

Abstract
Organelles are physically connected in membrane contact sites. The endoplasmic reticulum possesses three major receptors, VAP‐A, VAP‐B, and MOSPD2, which interact with proteins at the surface of other organelles to build contacts. VAP‐A, VAP‐B, and MOSPD2 contain an MSP domain, which binds a motif named FFAT (two phenylalanines in an acidic tract). In this study, we identified a non‐conventional FFAT motif where a conserved acidic residue is replaced by a serine/threonine. We show that phosphorylation of this serine/threonine is critical for non‐conventional FFAT motifs (named Phospho‐FFAT) to be recognized by the MSP domain. Moreover, structural analyses of the MSP domain alone or in complex with conventional and Phospho‐FFAT peptides revealed new mechanisms of interaction. Based on these new insights, we produced a novel prediction algorithm, which expands the repertoire of candidate proteins with a Phospho‐FFAT that are able to create membrane contact sites. Using a prototypical tethering complex made by STARD3 and VAP, we showed that phosphorylation is instrumental for the formation of ER‐endosome contacts, and their sterol transfer function. This study reveals that phosphorylation acts as a general switch for inter‐organelle contacts.
Keywords: cholesterol, inter‐organelle contact, lipid transfer protein, regulation, small linear motif
Subject Categories: Membrane & Intracellular Transport,
Phosphorylation of a non‐conventional FFAT motif promotes ER‐endosome membrane contact sites and sterol exchange.
Introduction
The endoplasmic reticulum (ER) is a membrane‐bound organelle primarily involved in protein and lipid synthesis. The ER consists of the nuclear envelope as well as tubes and sheets spreading throughout the cytosol up to the plasma membrane (PM) (Westrate et al, 2015). In the cytosol, the ER physically contacts other organelles including mitochondria, endosomes/lysosomes, autophagic structures, peroxisomes, lipid droplets, and the PM. These contacts, termed membrane contact sites (MCSs), correspond to close appositions, usually within 30 nm, of the ER membrane with the limiting membrane of another organelle; they do not result in the fusion of the two membranes (Levine & Loewen, 2006; Wu et al, 2018). MCSs are involved in major cellular processes such as lipid and calcium transport, and organelle positioning and dynamics (Wu et al, 2018; Prinz et al, 2019).
Contacts between the ER and another organelle require tether proteins that bridge their two membranes via protein–membrane or protein–protein interactions. The ER possesses three major receptors at its surface which allow the recruitment of proteins associated with the membrane of other organelles. These proteins named vesicle‐associated membrane protein‐associated proteins (VAP) A and B, and motile sperm domain‐containing protein 2 (MOSPD2), are members of the Major Sperm protein (MSP) domain‐containing family. VAP‐A, VAP‐B, and MOSPD2 are anchored in the ER membrane by a carboxyl‐terminal transmembrane domain with their MSP domain projecting into the cytosol. The MSP domains of VAP‐A/VAP‐B/MOSPD2 interact with small linear motifs named FFAT [two phenylalanines (FF) in an acidic tract (AT)] (Loewen et al, 2003; Murphy & Levine, 2016; Di Mattia et al, 2018). Contact sites are built by the direct binding between VAP‐A/VAP‐B/MOSPD2 at the ER surface and FFAT‐containing partners which are bound to another organelle (such as endosomes, mitochondria, peroxisomes, the Golgi) or the PM (Amarilio et al, 2005; Kawano et al, 2006; Rocha et al, 2009; De Vos et al, 2012; Alpy et al, 2013; Mesmin et al, 2013; Dong et al, 2016; Costello et al, 2017a, b; Di Mattia et al, 2018; Johnson et al, 2018; Kirmiz et al, 2018).
MCSs are functional structures that physically attach two distinct organelles to each other. Visualization of contacts by microscopy in live cells revealed that they are dynamic, with organelles associating and dissociating over time (Friedman et al, 2013; Valm et al, 2017). This observation implies that mechanisms regulating these processes must exist. While the molecular organization and function of MCSs are better understood, the regulation of the formation and disassembly of these structures remains quite unknown. In this study, we uncovered that some partners of VAP‐A/VAP‐B/MOSPD2 possess a special type of FFAT motif that can be phosphorylated on a discrete site, and that we consequently named Phospho‐FFAT. The phosphorylation of Phospho‐FFATs is essential for the interaction of VAP‐A/VAP‐B/MOSPD2 with their Phospho‐FFAT‐containing partners. This reveals the existence of a general molecular mechanism regulating the formation of inter‐organelle contact formation and function.
Results
Identification and functional characterization of a novel category of FFAT motifs potentially regulated by phosphorylation
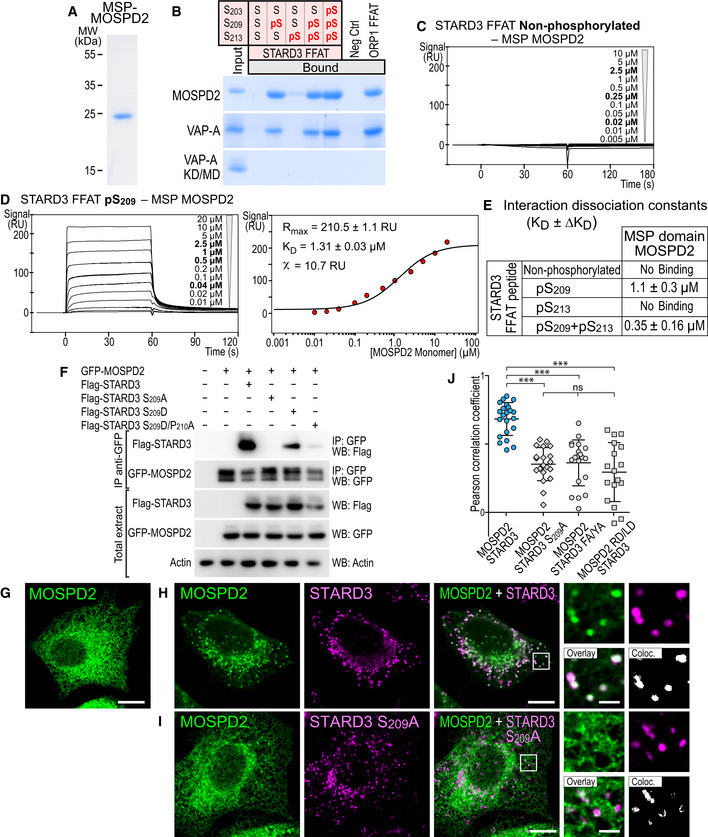
Conventional FFATs have seven core residues: E1F2F3D4A5×6E7, and an acidic flanking region (Fig 1A) (Loewen et al, 2003; Murphy & Levine, 2016). The residue at the fourth position is invariably acidic, either E or D; its substitution by an alanine precludes the interaction with the MSP domain (Loewen et al, 2003; Kawano et al, 2006). A survey of the literature revealed that some FFAT sequences differ from the conventional sequence: Their core FFAT motif contains a serine or a threonine residue at the 4th position instead of an acidic residue (Fig 1A). Because serine and threonine are polar amino acids that can gain a negative charge once their hydroxyl group is phosphorylated, we surmised that these residues might be phosphorylated to be able to bind the MSP domain. We identified six distinct VAP‐A/VAP‐B/MOSPD2 partners with an FFAT motif having this characteristic: STARD3 (steroidogenic acute regulatory (StAR)‐related lipid transfer domain‐containing 3), a late endosome (LE) protein involved in ER‐LE cholesterol transport; FIP200 also known as RB1‐inducible coiled‐coil protein 1 (RB1CC1; RBCC1), a cytosolic protein involved in autophagosome formation (Hara et al, 2008; Zhao et al, 2018); Mitoguardin2 (MIGA2), also known as FAM73B, a mitochondrial outer membrane protein involved in mitochondria dynamics (Huttlin et al, 2015; Murphy & Levine, 2016; Zhang et al, 2016; Freyre et al, 2019); PTPIP51 (protein tyrosine phosphatase‐interacting protein‐51, aka regulator of Microtubule dynamics protein 3, RMDN3; RMD3), a mitochondria protein involved in ER‐mitochondria contact formation and possessing two FFATs, a conventional one and a non‐conventional FFAT with a threonine at the 4th position (Stoica et al, 2014; Murphy & Levine, 2016; Huttlin et al, 2017; Di Mattia et al, 2018); and Kv2.1 and Kv2.2 (also known as potassium voltage‐gated channel subfamily B member 1 (KCNB1) and KCNB2, respectively) are voltage‐gated potassium channels involved in neuronal excitability and in the formation of ER‐PM contacts (Lim et al, 2000; Fox et al, 2015; Johnson et al, 2018; Kirmiz et al, 2018).
Figure 1. Identification of novel category of FFAT motifs regulated by phosphorylation.
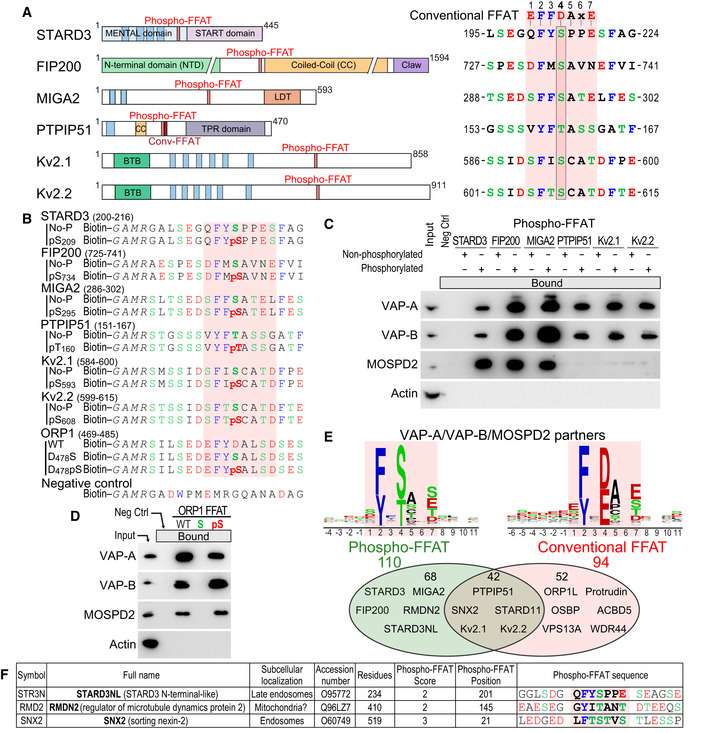
- Schematic representation of STARD3, FIP200, MIGA2, PTPIP51, KCNB1 (Kv2.1), and KCNB2 (Kv2.2) proteins and sequence of their Phospho‐FFAT motif (7 core residues and 4 upstream and downstream residues). Note that the 4th position of the FFAT (boxed) of these proteins is occupied by a serine or a threonine residue while conventional FFATs have an aspartic acid. Blue rectangles represent transmembrane helices. TPR: tetratrico peptide repeat; LDT: lipid droplet targeting domain; BTB: potassium channel tetramerization‐type BTB domain.
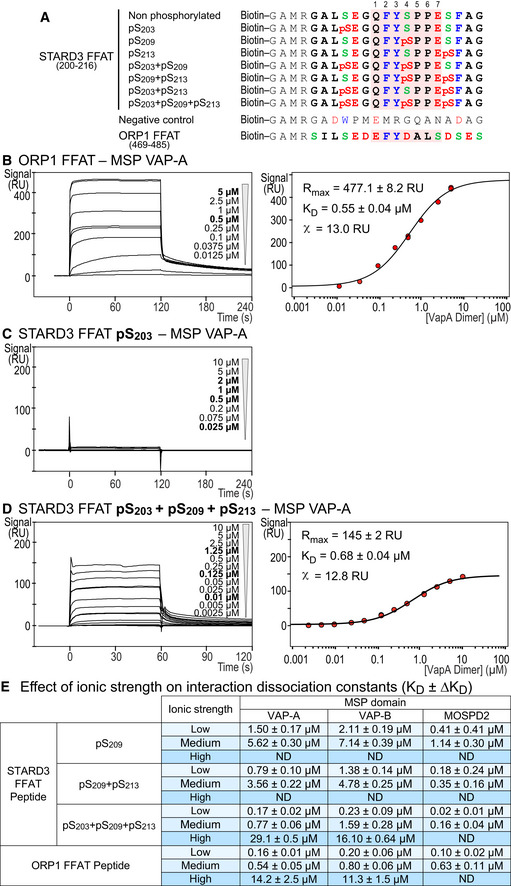
- Sequence of the peptides used for the pull‐down assays. The peptides are composed of an amino‐terminal biotin, a linker sequence and the FFAT sequence of STARD3 (residues 200–216), FIP200 (residues 725–741), MIGA2 (residues 286–302), PTPIP51 (residues 151–167), Kv2.1 (residues 584–600), Kv2.2 (residues 599–615), and ORP1 (residues 469–485) either without or with a phosphorylated residue (serine or threonine) at position 4 of the core FFAT motif. The negative control peptide is composed of a random sequence.
- Western blot analysis of proteins pulled down using the peptides described in (B). The input fraction corresponds to HeLa cell total protein extract. Bound proteins were analyzed using anti‐VAP‐A, anti‐VAP‐B and anti‐MOSPD2 antibodies. Actin was used as a loading control.
- Western blot analysis of proteins pulled down using peptides comprising WT and mutant (D478S and D478pS) ORP1 FFAT sequence peptides described in (B). Bound proteins were analyzed using anti‐VAP‐A, anti‐VAP‐B, and anti‐MOSPD2 antibodies. Actin was used as a loading control.
