

Abstract
Negative allosteric modulation of the metabotropic glutamate 5 (mGlu5) receptor has emerged as a potential strategy for the treatment of neurologic disorders. Despite the success in preclinical studies, many mGlu5 negative allosteric modulators (NAMs) that have reached clinical trials failed due to lack of efficacy. In this study, we provide a detailed in vitro pharmacological characterization of nine clinically and preclinically tested NAMs. We evaluated inhibition of l-glutamate–induced signaling with Ca2+ mobilization, inositol monophosphate (IP1) accumulation, extracellular signal–regulated kinase 1/2 (ERK1/2) phosphorylation, and real-time receptor internalization assays on rat mGlu5 expressed in HEK293A cells. Moreover, we determined association rates (kon) and dissociation rates (koff), as well as NAM affinities with [3H]methoxy-PEPy binding experiments. kon and koff values varied greatly between the nine NAMs (34- and 139-fold, respectively) resulting in long receptor residence times (>400 min) for basimglurant and mavoglurant, medium residence times (10–30 min) for AZD2066, remeglurant, and (RS)-remeglurant, and low residence times (<10 mins) for dipraglurant, F169521, F1699611, and STX107. We found that all NAMs inhibited l-glutamate–induced mGlu5 receptor internalization, generally with a similar potency to IP1 accumulation and ERK1/2 phosphorylation, whereas Ca2+ mobilization was less potently inhibited. Operational model of allosterism analyses revealed that dipraglurant and (RS)-remeglurant were biased toward (affinity) receptor internalization and away (cooperativity) from the ERK1/2 phosphorylation pathway, respectively. Our study is the first to measure mGlu5 NAM binding kinetics and negative allosteric modulation of mGlu5 receptor internalization and adds significant new knowledge about the molecular pharmacology of a diverse range of clinically relevant NAMs.
SIGNIFICANCE STATEMENT
The metabotropic glutamate 5 (mGlu5) receptor is important in many brain functions and implicated in several neurological pathologies. Negative allosteric modulators (NAMs) have shown promising results in preclinical models but have so far failed in human clinical trials. Here we provide the most comprehensive and comparative molecular pharmacological study to date of nine preclinically/clinically tested NAMs at the mGlu5 receptor, which is also the first study to measure ligand binding kinetics and negative allosteric modulation of mGlu5 receptor internalization.
Introduction
Glutamate is the main excitatory neurotransmitter in the brain, exerting its action by activating ionotropic glutamate receptors and metabotropic glutamate (mGlu) receptors. Belonging to class C G protein–coupled receptors (GPCRs), there are eight mGlu receptor subtypes, which are important in central nervous system functions, such as learning and locomotion (Aiba et al., 1994; Anwyl, 1999; Ayala et al., 2008; Niswender and Conn, 2010). Metabotropic glutamate receptor 5 (mGlu5) is classified as a group I mGlu receptor, together with mGlu1, and is mainly expressed on postsynaptic membranes, although it can also be found on presynaptic membranes and glial cells (Shigemoto et al., 1997; Aronica et al., 2003; Kuwajima et al., 2004; Leach and Gregory, 2017). mGlu5 is ubiquitously expressed throughout the brain, and is implicated in several pathologies, such that inhibitors of mGlu5 are potential therapeutics for Alzheimer’s disease, fragile X syndrome, Parkinson’s disease, and major depressive disorder (Huber et al., 2002; Michalon et al., 2012; Hughes et al., 2013; Hu et al., 2014; Nicoletti et al., 2015).
Due to high sequence similarity in the orthosteric binding site with other mGlu receptors, targeting allosteric binding pockets in the seven-transmembrane (7TM) domain of mGlu5 has emerged as a more promising drug discovery strategy (Harpsøe et al., 2015; Leach and Gregory, 2017). Allosteric modulators offer the possibility for spatiotemporal control by modulating receptor activity only in the presence of the orthosteric ligand. In the central nervous system, preservation of preexisting signaling patterns is crucial for many cognitive processes and is important in the maintenance of balanced long-term potentiation and long-term depression (Ayala et al., 2009). Negative allosteric modulators (NAMs) of mGlu5 diminish l-glutamate–induced receptor responses, and pharmacological inhibition of mGlu5 activity in animal models has led to the suggestion that mGlu5 inhibition is a viable therapeutic strategy for the treatment of major depressive disorder, fragile X syndrome, and l-DOPA–induced dyskinesia (Dolen et al., 2007; Michalon et al., 2012; Hughes et al., 2013; Lindemann et al., 2015). Multiple mGlu5 NAMs have progressed through to phase II clinical trials, however, have failed due to either lack of efficacy or concerns over adverse effects (Scharf et al., 2015; Barnes et al., 2018; Sebastianutto and Cenci, 2018).
The failure of mGlu5 NAMs to show efficacy in clinical studies raises questions about the translatability of results obtained in preclinical data and their power to predict drug behavior in humans (Berry-Kravis et al., 2018). Therefore, a detailed pharmacological characterization of these NAMs may provide a deeper insight into events that take place at the molecular level, which may be crucial to drug efficacy in vivo. Kinetics of compound binding has attracted increasing attention in pharmacological research, and studies on association rates (kon) and dissociation rates (koff) of GPCR ligands are on the rise (Klein Herenbrink et al., 2016; Doornbos et al., 2017; Strasser et al., 2017). The duration of biologic effect of a drug depends not only on the affinity of the drug for the receptor but also on the temporal stability of this ligand-protein complex. With this in mind, the dissociation rate of a drug from the receptor could be a valuable indicator of the duration of a biologic action of the drug in vivo (Tummino and Copeland, 2008; Lu and Tonge, 2010). Receptor residence time, calculated as 1/koff, has been shown to correlate with drug activity in vivo, in some cases increasing therapeutic effects, such as in the examples of the neurokinin 1 receptor antagonist aprepitant (Lindströmet al., 2007) and the muscarinic acetylcholine M3 receptor antagonist tiotropium (Dowling and Charlton, 2006). On the other hand, a faster dissociation rate was beneficial in the prevention of side effects associated with dopamine D2 receptor antagonism (Kapur and Seeman, 2001).
In this study, we performed a detailed in vitro pharmacological characterization of a range of clinically and preclinically tested mGlu5 NAMs (Fig. 1), as studied using four different functional assays in HEK293A cells expressing physiologic levels of rat mGlu5a (HEK293A-mGlu5-low) (Noetzel et al., 2012). This is the first study to measure mGlu5 NAM binding kinetics and negative modulation of l-glutamate–induced mGlu5 receptor internalization, demonstrates the importance of testing receptor kinetics and a range of pathway assays when profiling clinical candidates, and provides a molecular pharmacological basis to advance future drug development.
Fig. 1.

Inhibition of [3H]methoxy-PEPy binding to HEK293A-mGlu5-low cell membranes by mGlu5 NAMs. Data are means ± S.E.M. from three independent experiments performed in duplicate. All compounds completely displaced [3H]methoxy-PEPy binding. Veh. denotes vehicle (1% DMSO), which was kept constant for all concentrations of NAMs. Chemical structures for each ligand are shown, showing the chemical diversity sampled within this study. Error bars not shown lie within the dimensions of the symbol.
Materials and Methods
Materials.
3-fluoro-5-[5-[2-(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridin-2-yl]benzonitrile (STX107), 1-(3-chloro-4-fluoro-phenyl)-5-methyl-N-(2-methyl-4-pyridyl)triazole-4-carboxamide (referred to as F169521), 1-(3-chloro-4-fluoro-phenyl)-N-(2-chloro-4-pyridyl)-5-methyl-triazole-4-carboxamide (referred to as F1699611), (6-bromopyrazolo[1,5-a]pyrimidin-2-yl)-[(1R)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]methanone (remeglurant), (6-bromopyrazolo[1,5-a]pyrimidin-2-yl)-[(1RS)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]methanone [(RS)-remeglurant], methyl (3aR,4S,7aR)-4-hydroxy-4-[2-(3-methylphenyl)ethynyl]-3,3a,5,6,7,7a-hexahydro-2H-indole-1-carboxylate (mavoglurant), 6-fluoro-2-[4-(2-pyridinyl)-3-butyn-1-yl]-imidazo[1,2-a]pyridine (dipraglurant), 2-chloro-4-((1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-yl)ethynyl)pyridine (basimglurant), and 4-[5-[(1R)-1-[5-(3-chlorophenyl)-3-isoxazolyl]ethoxy]-4-methyl-4H-1,2,4-triazol-3-yl]pyridine (AZD2066) were obtained from H. Lundbeck A/S (Copenhagen, Denmark). [3H]3-methoxy5-(2-pyridinylethynyl)pyridine ([3H]methoxy-PEPy) was custom synthesized by Pharmaron (Manchester, UK). IP-One assay kit, Advanced phospho-ERK1/2 (Thr202/Tyr204) assay kit, and SNAP-Lumi4-Tb were purchased from Cisbio (Codolet, France). Probenecid, Pierce BCA Protein Assay kit, and Fluo-4 acetoxymethyl no wash kit were purchased from Thermo Fisher Scientific (Waltham, MA). Hanks’ balanced salt solution (HBSS), Dulbecco’s modified Eagle’s medium (DMEM) GlutaMAX-I, dialyzed fetal bovine serum (dFBS), and penicillin-streptomycin solution were purchased from Invitrogen (Carlsbad, CA). 2-Methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) was purchased from Tocris (Bristol, UK). MicroScint-20 was purchased from PerkinElmer (Waltham, MA). pcDNA3.1(+) plasmid encoding human β2-adrenoceptor with N-terminal Flag and SNAP tags (Flag-ST-β2AR) was previously described (Roed et al., 2014) pRK5 plasmids encoding rat mGlu5a with N-terminal hemagglutinin (HA) and SNAP tags (HA-ST-rmGlu5a) and excitatory amino acid transporter 3 (EAAT3) were gifts from Laurent Prézeau (Institut de Génomique Fonctionnelle, Montpellier, France) and previously described (Brabet et al., 1998; Doumazane et al., 2011). All of the other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture.
