

SUMMARY
Brain metastasis (br-met) develops in an immunologically-unique br-met niche. Central nervous system-native myeloid cells (CNS-myeloid) and bone marrow-derived myeloid cells (BMDM) cooperatively regulate brain immunity. The phenotypic heterogeneity and specific roles of these myeloid subsets in shaping the br-met niche to regulate br-met outgrowth have not been fully revealed. Applying multimodal single cell analyses, we elucidated a heterogeneous but spatially-defined CNS-myeloid response during br-met outgrowth. We found Ccr2+ BMDM minimally influenced br-met while CNS-myeloid promoted br-met outgrowth. Additionally, br-met-associated CNS-myeloid exhibited downregulation of Cx3cr1. Cx3cr1 knockout in CNS-myeloid increased br-met incidence, leading to an enriched interferon response signature and Cxcl10 upregulation. Significantly, neutralization of Cxcl10 reduced br-met, while rCxcl10 increased br-met and recruited VISTAHi PD-L1+ CNS-myeloid to br-met lesions. Inhibiting VISTA and PD-L1 signaling relieved immune suppression and reduced br-met burden. Our results demonstrate that loss of Cx3cr1 in CNS-myeloid triggers a Cxcl10-mediated vicious cycle, cultivating a br-met-promoting, immune-suppressive niche.
Graphical Abstract
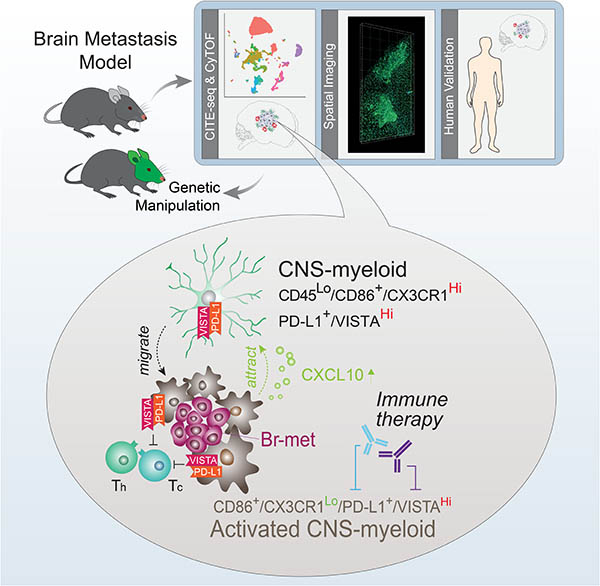
In Brief
A multimodal investigation into the tumor microenvironment of brain metastases in mouse models demonstrates the central role of CNS-resident myeloid populations in creating an immunosuppressive pro-metastatic microenvironment.
INTRODUCTION
Stephen Paget’s “seed and soil” hypothesis posited that metastatic success results from disseminated cancer cell (DCC)-metastatic niche interplay (Paget, 1989). The metastatic niche consists of unique organ-specific cellular and acellular components, which profoundly influence the progression of metastatic disease (Peinado et al., 2017; Quail and Joyce, 2013).
Brain metastasis (br-met) is frequently observed clinically. The brain is a complex organ and favorable soil for DCCs derived from non-CNS primary tumors (Achrol et al., 2019). To successfully outgrow in the brain, DCCs must interact with brain-resident cells and infiltrating immune cells - the br-met niche (Winkler, 2015). Such br-met niche cells include astrocytes, endothelial cells, and microglia, among others (Quail and Joyce, 2013; Winkler, 2015).
Once considered immune-privileged, the brain is now known to have an intricate, diverse immune presence during homeostasis and disease. Central nervous system-native myeloid cells (CNS-myeloid), namely microglia and Border Associated Macrophages (BAM), are the predominant immune population of the homeostatic brain and vitally contribute to brain homeostasis and diseases (Butovsky and Weiner, 2018; Quail and Joyce, 2013). During CNS pathologies, CNS-myeloid exhibit plasticity, playing either disease-resolving or disease-promoting roles (Butovsky and Weiner, 2018; Prinz and Priller, 2014; Prinz et al., 2019). Functioning non-redundantly to CNS-myeloid, peripheral bone marrow-derived myeloid cells (BMDM) may infiltrate the brain parenchyma and contribute to neuroinflammation (Prinz and Priller, 2014; Yamasaki et al., 2014).
Pivotal mediators of CNS diseases, CNS-myeloid and BMDM are implicated in br-met progression. Immune gene signatures from human br-mets have provided clinical evidence on the correlation between the br-met immune niche and br-met outcome (Fischer et al., 2019).
CNS-myeloid and BMDM have been observed in experimental br-met models (Benbenishty et al., 2019; Bowman et al., 2016). In experimental models, br-met-infiltrating myeloid cells reduce br-met by phagocytosis, but have the ability to facilitate br-met through MMP3 and Cathepsin S secretion (Benbenishty et al., 2019; Qiao et al., 2019; Sevenich et al., 2014). RNA-seq coupled with lineage tracing demonstrated stark transcriptional differences between CNS-myeloid and BMDM in glioma (Bowman et al., 2016). Recent studies have further performed compositional and bulk transcriptional profiling of human gliomas and br-met-associated leukocytes to describe a diverse br-met immune niche, implying its potential functional significance (Friebel et al., 2020; Klemm et al., 2020). Despite these substantial efforts, the differential contributions of CNS-myeloid and BMDM in regulating br-met have not yet been fully functionally or mechanistically resolved.
Single cell analyses have provided novel insights into brain immunity (Hammond et al., 2019; Keren-Shaul et al., 2017; Masuda et al., 2019; Mrdjen et al., 2018). In this study, combining single cell analyses with transgenic mouse models targeting and manipulating myeloid cells, we sought to elucidate the role of heterogeneous br-met-associated myeloid cell subsets in regulating br-met outgrowth. We found CNS-myeloid are most influential in promoting br-met by fostering immune suppression via negative immune checkpoints VISTA and PD-L1. Combinatorial targeting of these immune checkpoints presents a new opportunity for clinical management of br-met.
RESULTS
Myeloid Cells Infiltrate Br-mets and Display Spatially-Defined Morphological Patterns
Using a human br-met tissue array (Table S1), we observed hypertrophic Iba1+ myeloid cells surrounded and infiltrated human br-mets regardless of primary tumor origin (npatients = 37, ntumor origins = 14) (Figure 1A; Figure S1A), supporting prior observations (He et al., 2006; Pukrop et al., 2010). Iba1+ br-met-associated myeloid cells (Br.MAM) also accumulated around and within murine br-mets irrespective of brain anatomical location or br-met stage (Figure 1B–C; Figure S1B–D). Br.MAM density increased within br-mets relative to the naive brain (Figure 1C–D; Figure S1E). Despite morphological heterogeneity, most Br.MAM were hypertrophic with enlarged cell bodies and reduced protrusion features, indicative of activation and a robust response (Figure 1C–D) (Heindl et al., 2018). Principal component analysis (PCA) of the morphological features assessed resulted in two groups, with naive myeloid cells (NMC) assuming low morphology scores and Br.MAM assuming higher morphology scores, further suggesting their activation (Heindl et al., 2018) (Figure 1E). Spatially, Br.MAM morphology scores increased approaching br-met lesion borders and were highest within br-met lesions (Figure 1F).
Figure 1. Myeloid Cells Infiltrate Br-mets and are a Prominent, Diverse Component of the Br-mets Niche.
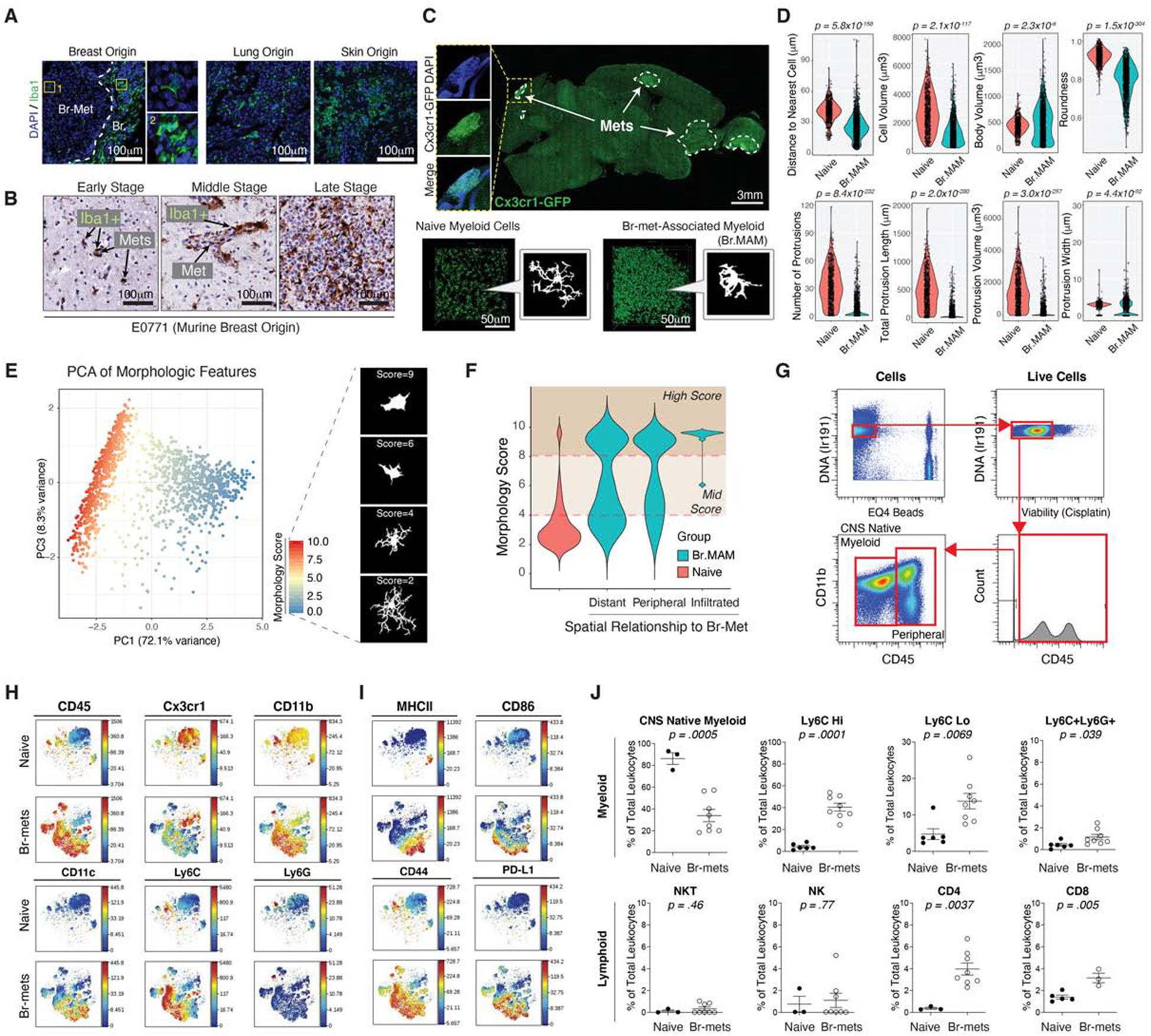
A) IF of myeloid cells in human br-mets.
B) IHC of myeloid cells in murine E0771 br-mets.
C) Fluorescence image of br-met-burdened Cx3cr1GFP/+ brain (top), 3D reconstruction and silhouettes of myeloid cells in the naive brain and in br-mets (bottom).
D) Quantifications of 3D morphological and spatial features of naive myeloid cells (NMC) and Br.MAM (≥ 650 myeloid cells per group; each dot represents 1 cell).
E) PCA based on morphologic features in (D) to assign morphology scores (left), silhouettes of myeloid cells of indicated morphology scores (each dot represents 1 cell) (right).
F) Morphology score distributions for NMC and Br.MAM grouped by proximity to br-mets.
G) Gating strategy to identify CD45+ cells in CyTOF data.
H-I) tSNEs comparing innate immune cell marker expression (H) and functional marker expression (I) in leukocytes in br-mets and the naive brain.
J) Frequency of indicated immune populations in naive and br-met-burdened brains as identified by manual gating (each dot represents 1 mouse).
Data in D and J analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean.
Mass Cytometry Identifies a Unique Br-met Immune Landscape
We next examined how br-mets altered the br-met immune landscape by mass cytometry (CyTOF). t-Distributed Stochastic Neighbor Embedding (tSNE) showed a contrast in surface marker expression between the naive brain and br-met (Figure 1G–I; Figure S1F–G). Immune cells from br-mets more highly expressed myeloid cell and functional markers (Figure 1H–I; Figure S1G). Manual gating indicated most naive brain leukocytes were homogenous CNS-myeloid (Figure 1J; Figure S1F), consistent with prior reports (Mrdjen et al., 2018). In contrast, br-mets were immunologically diverse, inclusive to CNS-myeloid and peripherally-derived immune cells, possessing a significant increase in all BMDM subsets and adaptive immune components compared to the naive brain (Figure 1J). Applying a gating scheme complementary to that which we used on mouse samples to human br-met CyTOF data (Friebel et al., 2020), we found human br-met samples were likewise infiltrated by a diverse array of myeloid and T cell subsets (Figure S1H–I).
Br.MAM Display Transcriptional Profiles Distinct from their Naive Counterparts
To reveal the molecular underpinnings of the two broad Br.MAM subsets in br-mets, we employed Cellular Indexing of Transcriptomes and Epitopes (CITE-seq) (Stoeckius et al., 2017), enabling transcriptome and surface marker profiling of naive and br-met-associated immune cells. Projecting sequenced cells from the two conditions on a UMAP showed unique clusters present only in br-mets (Figure 2A). Manual gating on DNA-barcoded antibodies identified CNS-myeloid and BMDM (Figure 2B; Figure S2A–B), the accuracy of which was confirmed by Tmem119 expression in CNS-myeloid and Itga4 expression in BMDM (Figure 2C) (Bowman et al., 2016). Reflecting CyTOF profiling, the ratio of CNS-myeloid to BMDM was higher in the naive brain than in br-mets (Figure 2C).
Figure 2. CITE-seq Reveals Br.MAM Subsets Display Transcriptional Profiles Distinct from their Naive Counterparts.
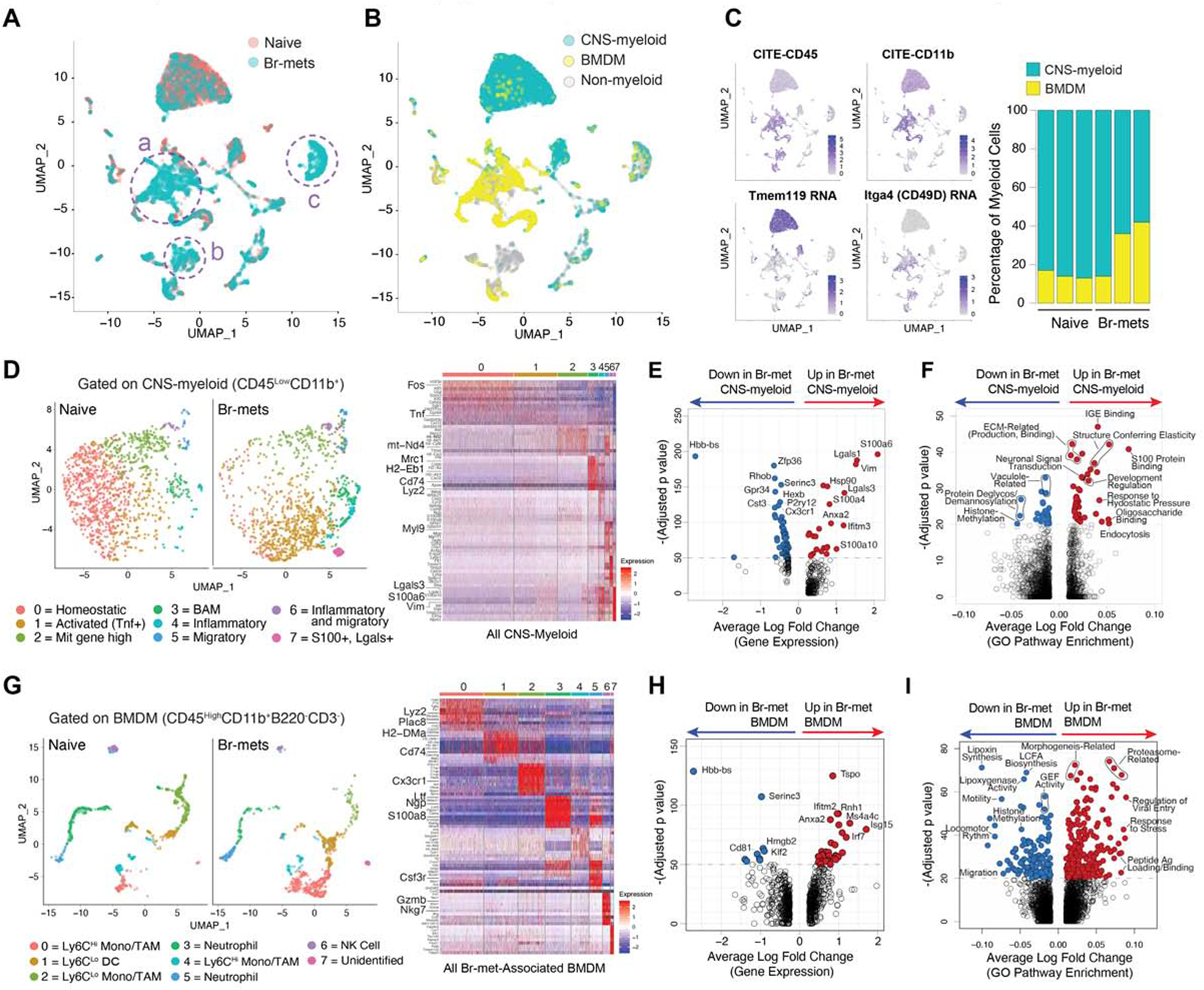
A) UMAP of cells from the naive brain and br-mets color coded by sample (naive or br-met).
