

Abstract
Temperature-sensitive (TS) missense mutants have been foundational for characterization of essential gene function. However, an unbiased approach for analysis of biochemical and biophysical changes in TS missense mutants within the context of their functional proteomes is lacking. We applied MS-based thermal proteome profiling (TPP) to investigate the proteome-wide effects of missense mutations in an application that we refer to as mutant thermal proteome profiling (mTPP). This study characterized global impacts of temperature sensitivity–inducing missense mutations in two different subunits of the 26S proteasome. The majority of alterations identified by RNA-Seq and global proteomics were similar between the mutants, which could suggest that a similar functional disruption is occurring in both missense variants. Results from mTPP, however, provide unique insights into the mechanisms that contribute to the TS phenotype in each mutant, revealing distinct changes that were not obtained using only steady-state transcriptome and proteome analyses. Computationally, multisite λ-dynamics simulations add clear support for mTPP experimental findings. This work shows that mTPP is a precise approach to measure changes in missense mutant–containing proteomes without the requirement for large amounts of starting material, specific antibodies against proteins of interest, and/or genetic manipulation of the biological system. Although experiments were performed under permissive conditions, mTPP provided insights into the underlying protein stability changes that cause dramatic cellular phenotypes observed at nonpermissive temperatures. Overall, mTPP provides unique mechanistic insights into missense mutation dysfunction and connection of genotype to phenotype in a rapid, nonbiased fashion.
Keywords: proteasome, protein-protein interaction, protein complex, protein stability, proteomics, mutant, mass spectrometry, protein structure, missense variant, systems biology, temperature-sensitive, thermal profiling
The ability to systematically assess the function of individual genes and gene products is critical for gaining a full picture of how the cell works (1). Much of our advancement in knowledge on biological processes and the underlying mechanisms is owed to the use of genetic methodologies that perturb normal gene function (discussed in Refs. 2 and 3). One such approach to study the function of essential genes is to use conditional sequence variants, such as temperature-sensitive (TS) mutants. TS mutants have been key tools for decades in the study of many essential biological processes including but not limited to characterization of the cell cycle (4–9), RNA polymerase II transcription (10–17), and autophagy (18–22), among others. TS mutants have distinct advantages in that they provide the ability to negatively regulate protein function without having to change the transcriptional context of the gene, add chemicals, or change media (23). Furthermore, by simply altering the temperature at which cells are grown, TS mutant proteins can be studied in a context in which they retain function (permissive temperature), lose function (nonpermissive temperature), or have partial function (semipermissive temperature) (24, 25).
Despite the long-term use of TS mutants, much is still left to discover in terms of the properties and mechanisms leading to temperature sensitivity. Many of the forward genetics screening processes that produce TS mutants result in multiple mutations, which can make determination of which mutations are causative and what mechanism is underlying the altered phenotype difficult. It has been predicted that the majority of phenotype-causing missense mutations in humans alter protein structure and lead to changes in protein stability or protein-protein interactions (PPIs) (26). This suggests that biophysical details behind a phenotype can likely be revealed by analyzing the cell-wide impacts of missense mutations on the proteome. For instance, an unbiased proteome-wide method for characterizing the global impact of temperature-sensitive mutations on PPIs would increase our ability to link genotype to phenotype in existing, uncharacterized mutants. It would also aid in the development of TS mutants for mechanistic studies of essential genes from any model system. More importantly, any approach developed to examine TS mutants could have broad applicability to studies on phenotype and/or disease-causing protein sequence variants in complex biological systems.
Current widely used approaches, such as mRNA sequencing and global proteomics, provide valuable insight on expression levels; however, they provide no insights into potential changes in protein structure and/or PPIs. Large-scale PPI analysis continues to develop using MS-based workflows (27–32). These methods have greatly increased our knowledge of PPIs but often require large amounts of starting material and involve other manipulations to the cell, such as introducing an epitope tag, or are reliant on antibodies that can differ widely in affinity and specificity (33, 34). Additionally, large-scale interactome studies have to this point been focused on the normal cellular sequence variant for each target protein (known as the bait), leaving protein sequence variants out of consideration. Studies that have incorporated the use of human protein sequence variants into their analysis have been limited by the use of expression systems that are unlikely to reflect the native proteome (26). A nontargeted, proteome-wide MS methodology to evaluate how sequence variants in a protein of interest could affect PPI networks would improve our ability to predict the phenotypic outcomes of genomic mutations and would be more scalable than affinity purification–based approaches. Additionally, broad surveys of protein stability could provide unique biophysical insights, which can be directly compared with complementary computational methods, such as molecular dynamics and multisite λ-dynamics simulations.
Nontargeted MS-based analyses have included approaches to characterize protein complex characteristics without affinity purification, including chromatography-based co-fractionation and thermal proximity co-aggregation (TPCA), which is a cellular thermal shift assay/TPP–related method (35–40). These methods have shown a great deal of promise but have not been fully developed for the investigation of disease-causing protein variants. From these studies, we propose that TPP-based analysis of protein thermal stability could have sufficient specificity and sensitivity to detect both large and small changes in PPI dynamics. Prior TPP experiments have revealed both direct and indirect consequences of on- and off-target drug and ligand binding through shifts in protein melt, suggesting that it could have broad applicability to discover biophysical changes in proteins in varying conditions (41–45). The principle that changes in protein context can be implied from alterations in thermal stability has also been used to study protein-binding metabolites or nucleic acids (43, 46–49), post-translational modifications (50–52), or changes in redox status (53). Additionally, TPP has been used to analyze protein complex behavior in steady-state conditions (40) and throughout different stages of the cell cycle (46, 54). Whereas effects of genetic perturbations such as knockouts have been described (55, 56), this technology has not yet been applied to study the impacts of missense mutations that lead to changes in cellular fitness. To explore the utility of thermal profiling-based methods for analysis of protein sequence variants that result in pronounced cellular fitness defects within the context of the proteome, we have performed mutant TPP analysis (mTPP) in TS mutant yeast using quantitative MS (57–59). Missense mutations could alter protein structure and lead to changes in PPIs within associated complexes and, in theory, affect the thermal stability of either the individual protein or the entire PPI network as a consequence of structural or PPI interface changes. Therefore, we hypothesized that mTPP could be applied to study the effects of missense mutations on the proteome to link phenotype-causing mutations to PPI networks. This work clearly shows that mutant TPP is a useful application of thermal profiling for studying genetic perturbations in diverse cellular systems, laying a foundation for novel characterization of the biophysical mechanisms within the context of native proteomes.
Results
Characterizing the global protein and RNA abundance effects of missense mutants in the 26S proteasome
For our model system in these experiments, we chose TS yeast strains with missense mutations within individual subunits of the 26S proteasome complex, an essential protein complex responsible for the majority of selective proteolysis occurring in the cell (60, 61). The yeast proteasome is a large multisubunit protein complex made up of 33 subunits that form two subcomplexes: a 19S regulatory particle and a 20S core complex. Subunits within the 19S regulatory particle recognize and selectively bind ubiquitylated substrates and deubiquitylate and unfold them (62). Substrates are then mechanically translocated into the proteolytic chamber of the 20S core for degradation (63, 64). Because the proteasome plays a major role in protein turnover and homeostasis, the large-scale changes that could occur in terms of protein abundance levels and protein post-translational modifications make it an intriguing model for these studies. One TS mutant was chosen from each of the subcomplexes of the proteasome: one representing the 19S regulatory particle (rpn5-ts) and the other representing the 26S proteasome core particle (pup2-ts).
Yeast strains were obtained from a repository of temperature-sensitive mutants the Hieter laboratory developed as a resource for studying essential genes (23, 24). Random missense mutations were introduced within an essential gene of interest with the resulting strains screened to identify mutations that caused a temperature-sensitive phenotype (23). The mutated essential genes are expressed from their native promoter and chromosomal context, allowing for analysis of missense mutation impact on the proteome without the confounding variable of altered protein expression levels. Additionally, the use of haploid yeast strains allows for the characterization of this approach in a genetic system that is not complicated by the presence of multiple alleles for a given gene of interest. These characteristics make these strains an ideal representative model for temperature sensitivity–inducing missense mutations. The initial global proteomics studies were performed at both the permissive (30 °C) and restrictive temperatures (37 °C) and revealed that there were extensive differences in the global proteome even at the permissive temperature. With these data in mind, all subsequent experiments were performed at the permissive temperature. Because the goal of these studies was to test whether mTPP was sufficient to identify changes in protein thermal stability of protein variants and their associated PPIs, it was reasoned that the less detrimental temperature would allow us to assess thermal stability changes without confounding changes in cellular biology. For instance, extensive changes in cellular signaling would be expected to occur as a consequence of proteasome dysfunction at restrictive temperatures in addition to the general changes that would occur as a consequence of heat stress.
Global proteomics and poly(A) RNA-Seq experiments were performed to gain a broad picture of the abundance changes occurring in these strains. Both mutant and WT cells were grown at permissive temperature (30 °C) prior to protein or RNA extraction. For the global proteomics, cells were subjected to the workflow presented in Fig. S1, and protein was extracted using 8 m urea and mechanical disruption. RNA was extracted using an established hot acid phenol method (65) prior to library preparation and sequencing on an Illumina system. A total of 3,889 proteins and 5,750 transcripts were quantified (Tables S1 and S2) across all three genotypes. In the pup2-ts mutant, 1,731 proteins changed (Fig. 1A), and 702 mRNA transcripts changed in abundance (Fig. 1B). In the rpn5-ts mutant, 1,997 proteins (Fig. 1C) and 539 mRNA transcripts (Fig. 1D) changed in abundance. Comparisons between protein and mRNA transcript abundance show a low, but positive, correlation (Fig. S2). It is becoming increasingly evident that the interplay across the different levels of gene expression is complex and that changes in protein abundance cannot be explained by changes in mRNA levels alone (66). The major role of the proteasome is to maintain proteostasis, and mutations in subunits that disrupt its degradation activity are likely to result in protein abundance changes that are not explained by mRNA transcript abundance alone. Additionally, the proteasome has been shown to play critical roles, both proteolytically and nonproteolytically, in the regulation of transcription and chromatin organization (67–69). The numerous roles of the proteasome across multiple stages of gene expression can have various effects on gene expression, further complicating the relationship between protein and mRNA abundance levels in proteasome mutants.
Figure 1.

Global changes in protein and mRNA abundance. Volcano plots of fold-change vs. -log10 p-value in pup2-ts/WT of protein (A) and mRNA transcripts (B) and in rpn5-ts/WT of protein (C) and mRNA transcripts (D). x axis, log2 -fold change of mutant/WT; y axis, −log10 p value or FDR. The significance threshold was set at p value or FDR ≤0.05. 3,862 data points are shown on each plot. Proteasome core subunits are indicated in blue, and regulatory subunits are indicated in orange. A, a total of 1,731 proteins significantly changed in pup2-ts, with 471 proteins decreasing and 1,260 proteins increasing in abundance. B, A total of 701 mRNA transcripts significantly changed in pup2-ts, with 109 transcripts decreasing and 592 transcripts increasing in abundance. C, A total of 1,997 proteins significantly changed in rpn5-ts, with 586 proteins decreasing and 1,411 proteins increasing in abundance. D, a total of 539 mRNA transcripts significantly changed in rpn5-ts, with 112 transcripts decreasing and 427 transcripts increasing in abundance.
As both rpn5-ts and pup2-ts are within the same protein complex, it was unsurprising that both global proteomics and RNA-Seq presented a large number of similar changes between both mutants. There was a 70% overlap in protein expression changes (Fig. S3) and a 52% overlap in mRNA expression changes (Fig. S4) in the two mutants. Both pup2-ts and rpn5-ts have increased abundance of many of the proteasome subunits (Fig. S5) as well as the transcription factor Rpn4 at both the mRNA and protein levels (Tables S1 and S2). The up-regulation at both the mRNA and protein level of the 26S proteasome is a known negative feedback mechanism for proteasome function that occurs through the transcription factor Rpn4. Rpn4 is a known substrate of the 26S proteasome and increases in abundance as a result of defective proteasome activity (70). The increase in Rpn4 abundance in both of these strains suggests that it is not being efficiently degraded, strongly indicating that the proteasome is not functioning properly. In an attempt to compensate for decreased proteasome activity in these strains, Rpn4 likely up-regulates the expression of the subunits of the proteasome to increase the number of proteasomes in the cell. Another strong indicator of proteasome malfunction is an increase of the proteasome chaperones Ump1 in both strains (Tables S1 and S2). Ump1 functions in assembly of the core proteasome and is degraded by the 20S proteasome prior to its binding the 19S regulatory particle and formation of the full 26S proteasome (71, 72). The large increase in Ump1 protein accumulation, 12.7-fold increase in protein relative to a 2.8-fold increase in mRNA, in pup2-ts may suggest that the TS mutations in Pup2 cause a proteasome assembly defect that exacerbates Ump1 accumulation (Tables S1 and S2). In rpn5-ts, the increase in Ump1 protein is comparable with the change in mRNA of UMP1 (2.5-fold increase in protein and 2.2-fold increase in mRNA), suggesting this may occur through a transcriptional response to the lack of proteasome activity. The increase in the chaperone Ump1 supports the likelihood of proteasome functional defects in these strains, consistent with previous work (73, 74). However, the large increase in Ump1 protein in pup2-ts suggests a potential defect in proper proteasome assembly that differs from the changes observed in rpn5-ts cells.
