distances and C-terminus to C-terminus distances (Supplementary Figure S2). In this figure, a close N-terminus to N-terminus distance signifies a docked orientation with the N-termini of the peptides facing each other, while a close C-terminus to C- terminus distance implies that the C-termini of the peptides are facing each other. In contrast, moderate N- to N- or C- to C-termini distances signify conformations in which N-terminus of one peptide faces the C-terminus of the other. The possible peptide orientations are shown as schematics in Supplementary Figure S2. Although the docking simulations generated a diverse set of bound peptide conformations, the N-terminal to N-terminal orientation arrangement of the peptide in the complex is the most populated one compared to other possible arrangements of N-terminal to C-terminal or C-terminal to C-terminal due to the binding energy is more favored to the N-terminal to N-terminal orientation (Supplementary Figure S2). Furthermore, this pose would be the pose of choice since the N-terminus of the peptide is connected to the transmembrane domain of CEACAM1-SF.
Figure 6. The ensemble of cytoplasmic domain of CEACAM1-SF conformations and its predicted binding mode to S100A10-AnxA2Nter pseudo dimer.

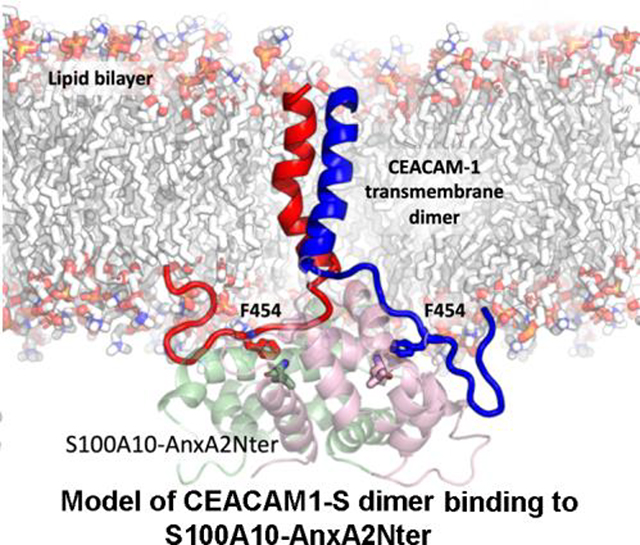
A: S100A10-AnxA2Nter (light blue) and cytoplasmic domain peptide of CEACAM1-SF (magenta). Positions of N- and C-termini and Phe454 are indicated. B: Molecular model of two (green and cyan) TM-cytoplasmic domains of CEACAM1-SF interactions with S100A10-AnxA2Nter pseudo dimer (lipid bilayer removed for clarity). Electrostatic surfaces (red for negative blue for positive). Phe454 and Thr457 are shown as space filling spheres to indicate their binding poses to one of the S100A10 dimers. Each Phe454 packs against L78 of S100A10. Thr457 is modeled as pT457 to indicate its proposed electrostatic repulsion. C: Predicted structure of the CEACAM1-SF dimer bound to S100A10-AnxA2Nter pseudo dimer in an embedded lipid bilayer. The contacting residues Phe454 of CEACAM1-SF are shown as sticks (EC domains of CEACAM1-SF not shown).
3.5. Model of transmembrane helical dimer of CEACAM1-SF bound to S100A10-AnxA2Nter fusion protein
Since the docking studies did not account for the close proximity of the plasma membrane that may potentially affect the proposed interaction, a model was generated in which the CEACAM1-SF peptide was attached to its transmembrane domain and inserted into a lipid bilayer. The structure of the CEACAM1-SF transmembrane dimer was modeled using a standard approach to visualize the binding of the actual C-terminus of a CEACAM1-SF transmembrane dimer to the S100A10-AnxA2Nter fusion protein (Figure 6B–C). The position of the key residue Phe-454 (numbering taken from the intact protein) was shown as a reference point. The fusion protein surface was color-coded based on the electrostatic potentials. This may be important when Thr-457 is phosphorylated, an event that has phenotypic consequences in a lumen formation assay [2]. As can be seen, the phosphorylation of Thr-457 may cause an electrostatic repulsion, preventing CEACAM1-SF to re-associate with AIIt.