B) UMAP color coded by myeloid subset (CNS-myeloid or BMDM).
C) Feature plots of indicated genes (left), CNS-myeloid to BMDM ratio in the naive brain and br-mets (right).
D) UMAPs of CNS-myeloid from the naive brain and br-mets color coded by transcriptional cluster (left), associated heatmap showing top differentially expressed genes (DEGs) among clusters (right).
E) Volcano plot of DEGs between br-met-associated and naive CNS-myeloid.
F) Volcano plot showing differentially expressed gene pathways (DEGPs) between br-met-associated and naive CNS-myeloid.
G) UMAPs of BMDM from the naive brain and br-mets color coded by transcriptional clusters (left), associated heatmap showing top DEGs between clusters (right).
H) Volcano plot of DEGs between br-met-associated and naive BMDM.
I) Volcano plot showing DEGPs between br-met-associated and naive BMDM.
Data in E, F, H, and I analyzed by Wilcoxon rank sum test.
See also Figures S2–3 and Table S2–3.
CNS-myeloid were composed of eight transcriptional clusters that shifted on the UMAP between conditions (Figure 2D; Table S2). BAM were restricted to cluster 3, while microglia of various transcriptional states composed the remaining clusters (Figure 2D). Cluster 0, marked by high expression of microglia homeostatic genes (Cx3cr1, Hexb, Jun), was enriched with naive microglia (Figure S2C; Table S2). Cluster 2 resembled Cluster 0 but had higher mitochondrial gene expression and slightly reduced microglia homeostatic gene expression, resembling a primed state (Figure 2D; Table S2). Clusters 1, 4, 5, 6, and 7 represented distinct microglia inflammatory states, all of which had reduced expression of microglia homeostatic genes (Figure 2D; Table S2). Cluster 3, composed of BAM, highly expressed antigen processing and presentation genes (H2-Aa, Ha-Ab1, Cd74) (Figure 2D; Table S2). Differential gene expression analysis between br-met-associated CNS-myeloid and naive CNS-myeloid identified br-met-associated CNS-myeloid upregulated pro-inflammatory genes (S100a4, S100a6, S100a10), anti-inflammatory genes (Lgals1, Lgals3, Ifitm3), and migration genes (Vim, Anxa) and downregulated microglia homeostatic genes (Cx3cr1, P2ry12, Hexb, Cst3) (Figure 2E; Table S2). Despite downregulating microglia homeostatic genes like Damage Associated Microglia (DAM) (Keren-Shaul et al., 2017), there were no other gene expression similarities between br-met-associated CNS-myeloid and DAM (Figure S2D). Lastly, br-met-associated CNS-myeloid were enriched for canonical macrophage functional pathways (endocytosis, IGE binding, ECM production and remodeling) and inflammatory pathways relative to their naive counterparts (Figure 2F; Table S2).
BMDM segregated into eight transcriptional clusters (Figure 2G), representing canonical BMDM subsets as inferred by RNA and surface marker expression (Figure S3A; Table S3). Stark transcriptional heterogeneity existed among and within canonically-identified BMDM, but did not represent M1/M2 polarization (Figure 2G; Figure S3B). Several BMDM clusters, including Clusters 0 (Ly6CHi monocytes), 1 (Ly6CLo DCs), 5 (neutrophils), and 7, were more abundant in br-mets relative to the naive brain, representing their influx to the brain and/or local expansion (Figure S2C). Genes related to inflammation (Tspo, Isg15, Ifitm2, Anxa2, Irf7) were upregulated, while Hbb-bs, Serinc3, Cd81, and Klf2 were downregulated in br-met-associated BMDM (Figure 2H, Table S3). Br-met-associated BMDM had higher enrichment of antigen processing and presentation and immune responsiveness pathways and lower enrichment of motility and lipoxin synthesis pathways (Figure 2I; Table S3). We further subsetted BMDM into three broad canonically-related categories for further analysis: Ly6CHi inflammatory monocytes, Ly6CLo patrolling monocytes, and neutrophils (Figure S3C–E). The transcriptional subclusters of each BMDM subset represented distinct canonical subtypes of BMDM and/or distinct transcriptional states (Figure S3C–E). Shifting of cluster proportions within each BMDM subpopulation was evident between the naive and br-met state (Figure S3C–E), suggesting cell infiltration and plasticity. Finally, we compared gene expression between conditions within each BMDM subset, finding all subsets increased expression of various inflammatory genes (Ifitm1, Il1b, S100a10, and Lgals1), while simultaneously displaying their own unique gene expression changes (Figure S3C–E).
To validate the human relevance of our observations, we analyzed publicly deposited naive CNS-myeloid (npatients = 3) and br-met (npatients = 3) scRNA-seq datasets (Geirsdottir et al., 2019; Laughney et al., 2020) (Figure 3A). As in mice, human br-met-associated CNS-myeloid downregulated microglia homeostatic genes (CX3CR1, CSF1R, TREM2) (Figure 3B–C; Table S4). Some of the top upregulated and downregulated genes in mouse br-met-associated CNS-myeloid showed similar trends in humans (upregulated: VIM, LGALS3, IFITM2, LYZ; downregulated: GFP34, CD81) (Figure 3B, D–E; Table S4). Cumulatively, these data point toward an active and diverse myeloid response to br-met and indicate our model is clinically relevant.
Figure 3. Human Br-met-associated CNS-myeloid are Unique from their Naive Counterparts.
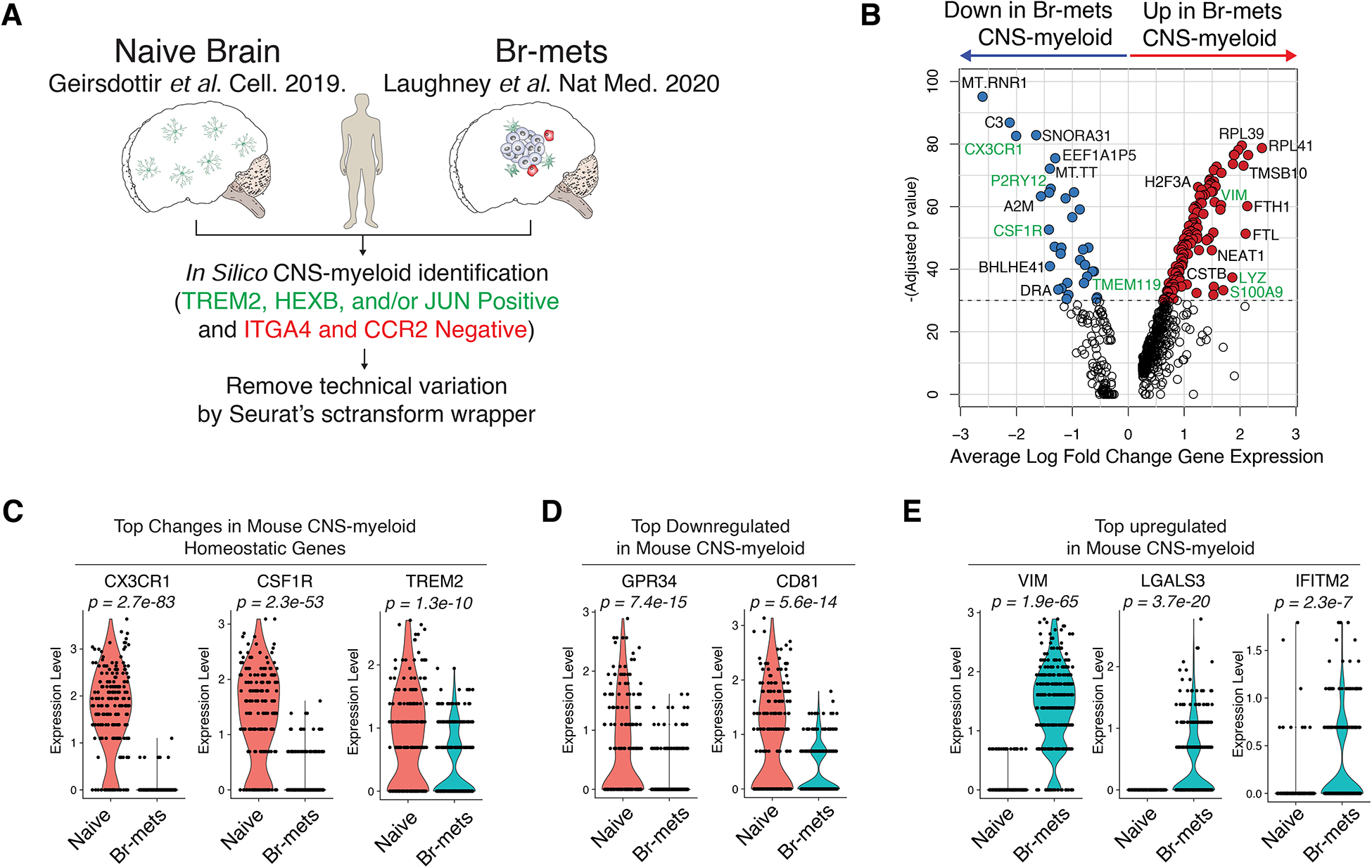
A) Human scRNA-seq analysis schematic.
B) Volcano plot of DEGs between br-met-associated and naive CNS-myeloid.
C-E) Violin plots of expression of CNS-myeloid homeostatic genes (C), top downregulated mouse genes (D), and top upregulated mouse genes (E) in br-met-associated and naive CNS-myeloid (each dot represents 1 cell).
All plots derived from pooling three biological replicates per experimental condition. Data analyzed by Wilcoxon rank sum test.
See also Table S4.
Br-met-Associated CNS-myeloid and BMDM Display Divergent and Convergent Transcriptomes
We next directly compared br-met-associated CNS-myeloid and BMDM. Projecting only br-met-associated myeloid cells on a tSNE showed a general segregation of the two populations with some overlap, particularly in Clusters 2 and 4 (Figure 4A). CNS-myeloid were enriched for microglia homeostatic genes and complement genes relative to BMDM (Figure 4B; Table S5). BMDM more highly expressed inflammatory genes (S100a11, Lgals, and Il1b) relative to CNS-myeloid (Figure 4B; Table S5). At the gene pathway level, CNS-myeloid were marked by migratory and regulatory gene signatures, while BMDM were enriched for cell adhesion and antigen presentation signatures (Figure 4C; Table S5).
Figure 4. Br.MAM Subsets Display Divergent and Convergent Phenotypes and Unique Spatial Distributions.
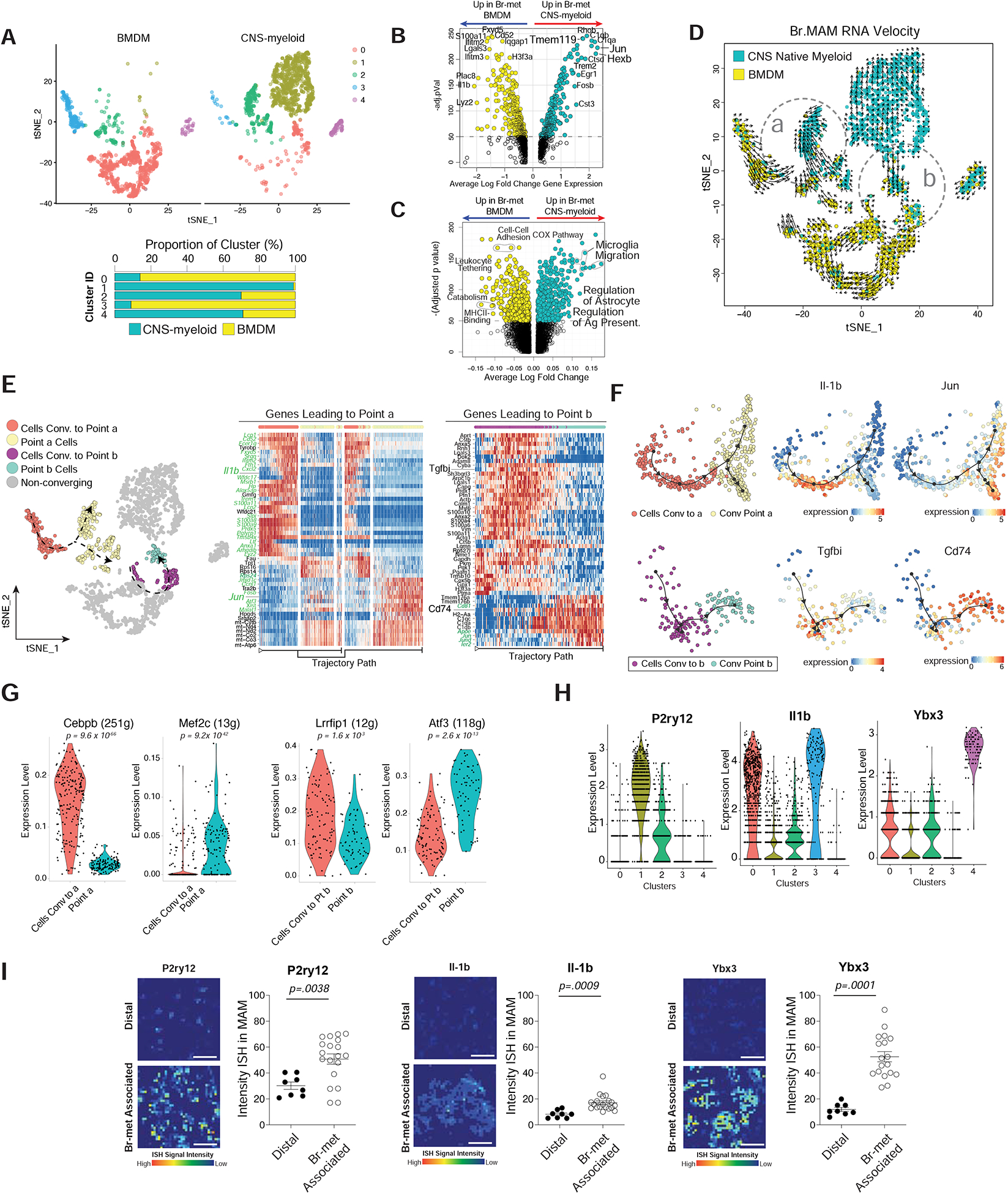
A) tSNE of Br.MAM color coded by transcriptional clusters (top), CNS-myeloid to BMDM ratio within each cluster (bottom).
B-C) Volcano plots showing DEGs (B) and DEGPs (C) between CNS-myeloid and BMDM during br-met.
D) tSNE overlaid with RNA velocity of Br.MAM.
E) tSNE annotated to show cells used for trajectory DEG analysis (left), heatmaps of trajectory DEGs related to point a (middle) and b (right). Genes in green font regulated by transcription factors (TFs) identified in panel (G).
F) Dimensional reduction representation of trajectory overlaid with indicated genes.
G) Violin plots of indicated TFs differentially expressed between cells converging and convergent point cells (each dot represents 1 cell).
H) Violin plots of indicated Br.MAM transcriptional cluster marker genes (each dot represents 1 cell).
I) Heatmaps of ISH intensity within myeloid cells associated with br-mets or distal to br-mets (left), associated quantification of ISH intensity within myeloid cells (each dot represents 1 field of view) (right).
Panels A-H derived from pooling of three biological replicates. Data in B, C, and G analyzed by Wilcoxon rank sum test. Data in I analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean.
See also Tables S5.