HEK293A cells stably expressing wild-type rat mGlu5a (HEK293A-mGlu5-low) were cultured in DMEM GlutaMAX-I supplemented with 10% dFBS, 1% penicillin-streptomycin, 16 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 500 μg/ml geneticin (G418). The cells were a gift from P. J. Conn (Vanderbilt University, Nashville, TN). The HEK293A cell line was cultured in DMEM GlutaMAX-I supplemented with 10% dFBS and 1% penicillin-streptomycin. Cells were maintained in a humidified incubator at 37°C and 5% CO2.
HEK293A Cell Membrane Preparation.
HEK293A-mGlu5-low cells were harvested and snap frozen on dry ice for 5 minutes, after which they were resuspended in ice-cold homogenization buffer (50 mM Tris-Cl, 0.9% NaCl, 10 mM EDTA, pH adjusted to 7.4). Cells were homogenized with a polytron in 3 × 30 second pulses, followed by centrifugation for 10 minutes at 1000g at 4°C. Next, the supernatant was centrifuged at 30,000g at 4°C for 60 minutes using a Sorvall Evolution RC ultracentrifuge (Thermo Fisher Scientific), and the pellet was resuspended in binding buffer (HBSS supplemented with 20 mM HEPES, 1.2 mM CaCl2, 2 mM NaHCO3, pH adjusted to 7.4). Protein concentration was quantified with Pierce BCA Protein Assay kit as per manufacturer’s instructions. Membranes were stored at – 80°C before use.
Inhibition Radioligand Binding.
HEK293A-mGlu5-low cell membranes were diluted in binding buffer. Compounds were diluted in binding buffer with 1% DMSO final concentration. Compounds, [3H]methoxy-PEPy (specific activity 85 Ci/mmol), and membranes (50 μg/well) were added to a transparent 96-well plate and incubated at room temperature while being shaken for 1 hour. Membranes were harvested on GF/C filter plates using a 96-well FilterMate harvester (PerkinElmer) to separate bound and free radioligand. Filter plates were dried at room temperature overnight before addition of MicroScint-20 scintillation liquid. Scintillation spectrometry was measured on a MicroBeta2 microplate counter (PerkinElmer) after incubation for 2 hours at room temperature.
Competition Association and Dissociation Binding.
HEK293A-mGlu5-low membranes (50 μg/well) diluted in buffer were added to a transparent 96-well plate. For the competition association binding experiments, compound and [3H]methoxy-PEPy mixture were added to the plate at different time points. For the dissociation binding experiments, membranes were preincubated with [3H]methoxy-PEPy for 1 hour at room temperature, after which 1 μM MPEP was added to each well in a reverse time course. At t = 0, membranes were harvested on GF/C filter plates using a 96-well FilterMate harvester (PerkinElmer), and filter plates were dried overnight at room temperature. Radioligand binding was determined by scintillation counting as described above.
Ca2+ Mobilization Assay.
HEK293A-mGlu5-low cells were seeded on a poly-d-lysine–coated black-walled clear-bottom Falcon 96-well plate (Corning Inc., Corning, NY) at a density of 40,000 cells per well 24 hours before the assay. On the day of the assay, cells were incubated for 1 hour at 37°C in assay buffer (HBSS buffer supplemented with 20 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2 with pH adjusted to 7.4) supplemented with 0.1% bovine serum albumin (BSA) followed by 3 hours in assay buffer supplemented with 10 U/ml glutamic-pyruvic transaminase (GPT), 10 mM pyruvic acid, and 0.1% BSA. Compounds were diluted in assay buffer supplemented with 2.5 mM probenecid to 1% final DMSO concentration. Fluo-4 acetoxymethyl cell permeant dye diluted in assay buffer supplemented with 2.5 mM probenecid was added to each well, and the plate was incubated for 1 hour at 37°C. After dye loading, wells were washed with assay buffer, and plates were preincubated with NAMs for 30 minutes at 37°C. Fluorescence was measured on a FlexStation 3 plate reader (Molecular Devices, San Jose, CA) at 37°C with a single addition of l-glutamate (320 nM l-glutamate final concentration corresponding to the EC80 concentration).
Inositol Monophosphate Accumulation Assay.
HEK293A-mGlu5-low cells were seeded on a poly-d-lysine–coated Falcon 96-well cell culture plate at a density of 25,000 cells per well 24 hours before the assay. On the day of the assay, plates were incubated at 37°C for 1 hour with wash buffer (HBSS buffer supplemented with 20 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2 with pH adjusted to 7.4) supplemented with 0.1% BSA followed by 3 hours in wash buffer supplemented with 10 U/ml GPT, 10 mM pyruvic acid, and 0.1% BSA. Compounds were diluted in assay buffer (HBSS buffer supplemented with 20 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2, 40 mM LiCl with pH adjusted to 7.4) to 1% final DMSO concentration. Cells were preincubated with compounds for 30 minutes at 37°C, after which 3.2 µM of l-glutamate final concentration (corresponding to the EC80 concentration) was added and the plate was incubated for 1 hour at 37°C. Cells were lysed with 30 µl lysis buffer (IP-One assay kit) for 30 minutes at room temperature. 10 μl of lysate and 10 µl of detection solution (IP-One assay kit) were transferred to a white 384-well plate and incubated at room temperature for 1 hour, as previously explained (Nørskov-Lauritsen et al., 2014). Fluorescence emission was measured at 615 and 665 nm using an EnVision 2104 Multilabel Reader (PerkinElmer) after excitation at 340 nm. Förster resonance energy transfer (FRET) ratios were calculated as 665 nm/615 nm, and inositol monophosphate (IP1) concentrations were obtained using the IP1 standard curve of the assay kit.
Extracellular Signal–Regulated Kinase 1/2 Phosphorylation Assay.
HEK293A-mGlu5-low cells were seeded on a poly-d-lysine–coated Falcon 96-well cell culture plate at a density of 25,000 cells per well. Eight hours after seeding, cell media was substituted with starvation media (DMEM GlutaMAX-I supplemented with 1% penicillin-streptomycin and 16 mM HEPES), and cells were starved for approximately 16 hours at 37°C and 5% CO2. On the day of the assay, plates were incubated at 37°C for 1 hour in assay buffer (HBSS buffer supplemented with 20 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2 with pH adjusted to 7.4) supplemented with 0.1% BSA followed by 3 hours in assay buffer supplemented with 10 U/ml GPT, 10 mM pyruvic acid, and 0.1% BSA. Compounds were diluted in assay buffer to 1% DMSO final concentration, and plates were preincubated with the compounds for 30 minute at 37°C prior to addition of 3.2 µM l-glutamate final concentration (corresponding to the EC80 concentration). After l-glutamate addition, cells were incubated for 5 minutes at 37°C. The assay was terminated by aspiration of compounds and addition of lysis buffer [Advanced phospho-ERK1/2 (Thr202/Tyr204) assay kit]. Phosphorylated extracellular signal–regulated kinase 1/2 (ERK1/2) was determined using the Advanced phospho-ERK1/2 (Thr202/Tyr204) assay, as per manufacturer’s instructions. Fluorescence was measured at 615 and 665 nm using an EnVision 2104 Multilabel Reader after excitation at 340 nm. FRET ratios were calculated as 665 nm/615 nm.
Receptor Internalization Assay.
