

Abstract
The health beneficial effects of walnut plentiful of n-3 polyunsaturated fatty acid had been attributed to its anti-inflammatory and anti-oxidative properties against various clinical diseases. Since we have published Fat-1 transgenic mice overexpressing 3-desaturase significantly mitigated Helicobacter pylori (H. pylori)-associated gastric pathologies including rejuvenation of chronic atrophic gastritis and prevention of gastric cancer, in this study, we have explored the underlying molecular mechanisms of walnut against H. pylori infection. Fresh walnut polyphenol extracts (WPE) were found to suppress the phosphorylation and nuclear translocation of signal transducer and activator of transcription 3 (STAT3) induced by H. pylori infection in RGM-1 gastric mucosal cells. Notably, H. pylori infection significantly decreased suppressor of cytokine signaling 1 (SOCS1), but WPE induced expression of SOCS1, by which the suppressive effect of walnut extracts on STAT3Tyr705 phosphorylation was not seen in SOCS1 KO cells. WPE induced significantly increased nuclear translocation nuclear translocation of PPAR-γ in RGM1 cells, by which PPAR-γ KO inhibited transcription of SOCS1 and suppressive effect of WPE on p-STAT3Tyr705 was not seen. WPE inhibited the expression of c-Myc and IL-6/IL-6R signaling, which was attenuated in the RGM1 cells harboring SOCS1 specific siRNA. Conclusively, WPE inhibits H. pylori-induced STAT3 phosphorylation in a PPAR-γ and SOCS1-dependent manner.
Keywords: Helicobacter pylori, walnut polyphenol extract, STAT3Tyr705, SOCS1, PPAR-γ
Introduction
Helicobacter pylori (H. pylori), a gram-negative bacterial pathogen living in stomach, has been implicated in development of gastric cancer through the development of precancerous lesions such as hypertrophic and atrophic gastritis, responsible for initiating and progressing mechanism implicated in gastric carcinogenesis.(1) Among mechanisms in H. pylori-carcinogenesis, including NF-κB based oxidative stress, cytokines mediated mutagenesis, apoptosis induced atrophic changes, and more carcinogenic actions,(2,3) it has been well documented as molecular pathomechanism that H. pylori infection causes the activation of signal transducer and activator of transcription 3 (STAT3), STAT3 phosphorylated on Tyr705 residue (p-STAT3Tyr705), forms a dimer, translocates to nucleus, and functions as transcription factor to regulate the target genes implicated in H. pylori-associated inflammation and carcinogenesis subsequent to IL-6 activation.(4–7) Though STAT3Ser727 phosphorylation can be pathogenically involved in oxidative stress and malignant transformation relevant to H. pylori infection since STAT3Ser727 localizes in mitochondria and associates with Ras dependent oncogenic transformation, p-STAT3Tyr705 had been revealed to be implicated in either Barrett’s esophagus and H. pylori-associated oncogenic inflammation.(8,9) CagA cytotoxin from H. pylori led to significant cytokine signaling pathways via MAPK activation and is responsible for gastric inflammation and carcinogenesis including increased cell proliferation, angiogenesis, inflammation, inhibition of immunocytes, and epithelial cell apoptosis.(10) Among these cytokines, IL-6, IL-6R, and gp130 with subsequent STAT3 activation are representationally dysregulated pathways in H. pylori infection.(11)
Therefore, agents or intervention to regulate IL-6 signaling seem to be of potential significance to solve unmet medical needs of H. pylori-associated gastric pathologies. Suppressor of cytokine signaling (SOCS) has been known to cope with oxidative stress and inflammatory cytokines relevant to H. pylori infection,(12,13) especially SOCS 1 that SOCS1,(14) named STAT induced STAT inhibitor (SSI) or JAK-binding protein (JAB), played immune regulation as well as inflammatory modulation, leaving the agents or drug having the potential of autoimmunity as well as cancer.(15,16)
Supported with our previous publications that n-3 polyunsaturated fatty acids (n-3 PUFAs) generating fat-1 transgenic mice showed significant rescuing from H. pylori-associated atrophic gastritis as well as gastric cancer,(17–19) we put hypothesis that walnut polyphenol extracts (WPE) containing abundant n-3 PUFAs can be food factor to ameliorate H. pylori-associated gastric inflammation and executed the current experiment to explore molecular mechanisms to limit H. pylori-associated IL-6 and their STAT3 activation before in vivo model of H. pylori-associated gastritis.
Materials and Methods
Materials
RPMI-1640 medium, fetal bovine serum, penicillin (FBS), streptomycin were products of GIBCO BRL (Grand Island, NY) and materials for culturing H. pylori were sheep blood agar, Gaspak and anaerobic jars (BD Biosciences, Sparks, MD). Primary antibody against actin was purchased from Sigma-Aldrich Co. (St. Louis, MO), antibodies for lamin B from Santa Cruz Biotechnology (Santa Cruz, CA), other antibodies for p-STAT3Tyr705, total STAT3 from Cell Signaling Technology (Beverly, MA), horseradish peroxidase (HRP)-conjugated secondary antibody from Pierce Biotechnology (Rockford, IL). DL-dithiothreitol (DTT), TRIzol, 4',6-diamidino-2-phenylindole (DAPI) from Invitrogen (Carlsbad, CA), and polyvinylidene difluoride (PVDF) membranes were supplied from Gelman laboratory (Ann Arbor, MI). The ECL chemiluminescent detection kit was purchased from LPS solution (Daejon, South Korea) and protein assay dye (Bradford) reagent was supplied by Bio-Rad Laboratories (Hercules, CA), bicinchonic acid (BCA) protein assay reagent was obtained from Pierce Biotechnology.
Preparation of WPE
Walnut polyphenol extract (WPE) from English walnuts (J. regia, California Walnut Commission) was prepared according to a previously described methanolic extraction method.(20) Briefly, after the walnuts were frozen for 24 h, the shelled kernels were finely ground and immersed in a solution of 75% acetone containing 526 µm/L sodium metabisulfite. The solution was subsequently purged with N2 to prevent oxidation and was incubated at 4°C. After 24 h, the solution was decanted, thereby resulting in a cold extract that was centrifuged at 8,000 × g for 10 min. The resulting supernatant was filtered using Whatman filter paper No. 2. To remove lipids from the sample, the acetone was removed under reduced pressure and methanol (50% aqueous, v/v) was added. After three consecutive hexane extractions, the extracts were lyophilized to a dry powder after removing the methanol to prevent oxidation. All of the prepared samples were stored at 80°C until needed.