To investigate the transcriptional plasticity of CNS-myeloid and BMDM during br-met, we performed RNA velocity (La Manno et al., 2018). RNA velocity showed CNS-myeloid and BMDM have trajectories directed within their own respective populations and toward each other (Figure 4D, point a and b). Using Dynverse trajectory inference (TI) analysis (Saelens et al., 2019), we examined genes correlated with cell trajectory that may drive convergence within (point b) and between (point a) populations (Figure 4E). Inflammatory genes (S100 family genes, Il1b, Tgfbi) were highly expressed along the trajectory of cells leading to both convergent points (Figure 4E). Upon arriving to point a, cells bifurcated into two separate trajectories that either upregulated microglia homeostatic genes (Jun, Fos) or highly expressed mitochondrial genes (Figure 4E). Cells converging to point b upregulated genes related to antigen presentation (CD74 and H2-Aa) and complement (C1qc, C1qa) (Figure 4E–F). Seeking transcription factors (TFs) that potentially regulate the cellular convergence, we performed SCENIC regulon analysis (Aibar et al., 2017). TFs Cepbp and Lrrfip1 were enriched in cells moving to point a and point b, respectively (Figure 4G). TFs Mef2c and Atf3 are enriched within point a and point b cells, respectively (Figure 4G). Many target genes of these TFs contribute to the convergence processes along the predicted trajectory (genes in green font on heatmaps in Figure 4E). Collectively, these results highlight myeloid plasticity and suggest there is an appreciable level of transcriptional convergence within and between CNS-myeloid and BMDM during br-met, the importance of which requires future investigation.
Br.MAM Sub-clusters Display Unique Spatial Distributions
The spatial distribution of immune cells determines their roles during disease (Keren-Shaul et al., 2017; Rivera and Bergers, 2013). We interrogated the spatial distribution of Br.MAM clusters by performing dual RNA in situ hybridization (RNA-ISH) and Iba1 immunofluorescence (IF). We selected marker genes for each cluster: Il-1b for Clusters 0 and 3, P2ry12 for Cluster 1, and Ybx3 for Cluster 4 (Figure 4H). All four transcriptional clusters were present nearer to br-mets rather than distal to br-mets (Figure 4I). Il-1b, marker of Clusters 0 and 3, was only slightly increased in myeloid cells near br-mets, indicating Clusters 0 and 3 are less spatially defined (Figure 4I). P2ry12, Cluster 1 marker, displayed a moderate spatial association with br-mets, while Ybx3, Cluster 4 marker, was expressed in Br.MAM in direct contact with br-mets (Figure 4I).
The General Myeloid Cell Population Promotes Br-met Outgrowth
To determine the functional effect of myeloid cells during br-met outgrowth, we used Cx3cr1CreERT/+:ROSA26iDTR/+ mice to deplete myeloid cells during br-met outgrowth (Figure 5A). We characterized Cx3cr1-driven reporter expression in splenocytes and analyzed Cx3cr1 protein expression in naive and br-met-associated brain leukocytes. All myeloid cells expressed the ZsGreen reporter or moderate to high levels Cx3cr1 (Figure S4). Few T cells or NK cells expressed the reporter or Cx3cr1, suggesting this model largely targets myeloid cells (Figure S4).
Figure 5. Depletion of the General Myeloid Population and CNS-myeloid Reduces Br-met.
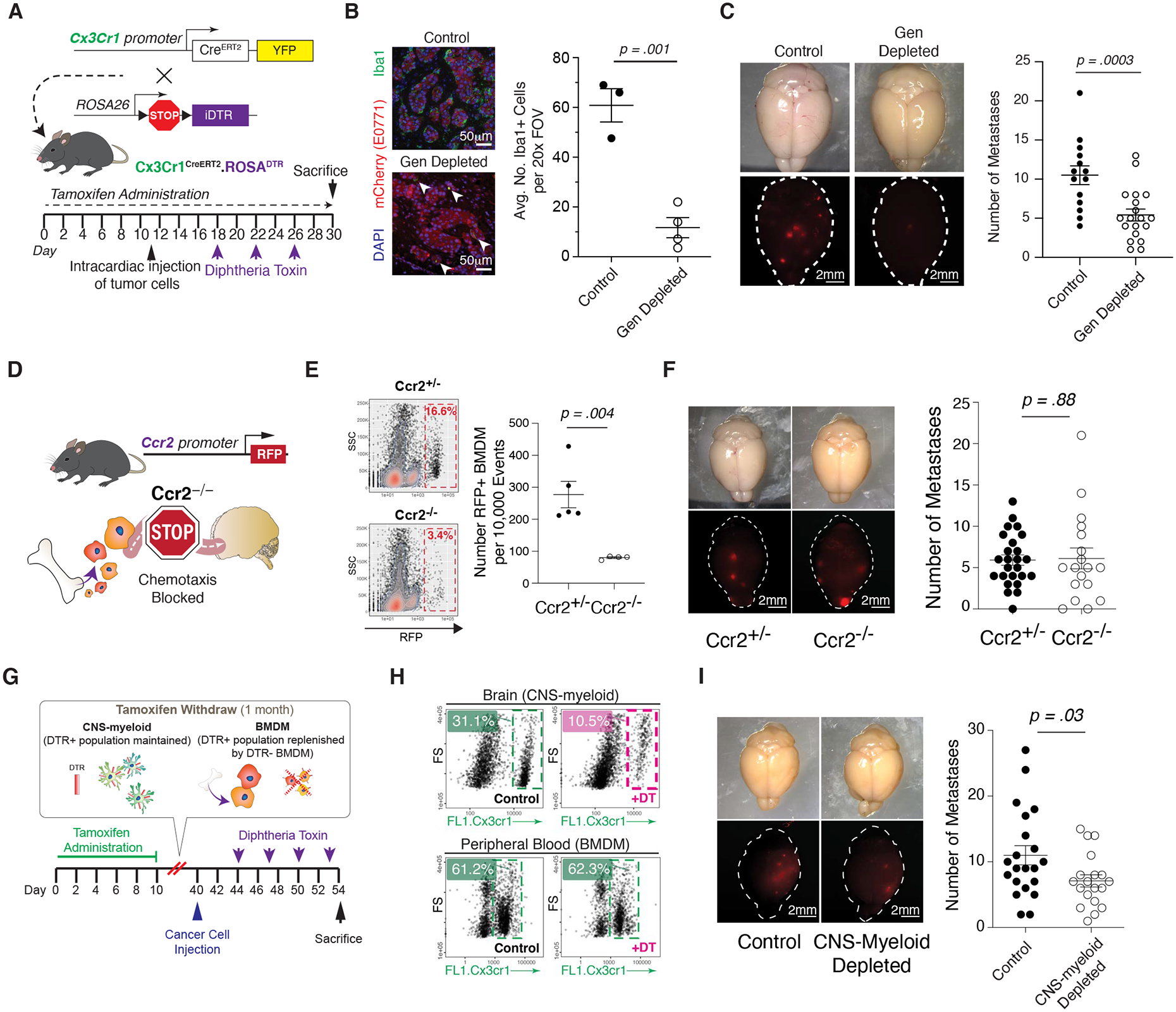
A) Schematic of Cx3cr1CreERT/+ROSAiDTR/+ mouse model.
B) IF of Br.MAM in control and myeloid-depleted mice (left), associated quantification of Iba1+ cells per 20x FOV in brains of control and myeloid-depleted mice during br-mets (each dot represents 1 mouse) (right).
C) Stereoscope images of br-mets in brains of control and myeloid-depleted mice (left), associated quantification of br-met number in control and myeloid-depleted mice (each dot represents 1 mouse; data pooled from two independent experiments with ≥3 mice per group) (right).
D) Schematic of Ccr2−/− mouse model.
E) Biaxial plots of RFP+ BMDM br-met infiltration in Ccr2+/− and Ccr2−/− mice (left), associated quantification (each dot represents 1 mouse; biaxial representative of ≥ 3 biological replicates per group) (right).
F) Stereoscope images of br-mets in brains of Ccr2+/− and Ccr2−/−mice (left), associated quantification of br-met number in Ccr2+/− and Ccr2−/− mice (each dot represents 1 mouse; data pooled from 3 independent experiments with ≥ 3 mice per group) (right).
G) Experimental schematic of CNS-myeloid-exclusive depletion.
H) Biaxial plots comparing Cx3cr1+ cell abundance in brain (top) and blood (bottom) of CNS-myeloid depleted mice relative to controls.
I) Stereoscope images of br-mets in brains of control and CNS-myeloid-depleted mice (left), associated quantification of br-met number in control and CNS-myeloid-depleted mice (each dot represents 1 mouse; quantification based on pooled data from 4 independent experiments with ≥ 4 mice per group) (right).
Data in B, C, E, F, and I analyzed by two-tailed student’s t test and error bars represent SEM, center represents mean.
See also Figures S4–5.
We administered Tamoxifen (TAM) and diphtheria toxin (DT) to these mice in a scheme that ensured thorough depletion of myeloid cells during br-met outgrowth, eliminating ~75% of myeloid cells in br-mets relative to control mice lacking the iDTR transgene (Figure 5A–B). Myeloid-depleted mice had reduced br-met burden and br-met proliferation (Figure 5C; Figure S5A), demonstrating a br-met-promoting role for the general myeloid population and corroborating previous reports (Andreou et al., 2017; Qiao et al., 2019). We also depleted myeloid cells prior to metastatic seeding and at the colonization stage of br-met (Figure S5B). In this depletion scheme, myeloid cells are depleted during the metastatic seeding stage, but microglia repopulate and newly-generated BMDM infiltrate br-mets by the metastatic outgrowth phase (Figure S5C) (Parkhurst et al., 2013). Depletion of myeloid cells during br-met colonization had no significant effect on endpoint br-met number (Figure S5D).
CNS-myeloid and Ccr2+ BMDM Have Differential Roles During Br-met Outgrowth
The presence of both CNS-myeloid and BMDM in br-mets prompted us to dissect the specific role of each myeloid subset on br-met outgrowth. First, we utilized a Ccr2 knockout mouse in which the Ccr2 gene is replaced by RFP (Figure 5D) to determine the contribution of BMDM on br-met outgrowth. Ccr2 knockout prevents the chemotaxis of Ccr2+ BMDM to sites of inflammation. Analysis of RFP expression in peripheral blood cells demonstrated that ~90%, ~18%, and <1% of Ly6CHi monocytes, Ly6CLo monocytes, and neutrophils, respectively, express RFP (Figure S5E). This data suggests that this model most significantly reduces br-met-associated Ly6CHi monocytes, the most abundant br-met-associated BMDM subset in mice (Figure 1J). While Ccr2 knockout in hosts reduced lung metastasis and glioma outgrowth (Chen et al., 2017; Lu and Kang, 2009), br-met burden and proliferation and overall survival in mice were not altered with Ccr2 knockout relative to Ccr2+/− controls despite the reduction of RFP+ BMDM brain infiltration (Figure 5E–F; Figure S5F–I).
To manipulate CNS-myeloid, we employed a temporally-segregated TAM and DT administration strategy as described previously to exclusively deplete CNS-myeloid in Cx3cr1CreERT/+:ROSA26iDTR/+ mice (Figure 5G) (Goldmann et al., 2016; Mundt et al., 2019; Parkhurst et al., 2013; Yona et al., 2013). This scheme depleted approximately two thirds of CNS-myeloid while sparing BMDM and minimally altering blood immune composition and peripheral immune cell composition in the brain (Figure 5H; Figure S5J). After depletion, remnant CNS-myeloid exhibited upregulation of apoptosis-related genes (Ctsd, Phlda1), ribosomal genes (Rpl10, Rpl10a), and Cx3cr1, and the downregulation of interferon-related genes (Isg15, Rsad, Ifit) and cytokines (Ccl7) (Figure S5K). Proportions of T cells remained similar between experimental conditions (Figure S5J), but within CD8+ T cells, the proportion of exhausted cells decreased and that of mildly cytotoxic T cells increased with CNS-myeloid depletion (Figure S5L–N). Most importantly, CNS-myeloid depletion reduced br-met burden (Figure 5I). These results collectively indicate that CNS-myeloid more prominently influence br-met outgrowth than BMDM.
Cx3cr1 Ablation Exacerbates Br-met
The fractalkine receptor, Cx3cr1, is highly expressed in steady-state microglia and maintains their homeostasis (Wolf et al., 2013). Cx3cr1 downregulation and its uncoupling from Cx3cl1 allow microglia to mount an inflammatory response, and genetic ablation of Cx3cr1 in mice can improve disease outcome (Cardona et al., 2006). Cx3cr1 was significantly downregulated in br-met-associated CNS-myeloid in our mouse model and human br-mets (Figures 2E and Figure 3C), which was reflected at the protein level in mice (Figure 6A). We questioned the functional implication of Cx3cr1 downregulation on regulating br-met by using Cx3cr1−/− mice (KO mice) (Figure S6A). Cx3cr1 knockout in hosts increased br-met number and proliferation and reduced host survival relative to Cx3cr1+/− (Het) hosts (Figure 6B–D).
Figure 6. Cx3cr1 Knockout Drives a CNS-myeloid Interferon Response that Promotes Br-met via Cxcl10.
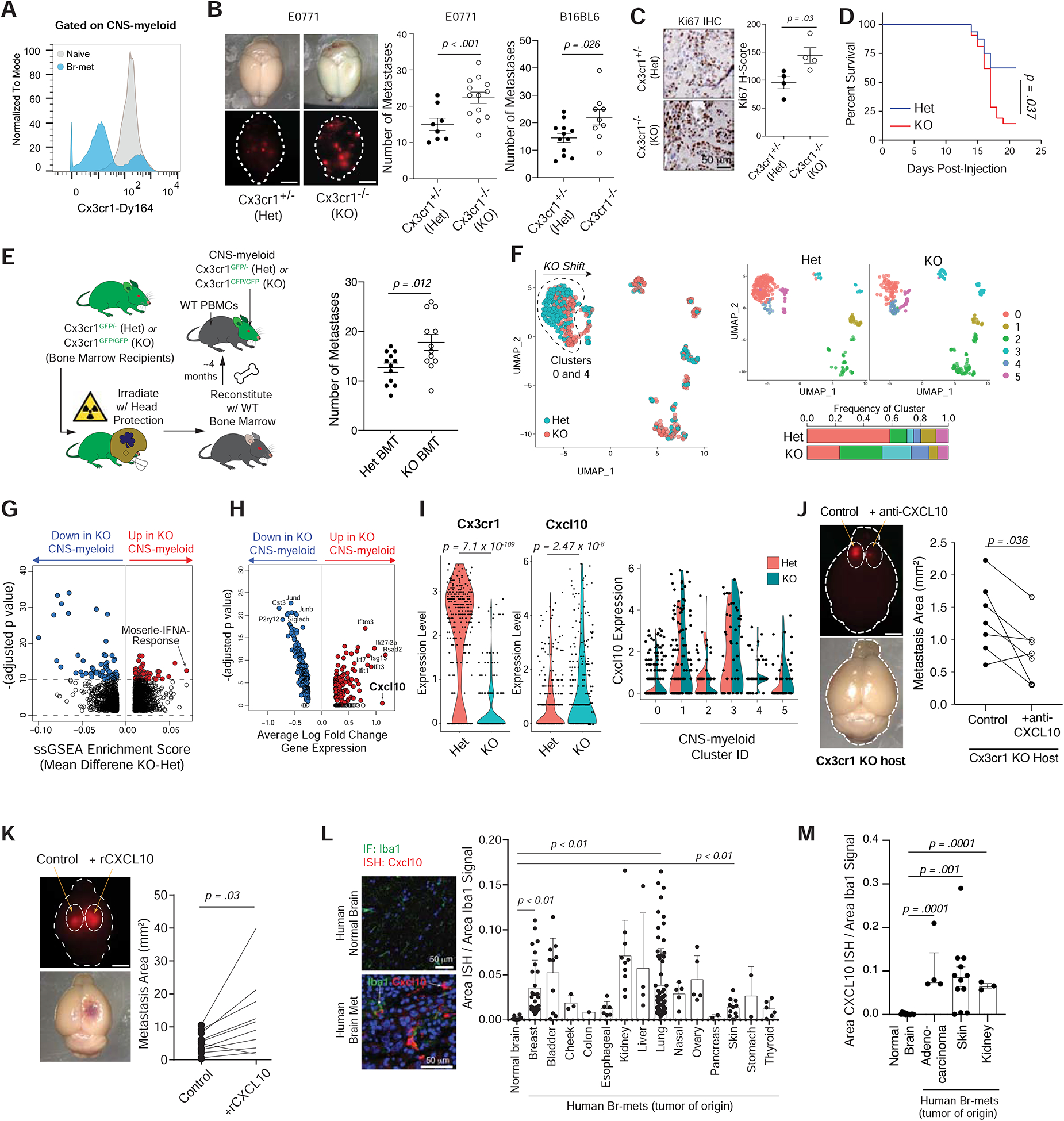
A) Histogram of Cx3cr1 protein expression in naive or br-met-associated CNS-myeloid.
