

Abstract
The Cdk8 kinase module (CKM) is a detachable Mediator subunit composed of cyclin C and one each of paralogs Cdk8/Cdk19, Med12/Med12L, and Med13/Med13L. Our previous RNA-Seq studies demonstrated that cyclin C represses a subset of hydrogen peroxide–induced genes under normal conditions but is involved in activating other loci following stress. Here, we show that cyclin C directs this transcriptional reprograming through changes in its promoter occupancy. Following peroxide stress, cyclin C promoter occupancy increased for genes it activates while decreasing at loci it represses under normal conditions. Promoter occupancy of other CKM components generally mirrored cyclin C, indicating that the CKM moves as a single unit. It has previously been shown that some cyclin C leaves the nucleus following cytotoxic stress to induce mitochondrial fragmentation and apoptosis. We observed that CKM integrity appeared compromised at a subset of repressed promoters, suggesting a source of cyclin C that is targeted for nuclear release. Interestingly, mTOR inhibition induced a new pattern of cyclin C promoter occupancy indicating that this control is fine-tuned to the individual stress. Using inhibitors, we found that Cdk8 kinase activity is not required for CKM movement or repression but was necessary for full gene activation. In conclusion, this study revealed that different stress stimuli elicit specific changes in CKM promoter occupancy correlating to altered transcriptional outputs. Finally, although CKM components were recruited or expelled from promoters as a unit, heterogeneity was observed at individual promoters, suggesting a mechanism to generate gene- and stress-specific responses.
Keywords: transcription, chromatin immunoprecipitation (ChIP), transcription promoter, oxidative stress, mammalian target of rapamycin (mTOR), cyclin-dependent kinase (CDK), cell death, transcription regulation
Cells exhibit multiple adaptive responses following exposure to cytotoxic agents. The failure to mitigate the cellular damage caused by these stressors can initiate regulated cell death (RCD) pathways (1). One common cause of cellular damage is reactive oxygen that can be derived either endogenously (e.g. elevated respiration, oxidase overexpression) or by environmental exposure to pro-oxidants (2), such as H2O2 or chemotherapeutics (3). An important early adaptation is transcriptional reprogramming, which directs many molecular processes within the cell. The transcriptional adaptations induced by oxidative stress include the up-regulation of both pro-death and prosurvival genes. For example, oxidative stress stimulates the tumor suppressor p53-dependent activation of genes that promote intrinsic RCD (iRCD) (4). In addition, genes encoding proteins mitigating the effects of oxidative stress are also induced (4). This system allows the cell to balance the amount of damage incurred with its ability to facilitate repairs. In contrast to oxidative stress, amino acid starvation inhibits mTOR, resulting in induction of several genes necessary for cell survival pathways including autophagy (5). However, prolonged mTOR inhibition represses cell growth eventually inducing cell death (6). Therefore, the balance between survival and death is influenced by the type, intensity, and duration of the stress that is relayed to the nucleus to impact transcription.
Our previous RNA-Seq studies identified the oxidative stress cyclin C-regulated transcriptome in mouse (Mus musculus) embryonic fibroblast (MEF) cells (7). Cyclin C was identified as a transcriptional repressor of meiotic and stress-response genes in the yeast Saccharomyces cerevisiae (8–11). However, mammalian cyclin C functions as both a transcriptional activator and a repressor, with an essential role in development (12–14). Cyclin C is a component of the Cdk8 kinase module (CKM), which is a detachable and transiently interacting subunit of the Mediator complex (15). The other members of the CKM include Cdk8 or Cdk19, Med12 or Med12L, and Med13 or Med13L (16). Cdk8/Cdk19, Med12/Med12L, and Med13/Med13L are paralogs with similar but not identical functions that are incorporated independently into CKMs, along with cyclin C, in a 1:1:1:1 stoichiometry (17). Given this dual role in transcription, it may not be surprising that genetic and epidemiological studies have provided evidence that the CKM can either stimulate or suppress tumorigenesis (18). For example, CDK8 overexpression stimulates colon and breast cancer progression (19, 20). Accordingly, Cdk8 inhibitors have been developed and tested in preclinical studies, demonstrating a positive impact within multidrug combination regimens (21). Conversely, deleting Ccnc (cyclin C) stimulates hyperplasia in thyroid and acute lymphoblastic leukemia mouse models (18, 22). Interestingly, there is evidence that the CKM regulates transcription in both a CDK8 kinase-dependent and kinase-independent manner (23). These studies indicate that the CKM plays a complicated role in transcriptional control responding to diverse inputs. However, how the CKM itself is controlled to mediate this dual transcriptional activity is unclear.
In addition to its nuclear role, ∼15–20% of cyclin C, but not Cdk8, translocates to the cytoplasm in response to oxidative stress (24). In addition to inactivating Cdk8 to alter transcription, cytoplasmic cyclin C directly binds both the dynamin-like GTPase Drp1 and the pro-apoptotic factor Bax to promote mitochondrial fragmentation and efficient iRCD execution, respectively (24–26). This mitochondrial role is conserved in yeast and our laboratory demonstrated that Med13 is required for cyclin C nuclear retention (27). To allow cyclin C nuclear release in response to oxidative stress in yeast, Med13 is degraded via the ubiquitin proteasome system (UPS) in a highly regulated process requiring multiple signaling pathways and Cdk8 activity (28, 29). Similarly, structural studies revealed that Med13/Med13L tether the CKM to the Mediator complex, suggesting that its role in retaining cyclin C in the nucleus is conserved (30). However, it is not clear how the pool of cyclin C is selected for nuclear release in mammalian cells and whether Med13/Med13L destruction is involved. Here, ChIP studies revealed that promoter recruitment or loss of cyclin C and the CKM directly correlated with its role as a transcriptional activator or repressor. This profile changed following starvation stress, indicating that cyclin C and CKM movements are precisely determined by the insult. Finally, CKM integrity was disrupted at a subset of repressed promoters suggesting a source for cyclin C nuclear release. Taken together, these findings are consistent with a model that the CKM is a dynamic complex that directs transcriptional repression and activation through changes in promoter occupancy at target genes.
Results
Cyclin C transcriptional regulation of oxidative stress-responsive genes
Our transcriptome analysis revealed that cyclin C mediates both positive and negative transcriptional control of genes induced by H2O2 exposure (7). To begin investigating this observation, we used Gene Ontology (31) to identify biological processes enriched in the data set for genes induced by H2O2 treatment. We chose to study two pathways, p53-induced genes and those required for autophagy as they represent stresses leading to cell death and survival, respectively. The p53 stress-response genes included Trp53inp1 (tumor protein p53–inducible nuclear protein 1), p21 (Cdkn1a; cyclin-dependent kinase inhibitor 1), and Jmy (junction-mediating and -regulatory protein) (labeled in figures in bold italic font). The autophagic genes chosen were Gabarap (GABA receptor–associated protein) and Prkaa2 (5′-AMP-activated protein kinase catalytic subunit α-2) (labeled in figures in underlined italic font). In addition, three genes, Adrm1 (proteasomal ubiquitin receptor Adrm1), Gtf2h1 (general transcription factor IIH subunit 1), and Crebrf (CREB3 regulatory factor), were chosen that were induced by H2O2 but are neither the p53-dependent nor autophagy-responsive genes (labeled in figures in italic font). Finally, Sall1 (Sal-like protein 1) and Col4a1 (Collagen α-1(IV) chain) were chosen as cyclin C–repressed and activated their transcription under nonstress conditions, respectively, but their transcript levels are not affected by H2O2 stress.