- Venn diagram of VAPs and/or MOSPD2 partners with FFAT scores between 0 and 3. Proteins with a Phospho‐FFAT and a conventional FFAT are shown in green and red, respectively. A total of 42 proteins possesses both a Phospho and a conventional FFAT. Consensus sequences of the 110 Phospho‐FFAT (left) and 94 conventional FFAT motifs (right) identified are shown as sequence logos.
- List of selected VAP‐A, VAP‐B, or MOSPD2 partners having a potential Phospho‐FFAT motif. For a full list, see Table EV2.
Data information: Acidic (D and E) or phosphorylated residues (pS and pT), alcoholic (S and T), and aromatic (F and Y) residues are in red, green, and blue, respectively; the other residues are in black.
Source data are available online for this figure.
To test whether these non‐conventional FFAT motifs are genuine binding sites, we examined the binding of the six proteins identified above to VAPs or MOSPD2. More precisely, we asked whether the phosphorylation of the 4th residue of their FFAT motifs was implicated in binding with the MSP domain. To this end, we carried out pull‐down assays of whole cell protein extracts using synthetic biotinylated peptides encompassing STARD3, FIP200, MIGA2, PTPIP51, Kv2.1, and Kv2.2 FFAT sequences whose 4th residue (serine or threonine) was either phosphorylated or not (Fig 1B). These peptides, and negative control peptide with a random sequence (Fig 1B), were attached to streptavidin beads, and incubated with HeLa cell protein extracts. Bound proteins were detected by SDS–PAGE followed by Western blot using antibodies against VAP‐A, VAP‐B, and MOSPD2 (Fig 1C). Peptides corresponding to the non‐phosphorylated FFAT motifs of STARD3, FIP200, MIGA2, PTPIP51, Kv2.1, and Kv2.2 retained none of the three proteins (Fig 1C). In contrast, VAP proteins were efficiently pulled down by all the peptides whose FFAT motif was phosphorylated (STARD3, FIP200, MIGA2, PTPIP51, Kv2.1, and Kv2.2). MOSPD2 interacted with the phosphorylated FFAT of STARD3, FIP200, and MIGA2, showing that the interaction of MOSPD2 with these FFATs requires phosphorylation. However, MOSPD2 did not interact with the phosphorylated FFAT of PTPIP51, Kv2.1, and Kv2.2. This result was surprising but not unexpected because the candidate proteins were primarily described as VAP‐A and B partners in the literature (Stoica et al, 2014; Murphy & Levine, 2016; Huttlin et al, 2017; Johnson et al, 2018; Kirmiz et al, 2018). This suggests that the landscape of MOSPD2 partners is different from that of VAP proteins.
We then asked whether a conventional FFAT motif could be converted into a phosphorylation‐dependent FFAT by replacing the acidic residue at the 4th position by a serine. To test this, we used the FFAT of oxysterol‐binding protein‐related protein 1 (OSBPL1A aka ORP1) (Loewen et al, 2003). We replaced D478 (position 4 of the FFAT sequence) by a serine and by a phosphorylated serine (Fig 1B). We then performed peptide pull‐down experiments. The FFAT of ORP1 efficiently pulled down VAP‐A and VAP‐B from the whole cell protein extract, whereas the non‐phosphorylated D478S mutant peptide did not (Fig 1D). Consistent with the idea that phosphorylation of the serine at position 4 of non‐conventional FFAT motifs is crucial for the binding, we found that the peptide with a phosphorylated serine was able to trap VAP‐A and VAP‐B.
Jointly, these data suggest that the interaction between VAP‐A/VAP‐B/MOSPD2 and several binding partners is activated by phosphorylation. To differentiate FFAT motifs characterized by a serine or a threonine in position 4 and switched on by phosphorylation from conventional ones, we named them Phospho‐FFATs.
In silico identification of Phospho‐FFAT motifs in the human proteome
In order to identify the human proteins possessing a Phospho‐FFAT, we used an in silico approach. We designed a position weight matrix strategy which has previously served to identify conventional FFATs in many proteins (Mikitova & Levine, 2012; Murphy & Levine, 2016; Slee & Levine, 2019). It allows the identification of motifs with variations around an ideal FFAT sequence. Proteins are ranked based on their best FFAT score, with a score 0 for an ideal FFAT sequence, and higher scores (up to 18.5) for distant motifs. We specifically focused on motifs which can be phosphorylated at position 4 of the core FFAT by imposing the presence of a serine or a threonine at that position. Among the 20,373 human proteins which were analyzed, 2,079 had a Phospho‐FFAT score below 3 (Table EV1). It is noteworthy that to be effective, a Phospho‐FFAT sequence has to be in the cytosolic part of the protein to be accessible to the MSP domain of VAP‐A/VAP‐B/MOSPD2, and is likely present in an unstructured part of the protein. Therefore, true Phospho‐FFATs likely represent only a subset of this list.
Because a large number of VAP‐A/VAP‐B/MOSPD2 partners have been identified by high‐throughput proteomics (Orchard et al, 2014; Oughtred et al, 2019), we took advantage of these resources to test the 427 potential partners listed in the Biogrid and IntAct databases. For this analysis, we also included the MOSPD2 partners identified by ourselves using a proteomic approach (Di Mattia et al, 2018). We screened the 488 known VAP‐A/VAP‐B/MOSPD2 partners for the presence of a candidate Phospho‐FFAT motif. We identified 110 VAP‐A/VAP‐B/MOSPD2 partners with a significant Phospho‐FFAT score (Table EV2). As expected, STARD3, FIP200, MIGA2, PTPIP51, Kv2.1, and Kv2.2 were attributed high scores by the algorithm, ranging from 0 to 2.5. Using another version of the algorithm allowing the identification of conventional FFATs, the two algorithms being mutually exclusive, 94 proteins were identified (Fig 1E). Interestingly, some proteins had two FFAT motifs, a conventional one and a Phospho‐FFAT (Table EV2). This analysis suggests that Phospho‐FFATs and conventional ones are equally distributed in the human proteome.
To further exploit this in silico approach, we sought for the presence of proteins containing a Phospho‐FFAT motif which were not identified by the original FFAT‐prediction algorithm (Mikitova & Levine, 2012; Murphy & Levine, 2016; Slee & Levine, 2019). We chose to describe three proteins of the list of 110 VAP‐A/VAP‐B/MOSPD2 partners (Table EV2) that are already characterized as being involved or potentially involved in the formation of MCSs (Fig 1F). One of these proteins is STARD3NL: The algorithm identified a Phospho‐FFAT motif that is consistent with the current knowledge about this protein. STARD3NL is a LE protein homologous to STARD3 (Alpy et al, 2002), involved in the formation of ER‐endosome contacts by interacting with VAP‐A/VAP‐B/MOSPD2 (Alpy et al, 2013; Di Mattia et al, 2018). The FFAT of STARD3NL is highly similar to that of STARD3, with all the characteristics of a Phospho‐FFAT, and therefore most likely requires phosphorylation to be active. Another example is RMDN2: This poorly characterized protein is homologous to PTPIP51 (aka RMDN3) notably in the amino‐terminal transmembrane region. Interestingly, this region is responsible for the addressing of PTPIP51 to mitochondria (Lv et al, 2006). This suggests that RMDN2 is also a mitochondrial protein; supporting this idea, RMDN2 was shown to be associated with the outer mitochondrial membrane in a high‐throughput proteomics study (Go et al, in preparation). The algorithm identified a unique potential Phospho‐FFAT motif in RMDN2 which is conserved with PTPIP51. This suggests that RMDN2 could be involved in ER‐mitochondria contacts regulated by phosphorylation. The last example is SNX2: The algorithm identified a Phospho‐FFAT motif in SNX2 (Fig 1F), a component of the retromer complex which is implicated in ER‐endosome contacts (Bonifacino & Rojas, 2006; Dong et al, 2016). Interestingly, Dong et al (2016) made a mutation analysis which is consistent with the Phospho‐FFAT motif identified here. Therefore, it is likely that SNX2’s interaction with VAP is regulated by phosphorylation.
To conclude, we developed an algorithm allowing the identification of candidate Phospho‐FFATs in an unbiased manner. By crossing the list with the repertoires of MSP domain binding partners, this algorithm revealed that Phospho‐FFATs are as common as conventional FFAT motifs in the human proteome.
Phosphorylation of the Phospho‐FFAT allows binding with VAP‐A and VAP‐B
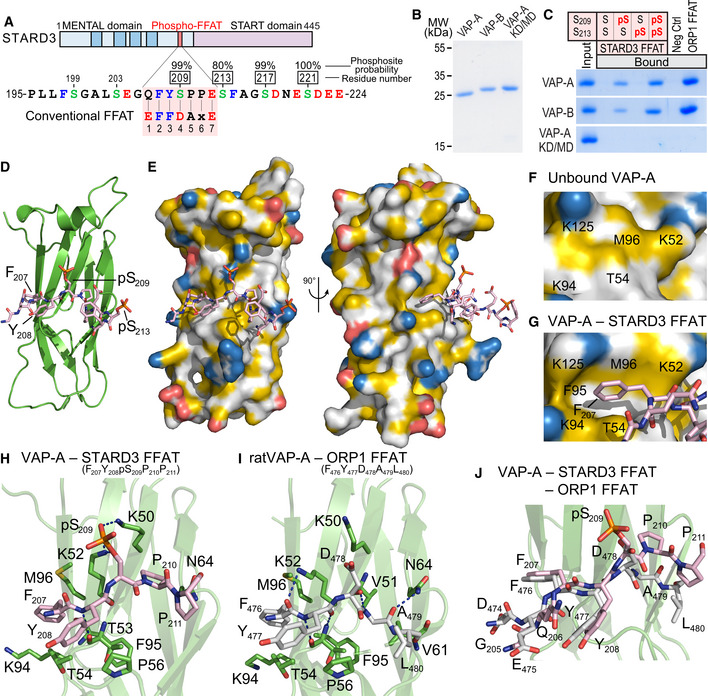
We selected one Phospho‐FFAT‐containing protein to study in‐depth its association mechanism. We chose STARD3, one of the 15 human START proteins, a family of lipid transfer proteins (LTP) involved in the intracellular transport of lipids. We selected STARD3 because we previously reported that STARD3 makes ER‐endosome contact sites by interacting with the ER proteins VAP‐A, VAP‐B, and MOSPD2 (Alpy et al, 2013; Di Mattia et al, 2018). These ER‐endosome contacts are active regions that allow STARD3‐mediated ER to endosome cholesterol transport (Alpy & Tomasetto, 2005; Wilhelm et al, 2017). The Phospho‐FFAT of STARD3 contains a serine residue at the 4th position (position 209 of the protein, hereafter referred to as S209; note that hereafter numberings of FFAT residues are labeled as subscript) (Figs 1A and 2A). To determine whether this particular residue is phosphorylated, we first interrogated the public database PhosphoSitePlus (Hornbeck et al, 2015)—which records post‐translational modifications in a comprehensive manner—for the presence of phosphorylation in STARD3. We found that a series of phosphorylations in human and mouse STARD3 were described in and around the FFAT motif, on S209, and on S213, S217, and S221 (Fig 2A). To confirm these observations, we sought for STARD3 phosphorylation in HeLa cells, given that STARD3 makes ER‐endosome contacts in these cells (Alpy et al, 2013). To this aim, we expressed the GST‐tagged STARD3 protein in HeLa cells, then purified the protein by affinity chromatography, and analyzed its phosphorylation by mass spectrometry. Once purified, the GST‐STARD3 protein was digested with trypsin and chymotrypsin, and subjected to ion trap liquid chromatography–tandem mass spectrometry (LC/MS/MS) analysis. Phosphorylation was detected on S209, S213, S217, and S221 residues (Figs 2A and EV1). Together, these data show that STARD3 is phosphorylated on several serine residues in the core of its FFAT motif and in its vicinity.
Figure 2. STARD3 has a non‐conventional FFAT motif which needs to be phosphorylated to form a complex with VAP proteins.
-
ASchematic representation of STARD3. Transmembrane helices in the MENTAL domain are in dark blue. The Phospho‐FFAT motif of STARD3 and the conventional FFAT motif sequences are aligned and highlighted in pink. Upper numbers correspond to the position of residues in STARD3. Lower numbers correspond to the position of residues in the FFAT sequence as described in (Loewen et al, 2003). Acidic (D and E), alcoholic (S and T), and aromatic (F and Y) residues are in red, green, and blue, respectively; the other residues are in black. Phosphorylated serines identified by LC/MS/MS are boxed, and the phosphosite probability determined with PhosphoRS is indicated.
-
BCoomassie Blue staining of the recombinant wild‐type MSP domains of VAP‐A and VAP‐B, and of the KD/MD mutant of VAP‐A, after SDS–PAGE.
-
CRecombinant MSP domains pulled down with peptides corresponding to the Phospho‐FFAT motif of STARD3 (unphosphorylated; monophosphorylated on S209 or S213; bi‐phosphorylated on S209 and S213), and with the control peptides were revealed by Coomassie Blue staining. The recombinant proteins subjected to the assay are showed in the input fraction.
-
D, EStructure of the MSP domain of VAP‐A in complex with a Phospho‐FFAT motif. Ribbon diagram (D) and surface representation (E) of the MSP domain of VAP‐A in complex with the Phospho‐FFAT of STARD3 depicted in stick model. The protein surface is colored according to the YRB scheme, showing hydrophobic, negatively and positively charged atoms in yellow, red, and blue, respectively; the other atoms are in white (Hagemans et al, 2015).