B) Stereoscope images of E0771 br-mets in brains of Cx3cr1+/− (Het) and Cx3cr1−/− (KO) mice (left), associated quantification of br-met number in Het and KO mice (each dot represents 1 mouse; quantification based on pooled data from 2 independent experiments with ≥ 3 mice per group) (middle). Quantification of B16BL6 br-mets that formed in Het and KO mice (each dot represents 1 mouse; quantification based on pooling of 3 independent experiments with ≥ 3 mice per group) (right).
C) Ki67 IHC of br-mets of Het and KO (left), associated Ki67 H-score quantification (each dot represents cumulative H score of all br-mets within one mouse) (right).
D) Survival plot of br-met-bearing Het and KO mice (Quantification based on 1 experiment).
E) Schematic bone marrow transplantation into Het and Hom hosts (left), associated quantification of br-met burden in Het BMT and Hom BMT hosts (each dot represents 1 mouse; quantification based on pooled data from 3 independent experiments with ≥ 3 mice per group) (right).
F) UMAP of CNS-myeloid color coded by genotype (left) and split by genotype and color coded by transcriptional cluster (top right), with stacked bar chart of cluster frequencies within each genotype (bottom right).
G) Volcano plot showing DEGPs between KO and Het CNS-myeloid.
H) Volcano plot showing DEGs between KO and Het CNS-myeloid.
I) Violin plots of Cx3cr1 and Cxcl10 expression in KO and Het CNS-myeloid (each dot represents 1 cell) (left and middle), and split violin plots showing Cxcl10 expression among genotypes within each transcriptional cluster (each dot represents 1 cell) (right).
J) Stereoscope images of br-mets in brains in which cancer cells were co-injected with HBSS (control, left hemisphere) or αCxcl10 (right hemisphere) in KO mice (each dot represents 1 mouse; quantification based on pooled data from 2 independent experiments with ≥ 3 mice per group).
K) Stereoscope images of br-mets in brains in which cancer cells were co-injected with HBSS (control, left hemisphere) or rCxcl10 (right hemisphere) (each dot represents 1 mouse; quantification based on pooled data from 2 independent experiments with ≥ 3 mice per group).
L) Dual Cxcl10 RNA-ISH/Iba1 IF in the naive human brain and human breast cancer br-mets samples (left), associated quantification of area of Cxcl10-ISH signal within myeloid cells. (each dot represents 1 field of view; Data derived from human tissue arrays; nnaive patient = 4; nbr-met patient = 37).
M) Quantification of area of Cxcl10-ISH signal within myeloid cells among normal human brain samples and human br-mets in independently acquired tissue (each dot represents 1 field of view; nnaive patient = 4; nbr-met patient = 5).
CITE-seq data derived from pooling two biological replicates per condition. Data in B, C, D, E, L, and M analyzed by two-tailed student’s t test and error bars represent SEM, except L in which error bars represent SD, center represents mean. Data in J and K analyzed by two-tailed paired student’s t test. Data in G and H analyzed by Wilcoxon rank sum test. Data in I analyzed by bimod likelihood-ratio test for single cell feature expression.
Cx3cr1 knockout caused minor immune compositional changes in the peripheral blood and naive brain but increased brain infiltration of monocytes during br-met (Figure S6B). Ly6CLo monocytes are Cx3cr1Hi (Figure S6C) and thus may be targeted by whole body Cx3cr1 knockout, which may alter their frequency or functionality in br-mets. To mitigate the potential effect of Cx3cr1 KO on BMDM, we performed bone marrow transplantation (BMT) on Cx3cr1 Het and KO mice to restrict Cx3cr1 knockout to CNS-myeloid (Figure 6E). During irradiation, we shielded the skulls of recipient mice to maintain the original Cx3cr1 genotype in CNS-myeloid and prevent blood brain barrier damage (Figure 6E). Four months post-BMT, the peripheral immune system was ~60% reconstituted (Figure S6D). KO BMT mice formed more br-mets than Het BMT mice (Figure 6E), phenocopying the effect of whole body Cx3cr1 knockout mice and strongly suggesting the effects of Cx3cr1 knockout are mostly attributable to its loss in CNS myeloid CNS-myeloid.
Cx3cr1 Knockout Promotes CNS-Myeloid Interferon Response
In naive mice, we found KO brain myeloid cells had a drastically increased protrusion number, thinner protrusions, and increased cell volume relative to Het myeloid cells (Figure S6E), suggesting their activation (Heindl et al., 2018). To gain insight into the molecular underpinnings of KO CNS-myeloid, we performed CITE-seq and focused our analyses to CNS-myeloid due to their more significant contribution to br-met via Cx3cr1 (Figure 5D–I; Figure 6E). Het and KO CNS-myeloid contributed to six transcriptional clusters (Figure 6F). Clusters 0 and 4 shifted on the UMAP between genotypes, and Cluster 2, 3, and 4 cells were more frequent in KO mice than Het mice (Figure 6F). The Moserle-IFNA-Response pathway was the most highly enriched gene pathway in KO CNS-myeloid relative to Het CNS-myeloid (Figure 6G; Table S6). Interferon-related genes (Cxcl10, Rsad2, Ifitm3, Ifit1, Irf7, Isg15) were consistently the most upregulated genes in KO CNS-myeloid (Figure 6H; Table S6). Interferon-induced gene Cxcl10 was the second most up-regulated gene in KO CNS-myeloid and was inversely correlated to Cx3cr1 (Figure 6I). Cxcl10 expression was increased among all CNS-myeloid transcriptional clusters except Cluster 2 (Figure 6I) and remained largely unchanged among BMDM subsets (Figure S6F–G).
Cxcl10 Promotes Br-met Outgrowth and is Highly Expressed in Human Br.MAM
To determine the influence of Cxcl10 on br-met, we first performed a loss of function experiment in which we intracranially co-injected αCxcl10 antibody with E0771.Br cells in a KO host. Brain hemispheres injected with cancer cells and αCxcl10 formed smaller br.met than control hemispheres injected with cancer cells and HBSS (Figure 6J). In a complimentary gain of function experiment, we intracranially co-injected recombinant Cxcl10 (rCxcl10) and E0771.Br cells. Brain hemispheres injected with cancer cells and rCxcl10 formed larger br.met than control hemispheres co-injected with cancer cells and HBSS (Figure 6K). Together, these studies indicate a causal role for Cxcl10 in promoting br-met outgrowth.
To confirm the relevance of Cxcl10 in human br-mets, we performed dual Cxcl10 RNA-ISH and Iba1 IF on human tissue (Table S1). Cxcl10 increased in Br.MAM in human br-mets (npatients = 37, ntumor origins = 14) relative to the naive human brain (npatients = 4) (Figure 6L; Figure S6H), which we confirmed in an independent group of br-met patients (npatients = 5, ntumor origins = 3) (Figure 6M; Table S1).
Cxcl10 recruits VISTAHi PD-L1+ CNS-myeloid to Suppress T Cells
We next sought to determine how Cxcl10 promoted br-met. rCxcl10 did not stimulate cancer cell proliferation, nor did E0771.Br express Cxcr3 (Figure S6I–J), suggesting Cxcl10 promotes br-met through tumor cell non-autonomous means.
Cxcl10 is an immune modulator (Liu et al., 2011). To explore Cxcl10 regulation of br-met immune composition, we performed CyTOF on control br-met-burdened brains and brains injected with tumor cells and rCxcl10. rCxcl10 co-injection increased myeloid cells marked by CD86, CD11c, and MHCII expression (Figure S7A). As Br.MAM accumulated within br-mets (Figure 1A–C; Figure 5I), we examined the spatial aspects of CD86+ CNS-myeloid in situ. CD86+ CNS-myeloid were nearly exclusively found within br-met lesions and ~3-fold more concentrated within br-mets than distal to br-mets (Figure 7A–B). To explore whether Cxcl10 could mediate CNS-myeloid recruitment, we performed an in vitro transwell migration assay. rCxcl10 enhanced SIM-A9 microglia migration across transwell inserts by ~50% relative to control wells (Figure 7C). Collectively, these findings suggest that Cxcl10 recruits a potentially pro-br-met CD86+ CNS-myeloid cells to br-met lesions.
Figure 7. Cxcl10 Recruits VISTAHi PD-L1+ CNS-myeloid to Suppress T Cells.
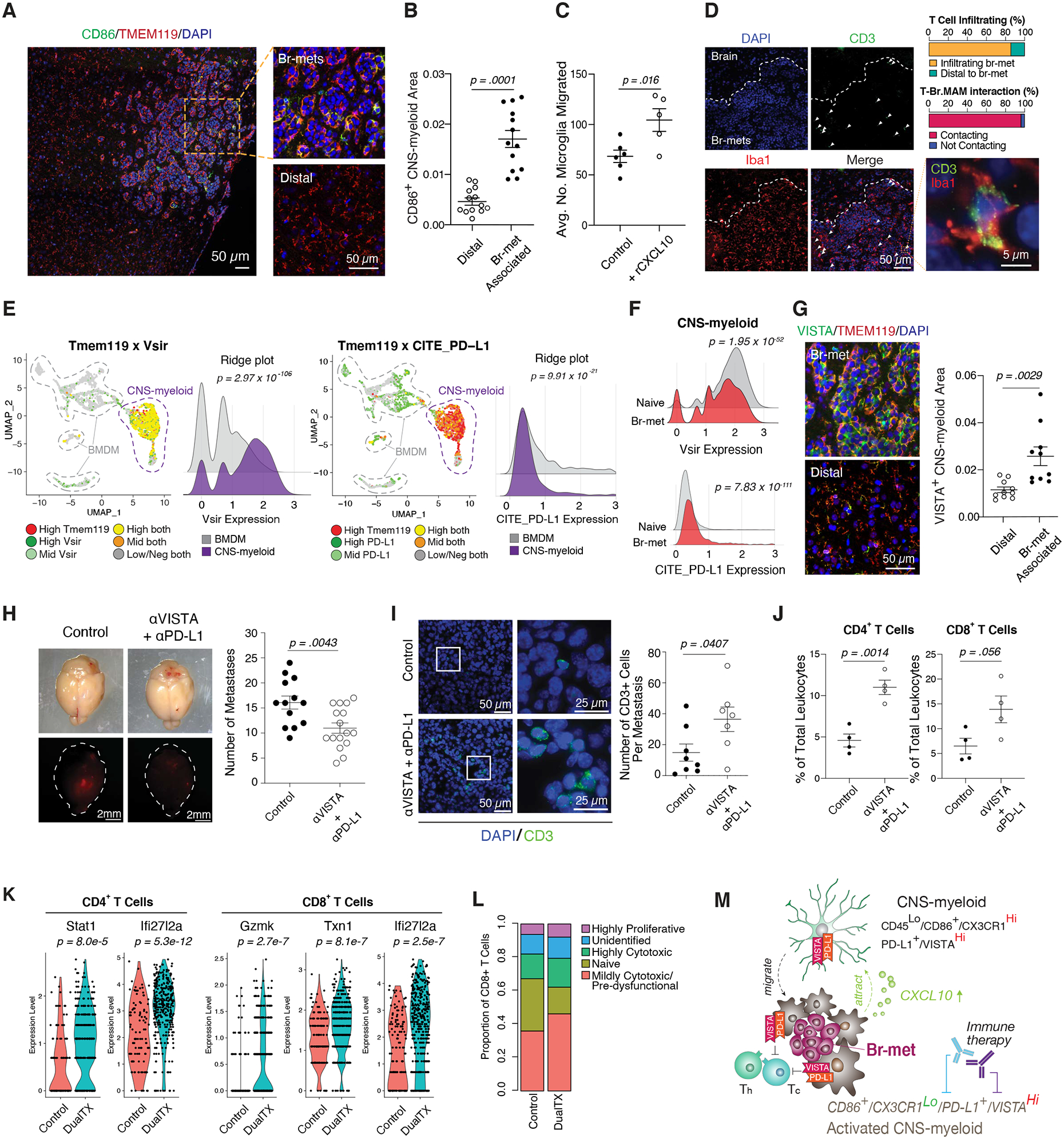
A) IF of CD86+ CNS-myeloid within br-met niche (left), magnified view of these cells within the lesion (top right) and distal to br-met (bottom right).
B) Quantification of area occupied by CD86+ CNS-myeloid within br-met lesions compared to distal to br-mets (each dot represents 1 br-met).
C) Quantification of number of SIM-A9 microglia migrated across transwell with rCxcl10 compared to control (each dot represents 1 well; quantification based on 1 experiment).
D) IF of T cell and myeloid cell spatial distribution within br-mets and associated quantifications of T cell infiltration and T cell-Br.MAM contact.
E) Feature plot and ridge plots showing Vsir RNA expression (left) and PD-L1 protein expression (right) in br-met-associated CNS-myeloid and BMDM.
F) Ridge plots comparing Vsir RNA expression (top) and PD-L1 protein expression (bottom) in the naive brain and br-met-burdened brain.
G) IF of VISTA+ CNS-myeloid within br-met lesions (top left) and distal to br-met (bottom left), associated quantification of area occupied by VISTA+ CNS-myeloid within br-mets compared to distal to br-mets (each dot represents 1 br-met) (right).
H) Stereoscope images of br-mets in brains of αVISTA + αPD-L1 treated mice and control mice (left), associated quantification of br-met number between treatment and control (each dot represents 1 mouse; quantification based on 2 independent experiments with ≥ 3 mice per group per experiment) (right).
I) IF of CD3+ T cells associated with E0771 br-mets in αVISTA + αPD-L1 treated mice and control mice (left), associated quantification of the number of CD3+ T cells per br-met (each dot represents 1 br-met) (right).
J) Quantification of proportion of CD4+ T cells (left) or CD8+ T cells (right) of all leukocytes in control mice and αVISTA + αPD-L1 treated mice. (Each dot represents 1 mouse).
K) Violin plots of T cell activation gene expression that significantly increased with αVISTA + αPD-L1 compared to control (each dot represents 1 cell).
L) Stacked bar chart showing proportions of T cell activation status in control mice and αVISTA + αPD-L1 treated mice.
M) Model of myeloid cell regulation of br-met outgrowth.
Data in E and F derived from pooling three biological replicates per condition. Data in K and L derived from pooling ≥ 4 biological replicates per condition. Data in B, C, G, H, I, and J analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean. Data in E, F, and K analyzed by Wilcoxon rank sum test.
See also Figure S7.
Tumor infiltration of myeloid cells that co-express CD86, CD11c, and MHCII is typically correlated with an anti-cancer response. Paradoxically, such a population correlated with increased br-met. We hypothesized intra-metastatic CD86+ CNS-myeloid could barricade T cell infiltration to br-met lesions and/or engage with and suppress T cells. T cells were not apparently barricaded from the br-met lesions, but >90% of br-met-associated T cells contacted Br.MAM (Figure 7D), suggesting myeloid-T cell engagement.
To explore the potential of CNS-myeloid suppression of T cells, we queried CD86+ Br.MAM gene set enrichment (Figure S7B–C). CD86+ Br.MAM upregulated numerous immunosuppressive pathways, including the PD-1/PD-L1 axis, CTLA-4 signaling, and FOXP3 targets (Figure S7C). Examination of negative immune checkpoint gene expression among all br-met niche cells showed CNS-myeloid expressed several checkpoint genes, including Vsir (VISTA) and Cd274 (PD-L1) (Figure S7D). Vsir was enriched in CNS-myeloid relative to other br-met niche cells (Figure 7E; Figure S7D). PD-L1 was expressed on CNS-myeloid and expressed slightly higher on BMDM (Figure 7E; Figure S7D), but was mostly present on Ly6CHi monocytes, which we demonstrated do not affect br-met outgrowth (Figure 5D–F). We additionally examined the expression dynamics and spatial features of VISTA and PD-L1. CNS-myeloid constitutively expressed Vsir highly (Figure 7F), whereas they expressed PD-L1 at a low level in the naive state and modestly upregulated PD-L1 during br-met (Figure 7F). Despite slight downregulation of Vsir during br-met (Figure 7F), VISTA+ CNS-myeloid density approximately doubled within the br-met lesions relative to regions distal to br-mets (Figure 7G). Cumulatively, this data suggests that CNS-myeloid could suppress T cells via VISTA and PD-L1.
Neutralization of VISTA and PD-L1 Reduces Br-met and Enhances T Cell Activity and Abundance
Given the non-redundant nature of immune suppression, we co-inhibited PD-L1 and VISTA signaling to determine their influence on br-met outgrowth. Co-treatment with αVISTA and αPD-L1 antibodies, but not single treatments, reduced br-met burden compared to control mice (Figure 7H; Figure S7E). αVISTA and αPD-L1 co-treatment increased CD3+ cell abundance and the frequency of T cell subsets (Figure 7I–J), and upregulated some T cell activation genes (Stat1, Gzmk, Ifi27l2a, and Txn1) and ribosomal genes (Figure 7K; Figure S7F). Within CD8+ T cells, dual therapy moderately increased the proportion of mildly cytotoxic cells and slightly increased the proportions of highly cytotoxic and proliferative cells (Figure 7L). CNS-myeloid exhibited no major transcriptional changes with dual therapy, but BMDM were marked by the upregulation of antigen presentation genes, Stat1, and Ifitm1, which may represent effects from the altered inflammatory milieu induced by the therapy (Figure S7G). While our data suggest αVISTA and αPD-L1 treatment reduces CNS-myeloid suppression of T cells in the br-met niche, the potential expression and functional contribution of these proteins on cells peripherally in lymphoid organs during br-met warrants future investigation.