We first confirmed the transcriptional changes observed with RNA-Seq by RT-qPCR analysis. As expected, the three gene sets exhibited various levels of induction in response to H2O2 treatment, whereas Sall1 and Col4a1 were not affected (Fig. 1A). Two patterns of transcription were observed for cyclin C–activated genes. For the p53-dependent set, cyclin C is required for steady-state levels of Trp53inp1 and p21 but accounted for only partial H2O2 induction (Fig. 1A, left side). The transcriptional analysis of nine additional H2O2-induced genes controlled by p53 identified seven that required cyclin C for activation and two that were repressed during normal growth (Fig. S1). The same patterns were observed in that cyclin C is required for maintaining both steady-state transcription and full H2O2-induced induction. A second pattern was observed for Adrm1 and Gabarap transcription in that cyclin C was required for both steady-state levels and any H2O2-mediated induction. For the cyclin C–repressed genes, two transcription patterns were again observed depending on p53 requirement. Deleting Ccnc resulted in a 4-fold derepression of Jmy mRNA that was not enhanced by H2O2 treatment (Fig. 1A, right side). However, H2O2-treated WT cells only displayed a 2-fold elevation in Jmy mRNA, suggesting that the pro-oxidant treatment only partially relieves cyclin C–dependent repression. Similar to Jmy regulation, two repressed genes (Dyrk3 and Noxa) exhibited elevated mRNA levels in the absence of cyclin C beyond that induced by H2O2 treatment in WT cells (Fig. S1). These findings suggest that H2O2 treatment does not elicit the full activation of these loci. A second profile was observed for autophagy-induced Prkaa2 and two additional repressed genes Gtf2h1 and Crebrf. These loci displayed mRNA levels in Ccnc−/− that matched WT H2O2-treated cells (Fig. 1A, right side). Moreover, the addition of H2O2 did not increase transcription in Ccnc−/− cells. These findings suggest that H2O2 treatment was able to remove all cyclin C–dependent repression.
Figure 1.

Cyclin C transcriptional regulation of oxidative stress-responsive genes. A, RT-qPCR analysis for the genes indicated in WT or Ccnc−/− MEF cultures before or after H2O2 treatment as indicated. The results are given as fold change from WT untreated control. The values are presented ± WT untreated control (Log2). B, ChIP analysis was quantified by qPCR for target promoter DNA that was first normalized to the input DNA control then compared with a nonspecific antibody control (GFP). The values are presented as percentages of input for a standard fraction of chromatin solution. Genes indicated in bold type are members of the p53 regulon. Genes indicated by underlining are also induced by autophagic conditions. Remaining loci represent other genes induced by H2O2 but are not members of the first two sets. C and D, RT-qPCR (C) and ChIP analysis (D) were conducted as just described for Angptl4. For all results, statistical significance is indicated by the following: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Next, ChIP analysis was used to monitor cyclin C promoter occupancy at these promoters before and after H2O2 treatment. For all genes, cyclin C demonstrated promoter enrichment over background in unstressed cells (Fig. 1B), suggesting that its control of the transcription at each locus is direct. Antibody specificity controls are shown in Fig. S2 for the Trp53inp1 locus. Following H2O2 exposure, there was a significant increase in promoter occupancy for the two p53-dependent genes activated by cyclin C (Trp53inp1 and p21) (Fig. 1B, left side). Previous reports demonstrated increased promoter occupancy at the p21 promoter by the CKM members cyclin C, Cdk8, and Med12 following Nutlin, but not UVC, treatment (32). In contrast, there was a significant decrease in promoter occupancy of cyclin C following oxidative stress for the repressed gene Jmy (Fig. 1B, right side). These results indicate that cyclin C is recruited to promoters when its transactivation ability is employed but removed when its repression function is no longer wanted. Similar increases in occupancy were observed for the activated autophagy Gabarap and independent Adrm1 genes. Likewise, loss in cyclin C occupancy was observed for the repressed genes Prkaa2, Gtf2h1, and Crebrf, consistent with removing CKM repression. No changes in promoter occupancy were observed for the H2O2-independent genes Col4a1 and Sall1. Taken together, these results are consistent with a model that cyclin C recruitment helps direct normal gene induction, whereas its removal is important for transcriptional derepression. To test whether this model is predictive, we examined cyclin C promoter occupancy for a member of an expression class that requires cyclin C for its steady state expression but is downregulated in response to H2O2 (Fig. 1C). According to our model, this gene set should exhibit loss of cyclin C occupancy from its promoter following stress. We tested one member of this group, Angptl4, and found that both cyclin C and Cdk8 were removed from the promoter following H2O2 treatment (Fig. 1D). Therefore, changes in CKM occupancy may be predictive from the transcriptional profile (repression vs. activation) and the transcriptional change after stress treatment.
The promoter occupancy of CKM components largely mirrors that of cyclin C
Our results indicate that cyclin C occupancy is increased when it serves a positive role in transcription but removed to relieve its repressor function. Biochemical studies purifying the CKM suggest that it is mostly found as an intact complex (15). Therefore, we next asked whether other components of the CKM also display similar dynamic changes in promoter occupancy. ChIP experiments were conducted using antibodies targeting three additional members of the CKM (Cdk8, Med13, and Med13L). Med13 and Med13L were chosen because their individual activities have not been clearly delineated. The results indicated significant promoter occupancy by each of the four members for each gene in WT unstressed cells for both cyclin C–activated (Fig. 2A) and –repressed (Fig. 2B) genes. Following H2O2 exposure, promoter occupancy patterns of the CKM members generally mirrored that observed for cyclin C for each of the induced genes Trp53inp1, p21, Gabarap, and Adrm1 (Fig. 2A). As expected, no change in promoter occupancy for any of these proteins was observed for H2O2-independent Col4a1. For the repressed genes Jmy and Gtf2h1, there was a significant decrease in promoter occupancy of each CKM member in response to oxidative stress similar to that observed for cyclin C (Fig. 2B). However, CKM promoter occupancy at Prkaa2 and Crebrf gave an intermediate result. Specifically, although cyclin C levels were diminished to nearly background levels at these promoters, significant levels of the remaining CKM members were detected (Fig. 2B, arrows). These findings indicate that cyclin C was separated from other CKM components at a subset of the Prkaa2 or Crebrf promoters. Interestingly, ChIP analysis showed that Cdk8 occupancy at the Trp53inp1 promoter was at the limits of detection in Ccnc−/− cells (Fig. S2). These results suggest a different fate for Cdk8 promoter occupancy when cyclin C is completely absent versus removed in response to stress. Together, these data indicate that promoter movements by cyclin C and other CKM components are largely similar, arguing that the CKM moves as a single unit to and from promoters. A potential caveat to this conclusion is the two repressed promoters, Prkaa2 and Crebrf, in which cyclin C and CKM removal appear disconnected.
Figure 2.