3.6. Peptide binding pose in complex with S100A10-AnxA2Nter
In order to find out if the N-terminal to N-terminal arrangement is a unique orientation in the complex with S100A10-AnxA2Nter fusion protein, paramagnetic relaxation enhancement experiments were performed. For this purpose, an N-terminal DOTA-SW peptide chelated with the non-paramagnetic lanthanide Y3+ was titrated to the 15N- S100A10-AnxA2Nter fusion protein to test whether addition of the DOTA group to SW would affect binding to the fusion protein. The CSPs of 15N- S100A10-AnxA2Nter in complex with Y3+-DOTA-SW vs Ac-SW shown in supplementary Figure S3 are very similar, and furthermore, the KD values of two complexes are closely similar (Table 2). This suggests that the Y3+-DOTA-SW binding mode to S100A10-AnxA2Nter is the same as Ac-SW. Next, the paramagnetic lanthanide Gd3+was added to DOTA-SW and titrated to 15N- S100A10-AnxA2Nter. The overlay of a small region of the 1H-15N HSQC spectra of free 15N- S100A10-AnxA2Nter (in blue color) and in the presence of Gd3+-DOTA-SW with a 74% concentration of 15N- S100A10-AnxA2Nter (in red color) is shown in Figure 7A. Due to the relaxation enhancement effect from paramagnetic Gd3+, the intensity of peaks from residues in proximity to Gd3+-DOTA were significantly reduced, or even reduced to baseline intensities. Peak intensity variations of S100A10-AnxA2Nter in the presence of two concentrations of Gd3+-DOTA-SW versus free S100A10-AnxA2Nter are shown in Figure 7B. The peak intensities represented in blue vs red color are from the complex in which the concentrations of Gd-DOTA-SW are 74% and 174% of S100A10-AnxA2Nter, respectively. As the concentration of Gd-DOTA-SW increased, the peak intensity of all residues is reduced further, with more peak intensities reduced to baseline levels. Under both conditions, the peak intensities with the greatest reduction are from the C-terminal residues of helix3, most of helix 4 and some residues in the loop between these two helices. This result supports the peptide binding pose with N-terminal to N-terminal as shown in Figure 7C, in which, the residues with peak intensity reduced by more than 60% compared to free S100A10-AnxA2Nter samples are highlighted in gold while the unassigned ones in blue as presented in the model of the complex.
Figure 7. Paramagnetic relaxation enhancement on 15N-S100A10-AnxA2Nter in complexes with Gd-DOTA-SW.

A: Selected 1H-15N HSQC spectra region of 15N-S100A10-AnxA2Nter in complex with Gd-DOTA-SW in which the concentration of Gd-DOTA-SW is 74% of 15N-S100A10-AnxA2Nter. The peaks colored in blue are from free 15N-S100A10-AnxA2Nter sample, and the peaks colored in red are from the complex. Some residues with significant intensity loss in complex are labeled. B: The peak intensity of S100A10-AnxA2Nter in complex with Gd-DOTA-SW (I) versus free S100A10-AnxA2Nter (IO) in two concentration of Gd-DOTA-SW. The data colored in blue and red are from the complex with molar ratio of 0.74:1 and 1.74:1 between Gd-DOTA-SW: S100A10-AnxA2Nter, respectively. C: Surface representation of S100A10-AnxA2Nter showing the bound CEACAM1-SF peptides as red cartoons and the position of the DOTA shown as cyan spheres. The positional density distribution of the two DOTA groups is shown as transparent red volume around the DOTA spheres. The residues in S100A10-AnxA2Nter which show more than 60% decrease in peak intensity in complex with Gd-DOTA-SW vs free S100A10-AnxA2Nter are colored orange and labeled. The peak intensity data for residues colored in blue in the proximity of DOTA group is not available due to either lack of chemical shift assignment or sever peak overlaps.