DISCUSSION
Defining the Immune Landscape of the Br-met Niche through Single Cell Analysis
Although myeloid cell subsets have been observed within br-mets (He et al., 2006; Zhang et al., 2015), their phenotypic and functional heterogeneity have not been fully revealed. Through single cell analyses, we characterized the compositional and transcriptional heterogeneity of br-met-associated leukocytes and modeled Br.MAM plasticity. We identified extensive compositional and transcriptional changes during br-met that implicated multiple immune cell types and molecular mediators in br-met regulation, supporting recent immune profiling studies in human brain tumors (Friebel et al., 2020; Klemm et al., 2020). Our single cell analyses refute the oversimplified binary view of myeloid polarization and echo a multi-dimensional model of myeloid cell activation (Xue et al., 2014). Additionally, our analyses uncovered the plastic nature of CNS-myeloid and BMDM during br-met (Figure 4). The plasticity we report reflect that of previous findings of BMDM becoming CNS-myeloid-like upon translocation to the brain (Bennett et al., 2018; Lavin et al., 2014), and also suggests the potential for CNS-myeloid to take on characteristics of infiltrating BMDM. Such dynamics and their functional relevance to br-met necessitate future in-depth study.
CNS-myeloid Foster an Immunosuppressive Br-met Niche
The br-met-promoting role of Cxcl10 that we report is seemingly paradoxical given the canonical role of Cxcl10 in recruiting disease-resolving T cells (Liu et al., 2011). To reconcile the differences, it is important to acknowledge that the brain is immunologically-unique and has evolved to be anti-inflammatory for neuro-protection (Deczkowska et al., 2018; Norris and Kipnis, 2019). The blood brain barrier and high expression of negative checkpoints, such as VISTA, in CNS-myeloid represent two such brain-specific anti-inflammatory evolutionary adaptations (Deczkowska et al., 2018; Norris and Kipnis, 2019). While VISTA expression in CNS-myeloid is similar between the naive state and br-met, VISTA+ CNS-myeloid density increases within br-met lesions. Although Cxcl10 potentially recruits T cells to br-mets, Cxcl10 can attract CNS-myeloid, which carry immunosuppressive proteins that dampen T cell activation. We posit that the potential anti-tumor benefit of Cxcl10-mediated T cell recruitment is counteracted by the accumulation of T cell-suppressive CNS-myeloid (Figure 7M). While this mechanism may confer benefits in autoimmune brain diseases, it undesirably fosters an immune suppressive niche for br-met to thrive.
In summary, we demonstrated that CNS-myeloid facilitate br-met via Cxcl10 signaling that fosters an immune suppressive niche. In light of our findings and recent clinical studies (Tawbi et al., 2018), further research on the immune suppressive capabilities of the br-met niche is necessary in order to design more effective immune therapy regimens for br-met patients.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to Lead Contact, Siyuan Zhang (szhang8@nd.edu).
Materials Availability
This study did not generate unique reagents.
Data and Code Availability
CITE-seq data is uploaded to GEO (GSE134285). All other data and code used to analyze data are available upon reasonable request for Lead Contact/corresponding author.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal studies were performed ethically and in compliance with IACUC protocol approved by the University of Notre Dame IACUC committee. C57BL/6 (000664/Black 6); B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ (021160/Cx3cr1CreER); C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J (007900/ROSA25iDTR); B6.129P-Cx3cr1tm1Litt/J (005582/CX3CR1-GFP); B6.129(Cg)-Ccr2tm2.1Ifc/J (017586/Ccr2 RFP); B6.Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J (007906/Ai6) mouse lines were purchased from Jackson Lab (Ben Harbor, ME). Cx3cr1CreER/+:R26iDTR/+ double transgenic mice were produced in-house by breeding Cx3cr1CreER with ROSA26iDTR mice. Cx3cr1CreER/+ mice, used as controls in general myeloid and CNS-myeloid depletion studies, were produced in-house by breeding Cx3cr1CreER mice to C57BL/6 mice. Cx3cr1GFP/+ mice were produced in-house by breeding Cx3cr1-GFP mice to C57BL/6 mice. Ccr2RFP/+ mice were obtained by breeding Ccr2 RFP mice to C57BL/6 mice. Mice were bred and maintained by Friemann Life Science Center (FLSC) at the University of Notre Dame, with the exception of some C57BL/6, which were bred and maintained for some time at Jackson Lab, but acclimated for a minimum of 1 week in FLSC prior to experimental use. All mice used in experiments were females aged from 2 months to 12 months. Females were exclusively used as breast cancer br-met is predominantly a female pathology. For αVISTA and αPD-L1 related experiments, mice were randomized after metastasis initiation but before administration of immune therapy. For experiments in which br-met-burdened brains and naive brains were compared, mice were randomized prior to metastasis initiation. For all other experiments, randomization was not possible as the comparison point was mouse genotype. No power analyses were used to predetermine sample sizes in mouse experiments. However, sample sizes were chosen based on prior literature using similar experimental paradigms.
Human Samples
Human brain metastasis (GL861) and normal brain (T174b) tissue arrays were purchased from US Biomax, Inc (Derwood, MD). Details of each array can be found on the Biomax website (brain metastasis array: https://www.biomax.us/tissue-arrays/Brain/GL861; normal brain array: https://www.biomax.us/tissue-arrays/Brain/T174b), and in Table S1. Additional human brain metastasis tissue sections were obtained from the Indiana University Melvin and Bren Simon Comprehensive Cancer Center Tissue Bank, details of which can be found in Table S1.
Cell Lines
E0771 cell line (female origin) was purchased from CH3 Biosystems (Amherst, NY, 940001-Vial), CMT 167 cell line (female origin) was purchased from Sigma Aldrich (10032302–1VL), and SIM-A9 (unknow sex origin as derived from P1 pups) were purchased from ATCC (ATCC-CRL-3265). B16BL6 cell line (male origin) was a gift from Dihua Yu’s lab (MD Anderson Cancer Center, Houston, TX). Brain seeking E0771 lines (E0771.Br) used in this study were generated by one round of in vivo selection for homing to the brain. E0771 cells were labeled with mCherry or ZsGreen by lentiviral transduction. E0771 cells were cultured in RPMI + 10% FBS + 1% Penicillin/Streptomycin, and CMT 167 and B16BL6 cells were cultured in DMEM High Glucose + 10% FBS + 1% Penicillin/Streptomycin. SIM-A9 cells were cultured in DMEM:F12 + 10% heat inactivated FBS + 5% heat inactivated horse serum. All cell lines were incubated at 37C, 5% CO2, humidity ~95%. Cell lines were tested and found negative for mouse pathogens by IDEXX BioResearch Inc. (IMPACT IV panel: PCR evaluation for: LCMV, MHV, MPV, MVM, Mycoplasma pulmonis, Mycoplasma sp., PVM, Sendai, TMEV).
METHOD DETAILS
Generation of Experimental Brain Metastases
For generation of brain metastasis in mice by cardiac injection, 2×105 cells were inoculated into the left cardiac ventricle of mice. Cardiac injections were used for experiments presented in Figures 1B–F; 4I, 5A–F; 6B–D (E0771 experiments only). For generation of brain metastasis by intracranial injection, only used in experiments related to Figures 6J and 6K in which anti-Cxcl10 or rCxcl10 needed to be directly injected to the metastatic lesion, approximately 5,000 cells were stereotaxically injected using a fine glass capillary needle and nanoliter injector (Nanoject II 3-000-204, Drummond Scientific Company, Broomall, PA) 1.25mm lateral and 1.5mm caudal relative to bregma, and 1.0mm deep. For generation of brain metastasis by carotid injection, 2–3×105 cells were injected into the carotid artery, which were used in all other experiments not listed above.
In Vivo Myeloid Depletion
To deplete the general Cx3cr1+ myeloid population in CX3CR1CreERT/+:R26iDTR/+ mice, mice were first administered 75mg kg−1 Tamoxifen (Sigma, T5648) dissolved in corn oil (Sigma, C8267) by intraperitoneal injection once daily for 10 consecutive days before tumor cell inoculation, and then daily thereafter, to induce genetic recombination. Mice were administered 1μg Diphtheria Toxin (Sigma, D0564) dissolved in 100μL sterile saline by intraperitoneal injection either (for outgrowth depletion) every other day beginning 7 days post-tumor cell inoculation or (for early depletion) three consecutive days prior to and one day following tumor cell injection. Tamoxifen and Diphtheria Toxin doses were reduced or skipped in some cases when mice were deemed too unhealthy and unable to handle further drug administration. For CNS-myeloid cell exclusive depletion, Tamoxifen and Diphtheria Toxin concentrations were identical to that used for general myeloid depletion, but the administration timings differed. Mice were treated with Tamoxifen for 10 consecutive days. A minimum of one month following Tamoxifen cessation, mice were injected with cancer cells, four days after which mice were administered Diphtheria Toxin every three days until experimental endpoint.
Bone Marrow Transplantation
Cx3cr1 Het and KO mice were irradiated twice, approximately 12 hours apart, receiving 525 rad per irradiation dose. Before irradiation in RS 2000 Biological Research Irradiator (Rad Source Technologies, Inc.), mice were anesthetized with ketamine/xylazine so that their heads could be positioned under a lead scaffold for the duration of irradiation to prevent the brain from receiving irradiation. Mice were reconstituted 24 hours after first radiation dose by retroorbital injection of approximately 5 million C57BL/6 wildtype donor-derived bone marrow cells supplemented with approximately 1 million C57BL/6 wildtype donor-derived T cells isolated from the spleen by pan T cell enrichment. Reconstituted mice were housed in sterile conditions for three months, after which reconstitution success was evaluated by flow cytometry, examining GFP expression in peripheral blood cell subsets relative to Cx3cr1 Het and KO counterparts that did not receive irradiation or BMT.
In Vivo Antibody Treatments and Cytokine Co-injection
For in vivo mouse recombinant Cxcl10 ligand (R&D Systems, 466-CR-010/CF) treatment, approximately 2.76ng of rCxcl10 dissolved in HBSS (Cytiva Life Sciences, SH30588.01) was co-injected with cancer cells. For in vivo goat anti-mouse Cxcl10 (R&D Systems, AF-466-SP) treatment of Cx3cr1-KO hosts, approximately 2.5μg of anti-Cxcl10 was co-injected with cancer cells. For dual immune checkpoint inhibition treatment, 300μg of armenian hamster anti-mouse VISTA (BioXCell, BE0310) and 20μg of rat anti-mouse PD-L1 (BioXCell, BE0101) were injected every two days starting three days following cancer cell injection. The same scheme was used for single immune checkpoint antibody blockade experiments.
In Vitro Ligand Treatments and MTT Assay
E0771.Br cells were plated at a density of 3,000 cells per well in a 96 well plate and treated with either rCxcl10 (R&D Systems,1245-GA-050) in triplicate with 1, 10, or 100ng of ligand per well. Following at least 48 hours of culture with ligand, an MTT assay was performed. Briefly, filtered 10mg/mL MTT solution (Sigma, M5655) was added to each well and incubated for 2 hours. After MTT incubation, SDS-based lysis buffer added for two hours until cells were solubilized. Finally, the plates were read on a plate reader at 570nm. For SIM-A9 microglia transwell migration assay, 50ng/mL rCxcl10 in media or media alone was placed below the 8um transwell insert (Corning, 3464) and SIM-A9 microglia were seeded on top of the transwell insert. 24 hours after seeding, non-migrated cells on the top of the membrane were removed with a cotton tip applicator, and the cells that had migrated to the opposite side of the membrane were fixed in 4% PFA, stained with DAPI (ThermoFisher Scientific, D1306), and 3 random fields of view per membrane were imaged. Subsequently, cells were counted and averaged to determine the number of migrated cells per well. The above in vitro studies were performed once.
Tissue Collection and Single Cell Preparation
For tissue collection for histological analysis, mice were anesthetized with isoflurane. Mice were then transcardially perfused with cold 1x PBS followed by cold 4% PFA. Tissue isolated for flow cytometry, CyTOF, or single cell RNA-seq analysis was collected identically with the exception that 4% PFA perfusion was excluded. Cells were isolated from mouse brains by digesting the brains into single cell suspensions and then enriched from other neural cells by density gradient centrifugation as follows: Brains from mice transcardially perfused with 1x PBS were extracted, minced with scissors, and triturated with a P1000 micropipette. The resulting brain tissue slurry was centrifuged at 300g for 2 minutes. The supernatant was removed and the pellet resuspended and processed as directed by the Miltenyi Biotec Neural Tissue Dissociation Kit (P) (Miltenyi Biotec, 130-092-628) or Multi-tissue Dissociation Kit I (Miltenyi Biotec, 130-110-201). Brains were enzymatically digested into a single cell suspension by rotating at 37C for approximately 25 minutes with trituration half way through incubation. The resulting cell suspension was strained through a 100μm cell filter as needed, diluted in 1x HBSS, and centrifuged at 300g for 10 minutes. The resulting supernatant was discarded and the pellet resuspended in 3mL 70% Percoll (GE Healthcare, 17-0891-02). The Percoll gradient and density layering was prepared (from bottom to top: 70%, 37%, 30%). Density gradient centrifugation was performed for 20 minutes at 2000rpm with no break. Myeloid cells were isolated from the buffy layer between the interface of the clear Percoll and red-colored Percoll. Following washing in 1x HBSS, the resultant cell suspension was processed as required for the desired application.
Flow Cytometry, CyTOF, and CITE-seq
Brain-derived cells were prepared for flow cytometry in a manner identical to that of their preparation for scRNA-seq through the Percoll gradient centrifugation step. Following gradient centrifugation, cells were washed in Cell Staining Buffer (Biolegend, 20201). For experiments relying on antibody staining, FC receptors were blocked by incubation with TruStain fcX in 100uL Cell Staining Buffer for 30 min at RT and then stained with one or a combination of the following antibodies: rat anti-mouse CD45–421 (Biolegend, 103133, 30-F11), rat anti-mouse CD4-PE (Biolegend, 100407, GK1.5), rat anti-mouse CD3–647 (Biolegend, 100209., 17A2), rat anti-mouse CD8-APC/Cy7 (Biolegend, 100713, 53-A7), rat anti-mouse Gr1–594 (Biolegend, 108448, Rb6–8C5), rat anti-mouse Ly6G-APC (Biolegend,127613, 1A8), rat anti-mouse Ly6C-PE (Biolegend, 128007, HK1.4), rat anti-mouse Ly6C-FITC (Biolegend, 128005, HK1.4), rat anti-mouse Ly6C-APC (Biolegend, 128015, HK1.4), rat anti-mouse/human CD11b-APC/Cy7 (Biolegend, 101225, M1/70), rat anti-mouse CD11c-PE/Dazzle (Biolegend, 117347, N418), rat anti-mouse/human CD11b-APC (Biolegend, 101212, M1/70), rat anti-mouse MHCII-APC/Cy7 (Biolegend, 107627, M5/114.15.2), rat anti-mouse F4/80-APC (Biolegend, 123116, BM8), rat anti-mouse NK1.1-APC/Cy7 (Biolegend, 108723, PK136), armenian hamster anti-mouse Cxcr3-APC (Biolegend, 126511, Cxcr3–173). Following staining, cells were washed in Cell Staining Buffer and then resuspended in Cell Staining Buffer, and finally run on either a BD Biosciences FACSAria III, BD LSRFortessa X-20, or Beckman Coulter FC500.
