

Abstract
Introduction:
Lung cancer incidence is higher among African Americans (AAs) compared with European Americans (EAs) in the United States, especially among men. While significant progress has been made profiling the genomic makeup of lung cancer in EAs, AAs continue to be underrepresented. Our objective was to chart the genome-wide landscape of somatic mutations in lung cancer tumors from African Americans.
Methods:
In this study, we used whole exome sequencing (WES) of 82 tumor and non-involved tissue pairs from AAs. Patients were selected from an ongoing case-control study conducted by the NCI and the University of Maryland.
Results:
Among all samples, we identified 178 significantly mutated genes (P <0.05), five of which passed the threshold for false discovery rate (FDR P<0.1). In lung adenocarcinoma (LUAD) tumors, mutation rates in STK11 (P=0.05) and RB1 (P=0.008) were significantly higher in AA LUAD tumors (25% and 13%, respectively) compared with TCGA EA samples (14% and 4%, respectively). In squamous cell carcinomas, mutation rates in STK11 (P=0.002) were significantly higher among AA (8%) than EA tumors from TCGA (1%). Integrated somatic mutation data with CIBERSORT data analysis revealed LUAD tumors from AAs carrying STK11 mutations have decreased interferon signaling.
Conclusions:
While a considerable degree of the somatic mutation landscape is shared between EAs and AAs, discrete differences in mutation frequency in potentially important oncogenes and tumor suppressors exist. Better understanding of the molecular basis of lung cancer in AA patients and leveraging this information to guide clinical interventions may help reduce disparities.
Keywords: lung cancer, NSCLC, whole exome sequencing, health disparity
Introduction
Of all racial and ethnic groups in the United States, African American men have the highest age-adjusted non-small cell lung cancer (NSCLC) incidence rates as well as the highest age-adjusted mortality rates1, 2. These trends persist despite lower tobacco exposure in terms of cigarettes smoked per day among African Americans (AAs) compared with European Americans2. Potential factors associated with these disparities include smoking behaviors—for example, disparities lessen when nicotine intake per cigarette is taken into account3—socioeconomic status, access to health care, and other factors2. In recent years, several large-scale genomic studies have begun to map differences in tumor biology between European Americans (EAs) and AAs4-9. These studies show that while a majority of tumor biology is shared between EAs and AAs, specific, and potentially actionable, differences exist.
Recent improvements in cancer survival have been largely due to advances in our understanding of cancer genomics its translation into targeted therapies. However, these studies have been dominated by research focused on populations of European or Asian descent10-12. Indeed, genomic studies of LUAD in European and Asian populations highlights population heterogeneity. For example, EGFR mutations are found more frequently among Asian populations, and several driver genes with low mutation frequency and specific mutational signatures have been identified10. In recent years, studies using targeted exome sequencing approaches to analyze African American populations have been published. These studies show that much of the somatic mutation landscape of NSCLC is shared between EAs and AAs5, 8, 9, but also that some notable differences can be found, especially in lung adenocarcinoma (LUAD), including JAK2 and PTPRT5. Despite these observations, ancestry differences in NSCLC genomics have yet to be systematically examined with whole exome sequencing (WES) in an AA cohort. A recent study by Lusk et al., conducted WES on a subset of lung tumors without known driver mutations9 the results of which suggested an unbiased WES assessment in an unselected sample set was warranted. To fill this knowledge gap, we used WES on matched tumor and non-involved tissue pairs.
Materials and Methods
Patient samples and DNA extraction
Patients were selected from an ongoing case-control study conducted by the NCI and the University of Maryland. Patients for this study were recruited between 1984 and 2013. At the time of surgery, a portion of the tumor specimen and non-involved adjacent lung tissue was flash frozen and stored at −80°C until needed. Clinical and pathological information was obtained from medical records, tumor boards and pathology reports (Table 1). Never smokers were defined as have smoked less than 100 cigarettes in their lifetime, former smokers were defined as individuals that quit smoking more than one year at the time of interview, while current smokers included individuals that continued to smoke and/or had quit within one year of interview. A participant’s sample was included if that patient was a candidate for surgery, gave informed consent, and, post pathological assessment, there was enough fresh frozen tissue for research analyses. Further, the participant needed to have matched genomic DNA available for comparison and the DNA extracted needed to meet sufficient quality control criteria to be included in the WES analysis.
Table 1:
Demographics of the study cohort (N = 82)
| Characteristics | AA |
|---|---|
| Age (years) | 63.5 ± 8.7 |
| Gender | |
| Female | 18 (22%) |
| Male | 64 (78%) |
| BMI (kg/m2) | |
| Underweight | 5 (6.1%) |
| Normal | 26 (31.7%) |
| Overweight | 19 (23.2%) |
| Obese | 12 (14.6%) |
| Unknown | 20 (24.4%) |
| Education | |
| Low | 26 (31.7%) |
| Medium | 26 (31.7%) |
| High | 6 (7.3%) |
| Unknown | 24 (29.3%) |
| Income | |
| Low | 17 (20.7%) |
| Medium | 27 (32.9%) |
| High | 7 (8.5%) |
| Unknown | 31 (37.8%) |
| Smoking Status | |
| Never | 3 (3.7%) |
| Former | 29 (35.4%) |
| Current | 48 (58.5%) |
| Unknown | 2 (2.4%) |
| Smoking Pack-years | |
| Low | 26 (31.7%) |
| Medium | 23 (28.0%) |
| High | 25 (30.5%) |
| Unknown | 8 (9.8%) |
| Menthol Use | |
| Yes | 26 (31.7%) |
| No | 12 (14.6%) |
| Unknown | 44 (53.7%) |
| Histology | |
| LUAD | 36 (43.9%) |
| LUSC | 39 (47.6%) |
| BAC | 4 (4.9%) |
| Other | 3 (3.7%) |
| Stage | |
| I | 49 (59.8%) |
| II | 23 (28.0%) |
| III | 5 (6.1%) |
| Unknown | 5 (6.1%) |
LUAD denotes lung adenocarcinoma, LUSC denotes lung squamous cell carcinoma, BAC denotes bronchioalveolar carcinoma
Education: low (Elementary school, middle school and 10th or 11th grade); medium (high School or GED, some college, technical school); high (College, professional school)
Income: low (under $15,000); medium ($15,000-60,000); high ($60,000 and above)
Pack-years of smoking: low (under 19 packyears smoked); medium (19.0-36.7 packyears smoked); high (greater than 36.7 packyears smoked)
BMI: underweight (under 18.5 kg/m2); normal (18.5-24.9 kg/m2); overweight (25.0-29.9 kg/m2); obese (greater than 30 kg/m2)
DNA was extracted from fresh, frozen macro-dissected primary lung tumor tissues using the Qiagen DNeasy Blood and Tissue kit spin column procedure, according to the manufacturer’s protocol (Qiagen), as previously described4. Isolated primary lung tumor DNA was initially quantified using a DS-11 spectrophotometer (DeNovix). Subsequent Qubit fluorometer analyses were performed to assess DNA integrity and ensure the presence of intact double-stranded DNA in all samples (Invitrogen). DNA with an A260-to-A280 ratio between 1.8 and 2.0, a minimum concentration of 12 ng μl−1 and a total concentration of 100 ng was used for further analysis.