Cell culture
RGM-1 cells from Prof. Hirofumi Matsui (Tsukuba Univ, Japan) were cultured in RPMI-1640 medium supplemented with 10% (v/v) FBS and 100 units/ml penicillin and 100 µg/ml streptomycin at 37°C in an incubator with humidified atmosphere of 95% O2/5% CO2.
Bacteria strain and infection condition
H. pylori (ATCC 43504) with the typical S shape, gram-negative rods, possessing the CagA and VacA were provided in a frozen state by ATCC. H. pylori ATCC 43504 strains were grown on tryptic soy agar with 5% sheep blood agar (BD Diagnostics) and Dent antibiotics supplement (Oxoid) at 37°C under microaerophilic conditions (Campy-Pak 273 System, BBL). RGM1 cells were incubated overnight in fresh serum- and antibiotic-free RPMI 274 medium and were infected with H. pylori at multiplicities of infection (MOI) of 50:1.
Western blot analysis
The cell lysates were prepared, and the protein concentration was measured as described previously 7. The equivalent amounts of proteins (10–30 µg) were subjected to electrophoresis on 8% or 12% SDS-polyacrylamide gel and transferred to PVDF membrane. The transferred proteins were blocked in 5% fat-free dry milk in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST) for 1 h at room temperature. Then, the membranes were incubated with primary antibodies in 3% fat-free dry milk in PBS overnight in 4°C. Membranes were washed followed by incubation with 1:3,000 dilution of respective HRP conjugated secondary antibodies for 1.5 h and again washed with PBST. Protein expressed was visualized with an ECL chemiluminescence detection kit.
Preparation of cytosolic and nuclear extracts
After H. pylori infection, cells were washed twice with ice-cold 1× PBS and scraped in 1 ml of PBS, followed by centrifugation at 1,700 × g for 5 min at 4°C. Pellets were resuspended in hypotonic buffer A [10 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT and 0.2 mM phenylmethylsulfonylfluoride (PMSF)] for 15 min on ice. Ten % Nonidet P-40 was then added to final concentration of 0.1% for less 3 than 5 min. The mixture was then centrifuged at 6,000 × g for 5 min at 4°C. Supernatant was collected as the cytosolic extract and stored at –80°C. The pellets were washed twice with hypotonic buffer A and resuspended again in hypertonic buffer C [20 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (pH 7.9), 20% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid, 0.5 mM DTT and 0.2 mM PMSF] for 1 h on ice and centrifuged at 18,000 × g for 15 min at 4°C. The supernatant containing nuclear proteins was collected and stored at –80°C. The protein concentrations of both fractions were determined by using the BCA protein assay reagent.
Confocal imaging analysis
Cells were infected with H. pylori for 12 h. After the cells were incubated with probe (1 nM) in prewarmed staining solution for 30 min, samples were fixed with cold 95% MeOH/5% acetic acid for 10 min at 4°C. Then samples were permeabilized with 0.2% Triton X-100 for 5 min at room temperature and blocked with 5% bovine serum albumin in PBST for 1 h at room temperature. Samples were incubated with primary antibody specific for phospho-STAT3Tyr705 overnight at 4°C, followed by incubation with fluorescein isothiocyanate-goat anti-rabbit IgG secondary antibody for 1 h at room temperature. Nuclear-staining was performed with DAPI for 5 min at room temperature. Images were assessed under a fluorescent microscopy.
Statistical analysis
Data from three independent experiments at least were expressed as the mean ± SD. The statistical significance of differences between two groups was evaluated using Student’s t test. Analysis was performed using Sigma plot (ver. 10). Statistical significance was accepted at p<0.05, unless otherwise indicated.
Results
Walnut extract inhibited H. pylori-induced IL-6 and additional inflammatory mediators including COX-2, c-Myc, tumor necrosis factor-alpha (TNF-α)
IL-6 is one of core mediators implicated in inflammatory and carcinogenic process in H. pylori associated gastric carcinogenesis via NF-κB, STAT3, and MAPK signaling pathways.(5) As seen in the Fig. 1A dealing with the changes of IL-6 mRNA along with H. pylori infection in AGS cells, H. pylori infection significantly increased IL-6 mRNA after 12 h and persisted up to 24 h. In this time, 24 h after 50 MOI H. pylori infection, increased IL-6 mRNA was significantly decreased with increasing doses of WPE (Fig. 1B). Additionally, we measured the levels of secreted IL-6 in supernatant by ELISA and found H. pylori infection, 50 MOI for 24 h, significantly increased IL-6 levels (p<0.01), and IL-6 levels were significantly decreased with WPE in a dose dependent manner (p<0.01, Fig. 1C). In addition to IL-6, H. pylori infection is also associated with significant increases in TNF-α, IFN-γ, and IL-8, after which walnut extracts also led to significant decreases of these inflammatory mediators (Fig. 1D). Especially, gastric diseases after H. pylori infection are closely associated with COX-2 and c-Myc as possible oncogenes in inflammation based gastric carcinogenesis. As seen in Fig. 1E, H. pylori infection significantly increased COX-2 and c-Myc expression after 6 h of H. pylori infection, but WPE significantly decreased c-Myc expressions (Fig. 1E). Taken all together, WPE can significantly decrease H. pylori-associated inflammatory mediators.
Fig. 1.

Changes of IL-6 and other inflammatory mediators after WPE. (A) Changes of IL-6 mRNA after H. pylori infection in RGM-1 cells RT-PCR for IL-6 mRNA. (B) Changes of IL-6 mRNA according to WPE in the presence of H. pylori infection. (C) IL-6 levels in the supernatants of RGM-1 cells according to walnut dose in the presence of H. pylori infection. *p<0.01 vs H. pylori alone. (D) Changes of inflammatory mediators RT-PCR for TNF-α, IFN-γ, IL-1β, and IL-8. (E) Western blot for COX-2 and c-Myc Expression after times of H. pylori infection. Time dependent increases of expression noted after 6 h (left) and changes of c-Myc after walnut administration in the presence of H. pylori (right).