4. Discussion
In the study of the complex between an AHANK peptide and S100A10-AnxA2Nter fusion protein, Dempsey et al. [10] used a 20-amino acid AHANK peptide (G5654-L5673). Their NMR CSP data showed two sets of cross peaks after formation of the complex, which indicated that the symmetry of S100A10-AnxA2Nter fusion protein broke after interaction with the AHANK peptide. This result is consistent with X-ray structures [10, 11]. One AHANK peptide covers the dimer interface and interacts with both helix 3, helix 4 and the C-terminus of the AnxA2 residues of each monomer. The two Phe residues (Phe-5658 and Phe-5666) on the same peptide chain are located in the hydrophobic pockets of two S100A10-AnxA2Nter dimers [11]. But when a shorter AHANK 15-amino acid (G5654-F5668) peptide was studied, or the 14-amino acid SMARCA3 (P26-F39) peptide was used to generate the complex, both X-ray structures showed that two peptides were bound to the S100A10-AnxA2Nter fusion protein in an N-terminus to N-terminus symmetrical pose [12]. In our study, the 15-amino peptide length of Ac-SW lacks additional Phe residues to cover the distance between the two hydrophobic pockets in the S100A10-AnxA2Nter fusion dimer. In fact, our NMR studies showed that only one set of CSPs was observed for 15N-S100A10-AnxA2Nter fusion dimer in complex with Ac-SW. In addition, the CSP trajectories form a straight line as shown for some representative residues in Supplementary Figure S4. All of these observations support the conclusion that two Ac-SW peptides symmetrically bind to the two binding pockets of the S100A10-AnxA2Nter fusion protein.
The paramagnetic relaxation enhancement and molecular dynamic studies showed that the N-terminus to N-terminus arrangement is a dominant pose for the CEACAM1-SF peptide interaction with the fusion protein. We further suggest that this pose has the biological function of bringing together two CEACAM1-SF cytoplasmic peptides to AIIt at the plasma membrane. This, in turn, would transmit a proximity signal to the extracellular domains that could now engage in cis-dimer formation. Furthermore, in order for CEACAM1 to form these cis-dimers, the transmembrane helices would have to have close contact, an interaction favored by the previously identified motif of 432GXXXG436 in the transmembrane α-helices of CEACAM1 [38].
It should be noted that although the membrane binding characteristics of AIIt depend on Ca2+, SA100A10 itself does not bind Ca2+ and the formation of the tetramer AIIt does not depend on Ca2+. Similarly, S100A10-AnxA2Nter lacks Ca2+ binding sites, and thus, addition of Ca2+ had no effect on CEACAM1 peptide binding in this study. Furthermore, the binding affinity of the CEACAM1 peptides (both long and short isoforms) to AIIt (this work) and to calmodulin [5] are similarly low (in the high μM to low mM range). Since this study was performed in solution, it does not include the proximity effect of two interactors at the membrane surface where a large concentration effect is observed due to the low volume characteristic of the membrane surface. In addition, low affinity interactions serve as a contact control mechanism in which the equilibrium between dimer and monomer formation can be fine-tuned in response to the Ca2+ concentration variation in cells. In this regard, the S100A10 protein in complex with AnxA2 is prevented from approaching the plasma membrane in the presence of high Ca2+ [18], while the opposite is true for Ca2+/calmodulin (Ca2+/CaM). Thus, high intracellular Ca2+ levels would cause the dissociation of CEACAM1 from AIIt, allow binding of Ca2+/CaM, pushing the equilibrium towards the monomer state. In accordance with this, it has been proposed that CEACAM1 can form trans-dimers between cells only in the monomer state and requires stabilization of the cytoplasmic domain with Ca2+/CaM [1, 39]. After Ca2+/CaM binds monomeric CEACAM1 [5, 6], CaMK2D could be recruited to the cytoplasmic domain, phosphorylating Thr-457 [40], effecting dissociation of Ca2+/CaM and subsequent recruitment of G-actin [2]. After that, polymerization of actin would stabilize the trans-dimer form, preventing recycling back to the AIIt associated dimeric form. A scheme summarizing these interactions is shown in Figure 8.
Figure 8. Scheme showing proposed steps controlling the conversion of cis- to trans-dimers in CEACAM1-SF.

1: The cis-dimer state is stabilized by the interaction of two short cytoplasmic domains with AIIt. According to other studies, only Annexin 2 (A2) interacts with the membrane in a calcium dependent manner. 2: When intracellular calcium (black dots) levels rise, AIIt undergoes a conformation change that pushes S100A10 away from the membrane, disrupting its interaction with CEACAM1-SF. 3: The cytoplasmic domain of CEACAM1 now binds to Ca2+/CaM (yellow). 4–5: CaMK2D (beige) is recruited to the Ca2+/CaM-CEACAM1-SF complex where it begins its autoactivation cycle and phosphorylates (red dots) CEACAM1-SF on Thr-457. 6: CaMK2D dissociates from CEACAM1-SF and allows G-actin (purple) binding. Not shown: G-actin polymerizes, stabilizing the formation of trans-dimers. (Ca2+ indicated by small black circles, phosphorylation by small red circles).