Whole exome sequencing and data processing
Whole exome sequencing was performed at the Cancer Genomics Research Laboratory, NCI Division of Cancer Epidemiology and Genetics (Gaithersburg, MD). Extracted DNA samples were used for library preparation using the NimbleGen SeqCap EZ Exome capture system with 64 Mb of exonic sequence targeted and the resulting post-capture enriched multiplexed sequencing libraries were used in cluster formation on an Illumina cBOT (Illumina, San Diego, CA, USA) and paired-end sequencing was performed with sequence across AHM5YYBBXX, BHMNLHBBXX, AHMCMTBBXX, BHM7M3BBXX, AHMCJHBBXX, AHMCFYBBXX flowcells on Illumina HiSeq following Illumina-provided protocols for 2x125 bp (HiSeq 2500) or 2x150bp (HiSeq 4000) paired-end sequencing.
Sequence reads were trimmed for adapters and low-quality bases using Trimmomatic software (version 0.33) and then aligned to the human hg19 reference genome using BWA mapping software (version 0.7.15)13. Duplicate reads were marked using Picard Tools, followed by re-alignment and base quality score recalibration using the Genome Analysis Toolkit (GATK) version 3.8.014.
Germline variant and ancestry analysis
Germline variants were called using GATK’s HaplotypeCaller15 in joint genotyping mode. Variants were then filtered for quality with the following criteria: QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < −12.5, ReadPosRankSum < −8.0 for SNPs; QD < 2.0, FS > 200.0, ReadPosRankSum < −20.0 for INDELs. For admixture analysis, INDELs and any SNPs that were not bi-allelic were removed, and the 1000 genomes phase III16 superpopulations were used as reference. We also excluded rare variants (≤0.05 frequency across all phase III 1000 genomes). In order to maximize the genetic comparisons by race, we examined each patient for African genetic ancestry (Methods) (Supplementary Table 1). We then used the tool Admixture v1.3.017 to estimate ancestry proportions for each of the 1000 genomes superpopulations (Supplementary Table 1). One patient that self-reported as AA had greater than 60% European ancestry and was therefore excluded from downstream analyses. Four more patients were also excluded from downstream analyses as two had tumor/non-tumor tissue mismatch pairs and the remaining two only had tumor tissue available. Thus, in total, the final study cohort consisted of 82 tumor-non-tumor tissue matched pairs.
Somatic variant analysis
Somatic variant calling was performed using muTect (v1.1.7)18, MuTect219, and Strelka (v2.9.0)20 in tumor-normal mode. Mutations called with at least two of these programs were considered in our study. Annotation of variants was performed using Ensembl’s Variant Effect Predictor (VEP) version 9221 and converted to Mutation Annotation Format (MAF) using the vcf2maf tool version 1.6.1622. A final set of somatic variants were generated using the following stringent filtering criteria. Commonly occurring variants annotated with a frequency of greater than 0.001 in the ExAC, gnomAD, or 1000 Genomes databases were excluded. Additional filtering steps to keep high quality variants included: 1) mutant allele frequency greater 0.05 in the tumor sample, 2) a count/depth of the mutant allele in non-tumor sample less than two; 3) a count/depth of the mutant allele in tumor sample greater than four; and 4) a total tumor sequencing depth greater than 100X. Finally, prior to all downstream analyses, variants in frequently mutated genes in exome data that are likely false positives were also removed23. Tumor mutation burden was defined as the number of somatic mutations in the coding region (Supplementary Table 2) and calculated as the total number of mutation counts divided by the size of the coding sequence region (64 Mb) of the NimbleGen SeqCap EZ Exome capture system. Mutation significance was performed using the MutSigCV algorithm24. The current version improves the background mutation rate estimation by pooling data from 'neighbor' genes in the covariate space and substantially reduces the number of false-positive findings. Tables with mutation data, per-sample coverage, gene covariables and mutation type were imported to the software. Genes with a Bonferroni-corrected p-value less than 0.1 were considered statistically significant (Supplementary Table 3).
TCGA data
Somatic mutation data for the TCGA-LUAD and LUSC dataset were retrieved using the TCGA mutations R package25, which provides pre-built objects using MAF files from the MC3 working group26. CIBERSORT data for LUAD and LUSC were extracted from the following paper27.
Mutational signature analysis
Mutational signatures in the targeted sequencing data were analyzed using R/Bioconductor package ‘MutationalPatterns’. The package covers a wide range of tools including mutational signatures, transcriptional and replicative strand bias, genomic distribution and association with genomic features. The reference mutation signatures were obtained from the COSMIC website (https://cancer.sanger.ac.uk/cosmic/signatures) for 65 signatures. These signatures were compared to the pattern of all possible single base substitutions in each sample independently. Etiologies were parsed from the HTML pages programmatically, the text was clustered, and then manually assigned an etiology based on the clustered descriptions. Cosine similarity is used as the comparison metric in Figure 3. For Supplementary Figure 4, the contribution of each known signature was computed as the optimal linear combination of mutational signatures that most closely reconstructs the mutation matrix for each sample28.
Data availability
The datasets generated during the current study have been uploaded to the dbGaP repository in compliance with the NIH Genomic Data Sharing Policy. Data can be accessed at dbGap study ID phs001895.
Results
Study cohort and estimation of genetic ancestry
We conducted WES on genomic DNA from 82 AA patients using tumor and non-tumor matched pairs. Most of these samples were lung squamous cell carcinomas (LUSC) (47.6%) and adenocarcinomas (LUAD) (43.9%). The median African ancestry was 85% (Supplementary Table 1). There were 64 males and 18 females with a mean age of 63.5 years. Over 58% and 3.7% of the patients were current and never smokers, respectively (Table 1).
