

Abstract
The incidence of diabetes, obesity, and metabolic diseases has reached an epidemic status worldwide. Insulin resistance is a common link in the development of these conditions, and hyperinsulinemia is a central hallmark of peripheral insulin resistance. However, how hyperinsulinemia leads to systemic insulin resistance is less clear. We now provide evidence that hyperinsulinemia promotes the release of soluble pro-inflammatory mediators from macrophages that lead to systemic insulin resistance. Our observations suggest that hyperinsulinemia induces sirtuin1 (SIRT1) repression and stimulates NF-κB p65 nuclear translocation and transactivation of NF-κB to promote the extracellular release of pro-inflammatory mediators. We further showed that low-dose naltrexone (LDN) abrogates hyperinsulinemia-mediated SIRT1 repression and prevents NF-κB p65 nuclear translocation. This, in turn, attenuates the hyperinsulinemia-induced release of pro-inflammatory cytokines and reinstates insulin sensitivity both in in vitro and in vivo diet-induced hyperinsulinemic mouse model. Notably, our data indicate that Sirt1 knockdown or inhibition blunts the anti-inflammatory properties of LDN in vitro. Using numerous complementary in silico and in vitro experimental approaches, we demonstrated that LDN can bind to SIRT1 and increase its deacetylase activity. Together, these data support a critical role of SIRT1 in inflammation and insulin resistance in hyperinsulinemia. LDN improves hyperinsulinemia-induced insulin resistance by reorienting macrophages toward anti-inflammation. Thus, LDN treatment may provide a novel therapeutic approach against hyperinsulinemia-associated insulin resistance.
Keywords: low-dose naltrexone, SIRT1, NF-κB, insulin resistance, sirtuin 1 (SIRT1), Toll-like receptor 4 (TLR4), insulin, cytokine induction, hyperinsulinemia, inflammation
The International Diabetes Federation estimates that ∼415 million individuals are living with diabetes, and as predicted by the World Health Organization, the number is expected to reach a staggering 642 million by 2040 (1). The development of type 2 diabetes mellitus (T2DM) is initially characterized by decreased insulin sensitivity, manifested by ensuing hyperglycemia and a compensatory response where pancreatic β-cells produce and secrete more insulin, and subsequently results in hyperinsulinemia (2).
Emerging data have suggested that insulin hypersecretion is not only an adaptive response to insulin resistance but may also be a primary defect (3, 4). β-cell hypersecretion is reported as a causal agent behind the development of hepatosteatosis, insulin resistance, and diabetes (3, 4).
A chronic low-grade inflammatory state (high plasma levels of numerous pro-inflammatory cytokines) is directly associated with the development of insulin resistance (7–9). Our study indicates that hyperinsulinemia provokes systemic inflammation to induce insulin resistance. Further, our observations suggest that hyperinsulinemia induces repression of SIRT1 and drives transactivation of NF-κB p65(a key protein), which promotes the synthesis of pro-inflammatory soluble mediators.
SIRT1, a mammalian ortholog of yeast silent information regulator 2, has emerged as a critical regulator of various metabolic and pathophysiological processes (10). SIRT1 regulates various biological processes by coordinating complex gene expression programs through the deacetylation of histones, transcription factors, and co-regulators (10, 11).
Naltrexone hydrochloride is an FDA-approved nonpeptide opioid antagonist that is often prescribed for the treatment of alcohol and opiate abuse. It was originally synthesized in 1963 and entered the heroin addiction clinical trial pipeline in 1973 (12). In 1984, the FDA approved it for heroin addiction under the brand named “Trexan,” and in 1995, the FDA approved a 50-mg dose of naltrexone (ReVia) for the treatment of alcohol addiction (13). Recently, naltrexone has gained a remarkable intervention in various neurological and autoimmune pathological conditions (14, 15). Naltrexone was synthesized as an orally active competitive opioid receptor antagonist; however, LDN exhibits paradoxical properties, including analgesia and anti-inflammatory actions (16). LDN simultaneously has an antagonistic effect on nonopioid receptors, including TLR4, that have not been reported at higher doses (16–19). Because insulin resistance is associated with a chronic low-grade inflammatory state, we sought to determine the role of LDN in the pathophysiology of insulin resistance associated with hyperinsulinemia in this study.
Here, using numerous complementary experimental approaches, we show the critical participation of SIRT1 in inflammation and insulin resistance associated with hyperinsulinemia. Our findings indicate that LDN rescues hyperinsulinemia-induced insulin resistance by attenuating NF-κB activity and blocking the release of pro-inflammatory mediators. Together, these data indicate that LDN improves insulin sensitivity by reorienting the macrophages toward anti-inflammation and that LDN may be used against insulin resistance, inflammation, and associated metabolic diseases.
Results
LDN treatment improves glucose tolerance and insulin sensitivity in HFD-induced hyperinsulinemic mice
We developed a short-term HFD-fed nonobese mouse model (Fig. 1A) to provide a deeper understanding of endogenous mechanisms that regulate insulin action in response to circulating hyperinsulinemia and study the effect of LDN on the insulin signaling axis in diet-induced hyperinsulinemic mice.
Figure 1.
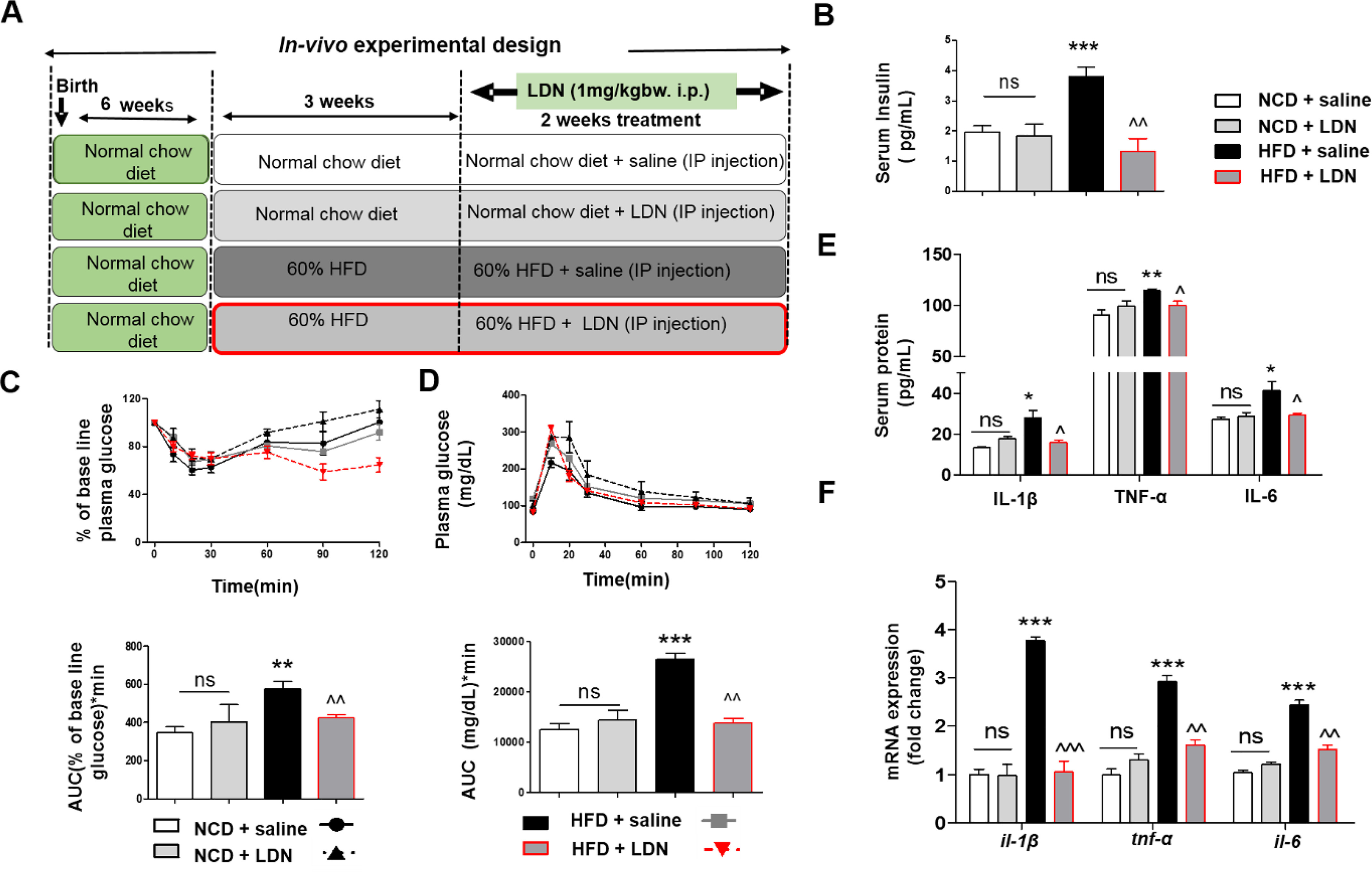
LDN prevents hyperinsulinemia and induction of pro-inflammatory cytokines release in HFD-induced hyperinsulinemic mice. A, schematic of protocols with time courses and time of LDN treatment and NCD or HFD exposures. B, fasting serum insulin levels of indicated mice groups. C, plasma glucose levels during ipITT of indicated mice groups (top) and AUC for glucose. D, plasma glucose level during ipGTT (top) of indicated mice groups with the area under the glucose curves in bottom panel. E, serum IL-1β, TNFα, and IL-6 levels of indicated mice groups. Cytokines levels were measured using Bio-Plex ProTM Mouse cytokine Standard 23 Plex Group 1 kit- (Bio-Rad, 171304070M) on a Bio-Plex-200 (Bio-Rad). F, quantitative mRNA expression of il-1β, tnf-α, and il-6 in purified ATMs from all group mice. Values are expressed as mean ± S.D. ***, P < 0.001; **, P < 0.01; *, P < 0.05 versus NCD + saline and ^^^, P < 0.001; ^^, P < 0.01; ^, P < 0.05 versus HFD + saline. (ANOVA followed by Bonferroni's Multiple Comparison).