For CyTOF experiments, brain-derived cells were prepared in a manner identical to that of their preparation for scRNA-seq through the Percoll gradient centrifugation step. Following gradient centrifugation, three brains were pooled together to comprise a single CyTOF sample and cells were washed and resuspended in Maxpar PBS (Fluidigm, 201058). Dead cells were labeled by incubation with .75 μM Cell-ID Cisplatin (Fluidigm, 201064) for 5 minutes and then washed in Maxpar Cell Staining Buffer (Fluidigm, 201068). FC receptors were blocked by incubation with TruStain fcX in 100 μL MaxPar Cell Staining Buffer for 30 min at RT. Cells were washed and then stained with a cocktail of metal-conjugated antibodies at RT for 30 minutes and then washed in MaxPar Cell Staining Buffer. Optimal concentrations were determined for each antibody by titration. The following pre-conjugated antibodies purchased from Fluidigm were used in this study: CD45–089Y (3089005B, 30-F11); Ly-6G-141Pr (3141008B, 1A8,); CD11c-142Nd (3142003B, N418); CD45R-144Nd (3144011B, RA3–6B2); CD4–145Nd (3145002B, RM4–5); CD11b-148Nd (3148003B, M1/70); CD44–150Nd (3150018B, IM7); CD25–151Eu (3151007B, 3C7); CD3e-152Sm (3152004B, 145–2C11); PD-L1–153Eu (3153016B, 10F.9G2); CTLA-4–154Sm (3154008B, UC10–4B9); PD-1–159Tb (3159024B, 29F.1A12); Ly-6C-162Dy (3162014B, HK1.4); CX3CR1–164Dy (3164023B, SA011F11); NK1.1–165Ho (3165018B, PK136); ckit-166Er (3166004B, 2B8); CD8a-168Er (3168003B, 53–6.7); CD86–172Yb (3172016B, GL1); I-A/I-E-209Bi (3209006B, M5/114.15.2). Cells were resuspended and fixed in 1.6% PFA prepared in MaxPar PBS for 20 minutes and then washed in MaxPar PBS. Nuclei were labeled by incubating fixed cells in 1:4000 Cell-ID Intercalator (Fluidigm, 201192B) dissolved in MaxPar Fix and Perm Buffer (Fluidigm, 201067) for 1h or overnight at 4C. Following nuclear labeling, cells were washed once in MaxPar Cell Staining Buffer and twice in MaxPar Water (Fluidigm, 201069). Samples were brought to 500,000 particulates/mL in MilliQ water containing .1x EQ beads (Fluidigm, 201078) and run in 450 μL injections on a CyTOF2 instrument.
For CITE-seq experiments, brain-derived cells were prepared in a manner identical to that of their preparation for scRNA-seq through the Percoll gradient centrifugation step. Following gradient centrifugation, samples were prepared for 10X Genomics Chromium (10X Genomics, PN-120223) as described in the CITE-seq and cell hashing protocol on the CITE-seq website (https://citeseq.files.wordpress.com/2019/02/cite-seq_and_hashing_protocol_190213.pdf). Briefly, samples were blocked by incubation with TruStain fcX in 50 μL cell staining buffer for 20 minutes on ice. Following block, samples were stained with the following Total-seq antibodies purchased from BioLegend:rat anti-CCR2/CD192 (150625, Biolegend, SA203G11); rat anti-CD117/c-kit (105843, Biolegend, 2B8); rat anti-CD11b (101265, Biolegend, M1/70); armenian hamster anti-CD11c (117355, Biolegend, N418); rat anti-CD172a/SIRPα (144033, Biolegend, P84); rat anti-CD25 (102055, Biolegend, PC61); rat anti-CD3 (100251, Biolegend, 17A2); rat anti-CD38 (102733, Biolegend, 90); rat anti-CD4 (100569, Biolegend, RM4–5); rat anti-CD44 (103045, Biolegend, IM7); rat anti-CD45 (103159, Biolegend, 30-F11); rat anti-CD45R/B220 (103263, Biolegend, RA3–6B2); rat anti-CD86 (105047, Biolegend, GL-1); rat anti-CD8a (100773, Biolegend, 53–6.7); mouse anti-CD90.1 (202547, Biolegend, OX-7); mouse anti-Cx3cr1 (149041, Biolegend, SA011F11); rat anti-F4/80 (123153, Biolegend, BM8); rat anti-I-A/I-E (107653, Biolegend, M5/114.15.2); rat anti-Ly6C (128047, Biolegend, HK1.4); rat anti-Ly6G (127655, Biolegend, 1A8); mouse anti-NK1.1 (108755, Biolegend, PK136); rat anti-PD-1 (109123, Biolegend, RMP1–30); rat anti-PD-L1 (153604, Biolegend, MIH6); rat anti-CD169/Siglec-1 (142425, Biolegend, 3D6.112); rat anti-Siglec-H (129615, Biolegend, 551); mouse anti-TMEM119 (853303, Biolegend, A16075D); mouse anti-XCR1 (148227, Biolegend, Zet); rat anti-CD24 (101841, Biolegend, M1/69); rat armenian hamster anti-CD103 (121437, Biolegend, 2e7); rat anti-CD49d (103623, Biolegend, R1–2); rat anti-mouse Hashtag 1–8 antibody (155801, Biolegend, M1/42; 30-F11). After 25 minutes of staining, samples were washed 4 times prior to loading on to the 10x Chromium.
Tissue Clearing
Tissue clearing was performed as previously described (Guldner et al., 2016). Briefly, tissues were collected as described above and were incubated in 4% PFA overnight at 4C. The next morning, the tissue sample was rinsed 3 times with 1x PBS, sliced into ~200μm sagittal sections, and then incubated in CUBIC1 (35% water; 25% urea; 25% N,N,N’,N’-Tetrakis(2-hydroxyproply)ethyl-enediamine [Alfa Aesar, L16280-AP], 99%; 15% triton-X) for a minimum of 3 days at 37C with gentle rotating. Following CUBIC1 incubation until the tissue was transparent to the eye, the sample was washed with multiple changes of 1x PBS over 24h at RT with gentle rotating. The sample was then incubated in 6μL DAPI:2mL 1x PBS for up to 24h at RT with gentle rotating. The DAPI-stained sample was washed for ~8h with 1x PBS at RT with gentle rotating. Finally, the tissue was incubated in CUBIC2 (50% sucrose; 25% urea; 15% water; 10% Triethanolamine[JT Baker, 02-004-534]) for a minimum of 1 day at RT before being imaged. CUBIC reagents were prepared as described in the original CUBIC protocol (Susaki et al., 2014).
Microscopy
Deep tissue imaging was performed on either an Olympus FV1000 or a Caliber ID RS-G4. While using the Olympus FV1000, either the confocal or two photon setting was used depending on which setting optimally excited the fluorophores present in the sample. On the Olympus FV1000, BrightZ setting was applied to increase signal as deeper areas of the sample were imaged. In all cases, a 25x objective (XLSLPLN25XGMP, Olympus USA; NA = 1.0 and WD = mm) was used on the Olympus FV1000. The Olympus FV1000 was equipped with filter set (460–500, 520–560, 525–625, 650–700 nm) and a mode-locked Ti:sapphire laser (Mai Tai DeepSee 690–1040 nm, Spectra-Physics). For the Caliber ID RS-G4, all images were acquired with a 20x objective. Imaging of 2D tissue slides was performed on an Olympus BX43 upright microscope for non-fluorescent histology and a Leica DM550 B inverted microscope for fluorescent histology. Gross imaging of whole brains was captured on a Leica M165 FC fluorescence stereoscope.
Tissue Immunohistochemistry
For immunofluorescence (IF), tissues were adhered to SuperFrost slides (VWR, 48311–703) and deparaffinized with two xylene washes. Tissue was rehydrated by washes with 100%, 90%, and 80% ethanol and ddH2O. Antigens were unmasked by boiling slides in 1x sodium citrate solution (2.94g sodium citrate trisodium salt dehydrate in 1L ddH2O, pH 6.0) either in a pressure cooker for approximately 20 minutes or in a beaker in a microwave for 10 minutes. Following antigen retrieval, tissues were blocked with 5% goat serum in TBST for one hour at RT, and then stained with primary antibody overnight in a humidifying chamber at 4C. Antibodies used for IF include: rabbit anti-Iba1 (Wako, polyclonal, 019–19741), rabbit anti-TMEM119 (Abcam, 28–3, Ab209064), guinea pig anti-TMEM119 (Synaptic Systems, polyclonal, 400–004), mouse anti-mCherry (Clontech, monoclonal, 632543), rabbit anti-CD3 (Abcam, SP7, ab16669), rabbit anti-CD86 (Cell Signaling Technologies, E5W6H; CST; 19589), rabbit anti-VISTA (Cell Signaling Technologies, D5L5T; CST; 54979). After overnight primary antibody incubation, slides were washed three times with TBST, and then incubated in secondary antibody for three hours in a humidifying chamber at RT. Secondary antibodies used include: Alexa Fluor 488 AffiniPure Goat Anti-Rabbit IgG, Fc fragment specific (Jackson Immuno Research, 111-545-008); Alexa Fluor 594 AffiniPure Goat Anti-Mouse IgG, Fcγ fragment specific (Jackson Immuno Research, 115-585-008); Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) (Jackson Immuno Research, 711-545-152); Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 (Invitrogen, A-21432); Alexa Fluor 594 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) (Jackson Immuno Research, 706-585-148). After secondary antibody incubation, slides were rinsed three times with TBST, stained with DAPI (Thermo Fisher Scientific, D1306), rinsed three times with TBST, and then mounted with Fluoromount G slide mounting media (Electron Microscopy Sciences, 17984–25). For DAB-based immunohistochemistry, samples were prepared similar to IF through antigen retrieval. After antigen retrieval, endogenous peroxidases were quenched with 3% hydrogen peroxide in tap water for 10 minutes. Next, samples were stained with the Vectastain Elite ABC-HRP Kit RTU (Vector Laboratories, PK-7200) per kit protocol. Primary antibodies used include: rabbit Ki-67 (Cell Signaling Technology, D3B5, 12202S). ImmPACT DAB (Vector Laboratories, SK-4105) was used as the peroxidase substrate and incubated on tissue until color developed (~30s).
RNAScope In Situ Hybridization with Co-immunostaining
Tissue sections were prepared using the RNAscope 2.5 manual assay following the manufacturer’s protocol for sample preparation and pretreatment (Advanced Cell Diagnostics, Document Number: 322452) and RNAscope 2.5 HD detection (Advanced Cell Diagnostics, Document Number: 322360-USM) with minor modifications. Modifications of the protocol include: extended tissue rehydration gradient; extension of boiling in target retrieval buffer to 20 minutes total, and omitting the Protease Plus incubation. Briefly, sample preparation and pretreatment included deparaffinization and rehydration, endogenous peroxidases quenching with RNAscope Hydrogen Peroxide (Advanced Cell Diagnostics, 322335), and targets retrieval by boiling in RNA Scope Target Retrieval Agent (Advanced Cell Diagnostics, 322000). Next, probes were hybridized in HybEZ Oven (Advanced Cell Diagnostics, 310010). Probes used include mouse P2ry12 (Advanced Cell Diagnostics, 317601), mouse Il1b (Advanced Cell Diagnostics, 316891), mouse Ybx3 (Advanced Cell Diagnostics, 538121), mouse Cxcl10 (Advanced Cell Diagnostics, 408921), and human Cxcl10 (Advanced Cell Diagnostics, 311851).
Amplifiers were hybridized and signal was detected using amplifiers and Fast Red reagents provided in RNAscope 2.5 HD Detection – RED kit (Advanced Cell Diagnostics, 322360). Washes between each step were performed with RNAscope Wash Buffer (Advanced Cell Diagnostics, 310091). To co-immunostain with ISH, following signal detection, samples were treated identically to IF samples starting with blocking in 5% goat serum.
scRNA-seq Library Preparation and Illumina RNA Sequencing
For CITE-seq experiments, 10x Genomics Chromium (10x Genomics, PN-120223) was used for single cell capture, cDNA libraries were prepared according to the standard CITE-seq (https://citeseq.files.wordpress.com/2019/02/cite-seq_and_hashing_protocol_190213.pdf) and 10x Genomics standard protocols. Kits necessary for library preparation included Chromium Single Cell 3’ Library and Gel Bead Kit v3 (10x Genomics, PN-10000092); Chromium Next GEM Single Cell 3’ GEM Library and Gel Bead Kit v3.1 (10x Genomics, PN-1000121); Chromium Chip B Single Cell Kit (10x Genomics, PN-1000074); Chromium i7 Multiplex Kit (10x Genomics, PN-120262). Primers required for library preparation include:
ADT_TruSeq_i7_UDI01 - CAAGCAGAAGACGGCATACGAGAACCGCGGGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA
ADT_TruSeq_i7_UDI04 - CAAGCAGAAGACGGCATACGAGTTGGACTTGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA
ADT-RPI-1 - CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA
ADT-RPI-2 - CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA
HTO_Nextera_i7_UDP01 - CAAGCAGAAGACGGCATACGAGATCGCTCAGTGTGACTGGAGTTCAGACGTGTGC
HTO_Nextera_i7_UDP02 - CAAGCAGAAGACGGCATACGAGATTATCTGACGTGACTGGAGTTCAGACGTGTGC
HTO_Nextera_i7_UDP03 - CAAGCAGAAGACGGCATACGAGATATATGAGAGTGACTGGAGTTCAGACGTGTGC
HTO_Nextera_i7_UDP04 - CAAGCAGAAGACGGCATACGAGATCTTATGGAGTGACTGGAGTTCAGACGTGTGC
HTO-N701 - CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTGACTGGAGTTCAGACGTGTGC
HTO-N702 - CAAGCAGAAGACGGCATACGAGATCTAGTACGGTGACTGGAGTTCAGACGTGTGC
The resulting ADT-/HTO-derived and mRNA-derived cDNA libraries were pooled and sequenced. 26 bp of cell barcode and UMI sequences and 91 bp RNA reads were generated with Illumina NovaSeq 6000.
2D Image Processing
For studies examining CD86 and VISTA spatial distribution, we first utilized an established method (He et al., 2019) to eliminate the noise in the images, then the pixel-level segmentation results were achieved by taking thresholds on the denoised images. Next, clustered nuclei were identified based on DBSCAN (Ester et al.) as the tumor regions, and the distal regions were defined as the regions which are 10+ μm away from the tumor regions. The pixel-level segmentation results along with the identified tumor and naive regions were reviewed by experts to ensure correctness.
3D Image Processing
All 3D image data was visualized using the volume or surfaces tool in Imaris software (Bitplane). For 3D image dataset processing, briefly, in the first step, we conducted image segmentation of tumors and microglia in 3D image stacks using the U-Net model (Ronneberger et al., 2015). To ensure segmentation accuracy, we selected and annotated the most representative 2D image slices in every 3D image and used the annotated 2D slices to train the U-Net model. Our training dataset contained 53 2D slices for glia cells and 98 slices for tumors. The U-Net segmentation produced voxel-level segmentation but did not group cell voxels into individual cells. To further acquire accurate instance-level segmentation of glial cells, we applied the method as previously described, based on the k-terminal cut algorithm (Yang et al., 2016), to disentangle connected glial cells in the U-Net segmentation into individual cells. Finally, all the results thus computed were verified by human experts to ensure correctness.
QUANTIFICATION AND STATISTICAL ANALYSIS
3D Image Analysis
MATLAB (Mathworks) was used to extract eight morphological features of each segmented microglia cell presented in Figure 1. Features were correlated with the distances between microglia and tumors. These distances were defined as the distances between the cell centers (the geometric center of each segmented cell body) and the tumor boundaries and were computed by the knn search function in MATLAB. Microglia cell bodies, protrusions, and total volumes were computed by multiplying the total number of voxels in each segmented cell by the voxel volume. The roundness of the microglia cell bodies was computed using MitoLoc (Vowinckel et al., 2015). The microglia cell density was estimated by the distance between each target microglia cell and its nearest neighboring microglia cell (as presented as distance to nearest neighbor in main figure), and alternatively by counting the numbers of the microglia cell bodies in 80μm × 80μm × 80μm cubes (as presented in supplemental figure). To quantitatively analyze microglia protrusions, we modeled each segmented microglia as a 3D grid graph and computed the shortest path tree from the cell center in this graph and then extracted protrusion tips by finding the voxels with the local maximal distances. To reduce false positives in our detection, we removed all the detected protrusion tips that were within 5 voxels to another longer protrusion. After that, the number of protrusions was computed as the number of protrusion tips and the lengths of protrusions were computed as the lengths of the shortest paths from the cell center to the protrusion tips along the graph. Finally, the average width of protrusions was computed by dividing the protrusion volume with the total protrusion length. To assign morphology scores, seven numeric features were measured to describe each cell. Six of these features (that is, all features except roundness) have heavily right-skewed distributions and thus were log(x+1) transformed. Further, many of these features are correlated, and thus principal component analysis was applied to create independent features. The first four principal components explain 99% of the variance in the data. Projecting data to these principal component directions, we have found that while the cells show no groupings in principal components 2 or 4, they form two distinct clusters in the space spanned by principal components 1 and 3, and these two clusters can be well separated by a straight line. Then a score was given to each cell. The size of the score was proportional to its distance to the separation line, and the sign of the score was determined by the side of the separation line the cell is on, which is positive for cancer-like cells and negative for non-cancer-like cells. Finally, the positive scores were scaled to range from 0 to 1, and the negative scores were scaled to range from −1 to 0. Differences in morphological features were analyzed for statistical significance using a two-tailed student’s T test. P values <.05 were considered statistically significant. 3D image data analysis was performed blinded.