In Figure 8 we considered the possibility that CEACAM1, whether in the cis- or trans-dimer state, could exist in clusters. If this is true, the recruitment of G-actin could allow actin to polymerize at these sites. Another prediction is that the N-terminal ecto-domain could form either parallel (cis-) or anti-parallel (trans-) dimers. This possibility was suggested by the x-ray crystal studies performed by Bonsor et al. [41]. The parallel arrangement of the N-domains was first reported by Tan et al. [42] who solved the x-ray structure of a soluble murine Ceacam1 expressed in glycosylation deficient CHO cells. The question of whether soluble CEACAM1 actually forms dimers in solution is a matter of controversy. Our own studies based on analytical ultracentrifugation on human CEACAM1 expressed in CHO cells provided evidence of only monomers [43]. Although a number of studies on CEACAM N-domains expressed in E. coli show clear evidence of dimer formation [41], their relevance may be called into question by a more recent NMR study that showed glycosylated soluble N-domain expressed in HEK293 cells does not form dimers [44]. However, since the glycosylation is away from the dimer interface, the role of glycosylation remains a mystery. It appears that the membrane arrangement of the entire molecule, including the interaction of the TM domains with the GXXXG motif [38], requires inside-out signaling to control the transition from cis- to trans-dimer formation.
Since CEACAM1 is often identified as a bacterial pathogen receptor (reviewed in [45]), it is worth asking if AIIt has also been identified with pathogen binding, perhaps linking the two findings. Indeed, while recent studies identified CEACAM1 as a receptor for H. pylori [46–48], a separate study identified AnxA2 as an H. pylori associated protein [49]. In that study, gastric cells infected with H. pylori before and after IFNγ treatment were lysed and analyzed by MALDI-TOF-MS on SELDI chips. Identification and upregulation of AnxA2 appeared to correlate with both H. pylori infection and IFNγ treatment. Since IFNγ expression is responsible for the upregulation of CEACAM1 [50–53], and CEACAM1 is a known receptor for H. pylori, these results suggest that a more detailed study of CEACAM1 in association with AIIt during H. pylori infection is warranted.
CONCLUSION
The short cytoplasmic domain of CEACAM1-SF, while only 12–14 amino acids in length, is capable of interacting with a number of cytoplasmic proteins that can affect cis- and trans-dimer formation at the cell surface N-terminal ectodomain. In this study we identify the binding mode within the cytoplasmic domain and its interaction with AIIt and the S100A10-AnxA2Nter pseudo complex by NMR, molecular dynamics, and modeling studies. We show that two cytoplasmic domain peptides interact with the S100A10-AnxA2Nter pseudo complex in an N-to N-terminal configuration. This finding may explain the basis of an inside-outside mediated complex formation that favors cis-dimer formation of CEACAM1 prior to its response to calcium signaling, that in turn, favors the disruption of the complex, thus leading to trans-dimer formation at the cell surface between two cells.
Supplementary Material
Highlights.
AIIt pseudodimer interaction with CEACAM1-S cytoplasmic domain was studied by NMR.
The CEACAM1-S cytoplasmic domain forms a dimer complex with the AIIt pseudodimer.
Amino acid Phe-450 of CEACAM1-S is the key residue that binds to AIIt pseudodimer.
This interaction may play a role in the cis-trans dimer formation of CEACAM1-S.
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are grateful to the support from NMR core, Dr. Yuelong Ma in synthetic and biopolymer chemistry core for the peptide synthesis. We thank Dr. Gary S. Shaw for sharing chemical shift assignment of S100A10-AnexA2Nter. The Mass Spectrometry & Proteomics Core facility of City of Hope was supported in part by the National Cancer Institute of the National Institutes of Health under award P30CA033572.
This research was supported by NIH grant P30 CA033572.