As shown in Fig. 1B, fasting serum insulin levels were higher in the HFD saline fed mice than in the normal chow diet (NCD) fed mice, indicating the development of hyperinsulinemic mice. Interestingly, fasting insulin levels decreased significantly after LDN treatment in the HFD group. Dynamic insulin tolerance tests (ipITT) and glucose tolerance tests (ipGTT) were carried out to measure the peripheral response to insulin and glucose, respectively, and to explore the in vivo efficacy of LDN on glucose homeostasis and whole-body insulin sensitivity. HFD-fed hyperinsulinemic mice displayed impaired insulin sensitivity (as evident by the higher area under the curve), compared with the NCD groups, and the insulin sensitivity returned to normal when HFD-fed mice were treated with LDN (Fig. 1C). A similar trend was observed for ipGTT, as the HFD-saline group exhibited an increased glucose excursion AUC, compared with mice on NCD, and it was significantly reduced in HFD-LDN mice (Fig. 1D).
Overall, we found that HFD-LDN mice were substantially more glucose tolerant than the HFD-saline group, and it had reduced basal insulin levels suggesting improved insulin sensitivity. Together, these findings indicate the potential new role of LDN in improving glucose tolerance and insulin sensitivity in HFD-induced hyperinsulinemic mice.
LDN suppresses hyperinsulinemia-induced pro-inflammatory cytokine release
Basal hyperinsulinemia is closely linked to pro-inflammatory conditions that lead to insulin resistance (20). Next, we tested the possible involvement of hyperinsulinemia in inducing the release of pro-inflammatory cytokines, and we determined the effects of LDN on the release of pro-inflammatory mediators in an HFD mouse model. Serum from HFD-induced hyperinsulinemic mice showed a significantly enhanced release of pro-inflammatory mediators IL-1β, TNFα, and IL-6. Interestingly, we found that LDN treatment attenuated hyperinsulinemia-induced serum IL-1β, TNFα, and IL-6 levels (Fig. 1E). These data suggest that diet-induced hyperinsulinemia resulted in the release of various pro-inflammatory mediators and LDN treatment significantly abrogates hyperinsulinemia-induced release of pro-inflammatory mediators in serum.
Adipose tissue macrophages (ATMs) are closely linked to an inflammatory condition that leads to insulin resistance (7). Increased accumulation of pro-inflammatory or classically activated M1 macrophages in adipose tissue is positively correlated with insulin resistance (21).
Therefore, we investigated the effect of HFD-induced hyperinsulinemia on cytokine expression in purified ATMs. Expression of pro-inflammatory markers, such as IL-1β, MCP-1, and CD11c, was induced (Fig. 1F and Fig. S1) in the HFD-saline group, but LDN treatment attenuated the expression profile of pro-inflammatory cytokine/chemokine that was induced by hyperinsulinemia and restored anti-inflammatory cytokine expression (Fig. S1). Taken together, these findings suggest that LDN inhibits hyperinsulinemia-induced inflammation.
Next, we tested the possible involvement of hyperinsulinemia-induced pro-inflammatory cytokines in the pathogenesis of insulin resistance by investigating the effect of hyperinsulinemia on macrophage cells. We challenged macrophage cells with a high dose of insulin (100 nm) for 24 h in the presence and absence of LDN (nontoxic low dose) and analyzed various parameters as discussed below (Fig. 2A). A nontoxic dose of LDN (5 μm) was selected for in vitro studies (Fig. S2). Our study revealed that a high dose of insulin induced an M1-like phenotype by elevating the levels of pro-inflammatory cytokines and chemokines (IL-1β, MCP-1, and CD11C) and LDN attenuated insulin-mediated M1 (IL-1β, MCP-1, and CD11C) cytokine mRNA expression (Fig. 2B and Fig. S3).
Figure 2.
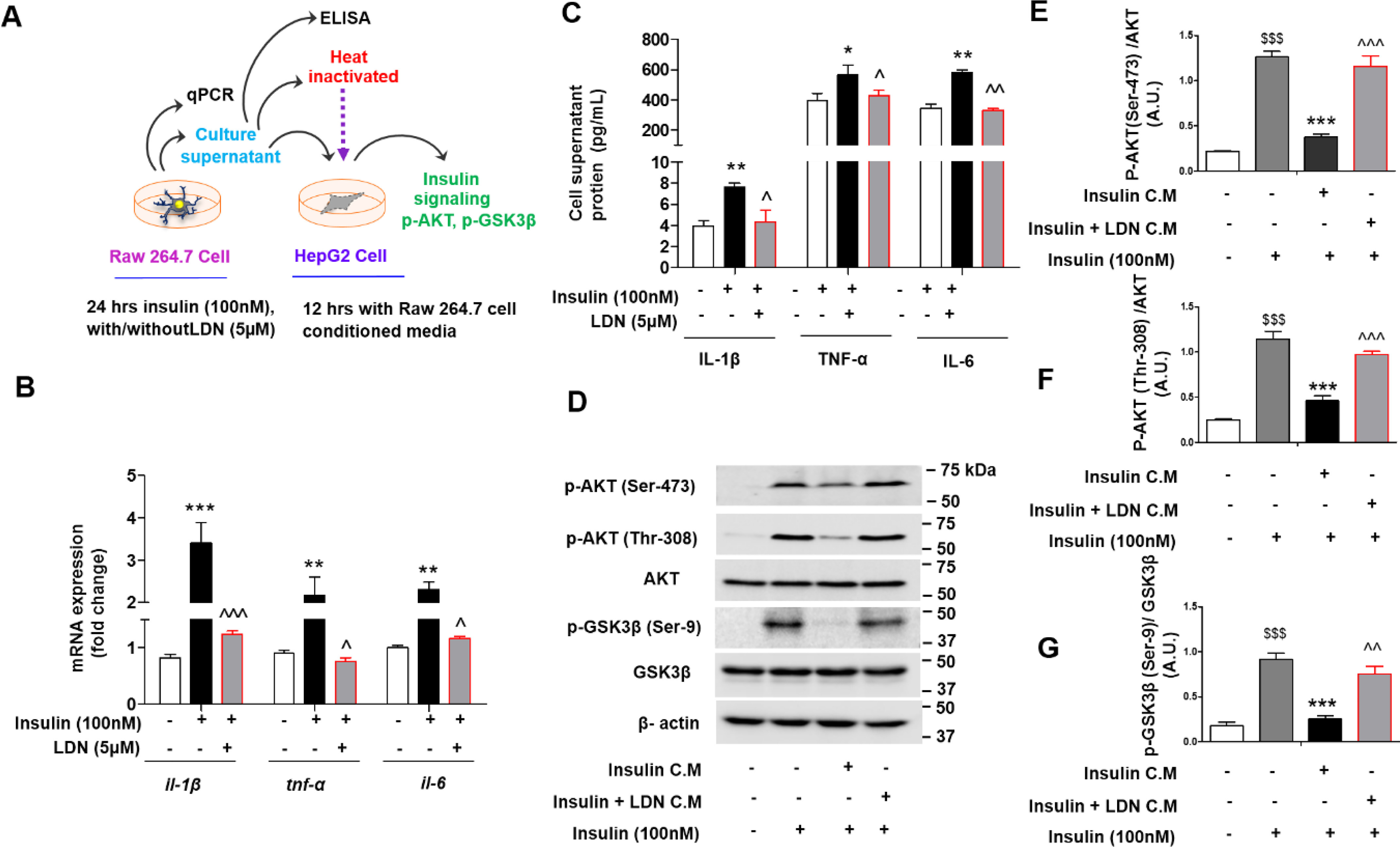
LDN inhibits inflammation and restored insulin sensitivity (surrogate markers AKT and GSK3β phosphorylation) in presence of hyperinsulinemia exposed conditioned media in vitro. A, schematic representing overall experimental design, and collection of conditioned media from murine macrophage (Raw 264.7) cells and treatment to hepatic (HepG2) cells. B and C, quantitative mRNA expression of indicated genes (il-1β, tnf-α, and il-6) (B) in murine macrophage cells, (C) levels of pro-inflammatory (IL-1β, TNFα, and IL-6) release in media from insulin challenged murine macrophage cells were measures using Bio-Plex ProTM mouse cytokine Standard 23 Plex Group 1 kit (Bio-Rad, 171304070M) on a Bio-Plex-200 (Bio-Rad). Values are expressed as mean ± S.D. ***, P < 0.001; **, P < 0.01; *, P < 0.05 versus control, and ^^^, P < 0.001; ^^, P < 0.01; ^, P < 0.05 versus insulin. (ANOVA followed by Bonferroni's Multiple Comparison). D–G, immunoblot (D) and (E–G) quantification for p-AKT (Ser-473), p-AKT(Thr-308), and p-GSK3β (Ser-9) phosphorylation status in HepG2 cell lysate treated with indicated conditioned media (C.M.) for 12 h followed by stimulated with or without 100 nm of insulin (last 15 min). Values are expressed as mean ± S.D. $$$P < 0.001 versus without insulin activation, ***, P < 0.001; **, P < 0.01; *, P < 0.05 versus insulin C.M. and ^^^, P < 0.001; ^^, P < 0.01; ^, P < 0.05 versus insulin + LDN C.M. $$$, P < 0.001; $$, P < 0.01; $, P < 0.05. (ANOVA followed by Bonferroni's Multiple Comparison).