Somatic mutation landscape in NSCLC from African Americans
In total, we detected 141,209 single nucleotide variants (SNV) and insertion and deletion events (Supplementary Table 1). The most common mutation type was a non-synonymous base change, consistent with previous reports11, 12 (Supplementary Table 4) (Supplementary Figure 1). The mutation burden per sample ranged from 0.02 to 59 per megabase (Mb) (median=3.2) and was consistent with previous studies of NSCLC (Supplementary Figure 1) (Supplementary Figure 2) (Supplementary Table 4). Using a recent definition for hypermutation (greater than 10 somatic single-nucleotide variants/Mb)29, there were 11 samples classified as “hypermutated”. One patient, a current smoker, had over 40 mutations/Mb. Known DNA repair genes, such as MSH3 and ERCC4, were among the mutated genes in this sample (Supplementary Table 2). The Ti/Tv ratios were as expected, with the most common base substitution bias toward cytosine (C) > adenine (A) transversions followed by cytosine (C) > thymine (T) transitions (Supplementary Figure 1), both of which are associated with exposure to cigarette smoking30.
Identification and characterization of somatic mutations in LUAD and LUSC
We initially used the MutSigCV algorithm to identify significantly mutated genes given the higher background mutation rate in NSCLC. We found 178 significantly mutated genes (p<0.05), five of which (TP53, STK11, RB1, CDKN2A, PIK3CG) had passed the threshold for false discovery rate (FDR P<0 .1) (Figure 1A, 1B) (Table 2) (Supplementary Table 3). TP53 was the most mutated gene with similar frequency in AAs and EAs (using TCGA data as reference) (Table 2). Given evidence for ethnicity-related EGFR mutations and that patients with exon 19 deletion and L858R mutation have a lower response rate to immunotherapy and greater response to receptor tyrosine kinase inhibitors31, we looked for the presence of EGFR mutations in the AA population and found one patient, a former smoker, with an E746-A750 deletion at exon 19 deletion. All KRAS mutations targeted codon 12 and 13 (Supplementary Table 4). Consistent with our previous findings5, the frequency of mutations in PTPRT and JAK2 genes was higher in AA LUAD tumors (17% and 14%, respectively) compared with TCGA EA patients (8% and 2%, respectively) (Table 2) (Supplementary Table 6).
Figure 1:
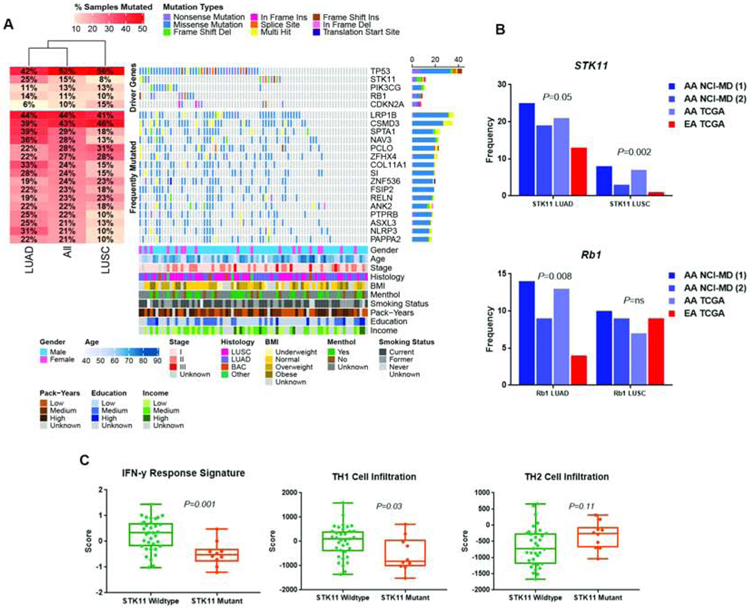
Somatic mutation landscape of lung cancer derived from whole exome sequencing of 82 lung cancers from African Americans. (A) Shown are genes with nonsynonymous and indel mutations of >10% frequencies. Mutant frequencies in the cohort are shown on the right. Demographic and lifestyle exposures are superimposed at the bottom of the oncoplot. (B) Enhanced mutation frequency of STK11 and RB1 in African Americans compared with European Americans. (C) Dysregulated interferon signaling among STK11 mutant tumors and infiltration of Th1 and Th2 cells. P-values determined using two-sided Student’s t tests.
Table 2:
Significant (top panel) and most frequently (bottom panel) mutated genes in lung cancer from African Americans
| AA LUAD | EA LUAD |
AA LUSC | EA LUSC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | NCI-MD [n=82] (AA) |
NCI-MD WES FF [n=36] |
NCI-MD Targeted FF [n=54] |
TCGA WES FF [n=52] |
TCGA WES FF [n=381] |
NCI-MD WES FF [n=39] |
NCI-MD Targeted FF [n=65] |
TCGA WES FF [n=27] |
TCGA WES FF [n=331] |
| TP53 | 0.52 | 0.42 | 0.46 | 0.65 | 0.49 | 0.56 | 0.68 | 0.93 | 0.83 |
| STK11 | 0.15 | 0.25 | 0.19 | 0.21 | 0.13 | 0.08 | 0.03 | 0.07 | 0.01 |
| PIK3CG | 0.13 | 0.11 | 0.06 | 0.10 | 0.06 | 0.13 | 0.14 | 0.07 | 0.09 |
| RB1 | 0.11 | 0.14 | 0.09 | 0.13 | 0.04 | 0.10 | 0.09 | 0.07 | 0.09 |
| CDKN2A | 0.10 | 0.06 | NA | 0.08 | 0.03 | 0.15 | 0.12 | 0.07 | 0.17 |
| LRP1B | 0.44 | 0.44 | 0.31 | 0.40 | 0.32 | 0.41 | 0.34 | 0.19 | 0.36 |
| CSMD3 | 0.43 | 0.39 | 0.35 | 0.52 | 0.38 | 0.46 | 0.42 | 0.56 | 0.43 |
| SPTA1 | 0.29 | 0.39 | NA | 0.35 | 0.23 | 0.18 | NA | 0.15 | 0.22 |
| NAV3 | 0.28 | 0.36 | NA | 0.31 | 0.19 | 0.13 | NA | 0.11 | 0.22 |
| PCLO | 0.28 | 0.22 | NA | 0.27 | 0.16 | 0.31 | NA | 0.11 | 0.18 |
| ZFHX4 | 0.27 | 0.22 | NA | 0.48 | 0.30 | 0.28 | NA | 0.22 | 0.28 |
| COL11A1 | 0.24 | 0.33 | NA | 0.31 | 0.19 | 0.15 | NA | 0.11 | 0.19 |
| SI | 0.24 | 0.28 | NA | 0.23 | 0.15 | 0.15 | NA | 0.11 | 0.18 |
| ZNF536 | 0.24 | 0.19 | NA | 0.29 | 0.20 | 0.23 | NA | 0.26 | 0.12 |
| FSIP2 | 0.23 | 0.22 | NA | NA | NA | 0.18 | NA | NA | NA |
| RELN | 0.23 | 0.19 | NA | 0.19 | 0.15 | 0.23 | NA | 0.15 | 0.18 |
| ANK2 | 0.22 | 0.22 | NA | 0.25 | 0.19 | 0.18 | NA | 0.15 | 0.14 |
| PTPRB | 0.22 | 0.25 | NA | 0.12 | 0.07 | 0.10 | NA | 0.07 | 0.13 |