Thermal proteome profiling can be applied to measure changes resulting from genetic perturbation
MS methods are widely utilized to measure changes in protein abundance, protein turnover rates, post-translational modifications (PTMs), and PPIs (75). Thermal proteome profiling (TPP) is an emerging method that is revolutionizing how MS is able to gain insights into the proteome. TPP has been used to study such events as target engagement, post-translational modifications, and cell cycle variation (40–58, 76); however, it has not yet been applied to biological questions regarding changes that occur as a consequence of single-gene missense mutations known to cause defects in cellular fitness through unknown mechanisms. We have applied TPP (Fig. 2A) (57, 77) to address the global effects of TS-inducing missense mutations on the proteome, which we refer to as mTPP.
Figure 2.

Mutant TPP workflow adaptations. A, equal amounts of protein from each lysate were subjected to six different temperature treatments, 35.0, 45.3, 50.1, 55.2, 60.7, and 74.9 °C, to induce protein denaturation. The soluble fractions from each treatment were digested in-solution with trypsin/Lys-C. The resulting peptides were labeled with isobaric mass tags (TMTsixplex™) and mixed by genotype prior to MS analysis. Resulting MS/MS data were analyzed using Proteome Discoverer™ 2.2 to identify and quantify abundance levels of peptides for each temperature treatment and each genotype. B, dot plots showing the abundance value for every protein detected from the MTPP experiments in WT, rpn5-ts, and pup2-ts. Box-and-whisker plots include minimum, maximum, and median. C, representative melt curves from a single replicate of the percentage of soluble protein following heat treatment are shown for selected protein complexes. Proteins isolated from WT are shown in gray, rpn5-ts in orange, and pup2-ts in teal. Shown here are curves of the RNA exosome (11 individual proteins), the 40S ribosome (39 individual proteins), and the chaperonin-containing T-complex (8 individual proteins). D, individual protein melt curves were created for every quantified protein and normalized using the TPP R package. Tm was calculated as the temperature at which 50% of the protein was denatured. Shown are one protein from each of the complexes in B.
Strains were grown at 30 °C as performed for the global proteomics and mRNA expression analyses shown in Fig. 1 and lysed in a nondenaturing lysis buffer containing 0.1% Tween. Cell lysate from each genotype was subjected to the workflow described in Fig. 2A for thermal profiling. Three biological replicates were prepared and were subjected to high-pH basic fractionation, resulting in 72 LC–MS/MS runs for each genotype. We detected and quantified a total of 4,073 proteins across three biological replicates for each of the three genotypes (Fig. 2B and Table S3). Data obtained from quantitative MS/MS experiments were used to make melt curves in two different ways (Fig. 2, C and D). Raw abundances from each temperature treatment were normalized to the ion abundance detected in the 45 °C sample and were plotted to visualize melt curves of protein complexes in mutant and WT (Fig. 2C). Melt curves of three different protein complexes, the RNA exosome, the 40S ribosome, and the chaperonin-containing T-complex, are shown in Fig. 2C. From these, we can see that proteins within a complex melt in a similar fashion, consistent with what has been previously shown in TPCA studies (40). The TPP R package (78) was used to generate normalized protein melt curves and calculate melt temperatures (Tm) for ∼3,400–3,600 proteins, depending on the replicate (numerical data for these in Table S4).
Average Tm value distributions were plotted for the proteomes of WT as compared with individual mutants (Fig. S6). Observation of average Tm in the range of 50–56 °C indicated that the vast majority of proteins denature in this region, which is comparable with what has been previously observed by TPP (42, 45, 46) and with other methods measuring Tm of the proteome (79). Similar to the global melt, average Tm for the individual protein complexes in Fig. 2C was ∼52–53 °C in WT and in both mutants. The largest variance relative to WT in the average Tm for the highlighted complexes was 0.32 °C (the calculated difference relative to WT for the exosome in rpn5-ts and the ribosome in pup2-ts) and was not statistically significant. Together, these data suggest that the mutations in rpn5-ts and pup2-ts do not have a large overall effect on global melt or the stability of the exosome, ribosome, or chaperonin protein complexes. Additionally, for the majority of individual proteins, such as those shown in Fig. 2D, we saw very similar Tm values between mutant and WT and tight fitting to a sigmoidal curve (as indicated by the r2 value). These data show a successful adaptation of the TPP workflow for mTPP with the cost-effective change of using only six temperature points, which is of particular advantage for screening studies. All biological replicate plots for the proteins shown in Fig. 2D are provided in Fig. S7.
mTPP measures the impact of single protein mutants on the stability of the global proteome and on protein-protein interactions
Changes in Tm (ΔTm) values were calculated by taking WTTm − mutantTm, thereby limiting calculations to proteins detected in both WT and mutant (Table S5). Further parsing was accomplished by limiting our data to melt curves with r2 values ≥0.9 and then by proteins that were detected in at least two of the three replicates, providing us with a final analysis of ΔTm in ∼2,050 proteins (Table S6). The 19S regulatory particle mutant, rpn5-ts, contains 13 missense mutations within Rpn5 (E347G, F402L, M171T, S200T, S416P, S54P, E294G, K147E, E214K, S295G, A328V, I353V, and Q342R), which confer temperature sensitivity at 37 °C (Fig. S8) (24). Structurally, these mutations are found around the exterior of Rpn5 (Fig. S9A) (80). The charge-change associated mutations are solvent-exposed, lessening their direct effects on the stability of rpn5-ts or the 19S regulatory particle, and many other mutations feature large-to-small changes in side-chain sizes, which collectively are expected to minimally perturb the secondary and tertiary folding of rpn5-ts, although they could impact protein-protein interactions between Rpn5 and non-19S-binding partners. Whole-proteome analysis of thermal stability found significant (defined by a 95% confidence interval) decreases in the thermal stability of 70 proteins and increases in the stability of 40 proteins (Fig. 3A and Table S7). Analysis of the melt curves of the subunits of the proteasome in rpn5-ts showed no changes in the 20S core relative to WT (Fig. 3B). At ∼55–60 °C, a flattening of the thermal melt curve was observed in both WT cells and rpn5-ts in raw melt curves. This observation could suggest a phased melting of the 20S core particle, perhaps representing dissociation of the subunits of the 20S complex followed by single subunit melting at slightly higher temperatures (Fig. 3B). Although this observation is interesting, following normalization, the curve flattening had minor impacts on sigmoidal curve fitting using the TPP package, which revealed that most of the 20 S subunits had average Tm values close to 58.5–61.5 °C with an S.D. of ∼0.58 across all three replicates. Despite the numerous missense mutations within RPN5, neither the thermal stability of Rpn5 itself nor of any subunits of the 19S regulatory particle were significantly affected (Fig. 3C). All replicate data for the raw melt curves for each proteasome subunits are provided in Fig. S10. Following normalization, thermal melt profiles for the proteasome subunits showed tight reproducibility across all replicates and genotypes, indicating that no changes occur in proteasome protein subunit stability in rpn5-ts (Fig. 3, D and E; normalized melt curves for all proteasome subunits are provided in Fig. S11). A common mechanism that is used to explain a temperature sensitivity phenotype is that elevated temperature may disrupt the structural integrity of a mutant protein. In fact, this is a general concept that is thought to explain temperature sensitivity in many conditional mutant organisms (81). However, the lack of an observed shift in stability of Rpn5 suggests that thermal sensitivity of this strain is not necessarily due to structural changes in Rpn5 at restrictive temperatures, but likely because of other PPI effects, changes in Rpn5 function, or compensation pathways that adjust for mutant protein function at the permissive temperatures.
Figure 3.

Mutations in Rpn5 do not affect the thermal stability of the proteasome. A, waterfall plots visualizing whole-proteome changes in melt temperature (Tm), WT − rpn5-ts. A total of 2,068 proteins were ordered according to change in Tm and plotted. Shown are median values across three biological replicates for proteins that were quantified in at least two replicates. Dotted lines signify a confidence interval of 95%. There were significant decreases in thermal stability of 70 proteins; 40 proteins had significant increases in thermal stability. B and C, representative melt curves from WT versus rpn5-ts for each of the 14 subunits of the 20S proteasome core (B) and the 19 subunits of the 19S regulatory particle (C) derived from data from one MTPP experiment. Each line represents an individual subunit. D and E, individual normalized melt curves of representative protein Pre10 from the 20S core (D) and Rpn2 from the 19S regulatory particle (E).
The 20S core particle mutant, pup2-ts, contains four missense mutations within Pup2 (C76R, L204Q, T113M, and S260P) and is unable to grow properly at 37 °C (Fig. S8) (24). In contrast to rpn5-ts, the pup2-ts mutations, with the exception of S260P, occur within the hydrophobic core of the subunit. These mutations include small-to-large side-chain size changes and a charge-change mutation, which suggests that these amino acid substitutions could be more disruptive to the stability of pup2-ts and, perhaps, to the larger 20S core particle (Fig. S9B) (82). mTPP analysis showed that a total of 22 proteins were thermally destabilized in the pup2-ts mutant (Fig. 4 and Table S7). Remarkably, of those 22 proteins, all 14 of the 20S core subunits were significantly destabilized, which was clearly observed in melt curve analysis (Fig. 5A). None of the regulatory particle subunits had altered thermal stability in pup2-ts (Fig. 5B), suggesting that the mutations in this protein likely affect proteasome activity by disrupting the core proteasome and not the regulatory particle. Normalization of the data via the TPP package resulted in similar findings showing significant destabilization of the 14 20S core particle subunits (Fig. 5, C and D). The specificity of these findings is striking, with nearly 64% of the destabilized proteins occurring within the same protein complex as the mutant protein. The normalized melt curves for each proteasome subunit in pup2-ts are provided in Fig. S11. Gene ontology analysis (83) of the destabilized protein group identified the gene ontology term proteasomal ubiquitin–independent protein catabolic process as the most enriched, with a -fold change enrichment value >100 and a false discovery rate of 4.25e−25. These findings clearly show that mTPP has a very high degree of selectivity to identify proteins with altered thermal properties and, in this case, their close PPI partners. Interestingly, 103 proteins were significantly stabilized in pup2-ts (Fig. 4). The pup2-ts–stabilized proteins could be either directly impacted by the pup2-ts mutation or due to secondary impacts of the mutations, such as disruption of proteasome function, leading to an accumulation of polyubiquitylation on these proteins or changes in their PPI network.
Figure 4.

Mutations in Pup2 disrupt the core proteasome. Waterfall plots visualize whole-proteome changes in melt temperature (Tm), WT − pup2-ts. A total of 2,046 proteins were ordered according to change in Tm and plotted. Shown are median values across three biological replicates for proteins that were quantified in at least two replicates. Dotted lines signify a confidence interval of 95%. There were significant decreases in thermal stability of 22 proteins; 103 proteins had significant increases in thermal stability.
Figure 5.

mTPP uncovers thermal destabilization of all 20S core subunits of the proteasome in pup2-ts mutant cells. A and B, representative raw data melt curves from WT versus pup2-ts for each of the 14 subunits of the 20S proteasome core (A) and the 19 subunits of the 19S regulatory particle (B) derived from data from the mTPP experiment. Each line represents an individual subunit. C and D, individual normalized melt curves of representative proteins Pre10 from the 20S core (C) and Rpn2 from the 19S regulatory particle (D).
Multiomics intersection analysis of mTPP with global proteome abundance and transcriptome data provides unique mechanistic insights into mutant dysfunction
Intersection analysis was used to visualize the total overlap between the mTPP, RNA-Seq, and global protein abundance data sets through the generation of Upset plots using the UpSetR package in R (84) and manual data set intersection analysis (Fig. 6). Integration of mTPP with proteomic and transcriptomic data can provide a clearer picture of the mechanisms linking genotype to phenotype and aid in mechanistic hypothesis generation. For example, a possible explanation for an increase in thermal stability in proteins in these mutants is through a change in the actions, abundance, and/or function of cellular chaperone proteins. There was an increase in the abundance of Ssa1 and Ssa2, two Hsp70 family members in both mutants: a ∼1.5-fold increase in pup2-ts and a ∼2-fold increase in rpn5-ts. Ssa1/2 have been shown to play a role in delivering misfolded protein to the proteasome during endoplasmic reticulum–associated degradation (ERAD) (85) Additionally, Hsp104, another chaperone linked to ERAD, was seen to increase in protein abundance 1.7- and 1.4-fold in pup2-ts and rpn5-ts, respectively (86). The up-regulation of cellular chaperones could be a compensation mechanism for defects in 26S proteasome function and could help explain why these cells are able to survive at permissive temperature despite altered proteasomal activity.
Figure 6.

Multiomics intersection analysis of pup2-ts and rpn5-ts. Upset plots visualize the overlap of gene products within the sets of significant changes measured in MTPP, global proteomics, and mRNA sequencing in pup2-ts (A) and rpn5-ts (B). An arrow indicates subset mentioned in the text.