Furthermore, we analyzed conditioned media proteins released from macrophages when challenged with high insulin in the presence and absence of LDN. Our results (Fig. 2C) showed a significant increase in IL-1β, TNFα, and IL-6 release in conditioned media from insulin-challenged macrophage cells, which was attenuated by LDN treatment.
To decipher hyperinsulinemia-induced aberrant release of pro-inflammatory cytokines involved in the development of insulin resistance, HepG2 cells were exposed to hyperinsulinemia-treated Raw 264.7 cell-conditioned media, and its downstream effects were analyzed. Phosphorylation of AKT (Ser-473), AKT (Thr-308), and GSK3β (Ser-9) were measured as a surrogate for insulin resistance. Interestingly, we found that conditioned media from hyperinsulinemia-treated macrophage cells blunted the phosphorylation of AKT (Ser-473), AKT (Thr-308), and GSK3β (Ser-9), compared with the conditioned media from non–insulin-treated macrophage cells, whereas conditioned media from the LDN-treated hyperinsulinemic group retained AKT (Ser-473), AKT (Thr-308), and GSK3β (Ser-9) phosphorylation (Fig. 2, D–G). Naltrexone has a t1/2 of 4–10 h (14), which excludes the possibility that the presence of naltrexone in conditioned media (after 24-h treatment) interferes with insulin sensitivity.
To delineate whether cytokines released by macrophages, or any other soluble factors, are responsible for inhibition of phosphorylation, we treated HepG2 cells with hyperinsulinemia-challenged macrophage-conditioned media after heat inactivation (Fig. 2A) and found that heat inactivation of conditioned media did not blunt AKT (Ser-473) phosphorylation in HepG2 cells (Fig. S4).
Together, these findings suggest that hyperinsulinemia induces the release of various pro-inflammatory mediators that are involved in the pathogenesis of insulin resistance and that LDN attenuated the hyperinsulinemia-mediated release of pro-inflammatory mediators and restored insulin sensitivity.
LDN suppresses hyperinsulinemia-induced cytosol-nucleus translocation of NF-κB
NF-κB is a key transcription factor that drives the pro-inflammatory phenotype. It is held quiescent in the cytoplasm in a complex with IκBα (22). In response to pro-inflammatory stimuli via TLR, IκBα is phosphorylated, thereby allowing NF-κB to translocate into the nucleus and activate the transcription of a cascade of pro-inflammatory cytokines and chemokines to induce inflammatory responses (23).
To investigate whether NF-κB transcription was regulated by hyperinsulinemia treatment, a construct containing a luciferase enzyme driven by an NF-κB promoter was transfected into macrophage cells. We observed that hyperinsulinemia increased the transactivation potential of NF-κB, and LDN prevented such transactivation (Fig. 3A). NF-κB activation also relies on posttranslational modifications of NF-κB subunits, such as RelA (p65) phosphorylation (24). In addition to the increased transactivation potential of NF-κB, an elevated level of p65 phosphorylation was also observed in hyperinsulinemic treatment compared with untreated cells, which were attenuated in the presence of LDN (Fig. 3B). Phosphorylation of the p65 subunit is required for the optimal p65-mediated transactivation potential of NF-κB.
Figure 3.
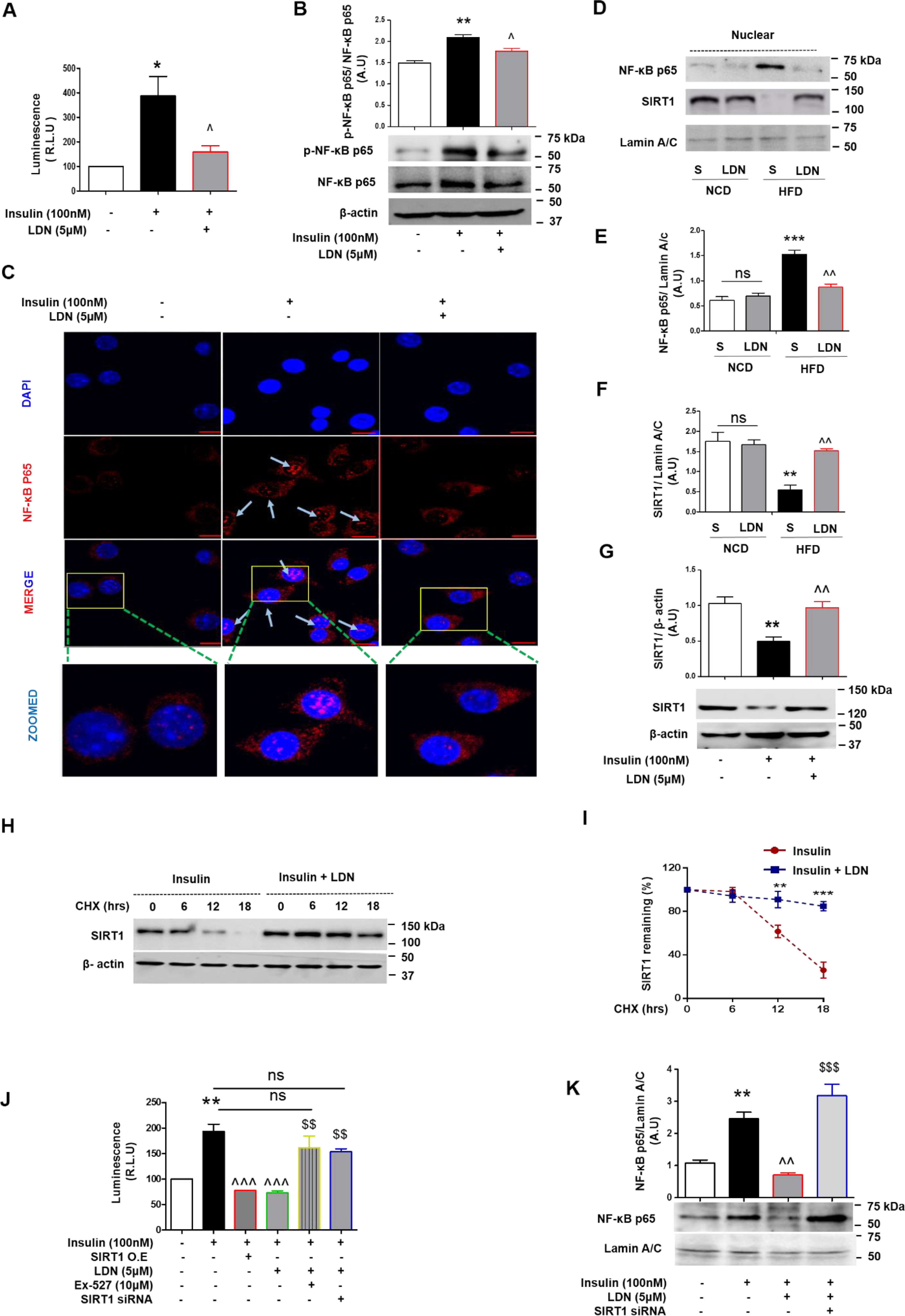
LDN specifically blocks hyperinsulin-stimulated nuclear localization NF-κB p65 and increases the stability of SIRT1 under hyperinsulinemia condition. A, relative light units (RLU) indicating luciferase activity after transient transfection into murine macrophage cells of NF-κB binding site promoter–luciferase reporter constructs followed by treatment with insulin (100 nm) and LDN (5 μm) for 24 h. Luminescence for NF-κB transcriptional activity was captured using steady glow reagent. B, immunoblot and quantification of p-NF-κB p65 in macrophage cells with insulin in the presence and absence of LDN for 24 h. C, murine macrophage cells were treated with LDN in the presence and absence of insulin (100 nm) for 6 h. After incubation, cells were fixed and incubated against NF-κB p65 as described under “Experimental procedures” and imaged by confocal microscopy. Image scale bar is 10 μm. D–F, IB (D) and (E and F) quantification of SIRT1 and NF-κB p65 expression and localization after completion food regime from NCD and HFD mice (S, saline; L, LDN). Representative Western blots of nuclear NF-κB p65 and SIRT1 level from liver nuclear extracts. Relative protein band density value of nuclear SIRT1 and NF-κB p65 was quantified by densitometry as the ratio of protein of interest to that of lamin A/C. Error bars show means ± S.D. G, IB and quantification of SIRT1 in murine macrophage cells treated with insulin in the presence and absence of LDN for 24 h. H, luciferase activity after transient transfection into murine macrophage cells of NF-κB binding site promoter-luciferase reporter and SIRT1 constructs followed by treatment with insulin, insulin-LDN, and insulin-LDN along with SIRT1 inhibitor (Ex-527) (10 μm). Values are expressed as mean ± S.D. ***, P < 0.001; **, P < 0.01; *, P < 0.05 versus control or (and) NCD-saline and ^^^, P < 0.001, ^^, P < 0.01, ^, P < 0.05 versus insulin or (and) HFD-saline. (ANOVA followed by Bonferroni's Multiple Comparison). H and I, immunoblot and quantification of SIRT1; immunoblot for endogenous SIRT1 levels in HepG2 cells pretreated with insulin and LDN for 24 h followed by CHX for indicated time points and protein levels of SIRT1 and β-actin as control were determined by IB. The SIRT1 band intensities relative to the 0 min time point were plotted (I). J, relative light units (RLU) indicating luciferase activity after transient transfection into murine macrophage cells of NF-κB binding site promoter-luciferase reporter followed by treatment with insulin, insulin-LDN, and insulin-LDN along with Sirt1 knockdown (Sirt1 siRNA). Values are expressed as mean ± S.D. **, P < 0.01, versus control, ^^^, P < 0.001, versus insulin and $$, P < 0.01 versus insulin-LDN (ANOVA followed by Bonferroni's Multiple Comparison). K, immunoblot and quantification of nuclear NF-κB p65 in macrophage cells treated with insulin in the presence and absence of LDN for 24 h and indicated conditions (with or without Sirt1 knockdown using Sirt1 specific siRNA). Indicate that Sirt1 is required for LDN to block hyperinsulin-stimulated NF-κB p65 nuclear localization. Values are expressed as mean ± S.D. **, P < 0.01, versus control; ^^, P < 0.01, versus insulin; and $$$, P < 0.001 versus insulin-LDN. (ANOVA followed by Bonferroni's Multiple Comparison).