STAT3 activations after H. pylori infection were significantly decreased with WPE
IL-6 led to STAT3 activation, which is closely intervened in inflammatory and carcinogenic events of H. pylori infection.(21,22) As seen in Fig. 2A, H. pylori infection is significantly associated with STAT3 activation, especially STAT3Tyr705. In this condition, WPE significantly inactivated STAT3 tyrosine phosphorylation at tyrosine 705 site (p<0.01). In order to confirm the intervention of STAT3 activation via nuclear translocation, we repeated the measurement of STAT3 in nuclear fraction, as anticipated, WPE significantly inhibited nuclear translocation of STAT3 (p<0.01, Fig. 2B). These findings were further confirmed by confocal imaging of STAT3 that walnut extract significantly inhibited nuclear translocation of STAT3 (Fig. 2C).
Fig. 2.
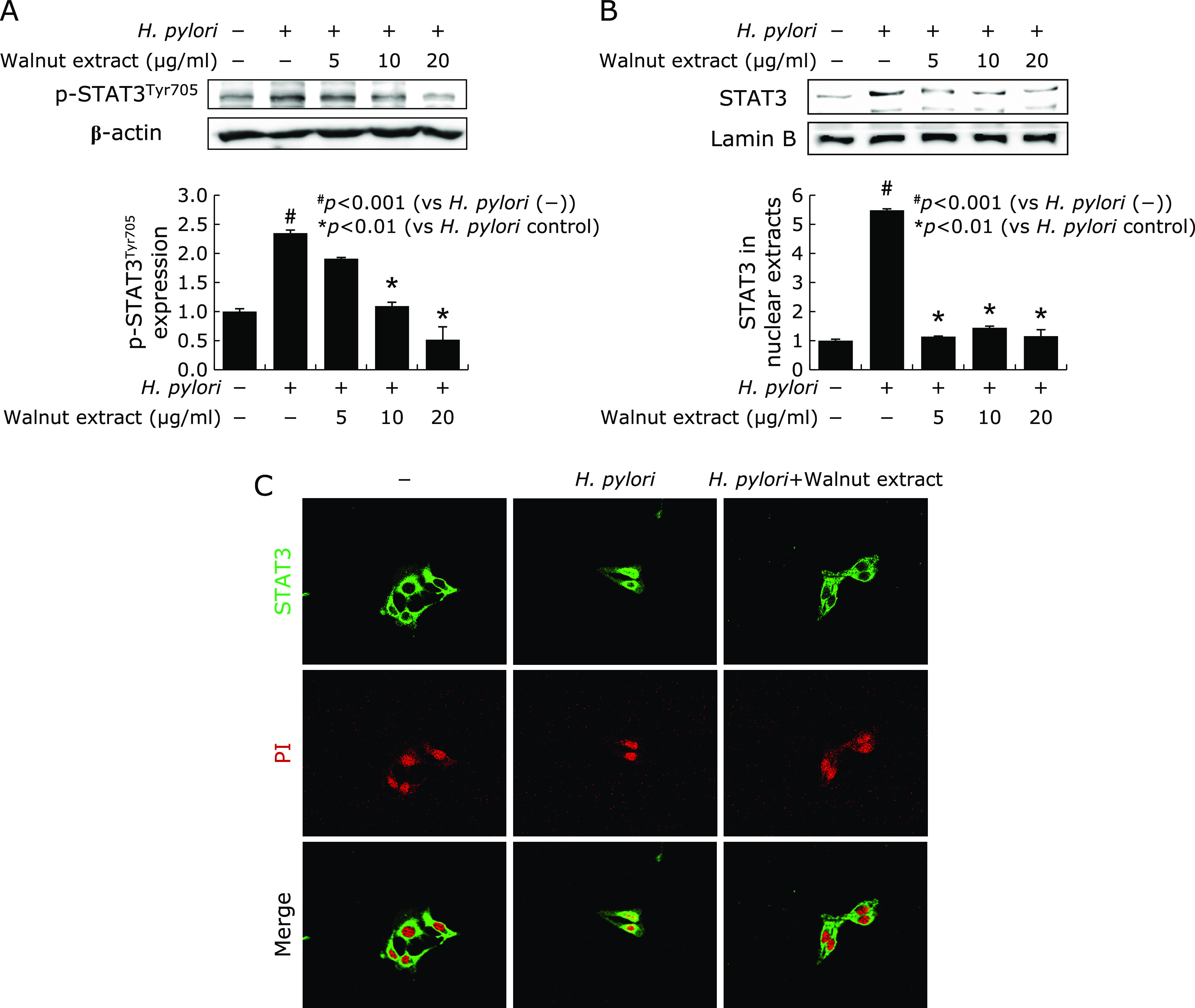
Inhibition of STAT3 activations with WPE in the presence of H. pylori infection. (A) p-STATTyr705 was measured in cytoplasm extract after WPE in the presence of H. pylori Increased p-STAT3Tyr705 after H. pylori infection was significantly decreased with 10 or 20 µg/ml WPE. *p<0.01. (B) Nuclear expression of STAT3 STAT3 nuclear translocation was significantly decreased with WPE in nuclear fraction of RGM-1 cells infected with H. pylori. (C) Confocal imaging of STAT3 and PI according to WPE in the presence of H. pylori STAT3 expression localized mainly in the cytoplasm of non-stimulated RGM-1 cells, but their expressions moved to nucleus after H. pylori infection. However, even in the presence of H. pylori, STATs expression localized in the cytoplasm with WPE administration.