Multiomics intersection analysis of the three -omics data sets revealed subcategories of interest (Fig. 6). There was little overlap between gene products that increased or decreased in parallel, and in some cases, there were some that changed in opposite ways (e.g. protein up/RNA down or protein up/destabilized), providing potential interesting candidates for further mechanistic studies. In the pup2-ts mutant, 14 of the 15 proteins seen to be both thermally destabilized and have increased mRNA. Intersects indicated in Fig. 6A by arrows are the subunits of the proteasome core, showing the strength of using mTPP in multiomics intersection analysis for identifying candidate protein subsets involved in the mechanism behind an observed phenotype. Aside from the core proteasome subunits, the additional member within this subset is the DNA damage–inducible protein 1 (Ddi1), a ubiquitin-binding protein that functions as a proteasome shuttle (reviewed in Ref. 87) that has recently been characterized as a ubiquitin-dependent protease (88). Ddi1 is destabilized in rpn5-ts as well (Table S7) and has been shown to interact with the proteasome via affinity purification MS (89). It is possible that the disruption in proteasome activity is impacting critical interactions between Ddi1 and the proteasome or its substrates, leading to Ddi1 destabilization. In rpn5-ts, one particular intersection that stood out is a gene that was thermally destabilized and decreased in abundance but had an increase in mRNA abundance, Tma17 (also known as Adc17), highlighted by an arrow in Fig. 6B. Tma17 is an important proteasome assembly chaperone that has been shown to help enhance lid assembly in response to stress in a regulatory mechanism independent of Rpn4 (90). An increase in TMA17 mRNA suggests that the cell may be trying to compensate for the destabilization and decreased abundance of this protein by up-regulating its mRNA transcript.
Analyzing the overlap of changes occurring in pup2-ts and rpn5-ts has the potential for discovery of general proteasome interactors as well as highlighting subunit-specific interactors. For example, the intersect in proteins that are increasing in abundance but whose mRNA levels are not increased in both mutants could identify candidate proteasome substrates. An Upset plot presenting the overlaps of like changes occurring in pup2-ts and rpn5-ts is shown in Fig. 7A. Almost 72% (1,117 of 1,554) of proteins that were found to increase in abundance using global proteomics were found to increase in both pup2-ts and rpn5-ts (Fig. 7B, abundance increased). A total of 363 mRNA transcripts were up-regulated in both pup2-ts and rpn5-ts; however, this was only ∼50% of the total increases observed in mRNA levels in both mutants (Fig. 7). Strikingly, there was a low percentage of proteins that had changes in thermal stability in the same direction in both mutants, six stabilized and three destabilized (Fig. 7), underscoring the remarkable selectivity and reproducibility of the mTPP approach. Compared with the commonly applied approaches of transcriptome and proteome abundance analysis, measuring the thermal stability of proteins using mTPP provided the most unique insights into the underlying impact of each proteasome subunit mutant on the biophysical state of the proteasome and the mechanism of missense mutation dysfunction.
Figure 7.

Comparing changes between rpn5-ts and pup2-ts. Shown is an upset plot of the overlap in changes between rpn5-ts (orange) and pup2-ts (teal). Gene products that are changing in the same way in both genotypes are indicated in gray. B, a stacked bar plot shows the percentage of each data set that is changing in the same direction in both mutants (gray) or changes unique to rpn5-ts (orange) or pup2-ts (teal).
Molecular modeling quantifies the contributions of individual missense mutations to destabilization of pup2-ts and the 20S proteasome
Following the observation that pup2-ts cells show destabilization of all of the proteins within the 20S core particle, molecular dynamics (MD) and multisite λ-dynamic (MSλD) simulations were performed to further investigate the structural and thermodynamic mechanisms of pup2-ts destabilization. MD simulations enable the dynamic motion of atoms in molecules to be modeled as a function of time, yielding atomistic insights into biophysical observables (91). From all-atom MD simulations of Pup2 and pup2-ts, isolated in solution, differences in root mean square fluctuation (ΔRMSF) of individual residues over the course of the simulation revealed that the C76R, T113M, and L204Q mutations induce several dynamic changes in the structure of Pup2 (Table S8 and Fig. S12). The S260P mutation was not included in the molecular model; this residue is positioned on the flexible C-terminal tail of pup2-ts, and no electron density for this region was captured in the crystal structure (82). Notably, large increases of dynamic fluctuations of ∼0.2–1.0 Å are observed in the two α-helices composed of residues 84–139, which contain the T113M mutation and sit directly adjacent to the C76R mutation. It is clear that these two small-to-large side-chain perturbations destabilize this region of Pup2 dramatically. In contrast, the L204Q mutation induces very few fluctuation changes. Extrapolating these findings onto the 1RYP crystal structure of the yeast 20S proteasome illustrates that Pup2 residues with the largest ΔRMSF line the internal cavity of the 20S core and form important PPIs with neighboring 20S α subunits (Fig. S12, C and D) (82). Inducing large dynamic fluctuations in this region of the 20S core particle is thus expected to weaken important interactions to neighboring subunits and disrupt the rigid structure of the core proteasome.
To further investigate the individual contributions of each mutation to Pup2 destabilization, MSλD calculations were performed. MSλD is a rigorous free-energy simulation method able to calculate changes in protein-binding affinity in response to point mutations (92, 93). Compared with other free-energy simulation methods, MSλD is highly efficient, requiring an order of magnitude less computation than standard approaches without loss of precision, and it facilitates sampling of different perturbations at multiple sites in a single simulation (92, 94–97). This offers the advantage of exploring nonadditive effects between multiple point mutations within a protein simultaneously (95). This study also represents the first application of MSλD to investigate protein-protein interactions. The mutations in pup2-ts, specifically C76R, T113M, and L204Q, were explored in a combinatorial fashion in both insolated and complexed states of Pup2. Relative free energies of binding (ΔΔGbind) between WT Pup2 and eight different combinations of the mutations, including the combination of all three found in pup2-ts, were computed using a standard thermodynamic cycle at 30 and 50 °C (Table 1 and Fig. S13) (98). The free-energy data in Table 1 fully support the experimental mTPP findings that the complete 20S core particle is destabilized by the incorporation of pup2-ts, with a computed ΔΔGbind of ∼3.8 kcal/mol. The most destabilizing perturbation is C76R, which introduces a large, charged residue into the hydrophobic core of Pup2 and causes several structural deformations in Pup2. Analysis of the MSλD trajectories shows that the Arg-76 can either protrude between residues 84–101 and 109–120 (referred to as “inward”) or between residues 66–74 and 84–101 (referred to as “outward”), as shown in Fig. 8. In both conformations, arginine disrupts the hydrophobic network of nearby valine, leucine, and isoleucine residues, opens up the core of the protein, and facilitates the penetration of nearby water molecules into the core of Pup2 (Fig. 8B). Interestingly, the T113M and L204Q mutations, alone, do not have a significant effect on the free energy of binding of Pup2 to the 20S core particle, with ΔΔGbind results of −0.20 kcal/mol or less. However, these residues play an important complementary role toward stabilizing the C76R mutation. Clear stabilizing, nonadditive, ΔΔGbind effects of −0.7 to −0.8 kcal/mol per residue are observed to reduce the singular C76R mutation ΔΔGbind from 5.15 to 3.79 kcal/mol in the full pup2-ts mutant. Structural observations help explain these effects. When Arg-76 protrudes inward, Met-113 can help fill the additional space introduced by Arg-76 as well as hydrogen-bond with water molecules that enter the core (Fig. 8A); when Arg-76 protrudes outward, Gln-205 is able to hydrogen-bond to the backbone of Gln-218 and thus stabilize large fluctuations in the bottom half of Pup2 (Fig. 8D). As temperature is increased to 50 °C, however, this nonadditive complementarity is reduced, and Pup2 is more fully destabilized by the C76R mutation. These effects likely contribute to the mTPP observed decrease in Tm for pup2-ts compared with WT Pup2. Collectively, these findings support that the C76R, T113M, and L204Q temperature-sensitive mutations in Pup2 destabilize the structure of Pup2 and weaken its ability to bind to the 20S core particle. They also reveal that the majority of these effects stem from the C76R mutation, with nonadditive, but complementary and stabilizing, effects from T113M and L204Q.
Table 1.
Relative free energies of binding (kcal/mol)
Changes in binding free energy (ΔΔGbind) for WT and eight mutant variants of Pup2 were calculated using multisite λ-dynamic simulations at two temperatures (30 and 50 °C)
| Residue identity |
ΔΔGbind at 30 °C | ΔΔGbind at 50 °C | ||
|---|---|---|---|---|
| 76 | 113 | 204 | ||
| C | T | L | 0.00 ± 0.81 | 0.00 ± 0.73 |
| C | T | Q | −0.01 ± 1.40 | −0.37 ± 1.20 |
| C | M | L | −0.20 ± 0.93 | −2.10 ± 0.26 |
| C | M | Q | −0.24 ± 0.88 | −1.48 ± 0.34 |
| R | T | L | 5.15 ± 0.53 | 6.07 ± 0.16 |
| R | T | Q | 4.46 ± 0.38 | 7.12 ± 0.24 |
| R | M | L | 4.34 ± 0.12 | 5.21 ± 0.25 |
| R | M | Q | 3.79 ± 0.23 | 6.27 ± 0.19 |
Figure 8.
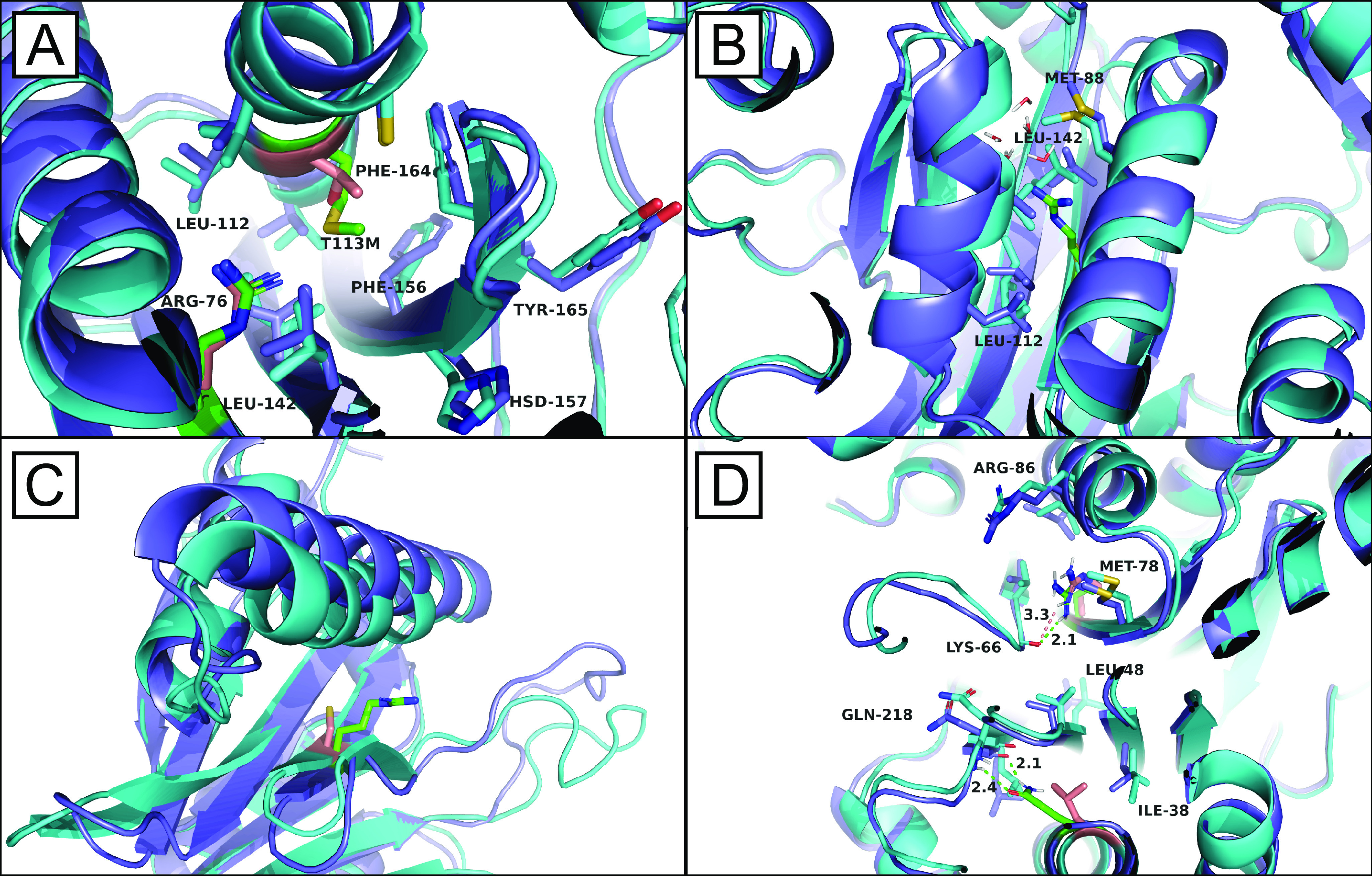
Structural observations from the MSλD trajectories. A, overlay of complex C76R Pup2 (cyan with residues 76 and 113 in peach) and complex C76R+T113M pup2-ts (purple with residues 76 and 113 in green). Arg-76 points inward, disrupting important hydrophobic interactions between Leu-144, Leu-112, Ile-144, Ile-70, and Val-116. Met-113, however, is able to occupy space adjacent to Arg-76 to help offset the disruption of this hydrophobic core in Pup2. B, overlay of complex native Pup2 (cyan) and complex C76R pup2-ts (purple with residue 76 in green). Nearby water molecules are able to penetrate into the core of Pup2 when Arg-76 protrudes inward and disrupts the hydrophobic network of nearby valine, leucine, and isoleucine residues. C, overlay of unbound native Pup2 (cyan with residue 76 in peach) and unbound C76R pup2-ts (purple with residue 76 in green). In addition to its inward conformation, Arg-76 is also observed to protrude outward, which induces large structural distortions in residues 84–101, the α-helix positioned above Arg-76, and 53–71, consisting of part of a β-sheet and a flexible loop. D, overlay of complex C76R Pup2 (cyan with residues 76 and 204 shown in peach) and complex C76R+L204Q pup2-ts (purple with residues 76 and 204 shown in green). As an interesting example of stabilizing complementarity, when Arg-76 points outward, Gln-204 (green) can hydrogen-bond to the backbone of Gln-218 to help stabilize nonnative fluctuations in the bottom half of Pup2.