Abbreviations
- CSP
Chemical shift perturbation
- AIIt
hetero-tetramer of S100A10 and AnxA2
- AnxA2
Annexin A2 S100A10-AnxA2Nter fusion: pseudo hetero-tetramer of S100A10 and AnxA2, S100A10-ENLYFQGD-AnxA2(1–13)
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Gray-Owen SD, Blumberg RS, CEACAM1: contact-dependent control of immunity, NatRev Immunol, 6 (2006) 433–446. [DOI] [PubMed] [Google Scholar]
- [2].Chen CJ, Kirshner J, Sherman MA, Hu W, Nguyen T, Shively JE, Mutation analysis of the short cytoplasmic domain of the cell-cell adhesion molecule CEACAM1 identifies residues that orchestrate actin binding and lumen formation, J Biol Chem, 282 (2007) 5749–5760. [DOI] [PubMed] [Google Scholar]
- [3].Schumann D, Chen CJ, Kaplan B, Shively JE, Carcinoembryonic antigen cell adhesion molecule 1 directly associates with cytoskeleton proteins actin and tropomyosin, J Biol Chem, 276 (2001) 47421–47433. [DOI] [PubMed] [Google Scholar]
- [4].Kirshner J, Schumann D, Shively JE, CEACAM1, a cell-cell adhesion molecule, directly associates with annexin II in a three-dimensional model of mammary morphogenesis, J Biol Chem, 278 (2003) 50338–50345. [DOI] [PubMed] [Google Scholar]
- [5].Ghazarian H, Hu W, Mao A, Nguyen T, Vaidehi N, Sligar S, Shively JE, NMR analysis of free and lipid nanodisc anchored CEACAM1 membrane proximal peptides with Ca(2+)/CaM, Biochim Biophys Acta Biomembr, 1861 (2019) 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Edlund M, Blikstad I, Obrink B, Calmodulin binds to specific sequences in the cytoplasmic domain of C-CAM and down-regulates C-CAM self-association, J Biol Chem, 271 (1996) 1393–1399. [DOI] [PubMed] [Google Scholar]
- [7].Hayes MJ, Shao D, Bailly M, Moss SE, Regulation of actin dynamics by annexin 2, EMBO J, 25 (2006) 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rescher U, Ruhe D, Ludwig C, Zobiack N, Gerke V, Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes, J Cell Sci, 117 (2004) 3473–3480. [DOI] [PubMed] [Google Scholar]
- [9].Bharadwaj A, Bydoun M, Holloway R, Waisman D, Annexin A2 heterotetramer: structure and function, Int J Mol Sci, 14 (2013) 6259–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dempsey BR, Rezvanpour A, Lee TW, Barber KR, Junop MS, Shaw GS, Structure of an asymmetric ternary protein complex provides insight for membrane interaction, Structure, 20 (2012) 1737–1745. [DOI] [PubMed] [Google Scholar]
- [11].Ozorowski G, Milton S, Luecke H, Structure of a C-terminal AHNAK peptide in a 1:2:2 complex with S100A10 and an acetylated N-terminal peptide of annexin A2, Acta Crystallogr D Biol Crystallogr, 69 (2013) 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Oh YS, Gao P, Lee KW, Ceglia I, Seo JS, Zhang X, Ahn JH, Chait BT, Patel DJ, Kim Y, Greengard P, SMARCA3, a chromatin-remodeling factor, is required for p11-dependent antidepressant action, Cell, 152 (2013) 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Johnsson N, Marriott G, Weber K, p36, the major cytoplasmic substrate of src tyrosine protein kinase, binds to its p11 regulatory subunit via a short amino-terminal amphiphatic helix, EMBO J, 7 (1988) 2435–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rety S, Sopkova J, Renouard M, Osterloh D, Gerke V, Tabaries S, Russo-Marie F, Lewit-Bentley A, The crystal structure of a complex of p11 with the annexin II N-terminal peptide, Nat Struct Biol, 6 (1999) 89–95. [DOI] [PubMed] [Google Scholar]
- [15].Rezvanpour A, Phillips JM, Shaw GS, Design of high-affinity S100-target hybrid proteins, Protein Sci, 18 (2009) 2528–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Spratt DE, Barber KR, Marlatt NM, Ngo V, Macklin JA, Xiao Y, Konermann L, Duennwald ML, Shaw GS, A subset of calcium-binding S100 proteins show preferential heterodimerization, FEBS J, 286 (2019) 1859–1876. [DOI] [PubMed] [Google Scholar]
- [17].Santamaria-Kisiel L, Shaw GS, Identification of regions responsible for the open conformation of S100A10 using chimaeric S100A11-S100A10 proteins, Biochem J, 434 (2011) 37–48. [DOI] [PubMed] [Google Scholar]