2D Image Analysis
In analyses examining cell number after cell CNS native myeloid or BMDM depletion, CD3+ T cell numbers with dual immune checkpoint therapy, or CD3+ T cell spatial localization and contact with Iba1+ myeloid cells, numbers of cells were counted either by using particle tool in ImageJ or FIJI or manual counting in ImageJ or FIJI (Schindelin et al., 2012; Schneider et al., 2012). Ki67 was assessed manually by performing an H-score. In all cases, statistical significance was assessed by using a two-tailed student’s T test and performed and visualized using GraphPad Prism (GraphPad Software). P values <.05 were considered statistically significant. 2D image data image analysis related to CCR2 experiments, CD86 IF, and VISTA IF were performed blinded, while all other 2D image data was not performed blinded.
Metastasis Burden and Size Analysis
mCherry+ br-mets were counted manually immediately upon extraction of brain from mice underneath a fluorescence stereo microscope. Metastasis size was quantified from fluorescent gross brain images by manually encircling metastases from a dorsal view of the brain in ImageJ or FIJI and subsequently using the analyze – measure command. Metastasis number and size were analyzed for statistical significance using a two-tailed student’s T test or, when appropriate, two-tailed paired student’s T test. P values <.05 were considered statistically significant. Statistical analyses were performed and visualized using GraphPad Prism (GraphPad Software). Br-met counts were not performed blinded.
Flow Cytometry and CyTOF Analysis
Flow cytometry and CyTOF data were gated, analyzed, and visualized using FlowJo software (BD) or Cytobank Premium (Beckman Coulter). Gating strategies are provided in main and supplemental figures of this paper. Cytobank applies the Barnes-Hut implementation of tSNE. Detailed descriptions of dimensionality reduction methods (viSNE and SPADE) applied by Cytobank, as well as references to the original publications establishing these methods, can be found at: https://support.cytobank.org/hc/en-us. Flow cytometry and CyTOF data were analyzed using a two-tailed student’s T test. P values <.05 were considered statistically significant. Statistical analyses were performed and visualized using GraphPad Prism (GraphPad Software). Flow cytometry and CyTOF analysis was not performed blinded.
CITE-seq Analysis
The raw base sequence calls generated from the sequencer were demultiplexed into sample-specific mRNA, ADT, and HTO FASTQ files with bcl2fastq through CellRanger 3.1.0 (10x Genomics). Raw FASTQ files were processed using Cellranger 3.1.0. We performed most filtering/normalization/scaling and dimensional reduction, clustering, and differential expression analyses using the R Studio and R package Seurat_3.1.1 (Ihaka and Gentleman, 1996; Satija et al., 2015). First, samples were demultiplexed into their sample of origin by the HTODemux command, which also removes multiplets. Next, RNA expression matrices were normalized and scaled using the SCTransform wrapper in Seurat. Low quality cells were filtered from the data by regressing on percent of mitochondria genes in this step. Normalized, scaled, and filtered data was then analyzed for significant principal components, underwent UMAP or tSNE dimensional reduction analysis, clustered, and analyzed for differentially expressed genes using Seruat. Inner workings of Seurat, including dimensionality reduction techniques, can be found on the following websites: https://rdrr.io/cran/Seurat/ and https://satijalab.org/seurat/. Most CITE-seq data analyzed by Wilcoxon rank sum test, with exceptions indicated in figure legends. Adjusted p values <.05 were considered statistically significant. GSVA (Hänzelmann et al., 2013) was performed on all high quality single cells, and results were uploaded into R studio and differential expression analyzed with Seurat package. SCENIC (Aibar et al., 2017) and Dyno trajectory analysis (Saelens et al., 2019) related to myeloid cell convergence was performed on subsets of cells with the most direct trajectories to their respective convergent points and cells within the convergent points. Dyno trajectory analysis was performed on selected cells as described here: https://dynverse.org/dyno/. CITE-seq analyses were not performed blinded.
Human scRNA-seq and CyTOF Data Analysis
Human scRNAseq datasets were derived from two previous publications (Geirsdottir et al., 2019; Laughney et al., 2020) and accessed through Gene Expression Omnibus (metastasis data sets: GSE123904; naive data sets: GSE134707). IBA1+ myeloid cells in each dataset were first identified as microglia (TREM2+, HEXB+, and/or JUN+ and ITGA4− and CCR2−) or BMDM (ITGA4+ and/or CCR2+). These genes were selected for identifying microglia and BMDM based on canonical definitions and because reads for these genes were present across all datasets. Microglia were used for further analysis as BMDM were too low in abundance across datasets for analysis. Individual patient datasets (6 total; 3 metastasis, 3 naïve) were combined and analyzed as one combined object in Seurat. Prior to differential gene expression analyses, technical variation was removed with Seurat’s sctransform wrapper. All human data was analyzed by Wilcoxon Rank Sum test. Adjusted p values <.05 were considered statistically significant. Human CyTOF data was derived from a previous publication (Friebel et al., 2020) and accessed through Mendeley Data (https://data.mendeley.com/datasets/jk8c3c3nmz/draft?a=c0a9d8dc-8ac2-4942-baf9-208de7a8c310). Data were uploaded to FlowJo and gated on singlets and live cells before immune cell gating, as described in Figure S2E. Proportions were derived by dividing the number of cells within the indicated population by all identified immune cells within the sample. Human scRNA-seq and CyTOF data were not analyzed blinded.
Supplementary Material
Figure S1. Myeloid Cells Respond to Br-mets, Related to Figure 1
A) IF of myeloid cells in human br-mets of indicated primary tumor origins.
B) IHC of myeloid cells in CMT.167 br-mets.
C) IF of CNS-myeloid (TMEM119+IBA1+) and BMDM (IBA1+TMEM119−) at different stages of br-mets.
D) Chart showing the ratio of the area of a br-met occupied by CNS-myeloid to that of BMDM.
Each dot represents the quantification for one br-met of the indicated area.
E) Quantification of the number of myeloid cells per volume in the naive brain and br-mets-burdened brain. Each dot represents one mouse (naive, n =3) or different z stack within a br-met (br-mets, n = 2 mice). Data analyzed by two tailed student’s t-test. Error bars represent SEM, center represents mean.
F) Manual gating strategy used to identify immune populations presented Figure 1J.
G) tSNE plots of indicated immune cell marker and functional marker expression in immune cells in the br-mets-burdened brain and the naive brain.
H) Manual gating strategy used to identify immune populations in human br-met CyTOF data presented in panel I.
I) Stacked bar charts of immune proportions in human br-met samples of indicated primary tumor origin.
Figure S2. CITE-seq Antibody-based Gating Strategy and DAM Gene Signature, Related to Figure 2
A) CITE-seq gating strategy for brain leukocytes.
B) CITE-seq gating strategy for blood leukocytes.
C) Proportions of naive or br-met-associated CNS-myeloid (left) or BMDM (right) contributing to each transcriptional cluster pictured in Figure 2D and 2G.
D) Violin plots of DAM stage 1 and stage 2 gene expression in CNS-myeloid split by sample origin (naive brain or br-met-burdened brain).
Data in A-D derived from pooled analysis of three biological replicates from each group.
Figure S3. BMDM are Heterogeneous and Distinct from their Naive Counterparts, Related to Figure 2
A) Joint analysis of RNA and cell surface marker expression in BMDM. Green: RNA expression; Red: Protein expression detected by CITE-antibodies.
B) Heatmap of M1/M2 polarization gene expression among Br.MAM.
C-E) Analysis of Ly6CLo (C), Ly6CHi (D), and Ly6C+Ly6G+ Neutrophil (E) BMDM subpopulations. UMAPs are split by condition (br-met or naive) and cells are color coded by transcriptional cluster. The associated stacked bar charts show proportion of transcriptional clusters within each condition. Heatmaps show the top differentially expressed genes among transcriptional clusters, with genes in large font being marker genes of a particular canonical cell subtype. Volcano plots show genes differentially expressed between the br-met and naive state within the indicated BMDM subset.
All data derived from pooled analysis of three biological replicates from each group.
Figure S4. Cx3cr1 Expression Among Leukocytes, Related to Figure 5
A) Stacked bar chart quantifying proportion of indicated splenocyte populations expressing ZsGreen reporter in Cx3cr1CreERT/+ROSAZsGreen/+ mice as determined by flow cytometry. Proportions represent the average of three biological replicates.
B) Gating strategy for data presented in (A).
C) Histograms of surface Cx3cr1 expression in brain infiltrating leukocytes in the naive and br-met-burdened brain as determined by CyTOF. Graphs representative of three biological replicates.
Figure S5. General Myeloid and CNS-myeloid Depletion Reduces Br-mets, Related to Figure 5
A) Ki67 IHC of br-mets of control and monocyte-depleted mice, scale bar = 40μm (left), and Ki67 H-score quantification of br-met cells in control and myeloid-depleted mice (each dot represents the cumulative H score of all br-mets examined within one mouse).
B) Timeline for early myeloid cell depletion experiment.
C) IF of myeloid cells in control mice and early myeloid-depleted mice three days following DT administration (top) and of myeloid cells at the experimental endpoint, scale bar = 100μm. (bottom).
D) Quantification of E0771 br-met number between control and early myeloid-depleted mice (each dot represents the total number of br-mets counted in one mouse; Experiment performed once).
E) Expression of RFP in indicated BMDM subsets in CCR2 mouse model. Proportions represents average of six biological replicates.
F) Representative cryo-immunofluorescence images of CCR2+ BMDM associated with E0771 br-mets in CCR2+/− mice and CCR2−/− mice, scale bar = 50μm.
G) Quantification of RFP+ BMDM associated with br-mets of CCR2+/− and CCR2−/− mice derived from cryo-immunofluorescence (each dot represents counts derived from one 40x FOV. All points derived from four mice in CCR2+/− group and three mice in CCR2−/− group).
H) Kaplan-Meier plots illustrating the survival in CCR2+/− and CCR2−/− mice with br-met.
I) Ki67 IHC of br-mets in CCR2+/− and CCR2−/− mice (left) and associated Ki67 H-score quantification (each dot represents the cumulative H score of all br-mets within one mouse), scale bar = 50μm (right).
J) Stacked bar charts showing immune cell proportions in the blood (left) and brain (right) of CNS-myeloid mice versus control mice as determined by CITE-seq. Blood data is derived from one mouse from each experimental condition and brain data is the combined analysis from two biological replicates from each experimental condition.
K) Volcano plot showing genes differentially expressed between remnant CNS-myeloid following depletion versus CNS-myeloid from non-depleted mice during br-met.
L) UMAPs of CD8+ T cells split by condition (CNS-myeloid depleted or non-depleted) and color coded by T cell activation status.
M) Feature plots showing the expression of T cell exhaustion markers.
N) Stacked bar charts showing CD8+ T cell activation status in the brains of CNS-myeloid cell-depleted mice versus non-depleted mice as determined by CITE-seq.
Data is the combined analysis from two biological replicates from each condition. Data in panels K, L, M, and N is the analysis from two biological replicates from each condition. Data in panels A, D, G, H, and I analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean. Differential expression is panel K analyzed by Wilcoxon rank sum test.
Figure S6. Cx3cr1 KO Promotes CNS-myeloid Interferon Response, Related to Figure 6
A) Schematic of Cx3cr1−/− mouse model for the genetic ablation of Cx3cr1.
B) Stacked bar charts of immune cell proportions in the blood (far left) and brain (far right) of Het and KO mice during naive state and br-mets. Each plot is the average of two mice.
C) Feature plots showing the expression and expression overlap of Ccr2, Ly6C, and Cx3cr1.
D) Representative biaxial plots showing GFP expression in Ly6CLo BMDM in non-BMT KO mice and BMT KO mice (top), and quantification of percent reconstitution among indicated immune cell populations in Het BMT and KO BMT mice (bottom).
E) Silhouettes of representative Cx3cr1+/− and Cx3cr1−/− myeloid cells in the naive brain (left), and associated violin plots comparing morphological features of myeloid cells in the brains of naive Cx3cr1+/− (Het) and Cx3cr1−/− (KO) mice.
F) Split violin plot showing Cxcl10 expression by genotype among indicated myeloid cell populations.
G) Volcano plot of genes differentially expressed between KO and Het BMDM during br-met. Plot based on pooling two replicates from each condition.
H) Cxcl10 RNA-ISH staining in myeloid cells in human br-met samples in br-met derived from various origins.
I) MTT assay quantification of in vitro treatment of E0771.Br cells with rCxcl10 (each dot represents 1 well; quantification based on 1 experiment).
J) Histograms showing Cxcr3 expression in indicated cell populations (based on 1 experiment). Data in panels E and I analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean.
Figure S7. Immune Checkpoint Expression in the Br-met Niche, Related to Figure 7
A) SPADE plots of indicated median marker expression (top) and fold change marker expression (bottom) in brain leukocytes in rCxcl10 co-injected br-met relative to control br.met.
B) tSNE plots highlighting CD86+ Br.MAM population.
C) Volcano plot showing DEGPs in between CD86+ Br.MAM and all other immune cells.
D) Violin plots of negative immune checkpoint gene expression among all br-met niche cells.
E) Br-met number quantification of mice treated with αPD-L1 alone (left) or αVISTA alone (right) compared to their respective controls. αPD-L1 single treatment performed one time; αVISTA single treatment performed three times.
F) Violin plots of ribosomal gene expression in CD8+ T cells (top) and CD4+ T cells (bottom) in control and αVISTA + αPD-L1 treated mice relative to controls.
G) Volcano plot showing DEGs between CNS-myeloid (left) or BMDM (right) with αVISTA + αPD-L1 compared to control.
Data in panel E analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean. Data in F and G analyzed by Wilcoxon rank sum test.
Table S1. Human Tissue Info, Related to Figure 1 and 6
Tab 1) Clinical information related to human br-met tissue array.
Tab 2) Clinical information related to normal human brain tissue array present in Figure 6L and 6M.
Tab 3) Clinical information related to normal human brain tissue array present in Figure 6M.
Table S2. CNS-myeloid DEGs and DEGPs, Related to Figure 2
Tab 1) DEGs of each transcriptional cluster of CNS-myeloid presented in Figure 2D.
Tab 2) DEGs of br-met-associated CNS-myeloid relative to naive CNS-myeloid presented in Figure 2E.
Tab 3) DEGPs of br-met-associated CNS-myeloid relative to naive CNS-myeloid presented in Figure 2F.
Table S3. BMDM DEGs and DEGPs, Related to Figure 2
Tab 1) DEGs of each transcriptional cluster of BMDM presented in Figure 2G.
Tab 2) DEGs of br-met-associated BMDM relative to naive BMDM presented in Figure 2H.
Tab 3) DEGPs of br-met-associated BMDM relative to naive BMDM presented in Figure 2I.
Table S4. Human CNS-myeloid DEGs, Related to Figure 3
DEGs of human br-met-associated CNS-myeloid relative to naive CNS-myeloid presented in Figure 3.
Table S5. CNS-myeloid vs BMDM DEGs and DEGPs, Related to Figure 4:
Tab 1) DEGs of br-met-associated CNS-myeloid relative to br-met-associated BMDM presented in Figure 4B.
Tab 2) DEGPs of br-met-associated CNS-myeloid relative to br-met-associated BMDM presented in Figure 4C.
Table S6: KO vs Het Differentially DEGs and DEGPs, Related to Figure 6:
Tab 1) DEGPs of KO CNS-myeloid relative to Het CNS-myeloid during br-mets presented in Figure 6G.
Tab 2) DEGs of KO CNS-myeloid relative to Het CNS-myeloid during br-mets presented in Figure 6H.
Highlights.