Discussion
Temperature sensitivity is generally thought to lead to loss of function of a mutant protein at restrictive temperatures due to decreased protein stability at higher temperatures. Our results suggest that this is not always the case and that the biophysical properties of TS proteins are likely mutation-specific, with unique mechanisms resulting in temperature sensitivity in pup2-ts and rpn5-ts. In the pup2-ts mutant, we calculated ΔTm values for over 2,000 proteins and remarkably only saw destabilization of 22 proteins, 14 of which are the 20S core proteasome itself (Fig. 4). These findings suggest that mutations in pup2-ts thermally destabilize Pup2 and that elevated temperatures can lead to a disruption of the stability of the entire proteasome core. This was well-supported by MD and MSλD simulations, which quantified the individual contributions of three pup2-ts mutations and demonstrated loss of binding affinity of pup2-ts to the 20S proteasome. These data also show the highly complementary nature of mTPP, MD, and MSλD, with mTPP providing experimental evidence in support of MD and MSλD findings. In summary, we conclude that the proteasome dysfunction in this mutant is most likely due to changes in folding and incorporation of Pup2 into the 20S core particle, leading to a defect in or a decreased efficiency of overall proteasome core assembly. As assembly of the regulatory particle occurs independently of the core (99), it is reasonable that mutations in the hydrophobic interior of Pup2 do not destabilize the 19S regulatory particle subunits alongside the 20S core. Interestingly, despite presenting with similar protein and RNA abundance alterations, the mutations in rpn5-ts do not appear to affect the thermal stability of Rpn5 or the 26S proteasome complex.
Mutations in rpn5-ts instead lead to temperature sensitivity via a different mechanism, likely disruption of other protein-protein interactions essential to proteasome function. The fact that many rpn5-ts mutations are close to the surface of the protein supports this notion. Other studies of the rpn5-ts mutant found that these mutations in Rpn5 cause nuclear mislocalization of the regulatory particle as well as chromosomal instability (73, 74). Of the 110 proteins mTPP identified to have stability changes in rpn5-ts, 50 are found within the nucleus, so it could be that the observed phenotype is due to the loss of essential nuclear interactions of the proteasome. Another possibility could be that the mutations in Rpn5 do not allow for proper maturation and function of the 26S proteasome (80, 100–108). The regulatory particle chaperone Tma17 was significantly destabilized and decreased in protein abundance, despite having an increase in mRNA abundance. Therefore, the mutations in rpn5-ts could prevent the proper interaction of Tma17 with the proteasome lid, leading to its destabilization. Due to the important role for Tma17 in increasing proteasome assembly when proteolysis capacity has been exceeded (109), the destabilization of Tma17 could be further crippling the proteasome defects in this mutant, and the increase in TMA17 transcript suggests the cell is trying to compensate for the destabilization and decreased abundance of this protein. Finally, it is possible that direct and/or indirect effects on protein PTMs, such as phosphorylation, could contribute to changes in stability, but phospho-TPP experiments suggest that these effects would likely be subtle (52).
Global protein and RNA abundance measurements from rpn5-ts and pup2-ts revealed a large number of possible candidates that could be causing phenotypic changes observed in the mutant strains. These experiments were performed at permissive temperature, indicating that even at temperatures that allow for normal cell growth, the mutations in rpn5-ts and pup2-ts are perturbing proteasome function. It remains a possibility that a number of the proteins that increase in abundance in both mutants as a consequence of proteasome perturbation are direct substrates of the proteasome. Additional studies measuring changes in PTMs could shine further light on these effects as well. For example, ubiquitylation in these mutant strains may help determine whether some of the changes in protein expression and/or stability in either of the proteasome mutants are due to accumulation of ubiquitylated proteins. Many of the alterations in global protein and RNA abundance were the same across the two mutants, which could lead to the conclusion that they have similar functional disruption. However, the additional data gained through mTPP on protein stability and potential PPI changes suggest that the rpn5-ts and pup2-ts mutants are altered through different mechanisms. These missense mutants contain relatively few proteins that are changed in thermal stability, most of which are unique to the mutant subunit studied, suggesting mTPP to be sensitive enough to measure mutation dependent shifts in thermal stability and protein-specific associations, even between two proteins within the same protein complex. As the mTPP protocol measures protein thermal stability at temperatures outside of physiological range, it allows for the exaggeration of biophysical effects that may be too small to cause catastrophic protein unfolding in live cells. Use of this application allows for the determination of mechanistic differences in the phenotypes of two mutant strains in the context of their native proteome that would be difficult, or perhaps impossible, to discover through other existing experimental methods in a high-throughput manner.
Our study produced quantitative melt curves for over 3,000 proteins across each of the three genotypes evaluated. Although these mutations affect overall proteostasis, only ∼100–130 proteins had a change in thermal stability, and there was not a global effect on protein thermal stability, with the average global melt temperature remaining similar to WT. There were few changes in thermal stability relative to abundance changes, which suggests that proteasome targets (many of them likely polyubiquitylated) may not have major changes in overall stability that could assist in the unfolding necessary for degradation by the 26S proteasome. mTPP identified possible PPIs that are unique to Pup2 and Rpn5 individually, as seen through proteins that change in thermal stability in only one of the two mutants, providing an avenue through which one can study the unique biophysical mechanisms that are occurring due to mutations in different proteins that lead to similar phenotypes. Finally, these data show that mTPP can be used to reproducibly measure global protein abundance from the low-temperature treatment conditions in yeast, removing the need for separate global proteomics experiments, consistent with what has previously been shown in TPP experiments in HeLa cells (46). Despite being isolated in two different buffers, correlation values were greater than 0.8 between experiments (Fig. S14A), and there was an 82% overlap in protein identification (Fig. S14B).mTPP is capable of measuring changes in the thermal stability of proteomes as a result of temperature sensitivity–inducing missense mutations without the need for any additional genetic manipulation or antibody-based interrogation. Considering the inherent challenges with antibody-based methods because of specificity concerns (34), methods that avoid dependence on antibodies are likely to increase the rigor and reproducibility of mutant protein interrogation. Additionally, the lack of a requirement of protein subunit epitope tagging, a genetic manipulation, allows for rapid interrogation of proteome-transcriptome-phenotype characterization. Epitope tagging is a key molecular biology tool for affinity purification followed by MS (AP-MS). Although AP-MS is a powerful approach for protein-protein interaction identification, it requires a large amount of starting material, which is challenging to obtain from many biological systems, including patient samples (110–112), and can require a substantial amount of time for optimization of the purification. Comparison of mTPP thermal stability changes with MD and MSλD was extremely useful to explain the experimental changes in protein thermal stability using existing three-dimensional structures for the 26S proteasome. However, mTPP can also provide biophysical insight in the absence of existing structural data for proteins of interest.
Not only are mutant proteins important molecular biology tools, but more than 100,000 genomic mutations have been linked to human diseases, of which ∼60% are predicted to affect protein stability and/or PPIs (26). Whereas genomic and transcriptomic sequencing data are abundant and readily accessible, changes in mRNA have been shown to explain only ∼40% of protein level changes (113). A full understanding of disease relies not only on genomic data, but also on defining the status of cellular proteins in the context of their abundance, post-translational modification status, PPIs, stability, and turnover rate. Previous studies that have obtained high-throughput data on disease-causing mutations, for instance, have relied on approaches, such as yeast two-hybrid and chaperone engagement studies, that are carried out in vitro (26). Although these approaches can provide unique molecular insights, they also rely on gene cloning and overexpression, which will artificially alter the balance of allele-specific (in diploid cells) and/or PPI partner expression, leading to confounding changes throughout the proteome. In addition to the capability of mTPP for the characterization of TS mutants for studies of normal biological mechanism, use of this method with disease-associated mutant proteins will help lay the foundation for a bioinformatic resource increasing understanding of disease mechanisms and addressing the pathogenicity of missense mutations.
Experimental procedures
Yeast strains and growth conditions for proteomics experiments
Temperature-sensitive yeast strains were obtained from the Hieter laboratory (23, 24). Two strains with missense mutations within the 26S proteasome, rpn5-ts and pup2-ts, were chosen for these experiments. Both mutant and WT cells were inoculated at an A600 = 0.3 and grown to an A600 = 0.8 in yeast extract, peptone, dextrose (YPD) medium at permissive temperature (30 °C). YPD was removed by filtration through a nitrocellulose membrane. Cells were flash-frozen with liquid nitrogen and stored at −80 °C to be used in subsequent sample preparation steps.
mTPP sample preparation
Cells for each genotype and biologically distinct replicate (i.e. three genotypes and three biological replicates, for a total of nine), were removed from nitrocellulose membrane using water, pelleted, and reconstituted in lysis buffer containing 40 mm Hepes-KOH (pH 7.5), 10% glycerol, 250 mm NaCl, 0.1% Tween 20, and fresh yeast protease inhibitors. Resulting suspensions were lysed using a mini bead beater and a vortex genie. After removing the beads, lysates were subjected to centrifugation (14,000 × g for 20 min) to pellet out cellular debris and to obtain a clear lysate. Protein concentration values for each resulting lysate were determined using the Bio-Rad protein assay (Bio-Rad), employing vendor-provided protocols. All lysates were then diluted to a protein concentration of 5 μg/μl for subsequent heat treatment. Six 50-μl aliquots for each genotype and replicate were then distributed across six PCR tubes and equilibrated at six temperature points—35.0, 45.3, 50.1, 55.2, 60.7, and 74.9 °C for 3 min in a thermocycler (Mastercycler Pro, Eppendorf, Hamburg, Germany) system as described elsewhere (57). Following heat treatment, lysates were centrifuged for 20 min at 4 °C to pellet out insoluble protein and then to decant the soluble fraction. A 20-μl aliquot from each heat-treated sample was subject to TCA-based protein precipitation overnight. Following centrifugation and acetone-based wash steps, resulting protein pellets were reconstituted in 30 μl of 8 m urea in Tris-HCl (pH 8.0). Samples were subjected to reduction of Cys-Cys bonds with 5 mm tris(2-carboxyethyl)phosphine hydrochloride and alkylation with 10 mm chloroacetamide to protect the reduced Cys residues. Samples were diluted to 2 m urea and were digested in-solution overnight using trypsin (Promega Corp., Madison, WI, USA) as described previously to derive peptides (111, 112, 114). Samples were desalted using a Sep-Pak® Vac 1 cc C18 Cartridge (Waters Corp., Milford, MA, USA) according to the vendor-provided vacuum manifold protocol that involves 1) priming the column, 2) loading the peptides onto the column, 3) washing the column to remove any buffers from the immobilized peptides, and 4) elution of immobilized peptides. This was performed to remove buffer additives and excess reagents as many lysis buffers are known to interfere with the TMT labeling reaction. Resulting peptide elutions were dried using a speed vacuum and subjected to TMT labeling using vendor-provided protocols. A TMTsixplexTM kit (Thermo Fisher Scientific, Waltham, MA, USA) was employed for the purpose, and, more specifically, channels TMT126, TMT127, TMT128, TMT129, TMT130, and TMT131 were used to label peptide solutions derived from the 35.0, 45.3, 50.1, 55.2, 60.7, and 74.9 °C temperature treatments, respectively. Labeling reactions were quenched, and the six labeled samples were mixed and were dried using a speed vacuum system. Dried samples were fractionated using reversed-phase fractionation columns (eight fractions) employing vendor-provided protocols (Pierce). The resulting eight fractions were dried using a speed vacuum system and resuspended in 0.1% formic acid (30 μl) prior to nano-LC–MS/MS analysis as described below.
mTPP data acquisition: Nano-LC–MS/MS
Nano-LC–MS/MS analyses were performed on a Q-Exactive Plus™ mass spectrometer (Thermo Fisher Scientific) coupled to an EASY-nLC™ HPLC system (Thermo Fisher Scientific). A 10-μl equivalent volume of the resuspended fractions from above was loaded using 300 bars as applied maximum pressure onto an in-house prepared reversed-phase column. Each reversed-phase column was prepared by pulling a 100 μm fused-silica column to carry a ∼5-μm tip for the nanospray using a P-2000 laser puller and then packing the capillary with C18 reverse-phase resin (particle size, 3-μm diameter; Dr. Maisch HPLC GmbH, Ammerbuch, Germany). The peptides were eluted using a varying mobile phase (MP) gradient from 95% phase A (formic acid/H2O, 0.1:99.9 (v/v)) to 24% phase B (formic acid/acetonitrile, 0.4:99.6 (v/v)) for 150 min, from 24% phase B to 35% phase B for 25 min, and then keeping the same MP composition for an additional 5 min at 400 nl/min to ensure elution of all peptides. Nano-LC mobile phase was introduced into the mass spectrometer using a Nanospray Flex Source (Proxeon Biosystems A/S, Odense, Denmark). The heated capillary temperature was kept at 275 °C, and ion spray voltage was kept at 2.5 kV. During peptide elution, the mass spectrometer method was operated in positive ion mode for 180 min, programmed to select the most intense ions from the full MS scan using a top 20 method. Additional parameters were as follows: microscans, 1; resolution, 70,000; AGC target, 3E6; maximum IT, 50 ms; number of scan ranges, 1; scan range, 400–1,600 m/z; and spectrum data type, “profile.” Then data-dependent MS/MS scans were performed with the following parameters: microscans, 1; resolution, 35,000; AGC target, 1E5; maximum IT, 64 ms; loop count, 20; MSX count, 1; isolation window, 0.7 m/z; fixed first mass, 100 m/z; NCE, 38.0; spectrum data type, “centroid.” The respective data-dependent settings were set with the following parameters: minimum AGC target, 1.00e3; intensity threshold, 1.6e4; apex trigger, “−”; charge exclusion, “1, 7, 8, >8”; multiple charge states, “all”; peptide match, “preferred”; exclude isotopes, “on”; dynamic exclusion, 30.0 s; if idle “pick others.” The data were recorded using Thermo Xcalibur (version 4.1.31.9) software (Thermo Fisher Scientific).