| ASXL3 | 0.21 | 0.25 | NA | 0.19 | 0.13 | 0.13 | NA | 0.04 | 0.08 |
| NLRP3 | 0.21 | 0.31 | NA | 0.15 | 0.10 | 0.10 | NA | 0.11 | 0.07 |
| PAPPA2 | 0.21 | 0.22 | NA | 0.27 | 0.16 | 0.10 | NA | 0.19 | 0.18 |
| PTPRT | 0.12 | 0.17 | 0.20 | 0.21 | 0.08 | 0.13 | 0.06 | 0.07 | 0.07 |
| JAK2 | 0.11 | 0.14 | 0.09 | 0.06 | 0.02 | 0.10 | 0.09 | 0.04 | 0.03 |
FF denotes fresh frozen, FFPE denotes formalin fixed paraffin-embedded, AA denotes African Americans, EA denotes European Americans, WES denotes whole exome sequencing, LUAD denotes lung adenocarcinoma, LUSC denotes lung squamous cell carcinoma
In LUAD, STK11 was mutated in 25% of LUAD tumors from AA patients in the NCI-MD samples compared with just 13% in TCGA EA samples (two-sample test of proportions P=0.05) (Figure 1B) (Table 2) (Supplementary Table 6). This increased mutation frequency was replicated in another dataset5 and in TCGA (Figure 1B). Similarly, LUSC samples from AAs had a higher STK11 mutation frequency in the NCI-MD tumors (8%) compared with EA tumors from TCGA (1%) (two-sample test of proportions (P=0.002)) (Figure 1B) (Table 2) (Supplementary Table 6). Again, this observation was validated in our previous dataset based on targeted sequencing5 and in TCGA (Table 2) (Figure 1B). Consistent with TCGA data for EAs, the spread of mutations was similar with no evidence of clear hot spots.
STK11 alterations have been identified as the most prevalent genomic driver of primary resistance to PD-1 axis inhibitors in the context of KRAS-mutant lung adenocarcinoma through the generation of an immune cold tumor microenvironment32, 33. While the co-occurrence of KRAS and STK11 alterations (mutation or amplification) is similar in both EAs and AAs (approximately 25%), we found that the majority of AA tumors with a STK11 alteration also carry KRAS mutations or amplifications (6/7). The co-occurrence of STK11 and KRAS variants in AAs was detected in both LUAD and LUSC (Supplementary Table 4). We integrated TCGA LUAD somatic mutation data with CIBERSORT data from the supplementary data of this paper27. As shown in Figure 1C, lung tumors from African Americans carrying STK11 mutations have decreased IFN-γ response signatures, a feature also indicative of a cold tumor microenvironment34. In accordance with decreased IFNγ signaling, we observed decreased Th1 cell infiltration and increased Th2 cell infiltration, with the strongest effects seen in STK11/KRAS altered cells (Supplementary Figure 3). These data suggest that, in African Americans, the somatic alterations in STK11 and KRAS are associated with an immune cold tumor microenvironment through decreased IFNγ dependent signaling, opposed to reduced cytotoxic T cell infiltration. This may be indicative of divergent STK11/KRAS-dependent inflammatory signaling in AAs. The concurrent reduction of IFNγ-dependent signaling as well as a Th2 skewed Th1/Th2 balance suggests not only a reduction in antitumor IFNγ-Th1 dependent signaling, but also an increase in protumor signaling by Th2 cells35.
RB1 and CDKN2A are also mutated at a higher frequency in LUAD among AAs compared with EAs (Figure 1B) (Table 2) (Supplementary Table 6). In AA patients with LUAD, RB1 was mutated at a frequency of 14% compared with 4% in EAs (P=0.008) (Table 2) (Figure 1B). This higher frequency was again confirmed in our other datasets5. CDKN2A mutations were also higher among AAs in this previous dataset, though these differences did not reach statistical significance. As observed in EAs, CDKN2A and RB1 mutations are primarily mutually exclusive in AAs, with 11/36 (31%) LUAD tumors carrying a mutation in either RB1 or CDKN2A. Among AAs with LUAD, PIK3CG was also significantly mutated in our dataset and in TCGA at a frequency of 11% and 10%, respectively, which is higher than the 6% observed among EAs (using TCGA as reference) (Table 2).
Additional recurrently mutated genes that did not reach a statistical significance by MutSigCV, but may functionally impact carcinogenesis, were also identified (Table 2). For example, mutation frequencies for genes in AAs with LUAD, supported in our other datasets, included SPTA1 (NCI-MD AA 39% and TCGA AA 35% vs. TCGA EA 23% (P=0.03), NAV3 (NCI-MD AA 36% and TCGA AA 31% vs. TCGA EA 19% (P=0.02)) and COL11A1 (NCI-MD AA 33% and TCGA AA 31% vs. TCGA EA 19% (P=0.04). SI (NCI-MD AA 28% and TCGA AA 23% vs. TCGA EA 15% (P=0.04)) and ASXL3 (NCI-MD AA 25% and TCGA AA 19% vs. TCGA EA 13% (P=0.05)). The mutation status of these genes was not associated with survival (Table 3). In AA LUSC tumors in the NCI-MD and TCGA data, the mutation frequency of ZNF536 (NCI-MD AA 23% and TCGA AA 26% vs. TCGA EA 12% (P=0.05)) was significantly higher compared with EA patients in TCGA.