WPE induced SOCS1 contributed to H. pylori-STAT3 activation
SOCS1 has been identified as negative regulator of STAT3 and we have found WPE can induce SOCS1 as anti-inflammatory mechanisms.(23) Therefore, under the hypothesis that WPE can provoke anti-inflammatory action via SOCS1 induction and subsequent STAT3 inactivation, we measured the changes of SOCS1 mRNA after H. pylori infection. As seen in Fig. 3A, host cells increased SOCS1 after 30 min H. pylori infection, but their levels were significantly decreased after 2 h. In this condition, WPE significantly preserved and induced SOCS1 in a dose dependent manner of WPE (p<0.01, Fig. 3A). These changes of SOCS1 mRNA were further validated with the expressions of SOCS1 via western blot. As seen in Fig. 3B, 20 mg/ml WPE significantly increased SOCS1 expressions after 2 h of treatment (p<0.01), signifying WPE significantly induced SOCS1. In order to verify this SOCS1 induction with WPE contributed to p-STAT3 inactivation, we generated SOCS1 KO cells and compared the changes of p-STAT3Tyr705 and c-Myc between Mock- and siSOCS1-treated cells after H. pylori infection (Fig. 3C). As resulted, p-STAT3Tyr705 and c-Myc increases after H. pylori infection was significantly decreased in Mock-transfected cells, while no changes in p-STAT3Tyr705 and c-Myc were noted in siSOCS1 transfected cells.
Fig. 3.

Cancellation of SOCS after H. pylori and restoring of SOCS1 with WPE. (A) Changes of SOCS1 mRNA with H. pylori infection RT-PCR for SOCS1 (left) and changes of SOCS mRNA after WPE (right). (B) Western blot of SOCS1 after WPE Statistically significant increases of SOCS1 at 2 h of WPE. *p<0.01. (C) STAT3 activation and the expression of c-Myc between Mock-transfected and siSOCS1 transfected RGM-1 WPE significantly decreased H. pylori-increased either p-STATTyr705 or c-Myc in Mock transfected RGM-1 cells, but these changes were abolished in siSOCS1-transfected cells.
WPE induced PPAR-γ led to significant inhibition of STAT3 activation via SOCS1 induction
In order to explain the contribution of SOCS1 to tackle H. pylori-associated STAT3 activation relevant to gastric inflammation, we checked the changes of peroxisome proliferator activated receptors-gamma (PPAR-γ), nuclear transcription factor of the steroid receptor superfamily, after WPE administration since we have preliminary data that WPE induced PPAR-γ. As seen in Fig. 4A, 30 min after WPE administration, PPAR-γ expressions were significantly increased (p<0.01). Since PPAR-γ is transcription factor existing in cytoplasm in order to transcript biological action, as seen in Fig. 4B, increasingly expressed in cytoplasm, but increasingly translocated in nucleus after WPE administration. In order to know the regulatory action of PPAR-γ activation in suppressing STAT3, we repeated the experiment to compare the STAT3Tyr705 phosphorylation between Mock-transfected and siPPAR-γ transfected cells according to WPE treatment. As result, WPE significantly decreased p-STAT3Tyr705 with WPE, but not in cells transfected with siPPAR-γ in spite of WPE administration (Fig. 4C) and these findings were further validated with the administration of BADGE, bisphenol A diglycidyl ether as PPAR-γ antagonist. As seen in Fig. 4D, activated STAT3Tyr705 after H. pylori infection was significantly inhibited with WPE administration, but not in cells treated with BADGE in spite of WPE co-administration, consistently suggesting that WPE is associated with PPAR-γ activation in inhibiting H. pylori-induced STAT3 activation.
Fig. 4.

Contribution of WPE-induced PPAR-γ on inactivation of STAT3. (A) Changes of PPAR-γ according WPE in RGM-1 cells. Significantly increased induction of PPAR-γ 30 min after WPE. *p<0.01. (B) Confocal imaging of PPAR-γ in RGM-1 cells. In normal RGM-1 cells, PPAR-γ localized mainly in the cytoplasm, its expression significantly increasingly increased in nucleus with WPE administration. (C) Changes of p-STAT3Tyr705. In Mock transfected RGM-1 cells, H. pylori infection led to increased activation of STAT3, but decreased with WPE administration. On the other hand, in cells transfected with siPPAR-γ, no changes of STAT3 were noted, signifying STAT3 inhibitory action of walnut was mediated with PPAR-γ. (D) Changes of p-STAT3Tyr705 H. pylori infection clearly increased STAT3 activation at Tyr705 site and its activation was significantly decreased with WPE. However, BADGE as PPAR-γ antagonist also abolished STAT3 inactivation effect of WPE, signifying STAT3 inhibitory action of WPE was mediated through PPAR-γ activation. (E) Since previous study showed SOCS1 regulated STAT3, RGM-1 cells transfected with PPAR-γ significantly decreased SOCS1 expression.
WPE regulated H. pylori-associated IL-6 and STAT3 activation via PPAR-γ activation and SOCS1 induction
Since previous data consistently showed PPAR-γ-transcribed SOCS1 is implicated anti-inflammatory action of WPE,(24) we compared the expression of SOCS1 between Mock transfected- and siPPAR-γ transfected cells in the expression of SOCS1 after WPE, as seen in Fig. 5A, SOCS1 mRNA was significantly decreased in siPPAR-γ transfected cells compared to Mock transfected cells. Next, we compared STAT3Tyr705 activation according to IL-6 and IL-6 KO condition. As noted in Fig. 5B, WPE significantly inhibited H. pylori-induced STAT3Tyr705 and c-Myc activation, but in cells transfected IL-6 or IL-6 receptor KO, no inhibition of STAT3Tyr705 was noted in spite of WPE administration and no induction of c-Myc induction.
Fig. 5.

WPE induced PPAR-γ to induce SOCS1 and simultaneously inactivated STAT3 via IL-6 inhibition. (A) WPE induced SOCS1 via PPAR-γ RGM-1 cells transfected with siPPAR-γ showed decreased expression of SOCS1 compared to Mock-transfected cells in the presence of WPE. (B) STAT3 and c-Myc expression according to IL-6 and IL-6 receptor. Cells transfected with either siIL-6 or siIL-6R showed no significant changes in STAT3 activation irrespective of WPE. Looking at c-Myc changes, WPE significantly decreased H. pylori-increased c-Myc in Mock-transfected cell, whereas no changes of c-Myc was noted in either siIL-6 or siIL-6 receptor.