Sample preparation for global quantitative proteomics comparison of mutant proteomes relative to WT
Three biological replicates of WT, pup2-ts, and rpn5-ts were prepared as explained above. Unlike the previous preparation, cells were lysed in 8 m urea using a mini bead beater and a vortex genie. A 50-μg equivalent of protein from each lysate was then subjected to reduction, alkylation, proteolytic digestion, and desalting as described above. Peptide samples were labeled using a 10plex kit (Thermo Fisher Scientific, Waltham, MA, USA). Specific labeling reagents corresponding to TMT127N, TMT127C, TMT128N, TMT128C, TMT129N, TMT129C, TMT130N, TMT130C, and TMT131 were respectively used to label the peptide solutions derived from three replicates from each genotype: WT, PUP2-TS, and RPN5-TS. Following reaction quenching, mixing, and the subsequent drying step, the resulting peptide mixture was fractionated as described, resulting in eight fractions. Each fraction was dried completely using a speed vacuum system and resuspended in 0.1% formic acid (30 μl) prior to the nano-LC–MS/MS analysis explained below.
Global proteomics data acquisition: Nano-LC–MS/MS
Nano-LC–MS/MS analyses were performed on an Orbitrap Fusion™ Lumos™ mass spectrometer (Thermo Fisher Scientific) coupled to an EASY-nLC™ HPLC system (Thermo Fisher Scientific). An 18-μl equivalent volume of the resuspended fractions was loaded onto an in-house prepared reversed phase column using 600 bars as the applied maximum pressure. Each reversed-phase column was prepared by pulling a 100-μm fused-silica column to carry a ∼5-μm tip for the nanospray using a P-2000 laser puller and then packing the capillary with a C18 reversed-phase resin (particle size, 3-μm diameter; Dr. Maisch HPLC GmbH). The peptides were eluted using a 180-min gradient increasing from 95% buffer A (0.1% formic acid in water) and 5% buffer B (0.1% formic acid in acetonitrile) to 25% buffer B at a flow rate of 400 nl/min. The peptides were eluted using a 180-min gradient increasing from 95% buffer A (0.1% formic acid in water) and 5% buffer B (0.1% formic acid in acetonitrile) to 25% buffer B at a flow rate of 400 nl/min. Nano-LC mobile phase was introduced into the mass spectrometer using a Nanospray Flex Source (Proxeon Biosystems A/S). During peptide elution, the heated capillary temperature was kept at 275 °C, and ion spray voltage was kept at 2.6 kV. The mass spectrometer method was operated in positive ion mode for 180 min, having a cycle time of 3 s for MS/MS acquisition. MS data were acquired using a data-dependent acquisition method that was programmed to have two data-dependent scan events following the first survey MS scan. During MS1, using a wide quadrupole isolation, survey scans were obtained with an Orbitrap resolution of 120,000 with vendor-defined parameters―m/z scan range, 375–1,500; maximum injection time, 50; AGC target, 4E5; microscans, 1; RF lens (%), 30; data type, “profile”; polarity, positive with no source fragmentation and to include charge states 2–7 for fragmentation. Dynamic exclusion for fragmentation was kept at 60 s. During MS2, the following vendor-defined parameters were assigned to isolate and fragment the selected precursor ions: isolation mode = quadrupole; isolation offset = off; isolation window = 0.7; multi-notch isolation = false; scan range mode = auto normal; FirstMass = 120; activation type = CID; collision energy (%) = 35; activation time = 10 ms; activation Q = 0.25; multistage activation = false; detector type = IonTrap; IonTrap scan rate = Turbo; maximum injection time = 50 ms; AGC target = 1E4; microscans = 1; data type = centroid. During MS3, daughter ions selected from neutral losses (e.g. H2O or NH3) of precursor ion CID during MS2 were subjected to further fragmentation using higher-energy C-trap dissociation to obtain TMT reporter ions and peptide-specific fragment ions using the following vendor-defined parameters: isolation mode = quadrupole; isolation window = 2; multi-notch isolation = true; MS2 isolation window (m/z) = 2; number of notches = 3; collision energy (%) = 65; orbitrap resolution = 50,000; scan range (m/z) = 100–500; maximum injection time = 105 ms; AGC target = 1E5; data type = centroid. The data were recorded using Thermo Scientific Xcalibur (version 4.1.31.9) software (Thermo Fisher Scientific).
Protein identification and quantification
Resulting RAW files were analyzed using Proteome Discoverer™ 2.2 (Thermo Fisher Scientific). The SEQUEST HT search engine was used to search against a yeast protein database from the UniProt sequence database (December 2015) containing 6,279 yeast protein and common contaminant sequences (FASTA file used available on ProteomeXchange under accession number PXD017223). Specific search parameters used were: trypsin as the proteolytic enzyme, peptides with a maximum of two missed cleavages, precursor mass tolerance of 10 ppm, and a fragment mass tolerance of 0.6 Da. Static modifications used for the search were 1) carbamidomethylation on cysteine residues and 2) TMTsixplex label on lysine residues and the N termini of peptides. Dynamic modifications used for the search were oxidation of methionine and acetylation of N termini. The percolator false discovery rate was set to a strict setting of 0.01 and a relaxed setting of 0.05. Values from both unique and razor peptides were used for quantification. For the mTPP experiments, no normalization setting was used for protein quantification; for the global protein abundance measurements, peptides were normalized by “total peptide amount.” Data are available via ProteomeXchange with identifiers PXD017222 and PXD017223.
TPP analysis and waterfall plots
The TPP package (version 3.12.0) (78) in R was used to generate normalized melt curves and to determine protein melt temperatures as described previously (77). Changes in Tm for each protein were calculated utilizing the function WT (Tm) – mutant (Tm), where WT (Tm) represents the median melt temperature of a protein determined in WT samples, and mutant (Tm) represents the median melt temperature of a protein determined in mutant samples. Resulting data processing was performed in R Studio (R Studio for Mac, version 1.1.456). Data were parsed down to proteins that were detected in at least two of the three replicates and had melt curves with r2 values >0.9. Proteins were ranked according to median change in Tm and ordered from the largest change (proteins that were destabilized in the mutant) to smallest change (proteins that were stabilized in the mutant). From the distribution of Tm changes in a single comparison, Tm changes that are out of the range, mean (Tm change) ± 2σ (where σ is the S.D.), were considered statistically significant Tm changes and identified as proteins destabilized or stabilized due to the mutation. Waterfall plots were created using ggplot2 (83).
RNA extraction
Both mutant and WT cells were inoculated at an A600 = 0.5 in YPD medium and permitted to recover for 90 min at permissive temperature (30 °C). Cells were pelleted, washed with water, and resuspended in 10 ml of acetate-EDTA buffer (50 mm sodium acetate, pH 5.2, 10 mm EDTA). RNA was isolated using a hot acid phenol method as described previously (65). In brief, 800 μl of 20% SDS and 10 ml of cold acid phenol were added to each sample in a Nalgene phenol-resistant tube. Samples were incubated for 10 min with rotation in a hybridization oven at 65 °C and cooled on ice for 5 min. The samples were then centrifuged at 10,000 rpm for 15 min, and the top phase was transferred to a 50-ml 5 PRIME Phase Lock Gel Tube (reference no. 2302870). 13 ml of chloroform was added, and samples were centrifuged at 3,000 rpm for 10 min; the remaining top phase was poured into a new phenol-resistant tube. One-tenth volume sodium acetate at pH 5.2 and an equal volume of isopropyl alcohol were added, and precipitated RNA was collected by centrifugation at 12,000 rpm for 45 min. The RNA pellet was washed with 70% EtOH and dried in a fume hood. RNA was resuspended with molecular biology grade water. Following purification, RNA was treated using the TURBO DNA-free kit (Thermo Fisher Scientific) to remove DNA contamination.
Library preparation and sequencing
The concentration and quality of total RNA samples were first assessed using an Agilent 2100 Bioanalyzer. A RIN (RNA integrity number) of five or higher was required to pass the quality control. Then 100 ng of RNA per sample were used to prepare dual-indexed strand-specific cDNA libraries using the KAPA mRNA HyperPrep kit (Roche Applied Science). The resulting libraries were assessed for quantity and size distribution using Qubit and an Agilent 2100 Bioanalyzer. Pooled libraries were utilized for clustering amplification on cBot using a HiSeq 3000/4000 PE Cluster Kit and sequenced with a 2 × 75-bp paired-end configuration on HiSeq4000 (Illumina) using a HiSeq 3000/4000 PE SBS Kit. A Phred quality score (Q score) was used to measure the quality of sequencing. More than 94% of the sequencing reads reached Q30 (99.9% base call accuracy).
Sequence alignment and gene counts
The sequencing data were first assessed using FastQC (version 0.11.5, Babraham Bioinformatics, Cambridge, UK) for quality control. All sequenced libraries were mapped to the yeast genome (UCSC sacCer3) using STAR RNA-seq aligner (version 2.5) (115) with the following parameter: “−outSAMmapqUnique 60.” The reads distribution across the genome was assessed using bamutils (from ngsutils version 0.5.9) (116). Uniquely mapped sequencing reads were assigned to S288C R64-2-1 20150113 annotated genes using featureCounts (subread version 1.5.1) (117) with the following parameters: “-s 2 –p –Q 10.” Genes with read count per million <0.5 in ≥4 of the samples were removed. The data were normalized using the TMM (trimmed mean of M values) method. Differential expression analysis was performed using edgeR (version 3.12.1) (118, 119). False discovery rate (FDR) was computed from p values using the Benjamini–Hochberg procedure. All files from the RNA-Seq experiments performed for this study have been uploaded to the Gene Expression Omnibus (GEO) under the accession number GSE143236.
Global abundance plots
Volcano plots and correlation plots were created in R Studio using ggplot2. Abundance values, -fold change ratios, and p values for the proteomics data were calculated by Proteome Discover™ 2.2 (Thermo Fisher Scientific). FPKM values, -fold change ratios and FDR values were calculated as described above. p value and FDR cutoff for significance was set at <0.05. Venn diagram was created using Venny version 2.1 (120). Abundance dot plots were created using GraphPad Prism version 6.00 for Windows. Upset plots were created using the UpSetR package in R Studio (84).
Molecular modeling computational details
System preparation
Initial starting coordinates for Pup2 and the 20S proteasome complex were obtained from the Protein Data Bank file 1RYP (82). The protein complex was set up similarly to Vilseck et al. (96), with only protein residues within a 27-Å radius around Pup2 included in the complex simulations. All protein backbone atoms beyond 17 Å from Pup2 were harmonically restrained to their crystallographic positions with a force constant of 10 kcal/mol·Å2. All side chains and protein residues within a 17-Å radius from Pup2, and including Pup2, remained fully flexible. Starting coordinates for isolated, unbound Pup2 were generated by extracting Pup2 (chain E) from the 1RYP complex. Residue flips for histidine, glutamine, and asparagine were assessed using the MolProbity webserver (121, 122), and protonation states of titratable residues were determined with the assistance of PROPKA (123). After examining the structures and considering the MolProbity and PROPKA results, the side chains of Glu-56 of chain L, His-16 of chain D, His-73 of chain E, and His-67 of chain M were protonated, and Lys-62 of chain F was deprotonated. All protein residues were modeled using the CHARMM36 all-atom force field for proteins (124, 125). Standard CHARMM36 patches of GLYP, NTER, and CTER were applied to native N- and C-terminal residues, and peptide fragments resulting from the truncation of the full 20S complex were capped with ACE and CT3 patches, respectively. The truncated complex and unbound Pup2 were then solvated with the TIP3P water model (126) using the CHARMM-GUI webserver (127) with a minimum of 10 Å of solvent from each face of the structure. This produced cubic water boxes with edge lengths of 125.0 Å (truncated complex) and 85 Å (Pup2). Na+ and Cl− ions were added to neutralize the net charge of the system and achieve an ionic strength of 100 mm NaCl. Prior to running MD or MSλD, the CHARMM molecular simulation package (version 44b1) was used to minimize each solvated system obtained from the CHARMM-GUI (128, 129). First, all protein atoms were harmonically restrained, whereas all ions and water molecules were relaxed with 200 steps of steepest-descent minimization, after which all restraints were removed and the entire system was subject to an additional 250–500 steps of steepest-descent minimization.