HEK293A cells were transiently transfected in 10-cm dishes (9 million cells/dish) with 24 µl Lipofectamine 2000, 1.2 μg EAAT3, 4.5 μg HA-ST-rmGlu5, and 3.9 µg pcDNA3.1(+) plasmids 48 hours before the assay. Twenty-four hours after transfection, cells were seeded on a white poly-d-lysine–coated Falcon 384-well culture plate (Corning Inc.) at a density of 20,000 cells per well, and plates were incubated for 24 hours at 37°C and 5% CO2. For determination of NAM interference with the internalization assay, HEK293A cells were transiently transfected with 1.33 µl/ml Lipofectamine 2000, 133 ng/ml Flag-ST-β2AR, and 400 ng/ml pcDNA3.1(+) and seeded directly in the plate at a density of 20,000 cells per well in 30 µl. On the day of the assay, plates were incubated for 2 hours at 37°C in assay buffer (HBSS buffer supplemented with 20 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2 with pH adjusted to 7.4) supplemented with 0.1% BSA followed by labeling of surface receptors with 100 nM SNAP-Lumi4-Tb diluted in assay buffer supplemented with 0.1% BSA for 1 hour at 37°C. After labeling, plates were washed twice with assay buffer supplemented with 0.1% BSA and twice with assay buffer. Compounds were diluted in assay buffer to 1% DMSO final concentration. Plates were preincubated with the compounds for 30 minutes at 37°C. Next, 50 µM fluorescein-O′-acetic acid, 11.6 µM l-glutamate (corresponding to the EC80 concentration), and 100 µM DL-threo-β-benzyloxyaspartic acid (DL-TBOA) were added. For NAM interference experiments, assay buffer or 100 µM isoprenaline was added at the same time as the compounds. Receptor internalization was measured using an EnVision 2104 Multilabel Reader every 6 minutes for 66 minutes at 37°C. The donor was excited at 340 nm, and donor and acceptor emissions were measured at 615 nm and 520 nm, respectively. Internalization ratios were calculated as 615 nm/520 nm. The assay method has been described previously (Levoye et al., 2015; Foster and Bräuner-Osborne, 2018). NAM interference was analyzed by calculating the area under the curve of 66-minute real-time isoprenaline internalization curves in presence of vehicle or NAMs after subtraction of the basal internalization. NAM concentrations that reduced the signal more than 25% compared with vehicle were excluded from further analysis.
Data Analysis.
Data were analyzed in GraphPad Prism software version 7 (GraphPad, San Diego, CA). For radioligand binding experiments, nonspecific binding was subtracted from each data point, and 100% was defined as the mean of the total specific radioligand binding. Inhibition binding data were fitted to a one-site binding function with the following equation:
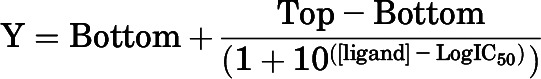 |
(1) |
where Y is the specific binding (%), Top and Bottom are the maximal and the minimal asymptotes, respectively, and IC50 is the concentration of ligand that induces a response midway between Top and Bottom and is reported as the negative logarithm within the text (pIC50). Obtained IC50 values were converted to KI estimates with the Cheng-Prusoff equation (Cheng and Prusoff, 1973), where the concentration of radioligand (1.9–2.1 nM) was slightly below the KD determined from saturation binding (see Table 1).
TABLE 1.
Affinity estimates and kinetic binding parameters for mGlu5 NAMs based on inhibition of [3H]methoxy-PEPy binding
Data are represented as means ± S.E.M. from indicated number (n) of independent experiments performed in duplicate.
| pKIa | n | konb (× 106) | koffc | Residence timed | n | pKDe | |
|---|---|---|---|---|---|---|---|
| M−1min−1 | min−1 | min | |||||
| [3H]methoxy-PEPy | 8.24 ± 0.09f | 4 | 14.2 ± 4.3 | 0.133 ± 0.006 | 7.6 ± 0.4 | 6 | 8.04 |
| AZD2066 | 8.53 ± 0.13 | 3 | 12.6 ± 1.3 | 0.045 ± 0.007 | 26.0 ± 6.2 | 5 | 8.50 |
| Basimglurant | 9.29 ± 0.06 | 3 | 13.6 ± 2.9 | 0.005 ± 0.002 | 491 ± 136 | 8 | 9.44 |
| Dipraglurant | 7.67 ± 0.11 | 3 | 33.8 ± 16.0 | 0.691 ± 0.235 | 2.2 ± 0.5 | 6 | 7.69 |
| F169521 | 7.21 ± 0.11 | 3 | 10.6 ± 3.2 | 0.471 ± 0.063 | 2.3 ± 0.4 | 4 | 7.35 |
| F1699611 | 7.58 ± 0.05 | 3 | 21.3 ± 7.4 | 0.496 ± 0.091 | 2.2 ± 0.4 | 4 | 7.63 |
| Mavoglurant | 8.10 ± 0.06 | 3 | 1.1 ± 0.2 | 0.006 ± 0.002 | 478 ± 157 | 8 | 8.26 |
| Remeglurant | 7.74 ± 0.07 | 3 | 4.5 ± 1.0 | 0.072 ± 0.016 | 19.9 ± 5.3 | 7 | 7.81 |
| (RS)-remeglurant | 7.48 ± 0.07 | 3 | 2.4 ± 0.5 | 0.078 ± 0.019 | 23.1 ± 11.9 | 5 | 7.48 |
| STX107 | 8.32 ± 0.08 | 3 | 37.9 ± 6.5 | 0.166 ± 0.011 | 6.2 ± 0.4 | 6 | 8.35 |
Negative logarithm of the equilibrium dissociation constant.
Association rate constant.
Dissociation rate constant.
Residence time is defined by 1/koff, where individual koff values approached zero for basimglurant and mavoglurant; these were limited to 0.001.
Negative logarithm of the equilibrium dissociation constant determined from kinetic parameters (koff/kon).
pKD derived from saturation binding paradigm.
koff of [3H]methoxy-PEPy was calculated with a one-phase exponential decay function using the following equation:
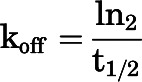 |
(2) |
kon of [3H]methoxy-PEPy was calculated using a single-phase association function with the following equation:
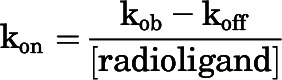 |
(3) |
where koff was calculated with eq. 2 and kon was calculated with an exponential association analysis.
Competition association binding data were fitted to the model for competitive binding (Motulsky and Mahan, 1984) to obtain kinetic rates for unlabeled ligand with the following equations:
 |
(4) |
where X represents time (minutes), L is the concentration of [3H]methoxy-PEPy, I is the concentration of the unlabeled ligand, and k1 (M−1min−1) and k2 (min−1) are the kon and koff rates of [3H]methoxy-PEPy, respectively. The kon and koff rates of [3H]methoxy-PEPy were calculated with eqs. 2 and 3 and fitted to the model to obtain the maximal number of receptors, or Bmax, and kon and koff rates of unlabeled ligand represented in the equation as k3 (M−1min−1) and k4 (min−1), respectively. Receptor residence time was calculated as 1/koff (minutes).
Data obtained from functional assays were normalized to 0% defined by the mean for the buffer value and 100% defined by the mean of the maximal orthosteric agonist response. Concentration-response curves were fitted to a four-parameter function with the following equation:
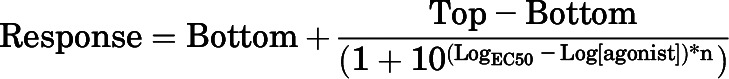 |
(5) |
where EC50 is the concentration of agonist that is required to give a half-maximal response and Top and Bottom are the maximal and minimal asymptotes, respectively, of the concentration-response curve. EC50 values are reported as negative logarithms (pEC50).
Allosteric modulation of glutamate-mediated responses was fitted to the following operational model of allosterism:
| (6) |
where [A] is the molar concentration of orthosteric ligand, [B] is the concentration of allosteric modulator, KA and KB are the equilibrium dissociation constants of the orthosteric ligand and allosteric modulator, respectively,  represents affinity cooperativity, and β is an empirical scaling factor defining the effect of a modulator on orthosteric efficacy. Parameters
represents affinity cooperativity, and β is an empirical scaling factor defining the effect of a modulator on orthosteric efficacy. Parameters 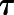 A and
A and 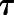 B represent the intrinsic ability of the orthosteric and allosteric ligand, respectively, to activate the receptor; Em is the maximal system response; and n represents the transducer slope. As validated previously (Gregory et al., 2012), we constrained the glutamate KA based on a previously determined affinity estimate from radioligand binding studies (Mutel et al., 2000); we also made the assumption that none of the NAMs modulated glutamate affinity (
B represent the intrinsic ability of the orthosteric and allosteric ligand, respectively, to activate the receptor; Em is the maximal system response; and n represents the transducer slope. As validated previously (Gregory et al., 2012), we constrained the glutamate KA based on a previously determined affinity estimate from radioligand binding studies (Mutel et al., 2000); we also made the assumption that none of the NAMs modulated glutamate affinity (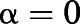 ) or had intrinsic efficacy (
) or had intrinsic efficacy (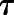 B = 0).
B = 0).
Real-time receptor internalization data were analyzed as area under the curve (AUC), and the data were normalized to 0% being the AUC of 1% DMSO and to 100% being the AUC of maximum l-glutamate activation. Normalized data of l-glutamate was fitted to a four-parameter function (eq. 5), whereas NAM data were fitted to the operational model of allosterism (eq. 6).
Fluorescence traces obtained with Ca2+ mobilization experiments were quantified in relative fluorescence units (RFUs) and represented as ΔRFU = RFUmax − RFUmin, where RFUmax is the peak value of agonist stimulation and RFUmin is the mean of the basal fluorescence that is measured for 20 seconds before agonist addition.
