

Abstract
Compensatory changes in energy expenditure occur in response to positive and negative energy balance, but the underlying mechanism remains unclear. Under low energy demand, the mitochondrial electron transport system is particularly sensitive to added energy supply (i.e. reductive stress), which exponentially increases the rate of H2O2 (JH2O2) production. H2O2 is reduced to H2O by electrons supplied by NADPH. NADP+ is reduced back to NADPH by activation of mitochondrial membrane potential–dependent nicotinamide nucleotide transhydrogenase (NNT). The coupling of reductive stress-induced JH2O2 production to NNT-linked redox buffering circuits provides a potential means of integrating energy balance with energy expenditure. To test this hypothesis, energy supply was manipulated by varying flux rate through β-oxidation in muscle mitochondria minus/plus pharmacological or genetic inhibition of redox buffering circuits. Here we show during both non-ADP– and low-ADP–stimulated respiration that accelerating flux through β-oxidation generates a corresponding increase in mitochondrial JH2O2 production, that the majority (∼70–80%) of H2O2 produced is reduced to H2O by electrons drawn from redox buffering circuits supplied by NADPH, and that the rate of electron flux through redox buffering circuits is directly linked to changes in oxygen consumption mediated by NNT. These findings provide evidence that redox reactions within β-oxidation and the electron transport system serve as a barometer of substrate flux relative to demand, continuously adjusting JH2O2 production and, in turn, the rate at which energy is expended via NNT-mediated proton conductance. This variable flux through redox circuits provides a potential compensatory mechanism for fine-tuning energy expenditure to energy balance in real time.
Keywords: mitochondrial metabolism, nicotinamide nucleotide transhydrogenase (NNT), energy metabolism, hydrogen peroxide (H2O2), bioenergetics, electron transport system (ETS), beta-oxidation, redox regulation, redox buffering circuits, hydrogen sulfide, nicotinamide nucleotide transhydrogenase
Body weight is remarkably stable in adults over long periods of time despite daily fluctuations in energy intake and expenditure (1). Consistent with homeostatic weight maintenance, energy expenditure rates increase in humans during periods of weight gain and decrease during periods of weight loss, well in excess of what can be accounted for by changes in fat free mass, the thermic effect of food, and fecal calorie loss (2, 3). Remarkably, even when body weight is held stable after active weight gain or loss, energy expenditure remains elevated or depressed, respectively (3). Changes in circulating thyroid hormone (4, 5), leptin (6), sympathetic tone (5), and skeletal muscle efficiency (7) have all been implicated, but the underlying mechanism(s) that accounts for these apparent compensatory changes in metabolic efficiency remains unresolved.
The ability of living cells to continuously respond to fluctuations in nutrient availability and energy demand depends on the interplay between metabolic and redox circuits. How these circuits connect and respond in real time is not clear. The dynamic equilibrium among opposing free energy driving forces across the inner mitochondrial membrane (i.e. ΔGredox – NAD+/NADH and FAD/FADH2, ΔGΔΨm – mitochondrial membrane potential, and ΔGATP – matrix [ATP]/[ADP]) provides a potential ideal mechanism for sensing cellular energy balance (8). This is obvious with respect to energy demand, where an increase in free [ADP] (i.e. decrease in mitochondrial ΔGATP) generates a corresponding increase in ATP synthase activity, an increase in proton (H+) conductance, a decrease in ΔGΔΨm, and an increase in electron flow and oxygen consumption (9). Less appreciated is the fact that the electron transport system (ETS) is equally suited to sense energy surplus, as an excess of NADH and/or FADH2 supply relative to demand, particularly at rest, increases the pressure head (i.e. ΔGredox), ΔGΔΨm, and thus reductive stress (i.e. oxidation potential) within the ETS, which exponentially increases the rate of mitochondrial H2O2 emission (10–13).
Mitochondrial JH2O2 emission reflects the balance between JH2O2 production and JH2O2 reduction to H2O. The latter is catalyzed by matrix thioredoxin and GSH peroxidases whose substrates, thioredoxin and GSH, are converted back to their reduced states by thioredoxin reductase and GSH reductase, respectively, using NADPH as the electron source (Fig. 1A). The reduction of NADP+ back to NADPH in turn is catalyzed, at least in part, by nicotinamide nucleotide transhydrogenase (NNT), an inner mitochondrial membrane protein that uses ΔΨm to establish and maintain a high NADPH/NADP+ ratio (14).
Figure 1.
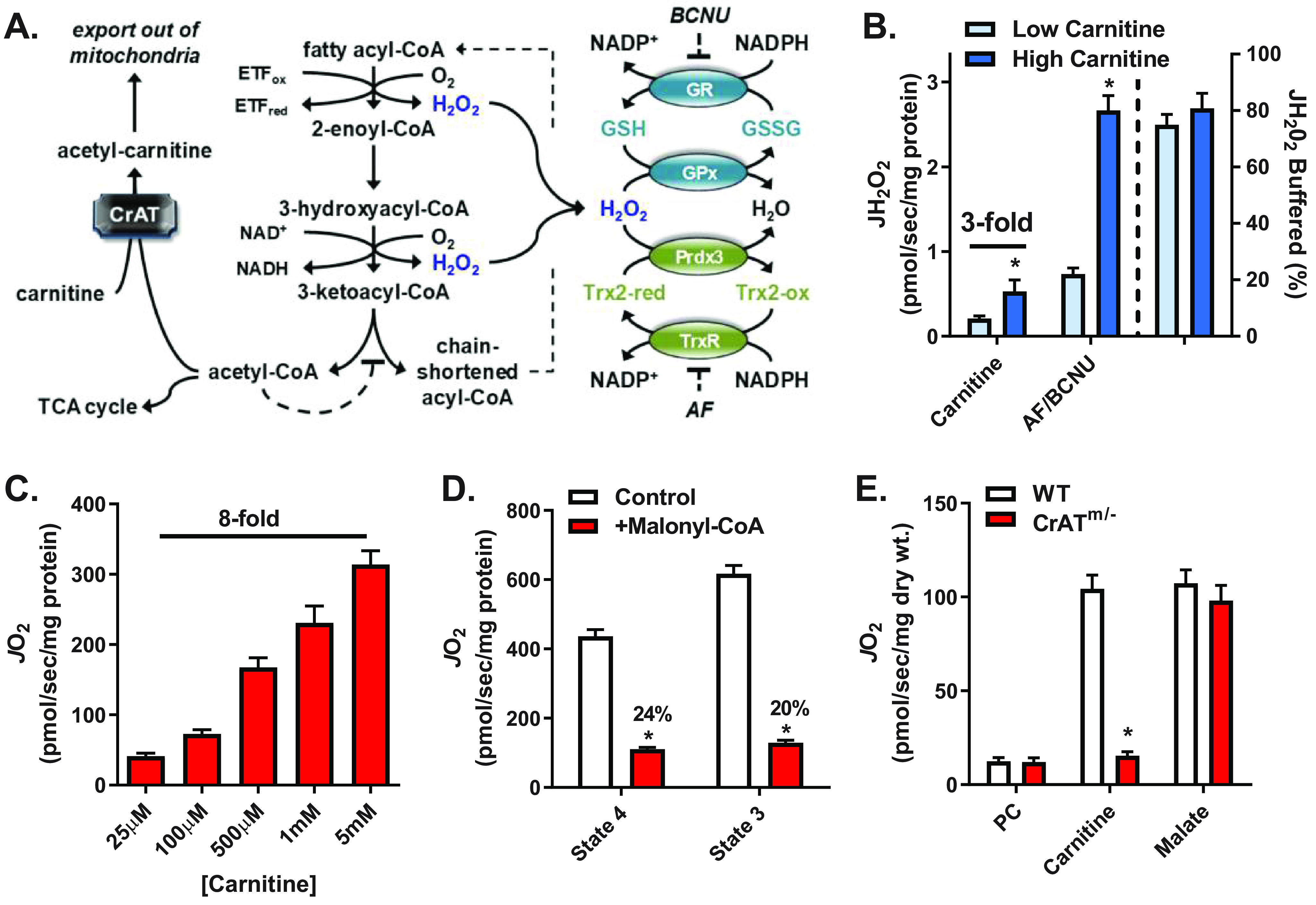
Increased flux through β-oxidation increases JO2, JH2O2 production, and electron flux through redox buffering circuits. A, schematic showing the mechanism by which carnitine via CrAT relieves acetyl-CoA–mediated inhibition of β-oxidation (left), sites of H2O2 generation from β-oxidation dehydrogenases (blue type, center), and the thioredoxin (Trx) and GSH redox buffering circuits (right). B, JH2O2 (left y axis) measured in mitochondria isolated from mouse skeletal muscle supported by PCoA (10 μm) and either low (25 μm) or high (5 mm) carnitine followed by the addition of AF/BCNU (1 μm/100 μm), inhibitors of Trx and GSH reductases, respectively. The percentage of JH2O2 buffered (right of dotted line, right y axis) reflects the percentage of JH2O2 produced but not emitted (i.e. (JH2O2 production (AF/BCNU rate) – JH2O2 emission (PCoA + carnitine rate))/JH2O2 production × 100). C, JO2 measured in mitochondria isolated from mouse skeletal muscle during basal (no ADP) respiration supported by PCoA (10 μm) and increasing concentrations of carnitine. D, PCoA plus carnitine (5 mm) supported JO2 during basal (state 4) or ADP-stimulated (state 3) respiration in the absence or presence of malonyl-CoA (100 μm), a CPT-1 inhibitor. E, JO2 measured in PmFBs from WT and CrATm/− mice during respiration supported by palmitoyl-carnitine (20 μm) followed by the additions of carnitine (5 mm) and malate (0.2 mm). All data are means ± S.E. (error bars); *, p < 0.05 versus corresponding control by unpaired t test; n = 5–13 mice/group.