Next, we examined NF-κB p65 subcellular localization, which was barely detectable in the nucleus of unstimulated macrophage cells. As expected, hyperinsulinemia increased nuclear NF-κB p65 localization, whereas LDN treatment blocked this shuttling of NF-κB to the nucleus from the cytosol (Fig. 3C). We also performed a Western blot analysis of liver nuclear extracts from experimental mice to determine the relative amount of NF-κB p65. Western blotting data showed increased nuclear NF-κB p65 in HFD-saline livers as compared with other experimental groups (Fig. 3, D and E). Taken together, these results suggest that NF-κB p65 is translocated into the nucleus in response to high insulin conditions, whereas LDN prevents NF-κB p65 nuclear translocation and blocks the hyperinsulin-stimulated phosphorylation of p65 subunit, which is required for optimal p65-mediated transactivation.
LDN treatment protects SIRT1 from hyperinsulinemia-induced down-regulation in the HFD mice model
SIRT1 is known to be a positive regulator of insulin signaling, and several studies have indicated the involvement of SIRT1 in insulin resistance and T2DM (25). SIRT1 activity and/or expression is reported to be down-regulated in different metabolically sensitive organs in diabetic and insulin-resistant rodent models (25).
To examine the effect of hyperinsulin and/or LDN on SIRT1, we measured the SIRT1 protein expression profile in hyperinsulinemic state in the liver, white adipose tissue lysates from NCD and HFD mice, and hyperinsulinemia-challenged macrophage cells in the presence and absence of LDN treatment. Interestingly, we observed that in the HFD-saline group, basal hyperinsulinemia resulted in significant repression of nuclear SIRT1 levels in the liver, whereas LDN treatment protected SIRT1 from such down-regulation (Fig. 3, D and F). Similar to liver tissue lysates, white adipose tissue lysates showed markedly reduced adipose nuclear SIRT1 levels in HFD-fed mice (with basal hyperinsulinemia), compared with the NCD group (Fig. S5). Interestingly, we observed that hyperinsulinemia significantly down-regulated SIRT1 protein levels in macrophages, whereas LDN treatment protected SIRT1 from such down-regulation (Fig. 3G).
We found that protein levels of endogenous SIRT1 decreased in hyperinsulinemic conditions, and conversely, LDN treatment maintained SIRT1 levels even under hyperinsulinemic conditions, which suggests that LDN may influence SIRT1 protein stability. To test this idea, endogenous SIRT1 protein levels were measured by time after inhibition of protein synthesis with cycloheximide (CHX). As shown in Fig. 3, levels of SIRT1 were reduced upon CHX treatment in the presence of insulin for 12 h (Fig. 3, H and I). Interestingly, although LDN treatment stabilized SIRT1 levels under insulin-treated conditions (Fig. 3, H and I), the SIRT1 mRNA levels did not significantly change under these conditions (Fig. S6).
Together, these results clearly illustrate that hyperinsulinemia-mediated SIRT1 degradation is abrogated in the presence of LDN.
SIRT1 is critical for LDN-mediated anti-inflammatory effects
To evaluate whether SIRT1 participates in regulating LDN-mediated attenuation of NF-κB activity, thereby down-regulating pro-inflammatory responses, we knocked down SIRT1 using murine sequence-specific siRNA in cultured mouse macrophage cells. Knockdown was specific, as shown on the immunoblot with an ∼90% reduction of SIRT1 (Fig. S7). Macrophage cells were transfected with an NF-κB/luciferase reporter construct and were treated with high insulin and LDN in the presence and absence of Sirt1 specific inhibitor (EX-527) or siRNA for Sirt1 and Sirt1 overexpression (SIRT1 O.E.). NF-κB transactivity showed a 2.5-fold increase when cells were treated with insulin as measured by luciferase assay (Fig. 3J), whereas, LDN and Sirt1 overexpression significantly attenuated hyperinsulinemia-induced NF-κB activity. Interestingly, LDN-mediated suppression of NF-κB promoter activity was absent when the cells were treated with EX-527 (Fig. 3J). Similar to Ex-527, in Sirt1 knockdown cells, the absence of Sirt1 significantly increased NF-κB promoter activity even in the presence of LDN (Fig. 3J). Similarly, to determine the role of Sirt1 in determining LDN efficacy, we treated macrophages with insulin in the presence and absence of LDN for 24 h and also with or without Sirt1 knockdown. Interestingly, we found that hyperinsulinemia increased NF-κB p65 nuclear localization, which was prevented by LDN; however, LDN was unable to attenuate NF-κB p65 nuclear localization when Sirt1 was silenced using Sirt1 siRNA (Fig. 3K). These observations indicate that Sirt1 is required for LDN to block hyperinsulin-stimulated nuclear localization of NF-κB p65 subunit and transactivation potential of NF-κB. To further characterize the potential role of SIRT1 in regulating LDN-mediated attenuation of pro-inflammatory gene expression, macrophage cells were treated with high insulin and LDN in the presence and absence of EX-527, siRNA specific for Sirt1, and analyzed for pro-inflammatory gene expression. As shown in Fig. S8, cells co-treated with LDN and EX-527 showed no change in pro-inflammatory gene expression profile as compared with the high insulin-treated group. Furthermore, macrophage cells with knocked down Sirt1 also abrogated the anti-inflammatory effect of LDN. Taken together, these results indicate that LDN specifically blocks hyperinsulin-stimulated nuclear localization of NF-κB p65, which is required for optimal p65-mediated transactivation potential of NF-κB and the LDN-stimulated anti-inflammatory phenotype depends on SIRT1.