Table 3:
Univariable relationship between somatic mutations with 5-year lung cancer specific survival
| All (N=82) | LUAD (N=36) | LUSC (N=39) | ||||
|---|---|---|---|---|---|---|
| Gene | HR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value |
| STK11 status | ||||||
| Wild Type STK11 | Reference | Reference | Reference | |||
| Mutant STK11 | 0.60 (0.21-1.70) | 0.33 | 0.82 (0.22-3.02) | 0.76 | 0.65 (0.09-4.86) | 0.67 |
| RB1 status | ||||||
| Wild Type RB1 | Reference | Reference | Reference | |||
| Mutant RB1 | 0.48 (0.17-1.36) | 0.17 | 0.31 (0.04-2.44) | 0.27 | 0.71 (0.20-2.44) | 0.58 |
| CDKN2A status | ||||||
| Wild Type CDKN2A | Reference | Reference | Reference | |||
| Mutant CDKN2A | 1.64 (0.64-4.23) | 0.30 | NA | NA | 2.93 (1.04-8.28) | 0.04 |
| PIK3CG status | ||||||
| Wild Type PIK3CG | Reference | Reference | Reference | |||
| Mutant PIK3CG | 0.68 (0.27-1.76) | 0.43 | 0.35 (0.04-2.7) | 0.31 | 0.76 (0.18-3.33) | 0.72 |
| PTPRT status | ||||||
| Wild Type PTPRT | Reference | Reference | Reference | |||
| Mutant PTPRT | 0.81 (0.38-1.73) | 0.59 | 0.38 (0.08-1.173) | 0.21 | 1.06 (0.35-3.23) | 0.92 |
| JAK2 Status | ||||||
| Wild Type JAK2 | Reference | Reference | Reference | |||
| Mutant JAK2 | 1.33 (0.63-2.84) | 0.46 | 0.57 (0.12-2.61) | 0.47 | 2.40 0.90-6.40 | 0.08 |
| LRP1B status | ||||||
| Wild Type LRP1B | Reference | Reference | Reference | |||
| Mutant LRP1B | 0.80 (0.41-1.56) | 0.51 | 0.45 (0.14-1.43) | 0.18 | 1.12 (0.43-3.40) | 0.72 |
| SPTA1 status | ||||||
| Wild Type SPTA1 | Reference | Reference | Reference | |||
| Mutant SPTA1 | 0.72 (0.37-1.39) | 0.33 | 0.63 (0.20-1.96) | 0.43 | 0.70 (0.26-1.85) | 0.47 |
| NAV3 status | ||||||
| Wild Type NAV3 | Reference | Reference | Reference | |||
| Mutant NAV3 | 0.91 (0.47-1.76) | 0.78 | 0.49 (0.13-1.80) | 0.28 | 0.78 (0.29-2.08) | 0.62 |
| COL11A1 status | ||||||
| Wild Type COL11A1 | Reference | Reference | Reference | |||
| Mutant COL11A1 | 0.90 (0.47-1.75) | 0.76 | 0.49 (0.15-1.62) | 0.24 | 0.96 (0.37-2.49) | 0.94 |
| SI status | ||||||
| Wild Type SI | Reference | Reference | Reference | |||
| Mutant SI | 0.69 (0.36-1.32) | 0.26 | 0.40 (0.12-1.33) | 0.14 | 0.93 (0.37-2.34) | 0.88 |
| ASXL3 status | ||||||
| Wild Type ASXL3 | Reference | Reference | Reference | |||
| Mutant ASXL3 | 1.70 (0.85-3.40) | 0.14 | 0.48 (0.10-2.19) | 0.34 | 4.49 (1.69-11.95) | 0.0026 |
| ZNF536 status | ||||||
| Wild Type ZNF536 | Reference | Reference | Reference | |||
| Mutant ZNF536 | 1.52 (0.77-3.02) | 0.23 | 0.91 (0.25-3.35) | 0.88 | 1.79 (0.67-4.81) | 0.25 |
HR denotes hazard ratio, CI denotes confidence interval
Finally, although high prevalence mutations are frequently focused on in somatic mutation studies, low frequency variants can also be of significance36, 37. We, therefore, looked for genes with a mutation frequency of at least 5% in AAs in both our dataset and TCGA and with 0% mutation prevalence in EAs, again using TCGA (Supplementary Table 6) and identified KRT9 as mutated in approximately 5% of LUAD tumors among AAs and 0% of LUADs among EAs. The gene product, keratin-9, is a type I cytokeratin expressed in terminally differentiated epidermal cells. Apart from melanoma, KRT9 is not frequently mutated in cancer.
Integration of CNV and somatic mutations
Oncogene and tumor suppressor gene function can be perturbed by both somatic mutation and somatic copy number change. We previously profiled somatic copy number alterations (SCNAs) in 62 of the samples for which we had WES data4 and integrated both datasets to get a more complete view of driver gene alterations across NSCLC in African Americans. As shown in Supplementary Figure 4, TP53 is the most frequently somatically altered gene in NSCLC among African Americans in both LUAD and LUSC. In LUAD, LRP1B, CDKN2A and STK11 are altered at frequencies of 46%, 41% and 38%, respectively. In LUSC, TP53 is again the most altered gene in African Americans (50%), followed by LRP1B and STK11 (44% for both). Interestingly, this combined analysis shows that overall, oncogenic activation of KRAS is somewhat similar in LUAD in both EA and AA, at approximately 33%. However, the mechanism of alteration, i.e., somatic mutation versus somatic copy number alteration (SNCA), differs by population, with KRAS amplification more common in AA than in EA.
Integration of mutation status, etiological exposures and mutational signatures
In an effort to further understand why some genes are mutated at a higher frequency among African Americans, mutation frequencies were correlated with demographic and etiological features such as sex, BMI, education, income and menthol cigarette use. No significant correlation was observed between the mutation status of STK11, RB1, CDKN2A, PIK3CG, PTPRT, JAK2 with these factors (Figure 2A) (Supplementary Table 7), but this may be partially due to the limited sample size when stratified comparisons were made.