Thus, in theory, anytime the rate of energy supply outpaces energy demand (i.e. nutrient overload), the resulting increase in JH2O2 production should induce a corresponding counterbalance increase in NNT-mediated energy expenditure. The purpose of the present study was to test this hypothesis by determining the extent to which flux rates through β-oxidation and the ETS during low rates of respiration influence overall/site-specific H2O2 production rates and whether JH2O2 production is coupled to NNT-mediated energy-consuming redox buffering circuits.
Results
Increased flux through β-oxidation increases JO2, JH2O2, and electron flux through redox buffering circuits
To examine whether mitochondrial JH2O2 emission may be coupled to compensatory changes in NNT-mediated energy expenditure, JH2O2 emission was measured by Amplex Ultra Red in mitochondria isolated from hind limb skeletal muscle of C57BL/6N (B6N) mice. Experiments were conducted under non-ADP-stimulated conditions (i.e. state 4 respiration) supported by palmitoyl-CoA in the presence of low (25 μm) or high (5 mm) carnitine. Transfer of long-chain fatty-acyl groups from CoA to carnitine by carnitine palmitoyltransferase-1 (CPT-1) is required for entry into the mitochondria and considered the rate-limiting step for β-oxidation (15, 16). We focused on fatty acid oxidation because it has multiple potential reductive stress points (i.e. sites of oxidant production), including dehydrogenation reactions within the β-oxidation pathway (17–19), the electron transfer flavoprotein-ubiquinone oxidoreductase (19–22), and within complexes I, II, and III of the ETS (19–23). Mitochondrial JH2O2 emission was 3-fold greater in the presence of high versus low carnitine (Fig. 1B). The subsequent addition of inhibitors to thioredoxin reductase (auranofin (AF)) and GSH reductase (carmustine, BCNU) induced a marked increase in JH2O2 detection under both carnitine concentrations. These findings demonstrate that H2O2 is produced at much higher rates than what is emitted and that the majority (∼70–80%) of H2O2 produced during β-oxidation is reduced to H2O (i.e. JH2O2 buffered (%)) by matrix redox buffering circuits (Fig. 1B, right y axis).
We next determined whether the carnitine-induced increase in JH2O2 production is associated with an increase in the rate of oxygen consumption (JO2), as measured by high-resolution respirometry (24). Progressive additions of carnitine from 25 μm to 5 mm in the presence of palmitoyl-CoA dose-dependently increased JO2 by 8-fold (Fig. 1C), with no further increase observed above 5 mm carnitine. Titration of carnitine in the presence of de-energized mitochondria (no substrate) or in the presence of NADH-generating substrates (e.g. 5 mm glutamate, 2 mm malate) did not affect JO2 (not shown), indicating the effect of carnitine was not due to a technical artifact or uncoupling. Inclusion of malonyl-CoA, an inhibitor of CPT-1, decreased maximal palmitoyl-CoA plus carnitine-supported JO2 by 75–80% (Fig. 1D), confirming that carnitine-induced flux through β-oxidation depends on CPT-1 activity.
In addition to facilitating transport into mitochondria, carnitine also accelerates β-oxidation flux by providing substrate for carnitine acetyltransferase (CrAT) (20, 24, 25), a mitochondrial enzyme that converts excess acetyl-CoA to membrane-permeant acetylcarnitine esters for export out of the mitochondria (26). Acetyl-CoA is a potent negative regulator of the β-ketoacyl-CoA thiolase reaction, the final step in β-oxidation (Fig. 1A) (27). To determine whether carnitine increases β-oxidation flux via CrAT, saponin-permeabilized skeletal muscle fiber bundles (PmFBs) were prepared from WT and muscle-specific CrAT knockout (CrATm/−) mice and studied during state 4 respiration supported by palmitoylcarnitine. In the absence of additional carnitine, JO2 was low in both WT and CrATm/− tissue (Fig. 1E). The addition of excess carnitine (5 mm) increased JO2 by ∼9-fold in PmFBs from WT mice but had no effect in PmFBs from CrATm/− mice. The subsequent addition of malate to facilitate entry of acetyl-CoA into the TCA cycle, and thus relieve acetyl-CoA–mediated inhibition of β-ketoacyl-CoA thiolase in CrATm/− mitochondria, increased JO2 by >8-fold, negating the difference between genotypes. These data confirm that carnitine addition accelerates CrAT activity, relieving acetyl-CoA–mediated product inhibition and thereby increasing flux through β-oxidation.
Integration of site-specific H2O2 production with redox buffering systems
To examine the integration of H2O2 production with the redox buffering systems, mitochondrial JH2O2 emission and JH2O2 production were measured during a series of substrate/inhibitor protocols designed to isolate electron leak to specific site(s) during β-oxidation. Electrons entering the Q-pool from β-oxidation have the potential to leak at the quinone-oxidizing sites of complex III (site IIIQo), complex I (site IQ), and complex II (site IIQ) (28). The flavin site in complex II (site IIF) also produces H2O2 during β-oxidation as a consequence of increased TCA cycle flux and release of oxaloacetate-mediated inhibition of complex II (Fig. 2A) (20, 29). During respiration supported by PCoA plus carnitine, the addition of malonate to block the forward reaction into complex II (i.e. site IIF) decreased H2O2 emission by ∼30% (Fig. 2B), leaving site IQ, site IIQ, site IIIQo, and the upstream β-oxidation dehydrogenases and flavoproteins as the potential remaining sources. The subsequent addition of AF/BCNU increased the rate of H2O2 detection by 5-fold (Fig. 2B), confirming the rate of H2O2 production greatly exceeds the rate of H2O2 emission and that flux through the redox buffering circuits accounts for the difference.
Figure 2.