CKM members maintain similar but not identical promoter occupancy patterns at target genes. ChIP analysis for the CKM components indicated are shown cyclin C–activated (A) and -repressed (B) loci before and following H2O2 treatment. Two genes whose mRNA expression is not altered by H2O2 (Col4a1 and Sall1) are shown. Arrows indicate genes displaying a disconnect between loss in cyclin C occupancy compared with the other CKM components. Data obtained from ChIP were compared with a nonspecific control antibody (GFP). For all results, statistical significance is indicated by the following: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
A subset of genes is regulated by cyclin C in response to both oxidative stress and mTOR inhibition
Previous studies have revealed regulatory cross-talk between the oxidative stress and autophagic signaling systems (33). Specifically, it has been reported that low-level oxidative stress induces autophagy (34), whereas amino acid starvation triggers a low-level oxidative stress response (35). To determine whether cyclin C exhibited coregulation of H2O2- and autophagy-induced genes, WT MEF cultures were treated with the mTOR inhibitor Torin1 (32). Similar to H2O2 stress, cyclin C was required for both steady-state and Torin1-induced transcription of Gabarap and Adrm1 (Fig. 3A, left side). In addition, two repressed genes, Prkaa2 and Crebrf, exhibited derepression following Torin1 treatment that was similar to those observed in Ccnc−/− cells, suggesting that removing cyclin C–dependent repression was sufficient for maximal induction (Fig. 3A, right side). Interestingly, one gene repressed by cyclin C, Gtf2h1, exhibited reduced mRNA levels following Torin1 treatment (Fig. 3A, arrow). These findings indicate that Torin1 and H2O2 treatment is processed similarly though cyclin C activity, although differences exist.
Figure 3.
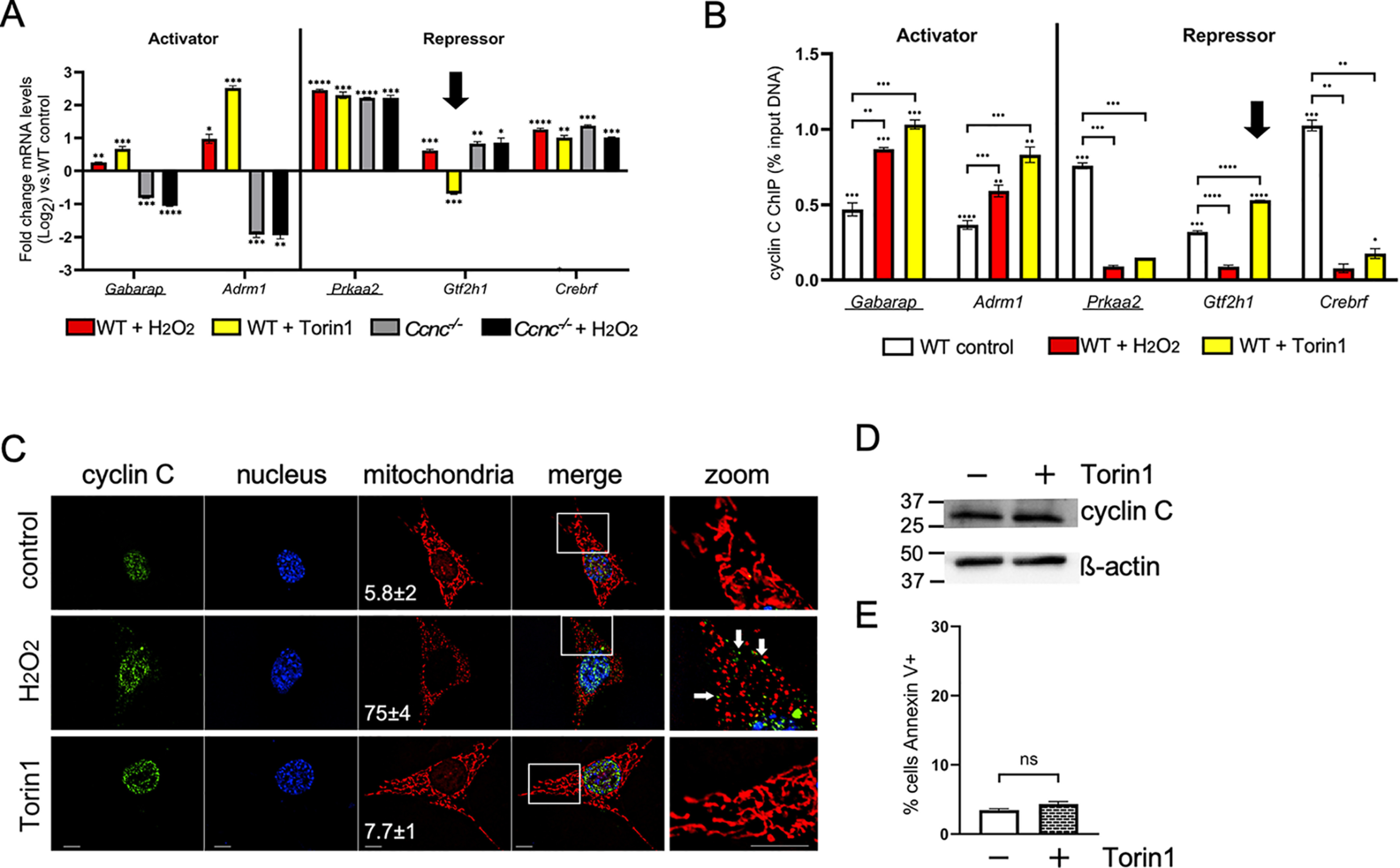
Cyclin C activates and represses an overlapping gene set induced following H2O2 and Torin1 treatment. A, mRNA levels for the indicated genes were determined by RT-qPCR following exposure to oxidative stress and Torin1. Genes known to be up-regulated under starvation conditions are underlined. Results from Fig. 1 for Ccnc−/− cells are shown for comparison. Arrow indicates Gtf2h1 repression following Torin 1 treatment. B, cyclin C ChIP analysis of the genes described in A. The arrow indicates the elevated occupancy of cyclin C at Gtf2h1 coincident with increased repression. C, subcellular localization of cyclin C was determined by immunocytochemistry. Mitochondria and nuclei were identified with MitoTracker Red and 4′,6-diamidino-2-phenylindole staining, respectively. The scales in the lower left corners of the images indicate 10 and 40 nm for the zoomed-in image. The percentage of the cells exhibiting mitochondrial fragmentation is indicated (n = 3, ± S.D). D, cyclin C levels in cultures following exposure to Torin1 (250 nm, 4 h). β-Actin was used as a loading control. Molecular mass markers (kDa) are indicated. E, the percentage of the population annexin V–positive following Torin1 treatment was determined by flow cytometry. Statistical significance is indicated by the following: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, not significant.
Similar to H2O2 stress, ChIP analysis following Torin1 exposure revealed increased cyclin C promoter occupancy for the induced genes Gabarap and Adrm1 (Fig. 3B, left side). In addition, cyclin C promoter occupancy was reduced at both Prkaa2 and Crebrf promoters. These results are consistent with our model that removing cyclin C from promoters is part of the derepression process. Although the impact of cyclin C is opposite, these data suggest the possibility that a common pathway is used to induce these loci following both H2O2 exposure and mTOR inhibition. However, Gtf2h1 mRNA levels decreased following Torin1 treatment (Fig. 3A, arrow), opposite to that observed for H2O2 stress. Because our results indicate that cyclin C is recruited to promoters demonstrating transcriptional induction, we would predict that its promoter occupancy would also increase following Torin1 treatment to establish additional Torin1-induced repression. ChIP analysis proved this prediction correct with a significant increase in cyclin C promoter occupancy following Torin1 treatment compared with untreated cells (Fig. 3B). Further analysis of the p53-mediated stress-response genes Trp53inp1, p21, and Jmy found no or little change in mRNA levels between control and Torin1-treated cells (Fig. S3A). Consistent with this result, cyclin C promoter occupancy also remained unchanged (Fig. S3B). Our ChIP results from the repressed Prkaa2 and Crebrf promoters suggested a possible source for nuclear released cyclin C following H2O2 treatment (Fig. 2B). Similarly, cyclin C is removed from these two promoters in response to Torin1 treatment. However, unlike H2O2 exposure, cyclin C remained nuclear following Torin1 treatment, and the mitochondria remained intact (Fig. 3C). Finally, there was no significant difference in cyclin C levels (Fig. 3D) or iRCD induction (Fig. 3E) in response to Torin1 treatment. These results suggest that not only is CKM promoter occupancy altered in response to a particular stimuli, its integrity may also be precisely controlled. These findings underscore the flexibility the cell has in employing cyclin C to fine-tune transcription and mitochondrial responses to various stressors.