LDN binds to SIRT1 and increases its deacetylase activity in vitro
We performed a blind docking experiment to investigate the possibility of naltrexone interactions with SIRT1. The docked complex of naltrexone binds to the main active site of SIRT1 with low binding energy of −10.94 kcal. The negative binding energy shows the spontaneous binding of naltrexone at the active site of SIRT1. The possibility of naltrexone binding at some other additional binding sites was also detected, but its minimum energy cluster showed the highest binding affinity for the main active site. Although no hydrogen bonding interaction was detected between naltrexone-SIRT1, the nearby close contact residues involved in hydrophobic interactions are Ala-262, Ser-265, Ile-270, Pro-271, Asp-272, Gln-345, Asn-346, Ile-347, and Asp-348 (Fig. 4, A and B). These close contact residues are responsible for holding naltrexone at the active site of SIRT1.
Figure 4.
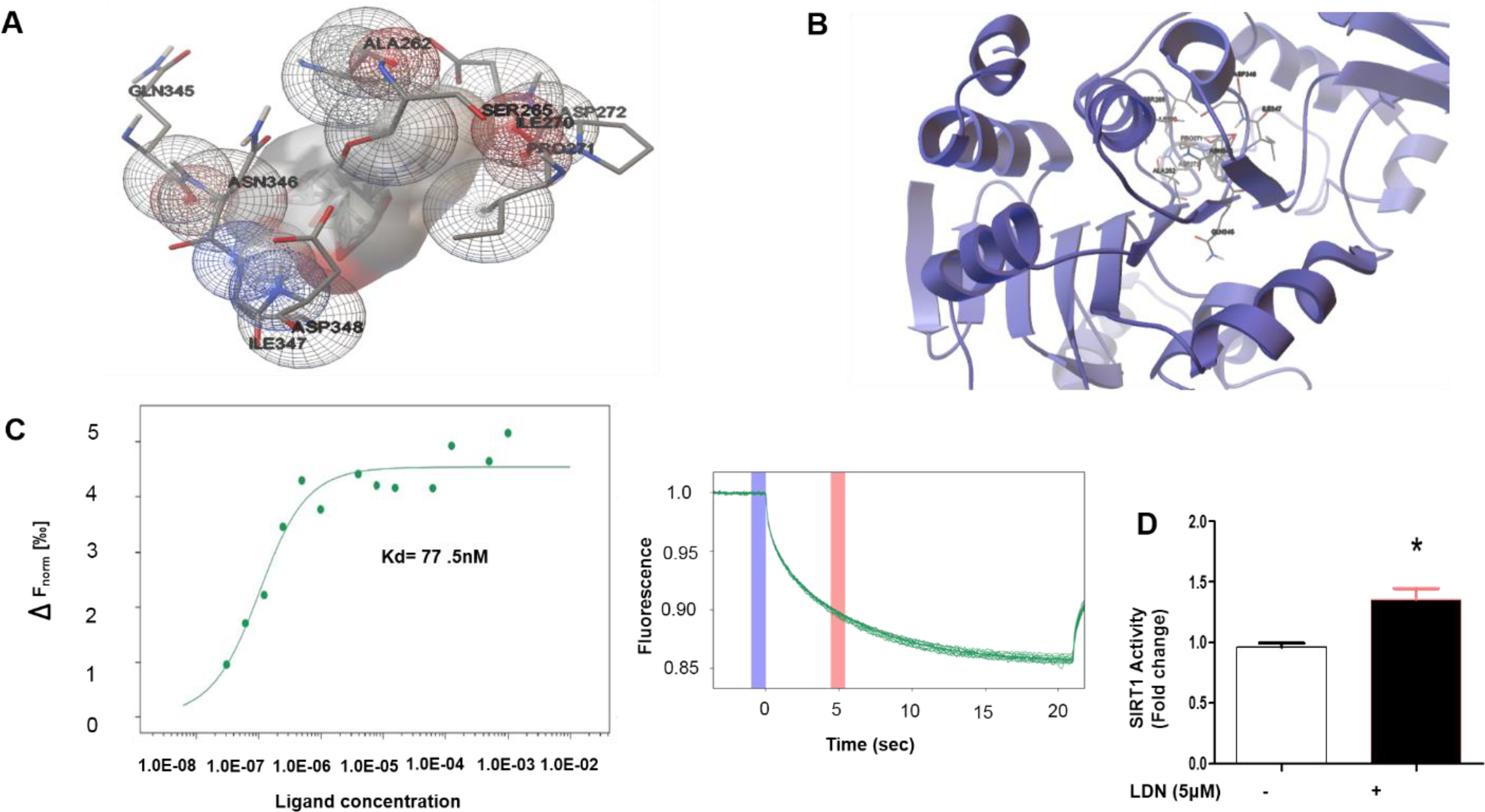
Naltrexone shows binding affinity for SIRT1 and increases SIRT1 deacetylate activity. A and B, the molecular interaction between naltrexone-SIRT1 protein complex. C, microscale thermophoresis data showing the binding of naltrexone upon addition of increasing concentrations of SIRT (1 mm to 3.05E-05 mm). The affinity (Kd 77.5 nm) was determined by fitting the data using MST principle with a signal to noise ratio of 13.46. The graph on the right exemplarily shows the thermophoretic running behavior of naltrexone upon SIRT1 binding at 40% MST power. D, naltrexone induces SIRT1 deacetylase activity, fluorescence intensity was measured at excitation 370 nm and emission 460 nm. Values are expressed as mean ± S.D. (n = 3). *, P < 0.05 as compare with control analyzed by t test.
To determine the interaction between SIRT1 and naltrexone and the equilibrium dissociation constant (Kd) of the naltrexone-SIRT1 complex, a series of microscale thermophoresis experiment measurements were recorded for fixed amounts of SIRT1 with an increasing amount of naltrexone. An S-shaped sigmoidal binding curve was obtained in the presence of the naltrexone-SIRT1 complex. From the experimental data, it was determined that the Kd = 77.5 nm for SIRT1 (Fig. 4C). This observation suggests a significant binding affinity between SIRT1 and naltrexone. Furthermore, SIRT1 deacetylase activity was evaluated using a deacetylase fluorometric assay kit (CS1040, Sigma-Aldrich). LDN treatment showed significantly higher SIRT1 deacetylase activity than the control (Fig. 4D). Taken together, our results provide direct evidence that naltrexone can physically interact with SIRT1 to induce deacetylate activity.
Discussion
Emerging evidence has indicated that macrophage polarization is a crucial step toward the pathogenesis of insulin resistance (26). Macrophages of the M1 phenotype act as pro-inflammatory mediators, and M1-like macrophage polarization is closely associated with obesity and diabetes (27), whereas macrophages of the M2 phenotype suppress inflammation (28). Moreover, the deletion of M1-like macrophages ameliorates insulin resistance in obese mice, and the reduction of M2-like macrophages predisposes lean mice to the development of insulin resistance (29).
In this study, we found a significantly increased basal insulin serum level in HFD-saline mice, compared with NCD mice, which demonstrates HFD-associated hyperinsulinemia development. The basal insulin levels of HFD-LDN mice were found to be significantly lower than those of HFD-saline mice, which confirms that LDN could prevent hyperinsulinemia. We also explored the in vivo efficacy of LDN in insulin sensitivity and glucose homeostasis. Interestingly, we found that LDN improved insulin sensitivity and glucose intolerance in HFD mice.