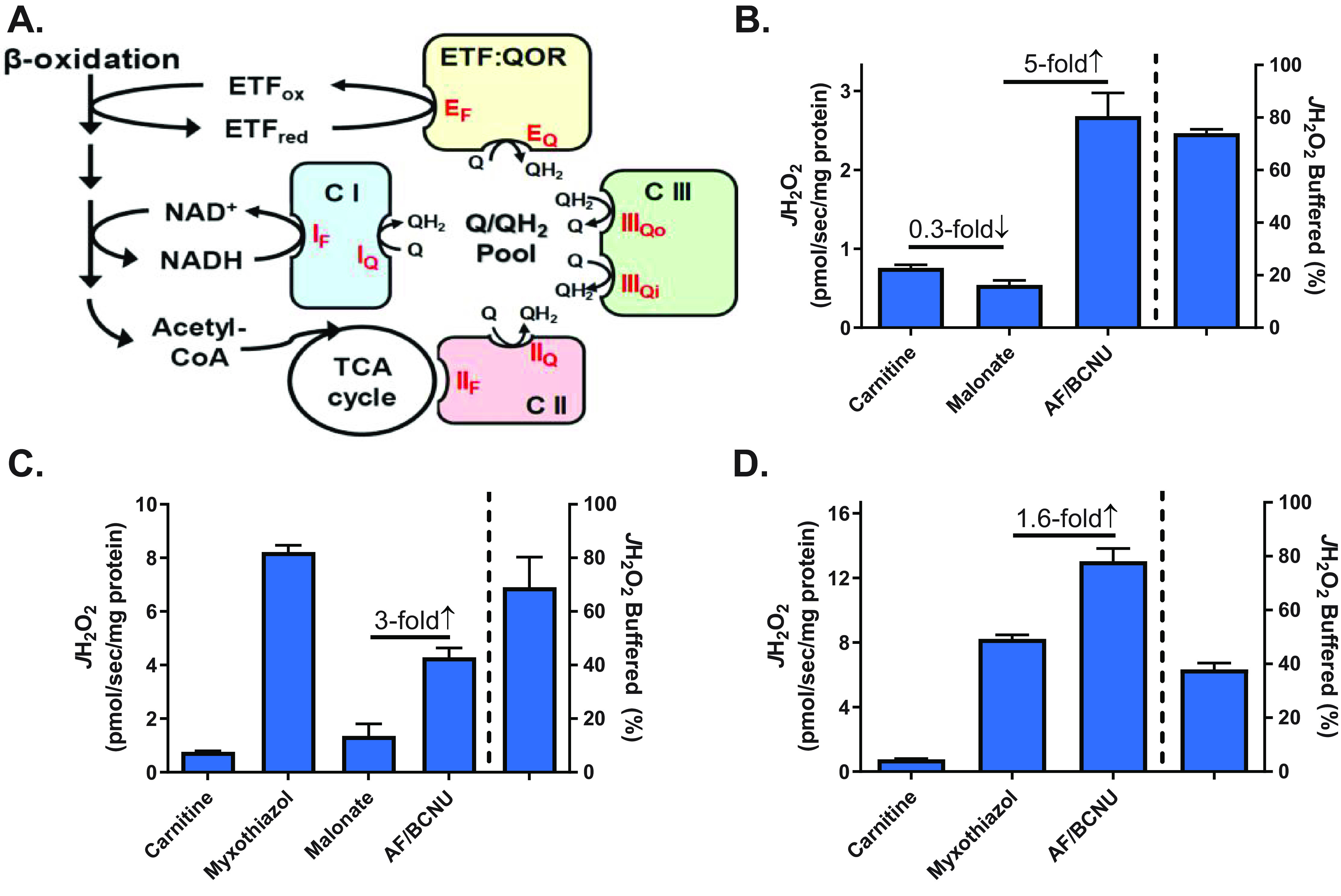
Integration of site-specific H2O2 production with redox buffering systems. A, schematic depicting specific flavin- and quinone-mediated sites of electron leak ( /H2O2 production, red type) within the β-oxidation pathway and ETS. B, JH2O2 (left y axis) measured in isolated mitochondria from mouse skeletal muscle during basal respiration supported by PCoA (10 μm) plus carnitine (5 mm) followed by the addition of malonate (site IIF inhibitor, 0.5 mm) and AF/BCNU (1 μm/100 μm). The percentage of JH2O2 buffered (right of dotted line, right y axis) is as defined in Fig. 1B. C, JH2O2 measured as in B followed by the addition of myxothiazol (site IIIQ inhibitor, 2 μm), malonate, and AF/BCNU. D, JH2O2 measured as in B followed by the addition of myxothiazol and AF/BCNU. All data are means ± S.E. (error bars); n = 5–6 mice/group.
Myxothiazol inhibits electron flow and H2O2 production at site IIIQo, causing upstream sites (IF, IIF, and the upstream β-oxidation enzymes) to become more reduced and dramatically increasing overall JH2O2 emission (Fig. 2C). The addition of malonate in the presence of myxothiazol decreased JH2O2 emission by nearly 90%, confirming reverse electron flow through complex II accounts for the majority of JH2O2 emission when forward electron flow into complex III is blocked during palmitoyl-CoA plus carnitine–supported respiration (20). The subsequent addition of AF/BCNU induced a 3-fold increase in JH2O2 emission, again indicating the majority of H2O2 produced (i.e. ∼70%) is reduced by the redox buffering circuits (Fig. 2C). In separate experiments, the addition of AF/BCNU after myxothiazol increased JH2O2 by only 1.6-fold (Fig. 2D), suggesting the efficiency of redox buffering circuits is limited at extremely high rates of H2O2 production. Taken together, these findings reveal that the rate of mitochondrial H2O2 emission elicited by β-oxidation is a stark underestimate of the actual rate of H2O2 production due to the capacity of the matrix redox buffering circuits to reduce H2O2 to H2O. The findings also suggest that the efficiency of redox buffering circuits is likely dependent on the integration and capacity of NADPH-generating sources.
Flux through NNT redox circuits contributes to energy expenditure
NNT uses the free energy of the mitochondrial protonmotive force to generate and maintain a highly reduced NADP+/NADPH ratio, which in turn generates and maintains highly reduced GSH (GSSG/GSH) and thioredoxin (Trx2ox/Trx2red) pools. Electrons are drawn from NADPH through the thioredoxin and GSH redox circuits to reduce H2O2 as a function of the demand imposed by JH2O2 production, which should therefore determine the rate of NNT-mediated proton conductance and thus energy expenditure. To test this hypothesis, proton conductance assays were carried out, similar to the approach used to define proton leak from uncoupling protein activity (12). In this assay, JO2 is measured during state 4 respiration supported by succinate/rotenone as ΔΨm is progressively decreased by titration of malonate, a complex II inhibitor. A difference in JO2 at the highest common ΔΨm reflects the difference in proton conductance into the matrix between experimental conditions. Experiments were conducted in the presence of two different β-oxidation flux/H2O2 production rates (induced by 25 μm versus 5 mm carnitine) and thus two different rates of demand on NNT. The higher carnitine concentration induced a leftward shift in the curve and a greater JO2 (1276 versus 1020 pmol/s/mg of protein) at the highest common ΔΨm (−154 mV) (Fig. 3A), consistent with a greater rate of proton conductance and energy expenditure when flux through β-oxidation is accelerated. Inclusion of AF/BCNU prevented the leftward shift in the proton conductance curve induced by high carnitine (Fig. 3B), confirming that increased electron flux through the thioredoxin and GSH redox buffering circuits accounted for the higher JO2.
Figure 3.