Discussion
From the current investigation, we reached to summary as shown in Fig. 6 that concerted actions of PPAR-γ induction, SOCS1 induction, and subsequent inactivation of IL-6-associated STAT3 signaling with walnut extract administration in H. pylori-infected non-transformed gastric mucosal cells, RGM-1 cells can ameliorate either gastric inflammation or gastric tumorigenesis. Though proven in in vitro cellular system, our study provides hint for further consideration to meet clinical application that dietary intake of walnut can be a way to rescue stomach from H. pylori infection.(25) Gastric inflammation/mutagenesis mediated by COX-2, IL-1β, IL-6, IL-8, IFN-γ, and c-Myc become basis for gastric tumorigenesis, so called “inflammation-dysplasia-carcinogenesis via chronic atrophic gastritis” pathway,(26,27) in which chronic dietary intervention can be anticipating strategy for prevention. Therefore, from our current study, we can conclude walnuts or its fresh phenolic extracts exerted significant rescuing action against H. pylori-associated gastric diseases including gastric cancer.
Fig. 6.

Schematic presentation how WPE afforded significant protection from H. pylori infection. WPE possessing anti-inflammatory, antioxidative, and anti-mutagenic actions induced PPAR-γ immediately after administration, 30 min. PPAR-γ significantly transcribed SOCS1, which exerted significant inhibitory action against STAT3. H. pylori infection led to increased IL-6/IL-6R/gp130, resulting in increased phosphorylation of STAT3Tyr705. These activations of STAT3 after H. pylori infection increased inflammatory mediators including COX-2, IL-1β, IL-6, and IFN-γ. Taken together, WPE contributed to inhibit inflammatory and mutagenic action of H. pylori via PPAR-γ activation, SOCS1 induction, and STAT3 inhibition.
As molecular mechanisms of preventive strategies achieved with WPE, first, we have focused on IL-6/JAK/STAT3 pathway because they are responsible for transcribing inflammatory and mutagenic mediators such as COX-2, IFN-γ, iNOS, IL-1β, and c-Myc relevant to H. pylori infection. STAT3 is a transcription factor activated by various external stimuli including cytokines and growth factors. Upon activation, STAT3 is phosphorylated on Tyr705 or phosphorylated on Ser727 and translocates to nucleus where it regulates expression of target genes involved in cell proliferation, survival.(28,29) Although phosphorylation of STAT3Tyr705 has been essential for its dimerization, nuclear translocation, transcriptional activity and oncogenic function, phosphorylated STATSer727 stimulated tumor growth by modulating the activity of complex I and the intracellular accumulation of reactive oxygen species (ROS). JAK1/STAT3 is known to be an upstream signaling of NF-κB activation producing IL-8/IL-1β/IL-2 and generating ROS after H. pylori infection.(6,30,31) Conclusively, H. pylori-induced STAT3 activation is mediated through ROS-induced upregulation of IL-6 expression, necessitating inhibiting STAT3 to mitigate gastric damages after H. pylori infection. Consequent to IL-6 generation after H. pylori infection, augmented gp130-mediated cytokine signaling should be blocked in order to prevent H. pylori-associated inflammation and carcinogenesis.(9,32) In addition to these epithelial components, though not studied in the current investigation, STAT3 imparted a profound influence on immune response to H. pylori infection,(33) for instance, inhibition of dendritic cell maturation via IL-10 mediated STAT3 activation to facilitate H. pylori infection,(34) triggering the expression of the bactericidal lectin REG3γ to allow H. pylori to manipulate host immunity in order to favor bacterial survival in the gastric mucosal niche.(35,36) Especially among H. pylori, also used in our study, H. pylori CagA activates STAT3 pathway in this propagation of gastric inflammation.(11) Since H. pylori-induced STAT3 activation is considered as significant signaling network in gastric cancer,(37–39) from the current study, WPE can contribute to significant inhibitory action of STAT3 via IL-6/gp130 inhibition.
PPAR-γ is a ligand-activated transcription factor. Since 15-deoxy-D12,14-prostaglandin J2 [15d-PGJ(2)] is a potent ligand for PPAR-γ, Cha B et al.(40,41) treated 15d-PGJ(2) in H. pylori-infected gastric epithelial cells and PPAR-γ agonist significantly inhibited the activations of NADPH oxidase and RANTES expression via either inhibiting JAK1/STAT3 or NF-κB, concluding that 15d-PGJ(2) as PPAR-γ agonist can be beneficial for the treatment of H. pylori-induced gastric inflammation. Our preliminary and background study before the current investigation, we have published several results that n-3 PUFAs was quite beneficial in limiting H. pylori infection. Our group published rather clear conclusions that n-3 PUFAs exerted convincing evidence that fat-1 transgenic mice producing optimal levels of n-3 PUFAs in the stomach or intestine significantly rescued from either H. pylori-associated gastropathy or nonsteroidal anti-inflammatory drug-induced gastrointestinal damages.(18,42) Thinking clinical application from the above study, authors et al felt need to administer n-3 PUFAs as dietary intervention.(19) Ji et al.(43) studied the molecular mechanism underlying anti-inflammatory effects of n-3 PUFA, docosahexaenoic acid (DHA) in his study, against H. pylori infection and drawn similar result shown in our current study of n-3 PUFA containing WPE that DHA inhibited H. pylori-induced STAT3 phosphorylation in a PPAR-γ dependent manner.