Figure 2:
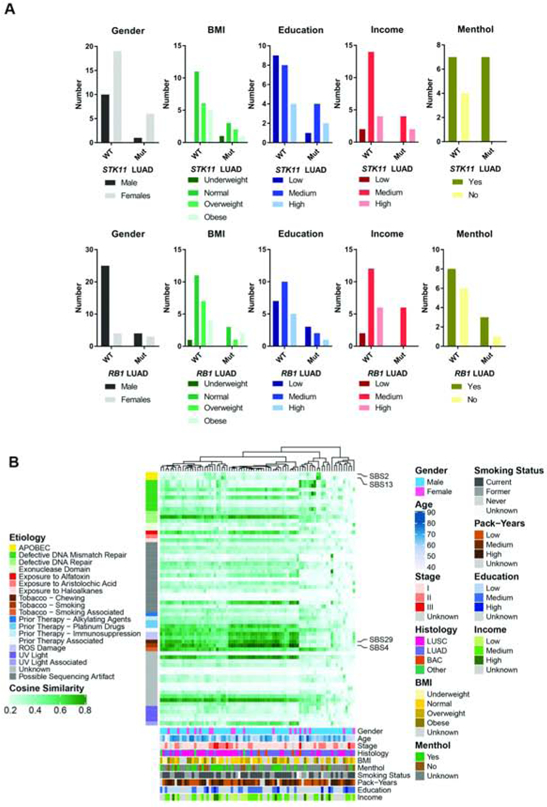
Contribution of mutational signatures to the somatic landscape of lung cancer in African Americans. Clustering of patients is shown based on the proportion of mutational signatures in each tumor. Mutational signatures are grouped as per their etiological origin (left). Demographic and lifestyle exposures are superimposed at the bottom of the oncoplot.
We also contextualized each SNV based on the 96 possible single base substitutions and known mutational signature patterns38 and questioned whether these signatures were enriched in tumors with specific somatic mutations. As expected for a NSCLC cohort of ever smokers, SBS4—which is tobacco associated—was the predominant signature among these tumors (Figure 2B) (Supplementary Table 8). SBS2 and SBS13, signatures attributed to increased activity of APOBEC family members, were also found. None of the SBS signatures had a clear or significant association with BMI, education, income, or menthol cigarette use (Supplementary Figures 5a and 5b).
The likelihood of acquiring a cancer-causing mutation is dependent on the underlying mutational processes39. We, therefore, integrated the SBS mutational signatures with the mutation status of genes enriched among African Americans. We hypothesized that the etiology of these SBS mutational signatures could indicate why certain mutational events occurred at a higher frequency in African Americans. The APOBEC gene signatures, SBS2 and SBS13, were significantly higher in JAK2, RB1, and PTPRT mutant tumors compared with wild-type tumors, with a stronger trend in LUAD (Supplementary Figure 6a and 6c). A previous paper highlighted links between APOBEC-induced mutagenesis and specific driver PIK3CA mutations across cancer types40 and we found significant consistent evidence of this association in our population also (Supplementary Figures 6b and 6d). While our data could be supporting evidence for a causative relationship between the APOBEC mutational activity and the acquisition of these driver mutations in African Americans, there is no statistical difference in the expression of APOBEC genes in tumor tissues from EA and AA (data not shown), no evidence of increased APOBEC signatures in AA versus EA overall5, 8, 41 and several other genes, where the mutation frequency is not enhanced in AA compared with EA, also had evidence for increased APOBEC activity (Supplementary Figure 6b and 6d). SBS4 was enriched in STK11 mutant tumors, with some evidence for SBS29—associated with smokeless tobacco—also enriched among these tumors (Supplementary Figure 6a and 6c).
Discussion
The overall goal of this research is to explore possible genetic differences in NSCLC mutations by race given that African American men have the highest incidence rate that is not fully explained by smoking behavior2, 3, 42. Here, we conducted an in-depth analysis of somatic mutations in African Americans using whole exome sequencing. We replicated our previous data using targeted exome sequencing that show increased JAK2 and PTPRT mutations among African Americans with LUAD. Further, we also present evidence that mutations in the tumor suppressor genes STK11, RB1 and CDKN2A are higher among African Americans, especially in LUAD, and replicated these observations in two independent datasets. Interestingly, we found that STK11 mutations, including in the context of somatically altered KRAS, were associated with a decreased interferon gamma signaling, consistent with previous data in European Americans32, 33. Further, we found high co-occurrence of STK11 with KRAS alterations. This may be relevant in the context of drugs targeting the immune system, in particular immune checkpoint inhibitors. However, given the fact that STK11 loss seems to increase response to platinum-compounds, how mutations of this gene modulate response to combined immune checkpoint inhibitor/chemotherapy treatment, which is increasingly offered to NSCLC patients43, will need to be systematically tested. While co-occurring mutations in STK11 and KEAP1 have been described in patient subsets44, 45, with possible consequences for chemotherapy and immunotherapy response, we did not detect co-occurring mutations in KEAP1 and STK11 in our cohort of AAs.
RB1 and CDKN2A mutations were generally mutually exclusive, suggesting that perturbation of this tumor suppressive pathway is important in LUAD among African Americans. Previous somatic mutation studies of NSCLC primarily used targeted gene or mutation-specific panels, and as such, did not cover the genes identified above8, 46-48. However, supplementary data from many of these studies show consistent results with those presented here, for STK1149, RB149, and JAK29. In other cases, our data were not consistent8 which could be due to different exposures related to geography, rates of admixture or methodologies.
The mechanism by which a gene is perturbed in cancer can be important. As noted earlier, an integrated analysis of somatic copy number and mutation analyses indicated that while, overall, oncogenic activation of KRAS is somewhat similar in EAs and AAs with LUAD, at approximately 33%, the mechanism of alteration, i.e., somatic mutation versus SNCA, differs by population with KRAS amplification more common in AAs than in EAs. Similarly, we found that mutations in CDKN2A and RB1 were more frequent among AAs but that copy number deletions were fewer4. It is possible that different underlying DNA repair4 or stress-response mechanisms50 underly the population divergence between somatic mutations and copy number changes. While inactivation of a pathway, independently of the underlining mechanism, manifests in a similar manner on tumor biology, one implication of these differences relates to genes that are co-deleted with a tumor suppressor, or not. For example, the co-deletion of MTAP with CDKN2A creates a synthetic lethal vulnerability to the MAT2A/PRMT5/RIOK1 axis and a potential novel therapeutic vulnerability51. Thus, patients with CDKN2A mutations, as opposed to CDKN2A deletion, might not respond to such a therapeutic approach. Further, within the guidance to develop immune checkpoint inhibitor treatment in the context of mutually altered KRAS and STK11 tumors, it will be important to consider the therapeutic indications of both somatic copy number and mutation changes.