Flux through NNT redox circuits contributes to energy expenditure. A, JO2 as a function of ΔΨm in mitochondria isolated from mouse skeletal muscle during basal respiration supported by PCoA (10 μm), succinate (10 mm), and either low (25 μm) or high (5 mm) carnitine. Note the difference in JO2 (numbers with arrows) at a given ΔΨm (dotted line), indicating difference in proton conductance (i.e. energy expenditure). B, JO2 measured as a function of ΔΨm as described in A in the presence of either low carnitine, high carnitine, or high carnitine plus AF/BCNU (0.1 μm/100 μm). C, JH2O2 (left y axis) measured in mitochondria isolated from skeletal muscle of C57BL/6N (+NNT) and C57BL/6NJ (−NNT) mice during respiration supported by PCoA (10 μm) and high (5 mm) carnitine. The percentage of JH2O2 buffered (right of dotted line, right y axis) is as defined in Fig. 1B. D, JO2 measured as a function of ΔΨm as described in A in mitochondria isolated from skeletal muscle of C57BL/6N (+NNT) and C57BL/6NJ (−NNT) mice. All data are means ± S.E. (error bars); *, p < 0.05 compared with B6N by unpaired t test; n = 6–12 mice/group.
To determine whether the greater proton conductance is mediated by NNT, skeletal muscle mitochondria were isolated from C57BL/6J (B6J) mice, which carry a naturally occurring, in-frame, five-exon deletion mutation within the Nnt gene, rendering the NNT protein nonfunctional (30, 31). Maximal β-oxidation flux (5 mm carnitine) induced a 2-fold greater JH2O2 emission in mitochondria from B6J mice compared with normal NNT-expressing B6N mice (Fig. 3C), consistent with the absence of flux through NNT-linked redox circuits in B6J mice. Indeed, when AF/BCNU was added to inhibit the thioredoxin/GSH redox circuits, JH2O2 production was not different between genotypes (i.e. JH2O2 production increased in B6N but not B6J mitochondria). As such, in the absence of NNT, mitochondria from B6J mice were able to buffer only ∼11% of the H2O2 produced (Fig. 3C), consistent with prior findings indicating NNT is the major supplier of NADPH with additional production coming from two NADP+-linked TCA enzymes, isocitrate dehydrogenase 2 and malic enzyme 3 (32). Finally, mitochondria from B6J mice displayed a rightward shift in the proton conductance curve and lower JO2 (1125 versus 1450 pmol/s/mg of protein in B6J versus B6N, respectively) at the highest common ΔΨm (−153 mV) under maximal β-oxidation flux/JH2O2 production conditions, confirming that NNT mediates the increase in proton conductance (Fig. 3D).
Proton conductance assays require specific substrate conditions (i.e. succinate, titration of malonate to progressively inhibit complex II) and are performed under a non-ADP-stimulated respiratory state (state 4). To assess the interplay between β-oxidation–induced JH2O2 production and flux through NNT-linked redox circuits under conditions that more closely model in vivo bioenergetics, mitochondria isolated from red skeletal muscle of B6N and B6J mice were subjected to an energy clamp system that incorporates the creatine kinase reaction to stepwise “clamp” ATP/ADP ratios at different values, and thus ΔGATP (i.e. the free energy backpressure on ATP synthase), over the entire range of ADP-stimulated respiration (33, 34). Respiration was again supported by PCoA (20 μm) and high carnitine (5 mm) to maximize reducing pressure from the β-oxidation pathway. Steady-state JO2 progressively decreased in mitochondria from B6N and B6J mice as ΔGATP became more negative (i.e. as JO2 approached state 4 values) (Fig. 4A), which generated a progressive increase in JH2O2 emission that was evident only in mitochondria from B6J mice (which lack NNT) (Fig. 4C). At the most negative ΔGATP (−66.9 kJ/mol), JH2O2 emission was 2-fold higher (Fig. 4F) and JO2 consumption was 18.6% lower (Fig. 4D) in B6J versus B6N mitochondria, consistent with the absence of an intact NNT-linked redox circuit in B6Js (Fig. 3D). The subsequent addition of BCNU/AF to inhibit redox circuit flux more than doubled the JH2O2 detected in B6N mitochondria (i.e. revealed the actual JH2O2 production), negating the difference between B6J and B6Ns (Fig. 4F). As expected, BCNU/AF decreased JO2 in mitochondria from both genotypes (Fig. 4D), whereas ΔΨm remained stable (Fig. 4, B and D).
Figure 4.
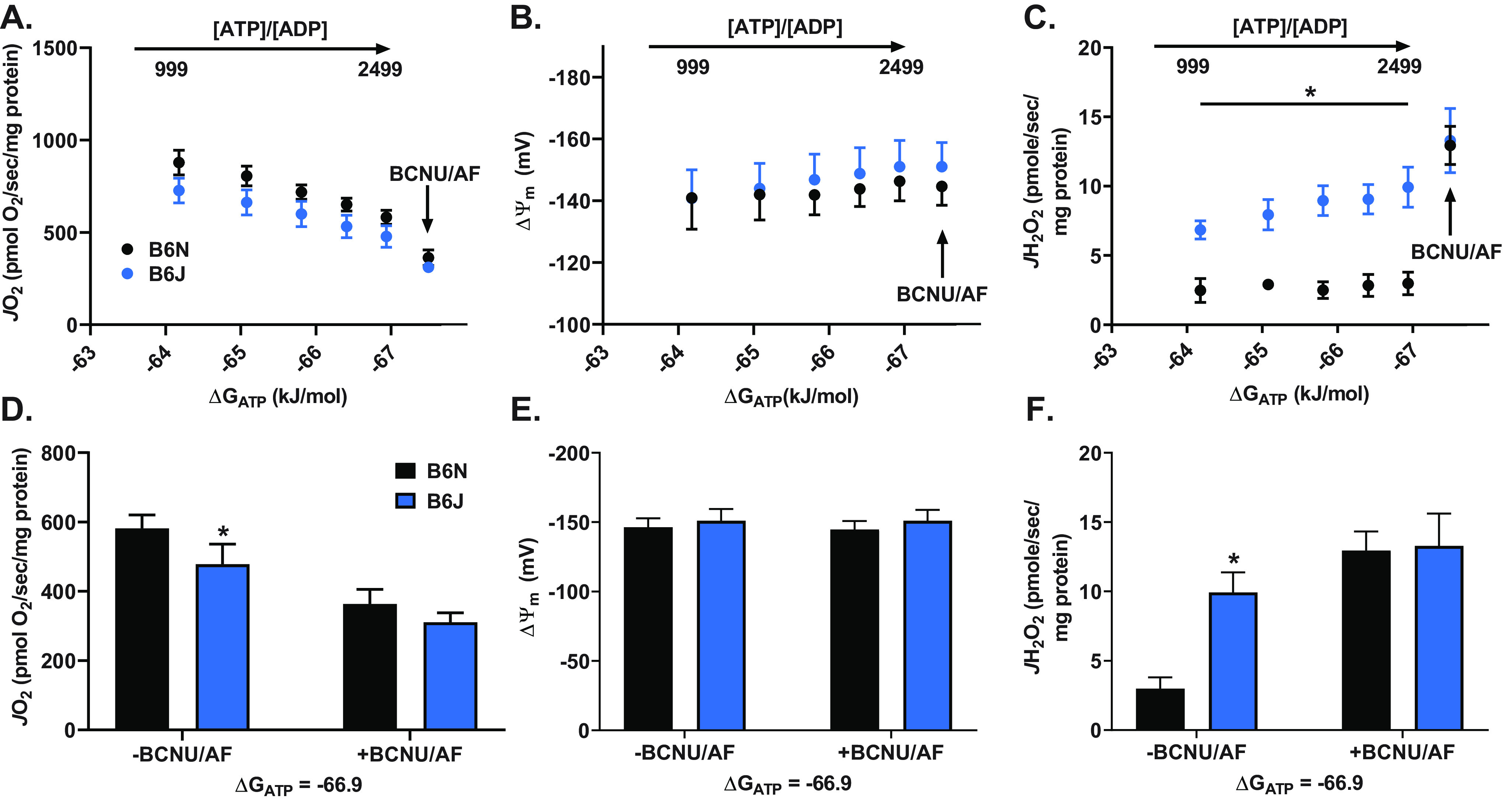
Flux through NNT contributes to energy expenditure during ΔGATP clamped respiration. A, JO2 versus clamped free energy of ATP hydrolysis (ΔGATP; increasing ATP/ADP free concentration ratio) in skeletal muscle mitochondria isolated from C57BL/6N (which express NNT) and C57BL/6J (which do not express NNT), supported by PCoA (20 μm) and carnitine (5 mm) −/+ BCNU/auranofin (0.1 μm/100 μm). B, ΔΨm versus clamped free energy of ATP hydrolysis (ΔGATP) measured under the same conditions. C, JH2O2 versus clamped free energy of ATP hydrolysis (ΔGATP) measured under the same conditions. D, pairwise comparison of JO2 between groups from A at the most negative ΔGATP (highest ATP/ADP ratio). E, pairwise comparison of ΔΨm between groups from A at the most negative ΔGATP. F, pairwise comparison of JH2O2 between groups from A at the most negative ΔGATP. All data are means ± S.E. (error bars); *, p < 0.05 compared with B6N using two-way ANOVA (repeated measures for A–C); n = 8 mice/group (JO2/JH2O2); n = 4 mice/group (ΔΨm).