In this investigation, with WPE administration, PPAR-γ transcribed SOCS significantly contributed to block STAT3 in H. pylori infection. Supported with our previous publication that anti-inflammatory signals of SOCS through STAT/JAK2 inactivation might be a key anti-inflammatory mechanism of probiotics, setting probiotics as a non-microbial strategy to H. pylori infection.(13) Generally, since SOCS1 not only participates in cell signaling, but also in ubiquitination mediated protein degradation process,(44–47) by which two extreme functions are possible, one is tumor growth, and the other is accelerated would healing.(48) Furthermore, focusing inflammatory regulation, anti-inflammatory as well as cytoprotection manifested as IFN-γ regulation and T cell differentiation prevails,(49,50) indicating a role in growth of gastrointestinal tissues, inflammatory bowel disease, gastritis, and cancer. The CIS (cytokine-inducible SH2 protein) and SOCS are a family of intracellular proteins, several of which have emerged as key physiological regulators of cytokine responses, including those that regulate the inflammatory systems.(51,52) SOCS, consisting of eight members (SOCS-1 to SOCS-7 and CIS), all sharing a central SH2 domain and a C-terminal SOCS box, expression of SOCS is induced by various cytokines, and its overexpression studies in various cell lines have demonstrated their inhibitory roles on JAK/STAT.(53) Expression of SOCS-1 inhibited both IL-6-induced receptor phosphorylation and STAT activation, acting in a classic negative feedback loop to regulate cytokine signal transduction.(54)
In spite of the IARC (Lyon)’s definition that H. pylori is the definite carcinogen of gastric cancer, the simple eradication of the bug is not enough to prevent resultant gastric cancer, and increasing microbial resistance further limits the eradication application.(55) Therefore, walnut, probiotics, and phytoceuticals as non-pathogenic microbial feed, can affect the host in a beneficial manner. However, the mechanism of their anti-inflammatory actions is still unclear. In the current study, we hypothesized that SOCS signaling could be a feasible anti-inflammatory mechanism of walnut against H. pylori infection. PPAR-γ/SOCS1/STAT3 pathway was documented as potential candidate. In the literature, six native Iranian plants including glycyrrhiza aspera, juglans regia, ligustrum vulgare, thymus kotschyanus, trachyspermum copticum, and xanthium brasilicum were determined as anti-H. pylori plants,(56) among which Juglans regia L. is walnut. In study showing the in vitro anti-H. pylori activity of some selected medicinal plants on clinical isolates of H. pylori, the extracts of Punica granatum and Juglans regia had remarkable anti-H. pylori activity with mean of inhibition zone diameter of 39 and 16 mm at 100 µg/disc, respectively.(57)
Walnut (Juglans regia L.) contains a complex array of natural compounds and phytochemicals, for instance, in the metabolite-profiling analysis, walnut caused a significant increase in several polyunsaturated fatty acids (PUFAs), including DHA and 9-oxo-10(E),12(E)-octadecadienoic acid (9-oxoODA), as well as kynurenic acid, that exhibits a wide range of health benefits, including anti-inflammatory, antioxidative, and regenerating actions, by which dietary walnut supplementation (14% walnut) showed significant effects in recovery from dextran sulfate sodium-induced colitis,(58) significant protection against fenitrothion- or malathion-mediated immunotoxicity,(59,60) enriching intestinal microbiome for improving health condition,(61) reducing telomere length,(62) ameliorating colitis and colitis-associated cancer,(63) suppressing colon cancer cell growth,(64) and regulating anti-cancer stem cells.(65) Though not touched in the current investigation, cancer stem cell markers including CD133, CD44, DLK1, and Notch1 as well as the β-catenin/p-GSK3β signaling pathway were significantly down-regulated and the self-renewal capacity of CSCs was suppressed. Taken together with our investigation, WPE can impose significant cancer preventive action as well as anti-cancer effects in H. pylori infection.
Author Contributions
Study concept and design: JMP and KBH; acquisition of data: YMH, JMA, and JMP; analysis and statistical analysis: SJH; interpretation of data: YMH, JMP and YJS; drafting of manuscript: YMH and KBH. All authors approved the final version of this manuscript to be published.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (No. NRF-2017R1C1B2009057 to YMH) and supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (116015-03-1-CG000).
Abbreviations
- CAG
chronic atrophic gastritis
- COX
cyclooxygenase
- H. pylori
Helicobacter pylori
- JAK
Janus kinase
- PPAR-γ
peroxisome proliferator-activated receptor gamma
- SOCS
suppressor of cytokine secretion
- STAT3
signal transducer and activator of transcription 3
- TNF-α
tumor necrosis factor-alpha
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Choi IJ, Kook MC, Kim YI, et al. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N Engl J Med 2018; 378: 1085–1095. [DOI] [PubMed] [Google Scholar]
- 2.Eid R, Moss SF. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2002; 346: 65–67. [PubMed] [Google Scholar]
- 3.Zullo A, Hassan C, Morini S. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2002; 346: 65–67. [PubMed] [Google Scholar]
- 4.Ishii Y, Shibata W, Sugimori M, et al. Activation of signal transduction and activator of transcription 3 signaling contributes to Helicobacter-associated gastric epithelial proliferation and inflammation. Gastroenterol Res Pract 2018; 2018: 9050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao S, Zhu C, Feng J, et al. Helicobacter hepaticus infection induces chronic hepatitis and fibrosis in male BALB/c mice via the activation of NF-kappaB, Stat3, and MAPK signaling pathways. Helicobacter 2020; 25: e12677. [DOI] [PubMed] [Google Scholar]