Results
Affinity, Association Rate, and Dissociation Rate of Binding for mGlu5 NAMs.
To date, all small molecule mGlu5 NAMs are thought to bind to a common pocket in the 7TM domain of the receptor, also known as the MPEP site (Harpsøe et al., 2015). Indeed, crystal structures of the 7TM domain of mGlu5 with different NAM chemotypes bound support this concept (Dore et al., 2014; Christopher et al., 2019). However, mGlu5 allosteric ligands, including NAMs, can possess complex binding isotherms, biased pharmacology, and differential effects in preclinical models (Trinh et al., 2018; Sengmany et al., 2019). Here we sought to undertake an in-depth assessment of the molecular pharmacological properties of diverse mGlu5 NAM chemotypes, including ligands that have progressed into clinical trials (basimglurant, mavoglurant, dipraglurant). We first measured the affinity (pKI) of each ligand based on inhibition of [3H]methoxy-PEPy binding to HEK293A-mGlu5-low cell membranes that express similar levels of mGlu5 as cortical astrocytes (Noetzel et al., 2012). All ligands fully displaced the radioligand, consistent with a competitive interaction with the MPEP binding site (Fig. 1). Of those tested, basimglurant had the highest affinity, and F169521 had the lowest, with KI estimates ranging from 0.5 to 62 nM (Table 1).
The binding kinetics of mGlu5 NAMs have not been previously described; however, for other GPCRs, ligand binding kinetics has improved predictions of in vivo efficacy (Copeland, 2016; Guo et al., 2016). To determine the kon and koff of these chemically diverse NAMs, we first determined the kon and koff rates of the radioligand [3H]methoxy-PEPy (Supplemental Fig. 1; Table 1). Kinetics of mGlu5 NAM binding was then measured using competition association experiments to generate time curves for [3H]methoxy-PEPy displacement (Fig. 2), which were globally analyzed to estimate kon and koff values for each ligand (Table 1). To confirm the robustness of our kinetic parameters, we compared the NAM affinities calculated from the kinetic parameters (pKD) to the values obtained with inhibition binding experiments (pKI) (Table 1). Here, we observed comparable affinity values across the two different experimental setups, with an R2 = 0.99 (Supplemental Fig. 2A).
Fig. 2.

Competition association binding of mGlu5 NAMs at HEK293A-mGlu5-low membranes. [3H]methoxy-PEPy specific binding measured at different time points in the absence or presence of indicated concentrations of different NAMs added simultaneously. Data are means ± S.E.M. from three to eight independent experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol. Curves shown are the best fit of the entire data set with eq. 4.
We observed a 34.5-fold difference in kon between mavoglurant and STX107, which were the NAMs with the slowest and the fastest association rate, respectively. Comparison of koff parameters revealed greater differences (138-fold) between the NAM with the slowest koff (basimglurant) and the NAM with the fastest koff (dipraglurant). These koff rates were further converted into residence times, calculated as the reciprocal value of the dissociation rate (1/koff) (Table 1). Based on these data, the NAMs could be grouped into three classes, where basimglurant and mavoglurant had slow koff/long receptor residence times (>400 minutes); medium residence times (10–30 minutes) for AZD2066, remeglurant, and (RS)-remeglurant; and fast koff/low residence times (<10 minutes) for dipraglurant, F169521, F1699611, and STX107. Furthermore, we investigated the linear relationship between kon and koff rates and the calculated NAM affinity pKD. Here, we observed a moderate correlation (R2 = 0.57) between affinity pKD and dissociation rate koff and no correlation between affinity pKD and association rate kon (Supplemental Fig. 2, B and C).
NAMs Inhibit Glutamate-Induced Ca2+ Mobilization, IP1 Accumulation, and ERK1/2 Phosphorylation.
We next measured the inhibitory effect of these NAM on mGlu5 activation of Ca2+ mobilization, IP1 accumulation, and ERK1/2 phosphorylation in response to an EC80 concentration of l-glutamate in HEK293A-mGlu5-low cells (Fig. 3). To diminish the effect of ambient/released glutamate, HEK293A cells were incubated with GPT before each assay (Sengmany et al., 2017). l-glutamate has greater potency in Ca2+ mobilization compared with IP1 accumulation and ERK1/2 phosphorylation assays (Supplemental Table 1); therefore different l-glutamate concentrations were employed to achieve ∼EC80 responses: 320 nM for Ca2+ mobilization and 3.2 μM for IP1 accumulation and ERK1/2 phosphorylation. As expected, all compounds inhibited l-glutamate–induced responses in all three functional assays. With the exception of (RS)-remeglurant in the ERK1/2 phosphorylation assay, all NAMs completely inhibited l-glutamate responses in all three measures of mGlu5 activity (Fig. 3). NAMs were consistently more potent at inhibiting l-glutamate stimulated IP1 accumulation and ERK1/2 phosphorylation than Ca2+ mobilization (Supplemental Table 2), with the exception of basimglurant, which had similar pIC50 values (within 2-fold) across all three measures.
Fig. 3.

Inhibition of l-glutamate–induced signaling in HEK293A-mGlu5-low cells. Effect of NAMs on Ca2+ mobilization response to 320 nM l-glutamate (A), IP1 accumulation in response to 3.2 μM l-glutamate (B), and ERK1/2 phosphorylation measured after 5-minute incubation with 3.2 μM l-glutamate (C). Data are means + S.E.M. from three to five independent experiments performed in triplicate. Error bars not shown lie within the dimensions of the symbol. Data were normalized to 0% as the mean of the vehicle response and 100% as the mean of maximal l-glutamate response.
NAMs Inhibit Glutamate-Induced mGlu5 Internalization.
Beyond acute activation of intracellular signal transduction pathways, allosteric ligands may also differentially influence receptor regulatory processes (Hellyer et al., 2019). Therefore, we next sought to assess the effect of mGlu5 NAMs on l-glutamate–induced receptor internalization. HEK293A cells were transiently transfected with SNAP-tagged mGlu5, as well as the EAAT3 glutamate transporter to reduce ambient glutamate. Transient mGlu5 expression resulted in ∼10 times higher mGlu5 levels compared with the stable cell line (4.6 pmol/mg vs. 0.3 pmol/mg, respectively; Supplemental Fig. 3). Introduction of a N-terminal SNAP tag did not affect orthosteric agonist potency for IP1 accumulation (data not shown). During the assay, glutamate transport was blocked with the nontransportable EAAT3 inhibitor DL-TBOA. This internalization assay relies on a time-resolved FRET technique that enables real-time measurement of receptor internalization with the help of a FRET donor Lumi4-Tb attached to the SNAP tag located at the N terminus of the receptor and a cell impermeant acceptor fluorescein-O′-acetic acid (Roed et al., 2014). There was appreciable (39% ± 4% of the AUC for 100 µM l-glutamate) increase in internalization when blocking glutamate transport with DL-TBOA without adding additional agonist (Fig. 4A). Upon stimulation with l-glutamate, we observed an increased response over unstimulated levels, indicative of agonist-induced mGlu5 internalization. Maximum mGlu5 internalization was reached around 60 minutes after stimulation with l-glutamate concentrations above 10 µM, in accordance with a previous study (Levoye et al., 2015). l-glutamate induced mGlu5 receptor internalization with a similar potency (Fig. 4B, pEC50: 5.47 ± 0.11; Supplemental Table 1) to that measured for IP1 accumulation in HEK293A-mGlu5-low cells (Supplemental Table 1). To account for ambient extracellular glutamate levels induced by inclusion of DL-TBOA in the assay, we refitted the glutamate concentration-response curve such that the bottom plateau was equal to that observed in the absence of DL-TBOA, removing responses for glutamate concentrations below the inflection point (dashed line in Fig. 4B). In doing so, we estimate that in the presence of DL-TBOA the ambient l-glutamate concentration is ∼0.9 µM.
Fig. 4.

l-glutamate–induced mGlu5 internalization and modulation by NAMs. (A) l-glutamate (added at t = 0) concentration-dependently increases mGlu5 internalization over time (based on FRET between fluorescein-O′-acetic acid and Lumi4-Tb–labeled SNAP-tagged mGlu5), achieving a maximal level within 60 minutes. DL-TBOA was included in these experiments to prevent glutamate transport and isolate changes in surface mGlu5 due to extracellular stimulation; however, this also results in apparent mGlu5 internalization over time in the absence of exogenously applied l-glutamate. In the absence of DL-TBOA, mGlu5 surface levels remain constant. (B) The area under the curve of real-time internalization ratios (ratio of donor and acceptor emissions) over 66 minutes were calculated to plot concentration-response curves. Cells were incubated with NAMs for 30 minutes prior to addition of a submaximal concentration (11.6 μM) of l-glutamate. The dashed line is the nonlinear fit of the l-glutamate response when the bottom plateau is constrained to equal the basal condition (no DL-TBOA, plotted here at −9) and glutamate concentrations below the EC50 excluded from the fit. (C and D) Data in panel B were normalized to the l-glutamate maximum response (100%) with the basal system response (no DL-TBOA) set to zero and fitted with an operational model of allosterism. In all panels, data are means + S.E.M. from three independent experiments performed in triplicate. Error bars not shown lie within the dimensions of the symbol.