-
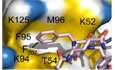
F, GClose‐up view of the hydrophobic pocket region in the unbound (F) and the Phospho‐FFAT‐bound (G) VAP‐A; residues of VAP‐A constituting the pocket are indicated.
-
HClose‐up view of the structure near the Phospho‐FFAT motif highlighting critical residues (in stick model) of human VAP‐A present in the binding interface.
-
IClose‐up view of the structure near the conventional FFAT of ORP1 as described in (PDBID: 1Z9O; Kaiser et al, 2005), highlighting critical residues of rat VAP‐A involved in the interaction. The nomenclature for rat VAP‐A residues is based on the UniProt sequence (Q9Z270) and not on the PDB file 1Z9O.
-
JSuperposition of the two structures shown in (H) and (I) showing the distinct conformations of the C‐terminal parts of each peptide.
Data information: Phosphorous, nitrogen, oxygen, and sulfur atoms are colored in orange, blue, red, and yellow, respectively. Carbon atoms are shown in green and gray/pink in the MSP domain and the FFAT motif, respectively. The numbers of the peptide residues are noted in subscript.
Source data are available online for this figure.
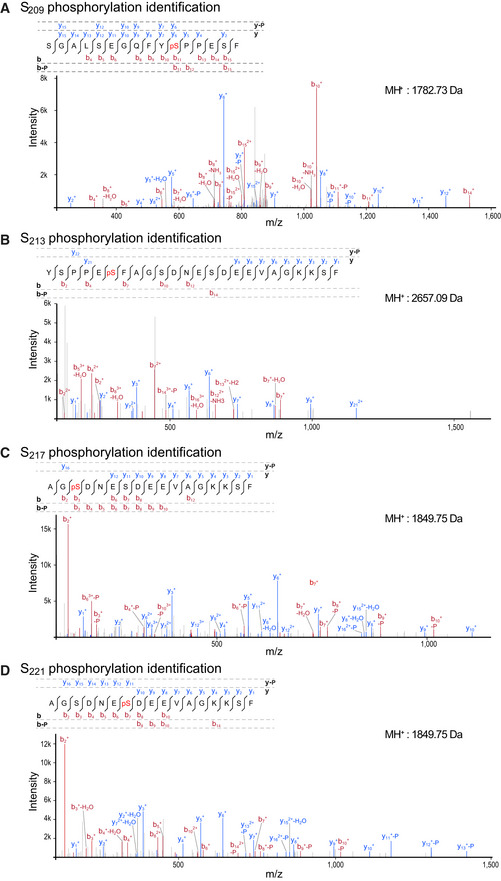
Figure EV1. MS/MS spectrum of peptides phosphorylated on S209, S213, S217, and S221 .
-
A–DMS/MS spectrum of peptides phosphorylated on S209 (A), S213 (B), S217 (C), and S221 (D). The molecular weight of peptides is indicated on the right. Spectra are an assembly of ions produced by collision‐induced dissociation of the precursor peptides. Fragmentation occurs preferentially at peptide bonds to generate b and y ions, which extend from the amino and carboxy terminus, respectively. The precursor peptide sequence and the different b and y ions identified in the spectrum are shown above. b and y ion peaks are shown in red and blue, respectively. b and y ions with the highest intensity are labeled on the spectrum. Neutral mass losses of H2O, NH3, and H3PO4 (P) are indicated. Analysis of y and b ion fragmentation patterns showed that S209 (A), S213 (B), S217 (C), and S221 (D) were phosphorylated in vivo.
We then explored whether VAP‐A and VAP‐B directly associate with the phosphorylated FFAT motif of STARD3 by in vitro binding assays using recombinant proteins and synthetic biotinylated peptides corresponding to the FFAT motif of STARD3. We produced in Escherichia coli and purified the MSP domains of VAP‐A and VAP‐B (Fig 2B). The MSP domain of VAP‐A, with a double K94D/M96D mutation (hereafter called KD/MD mutant), which is defective in binding the FFAT motif (Kaiser et al, 2005), was used as a negative control. These proteins were then incubated with streptavidin beads coupled with biotinylated peptides corresponding to either a control, or non‐phosphorylated or phosphorylated STARD3 FFAT motifs. We tested peptides bearing a phosphoserine either at position 209 or 213, or at both positions. We also included a peptide corresponding to the conventional FFAT motif of ORP1 (Loewen et al, 2003), and a random sequence of the same length, as positive and negative control, respectively (Fig EV3A). Retained proteins were eluted and analyzed by SDS–PAGE followed by Coomassie Blue staining (Fig 2C). The MSP domains of VAP‐A and VAP‐B interacted with the FFAT motif of ORP1, but did not interact with the non‐phosphorylated STARD3 FFAT motif (Fig 2C). Only the peptides with a phosphorylation on serine 209 retained the MSP domain of VAP‐A and VAP‐B. Indeed, the peptide with a single phosphorylation on S213 did not interact, while the ones with a phosphorylation on S209 or two phosphorylations, on S209 and S213, interacted with the recombinant MSP domains.
Figure EV3. The MSP domain of VAP‐A/VAP‐B/MOSPD2 binds the phosphorylated Phospho‐FFAT with an affinity in the micromolar range.
-
ASequence of the peptides used for SPR and the pull‐down assays. The peptides are composed of an amino‐terminal biotin, a linker sequence (GAMR) and the FFAT sequence of STARD3 (residues 200–216) either without phosphorylated serine or with combinations of phosphorylated serines (positions 203, 209, and 213 in STARD3 protein). The core FFAT sequence (residues 1–7) is highlighted in red. ORP1 FFAT peptide corresponding to residues 469–485 (Accession Number Q9BXW6‐1), and a random sequence (control peptide) are used as positive and negative control, respectively. Acidic (D and E) or phosphorylated serine (pS), alcoholic (S and T), and aromatic (F, W, and Y) residues are in red, green and blue, respectively; the other residues are in black.
-
B–DSPR analysis of the MSP domain of VAP‐A binding onto immobilized ORP1 FFAT peptide (B), pS203 (C) or pS203 + pS209 + pS213 (D) STARD3 FFAT peptides. Representative sensorgrams resulting from the interaction between the MSP domain of VAP‐A injected at different concentrations and the different STARD3 FFAT peptides are shown in (B left), (C), and (D left). Binding curves display the SPR signal (RU) as a function of time. Concentrations printed in bold indicate samples measured twice. Steady‐state analysis of the interaction between ORP1 FFAT (B right) and pS203 + pS209 + pS213 (D right) STARD3 FFAT peptide and the MSP domain of VAP‐A. Equilibrium responses (R eq) extracted from the left panel were plotted as a function of the dimeric MSP domain of VAP‐A concentration, and fitted with a Langmuir binding model.
-
EInteraction dissociation constants between the different FFAT peptides and the MSP domains of VAP‐A, VAP‐B, and MOSPD2. MSP concentrations are expressed as dimer for VAP‐A and VAP‐B, and monomer for MOSPD2. The different ionic strength correspond to the following buffers (pH 7.5): low: Tris–HCl 20 mM, NaCl 75 mM; medium: Tris–HCl 50 mM, NaCl 75 mM; high: Tris–HCl 50 mM, NaCl 300 mM. Buffers were supplemented with P20 (0.005% v/v). ND: not determined. Mean values of n independent experiments: n = 2 for pS209, pS209 + pS213, and S203 + pS209 + pS213 (high); 4 for pS203 + pS209 + pS213 (low), ORP1 (low and high), and pS203 + pS209 + pS213 (medium) with MOSPD2; 6 for pS203 + pS209 + pS213 (medium) with VAP‐A and VAP‐B; 12, 11, and 7 for ORP1 (medium) with VAP‐A, VAP‐B, and MOSPD2, respectively. Uncertainties are obtained from the standard deviation considering a t‐distribution coefficient for a risk factor of 32%.
Together, these experiments showed that phosphorylation of serine 209 is necessary and sufficient for the direct interaction of the STARD3 Phospho‐FFAT with the MSP domains of VAP‐A and VAP‐B.
Structural insight into the interaction between VAP‐A and a Phospho‐FFAT
To gain structural insights about the interaction between the MSP domain and a Phospho‐FFAT motif, we solved the crystal structure of the MSP domain of human VAP‐A in complex with a peptide corresponding to the human phosphorylated STARD3 FFAT (residues 197–216) (PDB ID: 6TQR). The structure was resolved to 1.85 Å (from anisotropic data—3 Å in the worst direction) by molecular replacement using the structure of rat VAP‐A with ORP1 FFAT [PDB ID 1Z9O (Kaiser et al, 2005)]. The asymmetric unit contains four copies of the MSP domain, two of which have the Phospho‐FFAT bound. The structure of human VAP‐A was strongly similar to that of rat VAP‐A (rmsd of 0.538 Å over 119 Cαs) (Fig 2D‐I). Overall, the first residues of the core motif of the Phospho‐FFAT bound the MSP domain similarly to the conventional FFAT motif of ORP1 (PDB ID: 1Z9O; Kaiser et al, 2005). Indeed, F207, the second residue of the FFAT motif, was bound to the hydrophobic pocket at the surface of the MSP domain constituted by aliphatic parts of the side chains of K52, T54, K94, M96, and K125 of VAP‐A (Fig 2G). The hydrogen bonds are also maintained between the main chain carbonyl and amide of Y208 of the peptide and the main chain of T53 of VAP‐A. The phosphorylated serine S209 forms ionic bonds with two lysines of VAP‐A, K50, and K52. Interestingly, the following core residue binds differently between the conventional and Phospho‐FFAT: In the conventional FFAT motif of ORP1, the residue in position 5 is an alanine (Fig 2I). The side chain of A479 sits in a hydrophobic pocket formed by V51, T53, V61, N64, and F95. Hydrogen bonds are also formed between the amide and carbonyl of A479 with the carbonyl of V51, and the side chain amide of N64, respectively. This conformation of the peptide allows L480, the 6th residue of the motif, to form a hydrogen bond between its carbonyl and the amide of V61, and a water‐mediated contact between its amide and the carbonyls of P56 and Y59. However, in the Phospho‐FFAT motif of STARD3, the residues in position 5 and 6 are both prolines (P210 and P211) (Fig 2H). This prevents P210 from contacting the carbonyl of V51 of VAP‐A. The side chain of P210 no longer sits in the hydrophobic pocket, but instead makes hydrophobic contacts with the side chains of K50 and N64. This prevents any of the other contacts seen for residues 5 and 6 of the conventional FFAT motif. The difference in position of peptide also lifts the phosphoserine in position 4 closer to the side chain amides of K50 and K52, allowing it to make strong contacts.
The presence of two Phospho‐FFAT‐bound and two unbound chains in the asymmetric unit revealed conformation changes between the bound and unbound domains (Appendix Fig S1). We noted modifications involving the VAP residue F95 which is buried in the hydrophobic core of the domain, and M96 residue at its surface. These changes notably result in an opening of the hydrophobic pocket which allows F207 of the Phospho‐FFAT to bind (Fig 2F and G, and Appendix Fig S1).
Interestingly, depending of the nature of the negatively charged residue in position 4 of the FFAT (aspartate or phosphorylated serine, in a conventional and Phospho‐FFAT motif, respectively), the interaction with the MSP domain of VAP‐A is different. Indeed, superposition of the FFAT motifs of STARD3 and ORP1 showed that the side chain of phosphorylated serine is longer than that of aspartate and forms an ionic bond with lysine K50 of VAP‐A (Fig 2H–J). To determine whether this ionic bond contributes to the interaction in vitro, we produced the recombinant MSP domain of VAP‐A in which K50 was replaced by a leucine (hereafter referred to as VAP‐A K50L). Then, we tested the interaction of this VAP‐A mutant with synthetic peptides corresponding to the phosphorylated FFAT of STARD3 or the conventional FFAT of ORP1 (Fig EV2A). Unlike wild‐type VAP‐A that interacted equally with the conventional and the Phospho‐FFAT, VAP‐A K50L only interacted with the conventional FFAT. Thus, the mutation of K50 in VAP‐A impedes the interaction of its MSP domain with the Phospho‐FFAT of STARD3, without affecting the ability of the protein to interact with the conventional FFAT of ORP1 in vitro. Given the high sequence identity (82%) between human VAP‐A and VAP‐B, we hypothesized that the interaction of FFAT motifs with each protein should be similar. Thus, we mutated the conserved lysine K43 in VAP‐B and tested the interaction of the mutant protein with the conventional and the Phospho‐FFAT in vitro (Fig EV2A). Similarly to VAP‐A K50L, VAP‐B K43L interacted with the conventional FFAT of ORP1, but not with the Phospho‐FFAT of STARD3, while wild‐type VAP‐B interacted equally with the two types of FFAT motifs.
Figure EV2. Interaction of VAP‐A, VAP‐B, and MOSPD2 with the Phospho‐FFAT of STARD3.