Molecular dynamics
MD simulations were run with OpenMM on graphic-processing units (130). For Pup2 and pup2-ts systems, four independent MD duplicates were run for 100 ns each. MD simulations were run in the isobaric-isothermal (NPT) ensemble at 30 °C and 1 atmosphere using Langevin dynamics, with a friction coefficient of 10 ps−1 and a Monte Carlo barostat (131). Periodic boundary conditions were employed with nonbonded cutoffs of 12 Å to truncate all long-range interactions. An integration time step of 2 fs was used, and trajectory frames were saved every 5,000 steps. Trajectories were then superimposed onto the minimized starting structure using VMD (132). Root mean square fluctuations were calculated as root mean square average distances between an atom and its mean position in a set of structures, using Equation 1.
| (Eq. 1) |
Over the course of Ns frames in each of the MD trajectories, xi coordinates of the backbone α carbon for each k residue in Pup2 were analyzed to calculate an RMSF for each residue (133). RMSF values were then averaged over the four duplicate runs for Pup2 and pup2-ts, respectively, and plotted in Fig. S12A. Root mean square deviations of all backbone α carbon atoms were then calculated with respect to the minimized starting structure to show global protein fluctuations as a function of time sampled.
| (Eq. 2) |
In this analysis, an RMSD is calculated for every frame by looping over all Na atoms with x0 reference and xi target coordinates (133). RMSDs between duplicate MD runs were again averaged and plotted in Fig. S12B.
Multisite λ-dynamics
MSλD calculations were performed in CHARMM using the domain decomposition (DOMDEC) module to enable the use of graphic-processing units (92, 93, 128, 129, 134). Simulations were run with a Nose–Hoover thermostat and Langevin pressure piston, with a friction coefficient of 20 ps−1, to maintain sampling within the NPT ensemble at 30 °C and 1 atmosphere (135–137). Trajectory frames were saved every 10,000 steps, and λ states were saved every 10 steps for computing relative free-energy differences. The SHAKE algorithm was employed to constrain all hydrogen-heavy atom bond lengths, and periodic boundary conditions were employed (138). The particle mesh Ewald method was used to compute all long-range electrostatic interactions(139–141), and all nonbonded Lennard–Jones long-range interactions were truncated at 10 Å, with force-switching to zero between 9 and 10 Å (142). With MSλD, the C76R, T113M, and L204Q pup2-ts mutations were simulated as a multiple-topology model, where all atomic components of native and mutant side chains were explicitly included, using the BLOCK facility within CHARMM. Following standard reported procedures for using MSλD (92–97), holonomic restraints were used to constrain λ between 0 and 1, thus maintaining physically relevant end points during the simulation (143). For calculating free-energy differences, a threshold of λ ≥ 0.99 was used as an approximation of the physical end state λ = 1.0. A soft-core Lennard Jones potential was used to scale all nonbonded interactions for the alchemical substituents by λ (94). Substituent dihedral angles were scaled by λ, whereas bonds, angles, and improper dihedral angles were not, as this was found previously to yield better sampling without causing conformations to become trapped in local energy minima (95, 97). The adaptive landscape flattening (ALF) algorithm was used to identify appropriate biasing potentials for MSλD (94, 95). ALF was run in two stages: first with several 100-ps simulations, followed by additional 1-ns simulations for bias refinement. The unbound Pup2 at 303.15 K ran 176 100-ps simulations followed by 18 1-ns simulations. ALF biases for the complex at 303.15 K were initialized with estimates from preliminary single-mutant simulations, and then 221 100-ps simulations and 14 1-ns simulations were run. Unfortunately, despite these great lengths of sampling with ALF and MSλD, converged biases that yielded consistent sampling of all alchemical end states were elusive. This can be attributed to the difficulty of exploring small-to-large perturbations within the hydrophobic core of Pup2. To circumvent this difficulty, the best biases from unbound and bound Pup2 were then used in one 5-ns production run followed by three sequential 25-ns production runs employing variable-bias replica exchange (VB-REX), an enhanced sampling algorithm (95). The unbound Pup2 at 323.15 K ran 500 100-ps simulations and 44 1-ns simulations, all using VB-REX for bias refinement. The bound Pup2 at 323.15 K ran 145 100-ps simulations using VB-REX for bias refinement. The best biases from unbound and bound Pup2 at 323.15 K were then used in three sequential 25-ns production runs employing VB-REX. Three to five duplicate runs with three replicas each were used in the 25-ns production iterations, and ALF was used in between iterations to compute biases for the next set of calculations. Ultimately, the last set of 25-ns production runs were used for computing differences in binding free energies between WT and mutant variants of Pup2. Thus, for the final free energies reported in Table 1, a total of 225 ns of sampling was performed for both solvated and complexed states of Pup2 at 303.15 K, and a total of 375 ns of sampling was performed for both the solvated and complexed state of Pup2 at 323.15 K. MSλD trajectories were then analyzed with the assistance of VMD and PyMOL (132, 144).
Data availability
All analyzed data sets used for this study are referenced in the article and included in the supporting information. The MS proteomics data have been deposited to the ProteomeXchange Consortium (145) via the PRIDE (146) partner repository with the data set identifiers PXD017223 (global) and PXD017222 (mTPP). The RNA-Seq data have been deposited to Gene Expression Omnibus (GEO) under the accession number GSE143236. Any code used for data analysis is available as referenced. Thermo Proteome Discoverer software is a commercial product; however, a free viewer is available for processed data files at https://thermo.flexnetoperations.com/.
Supplementary Material
Acknowledgments
We thank the current members of the Mosley laboratory (Whitney Smith-Kinnaman, Katlyn Hughes Burriss, Lynn Bedard, Dominique Baldwin, and Neil McCracken) and the Indiana University School of Medicine proteomics core (Emma Doud). We thank Brian G. Pulawski of the Vilseck laboratory. We also thank Maureen Harrington and Mark Goebl for helpful discussions of this work. We acknowledge the Indiana University Pervasive Technology Institute (supported by the Lilly Endowment, Inc.) for providing supercomputing and storage resources that have contributed to the research results reported within this paper. Acquisition of the IUSM Proteomics core instrumentation used for this project was provided by the Indiana University Precision Health Initiative. Reagents to carry out a portion of this work have graciously been provided via the Thermo Scientific TMT Research Award (to S. A. P. J.).
This article contains supporting information.
Author contributions—S. A. P. J. data curation; S. A. P. J., M. P. B., H. R. S. W., J. F. V., E. R S., J. Z. V., A. B. W., and A. L. M. formal analysis; S. A. P. J. and A. L. M. funding acquisition; S. A. P. J. validation; S. A. P. J., M. P. B., G. D. Q., H. R. S. W., J. F. V., E. R. S., J. Z. V., A. B. W., and A. L. M. investigation; S. A. P. J., M. P. B., J. Z. V., and A. L. M. visualization; S. A. P. J., G. D. Q., H. R. S. W., J. Z. V., and A. B. W. methodology; S. A. P. J. and A. B. W. writing-original draft; S. A. P. J., M. P. B., J. Z. V., A. B. W., and A. L. M. writing-review and editing; M. P. B., J. Z. V., A. B. W., and A. L. M. conceptualization; H. R. S. W. software; J. Z. V. and A. L. M. supervision; A. B. W. project administration.
Funding and additional information—This work was supported in part by National Institutes of Health Grant T32 HL007910 (to S. A. P. J.) and by the Showalter Research Trust (to A. L. M.). This work was also supported in part by the Indiana Clinical and Translational Sciences Institute, which is funded by National Institutes of Health Grant UL1TR002529, a National Center for Advancing Translational Sciences, Clinical, and Translational Sciences Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- TS
- temperature-sensitive
- PPI
- protein-protein interaction
- TPCA
- thermal proximity co-aggregation
- PTM
- post-translational modification
- TPP
- thermal proteome profiling
- mTTP
- mutant thermal proteome profiling
- ERAD
- endoplasmic reticulum–associated degradation
- MD
- molecular dynamics
- MSλD
- multisite λ-dynamic
- RMSF
- root mean square fluctuation
- RMSD
- root mean square deviation
- AP-MS
- affinity purification followed by MS
- MP
- mobile phase
- AGC
- automatic gain control
- CID
- collision-induced dissociation
- FDR
- false discovery rate
- ALF
- adaptive landscape flattening
- VB-REX
- variable-bias replica exchange.
References
- 1. Boone C. (2014) Yeast systems biology: our best shot at modeling a cell. Genetics 198, 435–437 10.1534/genetics.114.169128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nurse P., and Hayles J. (2019) Using genetics to understand biology. Heredity (Edinb.) 123, 4–13 10.1038/s41437-019-0209-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Winston F., and Koshland D. (2016) Back to the future: mutant hunts are still the way to go. Genetics 203, 1007–1010 10.1534/genetics.115.180596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hartwell L. H., Culotti J., and Reid B. (1970) Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. U. S. A. 66, 352–359 10.1073/pnas.66.2.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nurse P., Thuriaux P., and Nasmyth K. (1976) Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 146, 167–178 10.1007/BF00268085 [DOI] [PubMed] [Google Scholar]
- 6. Hartwell L. H., Mortimer R. K., Culotti J., and Culotti M. (1973) Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics 74, 267–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartwell L. H. (1971) Genetic control of the cell division cycle in yeast. II. Genes controlling DNA replication and its initiation. J. Mol. Biol. 59, 183–194 10.1016/0022-2836(71)90420-7 [DOI] [PubMed] [Google Scholar]
- 8. Lee M. G., and Nurse P. (1987) Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327, 31–35 10.1038/327031a0 [DOI] [PubMed] [Google Scholar]
- 9. Nurse P., and Bissett Y. (1981) Gene required in G1 for commitment to cell cycle and in G2 for control of mitosis in fission yeast. Nature 292, 558–560 10.1038/292558a0 [DOI] [PubMed] [Google Scholar]
- 10. Holstege F. C., Jennings E. G., Wyrick J. J., Lee T. I., Hengartner C. J., Green M. R., Golub T. R., Lander E. S., and Young R. A. (1998) Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95, 717–728 10.1016/S0092-8674(00)81641-4 [DOI] [PubMed] [Google Scholar]
- 11. Thompson C. M., and Young R. A. (1995) General requirement for RNA polymerase II holoenzymes in vivo. Proc. Natl. Acad. Sci. U. S. A. 92, 4587–4590 10.1073/pnas.92.10.4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hartzog G. A., Wada T., Handa H., and Winston F. (1998) Evidence that Spt4, Spt5, and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes Dev. 12, 357–369 10.1101/gad.12.3.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takagi Y., Calero G., Komori H., Brown J. A., Ehrensberger A. H., Hudmon A., Asturias F., and Kornberg R. D. (2006) Head module control of mediator interactions. Mol. Cell 23, 355–364 10.1016/j.molcel.2006.06.007 [DOI] [PubMed] [Google Scholar]
- 14. Winston F., Chaleff D. T., Valent B., and Fink G. R. (1984) Mutations affecting Ty-mediated expression of the HIS4 gene of Saccharomyces cerevisiae. Genetics 107, 179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gross C. A., Grossman A. D., Liebke H., Walter W., and Burgess R. R. (1984) Effects of the mutant σ allele rpoD800 on the synthesis of specific macromolecular components of the Escherichia coli K12 cell. J. Mol. Biol. 172, 283–300 10.1016/S0022-2836(84)80027-3 [DOI] [PubMed] [Google Scholar]
- 16. Nonet M., Scafe C., Sexton J., and Young R. (1987) Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol. Cell Biol. 7, 1602–1611 10.1128/mcb.7.5.1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cormack B. P., and Struhl K. (1992) The TATA-binding protein is required for transcription by all three nuclear RNA polymerases in yeast cells. Cell 69, 685–696 10.1016/0092-8674(92)90232-2 [DOI] [PubMed] [Google Scholar]
- 18. Kametaka S., Okano T., Ohsumi M., and Ohsumi Y. (1998) Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 273, 22284–22291 10.1074/jbc.273.35.22284 [DOI] [PubMed] [Google Scholar]
- 19. Shirahama K., Noda T., and Ohsumi Y. (1997) Mutational analysis of Csc1/Vps4p: involvement of endosome in regulation of autophagy in yeast. Cell Struct. Funct. 22, 501–509 10.1247/csf.22.501 [DOI] [PubMed] [Google Scholar]
- 20. Takeshige K., Baba M., Tsuboi S., Noda T., and Ohsumi Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311 10.1083/jcb.119.2.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thumm M., Egner R., Koch B., Schlumpberger M., Straub M., Veenhuis M., and Wolf D. H. (1994) Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 349, 275–280 10.1016/0014-5793(94)00672-5 [DOI] [PubMed] [Google Scholar]
- 22. Tsukada M., and Ohsumi Y. (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- 23. Kofoed M., Milbury K. L., Chiang J. H., Sinha S., Ben-Aroya S., Giaever G., Nislow C., Hieter P., and Stirling P. C. (2015) An updated collection of sequence barcoded temperature-sensitive alleles of yeast essential genes. G3 (Bethesda) 5, 1879–1887 10.1534/g3.115.019174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ben-Aroya S., Coombes C., Kwok T., O'Donnell K. A., Boeke J. D., and Hieter P. (2008) Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell 30, 248–258 10.1016/j.molcel.2008.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugaya K. (2018) Let's think again about using mammalian temperature-sensitive mutants to investigate functional molecules—the perspectives from the studies on three mutants showing chromosome instability. J. Cell. Biochem. 119, 7143–7150 10.1002/jcb.27205 [DOI] [PubMed] [Google Scholar]