Locus-specific release of cyclin C is regulated by the UPS
Under normal growth conditions, mammalian Med13 and Med13L exhibit a steady-state turnover mediated by the UPS (36). This study found that Med13 or Med13L degradation correlated with decreased CKM-Mediator association (36). In yeast, Med13 degradation in response to oxidative stress is also mediated by the UPS (27). Therefore, we tested whether H2O2-induced cyclin C promoter release and transcriptional derepression required the UPS. First, WT cells were treated with the proteasome inhibitor MG-132 prior to H2O2 treatment. Analysis of the mRNA levels from the cyclin C–repressed genes exhibited three different responses. The mRNA levels of the p53-regulated gene Jmy were unchanged compared with H2O2 treatment alone (Fig. 4A). Conversely, Gtf2h1 and Crebrf transcription was elevated above H2O2 treatment alone, suggesting that both stressors contributed to their increased transcription. Finally, H2O2-induced Prkaa2 mRNA induction was reduced upon MG-132 addition. This result suggested that MG-132 inhibited full derepression. ChIP analysis for the genes Jmy, Gtf2h1, and Crebrf showed a reduction in promoter occupancy following exposure to the combination treatment, comparable with that of oxidative stress alone (Fig. 4B). However, although cyclin C occupancy was reduced at the Prkaa2 promoter following treatment with both drugs, the level of reduction was significantly less than observed in H2O2-treated cells. These results are consistent with a model that MG-132 prevented Med13 and/or Med13L proteolysis, allowing at least partial retention of the CKM and reduced transcriptional repression. If correct, we would predict that Med13 and/or Med13L levels would be reduced in response to H2O2, but this effect was blunted by UPS inhibition. Consistent with this possibility, Western blotting analysis of Med13 and Med13L in extracts prepared from MEF cultures treated with H2O2 revealed an reduction of about 20% in protein levels compared with the untreated controls (Fig. 4C; quantified in Fig. 4D). However, this reduction was reversed upon addition of MG-132. Taken together, these results are consistent with the previous results from yeast and mammalian cells that cyclin C promoter release is associated with Med13/Med13L degradation. Importantly, not all promoters demonstrated elevated cyclin C retention following MG-132 treatment is consistent with our previous findings that the regulation of cyclin C occupancy and CKM integrity are controlled in a locus-specific manner.
Figure 4.

Release of cyclin C from promoters does not require Med13/Med13L degradation. A and B, RT-qPCR (A) and ChIP analysis (B) for the cyclin C–repressed genes indicated following H2O2/MG-132 treatment. ChIP data are shown as percentages of input DNA compared with a nonspecific control (GFP antibody). H2O2-treated WT control is from Fig. 1 to aid in comparing results. C, Med13 and Med13L levels were determined by Western blotting analysis in extracts prepared from MEF cultures following exposure to H2O2 and/or MG-132 as indicated. The β-actin concentrations were obtained using separate blots. The same β-actin blots are reproduced in both panels to allow easy comparison. D, the ratios of Med13 and Med13L signals to the β-actin internal control were calculated then compared to WT untreated control (n = 3). Statistical significance is indicated by the following: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Cyclin C regulates transcription both dependent and independent of Cdk8 kinase activity
Our yeast studies revealed that Cdk8 phosphorylation is required for Med13 destruction and subsequent cyclin C nuclear release into the cytoplasm (28). To test whether Cdk8 activity is required for the changes observed for the CKM promoter occupancy in mammalian cells, we directly compared transcription and promoter occupancy in Ccnc−/− mutants, and WT cells treated with the Cdk8 kinase inhibitor Senexin A (37). Following 24 h of Senexin A treatment, the mRNA levels of cyclin C–activated genes Trp53inp1, p21, Gabarap, and Adrm1 were reduced comparable with the Ccnc−/− cells (Fig. 5A, left side). These data indicate that before stress, steady-state transcription of these genes depends on Cdk8 kinase activity. Similarly, the p53-dependent genes Trp53inp1 and p21 were induced to levels similar to that observed in Ccnc−/− MEFs following H2O2 and Senexin A treatment (Fig. 5A). Moreover, the two additional cyclin C–activated genes, Gabarap and Adrm1, remained down-regulated following Senexin A treatment phenocopying the Ccnc−/− deletion results (Fig. 5A). These results indicate that Cdk8 kinase activity is required for both steady-state transcription and induction following H2O2 treatment although for differing levels depending on the locus. The examination of cyclin C promoter occupancy for these activated genes revealed that cyclin C recruitment to these promoters following H2O2 treatment did not require Cdk8 kinase activity (Fig. 5B, left side).
Figure 5.

Cyclin C transcriptional activation, but not repression, is dependent Cdk8 kinase activity. A and B, RT-qPCR (A) and ChIP (B) analyses are shown for the indicated genes following treatment with Senexin A and H2O2. Values for H2O2-treated cultures are from Fig. 1 to aid in comparisons. C, cyclin C levels in MEFs following Senexin A treatment (1 μm, 24 h), and Senexin A + H2O2 (0.4 mm, 4 h) were determined by Western blotting analysis. β-Actin was used as a loading control. D, the percentage of the population annexin V–positive following Torin1 treatment was determined by flow cytometry (n = 3). Statistical significance is indicated by the following: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Surprisingly, the two cyclin C–repressed genes, Jmy and Crebrf, were slightly down-regulated following Senexin A treatment compared with controls (Fig. 5A, right side). This is in contrast to Ccnc−/− MEFs that exhibited derepression of both genes. These results indicate that transcriptional repression of Jmy and Crebrf is independent of Cdk8 kinase activity. The up-regulation of Jmy and Crebrf following exposure to oxidative stress is associated with the promoter release of cyclin C. Compared with WT unstressed MEFs, ChIP analysis shows loss of cyclin C occupancy with Senexin A treatment (Fig. 5B, right side). However, subtle differences were observed with cyclin C release in Senexin A + H2O2-treated cells at the Crebrf promoter. Specifically, the gene Crebrf showed significantly more cyclin C at its promoter under these conditions (p = 0.02; Student's t test). However, the observed increase in cyclin C promoter occupancy for the gene Jmy was not statistically significant. These data indicate that although differences in cyclin C promoter occupancy are largely independent of Cdk8 kinase activity, its complete removal following H2O2 stress may be compromised. These differences are not due to changes in overall cyclin C levels following Senexin A addition (Fig. 5C). Interestingly, there was a significant decrease in iRCD efficiency in Senexin A–treated cells following exposure to oxidative stress (Fig. 5D). These results indicate that Cdk8 kinase activity was required for efficient iRCD. This reduction was not as severe as that observed in Ccnc−/− cells (Fig. 5D), suggesting differences exist between Senexin A treatment and Ccnc deletion for iRCD control. This difference may be due to the additional role of cyclin C at the mitochondria in oxidatively stressed cells (see “Discussion”). Taken together, these data demonstrate that Cdk8 kinase activity is required for full transcriptional activation, whereas repression appears independent of this function.