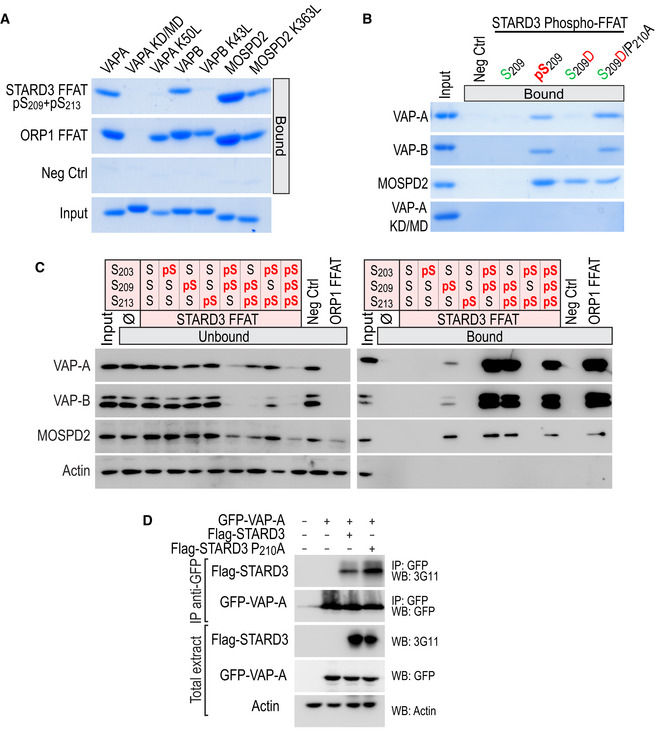
- Interaction of VAP‐A K50L, VAP‐B K43L, and MOSPD2 K363L mutants with conventional and Phospho‐FFAT motif. Wild‐type and mutant MSP domains were pulled down with phosphorylated STARD3 (pS209 + pS213), ORP1 FFAT peptides, and with the negative control peptide. The input fraction corresponds to the recombinant proteins used in the assay. Input and bound proteins were revealed by Coomassie Blue staining. Note that lysine K50 in VAP‐A and K43 in VAP‐B are required for the interaction with a Phospho‐FFAT, and not with a conventional FFAT.
- Interaction of VAP‐A, VAP‐B, MOSPD2 with phosphomimetic STARD3 Phospho‐FFAT motif. Wild‐type and mutant MSP domains were pulled down with unphosphorylated, phosphorylated (pS209), phosphomimetic (S209D and S209D/P210A), and with the negative control peptide. The input fraction corresponds to the recombinant proteins used in the assay. Input and bound proteins were revealed by Coomassie Blue staining. Acidic (D) or phosphorylated residues (pS), and alcoholic (S) residues are in red and green, respectively; the other residues are in black.
- STARD3 and VAP‐A/VAP‐B or MOSPD2 complex formation requires a unique phosphorylation of the Phospho‐FFAT motif. Western blot analysis of proteins pulled down using the peptides described in Fig EV3A. The input fraction corresponds to the HeLa cell total protein extract. The streptavidin beads were first coupled to the indicated biotinylated peptides, or left without peptide (ø). The soluble fraction after the incubation of the protein extract with the beads (Unbound; left), and proteins attached to the beads (Bound; right) were analyzed by Western blot using anti‐VAP‐A, anti‐VAP‐B, and anti‐MOSPD2 antibodies. Actin was used as a loading control.
- Immunoprecipitation (GFP‐Trap) experiments between GFP‐tagged VAP‐A and Flag‐tagged STARD3 (WT and P210A mutant). Approximatively 15 µg of total protein extracts was analyzed by Western blot using anti‐STARD3, anti‐GFP, and anti‐Actin antibodies. Immunoprecipitated material was analyzed using anti‐STARD3 and anti‐GFP antibodies.
Source data are available online for this figure.
This structural analysis showed that the overall binding mode of a conventional FFAT and a Phospho‐FFAT to VAP is different. While the first half of the core motif binds in an identical manner, it diverges in the other half with specific interactions being involved; more specifically, VAP‐A K50 and VAP‐B K43 residues are predominantly involved in binding the phosphorylated serine of the Phospho‐FFAT.
In vitro biophysical characterization of VAPs interaction with Phospho‐FFATs
To determine in a quantitative manner the binding affinities between VAP‐A and VAP‐B, and a conventional FFAT or a Phospho‐FFAT motif, we used the optical biosensing surface plasmon resonance (SPR) method. Either control, unphosphorylated or phosphorylated biotinylated peptides were immobilized onto a sensor chip and several concentrations of recombinant MSP domains were injected. A strong association of every MSP domain with the conventional FFAT motif of ORP1 was measured, as indicated by the interaction profiles typical for a domain/peptide interaction (Fig EV3B). In contrast, no binding was seen with the non‐phosphorylated FFAT of STARD3 (Fig 3A). Corroborating pull‐down assays (Fig 2C), phosphorylation of S209 in the peptide induced a dose‐dependent signal increase (Fig 3B), thus confirming that a unique phosphorylation of this residue is critical for the interaction. The sensorgrams reached a steady‐state response (R eq) during the association phase which allowed the determination of affinities. The values of R eq were fitted as a function of the MSP domain concentration assuming a Langmuir model. We have previously shown that the recombinant MSP domains of VAP‐A and VAP‐B form homodimers in solution (Di Mattia et al, 2018). Thus, the concentrations used to determine affinities correspond to those of VAP‐A and VAP‐B homodimers. Dissociation constants (K D) of 5.6 ± 0.3 and 7.1 ± 0.3 µM were determined by fitting the binding isotherms obtained for the interaction between the STARD3 FFAT pS209 and the MSP domain of VAP‐A and VAP‐B, respectively (Fig 3B).
Figure 3. The MSP domain binds the phosphorylated Phospho‐FFAT with an affinity in the micromolar range.
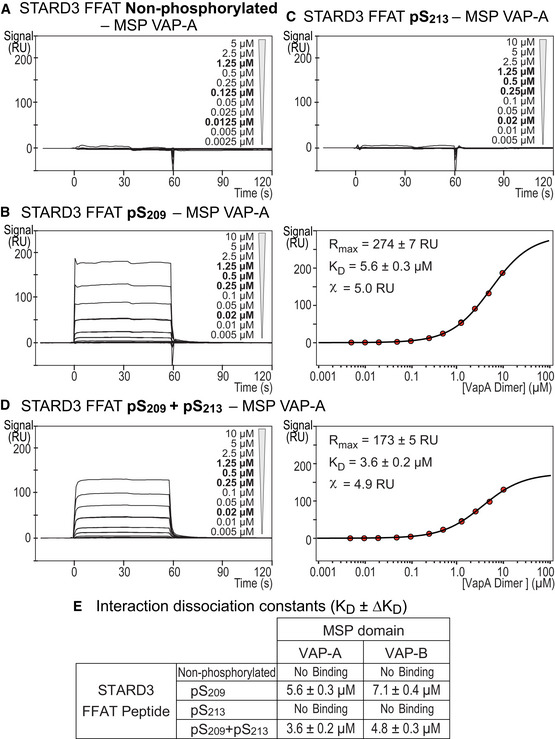
-
A–DSurface Plasmon resonance analysis of the MSP domain of VAP‐A binding onto immobilized unphosphorylated (A), monophosphorylated pS209 (B) and pS213 (C), and bi‐phosphorylated pS209 + pS213 (D) STARD3 FFAT peptides. Representative sensorgrams resulting from the interaction between the MSP domain of VAP‐A injected at different concentrations and the different FFAT peptides are shown in (A), (B left), (C), and (D left). Binding curves display the SPR signal (RU) as a function of time. Some samples were measured twice (concentrations printed in bold). Signal obtained for the negative control peptide immobilized on another flow cell is systematically subtracted, as well as the bulk effect recorded with buffer only. Steady‐state analysis of the interaction between pS209 (B right) and pS209 + pS213 (D right) STARD3 FFAT peptide, and the MSP domain of VAP‐A. Equilibrium responses (Req) extracted from the left panel in association phase were plotted as a function of the dimeric MSP domain of VAP‐A concentration, and fitted with a Langmuir binding model.
-
EDissociation constants between peptides corresponding to different kind of FFAT motifs and the MSP domains of VAP‐A and VAP‐B. The kinetic experiments were performed at 25°C in Tris–HCl 50 mM, pH 7.5, NaCl 75 mM buffer supplemented with 0.005% (v/v) surfactant polysorbate 20 (P20, GE Healthcare). Mean of n independent experiments: n = 2 for pS209 and pS209 + pS213; 3 for pS213; and 4 for the non‐phosphorylated FFAT. Uncertainties are obtained from the standard deviation considering a t‐distribution coefficient for a risk factor of 32%.
The presence of two phosphorylations on S209 and S213 slightly increased the affinity with the MSP domains (Fig 3D and E); this effect was further enhanced by the presence of three phosphorylations on S203, S209, and S213 (Fig EV3D and E). Moreover, we found that STARD3 FFAT peptides phosphorylated on pS213 only (Fig 3C) or pS203 only (Fig EV3C) did not interact with any of the MSP domains. These data established the crucial role of phosphorylation on S209 in the interaction process and showed that additional phosphorylation on residues in the vicinity of the core FFAT motif increased the affinity. The presence of several phosphorylated residues increases the number of negative charges in the FFAT peptide and increases interaction with the MSP domain. In agreement with the idea that electrostatic interactions play an important role, the interaction of the MSP domain of VAP‐A and VAP‐B with either a conventional FFAT or with a Phospho‐FFAT was found to depend on the ionic strength of the buffer used (Fig EV3E).
These experiments showed that phosphorylation of serine at the 4th position in the core FFAT motif is critical for the direct interaction of the Phospho‐FFAT with the MSP domains of VAP‐A and VAP‐B; in addition, the interaction occurs with a micromolar affinity, similar to that of a conventional FFAT motif.
Formation of the complex between STARD3 and VAPs requires phosphorylation of the Phospho‐FFAT motif
Having established a key role of phosphorylation on serine at position 4 of the Phospho‐FFAT of STARD3, the next step was to show that this phosphorylation was required for the assembly of the complex between STARD3 and VAP proteins.
First, we examined whether endogenous VAP‐A and VAP‐B could be isolated from whole cell protein extracts using synthetic biotinylated peptides corresponding to the FFAT motif of STARD3, either phosphorylated or not, as bait (Figs EV2, EV3 and EV2, EV3). We tested peptides bearing a phosphoserine either at position 209 or 213, or at both positions. In addition, to have a comprehensive understanding of the effect of phosphorylation near the FFAT motif, we included peptides containing a phosphoserine at position 203, even though it was not identified as being phosphorylated in vivo. To complete our series, we synthesized peptides with pS203 + pS209 and pS203 + pS213 combinations, as well as a peptide in which S203, S209, and S213 residues were phosphorylated. Each peptide was attached to streptavidin beads and incubated with HeLa cell protein extract. Bound proteins were eluted and analyzed by SDS–PAGE followed by Western blot using antibodies against VAP‐A and VAP‐B (Fig EV2C). As expected (Rocha et al, 2009; Di Mattia et al, 2018), the conventional ORP1 FFAT motif, used as positive control, efficiently pulled down VAP‐A and VAP‐B from the extract. Expected as well, the non‐phosphorylated FFAT motif of STARD3 did not pull‐down VAP‐A or VAP‐B (Fig EV2C). Consistent with the idea that phosphorylation at position 4 of the FFAT motif is crucial for the binding, we found that among the three monophosphorylated peptides, only the one with a phosphorylation on S209 was able to trap VAP‐A/VAP‐B. Moreover, we found that among peptides bearing a combination of phosphorylations (2 or 3 phosphoserines) only the ones having phosphorylated S209 efficiently pull‐down VAP‐A/VAP‐B. In line with the SPR experiments, we noted that the presence of another phosphoserine in addition to pS209 increased the interaction of the peptide with the VAP proteins, as indicated by the higher and lower amount of each protein in the bound and unbound fractions, respectively (Fig EV2C). These data confirmed the crucial role of the phosphorylation of S209, corresponding to the 4th position of the core FFAT motif of STARD3, for the interaction of this motif with its ER‐located VAP partners. Additional phosphorylations of S203 and S213 are not essential but strengthen the interaction.