Discussion
Linking JH2O2 production to NNT-mediated energy expenditure
Mitochondrial (and by extension cellular) redox circuits rely on both the GSSG/GSH (E0′ = −240 mV) and Trx2ox/Trx2red (E0′ = −230 mV) redox couples to protect against oxidants. The NADP+/NADPH redox couple, which possesses a lower standard midpoint potential (E0′ = −320 mV) serves as the source of electrons for both the GSH and thioredoxin redox couples. In vivo, NNT driven by ΔΨm maintains the NADP+/NADPH redox couple at a more negative (reduced) steady state (Eh = ∼−415 mV), which in turn holds both GSSG/GSH and Trx2ox/Trx2red in more reduced steady states (35). This resulting mitochondrial redox charge is distributed throughout the cell and thought to be responsible for maintaining ∼90% of the redox-sensitive thiols in the proteome in a reduced state (36, 37). Once the redox state of the proteome is established, it follows that the rate at which electrons are drawn from the reductive source (i.e. NADPH) through the redox circuits (i.e. Trxred and GSH) is determined by the rate of oxidative input into the circuit (i.e. the rate of H2O2 production and/or oxidation of redox-sensitive protein thiols). In isolated mitochondria under state 4 or low state 3 conditions, the ETS is extremely sensitive to forward reductive stress (i.e. ΔGredox), increasing JH2O2 production exponentially with even small increases in ΔΨm (10–12). The system is therefore poised to respond to energy supply outpacing energy demand by coupling the consequent rate H2O2 production directly to electron flux through redox buffering circuits linked to NNT and thus energy expenditure.
The objective of this study was to test this hypothesis by determining whether mitochondrial JH2O2 production stemming specifically from flux through β-oxidation is directly linked, via redox buffering circuits, to corresponding changes in NNT-mediated JO2. In vivo, mitochondrial JH2O2 production is thought to increase when flux rate through catabolic pathways, and thus reducing equivalent supply, outpaces metabolic demand (13). In isolated mitochondria, however, this is technically difficult to model due to the inability to recapitulate the same metabolic and redox free energy charges present in vivo. In the present study, we focused specifically on the β-oxidation pathway because flux through the pathway, and thus reducing equivalent supply to the ETS, can be experimentally accelerated by carnitine independent of the TCA cycle and/or ATP demand. The findings reveal that under non- or low-ATP demand states, JH2O2 production is directly related to the rate of flux through β-oxidation, that ∼80% of the H2O2 produced is reduced to H2O by electrons drawn through the thioredoxin and GSH redox circuits, and that flux through these redox circuits elicits a corresponding increase in NNT-mediated JO2 (i.e. energy expenditure). Together with a prior study showing a similar link between the pyruvate dehydrogenase complex and NNT (24), the data support the hypothesis that energy balance in mitochondria is continuously sensed and integrated, via JH2O2 production and NNT-linked redox buffering circuits, to compensatory changes in energy expenditure.
Potential contribution of NNT-linked redox buffering circuits to basal proton conductance
Basal proton conductance (i.e. state 4 respiration) in mitochondria is estimated to account for as much as 25% of resting metabolic rate (38), yet the molecular mechanism(s) responsible for what is commonly referred to as proton leak has remained enigmatic (12). Nonspecific proton conductance through the adenine nucleotide translocase (i.e. not associated with ATP/ADP exchange) reportedly accounts for ∼50% with the remainder generally attributed to nonspecific proton leak between the lipid bilayer and membrane-bound proteins (12, 39, 40). It is important to recognize that electron leak from the ETS to O2 to form superoxide, and its subsequent conversion to H2O2 by superoxide dismutase 2, is thermodynamically favorable under state 4 and low state 3 conditions (13). Thus, the coupling of JH2O2 production to NNT-mediated proton conductance via redox buffering circuits provides a potential additional mechanism contributing to resting metabolic rate (Fig. 5A). Consistent with this hypothesis, energy expenditure during the light cycle is lower in mice that lack NNT (B6J) as compared with genetically similar mice that express NNT (B6N) (24, 41, 42)
Figure 5.

Schematic showing the integration of JH2O2 with flux rates through NNT-linked redox buffering circuits. A, normal fuel supply. Blue lines indicate the drawing or “pull” of electrons by oxygen through the ETS and β-oxidation pathway and by H2O2 (derived from superoxide, not shown) through the redox buffering circuits from NADPH. Red lines indicate the “push” of electrons as a consequence of reductive stress (oxidation potential indicated by the gray gauges) leading to H2O2 production. JH+ represents the rate of proton conductance through NNT as a consequence of the rate of NAPDH oxidation. B, high fuel supply; as in A showing increase in reductive stress due to high fuel supply relative to demand increasing JH2O2 production, the rate of electron flux through redox buffering circuits, NNT-mediated proton conductance, and thus energy expenditure.
Potential physiological implications of NNT-linked redox buffering circuits
Whether flux through NNT-linked redox circuits contributes to energy expenditure in a physiologically meaningful way depends in part on the stoichiometric coupling of NNT (i.e. the number of H+ translocated across the mitochondrial membrane per pair of electrons transferred from NADH to NADP+ and the overall NADPH/O ratio). For ATP synthase, based on structural data from vertebrates (9), the H+/ATP stoichiometry of the proton turbine Fo component is well defined (∼2.7), and P/O ratios ranging from ∼2.0 to 2.8 have been empirically measured, depending on the experimental conditions (9, 43). NNT is also a complex protein arranged as a homodimer with each component containing a proton-translocating transmembrane domain and two nucleotide-binding domains in the matrix that mediate the electron transfer from NADH to NADP+ (44). Early studies using submitochondrial particles or liposomes containing reconstituted NNT suggested a H+/2e− stoichiometry of ∼1, which agrees with the roughly equivalent difference in redox poises of the NADP+/NADPH and NAD+/NADH couples and ΔΨm in mitochondria under basal conditions. However, the mechanism and stoichiometry underlying the coupling of H+ translocation to NADPH synthesis when the enzyme is actively generating NADPH remains unknown (44). NNT is thought to undergo significant protein conformational changes in response to changes in nucleotide binding (44, 45), which could alter the H+/2e− stoichiometry. For example, early work using submitochondrial particles suggested that NNT was much less efficient than ATP synthase (46). In the present study, JH2O2 production at the lowest clamped rate of respiration (ΔG = −66.9 kJ/mol) was ∼7.0 pmol/s/mg of protein higher in B6J than B6N mitochondria, which corresponded to ∼118.5 pmol/s/mg of protein lower JO2 in B6J versus B6N mitochondria (Fig. 4, D and F). Assuming this rate of H2O2 production drew the equivalent rate of NADPH oxidation and, in turn rate of NADPH resynthesis by NNT, the effective NADPH/O was ∼0.03, implying that when activated, NNT may be quite inefficient with respect to H+/2e− stoichiometry. There are of course a number of methodological limitations and caveats with such empirical data, including the possibility that factors other than the absence of NNT contribute to the difference in JO2 between B6J and B6N mitochondria. However, in contrast with ATP synthesis, where high efficiency is advantageous, a low efficiency of NADPH synthesis serves to counterbalance the very pressures responsible for increasing JH2O2 production (i.e. reductive stress from energy supply outpacing energy demand) (Fig. 5B).