Our analysis also identified PIK3CG as a gene significantly mutated in NSCLC in AAs. Data from TCGA support the increased mutation frequency of this gene in LUAD in African Americans but a recent analysis of adenocarcinomas using targeted sequencing did not8. Interestingly, population differences in the mutation frequency of this gene in Asian populations have been shown previously, where PIK3CG is mutated in ~30% of LUSCs52. PIK3CG encodes a protein that belongs to the PI3/PI4 family of kinases. It modulates extracellular signals, including those elicited by E-cadherin-mediated cell-cell adhesion and has been implicated in Notch signaling, stemness and migration in claudin-low breast cancer cells53. It has also been implicated in pancreatic cancer as an oncogene54, but data regarding NSCLC is sparse. Data from a few studies describe a low prevalence of mutations in PIK3CG in NSCLC55, including one that links mutations with poor overall survival56 and another that shows how knock-out of PIK3CG induces cell death52. Of note, recent data suggest that, in general, the PIK3CA pathway is less frequently altered in African Americans in both pan-cancer and NSCLC41. Further work on this gene in AAs is warranted.
We also identified several genes that have a higher mutation prevalence in NSCLC among AA patients compared to EA patients. For example, we found COL11A1 mutated at a higher frequency in African Americans with LUAD. COL11A1 is a collagen type XI α1 protein that encodes one of the two α chains of type XI collagen, a minor fibrillar collagen57. As a major component of the extracellular matrix (ECM), collagens are involved in the regulation of multiple biological processes, including cell proliferation, differentiation and migration57; it has also been implicated in NSCLC progression58. SPTA1 was mutated at a higher frequency among AAs with LUAD, compared with EAs. A reduction in SPTA1 protein expression was previously found in NSCLC59; it is a plasma membrane to the actin cytoskeleton and functions in the determination of cell shape, arrangement of transmembrane proteins, and organization of organelles. We found LRP1B (low-density lipoprotein receptor-related protein 1B) mutated in approximately 40% of LUAD samples. Mutations in this gene were previously reported in NSCLC, with some indications that it may be associated with response to immune checkpoint drugs due to a higher mutational burden observed among LRP1B mutant tumors60, 61. Of note, we also observed significantly increased tumor burden among LRP1B mutant tumors among African Americans (Supplementary Figure 7).
Endogenous biological processes and exogenous exposures contribute to the mutational landscape of tumors30, 62. We therefore assessed the mutational patterns of these tumors from African Americans and identified the same key mutational processes as seen in European Americans, i.e., SBS4 (tobacco-associated) and SBS2/13 (aberrant APOBEC activity). There were no significant associations between African ancestry and SBS signatures following correction for multiple testing, consistent with previous observations using targeted gene panels8. Although we had a limited sample size, we leveraged the available demographic and exposure data that we had on our participants but did not find significant associations between metrics of SES, such as income and education, with the genes we found enriched among African Americans. This analysis was likely impacted by power. To uncover the potential etiological processes driving the occurrence of mutations in JAK2, PTPRT, RB1, STK11, PIK3CG and CDKN2A, we also integrated somatic mutational signatures with the mutation status of these genes. Interestingly, SBS2 and SBS13, those associated with aberrant APOBEC signaling, were enriched in tumors with mutated RB1, JAK2, and PTPRT. This suggests that aberrant APOBEC activity in tumors from African Americans could drive the increased mutations of these genes. However, an analysis of APOBEC1 and 3 expression in data we had previously collected7 did not reveal consistent significant mRNA expression differences. A germline variant in cytidine deaminase, codon 70 (208G > A) that consists of a threonine instead of an alanine, is found mostly in African and Asian populations63, so it is possible that structural differences at the protein level could make a difference. However, we found several genes where mutation frequencies are not enriched in AAs, such as PIK3CA, RET, ARID1A, EGFR, NFE2L2 and SMARCA4, that also had increased SBS2 and SBS13 representation. Thus, additional studies should be conducted with greater sample size and demographic variables to further understand the potential etiological forces that contribute to these mutation differences in AAs. Of note, although our data do not point towards APOBEC and a role in disparities per se, the finding that these signatures are associated with the mutation profile of these genes in NSCLC is novel.
In summary, using whole exome sequencing, we extended our previous targeted exome analysis of NSCLC in African Americans, validated our previous observations and identified increased mutation frequency of several tumor suppressor genes in NSCLC. As before, most somatic mutation differences that we observe occur in LUAD. Future, larger studies with demographic, socioeconomic, and etiological data will be needed to assess the factors that contribute to these population differences in tumor biology as well as whether these differences in tumor biology relate to therapy outcomes, including ICI, at a gene level. Our data highlight the continuing importance of not only including minority populations in genomics research, but leveraging the inherent differences represented by race to contribute towards understanding observed differences in incidence, severity and treatment response. This will become especially useful as more is learned about how somatic mutations influence the tumor microenvironment, the immune response to carcinogenesis and treatment efficacy.
Supplementary Material
Acknowledgements
This work was supported by the Intramural Research Programs of the National Institute for Minority Health and Health Disparities and the Center for Cancer Research, National Cancer Institute. SRP was supported by 1R01CA239093 and a CINJ Health Equity Pilot Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. Ca-Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Ryan BM. Lung cancer health disparities. Carcinogenesis 2018;39:741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stram DO, Park SL, Haiman CA, et al. Racial/Ethnic Differences in Lung Cancer Incidence in the Multiethnic Cohort Study: An Update. J Natl Cancer Inst 2019;111:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinha S, Mitchell KA, Zingone A, et al. Higher prevalence of homologous recombination deficiency in tumors from African Americans versus European Americans. Nature Cancer 2020;1:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell KA, Nichols N, Tang W, et al. Recurrent PTPRT/JAK2 mutations in lung adenocarcinoma among African Americans. Nat Commun 2019;10:5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meaney CL, Mitchell KA, Zingone A, et al. Circulating Inflammation Proteins Associated With Lung Cancer in African Americans. J Thorac Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell KA, Zingone A, Toulabi L, et al. Comparative Transcriptome Profiling Reveals Coding and Noncoding RNA Differences in NSCLC from African Americans and European Americans. Clin Cancer Res 2017;23:7412–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell JD, Lathan C, Sholl L, et al. Comparison of Prevalence and Types of Mutations in Lung Cancers Among Black and White Populations. JAMA Oncol 2017;3:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lusk CM, Watza D, Dyson G, et al. Profiling the Mutational Landscape in Known Driver Genes and Novel Genes in African American Non-Small Cell Lung Cancer Patients. Clin Cancer Res 2019;25:4300–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Yang H, Teo ASM, et al. Genomic landscape of lung adenocarcinoma in East Asians. Nature genetics 2020;52:177–186. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poplin R, Ruano-Rubio V, DePristo MA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018:201178. [Google Scholar]
- 16.Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009;19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11 10 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–1817. [DOI] [PubMed] [Google Scholar]
- 21.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Memorial Sloan Kettering Cancer Center. vcf2maf. Available at https://github.com/mskcc/vcf2maf 2019.