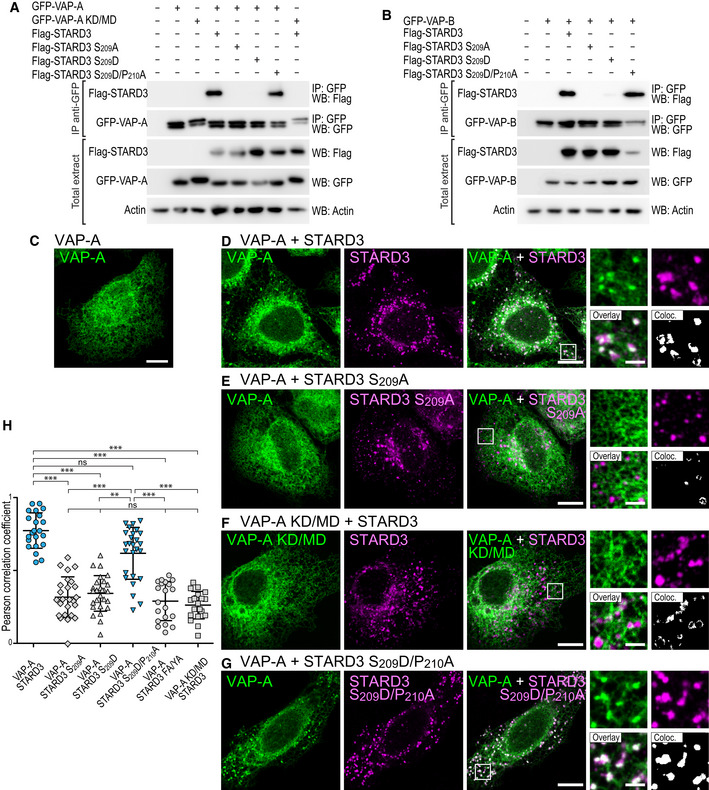
We then analyzed the ability of full‐length STARD3 to associate with VAP proteins. To test this, GFP‐tagged VAP‐A was co‐expressed in HeLa cells with wild‐type STARD3 or two mutants in which the S209 was replaced either by a non‐phosphorylatable residue, alanine (S209A), or a phosphomimetic residue, aspartate (S209D). The proteins were then immunoprecipitated using anti‐GFP antibodies. The GFP‐VAP‐A KD/MD mutant was used as a negative control. As we previously showed (Alpy et al, 2013), wild‐type STARD3 was co‐immunoprecipitated with VAP‐A (Fig 4A). In contrast, the STARD3 S209A mutant was not co‐precipitated (Fig 4A), suggesting that the interaction between VAP‐A and STARD3 requires a phosphorylatable serine at position 4 of the FFAT motif. Replacing S209 with the phosphomimetic aspartate residue did not, however, restore the VAP‐A/STARD3 interaction. The tridimensional structure of VAP‐A in complex with the Phospho‐FFAT of STARD3 shows that the proline residue in position 5 of the FFAT motif, by imposing a conformational rigidity in the peptide backbone, would prevent the aspartate residue from properly mimicking a phosphorylated serine, as it possesses a shorter side chain (Fig 2D–H). To circumvent this, we replaced this proline P210 by an alanine, and serine S209 by an aspartate, to generate the STARD3 S209D/P210A mutant. Remarkably, this mutant was co‐immunoprecipitated with VAP‐A (Fig 4A). The single mutant STARD3 P210A showed an improved binding with VAP‐A compared to WT STARD3 (Fig EV2D). In the context of the STARD3 Phospho‐FFAT motif, the presence of the P210 residue imposes a structural conformation enabling only the interaction with a phosphoserine at position 4. Thus, STARD3 requires a phosphorylatable serine in position 4 of its FFAT motif to interact with VAP‐A; structural constraints explain that a single phosphomimetic mutation does not restore the interaction, only the double mutation restores binding. To substantiate this result, we repeated the experiments with VAP‐B (Fig 4B). Like VAP‐A, VAP‐B bound to wild‐type STARD3, but not with its non‐phosphorylatable mutant STARD3 S209A (Fig 4B). VAP‐B, similarly to VAP‐A, did not interact with STARD3 S209D, while it interacted with STARD3 S209D/P210A. In agreement with these data, the recombinant MSP domain of VAP‐A and VAP‐B interacted with peptides corresponding to the STARD3 S209D/P210A mutant motif, while it did not interact with peptides having the STARD3 S209D mutant motif (Fig EV2B). Jointly, these results show that phosphorylation of the FFAT of STARD3 on its 4th position (S209) is indispensable for the protein to interact with its partners VAP‐A and VAP‐B. Structural constraints explain that a single phosphomimetic mutation does not restore the interaction, only a double mutation S209D/P210A restores binding. Moreover, additional phosphorylations of serine outside the core motif are not required for the interaction, but when S209 is phosphorylated, they increase the binding affinity.
Figure 4. In vivo, a unique phosphorylation of the Phospho‐FFAT motif allows STARD3 and VAP‐A/VAP‐B complex formation and the establishment of ER‐endosome contacts.
-
A, BBinding assays. Immunoprecipitation (GFP‐Trap) experiments between GFP‐tagged VAP‐A (WT and KD/MD mutant; A), VAP‐B (B) and Flag‐tagged STARD3 (WT and S209A, S209D, and S209D/P210A mutants). Please note that the mutations modify the FFAT and Phospho‐FFAT scores of this region of STARD3: compared to WT STARD3 (Phospho‐FFAT: 2.5; conventional FFAT: 6.5), the STARD3 S209A, mutant has both low Phospho‐FFAT and FFAT scores (6.5), the S209D mutant has a high conventional FFAT score (2.5) but a low Phospho‐FFAT score (6.5), and the double mutant has a conventional FFAT score of 1.5 and a Phospho‐FFAT score of 5.5. Approximatively 5 µg of total protein extracts was analyzed by Western blot using anti‐Flag, anti‐GFP, and anti‐Actin antibodies. Immunoprecipitated material was analyzed using anti‐Flag and anti‐GFP antibodies.
-
C–GRecruitment assays. GFP‐VAP‐A (C, D, E, G; green) and GFP‐VAP‐A KD/MD‐expressing cells (F; green) were left untransfected (C) or transfected with Flag‐STARD3 (D, F), Flag‐STARD3 S209A (E), and Flag‐STARD3 S209D/P210A (G), and labeled using anti‐Flag (magenta) antibodies. The subpanels on the right are higher magnification (3.5×) images of the area outlined in white. The Overlay panel shows merged green and magenta images. The Coloc panel displays a colocalization mask on which pixels where the green and the magenta channels co‐localize are shown in white. Scale bars: 10 µm. Inset scale bars: 2 µm.
-
HPearson’s correlation coefficients between VAP‐A (WT or KD/MD mutant) and STARD3 (WT or S209A, S209D, S209D/P210A, FA/YA mutants) staining are shown. Each dot represents a single cell (number of cells: VAP‐A–STARD3: 22; VAP‐A–STARD3 S209A: 25; VAP‐A–STARD3 S209D: 27; VAP‐A–STARD3 S209D/P210A: 26; VAP‐A–STARD3 FA/YA: 20; VAP‐A KD/MD–STARD3: 20, from at least three independent experiments). Means and error bars (SD) are shown. Kruskal–Wallis with Dunn’s multiple comparison test (**P < 0.01; ***P < 0.001).
Source data are available online for this figure.
Phosphorylation at the 4th residue of the Phospho‐FFAT motif of STARD3 is necessary for ER‐endosome contact formation in vivo
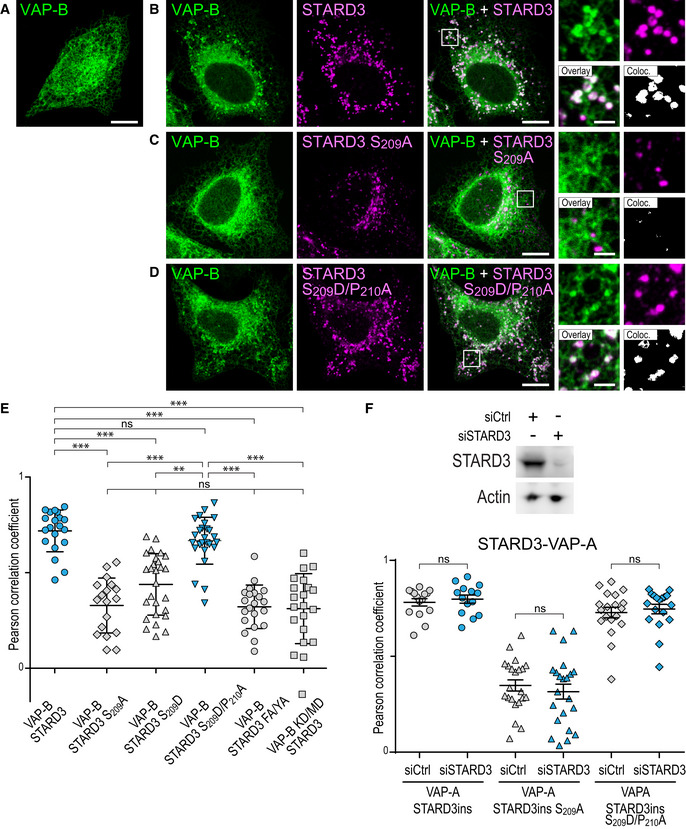
We previously reported that the creation of ER‐endosome contacts, arising from the interaction between STARD3 and VAP‐A or VAP‐B, induces a striking relocalization of these latter two proteins from a characteristic ER reticulated pattern to ER subdomains in contact with STARD3‐positive endosomes (Alpy et al, 2013; Wilhelm et al, 2017). In contrast, when a mutant STARD3 with the FFAT core deleted was expressed, neither VAP‐A or VAP‐B were relocated toward ER sub‐regions in contact with endosomes. Here, we performed similar experiments to address whether ER‐endosome contact formation depends on STARD3 FFAT phosphorylation. We expressed VAP‐A alone and in combination with either wild‐type STARD3, its non‐phosphorylatable counterpart S209A mutant, or its phosphomimetic S209D/P210A mutant. When expressed alone, VAP‐A was evenly present in the ER (Fig 4C). In the presence of wild‐type STARD3, VAP‐A was massively recruited toward endosomes (Fig 4D), with a positive correlation of VAP‐A and STARD3 signals (Fig 4H). In contrast, in the presence of the S209A mutant, VAP‐A remained uniformly distributed in the ER (Fig 4E), with no signal correlation (Fig 4H). This phenotype was similar to what is observed in cells expressing VAP‐A KD/MD or STARD3 F207A/Y208A (FFAT mutant named FA/YA) mutants, which are unable to form VAP/STARD3 complexes (Fig 4F and H; Alpy et al, 2013). Consistent with the biochemistry data, the phosphomimetic mutant STARD3 S209D/P210A induced the enrichment of VAP‐A around endosomes (Fig 4G), and fluorescent signals were as correlated as the ones measured with wild‐type STARD3 and VAP‐A (Fig 4H). The colocalization of STARD3 (WT and mutants) with VAP‐A was not affected by endogenous STARD3 protein (Fig EV4F). Jointly, these data showed that STARD3 phosphorylation on S209 allows the formation of ER‐endosome contacts in vivo.
Figure EV4. Effect of non‐phosphorylatable and phosphomimetic mutations of S209 on the formation of ER‐endosome contacts involving VAP‐B in vivo .
-
A–DGFP‐VAP‐B (A–D; green)‐expressing cells were left untransfected (A) or transfected with Flag‐STARD3 (B), Flag‐STARD3 S209A (C), and Flag‐STARD3 S209D/P210A (D), and labeled using anti‐Flag (magenta) antibodies. The subpanels on the right are higher magnification (3.5×) images of the area outlined in white. The Overlay panel shows merged green and magenta images. The Coloc panel displays a colocalization mask on which pixels where the green and the magenta channels co‐localize are shown in white. Scale bars: 10 µm. Inset scale bars: 2 µm.
-
EPearson’s correlation coefficients between VAP‐B (WT and KD/MD) and STARD3 (WT, S209A, S209D, S209D/P210A or FA/YA mutants) staining are shown. Each dot represents a single cell (number of cells: VAP‐B–STARD3: 20; VAP‐B–STARD3 S209A: 18; VAP‐B–STARD3 S209D: 27; VAP‐B–STARD3 S209D/P210A: 26; VAP‐B–STARD3 FA/YA: 21; VAP‐B KD/MD–STARD3: 21, from at least three independent experiments). Means and error bars (SD) are shown. Kruskal–Wallis with Dunn’s multiple comparison test (**P < 0.01; ***P < 0.001).
-
FTop: Western blot analysis of protein extracts from HeLa cells transfected with control siRNAs (siCtrl) and siRNAs targeting STARD3 (siSTARD3) using anti‐STARD3 and anti‐Actin antibodies. Bottom: After siRNA transfection, the cells were transfected with GFP‐VAP‐A and STARD3 (WT, STARD3 S209A, and STARD3 S209D/P210A). STARD3 expression vectors contained a cDNA with silent mutations rendering it insensitive to siRNAs. The cells were labeled using anti‐STARD3 antibodies. Pearson’s correlation coefficients between STARD3 (WT, STARD3 S209A, and STARD3 S209D/P210A) and VAP‐A staining are shown. Note that the colocalization between VAP‐A and STARD3 is similar in the presence and absence of endogenous STARD3. Means and error bars (SD) are shown. Kruskal–Wallis with Dunn’s multiple comparison test (ns: P ≥ 0.05).
Source data are available online for this figure.
Similar experiments were then performed with VAP‐B (Fig EV4A–E). Akin to VAP‐A, VAP‐B was evenly distributed in the ER when expressed alone, and in the presence of the non‐phosphorylatable STARD3 S209A mutant, while it was recruited around endosomes by wild‐type STARD3 and the phosphomimetic mutant STARD3 S209D/P210A (Fig EV4A–E).
Together, these data show that in vivo the phosphorylation of the STARD3 FFAT motif elicits the assembly of ER‐endosome contacts made by a complex between STARD3 and either VAP‐A or VAP‐B.
Phosphorylation of the FFAT motif is essential for STARD3 sterol transfer function in vitro
Having established a novel mechanism of complex formation for a protein bearing a Phospho‐FFAT motif like STARD3, we examined in vitro whether the phosphorylation of S209 within the non‐conventional FFAT motif was mandatory for STARD3 to be active, i.e., to connect the endosome and ER membranes, and to transfer sterol between them. To this end, we reconstituted the tethering complex in vitro with liposomes and recombinant proteins. First, we produced and purified recombinant proteins corresponding to the cytosolic part of STARD3 including the Phospho‐FFAT motif, the START domain, and an N‐terminal cysteine residue enabling the covalent attachment to liposomes doped with thiol‐reactive MPB‐PE lipid (Fig 5A). We produced this protein and a form constitutively phosphorylated on S209, termed thereafter cSTD3 and pS209 cSTD3, respectively. Because the production of recombinant proteins phosphorylated on a specific site is not possible using regular E. coli, we used a genomically recoded E. coli strain engineered to allow phosphoserine incorporation into recombinant proteins to produce pS209 cSTD3 (Park et al, 2011; Pirman et al, 2015). Recombinant proteins were analyzed by SDS–PAGE followed by SYPRO Orange staining. We observed one and two bands for cSTD3 and pS209 cSTD3, respectively (Fig 5B). To have a tool able to detect STARD3 phosphorylation in the FFAT motif core, we developed a phospho‐specific antibody recognizing STARD3 when phosphorylated on S209. (Appendix Fig S2). cSTD3 and pS209 cSTD3 were detected by the anti‐STARD3 antibody, but only the higher species in the pS209 cSTD3 sample was additionally detected by the anti‐phospho‐STARD3‐pS209 antibody, thus showing that a fraction of protein is phosphorylated on S209 (Fig 5B). As a confirmation, the analysis of pS209 cSTD3 by mass spectrometry showed that it was present as two major species corresponding to the expected masses of the phosphorylated (MW = 28,366 Da) and non‐phosphorylated (28,286 Da) proteins (Appendix Fig S3A). To confirm that the phosphorylation was indeed on S209, recombinant pS209 cSTD3 was digested with trypsin, and subjected to ion trap LC/MS/MS analysis (Appendix Fig S3B). This analysis confirmed that the phosphorylation site was on S209. Thus, we produced and purified a pS209 cSTD3 recombinant protein genuinely phosphorylated on S209.