Next, we investigated the effect of 30-minute preincubation with mGlu5 NAMs on receptor internalization induced by a submaximal concentration of l-glutamate (11.6 µM). High concentrations of AZD2066, remeglurant, (RS)-remeglurant, and STX107 interfered with the assay detection in a nonspecific manner (Supplemental Fig. 4), limiting the concentration ranges tested. From the real-time internalization traces (Supplemental Fig. 5), we calculated the AUC for each NAM (Fig. 4B). All NAMs reduced the l-glutamate–induced response to below the unstimulated condition (in the presence of DL-TBOA). AUC values were normalized to the maximum induced by l-glutamate (Fig. 4, C and D), with the baseline defined by the response in the absence of DL-TBOA. In contrast to results from acute signaling assays, none of the NAMs completely inhibited l-glutamate–induced mGlu5 internalization. NAM pIC50 values (Supplemental Table 2) for internalization were similar (within 3-fold) to those derived from Ca2+ mobilization assays, with the exception of basimglurant (10-fold lower) and dipraglurant (5-fold higher).
Quantification and Comparison of mGlu5 NAM Affinity and Cooperativity Estimates across Different Measures.
To quantify the affinity of NAMs (pKB) as well as apparent cooperativity with l-glutamate across the four functional measures of mGlu5 activity, we fitted the NAM titration curves in parallel with a control l-glutamate concentration-response curve to the operational model of allosterism (Gregory et al., 2012). To best fit the internalization data, the extrapolated curve with the true basal of the system was used, and the l-glutamate concentrations were corrected by subtracting the estimated level of ambient glutamate present. We first compared pKB values (Table 2) with pKI estimates obtained from equilibrium radioligand inhibition binding experiments (Fig. 5A). In all cases pKB values were lower than pKI estimates. However, we observed a high correlation between NAM pKB values in the Ca2+ mobilization (R2 = 0.82), IP1 accumulation (R2 = 0.95), and ERK1/2 phosphorylation (R2 = 0.87) assays to pKI values. On the other hand, we observed a weaker correlation of NAM pKB values in the real-time receptor internalization assay (R2 = 0.63) to pKI values. Indeed, linear regression of these data revealed that the slope for the internalization assay data was significantly different from 1 (Fig. 5A). To appreciate how each individual NAM compared across the four functional measures, we plotted the pKB estimates to visualize an affinity bias fingerprint (Fig. 5B).
TABLE 2.
Affinity estimates (pKB) for mGlu5 NAMs from functional assays
Collated data were fitted to the operational model of allosterism. Data represented as means ± S.E.M. from the indicated number (n) of independent experiments performed in triplicate.
| Ca2+ mobilization | IP1 accumulation | ERK1/2 phosphorylation | Receptor internalization | |||||
|---|---|---|---|---|---|---|---|---|
| pKB | n | pKB | n | pKB | n | pKB | n | |
| AZD2066 | 6.49 ± 0.15 | 3 | 7.82 ± 0.12 | 4 | 7.34 ± 0.14 | 3 | 6.75 ± 0.14 | 3 |
| Basimglurant | 8.33 ± 0.15 | 3 | 8.69 ± 0.13 | 4 | 8.66 ± 0.15 | 4 | 8.06 ± 0.12a | 3 |
| Dipraglurant | 5.83 ± 0.15 | 3 | 6.50 ± 0.12b | 4 | 6.67 ± 0.14b | 3 | 7.17 ± 0.13a,b | 3 |
| F169521 | 6.09 ± 0.15 | 3 | 6.19 ± 0.15 | 4 | 6.69 ± 0.14 | 3 | 6.24 ± 0.12 | 3 |
| F1699611 | 6.25 ± 0.15 | 3 | 7.08 ± 0.12b | 4 | 6.82 ± 0.14 | 3 | 6.71 ± 0.13 | 3 |
| Mavoglurant | 6.49 ± 0.15 | 3 | 7.55 ± 0.11b | 4 | 7.30 ± 0.14b | 3 | 7.04 ± 0.13 | 3 |
| Remeglurant | 6.09 ± 0.15 | 3 | 7.12 ± 0.12b | 4 | 7.09 ± 0.14b | 3 | 6.79 ± 0.13b | 3 |
| (RS)-remeglurant | 5.80 ± 0.15 | 3 | 6.54 ± 0.13b | 4 | 7.04 ± 0.13b | 3 | 6.33 ± 0.13c | 3 |
| STX107 | 6.97 ± 0.15 | 3 | 7.46 ± 0.11 | 4 | 7.52 ± 0.14 | 3 | 7.14 ± 0.19 | 3 |
Significantly different from estimate derived from IP1 accumulation assays, P < 0.05, one-way ANOVA with Tukey’s multiple comparisons post-test.
Significantly different from estimate derived from Ca2+ mobilization assays, P < 0.05, one-way ANOVA with Tukey’s multiple comparisons post-test.
Significantly different from estimate derived from ERK1/2 phosphorylation assays, P < 0.05, one-way ANOVA with Tukey’s multiple comparisons post-test.
Fig. 5.

Comparison of mGlu5 NAM affinity estimates between binding and functional measures. (A) Correlation plot for functional pKB estimates relative to inhibition binding derived pKI values for nine mGlu5 NAMs. In all cases, pKB estimates were lower than pKI values; however, there were significant correlations between binding pKI and pKB values from Ca2+ (R2 = 0.816; P = 0.0008), IP1 (R2 = 0.946; P < 0.0001), ERK1/2 phosphorylation (R2 = 0.866, P = 0.0003), and internalization (R2 = 0.631, P = 0.0106). The 95% confidence intervals of the slopes of the linear regressions for Ca2+ (black line), IP1 (gray line), and ERK1/2 phosphorylation (blue line) all included 1, but internalization (red line) was significantly different (slope = 0.70, 95% confidence interval = 0.44–0.96). Unity is represented by the dotted line. (B) Affinity estimates for each ligand across the four measures of mGlu5 function. *P < 0.05, one-way ANOVA Tukey’s multiple comparisons post-test.
A common fingerprint was evident across different scaffolds [AZD2066, dipraglurant, F1699611, mavoglurant, remeglurant, (RS)-remeglurant], which was that pKB estimates from Ca2+ mobilization assays were significantly lower (ranging from 4- to 21-fold) than those derived from IP1 accumulation and ERK1/2 phosphorylation studies. The exceptions to this fingerprint were basimglurant, STX107, and F169521, where pKB estimates were all within 4-fold of one another (Table 2). For the majority of NAMs, pKB from Ca2+ mobilization was similar to that derived from internalization assays; the exceptions were remeglurant and dipraglurant, where pKB values were higher for internalization than for Ca2+ (5- and 22-fold, respectively). Beyond apparent affinity, there was high negative cooperativity for the vast majority of compounds (Table 3). However, there were two notable exceptions. (RS)-remeglurant showed limited inhibition and therefore weaker negative cooperativity with l-glutamate for ERK1/2 phosphorylation relative to all other NAMs. In the internalization assay, the cooperativity with l-glutamate was limited for all NAMs. For (RS)-remeglurant, the limited cooperativity with l-glutamate between ERK1/2 phosphorylation and internalization was not significantly different (Student’s unpaired t test).
TABLE 3.
Cooperativity estimates (logβ) for mGlu5 NAMs from functional assays
Collated data were fitted to the operational model of allosterism. Data represented as means ± S.E.M. from the indicated number (n) of independent experiments performed in triplicate.