- 26. Sahni N., Yi S., Taipale M., Fuxman Bass J. I., Coulombe-Huntington J., Yang F., Peng J., Weile J., Karras G. I., Wang Y., Kovács I. A., Kamburov A., Krykbaeva I., Lam M. H., Tucker G., et al. (2015) Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660 10.1016/j.cell.2015.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huttlin E. L., Bruckner R. J., Paulo J. A., Cannon J. R., Ting L., Baltier K., Colby G., Gebreab F., Gygi M. P., Parzen H., Szpyt J., Tam S., Zarraga G., Pontano-Vaites L., Swarup S., et al. (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509 10.1038/nature22366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chick J. M., Munger S. C., Simecek P., Huttlin E. L., Choi K., Gatti D. M., Raghupathy N., Svenson K. L., Churchill G. A., and Gygi S. P. (2016) Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505 10.1038/nature18270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gavin A. C., Bösche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J. M., Michon A. M., Cruciat C. M., Remor M., Höfert C., Schelder M., Brajenovic M., Ruffner H., et al. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 10.1038/415141a [DOI] [PubMed] [Google Scholar]
- 30. Lambert J. P., Ivosev G., Couzens A. L., Larsen B., Taipale M., Lin Z. Y., Zhong Q., Lindquist S., Vidal M., Aebersold R., Pawson T., Bonner R., Tate S., and Gingras A. C. (2013) Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 10, 1239–1245 10.1038/nmeth.2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Go C. D., Knight J. D. R., Rajasekharan A., Rathod B., Hesketh G. G., Abe K. T., Youn J.-Y., Samavarchi-Tehrani P., Zhang H., Zhu L. Y., Popiel E., Lambert J.-P., Coyaud É., Cheung S. W. T., Rajendran D., et al. (2019) A proximity biotinylation map of a human cell. bioRxiv 10.1101/796391 [DOI]
- 32. Rolland T., Taşan M., Charloteaux B., Pevzner S. J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., Kamburov A., Ghiassian S. D., Yang X., Ghamsari L., Balcha D., et al. (2014) A proteome-scale map of the human interactome network. Cell 159, 1212–1226 10.1016/j.cell.2014.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yugandhar K., Gupta S., and Yu H. (2019) Inferring protein-protein interaction networks from mass spectrometry-based proteomic approaches: a mini-review. Comput. Struct. Biotechnol. J. 17, 805–811 10.1016/j.csbj.2019.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baker M. (2015) Reproducibility crisis: blame it on the antibodies. Nature 521, 274–276 10.1038/521274a [DOI] [PubMed] [Google Scholar]
- 35. Kirkwood K. J., Ahmad Y., Larance M., and Lamond A. I. (2013) Characterization of native protein complexes and protein isoform variation using size-fractionation-based quantitative proteomics. Mol. Cell Proteomics 12, 3851–3873 10.1074/mcp.M113.032367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drew K., Lee C., Huizar R. L., Tu F., Borgeson B., McWhite C. D., Ma Y., Wallingford J. B., and Marcotte E. M. (2017) Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 13, 932 10.15252/msb.20167490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Havugimana P. C., Hart G. T., Nepusz T., Yang H., Turinsky A. L., Li Z., Wang P. I., Boutz D. R., Fong V., Phanse S., Babu M., Craig S. A., Hu P., Wan C., Vlasblom J., et al. (2012) A census of human soluble protein complexes. Cell 150, 1068–1081 10.1016/j.cell.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kristensen A. R., Gsponer J., and Foster L. J. (2012) A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods 9, 907–909 10.1038/nmeth.2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wan C., Borgeson B., Phanse S., Tu F., Drew K., Clark G., Xiong X., Kagan O., Kwan J., Bezginov A., Chessman K., Pal S., Cromar G., Papoulas O., Ni Z., et al. (2015) Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344 10.1038/nature14877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tan C. S. H., Go K. D., Bisteau X., Dai L., Yong C. H., Prabhu N., Ozturk M. B., Lim Y. T., Sreekumar L., Lengqvist J., Tergaonkar V., Kaldis P., Sobota R. M., and Nordlund P. (2018) Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 359, 1170–1177 10.1126/science.aan0346 [DOI] [PubMed] [Google Scholar]
- 41. Becher I., Werner T., Doce C., Zaal E. A., Tögel I., Khan C. A., Rueger A., Muelbaier M., Salzer E., Berkers C. R., Fitzpatrick P. F., Bantscheff M., and Savitski M. M. (2016) Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 12, 908–910 10.1038/nchembio.2185 [DOI] [PubMed] [Google Scholar]
- 42. Reinhard F. B., Eberhard D., Werner T., Franken H., Childs D., Doce C., Savitski M. F., Huber W., Bantscheff M., Savitski M. M., and Drewes G. (2015) Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat. Methods 12, 1129–1131 10.1038/nmeth.3652 [DOI] [PubMed] [Google Scholar]
- 43. Huber K. V., Olek K. M., Müller A. C., Tan C. S., Bennett K. L., Colinge J., and Superti-Furga G. (2015) Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 12, 1055–1057 10.1038/nmeth.3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mateus A., Määttä T. A., and Savitski M. M. (2016) Thermal proteome profiling: unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 15, 13 10.1186/s12953-017-0122-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Savitski M. M., Reinhard F. B., Franken H., Werner T., Savitski M. F., Eberhard D., Martinez Molina D., Jafari R., Dovega R. B., Klaeger S., Kuster B., Nordlund P., Bantscheff M., and Drewes G. (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 10.1126/science.1255784 [DOI] [PubMed] [Google Scholar]
- 46. Becher I., Andres-Pons A., Romanov N., Stein F., Schramm M., Baudin F., Helm D., Kurzawa N., Mateus A., Mackmull M. T., Typas A., Muller C. W., Bork P., Beck M., and Savitski M. M. (2018) Pervasive protein thermal stability variation during the cell cycle. Cell 173, 1495–1507.e18 10.1016/j.cell.2018.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dziekan J. M., Yu H., Chen D., Dai L., Wirjanata G., Larsson A., Prabhu N., Sobota R. M., Bozdech Z., and Nordlund P. (2019) Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci. Transl. Med. 11, eaau3174 10.1126/scitranslmed.aau3174 [DOI] [PubMed] [Google Scholar]
- 48. Saei A. A., Beusch C. M., Sabatier P., Wells J. A., Chernobrovkin A., Rodin S., Näreoja K., Thorsell A.-G., Karlberg T., Cheng Q., Lundström S. L., Gaetani M., Végvári Á., Arnér E. S. J., Schüler H., et al. (2018) System-wide identification of enzyme substrates by thermal analysis (SIESTA). bioRxiv 10.1101/423418 [DOI] [PMC free article] [PubMed]
- 49. Sridharan S., Kurzawa N., Werner T., Günthner I., Helm D., Huber W., Bantscheff M., and Savitski M. M. (2019) Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 10, 1155 10.1038/s41467-019-09107-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang J. X., Lee G., Cavanaugh K. E., Chang J. W., Gardel M. L., and Moellering R. E. (2019) High throughput discovery of functional protein modifications by hotspot thermal profiling. Nat. Methods 16, 894–901 10.1038/s41592-019-0499-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Viéitez C., Busby B. P., Ochoa D., Mateus A., Galardini M., Jawed A., Memon D., Potel C. M., Vonesch S. C., Tu C. S., Shahraz M., Stein F., Steinmetz L. M., Savitski M. M., Typas A., et al. P (2019) Towards a systematic map of the functional role of protein phosphorylation. bioRxiv 10.1101/872770 [DOI]
- 52. Potel C. M., Kurzawa N., Becher I., Typas A., Mateus A., and Savitski M. M. (2020) Impact of phosphorylation on thermal stability of proteins. bioRxiv 10.1101/2020.01.14.903849 [DOI] [PubMed]
- 53. Sun W., Dai L., Yu H., Puspita B., Zhao T., Li F., Tan J. L., Lim Y. T., Chen M. W., Sobota R. M., Tenen D. G., Prabhu N., and Nordlund P. (2019) Monitoring structural modulation of redox-sensitive proteins in cells with MS-CETSA. Redox Biol. 24, 101168 10.1016/j.redox.2019.101168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dai L., Zhao T., Bisteau X., Sun W., Prabhu N., Lim Y. T., Sobota R. M., Kaldis P., and Nordlund P. (2018) Modulation of protein-interaction states through the cell cycle. Cell 173, 1481–1494.e13 10.1016/j.cell.2018.03.065 [DOI] [PubMed] [Google Scholar]
- 55. Mateus A., Bobonis J., Kurzawa N., Stein F., Helm D., Hevler J., Typas A., and Savitski M. M. (2018) Thermal proteome profiling in bacteria: probing protein state in vivo. Mol. Syst. Biol. 14, e8242 10.15252/msb.20188242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Banzhaf M., Yau H. C. L., Verheul J., Lodge A., Kritikos G., Mateus A., Hov A. K., Stein F., Wartel M., Pazos M., Solovyova A. S., Savitski M. M., den Blaauwen T., Typas A., and Vollmer W. (2019) The outer membrane lipoprotein Nlpl nucleates hydrolases within peptidoglycan multi-enzyme complexes in Escherichia coli. bioRxiv 10.1101/609503 [DOI] [PMC free article] [PubMed]
- 57. Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P., and Martinez Molina D. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 10.1038/nprot.2014.138 [DOI] [PubMed] [Google Scholar]
- 58. Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E. A., Dan C., Sreekumar L., Cao Y., and Nordlund P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 10.1126/science.1233606 [DOI] [PubMed] [Google Scholar]
- 59. Werner T., Sweetman G., Savitski M. F., Mathieson T., Bantscheff M., and Savitski M. M. (2014) Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 86, 3594–3601 10.1021/ac500140s [DOI] [PubMed] [Google Scholar]
- 60. Coux O., Tanaka K., and Goldberg A. L. (1996) Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801–847 10.1146/annurev.bi.65.070196.004101 [DOI] [PubMed] [Google Scholar]
- 61. Hilt W., and Wolf D. H. (1996) Proteasomes: destruction as a programme. Trends Biochem. Sci. 21, 96–102 10.1016/S0968-0004(96)10012-8 [DOI] [PubMed] [Google Scholar]
- 62. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 10.1146/annurev.biochem.78.081507.101607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Finley D., Ulrich H. D., Sommer T., and Kaiser P. (2012) The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360 10.1534/genetics.112.140467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bard J. A. M., Goodall E. A., Greene E. R., Jonsson E., Dong K. C., and Martin A. (2018) Structure and function of the 26S Proteasome. Annu. Rev. Biochem. 87, 697–724 10.1146/annurev-biochem-062917-011931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fox M. J., Gao H., Smith-Kinnaman W. R., Liu Y., and Mosley A. L. (2015) The exosome component Rrp6 is required for RNA polymerase II termination at specific targets of the Nrd1-Nab3 pathway. PLoS Genet. 11, e1004999 10.1371/journal.pgen.1004999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu Y., Beyer A., and Aebersold R. (2016) On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550 10.1016/j.cell.2016.03.014 [DOI] [PubMed] [Google Scholar]
- 67. Kinyamu H. K., Bennett B. D., Bushel P. R., and Archer T. K. (2020) Proteasome inhibition creates a chromatin landscape favorable to RNA Pol II processivity. J. Biol. Chem. 295, 1271–1287 10.1074/jbc.RA119.011174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kwak J., Workman J. L., and Lee D. (2011) The proteasome and its regulatory roles in gene expression. Biochim. Biophys. Acta 1809, 88–96 10.1016/j.bbagrm.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 69. Durairaj G., and Kaiser P. (2014) The 26S proteasome and initiation of gene transcription. Biomolecules 4, 827–847 10.3390/biom4030827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xie Y., and Varshavsky A. (2001) RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc. Natl. Acad. Sci. U. S. A. 98, 3056–3061 10.1073/pnas.071022298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ramos P. C., Höckendorff J., Johnson E. S., Varshavsky A., and Dohmen R. J. (1998) Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell 92, 489–499 10.1016/S0092-8674(00)80942-3 [DOI] [PubMed] [Google Scholar]
- 72. Burri L., Höckendorff J., Boehm U., Klamp T., Dohmen R. J., and Lévy F. (2000) Identification and characterization of a mammalian protein interacting with 20S proteasome precursors. Proc. Natl. Acad. Sci. U. S. A. 97, 10348–10353 10.1073/pnas.190268597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ben-Aroya S., Agmon N., Yuen K., Kwok T., McManus K., Kupiec M., and Hieter P. (2010) Proteasome nuclear activity affects chromosome stability by controlling the turnover of Mms22, a protein important for DNA repair. PLoS Genet. 6, e1000852 10.1371/journal.pgen.1000852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Peters L. Z., Karmon O., David-Kadoch G., Hazan R., Yu T., Glickman M. H., and Ben-Aroya S. (2015) The protein quality control machinery regulates its misassembled proteasome subunits. PLoS Genet. 11, e1005178 10.1371/journal.pgen.1005178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Smits A. H., and Vermeulen M. (2016) Characterizing protein-protein interactions using mass spectrometry: challenges and opportunities. Trends Biotechnol. 34, 825–834 10.1016/j.tibtech.2016.02.014 [DOI] [PubMed] [Google Scholar]
- 76. Ochoa D., Jarnuczak A. F., Viéitez C., Gehre M., Soucheray M., Mateus A., Kleefeldt A. A., Hill A., Garcia-Alonso L., Stein F., Krogan N. J., Savitski M. M., Swaney D. L., Vizcaíno J. A., Noh K. M., et al. (2020) The functional landscape of the human phosphoproteome. Nat. Biotechnol. 38, 365–373 10.1038/s41587-019-0344-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Franken H., Mathieson T., Childs D., Sweetman G. M., Werner T., Tögel I., Doce C., Gade S., Bantscheff M., Drewes G., Reinhard F. B., Huber W., and Savitski M. M. (2015) Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593 10.1038/nprot.2015.101 [DOI] [PubMed] [Google Scholar]