Figure 5. Phosphorylation of STARD3 in its Phospho‐FFAT motif governs membrane attachment and cholesterol transfer in in vitro reconstituted assays.
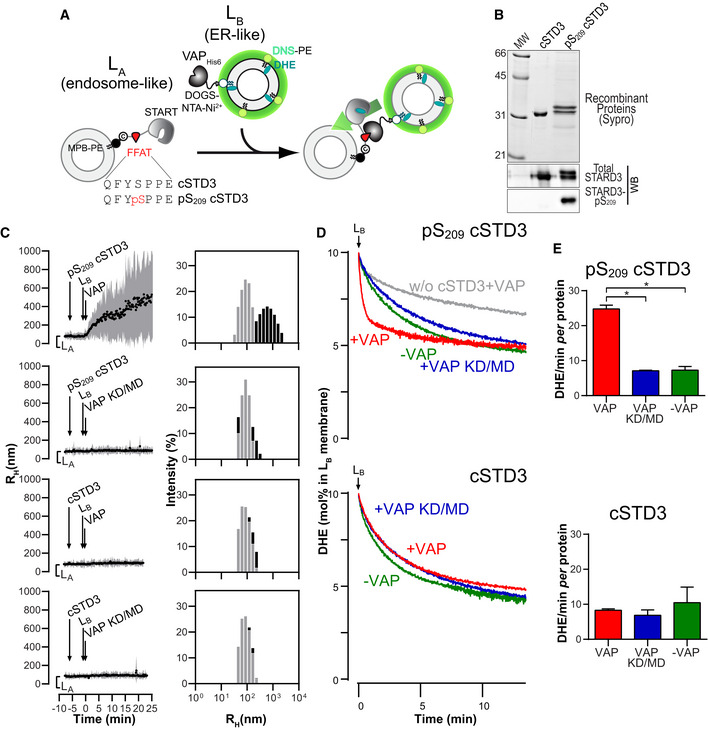
- Description of the experimental strategy. LA liposomes (endosome‐like) are decorated with cSTD3 or pS209 cSTD3 owing to covalent links with MPB‐PE (1.2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[4‐(p‐maleimidophenyl) butyramide]), and mixed with LB liposomes (ER‐like) covered by VAPHIS6 attached to DOGS‐NTA‐Ni2+ (1.2‐dioleoyl‐sn‐glycero‐3‐[(N‐(5‐amino‐1‐carboxypentyl) iminodiacetic acid) succinyl], nickel salt). For dehydroergosterol (DHE) transport experiment, LB liposomes also contain DHE and a dansyl‐phosphatidylethanolamine (DNS‐PE). The transport of DHE from LB to LA liposomes is followed by measuring the change in FRET from DHE to DNS‐PE.
- SDS–PAGE gel and Western blot analysis of purified cSTD3 and pS209 cSTD3 proteins. Top: The gel was stained with SYPRO Orange to visualize proteins and molecular weight markers. Bottom: Two similar gels were blotted onto nitrocellulose and analyzed for the presence of total and STARD3‐pS209 using specific antibodies.
- Aggregation assays. LA liposomes (50 µM total lipids) were incubated for 5 min with cSTD3 or pS209 cSTD3 (380 nM). Then, LB liposomes (50 µM total lipids) and VAP‐AHIS6 or VAP‐A KD/MDHIS6 (700 nM) were successively added. Left panels: mean radius (black dots) and polydispersity (shaded area) over time. Right panels: size distribution before (gray bars) and after the reaction (black bars).
- DHE transport assay. DOPC (1.2‐dioleoyl‐sn‐glycero‐3‐phosphocholine) liposomes (62.5 µM total lipids, LA) containing 3 mol% MPB‐PE were mixed with cSTD3 or pS209 cSTD3 (475 nM). After 5 min, liposomes (DOPC/DOGS‐NTA‐Ni2+/DNS‐PE/DHE liposomes 77.5/10/2.5/10 mol/mol, 62.5 µM total lipids, LB), covered or not with 1 µM of VAPHIS6 or VAP‐A KD/MDHIS6, were added. FRET between DHE and DNS‐PE in the LB liposomes diminishes as DHE is transported toward LA liposomes. The signal was converted into amount of DHE present in LB liposomes (in mol%).
- Initial DHE transport rate measured with cSTD3 or pS209 cSTD3 in the presence or absence of VAP‐A HIS6 or VAP‐A KD/MDHIS6. Data are represented as mean ± SEM (n = 3 for cSTD3‐VAP and n = 4 for all other data). Mann–Whitney test (*P < 0.05).
Source data are available online for this figure.
Next, to examine the tethering activity, cSTD3 and pS209 cSTD3 proteins were attached to one population of liposomes called hereafter LA liposomes. Liposome flotation assays confirmed that the two proteins were efficiently bound onto LA liposomes (Appendix Fig S3C–E) and that liposome‐bound pS209 cSTD3 remained phosphorylated (Appendix Fig S3F). Then, we examined whether LA liposomes bound with cSTD3 and pS209 cSTD3 were able to physically associate with another population of liposomes (called LB liposomes) covered with VAP‐A by dynamic light scattering (Fig 5B and C). To prepare LB liposomes, a recombinant VAP‐AHis6 protein with its carboxyl‐terminal transmembrane region deleted was anchored to the surface of liposomes thanks to the presence of NTA‐Ni2+ lipids (Wilhelm et al, 2017). When LA liposomes covered by pS209 cSTD3 were mixed with LB liposomes bearing VAP‐A, a rapid increase in the mean radius of liposomes (from ~ 90 nm up to 500 nm) occurred, indicating a connection between the two liposome populations (Fig 5C). This experiment was repeated with LB liposomes lacking VAP‐A, or covered with the VAP‐A KD/MD mutant, and in both cases, no aggregation was seen (Fig 5C). For the cSTD3 protein, no aggregation was observed even in the presence of a functional VAP (Fig 5C). These results indicated that a unique phosphorylation of the S209 serine in the Phospho‐FFAT motif of STARD3 allows the protein to tether membranes in a VAP‐dependent manner. It is noteworthy that the attachment of membranes does not provoke any fusion (Fig EV5) as measured by a standard FRET assay using an NBD‐PE/Rhod‐PE pair (Struck et al, 1981).
Figure EV5. The attachment of liposomes by pS209 cSTD3 does not induce membrane fusion.
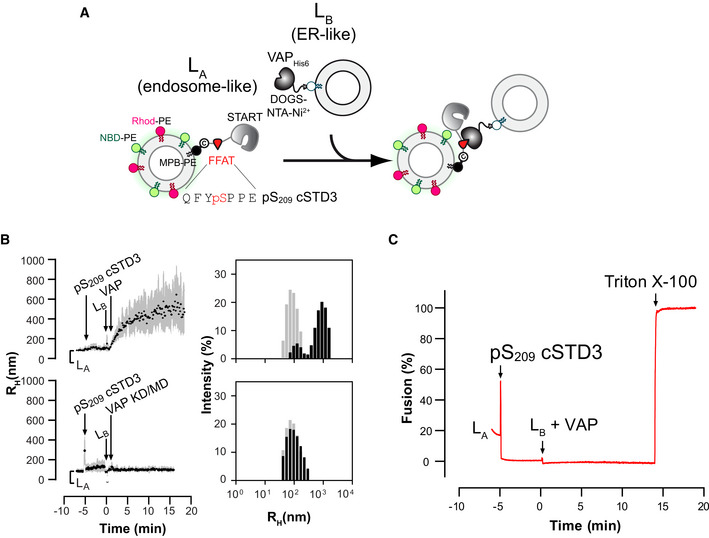
- Description of the experimental strategy. LA liposomes (endosome‐like) are decorated with pS209 cSTD3 owing to covalent links with MPB‐PE (1.2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[4‐(p‐maleimidophenyl) butyramide]), and mixed with LB liposomes (ER‐like) covered by VAPHIS6 attached to DOGS‐NTA‐Ni2+ (1.2‐dioleoyl‐sn‐glycero‐3‐[(N‐(5‐amino‐1‐carboxypentyl) iminodiacetic acid) succinyl]; nickel salt). LA liposomes contain the FRET pair NBD‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(7‐nitro‐2‐1,3‐benzoxadiazol‐4‐yl)) and Rhod‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl)). The fusion of LA and LB liposomes is followed by measuring an increase in NBD‐PE fluorescence, which is initially quenched due to FRET with Rhod‐PE. The percentage of fusion is equal to 100 × ((F − F 0)/(F max − F 0)) where F 0 is the signal measured before the addition of LB liposomes decorated with VAP‐AHis6, and F max is the signal measured after adding Triton X‐100 (1% v/v final concentration).
- Aggregation assays. LA liposomes (50 µM total lipids) were incubated for 5 min with pS209 cSTD3 (380 nM). Then, LB liposomes (50 µM total lipids) and VAP‐AHIS6 or VAP‐A (KD/MD)HIS6 (700 nM) were successively added. Left panels: mean radius (black dots) and polydispersity (shaded area) over time. Right panels: size distribution before (gray bars) and after the reaction (black bars).
- Fusion assay. DOPC (1.2‐dioleoyl‐sn‐glycero‐3‐phosphocholine) liposomes (62.5 µM total lipids, LA) containing 3 mol% MPB‐PE, 1% NBD‐PE, and 1% Rhod‐PE were mixed with pS209 cSTD3 (475 nM). After 5 min, liposomes (DOPC/DOGS‐NTA‐Ni2+ liposomes 90/10 mol/mol, 62.5 µM total lipids, LB), covered or not with 1 µM of VAPHIS6, were added. At the end of the experiment, Triton X‐100 was added (1% v/v final concentration) to disrupt liposomes and eliminate energy transfer, allowing the determination of the maximal fluorescence of NBD‐PE that would be measured for a complete membrane fusion.
Once demonstrated that pS209 cSTD3 protein allows for the tethering of two membranes, a further step was to study the kinetics of sterol transfer between them. To do so, we measured the intermembrane transfer of the fluorescent sterol dehydroergosterol (DHE) in real time by FRET (Fig 5D). LB liposomes including both DHE (10 mol%) and a second fluorescent lipid, dansyl‐phosphatidylethanolamine (DNS‐PE, 2.5 mol%), were covered with VAP‐A‐His6 and added to LA liposomes decorated with pS209 cSTD3 (Fig 5B). The transport of DHE from LB to LA liposomes was followed by measuring the decrease in energy transfer from DHE to DNS‐PE. A fast transport of DHE was observed within the first seconds, and DHE was entirely equilibrated between the two liposome populations after a few minutes. The initial DHE transport rate was 24.78 ± 1.07 DHE molecules/min per molecule of pS209 cSTD3 (Fig 5E). In contrast, when LB liposomes were covered by VAP‐A KD/MD or were naked, the transport rates were four‐time slower (7.10 ± 0.15 and 7.29 ± 1.04 DHE molecules/min per pS209 cSTD3, respectively). When cSTD3 was unphosphorylated, the transport rates measured in the presence or absence of functional VAP were similar (6.87 and 10.45 DHE molecules/min per cSTD3, respectively).
Jointly, these results showed that using in vitro reconstitution assays, phosphorylation of the Phospho‐FFAT motif of a liposome‐bound STARD3 allows tethering with VAP‐covered liposomes. The formation of this complex triggers a fast transfer of sterols between the two connected membranes. Thus, in a complete in vitro defined system, phosphorylation of STARD3’s Phospho‐FFAT drives membrane tethering by enabling STARD3/VAP complex formation that in turn allows the efficient transport of sterols mediated by the START domain.