- 6.Piao JY, Lee HG, Kim SJ, et al. Helicobacter pylori activates IL-6-STAT3 signaling in human gastric cancer cells: potential roles for reactive oxygen species. Helicobacter 2016; 21: 405–416. [DOI] [PubMed] [Google Scholar]
- 7.Jeong M, Park JM, Han YM, et al. Dietary prevention of Helicobacter pylori-associated gastric cancer with kimchi. Oncotarget 2015; 6: 29513–29526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel) 2019; 11: 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong H, Du W, Sun TT, et al. A positive feedback loop between STAT3 and cyclooxygenase-2 gene may contribute to Helicobacter pylori-associated human gastric tumorigenesis. Int J Cancer 2014; 134: 2030–2040. [DOI] [PubMed] [Google Scholar]
- 10.Howlett M, Menheniott TR, Judd LM, Giraud AS. Cytokine signalling via gp130 in gastric cancer. Biochim Biophys Acta 2009; 1793: 1623–1633. [DOI] [PubMed] [Google Scholar]
- 11.Bronte-Tinkew DM, Terebiznik M, Franco A, et al. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res 2009; 69: 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cha B, Kim KH, Matsui H, Kim H. Expression of suppressors of cytokine signaling-3 in Helicobacter pylori-infected rat gastric mucosal RGM-1cells. Ann N Y Acad Sci 2007; 1096: 24–28. [DOI] [PubMed] [Google Scholar]
- 13.Lee JS, Paek NS, Kwon OS, Hahm KB. Anti-inflammatory actions of probiotics through activating suppressor of cytokine signaling (SOCS) expression and signaling in Helicobacter pylori infection: a novel mechanism. J Gastroenterol Hepatol 2010; 25: 194–202. [DOI] [PubMed] [Google Scholar]
- 14.Sharma J, Larkin J 3rd. Therapeutic implication of SOCS1 modulation in the treatment of autoimmunity and cancer. Front Pharmacol 2019; 10: 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, Inflammation, and autoimmunity. Front Immunol 2012; 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. JAKSTAT 2013; 2: e24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee HJ, Han YM, An JM, et al. Role of omega-3 polyunsaturated fatty acids in preventing gastrointestinal cancers: current status and future perspectives. Expert Rev Anticancer Ther 2018; 18: 1189–1203. [DOI] [PubMed] [Google Scholar]
- 18.Han YM, Kim KJ, Jeong M, et al. Suppressed Helicobacter pylori-associated gastric tumorigenesis in Fat-1 transgenic mice producing endogenous ω-3 polyunsaturated fatty acids. Oncotarget 2016; 7: 66606–66622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JM, Kwon SH, Han YM, Hahm KB, Kim EH. Omega-3 polyunsaturated fatty acids as potential chemopreventive agent for gastrointestinal cancer. J Cancer Prev 2013; 18: 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson KJ, Teuber SS, Gobeille A, Cremin P, Waterhouse AL, Steinberg FM. Walnut polyphenolics inhibit in vitro human plasma and LDL oxidation. J Nutr 2001; 131: 2837–2842. [DOI] [PubMed] [Google Scholar]
- 21.Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting interleukin-6 signaling in clinic. Immunity 2019; 50: 1007–1023. [DOI] [PubMed] [Google Scholar]
- 22.Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018; 15: 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H, Yoshimura A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol 2011; 31: 980–985. [DOI] [PubMed] [Google Scholar]
- 24.Chang HY, Lee HN, Kim W, Surh YJ. Docosahexaenoic acid induces M2 macrophage polarization through peroxisome proliferator-activated receptor γ activation. Life Sci 2015; 120: 39–47. [DOI] [PubMed] [Google Scholar]
- 25.Jeong M, Park JM, Han YM, et al. Dietary intervention of artemisia and green tea extracts to rejuvenate Helicobacter pylori-associated chronic atrophic gastritis and to prevent tumorigenesis. Helicobacter 2016; 21: 40–59. [DOI] [PubMed] [Google Scholar]
- 26.Jencks DS, Adam JD, Borum ML, Koh JM, Stephen S, Doman DB. Overview of current concepts in gastric intestinal metaplasia and gastric cancer. Gastroenterol Hepatol (N Y) 2018; 14: 92–101. [PMC free article] [PubMed] [Google Scholar]
- 27.Piazuelo MB, Correa P. Gastric cancer: overview. Colomb Med (Cali) 2013; 44: 192–201. [PMC free article] [PubMed] [Google Scholar]
- 28.Balic JJ, Saad MI, Dawson R, et al. Constitutive STAT3 serine phosphorylation promotes Helicobacter-mediated gastric disease. Am J Pathol 2020; 190: 1256–1270. [DOI] [PubMed] [Google Scholar]
- 29.Balic JJ, Garama DJ, Saad MI, et al. Serine-phosphorylated STAT3 promotes tumorigenesis via modulation of RNA polymerase transcriptional activity. Cancer Res 2019; 79: 5272–5287. [DOI] [PubMed] [Google Scholar]
- 30.Cha B, Lim JW, Kim H. Jak1/Stat3 is an upstream signaling of NF-kappaB activation in Helicobacter pylori-induced IL-8 production in gastric epithelial AGS cells. Yonsei Med J 2015; 56: 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gobert AP, Wilson KT. Polyamine- and NADPH-dependent generation of ROS during Helicobacter pylori infection: a blessing in disguise. Free Radic Biol Med 2017; 105: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson CB, Judd LM, Menheniott TR, et al. Augmented gp130-mediated cytokine signalling accompanies human gastric cancer progression. J Pathol 2007; 213: 140–151. [DOI] [PubMed] [Google Scholar]
- 33.Menheniott TR, Judd LM, Giraud AS. STAT3: a critical component in the response to Helicobacter pylori infection. Cell Microbiol 2015; 17: 1570–1582. [DOI] [PubMed] [Google Scholar]
- 34.Rizzuti D, Ang M, Sokollik C, et al. Helicobacter pylori inhibits dendritic cell maturation via interleukin-10-mediated activation of the signal transducer and activator of transcription 3 pathway. J Innate Immun 2015; 7: 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KS, Kalantzis A, Jackson CB, et al. Helicobacter pylori CagA triggers expression of the bactericidal lectin REG3γ via gastric STAT3 activation. PLoS One 2012; 7: e30786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaebisch R, Mejías-Luque R, Prinz C, Gerhard M. Helicobacter pylori cytotoxin-associated gene A impairs human dendritic cell maturation and function through IL-10-mediated activation of STAT3. J Immunol 2014; 192: 316–323. [DOI] [PubMed] [Google Scholar]