Discussion
The CKM interacts with the Mediator complex to induce the transcription of genes responsive to several stress signaling pathways including p53-mediated (32), oxidative (7), or hypoxia (38). In addition, we found that cyclin C represses steady-state transcription of genes that are also induced by H2O2 treatment (7). However, mechanisms underlying how the CKM performs both positive and negative locus-specific roles in response to the same stress remain unclear. Because the CKM is a removable, substoichiometric component of Mediator, this report tested whether its gain or loss from promoters provided a regulatory mechanism to achieve this control. Using the CKM component cyclin C as a probe to monitor promoter occupancy, we examined both activated and repressed loci that respond to two stressors: H2O2-induced oxidative stress and starvation through mTOR inhibition. As observed in previous reports (32, 38), we found that cyclin C is recruited to promoters that require the CKM for stress-induced activation. In addition, we found that cyclin C occupancy also increases when additional repression is required. Conversely, cyclin C is removed from stress-induced genes repressed by the CKM under normal conditions. These patterns of cyclin C occupancy are specific to whether H2O2 or starvation stress was administered, suggesting that changes in CKM promoter occupancy may provide a strategy to support this complicated regulatory system. Our results also revealed that the other CKM members largely followed cyclin C promoter recruitment or release patterns, indicating that this complex moved as a unit, although exceptions were noted (see below). Given the relatively low concentration of CKM in the cell, these results suggest that the CKM being released from a promoter undergoing derepression has two fates. First, the intact CKM is recruited to a promoter requiring this complex for transcriptional activation (Fig. 6). Although speculative, this model takes into account early RNA polymerase II holoenzyme/Mediator purifications revealing that most of the CKM in the cell was associated with the Mediator and not part of a significant free pool (39). More comprehensive studies are required to determine whether the CKM removed from one promoter can be accounted for by occupancy increase at others. The other path involves dissolution of the CKM to allow cyclin C alone to translocate to the cytoplasm where it interacts with proteins at the mitochondria to induce fission and iRCD. To definitively establish this model, additional genes will need to be examined that are repressed by cyclin C and induced following H2O2 treatment. Taken together, these results reveal that the CKM exhibits remarkable locus-specific movement to and from promoters based on its transcriptional role. This heterogeneity in response observed at individual promoters suggests that this system offers the cell a myriad of combinations with which to combine transcriptional control with CKM fate.
Figure 6.

Model for CKM fate following H2O2 stress treatment. Examples of cyclin C–repressed (top panel) and activated (bottom panel) genes are shown. The stress signal induces CKM release from genes repressed under normal conditions. The CKM has two fates. Intact CKM leaving repressed promoters transits to loci to stimulate transcription (bottom panel). A subpopulation of CKMs dissolves allowing free cyclin C to exit the nucleus (top panel) and initiate mitochondrial fragmentation and iRCD via Drp1 and Bax recruitment, respectively.
CKM dysregulation is implicated in a wide variety of disorders and cancers (18). The finding that Med13 ablation leads to early embryonic lethality (40) suggested that Med13 and Med13L have at least partially independent regulatory roles. However, our analysis of cyclin C, Cdk8, Med13, and Med13L promoter occupancy found that the CKM generally moves as a unit both to and from promoters, although some differences were observed (Fig. 2). In addition, with the possible exception of the repressed Jmy locus, Med13 and Med13L were found at all promoters analyzed and behaved similarly with respect to promoter recruitment or removal. These results may suggest that, at least for this gene subset, Med13 and Med13L have similar functions regulating transcription. Therefore, another interpretation of the knockout results is that removing Med13 or Med13L may invoke a gene dosage-related phenotype. For example, recent studies found that MED13 or MED13L haploinsufficiency results in several overlapping, but not identical, developmental syndromes (41–46). Therefore, transcriptional control by Med13 and Med13L may be exquisitely sensitive to gene copy number. Eliminating both copies of Med13 or Med13L may be sufficient to disrupt embryonic transcriptional control sufficient to cause lethality. Analyzing the fate of mice heterozygous for both Med13 and Med13L may address this question.
Our previous results from yeast revealed that Med13 destruction is required for the nuclear release of cyclin C following oxidative stress (27). These results fit well with structural studies indicating that Med13 tethers the CKM to the Mediator (17). Additional yeast studies found that the CKM is also disrupted in response to many stressors including starvation (47). However, closer examination revealed that the fate of the CKM components changed depending on the stress. For example, in response to oxidative stress, the yeast Med13 is destroyed by the UPS, allowing cyclin C release into the cytoplasm to stimulate mitochondrial fragmentation and RCD prior to its destruction (28). In response to nitrogen starvation, cyclin C has a different fate. Rather than undergoing nuclear release, cyclin C is degraded before cytoplasmic relocalization, protecting the cell from its mitochondrial role promoting regulated cell death (48). Similar to the yeast results, we found that in response to H2O2 or Torin1 treatment, cyclin C is removed from promoters that it represses under normal growth conditions (Fig. 3B). However, several differences were observed between yeast and mammalian cells. First, cyclin C levels do not change significantly following H2O2 or starvation stress in murine cells. In addition, unlike yeast, in which the CKM is completely dismantled, this complex appears to remain mostly intact as it moves from one promoter to another. These differences may reflect the additional transcriptional responsibilities the CKM has in mammalian cells. In addition to stress or developmental responsive genes, RNA-Seq analysis also revealed a role for cyclin C in the transcriptional activation of genes involved in energy production and proliferation (7). Therefore, metazoans may have leveraged the requirement for cyclin C nuclear release with the increased role of the CKM in stress-independent transcription.
ChIP analysis revealed that CKM regulation was not always uniform. For example, H2O2 treatment resulted in nearly complete removal of cyclin C at the Prkaa2 and Crebrf promoters, but only partial reduction in Cdk8, Med13, or Med13L. This pattern was not observed for other repressed genes, Jmy and Gtf2h1. These results suggest that the CKM was partially dissolved at the Prkaa2 and Crebrf promoters. A potential explanation for these results is based on our finding of the mitochondrial role of cyclin C mentioned above. In the cytoplasm, cyclin C mediates mitochondrial fragmentation and programmed cell death in MEF cells through its interaction with mitochondrial fission proteins Drp1 and Bax, respectively (25, 26, 49). These observations may indicate that the CKM at the Prkaa2 and Crebrf promoters is disrupted to allow cyclin C nuclear release. In yeast, this release is triggered by Med13 destruction via the UPS. If a similar strategy is employed in MEFs, we would predict that MG-132 treatment would prevent CKM promoter release. In addition, failure to release the CKM would perhaps allow retention of its repressor function resulting in reduced transcription. These predictions were observed, albeit partially, with Prkaa2 in which cyclin C was retained and reduced H2O2-induced activation was observed following MG-132 treatment (Fig. 4). This partial response may be due to the heterogeneity of Med13/Med13L fate at these repressed promoters or simply a result of MG-132 treatment not completely inactivating the proteasome. Our finding that Med13 steady-state levels were increased after MG-132 treatment indicated that at least partial proteolysis protection was achieved. However, not all repressed genes behaved in this manner and even at the Prkaa2 promoter cyclin C still exhibited partial removal. These findings suggest that cyclin C release from promoters is facilitated by more than one mechanism that may be governed by locus-specific cues. Even at promoters that exhibit MG-132 sensitivity, there may be heterogeneity in the response. These findings suggest that the rules governing CKM movement and integrity are complex and may involve an intersection of signal transduction pathways, gene-specific transcription factors, and promoter chromatin contexts.