Phosphorylation of the FFAT motif is essential for STARD3 sterol transfer function in vivo
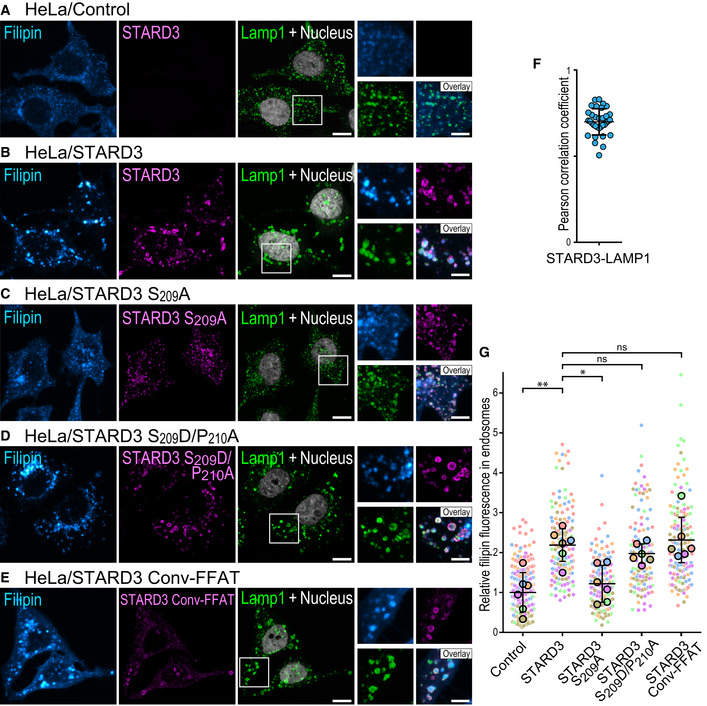
To substantiate these data, we used the fluorescent probe filipin to follow cholesterol distribution in cells (Appendix Fig S4). As previously described (Wilhelm et al, 2017), compared to control cells, HeLa cells expressing STARD3 accumulated cholesterol in LEs that were labeled with the Lamp1 marker (Fig 6A, B, F, and G). Next, to directly assess the contribution of S209 in the cholesterol accumulation phenotype, cholesterol was labeled in cells expressing the non‐phosphorylatable STARD3 S209A mutant. As expected from the fact that this mutant is unable to make ER–endosome contacts (Fig 4E), cholesterol did not accumulate in LEs of cells expressing STARD3 S209A (Fig 6C and G). In contrast, the phosphomimetic mutation S209D/P210A restored cholesterol accumulation at a level comparable to wild‐type STARD3 (Fig 6D and G). To further assess the effect of a non‐regulatable FFAT on STARD3, the STARD3 Conv‐FFAT mutant was constructed by replacing the Phospho‐FFAT of STARD3 by the conventional FFAT of the yeast transcriptional regulator Opi1p (EFFDASE) (Loewen et al, 2003). Cells expressing the STARD3 Conv‐FFAT mutant accumulated cholesterol in endosomes similarly to cells expressing the wild‐type STARD3 (Fig 6E and G).
Figure 6. STARD3‐mediated cholesterol transport in endosomes depends on S209 .
-
A–EHeLa/Ctrl (A), HeLa/STARD3 (B), HeLa/STARD3 S209A (C), HeLa/STARD3 S209D/P210A (D), and HeLa/STARD3 Conv‐FFAT (E) cells were labeled with anti‐STARD3 antibodies (magenta), anti‐Lamp1 antibodies (green) and with the fluorescent cholesterol probe filipin (Cyan Hot). Nuclei are shown in gray (TO‐PRO‐3). Higher magnifications images (2×) of the area outlined in white are shown on the right. The filipin, STARD3, and Lamp1 merged image is labeled Overlay. Scale bars: 10 µm. Inset scale bars: 5 µm.
-
FPearson’s correlation coefficients between STARD3 and Lamp1 staining in HeLa/STARD3 cells. Each dot represents a single cell (34 cells from 4 independent experiments). Mean and error bars (SD) are shown. Note that STARD3 and Lamp1 signals are highly correlated.
-
GRelative fluorescence intensity of intracellular filipin signals in endosomes of HeLa/Ctrl, HeLa/STARD3, HeLa/STARD3 S209A, HeLa/STARD3 S209D/P210A, and HeLa/STARD3 Conv‐FFAT cells. Data are displayed as a Superplot (Lord et al, 2020) showing the relative filipin fluorescence intensity in endosomes of individual cells (small circles) from 6 independent experiments (mean of each experiment shown as a large circle). Independent experiments are color‐coded. Means and error bars (SD) are shown as black bars. Kruskal–Wallis with Dunn’s multiple comparison test (*P < 0.05; **P < 0.01; n = 6 independent experiments).
Together, these data show that the phosphorylation of S209 is necessary for STARD3 function in cholesterol transport toward LEs. Of interest, replacement of a Phospho‐FFAT with a phosphomimetic mutant or a conventional FFAT motif, transforming it into a constitutively active motif, restored the transport function of STARD3. These results support the idea that the regulation by phosphorylation of the Phospho‐FFAT of STARD3 is a way to mitigate the effect of the protein on sterol distribution.
MOSPD2 interacts with conventional and Phospho‐FFAT motifs
We recently identified the MOSPD2 protein as a third member of the VAP family (Di Mattia et al, 2018). To study more precisely the binding characteristics of MOSPD2 with Phospho‐FFAT motifs, we produced in E. coli and purified the MSP domain of MOSPD2 (Fig 7A). Next, the recombinant MSP domain of MOSPD2 was incubated with streptavidin beads coupled to biotinylated peptides as described before for VAP. Bound proteins were eluted and analyzed by SDS–PAGE followed by Coomassie Blue staining (Fig 7B). Similarly to VAPs, the MSP domain of MOSPD2 did not interact with the non‐phosphorylated FFAT of STARD3, but interacted with peptides with a phosphorylation on S209 (Fig 7B). We used SPR to determine the interaction dissociation constants between the different peptides and the MSP domain of MOSPD2. The concentrations used to determine affinities corresponded to a monomer for MOSPD2 since we have previously shown that the recombinant MSP domain of the protein is monomeric (Di Mattia et al, 2018). The kinetic profile of the MSP domain of MOSPD2 was similar to those of VAPs (Fig 7C and D), with one obligate phosphorylation site on S209, and additional phosphorylation increasing the binding affinity (from 1.1 ± 0.3 µM for pS209, to 0.35 ± 0.16 µM for pS209 + pS213 for instance) (Fig 7E). Interestingly, the affinity for a conventional FFAT was similar for MOSPD2, VAP‐A, and VAP‐B, as previously reported (Di Mattia et al, 2018), while MOSPD2 had a slightly higher affinity for Phospho‐FFAT compared to VAP‐A and VAP‐B (Figs 3, EV3, 7). Together, these data show that MOSPD2 interacts in the micromolar affinity range with the Phospho‐FFAT motif only when its 4th residue is phosphorylated.
Figure 7. MOSPD2 interacts with the Phospho‐FFAT motif in a phosphorylation‐dependent manner.
-
ACoomassie Blue staining of the recombinant MSP domain of MOSPD2 after SDS–PAGE.
-
BRecombinant MSP domains pulled down with STARD3 FFAT peptides (unphosphorylated, monophosphorylated on S209 or S213, bi‐phosphorylated on S209 and S213, or tri‐phosphorylated on S203, S209 and S213 and control peptides were revealed by Coomassie Blue staining. The recombinant proteins subjected to the assay are shown in the input fraction.
-
C, DSPR analysis of the MSP domain of MOSPD2 binding onto immobilized unphosphorylated (C) and pS209 (D) STARD3 FFAT. Representative sensorgrams resulting from the interaction between the MSP domain of MOSPD2 injected at different concentrations and the different FFAT peptides are shown in (C) and (D left). Binding curves display the SPR signal (RU) as a function of time. Concentrations printed in bold indicate samples measured twice. Signal obtained for the negative control peptide immobilized on another flow cell is systematically subtracted, as well as the bulk effect recorded with buffer only. (D right) Steady‐state analysis of the interaction between pS209 STARD3 FFAT peptide and the MSP domain of MOSPD2. Equilibrium responses (Req) extracted from the left panel were plotted as a function of the monomeric concentration of the MSP domain of MOSPD2, and fitted with a 1:1 binding model. The experiments were performed at 25°C in Tris–HCl 50 mM, pH 7.5, NaCl 75 mM buffer supplemented with 0.005% (v/v) surfactant polysorbate 20 (P20, GE Healthcare). Mean of 2 independent experiments. Uncertainties are obtained from the standard deviation considering a t‐distribution coefficient for a risk factor of 32%.
-
EInteraction dissociation constants between the different STARD3 FFAT peptides and the MSP domains of MOSPD2.
-
FImmunoprecipitation (GFP‐Trap) experiments between GFP‐tagged MOSPD2 and Flag‐tagged STARD3 (WT and S209A, S209D, and S209D/P210A mutants). Approximatively 5 µg of total protein extracts was analyzed by Western blot using anti‐Flag, anti‐GFP and anti‐Actin antibodies. Immunoprecipitated material was analyzed using anti‐Flag and anti‐GFP antibodies.
-
G–IGFP‐MOSPD2‐expressing cells (green) were left untransfected (G) or transfected with Flag‐STARD3 (H) and Flag‐STARD3 S209A (I), and labeled using anti‐Flag (magenta) antibodies. The subpanels on the right are higher magnification (3.5×) images of the area outlined in white. The Overlay panel shows merged green and magenta images. The Coloc panel displays a colocalization mask on which pixels where the green and the magenta channels co‐localize are shown in white. Scale bars: 10 µm.
-
JPearson’s correlation coefficients between MOSPD2 (WT or RD/LD) and STARD3 (WT, S209A, or FA/YA) staining are shown. Each dot represents a single cell (number of cells: MOSPD2–STARD3: 22; MOSPD2– STARD3 S209A: 21; MOSPD2–STARD3 FA/YA: 20; MOSPD2 RD/LD–STARD3: 17, from at least three independent experiments). Means and error bars (SD) are shown. Kruskal–Wallis with Dunn’s multiple comparison test (***P < 0.001).
Source data are available online for this figure.
We characterized the interaction between a phospho‐FFAT and MOSPD2 in vivo. To do so, we immunoprecipitated GFP‐tagged MOSPD2 from cells co‐expressing the wild‐type and mutant STARD3 proteins (Fig 7F). Wild‐type STARD3 was co‐immunoprecipitated with MOSPD2. In contrast, the non‐phosphorylatable STARD3 mutant S209A was not co‐precipitated (Fig 7F). We showed before that the single phosphomimetic mutation S209D was not sufficient to restore the interaction between STARD3 and VAP‐A or VAP‐B, while the double‐mutation S209D/P210A restored binding (Fig 4A and B). MOSPD2 behaved differently: It associated, yet only moderately, with the single S209D mutant, and almost not with the S209D/P210A double mutant (Fig 7F); one reason could be that the tridimensional structure of the MSP domain of MOSPD2 differs from the one of VAPs.
We next addressed whether in cells MOSPD2 was forming contact sites when co‐expressed with STARD3. We imaged MOSPD2 recruitment on endosomes. Since the phosphomimetic mutant of STARD3 could not fully restore the interaction in vitro, we only tested the effect of the non‐phosphorylatable mutant STARD3 S209A (Fig 7I). Consistently, while wild‐type STARD3 expression induced a strong enrichment of MOSPD2 around endosomes, MOSPD2 remained evenly distributed along the whole ER in the presence of the STARD3 S209A mutant (Fig 7G–J). Similarly, MOSPD2 with the R404D/L406D double mutation (named RD/LD) which is defective in binding FFAT motifs remained uniformly localized in the ER (Fig 7J).
Together, these data showed that the interaction of the MSP domain of MOSPD2 with the Phospho‐FFAT motif depends on the critical phosphorylation of its central serine (S209 in STARD3), similarly to the MSP domain of VAP‐A and VAP‐B. They also unveiled specific features of MOSPD2 in terms of affinity and mode of interaction.
Structural characterization of the MSP domain of MOSPD2 in complex with a conventional FFAT and a Phospho‐FFAT
The 3D structure of the MSP domain of human MOSPD2 has not been previously characterized. Moreover, interactions studies presented before suggest that structural differences may exist between MOSPD2 and VAP MSP domains. To document the binding mode of MOSPD2 with the two types of FFAT motifs, we solved the structure of the MSP domain of MOSPD2 unbound (PDB ID: 6TQT) (Fig 8A), and in complex with peptides corresponding to a conventional FFAT (ORP1) (PDB ID: 6TQS) (Fig 8B) and a Phospho‐FFAT (STARD3) (PDB ID: 6TQU) (Fig 8C). The structure of the unbound state of the domain was resolved to 1.5 Å. The structure was broadly similar to that of rat VAP‐A (PDB ID 1Z9L; (Kaiser et al, 2005); rmsd of 2.382 Å over 104 Cαs). The main differences are seen in a flexible loop (residues 337–346 in human MOSPD2, residues 24–33 in VAP‐A), and in the α‐helix, which contains an extra turn at the N‐terminal in MOSPD2. Besides, sequence analyses show that the residues that are conserved between the VAPs and MOSPD2 are mostly found in the region which interacts with FFATs (Appendix Fig S5). In this binding interface, two main differences are present: (i) An asparagine and a serine are swapped between the VAP proteins and MOSPD2: N64 and S65 in VAP‐A (N57 and S58 in VAP‐B) are replaced with S377 and N378 in MOSPD2, respectively; (ii) K52 in VAP‐A is replaced with an arginine, R365.
Figure 8. Structure of the MSP domain of MOSPD2 in its unbound form, and in complex with a conventional FFAT and a Phospho‐FFAT.