- [18].Illien F, Finet S, Lambert O, Ayala-Sanmartin J, Different molecular arrangements of the tetrameric annexin 2 modulate the size and dynamics of membrane aggregation, Biochim Biophys Acta, 1798 (2010) 1790–1796. [DOI] [PubMed] [Google Scholar]
- [19].Prantner AM, Sharma V, Garbow JR, Piwnica-Worms D, Synthesis and characterization of a Gd-DOTA-D-permeation peptide for magnetic resonance relaxation enhancement of intracellular targets, Mol Imaging, 2 (2003) 333–341. [DOI] [PubMed] [Google Scholar]
- [20].Schanda P, Brutscher B, Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds, J Am Chem Soc, 127 (2005) 8014–8015. [DOI] [PubMed] [Google Scholar]
- [21].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J Biomol NMR, 6 (1995) 277–293. [DOI] [PubMed] [Google Scholar]
- [22].Johnson BA, Blevins RA, NMR View: A computer program for the visualization and analysis of NMR data, J Biomol NMR, 4 (1994) 603–614. [DOI] [PubMed] [Google Scholar]
- [23].Lee W, Tonelli M, Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy, Bioinformatics, 31 (2015) 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Williamson MP, Using chemical shift perturbation to characterise ligand binding, Prog Nucl Magn Reson Spectrosc, 73 (2013) 1–16. [DOI] [PubMed] [Google Scholar]
- [25].Wang JT, Xu X, Alontaga AY, Chen Y, Liu Y, Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction, Cell reports, 7 (2014) 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Case RMBDA, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L and Kollman PA, AMBER 2016, University of California, San Francisco, (2016). [Google Scholar]
- [27].Nguyen H, Roe DR, Simmerling C, Improved Generalized Born Solvent Model Parameters for Protein Simulations, J Chem Theory Comput, 9 (2013) 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J Chem Theory Comput, 11 (2015) 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shao O, Zhu WL, Assessing AMBER force fields for protein folding in an implicit solvent, Phys Chem Chem Phys, 20 (2018) 7206–7216. [DOI] [PubMed] [Google Scholar]
- [30].Hopkins CW, Le Grand S, Walker RC, Roitberg AE, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning, J Chem Theory Comput, 11 (2015) 1864–1874. [DOI] [PubMed] [Google Scholar]
- [31].Neria E, Fischer S, Karplus M, Simulation of activation free energies in molecular systems, J Chem Phys, 105 (1996) 1902–1921. [Google Scholar]
- [32].Hunenberger P, Thermostat algorithms for molecular dynamics simulations, Adv Polym Sci, 173 (2005) 105–147. [Google Scholar]
- [33].Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR, Molecular-Dynamics with Coupling to an External Bath, J Chem Phys, 81 (1984) 3684–3690. [Google Scholar]
- [34].Polyansky AA, Volynsky PE, Efremov RG, Multistate organization of transmembrane helical protein dimers governed by the host membrane, J Am Chem Soc, 134 (2012) 14390–14400. [DOI] [PubMed] [Google Scholar]
- [35].Polyansky AA, Chugunov AO, Volynsky PE, Krylov NA, Nolde DE, Efremov RG, PREDDIMER: a web server for prediction of transmembrane helical dimers, Bioinformatics, 30 (2014) 889–890. [DOI] [PubMed] [Google Scholar]
- [36].Russ WP, Engelman DM, The GxxxG motif: a framework for transmembrane helix-helix association, J Mol Biol, 296 (2000) 911–919. [DOI] [PubMed] [Google Scholar]
- [37].Lu R, Niesen MJ, Hu W, Vaidehi N, Shively JE, Interaction of actin with carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) receptor in liposomes is Ca2+- and phospholipid-dependent, J Biol Chem, 286 (2011) 27528–27536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Patel PC, Lee HS, Ming AY, Rath A, Deber CM, Yip CM, Rocheleau JV, Gray-Owen SD, Inside-out signaling promotes dynamic changes in the carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1) oligomeric state to control its cell adhesion properties, J Biol Chem, 288 (2013) 29654–29669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Klaile E, Vorontsova O, Sigmundsson