Our results are consistent with current models of the positive role for the CKM through one or more reported targets of Cdk8, including histone H3 or Cdk9 (P-TEFb) (47, 50). The differences observed in the requirement of cyclin C for promoter activation may be due to the presence of locus-specific transactivators. For example, the p53-dependent response network only partly requires cyclin C for full gene activation in H2O2-stressed cells. Conversely, Adrm1 and Gabarap require cyclin C for both steady-state and stress-induced transcription. Many of the genes induced by cyclin C following H2O2 treatment mediate iRCD. Together with the finding that Cdk8 kinase activity is required for transactivation, it is not surprising that Senexin A significantly reduced iRCD in response to oxidative stress. Interestingly, deleting Ccnc resulted in a substantial reduction in iRCD efficiency in response to H2O2-induced oxidative stress treatment (Fig. 5D). The difference between the iRCD impact of deleting Ccnc and inhibiting Cdk8 may be due to the two, separate contributions of cyclin C. Similar to Senexin A treatment, Ccnc ablation inactivates Cdk8. However, the additional cytoplasmic role of cyclin C at the mitochondria may represent the additional impact on iRCD efficiency. Therefore, Senexin A treatment may demonstrate the transcriptional aspect of cyclin C–Cdk8–dependent iRCD. We have previously demonstrated that cyclin C nuclear release is not dependent on Cdk8 activity (24). Therefore, deleting Ccnc removes both functions, resulting in more cellular protection from H2O2 treatment. This conclusion may be a bit simplistic because deleting Ccnc also removes the repressor function of the CKM. The contribution of the repressor aspect of cyclin C function to iRCD efficiency has yet to be separated from its other activities. Taken together, these findings reveal the contribution of cyclin C at multiple levels of the oxidative stress-response pathway.
Experimental procedures
Cell culture
Ccnc+/+ (WT) and Ccnc−/− (24) immortalized MEF cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C and 5% CO2. WT and Ccnc−/− MEF cultures were grown to 75% confluence and treated as follows. Oxidative stress included exposure to 0.4 mm H2O2 (Sigma–Aldrich, CAS: 7722-84-1) in Dulbecco's modified Eagle's medium without fetal bovine serum for 4 h. The cells were treated with 250 nm Torin1 (Santa Cruz Biotechnology; CAS: 1222998-36-8) for 4 h. The cells were treated with 1 μm Senexin A (Tocris; CAS: 1780390-76-2) for 24 h. The cells were treated with 5 μm MG-132 (Calbiochem; CAS: 133407-82-6) for 1 h before oxidative stress treatment. The treatment protocols above were completed for 24 h for iRCD assay analysis. All experiments were conducted with three independent trials conducted in triplicate for each condition.
Immunocytochemistry/immunofluorescence
MEFs were cultured as described above on coverslips and then fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, blocked with 2% BSA, and incubated with primary antibody (1:2000 dilution of anti–cyclin C–Bethyl-A301-989A). Following a washing step, the coverslips were incubated with secondary antibody (1:2000 dilution of anti-rabbit Alexa Fluor 488; Invitrogen; A-11008) and washed again before coverslips were mounted with 4′,6-diamidino-2-phenylindole–containing medium (Vector Laboratory). The images were acquired with a Nikon Eclipse 90i microscope equipped with a Retiga Exi charge-coupled device camera and NIS software for data analysis. Mitochondrial staining was completed using MitoTracker Red CMXRos 30 min before fixing, as described by the manufacturer (Molecular Probes). Quantitation was accomplished with three independent cultures, with 200 cells counted per sample. All microscopy experiments were conducted with three independent experiments conducted in triplicate for each condition.
Western blotting analysis
Total cell lysate was prepared from cultured cells, as described above, in RIPA buffer (150 mm NaCl, 1% Igepal CA-630, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris-HCl, pH 8, 5 mm EDTA, 20 mm NaF, 0.2 mm sodium orthovanadate, and Sigma mammalian protease inhibitors) for 30 min with mild agitation at 4 °C. Lysates were centrifuged at 13,000 × g for 10 min at 4 °C. Protein concentrations were determined by the Bradford protein assay (Bio-Rad), and 30 μg of total protein was used for analysis. Western blotting analysis for cyclin C was conducted with rabbit polyclonal primary antibody (1:2500 dilution) directed against cyclin C (Bethyl-A301-989A) and anti-rabbit secondary antibody (1:5000 dilution) (Abcam; ab6702). Western blotting analysis for Med13 and Med13L were conducted with mouse mAb (1:2500 dilution) directed against Med13 (Santa Cruz Biotechnology; sc515557) and anti-mouse secondary antibody (1:5000 dilution) (Abcam; ab6708); and rabbit polyclonal primary antibody (1:2500 dilution) directed against Med13L (Bethyl-A302-421A) and anti-rabbit secondary antibody (1:5000 dilution) (Abcam; ab6702). Western blots were incubated with CDP-STAR (CDP*) substrate, and signals were visualized using an iBright FL1500 imaging system (Thermo). β-Actin (1:2500 dilution) (Abcam-ab8227) was used as loading controls. All Western blotting analyses were conducted with three independent experiments conducted in triplicate for each condition. The quantitation of Med13 and Med13L protein signals were completed using iBright Analysis software.
Flow cytometry iRCD assays
MEFs were seeded in culture conditions described above in 12-well plates at a density of 5 × 104 cells and grown for 24 h; before stress application for 24 h, as described above. Annexin V assays were conducted following the manufacturer's instructions (BD Biosciences), using a BD AccuriTM C6 flow cytometer (BD Biosciences). All iRCD experiments were conducted with three independent experiments conducted in triplicate for each condition.
ChIP assays
WT and Ccnc−/− MEF cultures treated as described above were cross-linked with 1% formaldehyde for 10 min at room temperature. The cross-linking reaction was quenched by the addition of 125 mm final concentration glycine. The cells were washed with cold PBS (containing Sigma mammalian protease inhibitors); lysed with RIPA buffer (150 mm NaCl, 1% Igepal CA-630, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris-HCl, pH 8, 5 mm EDTA, 20 mm NaF, 0.2 mm sodium orthovanadate, and Sigma mammalian protease inhibitors); and subjected to sonication to produce DNA fragments of <500 bp. For immunoprecipitation, 1 μg of antibody (α-cyclin C (Bethyl-A301-989A), α-Cdk8 (Santa Cruz Biotechnology; sc1521), α-Med13 (Santa Cruz Biotechnology; sc515557), α-Med13L (Bethyl-A302-421A), or α-GFP (Takara Bio; 632375)), was added to samples and incubated for 8 h at 4 °C. The α-GFP antibody served as a nonspecific control. The lysates were then incubated with magnetic Dynabeads (protein G) (Invitrogen; 10004D) to collect immunoprecipitated samples. Following incubation, the beads were washed twice with RIPA buffer, four times with ChIP wash buffer (100 mm Tris-HCl, pH 8.5, 500 mm LiCl, 1% (v/v) Nonidet P-40, 1% (w/v) deoxycholic acid), twice with RIPA buffer again, and twice with 1× TE (10 mm Tris-HCl, pH 8.0, 1 mm EDTA). Immunocomplexes were eluted for 10 min at 65 °C with 1% SDS, and cross-linking was reversed by adding NaCl to a final concentration of 200 mm and incubating for 5 h at 65 °C. DNA was purified for each sample, and a fraction (1/100) was used in qPCR amplification using ThermoFisherTM PowerSYBRTM Green PCR Master Mix and a StepOneTM real-time PCR system. Primers specific to each individual gene promoter region were designed for quantification of the signal via the percentage input method. Primer sequences for ChIP qPCR analysis can be found in Table S1. ChIP assays were conducted with three independent preparations in triplicate for statistical analysis via Student's t test.