In conclusion, this study reveals the degree to which mitochondrial bioenergetics couples energy/redox balance to energy expenditure, presumably to minimize disturbances to redox homeostasis throughout the proteome. In vivo, this implies that electron flux through redox buffering circuits enables adjustments in energy expenditure to changes in energy balance in real time, using redox potential within pivotal redox reactions as both the sensor and initiator of the counterbalance response. The cycling of electrons through redox buffering circuits could thus account for a significant portion of resting metabolic rate as well as contribute to the compensatory changes in energy expenditure observed in humans under positive and negative steady-state energy balance conditions (2). The identification of how redox state sensing mechanisms are coupled to compensatory changes in flux through mitochondrial redox buffering circuits also provides potential new avenues for therapeutic development to treat diseases stemming from chronic metabolic imbalance.
Experimental procedures
Animals
Male C57Bl/6NJ (B6N) and C57Bl/6J (B6J) mice (Jackson Laboratories (Bar Harbor, ME, USA), 005304 and 00664, respectively) were bred and housed in a temperature-controlled (22 °C) facility with a 12-h light/dark cycle. Mice were fed a standard, low-fat pellet diet (Purina ProLab ISOPro RMH 3000, St. Louis, MO, 5P76). At 12–20 weeks of age, mice were anesthetized with intraperitoneal ketamine/xylazine (18/2 mg/ml) injection (5 µl/g body weight) prior to tissue collection and cervical dislocation. All animal procedures were approved by the East Carolina University Institutional Animal Care and Use Committee.
Mitochondrial isolation
Differential centrifugation was used to isolate skeletal muscle mitochondria as performed previously (47) but with a few modifications. In brief, whole gastrocnemius, tibialis anterior, and quadriceps muscles from both hind limbs were dissected and placed immediately in a Petri dish containing 8 ml of ice-cold mitochondrial isolation medium (MIM): 300 mm sucrose (Thermo Fisher Scientific, BP2201), 10 mm HEPES (Sigma, H3375), and 1 mm EGTA (Sigma, E4378). Muscles were transferred to a separate Petri dish and finely minced with scissors while kept on ice. The minced muscle was homogenized in 8 ml of ice-cold MIM + 0.5 mg/ml BSA (Sigma, A3803) until consistent (∼8 passes) using a tight-fitting Teflon glass homogenizer. The homogenate was centrifuged for 10 min at 800 × g and 4 °C. The resulting supernatant was transferred to an Oakridge tube and centrifuged at 12,000 × g and 4 °C for an additional 10 min. The supernatant was discarded, and the pellet was rinsed once in 1 ml of MIM. The pellet was gently resuspended in 150 µl of MIM. The resulting mitochondrial preparation was diluted 50× prior to measuring the mitochondrial protein concentration (Pierce BCA Protein Assay, Thermo Fisher Scientific, 23225).
PmFB preparation
This technique was performed as described previously (48). Briefly, portions of red gastrocnemius muscle were dissected and immediately placed in ice-cold buffer X—50 mm K-MES (Sigma, M0895), 7.23 mm K2EGTA (100 mm stock solution prepared by measuring 100 mm EGTA with 200 mm KOH (Sigma, 221473), pH 7.1), 2.77 mm CaK2EGTA (100 mm stock solution prepared by dissolving 2.002 g of CaCO3 (Sigma, C4830) in 200 ml of K2EGTA, pH 7.1), 20 mm imidazole (Sigma, I2399), 20 mm taurine (Sigma, T8691), 5.7 mm ATP (Sigma, A6419), 14.3 mm phosphocreatine (Sigma, P7936), and 6.56 mm MgCl2-6H2O (Sigma, M2670), pH 7.1—for fiber separation with needle-tip forceps viewed under a microscope. Separated fiber bundles were then permeabilized to give access to the mitochondria by incubating on a rocker in buffer X containing 30 µg/ml saponin (Sigma, S4521) for 30 min at 4 °C. The PmFBs were then transferred to Buffer Z—105 mm K-MES, 30 mm KCl (Sigma, P9541), 1 mm EGTA, 10 mm K2HPO4 (Sigma, P5655), 5 mm MgCl2-6H2O, 0.5 mg/ml BSA, pH 7.2—and incubated on the rocker at 4 °C for at least 20 min or until experimentation (<45 min). At the conclusion of the experiment, fibers were rinsed in distilled H2O, placed in empty tubes with perforated caps, and freeze-dried to determine dry weights.
Model JO2, ΔΨm, and JH2O2 emission/production measurements
High-resolution respirometers (O2K, OROBOROS Innsbruck, Austria) were used to measure JO2. All experiments were carried out at 37 °C in a 2-ml reaction volume with continuous stirring. A conditioned tetraphenylphosphonium (TPP)-selective electrode integrated to an O2K respirometer was used to measure ΔΨm simultaneous with JO2. The JH2O2 emission/production was measured using the Amplex UltraRed/horseradish peroxidase fluorescence system in FluoroMax/Fluorolog spectrofluorometers (HORIBA Jobin Yvon, Edison, NJ). All JO2 and JH2O2 experiments were carried out at 37 °C in buffer Z with continuous stirring. Experiments with PmFBs were supplemented with 20 mm creatine monohydrate (Sigma, C3630) and 25 μm blebbistatin (Sigma, B0560). For experiments measuring JO2 as a function of ΔΨm, palmitoyl-CoA (10 μm; Sigma, P9716), l-carnitine (25 μm or 5 mm; Sigma, C0283), TPP (1.1 μm; Sigma, 87890), carboxyatractyloside (1.5 μm; Sigma, C4992), rotenone (5 μm; Sigma, R8875), GDP (500 μm; Sigma, G7127), and succinate (10 mm, pH 7.1; Sigma, S3674) were included in the experiment buffer during the background period. Upon stabilization, serial additions of TPP (1.28, 1.38, 1.47, 1.56, and 1.66 μm) were added to create a standard curve, followed by the addition of 150 µg of mitochondria. JO2 was measured in response to progressive lowering of ΔΨm by titration (0.5–5 mm) of the complex II inhibitor malonate (pH 7.1; Sigma, M1296). All JH2O2 experiments were performed in buffer Z supplemented with Amplex UltraRed (5 μm; Invitrogen, A36006), horseradish peroxidase (1 unit/ml; Sigma, P8375), and Cu-Zn superoxide dismutase (25 units/ml; Sigma, S7446). Other reagents used during these assays include palmitoyl-carnitine (Sigma, P1645), ADP (pH 7.1; Sigma, A5285), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (Sigma, C2920), myxothiazol (Sigma, T5580), malate (pH 7.1; Sigma, M7397), glutamate (pH 7.1; Sigma, G5889), malonyl-CoA (Sigma, M4263), AF (Sigma, A6733), and BCNU (Sigma, C0400).