| Ca2+ mobilization | IP1 accumulation | ERK1/2 phosphorylation | Receptor internalization | |||||
|---|---|---|---|---|---|---|---|---|
| logβ | n | logβ | n | logβ | n | logβ | n | |
| AZD2066 | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.30 ± 0.04 | 3 |
| Basimglurant | full NAM | 3 | full NAM | 4 | full NAM | 4 | −0.41 ± 0.06 | 3 |
| Dipraglurant | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.32 ± 0.04 | 3 |
| F169521 | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.44 ± 0.07 | 3 |
| F1699611 | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.28 ± 0.03 | 3 |
| Mavoglurant | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.26 ± 0.03 | 3 |
| Remeglurant | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.39 ± 0.05 | 3 |
| (RS)-remeglurant | full NAM | 3 | full NAM | 4 | −0.24 ± 0.04 | 3 | −0.41 ± 0.07 | 3 |
| STX107 | full NAM | 3 | full NAM | 4 | full NAM | 3 | −0.30 ± 0.05 | 3 |
Discussion
Several mGlu5 NAMs have been tested in preclinical studies and clinical trials for different indications, but none has been approved for clinical use due to lack of efficacy or adverse effects. Central nervous system adverse effects, e.g., dizziness and attention deficits (Rohof et al., 2012) are associated with mGlu5 NAMs as well as psychoactive potential for AZD2066, which has been described as a class effect (Swedberg and Raboisson, 2014). For example, mavoglurant has entered clinical trials several times but failed to show efficacy in the treatment of fragile X syndrome (ClinicalTrials.gov NCT01357239; NCT01253629) and l-DOPA–induced dyskinesia (Trenkwalder et al., 2016). Basimglurant, a very potent mGlu5 NAM with long half-life in rats, has reached phase II clinical trials for the treatment of fragile X syndrome and as an adjunctive therapy in major depressive disorder (Jaeschke et al., 2015; Lindemann et al., 2015). Nevertheless, basimglurant did not show significant improved efficacy in patients with major depressive disorder and fragile X syndrome when compared with placebo (Quiroz et al., 2016; Youssef et al., 2018). Interestingly, dipraglurant showed efficacy in phase II clinical trials for the treatment of l-DOPA–induced dyskinesia in patients with Parkinson’s disease, granting it a status of an orphan drug by the US Food and Drug Administration and progression to phase III clinical trials (Emmitte, 2017). Despite these multiple clinical studies targeting mGlu5, there is currently a lack of comparative molecular pharmacological data on these NAM compounds, which could potentially explain some of the differences observed in clinical studies and inspire future drug design. Accordingly, we have performed a comprehensive pharmacological characterization of nine preclinically/clinically tested NAMs: AZD2066, basimglurant, dipraglurant, F169521, F1699611, mavoglurant, remeglurant, (RS)-remeglurant, and STX107 (Fig. 1).
Ligand binding kinetics are becoming increasingly recognized as a critical factor in drug development (Copeland, 2016; Guo et al., 2016). Here we showed that all of the mGlu5 NAMs fully displaced the radioligand [3H]methoxy-PEPy (Fig. 2), with affinity estimates comparable to previous studies where available (Kågedalet al., 2013; Dore et al., 2014; Lindemann et al., 2015; Westmark et al., 2018). For the first time, we determined the kinetic rates of association and dissociation for the NAMs. The observed koff values were spread over two orders of magnitude, whereas kon values ranged within one and a half orders of magnitude. There was a correlation between the affinity and the dissociation rate, but not the association rate (Supplemental Fig. 2), suggesting that affinity is koff- rather than kon-driven for the NAMs used in this study. We identified basimglurant and mavoglurant as NAMs with residence times longer than 7 hours, which is more than 200-fold higher than the NAMs with the lowest residence time, dipraglurant and F1699611. These data are intriguing, especially as there are few reports regarding pharmacokinetics of mGlu5 NAMs. The notable exception is that studies in rodents and clinical data in healthy subjects show that basimglurant has a much longer half-life relative to mavoglurant (Levenga et al., 2011; Gantois et al., 2013; Walles et al., 2013; Fowler et al., 2017). In our data, the association rate of basimglurant is 10-fold higher than mavoglurant; therefore kon rather than koff may be one contributing factor to the longer half-life of basimglurant. In the clinic, there are few examples where different mGlu5 NAMs have been assessed for the same indication. In patients with fragile X syndrome, neither basimglurant nor mavoglurant showed efficacy in reversing behavioral deficits (Berry-Kravis et al., 2016; Youssef et al., 2018). For L-dopa-induced dyskinesia in patients with Parkinson’s disease, although mavoglurant was no better than placebo and associated with more adverse events (Trenkwalder et al., 2016), dipraglurant, which has a much lower residence time, was well tolerated and improved dyskinesia (Tison et al., 2016). Further studies exploring the pharmacokinetics/pharmacodynamics relationships of mGlu5 NAMs are required to establish if ligand kinetics contributes to preclinical/clinical efficacy.
We used the operational model of allosterism to determine functional pKB affinity estimates for the NAMs. Generally, the rank-order of NAM affinities were similar across all measures. Basimglurant had the highest affinity in radioligand displacement and all four functional assays, consistent with previously reported pharmacology (Lindemann et al., 2015). We speculate that basimglurant recognizes a larger complement and/or more stable inactive receptor conformations than the other NAMs, which gives rise to its higher affinity. Dipraglurant, F16952, F1699611, and (RS)-remeglurant grouped as the NAMs with the lowest affinity in these five assays with the exception of the internalization assay. In the internalization assay, dipraglurant showed relatively high affinity, ranking second only to basimglurant. NAMs generally displayed higher affinity in the IP1 and ERK1/2 assays compared with Ca2+ mobilization and internalization assays (Fig. 5A). Accordingly, there was a strong correlation between affinities obtained by radioligand displacement and in the functional assays, although it was weakest for receptor internalization (R2 = 0.631 for internalization vs. R2 ≥ 0.816 for the other assays; Fig. 5A). These data highlight dipraglurant as an NAM with affinity bias toward the internalization pathway and are consistent with our previous report, where the apparent affinity of dipraglurant was dependent on the mGlu5 signaling response and cell type measured (HEK293A-mGlu5-low vs. mouse cortical neurons) (Sengmany et al., 2019). Dipraglurant also had the shortest receptor residence time (Table 1), which could potentially be a cause of the observed bias as binding kinetics have been previously shown to influence apparent bias of the dopamine D2 receptor (Klein Herenbrink et al., 2016). However, as F169521 and F1699611 displayed very similar receptor residence times without showing bias toward the internalization pathway, this explanation appears less likely. The bias could instead be caused by other mechanisms such as receptor conformational–driven bias, but more studies are needed to elucidate the mechanism.
All NAMs fully inhibited l-glutamate activation of the mGlu5 receptor in the Ca2+ mobilization and IP1 accumulation functional assays (Fig. 3, A and B). All NAMs, except (RS)-remeglurant, also fully inhibited ERK1/2 phosphorylation, whereas none of the NAMs fully inhibited receptor internalization (Fig. 3C; Fig. 4). Accordingly, the logβ cooperativity estimates derived from the operational model of allosterism showed strong negative cooperativity for NAMs, with full inhibition in the functional assays but weaker logβ values for (RS)-remeglurant in the ERK1/2 phosphorylation and for all NAMs in the internalization assay (Table 3). Given that all NAMs showed a similar partial inhibitory profile in the internalization assay, where transient expression yielded 10 times higher mGlu5 levels, and that these experiments were performed in the presence of the glutamate transporter inhibitor DL-TBOA to prevent cellular uptake of exogenous l-glutamate during the experiment, we cannot rule out that the partial inhibition is (in part) caused by this experimental condition. However, (RS)-remeglurant has a unique profile in the ERK1/2 phosphorylation assay, where it had weaker negative cooperativity than the other NAMs, indicating bias away from this pathway. It is very interesting to note the (R)-enantiomer remeglurant did not show this bias profile, suggesting that the bias is driven by the presumably less potent (S)-enantiomer. Unfortunately, (S)-remeglurant was unavailable and thus was not tested directly in the present study.
The different magnitudes of negative cooperativity by structurally diverse NAMs suggests that the different chemotypes stabilize distinct complements of receptor conformations, rather than a single inactive receptor state. Stabilization of different inactive conformations by different NAMs is consistent with limited structure-function analyses of the common class C GPCR allosteric binding pocket, where single point mutations can engender a “switch” in modulator cooperativity from positive to negative/neutral or vice versa in a chemotype-dependent manner (Petrel et al., 2004; Hu et al., 2006; Fukuda et al., 2009; Gregory et al., 2013, 2014).
Overall, this study has significantly increased our knowledge of the molecular pharmacological profiles of nine clinically and preclinically tested mGlu5 NAMs. Unfortunately, the NAMs have not been tested in comparative (pre-)clinical studies, so it is not possible to use our data to rationalize the lack of animal to human translation. Our results show that the affinities and residence times span two orders of magnitude. We also show that kinetic binding parameters kon and koff are not well correlated with binding affinities. Binding kinetics are becoming increasingly recognized as important parameters in drug development programs as, e.g., the ligand-receptor residence time can have profound effect on the pharmacodynamic effect in vivo (Copeland, 2016; Guo et al., 2016). The large differences in binding kinetics of clinically relevant NAMs tested in this study underscore the importance of taking this into consideration in future drug development programs. Finally, we show that dipraglurant and (RS)-remeglurant are biased toward the receptor internalization (i.e., relatively high affinity) and away from the ERK1/2 phosphorylation pathway (i.e., relatively low negative cooperativity), which emphasizes the importance of using a range of pathway assays when profiling clinical candidates to assess their potential signaling bias.
Acknowledgments
We thank Dr. Morten Jørgensen and Dr. Søren Møller Nielsen (H. Lundbeck A/S, Denmark) for providing the NAMs tested in this study. We thank Dr. Laurent Prézeau (Institut de Génomique Fonctionnelle, Montpellier, France) for providing the HA-ST-rmGlu5a plasmid.