RT-qPCR analysis
Total RNA was prepared from the cell samples using Monarch® total RNA miniprep kit. On-column DNase treatment was performed to eliminate contaminating DNA during RNA extraction. Total RNA (500 ng) was converted to cDNA using the ThermoFisherTM Maxima cDNA synthesis kit. The cDNA from each sample (1/100 dilution) was subjected to qPCR amplification using ThermoFisherTM PowerSYBRTM Green PCR Master Mix and a StepOneTM real-time PCR system. These assays were conducted with three independent preparations assayed in duplicate. Gapdh was used as the internal standard for comparative (ΔΔCT) quantitation (51). Statistical significance was determined via Student's t test analysis. Primer sequences for RT-qPCR analysis can be found in Table S1.
Data availability
All data are contained with the article.
Supplementary Material
Acknowledgments
We thank Michael Law and David Stillman for helpful discussions and comments on this manuscript.
This article contains supporting information.
Author contributions—D. C. S. data curation; D. C. S. formal analysis; D. C. S. methodology; D. C. S. writing-original draft; K. F. C. and R. S. conceptualization; K. F. C. funding acquisition; R. S. project administration; R. S. writing-review and editing.
Funding and additional information—This work was supported by National Institutes of Health Grants GM113196 (to K. F. C.) and GM113052 (to R. S.). This work was also supported by funds from the W. W. Smith Foundation (to K. F. C.) and the New Jersey Camden Health Initiative (to R. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- CKM
- Cdk8 kinase module
- RCD
- regulated cell death
- iRCD
- intrinsic RCD
- MEF
- mouse embryonic fibroblast
- UPS
- ubiquitin–proteasome system
- qPCR
- quantitative PCR.
References
- 1. Fulda S., Gorman A. M., Hori O., and Samali A. (2010) Cellular stress responses: cell survival and cell death. Int J. Cell Biol. 2010, 214074 10.1155/2010/214074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Avery S. V. (2011) Molecular targets of oxidative stress. Biochem. J. 434, 201–210 10.1042/BJ20101695 [DOI] [PubMed] [Google Scholar]
- 3. Martins N. M., Santos N. A., Curti C., Bianchi M. L., and Santos A. C. (2008) Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J. Appl. Toxicol. 28, 337–344 10.1002/jat.1284 [DOI] [PubMed] [Google Scholar]
- 4. Siauciunaite R., Foulkes N. S., Calabrò V., and Vallone D. (2019) Evolution shapes the gene expression response to oxidative stress. Int. J. Mol. Sci. 20, 2040 10.3390/ijms20082040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schworer C. M., and Mortimore G. E. (1979) Glucagon-induced autophagy and proteolysis in rat liver: mediation by selective deprivation of intracellular amino acids. Proc. Natl. Acad. Sci. U.S.A. 76, 3169–3173 10.1073/pnas.76.7.3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hua H., Kong Q., Zhang H., Wang J., Luo T., and Jiang Y. (2019) Targeting mTOR for cancer therapy. J. Hematol. Oncol. 12, 71 10.1186/s13045-019-0754-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stieg D. C., Chang K.-T., Cooper K. F., and Strich R. (2019) Cyclin C regulated oxidative stress responsive transcriptome in Mus musculus embryonic fibroblasts. G3 (Bethesda) 9, 1901–1908 10.1534/g3.119.400077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strich R., Slater M. R., and Esposito R. E. (1989) Identification of negative regulatory genes that govern the expression of early meiotic genes in yeast. Proc. Natl. Acad. Sci. U.S.A. 86, 10018–10022 10.1073/pnas.86.24.10018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cooper K. F., Mallory M. J., Smith J. B., and Strich R. (1997) Stress and developmental regulation of the yeast C-type cyclin Ume3p (Srb11p/Ssn8p). EMBO J. 16, 4665–4675 10.1093/emboj/16.15.4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Surosky R. T., Strich R., and Esposito R. E. (1994) The yeast UME5 gene regulates the stability of meiotic mRNAs in response to glucose. Mol. Cell. Biol. 14, 3446–3458 10.1128/mcb.14.5.3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holstege F. C., Jennings E. G., Wyrick J. J., Lee T. I., Hengartner C. J., Green M. R., Golub T. R., Lander E. S., and Young R. A. (1998) Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95, 717–728 10.1016/S0092-8674(00)81641-4 [DOI] [PubMed] [Google Scholar]
- 12. Loncle N., Boube M., Joulia L., Boschiero C., Werner M., Cribbs D. L., and Bourbon H. M. (2007) Distinct roles for Mediator Cdk8 module subunits in Drosophila development. EMBO J. 26, 1045–1054 10.1038/sj.emboj.7601566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li N., Fassl A., Chick J., Inuzuka H., Li X., Mansour M. R., Liu L., Wang H., King B., Shaik S., Gutierrez A., Ordureau A., Otto T., Kreslavsky T., Baitsch L., et al. (2014) Cyclin C is a haploinsufficient tumour suppressor. Nat. Cell Biol. 16, 1080–1091 10.1038/ncb3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westerling T., Kuuluvainen E., and Mäkelä T. P. (2007) Cdk8 is essential for preimplantation mouse development. Mol. Cell. Biol. 27, 6177–6182 10.1128/MCB.01302-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bourbon H. M. (2008) Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 36, 3993–4008 10.1093/nar/gkn349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Borggrefe T., Davis R., Erdjument-Bromage H., Tempst P., and Kornberg R. D. (2002) A complex of the Srb8, -9, -10, and -11 transcriptional regulatory proteins from yeast. J. Biol. Chem. 277, 44202–44207 10.1074/jbc.M207195200 [DOI] [PubMed] [Google Scholar]
- 17. Allen B. L., and Taatjes D. J. (2015) The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 16, 155–166 10.1038/nrm3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clark A. D., Oldenbroek M., and Boyer T. G. (2015) Mediator kinase module and human tumorigenesis. Crit. Rev. Biochem. Mol Biol 50, 393–426 10.3109/10409238.2015.1064854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eirew P., Steif A., Khattra J., Ha G., Yap D., Farahani H., Gelmon K., Chia S., Mar C., Wan A., Laks E., Biele J., Shumansky K., Rosner J., McPherson A., et al. (2015) Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–426 10.1038/nature13952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Firestein R., Bass A. J., Kim S. Y., Dunn I. F., Silver S. J., Guney I., Freed E., Ligon A. H., Vena N., Ogino S., Chheda M. G., Tamayo P., Finn S., Shrestha Y., Boehm J. S., et al. (2008) CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 455, 547–551 10.1038/nature07179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Giovanni C., Novellino E., Chilin A., Lavecchia A., and Marzaro G. (2016) Investigational drugs targeting cyclin-dependent kinases for the treatment of cancer: an update on recent findings (2013-2016). Expert Opin. Investig. Drugs 25, 1215–1230 10.1080/13543784.2016.1234603 [DOI] [PubMed] [Google Scholar]