We found the possible involvement of hyperinsulinemia-induced pro-inflammatory cytokines in the pathogenesis of insulin resistance where conditioned media from the hyperinsulinemic group blunted the phosphorylation of AKT (Ser-473), AKT (Thr-308), and GSK3β (Ser-9) which was abrogated in the presence of LDN.
The current study reports an association between hyperinsulinemia and systemic insulin resistance by promoting the release of soluble mediators from peripheral organs, such as adipose tissue and the liver. Hyperinsulinemia, which occurs early during the development of T2DM, down-regulates SIRT1, promotes NF-κB nuclear localization, and activates TLR4 dependent pro-inflammation, which may ultimately lead to insulin resistance.
Here, our data indicate a vicious cycle in which hyperinsulinemia-induced repression of SIRT1 also results in nuclear localization of NF-κB p65, thus maintaining inflammation and insulin resistance, which in turn leads to hyperinsulinemia.
Moreover, this study also revealed an unexpected role of LDN as an anti-inflammatory agent by maintaining SIRT1 protein expression under hyperinsulinemic conditions, and LDN treatment prevents NF-κB nuclear translocation, thereby markedly attenuating hyperinsulinemia-induced release of pro-inflammatory modulators and reducing inflammation-induced systemic insulin resistance in an HFD-induced hyperinsulinemic mouse model. Although SIRT1 is considered a nuclear protein, cytosolic localization of SIRT1 is also well documented (30). To modulate SIRT1 activity, naltrexone must first reach inside the cells and then the nucleus. Interestingly, Cheng et al. (31) has shown that naltrexone can passively enter cells and even in the nucleus. At this juncture, we can speculate that LDN can get inside the cells and even get inside the nucleus to modulate SIRT1 stability. In this context, LDN and SIRT1 crosstalk can be explored further to ameliorate inflammation-induced diseases.
However, the use of anti-inflammatory agents has a very modest effect on improving insulin sensitivity, as reported by the number of studies (32, 33). The present studies suggest that LDN prevents hyperinsulinemia and induces pro-inflammatory cytokine release. LDN improves glucose tolerance and insulin sensitivity in HFD-induced hyperinsulinemic mice. Moreover, our studies indicate that LDN specifically blocks hyperinsulin-stimulated nuclear localization of NF-κB p65, which is required for optimal p65-mediated transactivation potential of NF-κB. Our findings suggest that SIRT1 plays a critical role in hyperinsulin-induced NF-κB regulation, because SIRT1 overexpression could decrease NF-κB reporter activity, whereas a SIRT1 inhibitor or knockdown of SIRT1 enhances it. Finally, our data suggest that LDN may bind to SIRT1 protein and increase its deacetylase activity. Based on these findings, we propose that LDN, an FDA-approved nonpeptide opioid antagonist, may be used as an anti-inflammatory drug in the treatment for insulin resistance.
Experimental procedures
Drugs and chemicals
A low pharmacological dose of naltrexone hydrochloride from MP Biomedicals was used (34–36).
Cell lines and culture treatment
Human hepatocellular carcinoma (HepG2) and Mus musculus macrophage (Raw 264.7) cell lines were maintained in regular DMEM and RPMI media supplemented with 10% FBS and 1% penicillin-streptomycin in 5% CO2 humid incubator maintained at 37°C. HepG2 and Raw 264.7 cells were obtained from Sigma-Aldrich and ATCC, respectively. Transfection experiments were performed using Lipofectamine 3000 (Thermo Scientific) or Lipofectamine RNAimax (Thermo Scientific) for plasmid or siRNA, respectively, strictly following the manufacturer's protocol. SIRT1 specific inhibitor (EX-527) was pretreated for 2 h wherever required.
Cell viability
To determine the nontoxic dose of the drug, cell viability assay was carried out in Raw 264.7 cells using MTT dye (3-(4,5-dimethyl thiazol-2yl)-2,5-diphenyl tetrazolium bromide). Cells were seeded in 96-well plates and allowed to adhere overnight. Further, cells were treated with varying doses (0, 2, 4, 6, 8, 10, 20, 40 μm) of the drug for 24 h. Post treatment, 10 µl of MTT (5 mg/ml stock in PBS) was added to all the wells. The formazan crystals thus formed were solubilized in 200 µl DMSO. The absorbance was recorded at 570 nm using a plate reader (Bio-Rad, model no: iMark and serial no: 18395) (37).
RNA isolation and gene expression profile
Raw 264.7 cultured cells were treated with 5 μm of LDN with or without 100 nm insulin for 24 h. RNA was isolated from the cells using RNA-Xpress reagent (HiMedia) and 1 µg of RNA was reverse-transcribed (using iScript cDNA Synthesis kit, 170-8891, Bio-Rad). qPCR was carried out following standard procedures using SYBR Green (Bio-Rad) mouse primers indicated in Table S2. Expression levels were calculated using the 2-ΔΔCT method with 18S rRNA as internal control (38).
Immunoblot analysis
Raw 264.7 cultured cells were incubated with LDN (0 and 5 μm) in the presence or absence of insulin (100 nm) for 24 h and protein expression of various insulin receptor downstream (SIRT1, p-NF-κB p65) was studied. Furthermore, the phosphorylation level of AKT (Ser-473), AKT (Thr-308), and GSK3β (Ser-9) were evaluated by treating half-diluted condition media (supernatant from insulin-challenged Raw 264.7 cells (Fig. 1B), and serum from all group of mice) to HepG2 cells for 12 h. Cells were lysed by RIPA buffer containing 1% protease-phosphatase inhibitors. Protein concentration was determined by BCA assay reagent as described in the manufacturer's (Thermo Scientific) manual. 40–60 µg of protein was loaded for 10% SDS-PAGE and electro-blotted on to PVDF membranes and incubated in 5% milk blocking solution for 2 h at room temperature. The membrane was probed against the primary antibody (1:2000 diluted in TBST). After washing with TBST, the membrane was incubated with HRP-conjugated goat anti-rabbit/mouse IgG for 2 h and visualized by chemiluminescence.
NF-κB reporter assay
Raw 264.7 cells seeded in 24-well plates were transfected with an NF-κB specific firefly luciferase reporter plasmid, a negative control containing noninducible firefly luciferase reporter and a positive control containing a constitutively expressing GFP and firefly luciferase construct using lipofectamine p3000 transfection reagent (Invitrogen). After 4 h of transfection, the transfection media was replaced with complete media and incubated for another 30 h. Further, the cells were scraped and seeded in white 96-well plates, allowed to adhere properly overnight, and treated with LDN (0 and 5 μm) with or without 100 nm of insulin for 24 h. The resulting cells were subsequently used for NF-κB activity measurement by adding steady Glo reagent (equal to the volume of media) and measuring the luminescence after 5 min of incubation at room temperature.
Immunocytochemistry
Raw 264.7 cells cultured in poly l-lysine–coated chamber slide was treated with LDN (0 and 5 μm) in the presence or absence of 100 nm insulin for 6 h. After treatment, cells were washed with PBS and fixed in chilled methanol for 10–15min at 4°C. Further cells were washed with PBST (0.05% Tween 20 in PBS) to remove traces of methanol. Blocking buffer (2% FBS in PBST) was added and allowed to shake moderately for 2 h at room temperature. Next cells were incubated with primary antibody (1:500 dilution) overnight at 4°C. After washing with PBST (0.1% Tween-20), 2 h incubation with Alexa Fluor secondary antibodies (Table S1) at 1:1000 dilution was done. Slides containing cells were washed three times with PBST and mounted in DAPI mounting media (Vectashield, cat. no. H1200) within the dark. Images were recorded by using confocal microscopy (Nikon ECLIPSE-Ti, class 4 laser product IEC/EN60825-1:2007) at excitation 490 nm and emission 540 nm.
SIRT1 degradation assay
The cells were pretreated with 100 nm of insulin for 24 h with or without LDN and then treated with 10 μm CHX for the indicated duration of time as mentioned in the figure legend. The endogenous SIRT1 expression level was evaluated by immunoblotting. The SIRT1 band intensities relative to the 0 min time point were plotted.
siRNA transfection
Raw 264.7 cells were transfected with 20 nm scramble siRNA or siRNA sirt1 in serum and antibiotic-free media.