Myeloid cells in brain metastases are compositionally and transcriptionally diverse
CNS-native myeloid cells (CNS-myeloid) are key myeloid promoters of brain metastasis
Cxcl10 mediates recruitment of immune-suppressive CNS-myeloid to brain metastasis
Blocking VISTA and PD-L1 signaling reduces brain metastasis outgrowth
ACKNOWLEDGEMENTS
This work was funded by NIH R01 CA194697-01 (S.Z.), NIH R01 CA222405 - 01A1 (S.Z.), Notre Dame CRND Catalyst Award (S.Z. and I.H.G.), NIH CTSI core facility pilot grants (S.Z.), NSF CCF-1617735 (D.Z.C.). We would like to acknowledge and thank the Dee Family endowment (S.Z.). For insightful discussion and technical assistance, we thank Zhang Lab members, Vytas Bindokas, Ph.D., Samuel W. Brady, Ph.D., Kevin Brown, Ph.D., Andrea Gunawan, M.S., Harikrishna Nakshatri, Ph.D., and Charles R. Tessier, Ph.D. We are grateful for the use of the following core facilities: Notre Dame Genomics and Bioinformatics Core Facility, Notre Dame Freimann Life Sciences Center, Harper Cancer Research Institute Biorepository, Indiana University School of Medicine South Bend Imaging and Flow Cytometry Core, University of Chicago Integrated Light Microscopy Facility, Indiana University Simon Cancer Center Core Facility, Michigan State University Genomics Core.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L, Peters S, Arvold ND, Harsh GR, Steeg PS, et al. (2019). Brain metastases. Nat. Rev. Dis. Primer 5, 5. [DOI] [PubMed] [Google Scholar]
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreou KE, Soto MS, Allen D, Economopoulos V, de Bernardi A, Larkin JR, and Sibson NR (2017). Anti-inflammatory Microglia/Macrophages As a Potential Therapeutic Target in Brain Metastasis. Front. Oncol 7, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbenishty A, Gadrich M, Cottarelli A, Lubart A, Kain D, Amer M, Shaashua L, Glasner A, Erez N, Agalliu D, et al. (2019). Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation. PLoS Biol. 17, e2006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, and Barres BA (2018). A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, Dhara S, Simpson K, Gardner EE, Iacobuzio-Donahue CA, et al. (2016). Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 17, 2445–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, and Weiner HL (2018). Microglial signatures and their role in health and disease. Nat. Rev. Neurosci 19, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci 9, 917–924. [DOI] [PubMed] [Google Scholar]
- Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, Rasmussen R, Dwivedi B, Seby S, Wolf SA, et al. (2017). Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 77, 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Amit I, and Schwartz M (2018). Microglial immune checkpoint mechanisms. Nat. Neurosci 21, 779–786. [DOI] [PubMed] [Google Scholar]
- Ester M, Kriegel H-P, and Xu X 1996. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Simoudis E, Han J, and Fayyad U, eds. (United States: AAAI Press; ). pp. 226–231. [Google Scholar]
- Fischer GM, Jalali A, Kircher DA, Lee W-C, McQuade JL, Haydu LE, Joon AY, Reuben A, de Macedo MP, Carapeto FCL, et al. (2019). Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 9, 628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, Regli L, Weller M, Greter M, Tugues S, et al. (2020). Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181, 1626–1642.e20. [DOI] [PubMed] [Google Scholar]
- Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, Balic A, Giladi A, Sheban F, Dutertre C-A, et al. (2019). Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 179, 1609–1622.e16. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol 17, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldner IH, Yang L, Cowdrick KR, Wang Q, Alvarez Barrios WV, Zellmer VR, Zhang Y, Host M, Liu F, Chen DZ, et al. (2016). An Integrative Platform for Three-dimensional Quantitative Analysis of Spatially Heterogeneous Metastasis Landscapes. Sci. Rep 6, 24201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He BP, Wang JJ, Zhang X, Wu Y, Wang M, Bay B-H, and Chang AY-C (2006). Differential reactions of microglia to brain metastasis of lung cancer. Mol. Med. Camb. Mass 12, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Cai D, Zhou J, Luo J, and Chen S-L (2019). Adaptive Weighting Depth-variant Deconvolution of Fluorescence Microscopy Images with Convolutional Neural Network. ArXiv190703217 Cs Eess. [Google Scholar]
- Heindl S, Gesierich B, Benakis C, Llovera G, Duering M, and Liesz A (2018). Automated Morphological Analysis of Microglia After Stroke. Front. Cell. Neurosci 12, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, and Gentleman R (1996). R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat 5, 299–314. [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, Brouland J-P, Iacobuzio-Donahue CA, Brennan C, Tabar V, et al. (2020). Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée V-P, Xie Y, Masilionis I, Carr AJ, Kottapalli S, et al. (2020). Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Guo S, and Stiles JK (2011). The emerging role of CXCL10 in cancer (Review). Oncol. Lett 2, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, and Kang Y (2009). Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem 284, 29087–29096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, null, Scheiwe C, Nessler S, Kunz P, van Loo G, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. [DOI] [PubMed] [Google Scholar]
- Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, Lelios I, Heppner FL, Kipnis J, Merkler D, et al. (2018). High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 599. [DOI] [PubMed] [Google Scholar]
- Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, and Becher B (2019). Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol 4. [DOI] [PubMed] [Google Scholar]
- Norris GT, and Kipnis J (2019). Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med 216, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, and Gan W-B (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, et al. (2017). Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317. [DOI] [PubMed] [Google Scholar]
- Prinz M, and Priller J (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci 15, 300–312. [DOI] [PubMed] [Google Scholar]
- Prinz M, Jung S, and Priller J (2019). Microglia Biology: One Century of Evolving Concepts. Cell 179, 292–311. [DOI] [PubMed] [Google Scholar]
- Pukrop T, Dehghani F, Chuang H-N, Lohaus R, Bayanga K, Heermann S, Regen T, Van Rossum D, Klemm F, Schulz M, et al. (2010). Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 58, 1477–1489. [DOI] [PubMed] [Google Scholar]
- Qiao S, Qian Y, Xu G, Luo Q, and Zhang Z (2019). Long-term characterization of activated microglia/macrophages facilitating the development of experimental brain metastasis through intravital microscopic imaging. J. Neuroinflammation 16, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera LB, and Bergers G (2013). Location, location, location: macrophage positioning within tumors determines pro- or antitumor activity. Cancer Cell 24, 687–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneberger O, Fischer P, and Brox T (2015). U-Net: Convolutional Networks for Biomedical Image Segmentation In Medical Image Computing and Computer-Assisted Intervention – MICCAI 2015, Navab N, Hornegger J, Wells WM, and Frangi AF, eds. (Cham: Springer International Publishing; ), pp. 234–241. [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, and Saeys Y (2019). A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, et al. (2014). Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol 16, 876–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, Yokoyama C, Onoe H, Eguchi M, Yamaguchi S, et al. (2014). Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739. [DOI] [PubMed] [Google Scholar]
- Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et al. (2018). Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N. Engl. J. Med 379, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vowinckel J, Hartl J, Butler R, and Ralser M (2015). MitoLoc: A method for the simultaneous quantification of mitochondrial network morphology and membrane potential in single cells. Mitochondrion 24, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler F (2015). The brain metastatic niche. J. Mol. Med. Berl. Ger 93, 1213–1220. [DOI] [PubMed] [Google Scholar]
- Wolf Y, Yona S, Kim K-W, and Jung S (2013). Microglia, seen from the CX3CR1 angle. Front. Cell. Neurosci 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, et al. (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med 211, 1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhang Y, Guldner IH, Zhang S, and Chen DZ (2016). 3D Segmentation of Glial Cells Using Fully Convolutional Networks and k-Terminal Cut In Medical Image Computing and Computer-Assisted Intervention – MICCAI 2016, Ourselin S, Joskowicz L, Sabuncu MR, Unal G, and Wells W, eds. (Cham: Springer International Publishing; ), pp. 658–666. [Google Scholar]
- Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang W-C, Li P, Li M, Wang X, Zhang C, et al. (2015). Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Myeloid Cells Respond to Br-mets, Related to Figure 1
A) IF of myeloid cells in human br-mets of indicated primary tumor origins.
B) IHC of myeloid cells in CMT.167 br-mets.
C) IF of CNS-myeloid (TMEM119+IBA1+) and BMDM (IBA1+TMEM119−) at different stages of br-mets.
D) Chart showing the ratio of the area of a br-met occupied by CNS-myeloid to that of BMDM.
Each dot represents the quantification for one br-met of the indicated area.
E) Quantification of the number of myeloid cells per volume in the naive brain and br-mets-burdened brain. Each dot represents one mouse (naive, n =3) or different z stack within a br-met (br-mets, n = 2 mice). Data analyzed by two tailed student’s t-test. Error bars represent SEM, center represents mean.
F) Manual gating strategy used to identify immune populations presented Figure 1J.
G) tSNE plots of indicated immune cell marker and functional marker expression in immune cells in the br-mets-burdened brain and the naive brain.
H) Manual gating strategy used to identify immune populations in human br-met CyTOF data presented in panel I.
I) Stacked bar charts of immune proportions in human br-met samples of indicated primary tumor origin.
Figure S2. CITE-seq Antibody-based Gating Strategy and DAM Gene Signature, Related to Figure 2
A) CITE-seq gating strategy for brain leukocytes.
B) CITE-seq gating strategy for blood leukocytes.
C) Proportions of naive or br-met-associated CNS-myeloid (left) or BMDM (right) contributing to each transcriptional cluster pictured in Figure 2D and 2G.
D) Violin plots of DAM stage 1 and stage 2 gene expression in CNS-myeloid split by sample origin (naive brain or br-met-burdened brain).
Data in A-D derived from pooled analysis of three biological replicates from each group.
Figure S3. BMDM are Heterogeneous and Distinct from their Naive Counterparts, Related to Figure 2
A) Joint analysis of RNA and cell surface marker expression in BMDM. Green: RNA expression; Red: Protein expression detected by CITE-antibodies.
B) Heatmap of M1/M2 polarization gene expression among Br.MAM.
C-E) Analysis of Ly6CLo (C), Ly6CHi (D), and Ly6C+Ly6G+ Neutrophil (E) BMDM subpopulations. UMAPs are split by condition (br-met or naive) and cells are color coded by transcriptional cluster. The associated stacked bar charts show proportion of transcriptional clusters within each condition. Heatmaps show the top differentially expressed genes among transcriptional clusters, with genes in large font being marker genes of a particular canonical cell subtype. Volcano plots show genes differentially expressed between the br-met and naive state within the indicated BMDM subset.
All data derived from pooled analysis of three biological replicates from each group.
Figure S4. Cx3cr1 Expression Among Leukocytes, Related to Figure 5
A) Stacked bar chart quantifying proportion of indicated splenocyte populations expressing ZsGreen reporter in Cx3cr1CreERT/+ROSAZsGreen/+ mice as determined by flow cytometry. Proportions represent the average of three biological replicates.
B) Gating strategy for data presented in (A).
C) Histograms of surface Cx3cr1 expression in brain infiltrating leukocytes in the naive and br-met-burdened brain as determined by CyTOF. Graphs representative of three biological replicates.
Figure S5. General Myeloid and CNS-myeloid Depletion Reduces Br-mets, Related to Figure 5
A) Ki67 IHC of br-mets of control and monocyte-depleted mice, scale bar = 40μm (left), and Ki67 H-score quantification of br-met cells in control and myeloid-depleted mice (each dot represents the cumulative H score of all br-mets examined within one mouse).
B) Timeline for early myeloid cell depletion experiment.
C) IF of myeloid cells in control mice and early myeloid-depleted mice three days following DT administration (top) and of myeloid cells at the experimental endpoint, scale bar = 100μm. (bottom).
D) Quantification of E0771 br-met number between control and early myeloid-depleted mice (each dot represents the total number of br-mets counted in one mouse; Experiment performed once).
E) Expression of RFP in indicated BMDM subsets in CCR2 mouse model. Proportions represents average of six biological replicates.
F) Representative cryo-immunofluorescence images of CCR2+ BMDM associated with E0771 br-mets in CCR2+/− mice and CCR2−/− mice, scale bar = 50μm.
G) Quantification of RFP+ BMDM associated with br-mets of CCR2+/− and CCR2−/− mice derived from cryo-immunofluorescence (each dot represents counts derived from one 40x FOV. All points derived from four mice in CCR2+/− group and three mice in CCR2−/− group).
H) Kaplan-Meier plots illustrating the survival in CCR2+/− and CCR2−/− mice with br-met.
I) Ki67 IHC of br-mets in CCR2+/− and CCR2−/− mice (left) and associated Ki67 H-score quantification (each dot represents the cumulative H score of all br-mets within one mouse), scale bar = 50μm (right).
J) Stacked bar charts showing immune cell proportions in the blood (left) and brain (right) of CNS-myeloid mice versus control mice as determined by CITE-seq. Blood data is derived from one mouse from each experimental condition and brain data is the combined analysis from two biological replicates from each experimental condition.
K) Volcano plot showing genes differentially expressed between remnant CNS-myeloid following depletion versus CNS-myeloid from non-depleted mice during br-met.
L) UMAPs of CD8+ T cells split by condition (CNS-myeloid depleted or non-depleted) and color coded by T cell activation status.
M) Feature plots showing the expression of T cell exhaustion markers.
N) Stacked bar charts showing CD8+ T cell activation status in the brains of CNS-myeloid cell-depleted mice versus non-depleted mice as determined by CITE-seq.
Data is the combined analysis from two biological replicates from each condition. Data in panels K, L, M, and N is the analysis from two biological replicates from each condition. Data in panels A, D, G, H, and I analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean. Differential expression is panel K analyzed by Wilcoxon rank sum test.
Figure S6. Cx3cr1 KO Promotes CNS-myeloid Interferon Response, Related to Figure 6
A) Schematic of Cx3cr1−/− mouse model for the genetic ablation of Cx3cr1.
B) Stacked bar charts of immune cell proportions in the blood (far left) and brain (far right) of Het and KO mice during naive state and br-mets. Each plot is the average of two mice.
C) Feature plots showing the expression and expression overlap of Ccr2, Ly6C, and Cx3cr1.
D) Representative biaxial plots showing GFP expression in Ly6CLo BMDM in non-BMT KO mice and BMT KO mice (top), and quantification of percent reconstitution among indicated immune cell populations in Het BMT and KO BMT mice (bottom).
E) Silhouettes of representative Cx3cr1+/− and Cx3cr1−/− myeloid cells in the naive brain (left), and associated violin plots comparing morphological features of myeloid cells in the brains of naive Cx3cr1+/− (Het) and Cx3cr1−/− (KO) mice.
F) Split violin plot showing Cxcl10 expression by genotype among indicated myeloid cell populations.
G) Volcano plot of genes differentially expressed between KO and Het BMDM during br-met. Plot based on pooling two replicates from each condition.
H) Cxcl10 RNA-ISH staining in myeloid cells in human br-met samples in br-met derived from various origins.
I) MTT assay quantification of in vitro treatment of E0771.Br cells with rCxcl10 (each dot represents 1 well; quantification based on 1 experiment).
J) Histograms showing Cxcr3 expression in indicated cell populations (based on 1 experiment). Data in panels E and I analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean.
Figure S7. Immune Checkpoint Expression in the Br-met Niche, Related to Figure 7
A) SPADE plots of indicated median marker expression (top) and fold change marker expression (bottom) in brain leukocytes in rCxcl10 co-injected br-met relative to control br.met.
B) tSNE plots highlighting CD86+ Br.MAM population.
C) Volcano plot showing DEGPs in between CD86+ Br.MAM and all other immune cells.
D) Violin plots of negative immune checkpoint gene expression among all br-met niche cells.
E) Br-met number quantification of mice treated with αPD-L1 alone (left) or αVISTA alone (right) compared to their respective controls. αPD-L1 single treatment performed one time; αVISTA single treatment performed three times.
F) Violin plots of ribosomal gene expression in CD8+ T cells (top) and CD4+ T cells (bottom) in control and αVISTA + αPD-L1 treated mice relative to controls.
G) Volcano plot showing DEGs between CNS-myeloid (left) or BMDM (right) with αVISTA + αPD-L1 compared to control.
Data in panel E analyzed by two-tailed student’s t test, error bars represent SEM, center represents mean. Data in F and G analyzed by Wilcoxon rank sum test.
Table S1. Human Tissue Info, Related to Figure 1 and 6
Tab 1) Clinical information related to human br-met tissue array.
Tab 2) Clinical information related to normal human brain tissue array present in Figure 6L and 6M.
Tab 3) Clinical information related to normal human brain tissue array present in Figure 6M.
Table S2. CNS-myeloid DEGs and DEGPs, Related to Figure 2
Tab 1) DEGs of each transcriptional cluster of CNS-myeloid presented in Figure 2D.
Tab 2) DEGs of br-met-associated CNS-myeloid relative to naive CNS-myeloid presented in Figure 2E.
Tab 3) DEGPs of br-met-associated CNS-myeloid relative to naive CNS-myeloid presented in Figure 2F.
Table S3. BMDM DEGs and DEGPs, Related to Figure 2
Tab 1) DEGs of each transcriptional cluster of BMDM presented in Figure 2G.
Tab 2) DEGs of br-met-associated BMDM relative to naive BMDM presented in Figure 2H.
Tab 3) DEGPs of br-met-associated BMDM relative to naive BMDM presented in Figure 2I.
Table S4. Human CNS-myeloid DEGs, Related to Figure 3
DEGs of human br-met-associated CNS-myeloid relative to naive CNS-myeloid presented in Figure 3.
Table S5. CNS-myeloid vs BMDM DEGs and DEGPs, Related to Figure 4:
Tab 1) DEGs of br-met-associated CNS-myeloid relative to br-met-associated BMDM presented in Figure 4B.
Tab 2) DEGPs of br-met-associated CNS-myeloid relative to br-met-associated BMDM presented in Figure 4C.
Table S6: KO vs Het Differentially DEGs and DEGPs, Related to Figure 6:
Tab 1) DEGPs of KO CNS-myeloid relative to Het CNS-myeloid during br-mets presented in Figure 6G.
Tab 2) DEGs of KO CNS-myeloid relative to Het CNS-myeloid during br-mets presented in Figure 6H.
Data Availability Statement
CITE-seq data is uploaded to GEO (GSE134285). All other data and code used to analyze data are available upon reasonable request for Lead Contact/corresponding author.