Abbreviations
- 7TM
seven-transmembrane
- [3H]methoxy-PEPy
[3H]3-methoxy5-(2-pyridinylethynyl)pyridine
- AUC
area under the curve
- AZD2066
4-[5-[(1R)-1-[5-(3-chlorophenyl)-3-isoxazolyl]ethoxy]-4-methyl-4H-1,2,4-triazol-3-yl]pyridine
- basimglurant
2-chloro-4-((1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-yl)ethynyl)pyridine
- BSA
bovine serum albumin
- dFBS
dialyzed fetal bovine serum
- dipraglurant
6-fluoro-2-[4-(2-pyridinyl)-3-butyn-1-yl]-Imidazo[1,2-a]pyridine
- DL-TBOA
DL-threo-β-benzyloxyaspartic acid
- DMEM
Dulbecco’s modified Eagle’s medium
- EAAT3
excitatory amino acid transporter 3
- ERK1/2
extracellular signal–regulated kinase 1/2
- F169521
1-(3-chloro-4-fluoro-phenyl)-5-methyl-N-(2-methyl-4-pyridyl)triazole-4-carboxamide
- F1699611
1-(3-chloro-4-fluoro-phenyl)-N-(2-chloro-4-pyridyl)-5-methyl-triazole-4-carboxamide
- FRET
Förster resonance energy transfer
- GPCR
G protein–coupled receptor
- GPT
glutamic-pyruvic transaminase
- HA
hemagglutinin
- HBSS
Hanks’ balanced salt solution
- IP1
inositol monophosphate
- koff
dissociation rate
- kon
association rate
- mavoglurant
methyl (3aR,4S,7aR)-4-hydroxy-4-[2-(3-methylphenyl)ethynyl]-3,3a,5,6,7,7a-hexahydro-2H-indole-1-carboxylate
- mGlu
metabotropic glutamate
- MPEP
2-methyl-6-(phenylethynyl)pyridine hydrochloride
- NAM
negative allosteric modulator
- RFU
relative fluorescence unit
- remeglurant
(6-bromopyrazolo[1,5-a]pyrimidin-2-yl)-[(1R)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]methanone
- (RS)-remeglurant
(6-bromopyrazolo[1,5-a]pyrimidin-2-yl)-[(1RS)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]methanone
- STX107
3-fluoro-5-[5-[2-(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridin-2-yl]benzonitrile
Authorship Contributions
Participated in research design: Arsova, Vedel, Hansen, Foster, Gregory, Bräuner-Osborne.
Conducted experiments: Arsova, Møller.
Contributed new reagents or analytical tools: Hansen.
Performed data analysis: Arsova, Møller, Gregory.
Wrote or contributed to drafting the manuscript: Arsova, Møller, Gregory, Bräuner-Osborne.
Editing and approval of final manuscript: All authors.
Footnotes
A.A. acknowledges financial support from the University of Copenhagen, Oticon Foundation, and Torben and Alice Frimodts Foundation. H.B.-O. acknowledges financial support from the Augustinus Foundation and the Lundbeck Foundation. This project received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Grant 797497 (T.C.M.). This work was supported by the National Health & Medical Research Council of Australia (NHMRC): Project Grants APP1084775 (K.J.G.) and APP1127322 (K.J.G.). K.J.G. is supported by an Australian Research Council Future Fellowship: FT170100392. J.L.H. is an employee and shareholder of Novo Nordisk A/S.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. (1994) Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell 79:365–375. [DOI] [PubMed] [Google Scholar]
- Anwyl R. (1999) Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev 29:83–120. [DOI] [PubMed] [Google Scholar]
- Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, Troost D. (2003) Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci 17:2106–2118. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, et al. (2009) mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34:2057–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Niswender CM, Luo Q, Banko JL, Conn PJ. (2008) Group III mGluR regulation of synaptic transmission at the SC-CA1 synapse is developmentally regulated. Neuropharmacology 54:804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SA, Sheffler DJ, Semenova S, Cosford NDP, Bespalov A. (2018) Metabotropic glutamate receptor 5 as a target for the treatment of depression and smoking: robust preclinical data but inconclusive clinical efficacy. Biol Psychiatry 83:955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, Brinkman M, Rerat K, Koumaras B, Zhu L, et al. (2016) Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med 8:321ra5. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis EM, Lindemann L, Jonch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, et al. (2018) Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Discov 17:280–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabet I, Parmentier ML, De Colle C, Bockaert J, Acher F, Pin JP. (1998) Comparative effect of L-CCG-I, DCG-IV and gamma-carboxy-l-glutamate on all cloned metabotropic glutamate receptor subtypes. Neuropharmacology 37:1043–1051. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- Christopher JA, Orgovan Z, Congreve M, Dore AS, Errey JC, Marshall FH, Mason JS, Okrasa K, Rucktooa P, Serrano-Vega MJ, et al. (2019) Structure-based optimization strategies for G protein-coupled receptor (GPCR) allosteric modulators: a case study from analyses of new metabotropic glutamate receptor 5 (mGlu5) X-ray structures. J Med Chem 62:207–222. [DOI] [PubMed] [Google Scholar]
- Copeland RA. (2016) The drug-target residence time model: a 10-year retrospective. Nat Rev Drug Discov 15:87–95. [DOI] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. (2007) Correction of fragile X syndrome in mice. Neuron 56:955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doornbos MLJ, Cid JM, Haubrich J, Nunes A, van de Sande JW, Vermond SC, Mulder-Krieger T, Trabanco AA, Ahnaou A, Drinkenburg WH, et al. (2017) Discovery and kinetic profiling of 7-Aryl-1,2,4-triazolo[4,3-a]pyridines: positive allosteric modulators of the metabotropic glutamate receptor 2. J Med Chem 60:6704–6720. [DOI] [PubMed] [Google Scholar]
- Doré AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B, et al. (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511:557–562. [DOI] [PubMed] [Google Scholar]
- Doumazane E, Scholler P, Zwier JM, Trinquet E, Rondard P, Pin JP. (2011) A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J 25:66–77. [DOI] [PubMed] [Google Scholar]
- Dowling MR, Charlton SJ. (2006) Quantifying the association and dissociation rates of unlabelled antagonists at the muscarinic M3 receptor. Br J Pharmacol 148:927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmitte KA. (2017) mGlu5 negative allosteric modulators: a patent review (2013 - 2016). Expert Opin Ther Pat 27:691–706. [DOI] [PubMed] [Google Scholar]
- Foster SR, Bräuner-Osborne H. (2018) Investigating internalization and intracellular trafficking of GPCRs: new techniques and real-time experimental approaches. Handb Exp Pharmacol 245:41–61. [DOI] [PubMed] [Google Scholar]
- Fowler S, Guerini E, Qiu N, Cleary Y, Parrott N, Greig G, Mallalieu NL. (2017) Low potential of basimglurant to be involved in drug-drug interactions: influence of non-Michaelis-Menten P450 kinetics on fraction metabolized. J Pharmacol Exp Ther 360:164–173. [DOI] [PubMed] [Google Scholar]
- Fukuda J, Suzuki G, Kimura T, Nagatomi Y, Ito S, Kawamoto H, Ozaki S, Ohta H. (2009) Identification of a novel transmembrane domain involved in the negative modulation of mGluR1 using a newly discovered allosteric mGluR1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4H-chromen-4-one. Neuropharmacology 57:438–445. [DOI] [PubMed] [Google Scholar]
- Gantois I, Pop AS, de Esch CE, Buijsen RA, Pooters T, Gomez-Mancilla B, Gasparini F, Oostra BA, D’Hooge R, Willemsen R. (2013) Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav Brain Res 239:72–79. [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Malosh C, Mendenhall JL, Zic JZ, Bates BS, Noetzel MJ, Squire EF, Turner EM, Rook JM, et al. (2014) Identification of specific ligand-receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem Neurosci 5:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J, Conn PJ. (2013) Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol 83:991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, et al. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol Pharmacol 82:860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Heitman LH, IJzerman AP. (2016) The added value of assessing ligand-receptor binding kinetics in drug discovery. ACS Med Chem Lett 7:819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsøe K, Isberg V, Tehan BG, Weiss D, Arsova A, Marshall FH, Bräuner-Osborne H, Gloriam DE. (2015) Selective negative allosteric modulation of metabotropic glutamate receptors - a structural perspective of ligands and mutants. Sci Rep 5:13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellyer SD, Albold S, Sengmany K, Singh J, Leach K, Gregory KJ. (2019) Metabotropic glutamate receptor 5 (mGlu5 )-positive allosteric modulators differentially induce or potentiate desensitization of mGlu5 signaling in recombinant cells and neurons. J Neurochem 151:301–315. [DOI] [PubMed] [Google Scholar]
- Hu J, Jiang J, Costanzi S, Thomas C, Yang W, Feyen JH, Jacobson KA, Spiegel AM. (2006) A missense mutation in the seven-transmembrane domain of the human Ca2+ receptor converts a negative allosteric modulator into a positive allosteric modulator. J Biol Chem 281:21558–21565. [DOI] [PubMed] [Google Scholar]