Creatine kinase energetic clamp
Experiments utilizing the creatine kinase energetic clamp technique were performed as described previously (33, 34, 49). Briefly, JO2 and ΔΨm were measured simultaneously in O2K respirometers under increasing values of clamped ATP-free energy using known amounts of creatine (Cr), phosphocreatine (PCr), and ATP in the presence of excess creatine kinase (CK). ΔGATP was calculated using the equilibrium constant of the CK reaction (i.e. KCK) from the equation where ΔG′oATP is the standard apparent transformed Gibbs energy (at specific pH, ionic strength, free magnesium, and pressure), R is the gas constant (8.3145 J/kmol), and T is the temperature in Kelvin (310.15). Experiments were performed in Buffer Z with the exception that the KCl was replaced by sucrose (10 mm) to prevent the ionic strength from increasing too high from counterion equivalents in the PCr additions. Buffer Z was also supplemented with ATP (5 mm), Cr (5 mm), PCr (1 mm), CK (20 units/ml), PCoA (20 μm), and carnitine (5 mm). PCr was sequentially added to progressively increase ΔGATP. Both ΔG′oATP and K′CK were calculated at each titration step to account for changes in buffer ionic strength and free magnesium (33). JH2O2 was measured fluorometrically in parallel experiments under identical buffer and CK clamped conditions.
| (Eq. 1) |
Statistics
Data are presented as means ± S.E. The data exhibited normal distribution and were analyzed by unpaired Student's t tests with significance set at p < 0.05. Microsoft Excel and GraphPad Prism were used for statistical tests and data presentation.
Data availability
All raw data used to generate the data figures are available upon request from Dr. P. Darrell Neufer (neuferp@ecu.edu).
Acknowledgments
We thank Deborah Muoio, Ph.D., for providing access to muscle tissue from CrATm/− mice.
Author contributions—C. D. S., C. A. S., C.-T. L., K. H. F.-W., and P. D. N. conceptualization; C. D. S., C. A. S., and C.-T. L. data curation; C. D. S., C. A. S., C.-T. L., and K. H. F.-W. formal analysis; C. D. S. and P. D. N. supervision; P. D. N. funding acquisition; C. D. S., C. A. S., C.-T. L., K. H. F.-W., and P. D. N. methodology; C. D. S., C. A. S., and P. D. N. writing-original draft; C. D. S., C. A. S., C.-T. L., K. H. F.-W., and P. D. N. writing-review and editing.
Funding and additional information—This work was supported by National Institutes of Health Grants R01 DK096907 and R01 DK110656 (to P. D. N.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- ΔG
- free energy
- ΔΨm
- mitochondrial membrane potential
- AF
- auranofin
- BCNU
- bis-chloroethylnitrosourea
- B6N
- C57BL/6NJ
- B6J
- C57BL/6J
- CPT-1
- carnitine palmitoyltransferase-1
- CrAT
- carnitine acetyltransferase
- ETS
- electron transport system
- JH2O2
- rate of hydrogen peroxide
- JO2
- rate of oxygen consumption
- NNT
- nicotinamide nucleotide transhydrogenase
- PmFB
- permeabilized fiber bundle
- Trx2
- thioredoxin 2
- TCA
- tricarboxylic acid
- MIM
- mitochondrial isolation medium
- TPP
- tetraphenylphosphonium
- Cr
- creatine
- PCr
- phosphocreatine
- CK
- creatine kinase.
References
- 1. Belanger A. J., Cupples L. A., and D'Agostino R. B. (1988) Means at each examination and inter-examination consistency of specified characteristics: Framingham Heart Study, 30-year follow up. in The Framingham Study: An Epidemiological Investigation of Cardiovascular Disease (Kannel W. B., Wolf P. A., and Garrison R. J., eds) Government Publishing Office, Washington, D.C. [Google Scholar]
- 2. Leibel R. L., Rosenbaum M., and Hirsch J. (1995) Changes in energy expenditure resulting from altered body weight. N. Engl. J. Med. 332, 621–628 10.1056/NEJM199503093321001 [DOI] [PubMed] [Google Scholar]
- 3. Rosenbaum M., Hirsch J., Gallagher D. A., and Leibel R. L. (2008) Long-term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am. J. Clin. Nutr. 88, 906–912 10.1093/ajcn/88.4.906 [DOI] [PubMed] [Google Scholar]
- 4. Rosenbaum M., Goldsmith R. L., Haddad F., Baldwin K. M., Smiley R., Gallagher D., and Leibel R. L. (2018) Triiodothyronine and leptin repletion in humans similarly reverse weight-loss-induced changes in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 315, E771–E779 10.1152/ajpendo.00116.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosenbaum M., Hirsch J., Murphy E., and Leibel R. L. (2000) Effects of changes in body weight on carbohydrate metabolism, catecholamine excretion, and thyroid function. Am. J. Clin. Nutr. 71, 1421–1432 10.1093/ajcn/71.6.1421 [DOI] [PubMed] [Google Scholar]
- 6. Rosenbaum M., Goldsmith R., Bloomfield D., Magnano A., Weimer L., Heymsfield S., Gallagher D., Mayer L., Murphy E., and Leibel R. L. (2005) Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J. Clin. Invest. 115, 3579–3586 10.1172/JCI25977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldsmith R., Joanisse D. R., Gallagher D., Pavlovich K., Shamoon E., Leibel R. L., and Rosenbaum M. (2010) Effects of experimental weight perturbation on skeletal muscle work efficiency, fuel utilization, and biochemistry in human subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R79–R88 10.1152/ajpregu.00053.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Willis W. T., Jackman M. R., Messer J. I., Kuzmiak-Glancy S., and Glancy B. (2016) A Simple hydraulic analog model of oxidative phosphorylation. Med. Sci. Sports Exerc. 48, 990–1000 10.1249/01.mss.0000487975.15186.13, 10.1249/MSS.0000000000000884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nicholls D. G., and Ferguson S. J. (2013) Bioenergetics, 4th Ed., Elsevier Ltd., London [Google Scholar]
- 10. Korshunov S. S., Skulachev V. P., and Starkov A. A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18 10.1016/S0014-5793(97)01159-9 [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., Fiskum G., and Schubert D. (2002) Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80, 780–787 10.1046/j.0022-3042.2002.00744.x [DOI] [PubMed] [Google Scholar]
- 12. Divakaruni A. S., and Brand M. D. (2011) The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 26, 192–205 10.1152/physiol.00046.2010 [DOI] [PubMed] [Google Scholar]
- 13. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rydstrom J. (2006) Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 1757, 721–726 10.1016/j.bbabio.2006.03.010 [DOI] [PubMed] [Google Scholar]
- 15. McGarry J. D., and Brown N. F. (1997) The mitochondrial carnitine palmitoyltransferase system: from concept to molecular analysis. Eur. J. Biochem. 244, 1–14 10.1111/j.1432-1033.1997.00001.x [DOI] [PubMed] [Google Scholar]
- 16. Park E. A., and Cook G. A. (1998) Differential regulation in the heart of mitochondrial carnitine palmitoyltransferase-I muscle and liver isoforms. Mol. Cell. Biochem. 180, 27–32 10.1023/A:1006874503379 [DOI] [PubMed] [Google Scholar]
- 17. Kakimoto P. A. H. B., Tamaki F. K., Cardoso A. R., Marana S. R., and Kowaltowski A. J. (2015) H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol. 4, 375–380 10.1016/j.redox.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schönfeld P., and Wojtczak L. (2012) Brown adipose tissue mitochondria oxidizing fatty acids generate high levels of reactive oxygen species irrespective of the uncoupling protein-1 activity state. Biochim. Biophys. Acta 1817, 410–418 10.1016/j.bbabio.2011.12.009 [DOI] [PubMed] [Google Scholar]
- 19. Tahara E. B., Navarete F. D. T., and Kowaltowski A. J. (2009) Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 46, 1283–1297 10.1016/j.freeradbiomed.2009.02.008 [DOI] [PubMed] [Google Scholar]