- 22. Jezek J., Wang K., Yan R., Di Cristofano A., Cooper K. F., and Strich R. (2019) Synergistic repression of thyroid hyperplasia by cyclin C and Pten. J. Cell Sci. 132, jcs230029 10.1242/jcs.230029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menzl I., Zhang T., Berger-Becvar A., Grausenburger R., Heller G., Prchal-Murphy M., Edlinger L., Knab V. M., Uras I. Z., Grundschober E., Bauer K., Roth M., Skucha A., Liu Y., Hatcher J. M., et al. (2019) A kinase-independent role for CDK8 in BCR-ABL1+ leukemia. Nat. Commun. 10, 4741 10.1038/s41467-019-12656-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang K., Yan R., Cooper K. F., and Strich R. (2015) Cyclin C mediates stress-induced mitochondrial fission and apoptosis. Mol. Biol. Cell 26, 1030–1043 10.1091/mbc.E14-08-1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ganesan V., Willis S. D., Chang K. T., Beluch S., Cooper K. F., and Strich R. (2019) Cyclin C directly stimulates Drp1 GTP affinity to mediate stress-induced mitochondrial hyper-fission. Mol. Biol. Cell 30, 302–311 10.1091/mbc.E18-07-0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jezek J., Chang K. T., Joshi A. M., and Strich R. (2019) Mitochondrial translocation of cyclin C stimulates intrinsic apoptosis through Bax recruitment. EMBO Rep. 20, e47425 10.15252/embr.201847425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khakhina S., Cooper K. F., and Strich R. (2014) Med13p prevents mitochondrial fission and programmed cell death in yeast through nuclear retention of cyclin C. Mol. Biol. Cell 25, 2807–2816 10.1091/mbc.E14-05-0953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stieg D. C., Willis S. D., Ganesan V., Ong K. L., Scuorzo J., Song M., Grose J., Strich R., and Cooper K. F. (2018) A complex molecular switch directs stress-induced cyclin C nuclear release through SCF(Grr1)-mediated degradation of Med13. Mol. Biol. Cell 29, 363–375 10.1091/mbc.E17-08-0493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willis S. D., Stieg D. C., Ong K. L., Shah R., Strich A. K., Grose J. H., and Cooper K. F. (2018) Snf1 cooperates with the CWI MAPK pathway to mediate the degradation of Med13 following oxidative stress. Microb. Cell 5, 357–370 10.15698/mic2018.08.641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsai K. L., Sato S., Tomomori-Sato C., Conaway R. C., Conaway J. W., and Asturias F. J. (2013) A conserved Mediator-CDK8 kinase module association regulates Mediator–RNA polymerase II interaction. Nat. Struct. Mol. Biol. 20, 611–619 10.1038/nsmb.2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., et al. (2000) Gene ontology: tool for the unification of biology: the Gene Ontology Consortium. Nat. Genet. 25, 25–29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Donner A. J., Szostek S., Hoover J. M., and Espinosa J. M. (2007) CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 27, 121–133 10.1016/j.molcel.2007.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Filomeni G., De Zio D., and Cecconi F. (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388 10.1038/cdd.2014.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Filomeni G., Desideri E., Cardaci S., Rotilio G., Ciriolo M. R. (2010) Under the ROS…thiol network is the principal suspect for autophagy commitment. Autophagy 6, 999–1005 10.4161/auto.6.7.12754 [DOI] [PubMed] [Google Scholar]
- 35. Chen Y., Azad M. B., and Gibson S. B. (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 16, 1040–1052 10.1038/cdd.2009.49 [DOI] [PubMed] [Google Scholar]
- 36. Davis M. A., Larimore E. A., Fissel B. M., Swanger J., Taatjes D. J., and Clurman B. E. (2013) The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 27, 151–156 10.1101/gad.207720.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Porter D. C., Farmaki E., Altilia S., Schools G. P., West D. K., Chen M., Chang B. D., Puzyrev A. T., Lim C. U., Rokow-Kittell R., Friedhoff L. T., Papavassiliou A. G., Kalurupalle S., Hurteau G., Shi J., et al. (2012) Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. U.S.A. 109, 13799–13804 10.1073/pnas.1206906109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galbraith M. D., Allen M. A., Bensard C. L., Wang X., Schwinn M. K., Qin B., Long H. W., Daniels D. L., Hahn W. C., Dowell R. D., and Espinosa J. M. (2013) HIF1A employs CDK8-Mediator to stimulate RNAPII elongation in response to hypoxia. Cell 153, 1327–1339 10.1016/j.cell.2013.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maldonado E., Shiekhattar R., Sheldon M., Cho H., Drapkin R., Rickert P., Lees E., Anderson C. W., Linn S., and Reinberg D. (1996) A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Nature 381, 86–89 10.1038/381086a0 [DOI] [PubMed] [Google Scholar]
- 40. Miao Y. L., Gambini A., Zhang Y., Padilla-Banks E., Jefferson W. N., Bernhardt M. L., Huang W., Li L., and Williams C. J. (2018) Mediator complex component MED13 regulates zygotic genome activation and is required for postimplantation development in the mouse. Biol. Reprod. 98, 449–464 10.1093/biolre/ioy004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calpena E., Hervieu A., Kaserer T., Swagemakers S. M. A., Goos J. A. C., Popoola O., Ortiz-Ruiz M. J., Barbaro-Dieber T., Bownass L., Brilstra E. H., Brimble E., Foulds N., Grebe T. A., Harder A. V. E., Lees M. M., et al. (2019) De novo missense substitutions in the gene encoding CDK8, a regulator of the mediator complex, cause a syndromic developmental disorder. Am. J. Hum. Genet. 104, 709–720 10.1016/j.ajhg.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adegbola A., Musante L., Callewaert B., Maciel P., Hu H., Isidor B., Picker-Minh S., Le Caignec C., Delle Chiaie B., Vanakker O., Menten B., Dheedene A., Bockaert N., Roelens F., Decaestecker K., et al. (2015) Redefining the MED13L syndrome. Eur. J. Hum. Genet. 23, 1308–1317 10.1038/ejhg.2015.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Haelst M. M., Monroe G. R., Duran K., van Binsbergen E., Breur J. M., Giltay J. C., and van Haaften G. (2015) Further confirmation of the MED13L haploinsufficiency syndrome. Eur. J. Hum. Genet. 23, 135–138 10.1038/ejhg.2014.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Snijders Blok L., Hiatt S. M., Bowling K. M., Prokop J. W., Engel K. L., Cochran J. N., Bebin E. M., Bijlsma E. K., Ruivenkamp C. A. L., Terhal P., Simon M. E. H., Smith R., Hurst J. A., Study D. D. D., McLaughlin H., et al. (2018) De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum. Genet. 137, 375–388 10.1007/s00439-018-1887-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamamoto T., and Shimojima K. (2015) A novel MED12 mutation associated with non-specific X-linked intellectual disability. Hum. Genome Var. 2, 15018 10.1038/hgv.2015.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nizon M., Laugel V., Flanigan K. M., Pastore M., Waldrop M. A., Rosenfeld J. A., Marom R., Xiao R., Gerard A., Pichon O., Le Caignec C., Gerard M., Dieterich K., Truitt Cho M., McWalter K., et al. (2019) Variants in MED12L, encoding a subunit of the mediator kinase module, are responsible for intellectual disability associated with transcriptional defect. Genet. Med. 21, 2713–2722 10.1038/s41436-019-0557-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Donner A. J., Ebmeier C. C., Taatjes D. J., and Espinosa J. M. (2010) CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 17, 194–201 10.1038/nsmb.1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Willis S. D., Hanley S. E., Beishke T., Tati P. D., and Cooper K. F. (2020) Ubiquitin–proteasome-mediated cyclin C degradation promotes cell survival following nitrogen starvation. Mol. Biol. Cell 31, 1015–1031 10.1091/mbc.E19-11-0622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cooper K. F., Khakhina S., Kim S. K., and Strich R. (2014) Stress-induced nuclear-to-cytoplasmic translocation of cyclin C promotes mitochondrial fission in yeast. Dev. Cell 28, 161–173 10.1016/j.devcel.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Knuesel M. T., Meyer K. D., Donner A. J., Espinosa J. M., and Taatjes D. J. (2009) The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell. Biol. 29, 650–661 10.1128/MCB.00993-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained with the article.