Animals and treatments
C57BL/6 mice (Male, 6 weeks old) were procured from the animal house of Indian Institute of Toxicology Research (IITR), Lucknow, India. All experiments were done following the guidelines prescribed by and with the approval of the Animal Ethics Committees of CSIR-IITR. Animals were housed under standardized conditions in the animal facility with free access to food and water, maintaining a constant temperature of 23°C ± 1°C, relative humidity 55% ± 10%, and 12:12 h light and dark cycle throughout experimentation. Mice were randomly divided into four groups. All animals were kept ad libitum with free access for water and food (normal chow diet and HFD group on HFD) for the entire experimental duration. To generate HFD-hyperinsulinemic animals, 6-week-old C57Bl/6J male mice were fed for 3 weeks a diet containing 60% calories as lipids (Altromin). Controls were male littermates fed normal chow diet in parallel.
Group I (NCD-saline)
Mice were on normal chow diet. Saline was injected intraperitoneally (i.p.) every day starting from 4th week until 5th week (end of the experiment).
Group II (NCD-LDN)
Mice were on normal chow diet subjected to LDN treatment (1 mg/kg bw i.p.) every day for 2 weeks (4th and 5th week) (34–36).
Group III (HFD-saline)
Mice were on high fat diet (a diet containing 60% calories as lipids (altromin)). Saline was injected intraperitoneally (i.p.) every day starting from 4th week until 5th week (end of the experiment).
Group IV (HFD-LDN)
Mice were on high fat diet (a diet containing 60% calories as lipids (altromin)). LDN treatment (34–36) (1 mg/kg bw i.p.) was done every day for 2 weeks (4th and 5th week) starting from the 4th week until the 5th week (end of the experiment).
Metabolic parameters analysis
The animals were prescreened for elevated blood glucose levels to ensure homogeneity in the test animals. Blood glucose level was measured by drawing the blood sample from the tail tip by using a commercial Glucometer (T.S Contour, Bayer's). All the mice fasted for 6 h with free access to normal water and peripheral response to glucose was measured by ipGTT injected with 2g/kg bw i.p. of d-glucose. Insulin sensitivity was examined by the ITT in 6-h fasted mice challenged with 0.5 units/kg i.p. insulin (39). The results were expressed by calculating the percentage of area under the glucose and basal insulin curve (AUC) using GraphPad Prism.
ATMs isolation and analysis
Mice were euthanized chemically and epididymal fat was processed for isolation of ATMs. 1 g of adipose fat was rinsed in PBS and minced to small pieces in HEPES-DMEM buffer containing 10 mg/ml BSA. The suspension was centrifuged at 1000 × g for 10 min and the resultant supernatant was pipetted off to fresh tubes. 1 mg/ml of collagenase type IV and 50 units/ml DNase-II were added to this suspension and incubated at 37°C for 45 min with moderate shaking, filtered through 250-micron filter, and the resultant solution was centrifuged again at 1000 × g for 10 min. Floating cells contained adipocyte, and pellet are stromal vascular cells. Red blood cell lysis buffer was added gently to disrupt the sedimented pellets and centrifuged at 1000 × g for 10 min at 4°C. Fat macrophage cells were isolated by using BD IMag anti-mouse CD11b+ magnetic beads through positive selection under the magnetic field. The percentage purity of macrophages isolation was determined by the FACS CANTO II flow cytometer using APC-tagged CD11b mAb. The isolated macrophages were processed for RNA isolation, cDNA synthesis, and various M1-M2 markers were evaluated using real-time PCR.
Serum ELISA
Serum insulin level was measured using insulin ELISA (Thermo Scientific). Serum IL-1Β, TNFα, and IL-6 were measured using Bio-Plex ProTM Mouse cytokine Standard 23 Plex Group 1 kit (Bio-Rad, 171304070M) on a Bio-Plex-200 (Bio-Rad) following the manufacturer's instructions.
Docking simulation
3D information of naltrexone (DB00704) extracted from the DrugBank database and SIRT1 information (PDB ID: 4ZZI) was downloaded from the Protein Data Bank. Autodock-4.2, an open-source software, was used to understand protein–ligand interaction. The molecular docking of selected ligand-compound against the 3D structure of the protein was performed using ADT GUI of Autodock v4.2. Ligand preparation involved the addition of Gasteiger charges. Naltrexone has four active torsions present. Additional water molecules were removed, polar hydrogen atoms were added during receptor preparation. The whole macromolecule-centered 3D grid of 126 × 126 × 126 with the spacing of 0.375 Å was created for a grid search. The conformational search of the ligand was performed by applying the Lamarckian genetic algorithm. The other docking parameters selected were 10 GA runs, the population size of 150, maximum energy evaluations in the range of 250,000, the maximum number of generations 27,000, mutation rate 0.02, crossover rate 0.8, and all other parameters were set to the default values of the software. ADT GUI was used for visualization of our docking results.
MicroScale thermophoresis (MST) measurements
MST technology was used to determine the binding affinity between LDN and SIRT1 (5, 6). For MST measurements, purified N-His6–tagged SIRT1 from Sigma-Aldrich (fluorescent binding partner) considered as target protein was His labeled and kept constant (50 nm). The concentration of nonfluorescent binding partner (LDN as a ligand was varied from 1 mm to 3.05E-05 mm) was placed into the instrument (Monolith NT.115, NanoTemper). All the dilutions were loaded using standard capillaries and measured by stranded protocol. The samples were measured by standard protocols at 40% MST power. The changes of the fluorescent thermophoresis signaling were plotted against the concentration of the serially diluted LDN. Kd value was determined using NanoTemper analysis software (NT analysis 1.5.37).
Statistical analysis
All results are expressed as mean ± S.D. Statistical significance was assessed by Student's t test (unpaired two-tailed) or ANOVA followed by Post-Bonferroni's multiple comparisons whenever required and analyzed using GraphPad Prism (version 5; GraphPad Software). A p value of less than 0.05 was considered to be statistically significant.
Data availability
The datasets supporting the conclusions of this article are included within the article and its supporting information.
Supplementary Material
Acknowledgments
We sincerely thank the BioX Centre of IIT Mandi for the use of different analytical instruments and Dr. Manoj Raje, CSIR-IMTECH, Chandigarh, India, for allowing us to use NanoTemper facility. Thanks to CSIR-IITR for animal research facility, and A. C. thanks the Department of Science & Technology (DST) for the INSPIRE fellowship. We thank members of the P. M. Lab for the useful discussions.
This article contains supporting information.
Author contributions—A. C. and P. M. data curation; A. C. software; A. C., K. G., and P. M. formal analysis; A. C. and A. K. K. validation; A. C., K. G., A. K. K., S. K., M. K. Y., and P. M. investigation; A. C. and P. M. visualization; A. C., K. G., A. K. K., and P. M. methodology; A. C. and P. M. writing-original draft; A. C., K. G., S. K., M. K. Y., D. G., and P. M. writing-review and editing; D. G. and P. M. supervision; P. M. conceptualization; P. M. resources; P. M. funding acquisition; P. M. project administration.
Funding and additional information—This work was supported by Science and Engineering Research Board (SERB), Grant ECR/2015/000165 (to P. M.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- T2DM
- type 2 diabetes mellitus
- LDN
- low-dose naltrexone
- HFD
- high-fat diet
- NCD
- normal chow diet
- ITT
- insulin tolerance tests
- GTT
- glucose tolerance tests
- AUC
- area under the curve
- ATM
- adipose tissue macrophage
- CHX
- cycloheximide
- bw
- body weight
- MST
- MicroScale thermophoresis
- ANOVA
- analysis of variance
- IB
- immunoblot.