- 37.Zhao J, Dong Y, Kang W, et al. Helicobacter pylori-induced STAT3 activation and signalling network in gastric cancer. Oncoscience 2014; 1: 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun TT, Tang JY, Du W, et al. Bidirectional regulation between TMEFF2 and STAT3 may contribute to Helicobacter pylori-associated gastric carcinogenesis. Int J Cancer 2015; 136: 1053–1064. [DOI] [PubMed] [Google Scholar]
- 39.Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in gastric cancer. Expert Opin Ther Targets 2012; 16: 889–901. [DOI] [PubMed] [Google Scholar]
- 40.Cha B, Lim JW, Kim KH, Kim H. 15-deoxy-D12,14-prostaglandin J2 suppresses RANTES expression by inhibiting NADPH oxidase activation in Helicobacter pylori-infected gastric epithelial cells. J Physiol Pharmacol 2011; 62: 167–174. [PubMed] [Google Scholar]
- 41.Cha B, Kim KH, Kim H. 15-Deoxy-delta 12, 14,-prostaglandin J2 suppresses nuclear factor-kappaB-mediated apoptosis of Helicobacter pylori-infected gastric epithelial cells. Ann N Y Acad Sci 2009; 1171: 457–463. [DOI] [PubMed] [Google Scholar]
- 42.Han YM, Park JM, Kang JX, et al. Mitigation of indomethacin-induced gastrointestinal damages in fat-1 transgenic mice via gate-keeper action of ω-3-polyunsaturated fatty acids. Sci Rep 2016; 6: 33992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji HG, Piao JY, Kim SJ, et al. Docosahexaenoic acid inhibits Helicobacter pylori-induced STAT3 phosphorylation through activation of PPARγ. Mol Nutr Food Res 2016; 60: 1448–1457. [DOI] [PubMed] [Google Scholar]
- 44.Ying J, Qiu X, Lu Y, Zhang M. SOCS1 and its potential clinical role in tumor. Pathol Oncol Res 2019; 25: 1295–1301. [DOI] [PubMed] [Google Scholar]
- 45.Yeganeh M, Gui Y, Kandhi R, et al. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene 2016; 35: 4200–4211. [DOI] [PubMed] [Google Scholar]
- 46.Beaurivage C, Champagne A, Tobelaim WS, Pomerleau V, Menendez A, Saucier C. SOCS1 in cancer: an oncogene and a tumor suppressor. Cytokine 2016; 82: 87–94. [DOI] [PubMed] [Google Scholar]
- 47.Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci 2000; 113 (Pt 16): 2813–2819. [DOI] [PubMed] [Google Scholar]
- 48.Chevrier M, Bobbala D, Villalobos-Hernandez A, et al. Expression of SOCS1 and the downstream targets of its putative tumor suppressor functions in prostate cancer. BMC Cancer 2017; 17: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ilangumaran S, Rottapel R. Regulation of cytokine receptor signaling by SOCS1. Immunol Rev 2003; 192: 196–211. [DOI] [PubMed] [Google Scholar]
- 50.Hanada T, Kinjyo I, Inagaki-Ohara K, Yoshimura A. Negative regulation of cytokine signaling by CIS/SOCS family proteins and their roles in inflammatory diseases. Rev Physiol Biochem Pharmacol 2003; 149: 72–86. [DOI] [PubMed] [Google Scholar]
- 51.Inagaki-Ohara K, Hanada T, Yoshimura A. Negative regulation of cytokine signaling and inflammatory diseases. Curr Opin Pharmacol 2003; 3: 435–442. [DOI] [PubMed] [Google Scholar]
- 52.Greenhalgh CJ, Miller ME, Hilton DJ, Lund PK. Suppressors of cytokine signaling: relevance to gastrointestinal function and disease. Gastroenterology 2002; 123: 2064–2081. [DOI] [PubMed] [Google Scholar]
- 53.Larsen L, Röpke C. Suppressors of cytokine signalling: SOCS. APMIS 2002; 110: 833–844. [DOI] [PubMed] [Google Scholar]
- 54.Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997; 387: 917–921. [DOI] [PubMed] [Google Scholar]
- 55.Herrero R, Park JY, Forman D. The fight against gastric cancer - the IARC Working Group report. Best Pract Res Clin Gastroenterol 2014; 28: 1107–1114. [DOI] [PubMed] [Google Scholar]
- 56.Nariman F, Eftekhar F, Habibi Z, Falsafi T. Anti-Helicobacter pylori activities of six Iranian plants. Helicobacter 2004; 9: 146–151. [DOI] [PubMed] [Google Scholar]
- 57.Hajimahmoodi M, Shams-Ardakani M, Saniee P, et al. In vitro antibacterial activity of some Iranian medicinal plant extracts against Helicobacter pylori. Nat Prod Res 2011; 25: 1059–1066. [DOI] [PubMed] [Google Scholar]
- 58.Nakanishi M, Matz A, Klemashevich C, Rosenberg DW. Dietary walnut supplementation alters mucosal metabolite profiles during DSS-induced colonic ulceration. Nutrients 2019; 11: 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu H, Wan Y, Wang Y, et al. Walnut polyphenol extract protects against fenitrothion-induced immunotoxicity in murine splenic lymphocytes. Nutrients 2018; 10: 1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao Y, Fan C, Zhang A, et al. Walnut polyphenol extract protects against malathion- and chlorpyrifos-induced immunotoxicity by modulating TLRx-NOX-ROS. Nutrients 2020; 12: 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bamberger C, Rossmeier A, Lechner K, et al. A walnut-enriched diet affects gut microbiome in healthy caucasian subjects: a randomized, controlled trial. Nutrients 2018; 10: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shin PK, Zoh Y, Choi J, Kim MS, Kim Y, Choi SW. Walnut phenolic extracts reduce telomere length and telomerase activity in a colon cancer stem cell model. Nutr Res Pract 2019; 13: 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koh SJ, Choi YI, Kim Y, et al. Walnut phenolic extract inhibits nuclear factor kappaB signaling in intestinal epithelial cells, and ameliorates experimental colitis and colitis-associated colon cancer in mice. Eur J Nutr 2019; 58: 1603–1613. [DOI] [PubMed] [Google Scholar]
- 64.Lee J, Kim YS, Lee J, et al. Walnut phenolic extract and its bioactive compounds suppress colon cancer cell growth by regulating colon cancer stemness. Nutrients 2016; 8: 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chung J, Kim YS, Lee J, Lee JH, Choi SW, Kim Y. Compositional analysis of walnut lipid extracts and properties as an anti-cancer stem cell regulator via suppression of the self-renewal capacity. Food Sci Biotechnol 2016; 25: 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]