- 23.Shyr C, Tarailo-Graovac M, Gottlieb M, et al. FLAGS, frequently mutated genes in public exomes. BMC Med Genomics 2014;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayakonda A TCGAmutations: Pre-compiled somatic mutations from TCGA cohorts. R package 2019;version 0.2.0. [Google Scholar]
- 26.Ellrott K, Bailey MH, Saksena G, et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 2018;6:271–281 e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorsson V, Gibbs DL, Brown SD, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blokzijl F, Janssen R, van Boxtel R, et al. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med 2018;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell BB, Light N, Fabrizio D, et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017;171:1042–1056 e1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354:618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hastings K, Yu HA, Wei W, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann Oncol 2019;30:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov 2018;8:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rizvi H, Sanchez-Vega F, La K, et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018;36:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonaventura P, Shekarian T, Alcazer V, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Frontiers in immunology 2019;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao P, Bu X, Wei X, et al. Dendritic cell immunotherapy combined with cytokine-induced killer cells promotes skewing toward Th2 cytokine profile in patients with metastatic non-small cell lung cancer. Int Immunopharmacol 2015;25:450–456. [DOI] [PubMed] [Google Scholar]
- 36.Craig DW, O'Shaughnessy JA, Kiefer JA, et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther 2013;12:104–116. [DOI] [PubMed] [Google Scholar]
- 37.Lazzari C, Spitaleri G, Catania C, et al. Targeting ALK in patients with advanced non small cell lung cancer: biology, diagnostic and therapeutic options. Crit Rev Oncol Hematol 2014;89:358–365. [DOI] [PubMed] [Google Scholar]
- 38.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 2013;3:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Temko D, Tomlinson IPM, Severini S, et al. The effects of mutational processes and selection on driver mutations across cancer types. Nat Commun 2018;9:1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGranahan N, Favero F, de Bruin EC, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015;7:283ra254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan J, Hu Z, Mahal BA, et al. Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer cell 2018;34:549–560 e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haiman CA, Stram DO, Wilkens LR, et al. Ethnic and racial differences in the smoking-related risk of lung cancer. N Engl J Med 2006;354:333–342. [DOI] [PubMed] [Google Scholar]
- 43.Bonanno L, Zulato E, Pavan A, et al. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 2017;376:2109–2121. [DOI] [PubMed] [Google Scholar]
- 45.Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araujo LH, Lammers PE, Matthews-Smith V, et al. Somatic Mutation Spectrum of Non-Small-Cell Lung Cancer in African Americans: A Pooled Analysis. J Thorac Oncol 2015;10:1430–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bollig-Fischer A, Chen W, Gadgeel SM, et al. Racial diversity of actionable mutations in non-small cell lung cancer. J Thorac Oncol 2015;10:250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Araujo LH, Timmers C, Bell EH, et al. Genomic Characterization of Non-Small-Cell Lung Cancer in African Americans by Targeted Massively Parallel Sequencing. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:1966–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kytola V, Topaloglu U, Miller LD, et al. Mutational Landscapes of Smoking-Related Cancers in Caucasians and African Americans: Precision Oncology Perspectives at Wake Forest Baptist Comprehensive Cancer Center. Theranostics 2017;7:2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hastings PJ, Lupski JR, Rosenberg SM, et al. Mechanisms of change in gene copy number. Nat Rev Genet 2009;10:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marjon K, Cameron MJ, Quang P, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep 2016;15:574–587. [DOI] [PubMed] [Google Scholar]
- 52.Zhang P, Kang B, Xie G, et al. Genomic sequencing and editing revealed the GRM8 signaling pathway as potential therapeutic targets of squamous cell lung cancer. Cancer Lett 2019;442:53–67. [DOI] [PubMed] [Google Scholar]
- 53.Zhang S, Chung WC, Wu G, et al. Manic fringe promotes a claudin-low breast cancer phenotype through notch-mediated PIK3CG induction. Cancer Res 2015;75:1936–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang S, Liu J, Xu K, et al. Notch signaling via regulation of RB and p-AKT but not PIK3CG contributes to MIA PaCa-2 cell growth and migration to affect pancreatic carcinogenesis. Oncol Lett 2018;15:2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grosse C, Soltermann A, Rechsteiner M, et al. Oncogenic driver mutations in Swiss never smoker patients with lung adenocarcinoma and correlation with clinicopathologic characteristics and outcome. PLoS One 2019;14:e0220691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qian J, Zhao S, Zou Y, et al. Genomic Underpinnings of Tumor Behavior in In Situ and Early Lung Adenocarcinoma. Am J Respir Crit Care Med 2020;201:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prockop DJ, Kivirikko KI, Tuderman L, et al. The biosynthesis of collagen and its disorders (first of two parts). N Engl J Med 1979;301:13–23. [DOI] [PubMed] [Google Scholar]
- 58.Shen L, Yang M, Lin Q, et al. COL11A1 is overexpressed in recurrent non-small cell lung cancer and promotes cell proliferation, migration, invasion and drug resistance. Oncol Rep 2016;36:877–885. [DOI] [PubMed] [Google Scholar]
- 59.Fahrmann JF, Grapov D, Phinney BS, et al. Proteomic profiling of lung adenocarcinoma indicates heightened DNA repair, antioxidant mechanisms and identifies LASP1 as a potential negative predictor of survival. Clin Proteomics 2016;13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen H, Chong W, Wu Q, et al. Association of LRP1B Mutation With Tumor Mutation Burden and Outcomes in Melanoma and Non-small Cell Lung Cancer Patients Treated With Immune Check-Point Blockades. Frontiers in immunology 2019;10:1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frances A, Cordelier P. The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy? Mol Ther 2020;28:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study have been uploaded to the dbGaP repository in compliance with the NIH Genomic Data Sharing Policy. Data can be accessed at dbGap study ID phs001895.