K, Muller MM, Singer BB, Ofverstedt LG, Svensson S, Skoglund U, Obrink B, The CEACAM1 N-terminal Ig domain mediates cis- and trans-binding and is essential for allosteric rearrangements of CEACAM1 microclusters, J Cell Biol, 187 (2009) 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nguyen T, Chen CJ, Shively JE, Phosphorylation of CEACAM1 molecule by calmodulin kinase IID in a three-dimensional model of mammary gland lumen formation, J Biol Chem, 289 (2014) 2934–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bonsor DA, Gunther S, Beadenkopf R, Beckett D, Sundberg EJ, Diverse oligomeric states of CEACAM IgV domains, Proc Natl Acad Sci U S A, 112 (2015) 13561–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tan K, Zelus BD, Meijers R, Liu JH, Bergelson JM, Duke N, Zhang R, Joachimiak A, Holmes KV, Wang JH, Crystal structure of murine sCEACAM1a[1,4]: a coronavirus receptor in the CEA family, EMBO J, 21 (2002) 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schumann D, Huang J, Clarke PE, Kirshner J, Tsai SW, Schumaker VN, Shively JE, Characterization of recombinant soluble carcinoembryonic antigen cell adhesion molecule 1, Biochem Biophys Res Commun, 318 (2004) 227–233. [DOI] [PubMed] [Google Scholar]
- [44].Zhuo Y, Yang JY, Moremen KW, Prestegard JH, Glycosylation alters dimerization properties of a cell-surface signaling protein, Carcinoembryonic Antigen-related Cell Adhesion Molecule 1 (CEACAM1), The Journal of Biological Chemistry 291 (2016) 20085–20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zimmermann W, Kammerer R, Coevolution of paired receptors in Xenopus carcinoembryonic antigen-related cell adhesion molecule families suggests appropriation as pathogen receptors, BMC Genomics, 17 (2016) 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Koniger V, Holsten L, Harrison U, Busch B, Loell E, Zhao Q, Bonsor DA, Roth A, Kengmo-Tchoupa A, Smith SI, Mueller S, Sundberg EJ, Zimmermann W, Fischer W, Hauck CR, Haas R, Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA, Nat Microbiol, 2 (2016) 16188. [DOI] [PubMed] [Google Scholar]
- [47].Javaheri A, Kruse T, Moonens K, Mejias-Luque R, Debraekeleer A, Asche CI, Tegtmeyer N, Kalali B, Bach NC, Sieber SA, Hill DJ, Koniger V, Hauck CR, Moskalenko R, Haas R, Busch DH, Klaile E, Slevogt H, Schmidt A, Backert S, Remaut H, Singer BB, Gerhard M, Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs, Nat Microbiol, 2 (2016) 16189. [DOI] [PubMed] [Google Scholar]
- [48].Moonens K, Hamway Y, Neddermann M, Reschke M, Tegtmeyer N, Kruse T, Kammerer R, Mejias-Luque R, Singer BB, Backert S, Gerhard M, Remaut H, Helicobacter pylori adhesin HopQ disrupts trans dimerization in human CEACAMs, EMBO J, 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Das S, Sierra JC, Soman KV, Suarez G, Mohammad AA, Dang TA, Luxon BA, Reyes VE, Differential protein expression profiles of gastric epithelial cells following Helicobacter pylori infection using ProteinChips, J Proteome Res, 4 (2005) 920–930. [DOI] [PubMed] [Google Scholar]
- [50].Takahashi H, Kobayashi H, Hashimoto Y, Matsuo S, Iizuka H, Interferon-gamma-dependent stimulation of Fas antigen in SV40-transformed human keratinocytes: modulation of the apoptotic process by protein kinase C, J Invest Dermatol, 105 (1995) 810–815. [DOI] [PubMed] [Google Scholar]
- [51].Chen CJ, Lin TT, Shively JE, Role of interferon regulatory factor-1 in the induction of biliary glycoprotein (cell CAM-1) by interferon-gamma, J Biol Chem, 271 (1996) 28181–28188. [DOI] [PubMed] [Google Scholar]
- [52].Gencheva M, Chen CJ, Nguyen T, Shively JE, Regulation of CEACAM1 transcription in human breast epithelial cells, BMC Mol Biol, 11 (2010) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dery KJ, Silver C, Yang L, Shively JE, Interferon regulatory factor 1 and a variant of heterogeneous nuclear ribonucleoprotein L coordinately silence the gene for adhesion protein CEACAM1, J Biol Chem, 293 (2018) 9277–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.