- Hu NW, Nicoll AJ, Zhang D, Mably AJ, O’Malley T, Purro SA, Terry C, Collinge J, Walsh DM, Rowan MJ. (2014) mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat Commun 5:3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99:7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ZA, Neal SJ, Smith DL, Sukoff Rizzo SJ, Pulicicchio CM, Lotarski S, Lu S, Dwyer JM, Brennan J, Olsen M, et al. (2013) Negative allosteric modulation of metabotropic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression. Neuropharmacology 66:202–214. [DOI] [PubMed] [Google Scholar]
- Jaeschke G, Kolczewski S, Spooren W, Vieira E, Bitter-Stoll N, Boissin P, Borroni E, Büttelmann B, Ceccarelli S, Clemann N, et al. (2015) Metabotropic glutamate receptor 5 negative allosteric modulators: discovery of 2-chloro-4-[1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-ylethynyl]pyridine (basimglurant, RO4917523), a promising novel medicine for psychiatric diseases. J Med Chem 58:1358–1371. [DOI] [PubMed] [Google Scholar]
- Kågedal M, Cselényi Z, Nyberg S, Raboisson P, Ståhle L, Stenkrona P, Varnäs K, Halldin C, Hooker AC, Karlsson MO. (2013) A positron emission tomography study in healthy volunteers to estimate mGluR5 receptor occupancy of AZD2066 - estimating occupancy in the absence of a reference region. Neuroimage 82:160–169. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P. (2001) Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: a new hypothesis. Am J Psychiatry 158:360–369. [DOI] [PubMed] [Google Scholar]
- Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, et al. (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7:10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima M, Hall RA, Aiba A, Smith Y. (2004) Subcellular and subsynaptic localization of group I metabotropic glutamate receptors in the monkey subthalamic nucleus. J Comp Neurol 474:589–602. [DOI] [PubMed] [Google Scholar]
- Leach K, Gregory KJ. (2017) Molecular insights into allosteric modulation of Class C G protein-coupled receptors. Pharmacol Res 116:105–118. [DOI] [PubMed] [Google Scholar]
- Levenga J, Hayashi S, de Vrij FM, Koekkoek SK, van der Linde HC, Nieuwenhuizen I, Song C, Buijsen RA, Pop AS, Gomezmancilla B, et al. (2011) AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis 42:311–317. [DOI] [PubMed] [Google Scholar]
- Levoye A, Zwier JM, Jaracz-Ros A, Klipfel L, Cottet M, Maurel D, Bdioui S, Balabanian K, Prézeau L, Trinquet E, et al. (2015) A broad G protein-coupled receptor internalization assay that combines SNAP-Tag labeling, diffusion-enhanced resonance energy transfer, and a highly emissive terbium cryptate. Front Endocrinol (Lausanne) 6:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann L, Porter RH, Scharf SH, Kuennecke B, Bruns A, von Kienlin M, Harrison AC, Paehler A, Funk C, Gloge A, et al. (2015) Pharmacology of basimglurant (RO4917523, RG7090), a unique metabotropic glutamate receptor 5 negative allosteric modulator in clinical development for depression. J Pharmacol Exp Ther 353:213–233. [DOI] [PubMed] [Google Scholar]
- Lindström E, von Mentzer B, Påhlman I, Ahlstedt I, Uvebrant A, Kristensson E, Martinsson R, Novén A, de Verdier J, Vauquelin G. (2007) Neurokinin 1 receptor antagonists: correlation between in vitro receptor interaction and in vivo efficacy. J Pharmacol Exp Ther 322:1286–1293. [DOI] [PubMed] [Google Scholar]
- Lu H, Tonge PJ. (2010) Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol 14:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky HJ, Mahan LC. (1984) The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol 25:1–9. [PubMed] [Google Scholar]
- Mutel V, Ellis GJ, Adam G, Chaboz S, Nilly A, Messer J, Bleuel Z, Metzler V, Malherbe P, Schlaeger EJ, et al. (2000) Characterization of [(3)H]Quisqualate binding to recombinant rat metabotropic glutamate 1a and 5a receptors and to rat and human brain sections. J Neurochem 75:2590–2601. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Ngomba RT, Gradini R, Battaglia G. (2015) Metabotropic glutamate receptors as drug targets: what’s new? Curr Opin Pharmacol 20:89–94. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, et al. (2012) Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol 81:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørskov-Lauritsen L, Thomsen AR, Bräuner-Osborne H. (2014) G protein-coupled receptor signaling analysis using homogenous time-resolved Förster resonance energy transfer (HTRF®) technology. Int J Mol Sci 15:2554–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M. (2004) Positive and negative allosteric modulators of the Ca2+-sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J Biol Chem 279:18990–18997. [DOI] [PubMed] [Google Scholar]
- Quiroz JA, Tamburri P, Deptula D, Banken L, Beyer U, Rabbia M, Parkar N, Fontoura P, Santarelli L. (2016) Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry 73:675–684. [DOI] [PubMed] [Google Scholar]
- Roed SN, Wismann P, Underwood CR, Kulahin N, Iversen H, Cappelen KA, Schäffer L, Lehtonen J, Hecksher-Soerensen J, Secher A, et al. (2014) Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol Cell Endocrinol 382:938–949. [DOI] [PubMed] [Google Scholar]
- Rohof WO, Lei A, Hirsch DP, Ny L, Astrand M, Hansen MB, Boeckxstaens GE. (2012) The effects of a novel metabotropic glutamate receptor 5 antagonist (AZD2066) on transient lower oesophageal sphincter relaxations and reflux episodes in healthy volunteers. Aliment Pharmacol Ther 35:1231–1242. [DOI] [PubMed] [Google Scholar]
- Scharf SH, Jaeschke G, Wettstein JG, Lindemann L. (2015) Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr Opin Pharmacol 20:124–134. [DOI] [PubMed] [Google Scholar]
- Sebastianutto I, Cenci MA. (2018) mGlu receptors in the treatment of Parkinson’s disease and l-dopa-induced dyskinesia. Curr Opin Pharmacol 38:81–89. [DOI] [PubMed] [Google Scholar]
- Sengmany K, Hellyer SD, Albold S, Wang T, Conn PJ, May LT, Christopoulos A, Leach K, Gregory KJ. (2019) Kinetic and system bias as drivers of metabotropic glutamate receptor 5 allosteric modulator pharmacology. Neuropharmacology 149:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengmany K, Singh J, Stewart GD, Conn PJ, Christopoulos A, Gregory KJ. (2017) Biased allosteric agonism and modulation of metabotropic glutamate receptor 5: implications for optimizing preclinical neuroscience drug discovery. Neuropharmacology 115:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, et al. (1997) Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17:7503–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Seifert R. (2017) Binding kinetics and pathways of ligands to GPCRs. Trends Pharmacol Sci 38:717–732. [DOI] [PubMed] [Google Scholar]
- Swedberg MD, Raboisson P. (2014) AZD9272 and AZD2066: selective and highly central nervous system penetrant mGluR5 antagonists characterized by their discriminative effects. J Pharmacol Exp Ther 350:212–222. [DOI] [PubMed] [Google Scholar]
- Tison F, Keywood C, Wakefield M, Durif F, Corvol JC, Eggert K, Lew M, Isaacson S, Bezard E, Poli SM, et al. (2016) A phase 2A trial of the novel mGluR5-negative allosteric modulator dipraglurant for levodopa-induced dyskinesia in Parkinson’s disease. Mov Disord 31:1373–1380. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C, Stocchi F, Poewe W, Dronamraju N, Kenney C, Shah A, von Raison F, Graf A. (2016) Mavoglurant in Parkinson’s patients with l-dopa-induced dyskinesias: two randomized phase 2 studies. Mov Disord 31:1054–1058. [DOI] [PubMed] [Google Scholar]
- Trinh PNH, May LT, Leach K, Gregory KJ. (2018) Biased agonism and allosteric modulation of metabotropic glutamate receptor 5. Clin Sci (Lond) 132:2323–2338. [DOI] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. (2008) Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 47:5481–5492. [DOI] [PubMed] [Google Scholar]
- Walles M, Wolf T, Jin Y, Ritzau M, Leuthold LA, Krauser J, Gschwind HP, Carcache D, Kittelmann M, Ocwieja M, et al. (2013) Metabolism and disposition of the metabotropic glutamate receptor 5 antagonist (mGluR5) mavoglurant (AFQ056) in healthy subjects. Drug Metab Dispos 41:1626–1641. [DOI] [PubMed] [Google Scholar]
- Westmark PR, Dekundy A, Gravius A, Danysz W, Westmark CJ. (2018) Rescue of Fmr1KO phenotypes with mGluR5 inhibitors: MRZ-8456 versus AFQ-056. Neurobiol Dis 119:190–198. [DOI] [PubMed] [Google Scholar]
- Youssef EA, Berry-Kravis E, Czech C, Hagerman RJ, Hessl D, Wong CY, Rabbia M, Deptula D, John A, Kinch R, et al. FragXis Study Group (2018) Effect of the mGluR5-NAM basimglurant on behavior in adolescents and adults with fragile X syndrome in a randomized, double-blind, placebo-controlled trial: FragXis phase 2 results. Neuropsychopharmacology 43:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]