- 78. Childs D., Kurzawa N., Franken H., Doce C., Savitski M., and Huber W. (2018) TPP: Analyze thermal proteome profiling (TPP) experiments, R package version 3.10.0 [Google Scholar]
- 79. Leuenberger P., Ganscha S., Kahraman A., Cappelletti V., Boersema P. J., von Mering C., Claassen M., and Picotti P. (2017) Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 355, eaai7825 10.1126/science.aai7825 [DOI] [PubMed] [Google Scholar]
- 80. Unverdorben P., Beck F., Śledź P., Schweitzer A., Pfeifer G., Plitzko J. M., Baumeister W., and Förster F. (2014) Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. U. S. A. 111, 5544–5549 10.1073/pnas.1403409111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lovato T. L., Adams M. M., Baker P. W., and Cripps R. M. (2009) A molecular mechanism of temperature sensitivity for mutations affecting the Drosophila muscle regulator myocyte enhancer factor-2. Genetics 183, 107–117 10.1534/genetics.109.105056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Groll M., Ditzel L., Löwe J., Stock D., Bochtler M., Bartunik H. D., and Huber R. (1997) Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 386, 463–471 10.1038/386463a0 [DOI] [PubMed] [Google Scholar]
- 83. Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., and Thomas P. D. (2017) PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 10.1093/nar/gkw1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Conway J. R., Lex A., and Gehlenborg N. (2017) UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 10.1093/bioinformatics/btx364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nakatsukasa K., Huyer G., Michaelis S., and Brodsky J. L. (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132, 101–112 10.1016/j.cell.2007.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Doonan L. M., Guerriero C. J., Preston G. M., Buck T. M., Khazanov N., Fisher E. A., Senderowitz H., and Brodsky J. L. (2019) Hsp104 facilitates the endoplasmic-reticulum-associated degradation of disease-associated and aggregation-prone substrates. Protein Sci. 28, 1290–1306 10.1002/pro.3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Saeki Y. (2017) Ubiquitin recognition by the proteasome. J. Biochem. 161, 113–124 10.1093/jb/mvw091 [DOI] [PubMed] [Google Scholar]
- 88. Yip M. C. J., Bodnar N. O., and Rapoport T. A. (2020) Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. U. S. A. 117, 7776–7781 10.1073/pnas.1902298117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Guerrero C., Milenkovic T., Przulj N., Kaiser P., and Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U. S. A. 105, 13333–13338 10.1073/pnas.0801870105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hanssum A., Zhong Z., Rousseau A., Krzyzosiak A., Sigurdardottir A., and Bertolotti A. (2014) An inducible chaperone adapts proteasome assembly to stress. Mol. Cell 55, 566–577 10.1016/j.molcel.2014.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Adcock S. A., and McCammon J. A. (2006) Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 106, 1589–1615 10.1021/cr040426m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Knight J. L., and Brooks C. L. (2011) Multisite λ dynamics for simulated structure–activity relationship studies. J. Chem. Theory Comput. 7, 2728–2739 10.1021/ct200444f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kong X., and Brooks C. L. (1996) λ-Dynamics: a new approach to free energy calculations. J. Chem. Phys. 105, 2414–2423 10.1063/1.472109 [DOI] [Google Scholar]
- 94. Hayes R. L., Armacost K. A., Vilseck J. Z., and Brooks C. L. (2017) Adaptive landscape flattening accelerates sampling of alchemical space in multisite λ dynamics. J. Phys. Chem. B 121, 3626–3635 10.1021/acs.jpcb.6b09656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hayes R. L., Vilseck J. Z., and Brooks C. L. (2018) Approaching protein design with multisite λ dynamics: accurate and scalable mutational folding free energies in T4 lysozyme. Protein Sci. 27, 1910–1922 10.1002/pro.3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vilseck J. Z., Armacost K. A., Hayes R. L., Goh G. B., and Brooks C. L. (2018) Predicting binding free energies in a large combinatorial chemical space using multisite λ dynamics. J. Phys. Chem. Lett. 9, 3328–3332 10.1021/acs.jpclett.8b01284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Vilseck J. Z., Sohail N., Hayes R. L., and Brooks C. L. (2019) Overcoming challenging substituent perturbations with multisite λ-dynamics: a case study targeting β-secretase 1. J. Phys. Chem. Lett. 10, 4875–4880 10.1021/acs.jpclett.9b02004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kollman P. (1993) Free energy calculations: applications to chemical and biochemical phenomena. Chem. Rev. 93, 2395–2417 10.1021/cr00023a004 [DOI] [Google Scholar]
- 99. Tomko R. J. Jr., and Hochstrasser M. (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 82, 415–445 10.1146/annurev-biochem-060410-150257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chen S., Wu J., Lu Y., Ma Y. B., Lee B. H., Yu Z., Ouyang Q., Finley D. J., Kirschner M. W., and Mao Y. (2016) Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. U. S. A. 113, 12991–12996 10.1073/pnas.1614614113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Eisele M. R., Reed R. G., Rudack T., Schweitzer A., Beck F., Nagy I., Pfeifer G., Plitzko J. M., Baumeister W., Tomko R. J. Jr., and Sakata E. (2018) Expanded coverage of the 26S proteasome conformational landscape reveals mechanisms of peptidase gating. Cell Rep. 24, 1301–1315.e5 10.1016/j.celrep.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Guo Q., Lehmer C., Martinez-Sanchez A., Rudack T., Beck F., Hartmann H., Perez-Berlanga M., Frottin F., Hipp M. S., Hartl F. U., Edbauer D., Baumeister W., and Fernandez-Busnadiego R. (2018) In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell 172, 696–705 10.1016/j.cell.2017.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Huang X., Luan B., Wu J., and Shi Y. (2016) An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 23, 778–785 10.1038/nsmb.3273 [DOI] [PubMed] [Google Scholar]
- 104. Luan B., Huang X., Wu J., Mei Z., Wang Y., Xue X., Yan C., Wang J., Finley D. J., Shi Y., and Wang F. (2016) Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proc. Natl. Acad. Sci. U. S. A. 113, 2642–2647 10.1073/pnas.1601561113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Matyskiela M. E., Lander G. C., and Martin A. (2013) Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol. 20, 781–788 10.1038/nsmb.2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Śledź P., Unverdorben P., Beck F., Pfeifer G., Schweitzer A., Förster F., and Baumeister W. (2013) Structure of the 26S proteasome with ATP-S bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc. Natl. Acad. Sci. U. S. A. 110, 7264–7269 10.1073/pnas.1305782110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wehmer M., Rudack T., Beck F., Aufderheide A., Pfeifer G., Plitzko J. M., Förster F., Schulten K., Baumeister W., and Sakata E. (2017) Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. U. S. A. 114, 1305–1310 10.1073/pnas.1621129114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhu Y., Wang W. L., Yu D., Ouyang Q., Lu Y., and Mao Y. (2018) Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun. 9, 1360 10.1038/s41467-018-03785-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Marshall R. S., and Vierstra R. D. (2019) Dynamic regulation of the 26S proteasome: from synthesis to degradation. Front. Mol. Biosci. 6, 40 10.3389/fmolb.2019.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dronamraju R., Kerschner J. L., Peck S. A., Hepperla A. J., Adams A. T., Hughes K. D., Aslam S., Yoblinski A. R., Davis I. J., Mosley A. L., and Strahl B. D. (2018) Casein kinase II phosphorylation of Spt6 enforces transcriptional fidelity by maintaining Spn1-Spt6 interaction. Cell Rep. 25, 3476–3489.e5 10.1016/j.celrep.2018.11.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mosley A. L., Florens L., Wen Z., and Washburn M. P. (2009) A label free quantitative proteomic analysis of the Saccharomyces cerevisiae nucleus. J. Proteomics 72, 110–120 10.1016/j.jprot.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mosley A. L., Sardiu M. E., Pattenden S. G., Workman J. L., Florens L., and Washburn M. P. (2011) Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol. Cell. Proteomics 10, M110.000687 10.1074/mcp.m110.000687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- 114. Smith-Kinnaman W. R., Berna M. J., Hunter G. O., True J. D., Hsu P., Cabello G. I., Fox M. J., Varani G., and Mosley A. L. (2014) The interactome of the atypical phosphatase Rtr1 in Saccharomyces cerevisiae. Mol. Biosystems 10, 1730–1741 10.1039/c4mb00109e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., and Gingeras T. R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Breese M. R., and Liu Y. (2013) NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29, 494–496 10.1093/bioinformatics/bts731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liao Y., Smyth G. K., and Shi W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 118. Robinson M. D., McCarthy D. J., and Smyth G. K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. McCarthy D. J., Chen Y., and Smyth G. K. (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Oliveros J. C. (2007) Venny: an interactive tool for comparing lists with Venn's diagrams, Centro Nacional de Biotecnología, Madrid [Google Scholar]
- 121. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic acids research 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Olsson M. H., Søndergaard C. R., Rostkowski M., and Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 124. Best R. B., Mittal J., Feig M., and MacKerell A. D. Jr (2012) Inclusion of many-body effects in the additive CHARMM protein CMAP potential results in enhanced cooperativity of α-helix and β-hairpin formation. Biophys. J. 103, 1045–1051 10.1016/j.bpj.2012.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 127. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 128. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., and Karplus M. (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 10.1002/jcc.540040211 [DOI] [Google Scholar]
- 129. Brooks B. R., Brooks C. L., Mackerell A. D., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Eastman P., Swails J., Chodera J. D., McGibbon R. T., Zhao Y., Beauchamp K. A., Wang L. P., Simmonett A. C., Harrigan M. P., Stern C. D., Wiewiora R. P., Brooks B. R., and Pande V. S. (2017) OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 13, e1005659 10.1371/journal.pcbi.1005659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hünenberger P. H. (2005) Thermostat algorithms for molecular dynamics simulations. in Advanced Computer Simulation: Approaches for Soft Matter Sciences I (Holm C., and Kremer K., eds) pp 105–149, Springer, Berlin [Google Scholar]
- 132. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 133. Kuzmanic A., and Zagrovic B. (2010) Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 98, 861–871 10.1016/j.bpj.2009.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hynninen A. P., and Crowley M. F. (2014) New faster CHARMM molecular dynamics engine. J. Comput. Chem. 35, 406–413 10.1002/jcc.23501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hoover W. G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 31, 1695–1697 10.1103/physreva.31.1695 [DOI] [PubMed] [Google Scholar]
- 136. Feller S. E., Zhang Y., Pastor R. W., and Brooks B. R. (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 10.1063/1.470648 [DOI] [Google Scholar]
- 137. Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 10.1063/1.447334 [DOI] [Google Scholar]
- 138. Ryckaert J.-P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 139. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 140. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 141. Wallace J. A., and Shen J. K. (2012) Charge-leveling and proper treatment of long-range electrostatics in all-atom molecular dynamics at constant pH. J. Chem. Phys. 137, 184105 10.1063/1.4766352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Steinbach P. J., and Brooks B. R. (1994) New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15, 667–683 10.1002/jcc.540150702 [DOI] [Google Scholar]
- 143. Knight J. L., and Brooks C. L. 3rd (2011) Applying efficient implicit nongeometric constraints in alchemical free energy simulations. J. Comput. Chem. 32, 3423–3432 10.1002/jcc.21921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 2.3.0, Schrödinger, LLC, New York [Google Scholar]
- 145. Deutsch E. W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D. S., Bernal-Llinares M., Okuda S., Kawano S., Moritz R. L., Carver J. J., Wang M., Ishihama Y., Bandeira N., et al. (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106 10.1093/nar/gkw936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yılmaz Ş., Tiwary S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All analyzed data sets used for this study are referenced in the article and included in the supporting information. The MS proteomics data have been deposited to the ProteomeXchange Consortium (145) via the PRIDE (146) partner repository with the data set identifiers PXD017223 (global) and PXD017222 (mTPP). The RNA-Seq data have been deposited to Gene Expression Omnibus (GEO) under the accession number GSE143236. Any code used for data analysis is available as referenced. Thermo Proteome Discoverer software is a commercial product; however, a free viewer is available for processed data files at https://thermo.flexnetoperations.com/.