- 20. Perevoshchikova I. V., Quinlan C. L., Orr A. L., Gerencser A. A., and Brand M. D. (2013) Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 61, 298–309 10.1016/j.freeradbiomed.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodrigues J. V., and Gomes C. M. (2012) Mechanism of superoxide and hydrogen peroxide generation by human electron-transfer flavoprotein and pathological variants. Free Radic. Biol. Med. 53, 12–19 10.1016/j.freeradbiomed.2012.04.016 [DOI] [PubMed] [Google Scholar]
- 22. Seifert E. L., Estey C., Xuan J. Y., and Harper M. E. (2010) Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 285, 5748–5758 10.1074/jbc.M109.026203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. St-Pierre J., Buckingham J. A., Roebuck S. J., and Brand M. D. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790 10.1074/jbc.M207217200 [DOI] [PubMed] [Google Scholar]
- 24. Fisher-Wellman K. H., Lin C. T., Ryan T. E., Reese L. R., Gilliam L. A., Cathey B. L., Lark D. S., Smith C. D., Muoio D. M., and Neufer P. D. (2015) Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem. J. 467, 271–280 10.1042/BJ20141447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muoio D. M., Noland R. C., Kovalik J. P., Seiler S. E., Davies M. N., DeBalsi K. L., Ilkayeva O. R., Stevens R. D., Kheterpal I., Zhang J., Covington J. D., Bajpeyi S., Ravussin E., Kraus W., Koves T. R., et al. (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 15, 764–777 10.1016/j.cmet.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jogl G., Hsiao Y. S., and Tong L. (2004) Structure and function of carnitine acyltransferases. Ann. N. Y. Acad. Sci. 1033, 17–29 10.1196/annals.1320.002 [DOI] [PubMed] [Google Scholar]
- 27. Schulz H. (1994) Regulation of fatty acid oxidation in heart. J. Nutr. 124, 165–171 10.1093/jn/124.2.165 [DOI] [PubMed] [Google Scholar]
- 28. Brand M. D. (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 100, 14–31 10.1016/j.freeradbiomed.2016.04.001 [DOI] [PubMed] [Google Scholar]
- 29. Quinlan C. L., Perevoshchikova I. V., Hey-Mogensen M., Orr A. L., and Brand M. D. (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312 10.1016/j.redox.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toye A. A., Lippiat J. D., Proks P., Shimomura K., Bentley L., Hugill A., Mijat V., Goldsworthy M., Moir L., Haynes A., Quarterman J., Freeman H. C., Ashcroft F. M., and Cox R. D. (2005) A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48, 675–686 10.1007/s00125-005-1680-z [DOI] [PubMed] [Google Scholar]
- 31. Freeman H. C., Hugill A., Dear N. T., Ashcroft F. M., and Cox R. D. (2006) Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 55, 2153–2156 10.2337/db06-0358 [DOI] [PubMed] [Google Scholar]
- 32. Dey S., Sidor A., and O'Rourke B. (2016) Compartment-specific control of reactive oxygen species scavenging by antioxidant pathway enzymes. J. Biol. Chem. 291, 11185–11197 10.1074/jbc.M116.726968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fisher-Wellman K. H., Davidson M. T., Narowski T. M., Lin C. T., Koves T. R., and Muoio D. M. (2018) Mitochondrial diagnostics: a multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Rep. 24, 3593–3606.e10 10.1016/j.celrep.2018.08.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glancy B., Willis W. T., Chess D. J., and Balaban R. S. (2013) Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809 10.1021/bi3015983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Go Y. M., and Jones D. P. (2008) Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 1780, 1273–1290 10.1016/j.bbagen.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Go Y.-M., and Jones D. P. (2013) The redox proteome. J. Biol. Chem. 288, 26512–26520 10.1074/jbc.R113.464131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones D. P., and Sies H. (2015) The redox code. Antioxid. Redox Signal. 23, 734–746 10.1089/ars.2015.6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rolfe D. F., Newman J. M., Buckingham J. A., Clark M. G., and Brand M. D. (1999) Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. Am. J. Physiol. 276, C692–C699 10.1152/ajpcell.1999.276.3.C692 [DOI] [PubMed] [Google Scholar]
- 39. Brand M. D., Pakay J. L., Ocloo A., Kokoszka J., Wallace D. C., Brookes P. S., and Cornwall E. J. (2005) The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362 10.1042/BJ20050890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brookes P. S., Buckingham J. A., Tenreiro A. M., Hulbert A. J., and Brand M. D. (1998) The proton permeability of the inner membrane of liver mitochondria from ectothermic and endothermic vertebrates and from obese rats: correlations with standard metabolic rate and phospholipid fatty acid composition. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 119, 325–334 10.1016/S0305-0491(97)00357-X [DOI] [PubMed] [Google Scholar]
- 41. Fisher-Wellman K. H., Ryan T. E., Smith C. D., Gilliam L. A., Lin C. T., Reese L. R., Torres M. J., and Neufer P. D. (2016) A direct comparison of metabolic responses to high fat diet in C57BL/6J and C57BL/6NJ mice. Diabetes 65, 3249–3261 10.2337/db16-0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simon M. M., Greenaway S., White J. K., Fuchs H., Gailus-Durner V., Wells S., Sorg T., Wong K., Bedu E., Cartwright E. J., Dacquin R., Djebali S., Estabel J., Graw J., Ingham N. J., et al. (2013) A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 14, R82 10.1186/gb-2013-14-7-r82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lark D. S., Torres M. J., Lin C. T., Ryan T. E., Anderson E. J., and Neufer P. D. (2016) Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am. J. Physiol. Cell Physiol. 311, C239–C245 10.1152/ajpcell.00124.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Q., Padayatti P. S., and Leung J. H. (2017) Proton-translocating nicotinamide nucleotide transhydrogenase: a structural perspective. Front. Physiol. 8, 1089 10.3389/fphys.2017.01089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jackson J. B., Leung J. H., Stout C. D., Schurig-Briccio L. A., and Gennis R. B. (2015) Review and hypothesis: New insights into the reaction mechanism of transhydrogenase: swivelling the dIII component may gate the proton channel. FEBS Lett. 589, 2027–2033 10.1016/j.febslet.2015.06.027 [DOI] [PubMed] [Google Scholar]
- 46. Dontsov A. E., Grinius L. L., Jasaitis A. A., Severina I. I., and Skulachev V. P. (1972) A study on the mechanism of energy coupling in the redox chain. I. Transhydrogenase: the fourth site of the redox chain energy coupling. J. Bioenerg. 3, 277–303 10.1007/BF01515975 [DOI] [PubMed] [Google Scholar]
- 47. Lark D. S., Reese L. R., Ryan T. E., Torres M. J., Smith C. D., Lin C. T., and Neufer P. D. (2015) Protein kinase A governs oxidative phosphorylation kinetics and oxidant emitting potential at complex I. Front. Physiol. 6, 332 10.3389/fphys.2015.00332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anderson E. J., Lustig M. E., Boyle K. E., Woodlief T. L., Kane D. A., Lin C. T., Price J. W. 3rd, Kang L., Rabinovitch P. S., Szeto H. H., Houmard J. A., Cortright R. N., Wasserman D. H., and Neufer P. D. (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581 10.1172/JCI37048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goldberg E. J., Buddo K. A., McLaughlin K. L., Fernandez R. F., Pereyra A. S., Psaltis C. E., Lin C. T., Hagen J. T., Boykov I. N., Nguyen T. K., Gowdy K. M., Ellis J. M., Neufer P. D., McClung J. M., and Fisher-Wellman K. H. (2019) Tissue-specific characterization of mitochondrial branched-chain keto acid oxidation using a multiplexed assay platform. Biochem. J. 476, 1521–1537 10.1042/BCJ20190182 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw data used to generate the data figures are available upon request from Dr. P. Darrell Neufer (neuferp@ecu.edu).