References
- 1. International Diabetes Foundation. (2009) IDF Diabetes Atlas, 4th Ed., International Diabetes Foundation, Brussels, Belgium [Google Scholar]
- 2. Kahn S. E. (2001) The importance of β-cell failure in the development and progression of type 2 diabetes. J. Clin. Endocrinol. Metab. 86, 4047–4058 10.1210/jcem.86.9.7713 [DOI] [PubMed] [Google Scholar]
- 3. Nankervis A., Proietto J., Aitken P., and Alford F. (1985) Hyperinsulinaemia and insulin insensitivity: Studies in subjects with insulinoma. Diabetologia 28, 427–431 10.1007/BF00280885 [DOI] [PubMed] [Google Scholar]
- 4. Sbraccia P., D'Adamo M., Leonetti F., Caiola S., Iozzo P., Giaccari A., Buongiorno A., and Tamburrano G. (1996) Chronic primary hyperinsulinaemia is associated with altered insulin receptor mRNA splicing in muscle of patients with insulinoma. Diabetologia 39, 220–225 10.1007/BF00403966 [DOI] [PubMed] [Google Scholar]
- 5. Fisher E., Zhao Y., Richardson R., Janik M., Buell A. K., Aigbirhio F. I., and Tóth G. (2017) Detection and characterization of small molecule interactions with fibrillar protein aggregates using microscale thermophoresis. ACS Chem. Neurosci. 8, 2088–2095 10.1021/acschemneuro.7b00228 [DOI] [PubMed] [Google Scholar]
- 6. Wienken C. J., Baaske P., Rothbauer U., Braun D., and Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 1–7 10.1038/ncomms1093 [DOI] [PubMed] [Google Scholar]
- 7. Blüher M. (2012) Clinical relevance of adipokines. Diabetes Metab. J. 36, 317–327 10.4093/dmj.2012.36.5.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shin K. C., Hwang I., Choe S. S., Park J., Ji Y., Kim J. I., Lee G. Y., Choi S. H., Ching J., Kovalik J.-P., and Kim J. B. (2017) Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat. Commun. 8, 1087 10.1038/s41467-017-01232-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., and Ferrante A. W. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 10.1172/JCI200319246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma M. C., Chiu T. J., Lu H. I., Huang W. T., Lo C. M., Tien W. Y., Lan Y. C., Chen Y. Y., Chen C. H., and Li S. H. (2018) SIRT1 overexpression is an independent prognosticator for patients with esophageal squamous cell carcinoma. J. Cardiothorac. Surg. 13, 25 10.1186/s13019-018-0718-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X., Zhang S., Blander G., Tse J. G., Krieger M., and Guarente L. (2007) SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell. 28, 91–106 10.1016/j.molcel.2007.07.032 [DOI] [PubMed] [Google Scholar]
- 12. Cornish J. W., Metzger D., Woody G. E., Wilson D., McLellan A. T., Vandergrift B., and O'Brien C. P. (1997) Naltrexone pharmacotherapy for opioid dependent federal probationers. J. Subst. Abuse Treat. 14, 529–534 10.1016/S0740-5472(97)00020-2 [DOI] [PubMed] [Google Scholar]
- 13. Oser C. B., and Roman P. M. (2007) Organizational-level predictors of adoption across time: Naltrexone in private substance-use disorders treatment centers. J. Stud. Alcohol Drugs 68, 852–861 10.15288/jsad.2007.68.852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toljan K., and Vrooman B. (2018) Low-dose naltrexone (LDN)—review of therapeutic utilization. Med. Sci. 6, 82 10.3390/medsci6040082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weerts E. M., Kim Y. K., Wand G. S., Dannals R. F., Lee J. S., Frost J. J., and McCaul M. E. (2008) Differences in δ- and μ-opioid receptor blockade measured by positron emission tomography in naltrexone-treated recently abstinent alcohol-dependent subjects. Neuropsychopharmacology 33, 653–665 10.1038/sj.npp.1301440 [DOI] [PubMed] [Google Scholar]
- 16. Younger J., Parkitny L., and McLain D. (2014) The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin. Rheumatol. 33, 451–459 10.1007/s10067-014-2517-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Zhang Y., Peng Y., Hutchinson M. R., Rice K. C., Yin H., and Watkins L. R. (2016) Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br. J. Pharmacol. 173, 856–869 10.1111/bph.13394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutchinson M. R., Zhang Y., Brown K., Coats B. D., Shridhar M., Sholar P. W., Patel S. J., Crysdale N. Y., Harrison J. A., Maier S. F., Rice K. C., and Watkins L. R. (2008) Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: Involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 28, 20–29 10.1111/j.1460-9568.2008.06321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cant R., Dalgleish A. G., and Allen R. L. (2017) Naltrexone inhibits IL-6 and TNFα production in human immune cell subsets following stimulation with ligands for intracellular toll-like receptors. Front. Immunol. 8, 809 10.3389/fimmu.2017.00809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kappert K., Meyborg H., Clemenz M., Graf K., Fleck E., Kintscher U., and Stawowy P. (2008) Insulin facilitates monocyte migration: A possible link to tissue inflammation in insulin-resistance. Biochem. Biophys. Res. Commun. 365, 503–508 10.1016/j.bbrc.2007.11.006 [DOI] [PubMed] [Google Scholar]
- 21. Surmi B. K., and Hasty A. H. (2008) Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol. 3, 545–556 10.2217/17460875.3.5.545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cohen L., Henzel W. J., and Baeuerle P. A. (1998) IKAP is a scaffold protein of the IκB kinase complex. Nature 395, 292–296 10.1038/26254 [DOI] [PubMed] [Google Scholar]
- 23. Yamaoka S., Courtois G., Bessia C., Whiteside S. T., Weil R., Agou F., Kirk H. E., Kay R. J., and Israël A. (1998) Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell 93, 1231–1240 10.1016/S0092-8674(00)81466-X [DOI] [PubMed] [Google Scholar]
- 24. Viatour P., Merville M.-P., Bours V., and Chariot A. (2005) Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 30, 43–52 10.1016/j.tibs.2004.11.009 [DOI] [PubMed] [Google Scholar]
- 25. Li Y., Xu S., Giles A., Nakamura K., Lee J. W., Hou X., Donmez G., Li J., Luo Z., Walsh K., Guarente L., and Zang M. (2011) Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 25, 1664–1679 10.1096/fj.10-173492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olefsky J. M., and Glass C. K. (2010) Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246 10.1146/annurev-physiol-021909-135846 [DOI] [PubMed] [Google Scholar]
- 27. Gordon S. (2003) Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 10.1038/nri978 [DOI] [PubMed] [Google Scholar]
- 28. Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Eagle A. R., Vats D., Brombacher F., Ferrante A. W., and Chawla A. (2007) Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patsouris D., Li P. P., Thapar D., Chapman J., Olefsky J. M., and Neels J. G. (2008) Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 8, 301–309 10.1016/j.cmet.2008.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bai W., and Zhang X. (2016) Nucleus or cytoplasm? The mysterious case of SIRT1's subcellular localization. Cell Cycle 15, 3337–3338 10.1080/15384101.2016.1237170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng F., McLaughlin P. J., Banks W. A., and Zagon I. S. (2009) Passive diffusion of naltrexone into human and animal cells and upregulation of cell proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R844–R852 10.1152/ajpregu.00311.2009 [DOI] [PubMed] [Google Scholar]
- 32. Larsen C. M., Faulenbach M., Vaag A., Vølund A., Ehses J. A., Seifert B., Mandrup-Poulsen T., and Donath M. Y. (2007) Interleukin-1–receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526 10.1056/NEJMoa065213 [DOI] [PubMed] [Google Scholar]
- 33. Stanley T. L., Zanni M. V., Johnsen S., Rasheed S., Makimura H., Lee H., Khor V. K., Ahima R. S., and Grinspoon S. K. (2011) TNF-α antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J. Clin. Endocrinol. Metab. 96, E146–E150 10.1210/jc.2010-1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cabanas H., Muraki K., Staines D., and Marshall-Gradisnik S. (2019) Naltrexone restores impaired transient receptor potential melastatin 3 ion channel function in natural killer cells from myalgic encephalomyelitis/chronic fatigue syndrome patients. Front. Immunol. 10, 2545 10.3389/fimmu.2019.02545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kao J. H., Gao M. J., Yang P. P., Law P. Y., Loh H. H., and Tao P. L. (2015) Effect of naltrexone on neuropathic pain in mice locally transfected with the mutant μ-opioid receptor gene in spinal cord. Br. J. Pharmacol. 172, 630–641 10.1111/bph.12790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tanaka K., Kondo H., Hamamura K., and Togari A. (2019) Systemic administration of low-dose naltrexone increases bone mass due to blockade of opioid growth factor receptor signaling in mice osteoblasts. Life Sci. 224, 232–240 10.1016/j.lfs.2019.03.069 [DOI] [PubMed] [Google Scholar]
- 37. Dogra S., Kar A. K., Girdhar K., Daniel P. V., Chatterjee S., Choubey A., Ghosh S., Patnaik S., Ghosh D., and Mondal P. (2019) Zinc oxide nanoparticles attenuate hepatic steatosis development in high-fat-diet fed mice through activated AMPK signaling axis. Nanomedicine 17, 210–222 10.1016/j.nano.2019.01.013 [DOI] [PubMed] [Google Scholar]
- 38. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2 C T method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 39. Vineeth Daniel P., Kamthan M., Gera R., Dogra S., Gautam K., Ghosh D., and Mondal P. (2019) Chronic exposure to Pb2+ perturbs ChREBP transactivation and coerces hepatic dyslipidemia. FEBS Lett. 593, 3084–3097 10.1002/1873-3468.13538 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its supporting information.