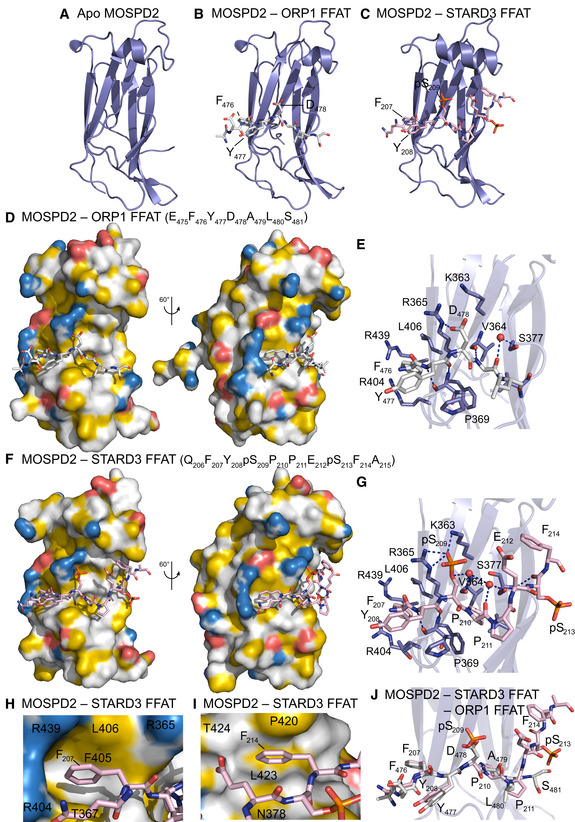
-
A–CRibbon diagram of the MSP domain of MOSPD2 in its unbound form (A), in complex with the conventional FFAT of ORP1 (B), and in complex with the Phospho‐FFAT of STARD3 (C).
-
DSurface representation of the MSP domain of MOSPD2 in complex with the conventional FFAT of ORP1.
-
EClose‐up view of the structure near the conventional FFAT motif highlighting critical residues (in stick model) of human MOSPD2 present in the binding interface.
-
FSurface representation of the MSP domain of MOSPD2 in complex with the phosphorylated FFAT of STARD3.
-
GClose‐up view of the structure near the Phospho‐FFAT motif highlighting critical residues (in stick model) of human MOSPD2 present in the binding interface.
-
H, IClose‐up view of the hydrophobic pockets interacting with the 2nd residue (F207) (H) and the 9th residue (F214) (I) of the Phospho‐FFAT of STARD3; residues of MOSPD2 constituting the pockets are indicated.
-
JSuperposition of the structures shown in (E) and (G) showing the similar trajectories of the peptides.
Data information: Phosphorous, nitrogen, and oxygen atoms are colored in orange, blue, and red, respectively. Carbon atoms are shown in slate blue, gray, and rose in the MSP domain, the classic FFAT motif, and the Phospho‐FFAT motif, respectively. (D, F, H, I): The protein surface is colored according to the YRB scheme, showing hydrophobic, negatively and positively charged atoms in yellow, red, and blue, respectively; the other atoms are in white (Hagemans et al, 2015). (E, G): Red spheres are water molecules.
The 3D structure of the MSP domain of rat VAP‐A and human MOSPD2 bound with the conventional FFAT motif of ORP1 (Fig 8D) are similar. Superposition of the domains showed a few changes in the binding interface, with almost all contacts being maintained. Since N64 in VAP‐A is replaced with S377 in MOSPD2, the direct hydrogen bond between side chain of N64 and the main chain carbonyl of A479 in the peptide is replaced with a water‐mediated contact. K52 in VAP‐A is replaced with R365, which makes a hydrogen bond with D478 of the peptide (Fig 8E).
However, a different picture is seen when comparing the structures of human VAP‐A and MOSPD2 bound to the phosphorylated FFAT of STARD3 (Fig 8F and G). In the MOSPD2 complex, residues corresponding to the 5th and 6th positions of the phospho‐FFAT (P210 and P211) maintain broadly similar orientations than their cognate residues in the conventional FFAT (A479 and L480). However, a hydrogen bond cannot be maintained between P210 of the Phospho‐FFAT and the carbonyl of V364 of the MSP domain. In the conventional FFAT, a water‐mediated contact exists between the amide of position 6 (L480 in ORP1) and the carbonyls of P369 and Y372 in the MSP domain. These contacts, again, cannot be maintained with the Phospho‐FFAT where a proline is in position 6 of the motif (Fig 8J). Another difference is that more residues carboxyl‐terminal to the FFAT core sequence can be seen bound to the MSP domain in MOSPD2 than in VAP‐A. This can be explained by specific MOSPD2 features. Firstly, as previously noted, N64 in human VAP‐A is replaced with S377 in MOSPD2. This shorter side chain allows the carbonyl and amide of pS213 one residue after the end of the phosphorylated FFAT motif to form hydrogen bonds with the amide and hydroxyl of S377. The larger side chain of N64 in VAP‐A would prevent these contacts from forming. Secondly, MOSPD2 possesses one extra turn at the N‐term of the α‐helix, which creates a hydrophobic pocket composed of N378, P420, L423, and T424 into which F214, two residues after the FFAT motif of STARD3, can bind (Fig 8I). The lack of this extension in VAP‐A and N378 being replaced by S65 in VAP‐A would prevent a hydrophobic residue from binding here. It should be noted that the contacts between pS213 and F214 are only maintained in one copy of the complex in our structure. In the second copy, F214 makes crystal contacts with a symmetry‐related molecule.
Finally, the structural differences seen between the unbound and complexed forms of VAP‐A are not seen between the unbound and complexed forms of MOSPD2, with F405 (F95 in VAP‐A) maintained in the same position as in the complexed form of VAP‐A. M96 in human VAP‐A is replaced in MOSPD2 with a leucine, L406, in the tp rotamer, similar to the conformation of M96 in complexed VAP‐A (Fig 8H).
We next assessed whether K363 in MOSPD2, which is at the same position as K50 in VAP‐A and K43 in VAP‐B, had a predominant role in binding the Phospho‐FFAT. K363 of MOSPD2 was mutated in leucine (K363L mutant), and the interaction of the mutant protein with the different peptides was assessed. Unlike VAP‐A and VAP‐B, the MOSPD2 K363L mutation only partially affected the binding of both the conventional and the Phospho‐FFAT motif (Fig EV2A).
Altogether these structural and functional data show that MOSPD2 binds conventional and Phospho‐FFAT on a similar interface as VAP‐A, but MOSPD2 has unique and specific features which increase the binding interface, in particular a hydrophobic pocket binding the 9th residue of the FFAT motif.
Discussion
Distinct organelles are physically attached by membrane contact sites (Loewen et al, 2003; Wu et al, 2018). These connections must be reversible, because it is known that organelles associate and dissociate over time (Friedman et al, 2011, 2013; Valm et al, 2017). Such a dynamics implies that there are molecular mechanisms controlling association and dissociation (Alli‐Balogun & Levine, 2019). The ER makes physical connections with nearly all organelles (Balla et al, 2019). To date, no clear molecular mechanism was reported to explain a controlled and/or reversible association, except for the yeast mitochondria‐vacuole contacts which were shown to be disrupted by phosphorylation of Vps39 (Hönscher et al, 2014). In our study, we focused on contacts made by the ER and other organelles. The ER uses VAP‐A/VAP‐B/MOSPD2, three distinct membrane‐bound proteins, as receptors on its surface to bind proteins from other organelles (Murphy & Levine, 2016; Di Mattia et al, 2018). The interaction between VAP‐A/VAP‐B/MOSPD2 and their partners allows the formation of contacts between the ER and the other organelles of the cell, by binding a short linear motif, named FFAT, present in protein partners that are attached or integral to other organelle membranes. This single molecular mechanism explains a large part of the formation of contact sites with the ER. How these are controlled remains unclear.
Many proteins binding to VAP‐A/VAP‐B/MOSPD2 have been identified over the last years (Huttlin et al, 2015; Murphy & Levine, 2016; Di Mattia et al, 2018). A number of these proteins do not have a conventional FFAT motif; however, their sequences suggested the presence of another kind of FFAT motif that could be activated by phosphorylation, that we defined here as a Phospho‐FFAT motif. The presence of a phosphorylatable serine/threonine residue at a key position of the motif supported the idea that phosphorylation governs the interaction with VAP‐A/VAP‐B/MOSPD2. Moreover, the presence of a phosphorylatable residue within the core motif would serve as a reversible switch for forming contact sites. Modifying the FFAT search algorithm into a Phospho‐FFAT search one allowed the identification of many candidate proteins in the human proteome. Interestingly, by crossing the list with proteomics data, we found that Phospho‐FFAT motifs are as equally distributed as conventional FFAT motifs. In addition, both motifs coexist in about half of the candidates. Taking the complex formed by STARD3 and VAP‐A/VAP‐B/MOSPD2 as a model, we functionally characterized Phospho‐FFAT motifs. We showed that in the absence of phosphorylation, the motif is not recognized by VAP‐A/VAP‐B/MOSPD2; phosphorylation on the 4th residue switches the motif on and allows its recognition by VAP‐A/VAP‐B/MOSPD2, triggering the formation of ER‐endosome contact and enabling cholesterol transfer (Fig 9).
Figure 9. Phosphorylation acts as a switch for inter‐organelle contact formation.
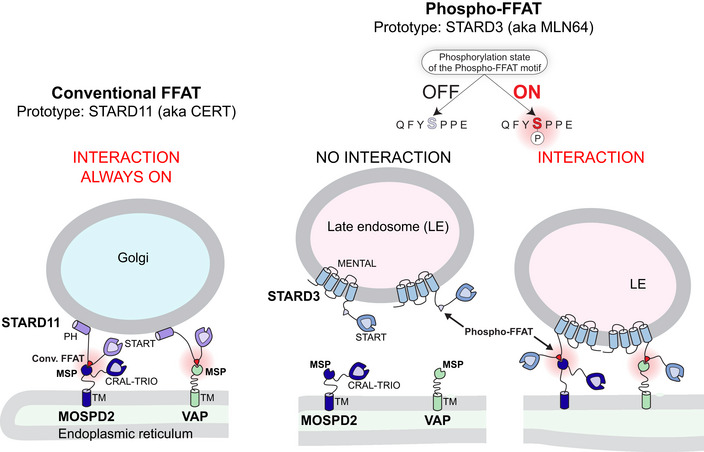
Schematic representation of the two types of FFATs: (left) conventional FFATs (illustrated here with STARD11/CERT) which allow the formation of a stable complex between VAPs/MOSPD2 and thus the formation of MCSs; (right) a novel type of FFATs that we named Phospho‐FFATs (illustrated here with STARD3), which strictly depend on phosphorylation to be active. Thus, phosphorylation acts as a switch mechanism to turn on the interaction between VAPs/MOSPD2 and their partners possessing a Phospho‐FFAT, and thus membrane contact site formation.
Phosphorylation modifies the chemical properties of a protein, modulating its charge (phosphorylated serine contains two negative charges at physiological pH), and sometimes its shape. The FFAT motif was defined as a core sequence flanked by an acidic tract. These adjacent D/E residues bear negative charges that are crucial during the process of recognition of the FFAT sequence by the MSP domain. Nuclear magnetic resonance studies revealed that the interaction of the FFAT with the MSP domain occurs in two steps: First, the acidic residues make non‐specific electrostatic interactions with the electropositive surface of the MSP domain (Furuita et al, 2010). Then, this intermediate complex is stabilized by specific interactions, notably by the phenylalanine in the 2nd position of the FFAT core. Most importantly, higher numbers of negative charges (including phosphorylation) in the acidic tract, by affecting the formation of the intermediate complex, increase the apparent binding affinity of the MSP domain for the FFAT motif (Furuita et al, 2010). This was demonstrated for STARD11 (also known as CERT) which is phosphorylated on S315 located in the acidic tract of its conventional FFAT motif. Phosphorylation of this serine enhances the interaction with VAP and favors its ceramide transport activity in ER‐Golgi contacts (Kumagai et al, 2014). In the case of STARD3, we found that the kinetics of sterol transport in vitro were similar when the START domain of STARD3 was associated with a conventional FFAT motif (Wilhelm et al, 2017), or with its own Phospho‐FFAT motif bearing a single phosphorylation on the fourth core residue S209. We can then speculate that in the case of STARD3, additional phosphorylations in the vicinity of the Phospho‐FFAT could increase the speed of sterol transport; unfortunately, we could not test this as the production in E. coli of the protein with several phosphorylated serines was unsuccessful. However, we did show that additional phosphorylations, such as the one on S213, increase the affinity of VAP for the FFAT motif. Thus, phosphorylations are involved in two different types of regulation: Phosphorylation can act as a switch to turn on and off the interaction, and it can act as a modulator to increase the affinity between VAP‐A/VAP‐B/MOSPD2 and their partners, and possibly fine‐tune the function of the complex. An opposite function of phosphorylation on the FFAT motif can be envisaged; indeed, a phosphomimetic residue replacing the serine at position 5 of the FFAT of AKAP220 protein was shown to inhibit the binding to VAPs, which is consistent with the existence of an inhibitory role of phosphorylation on serines/threonines at position 5 of the motif (Mikitova & Levine, 2012).
Proteins involved in inter‐organelle contacts have targeting determinants for two distinct organelles to bridge them (Alli‐Balogun & Levine, 2019). Thus, the regulation of these targeting determinants can theoretically modulate MCS formation. For instance, OSBP is targeted to the Golgi by the affinity of its PH domain for PI(4)P (phosphatidylinositol 4‐phosphate), and to the ER by its FFAT motif which interacts with VAP (Mesmin et al, 2013). The regulation of PI(4)P levels affects OSBP’s ability to bind to the Golgi (Mesmin et al, 2017), and thus to generate ER‐Golgi contacts. Interestingly, protein phosphorylation can also regulate PI(4)P recognition (Kumagai et al, 2007; Sugiki et al, 2018). In the case of STARD3, only the binding with VAPs or MOSPD2 can be regulated; the other targeting determinant, the anchorage in the endosome membrane, is constitutive. It is intriguing to note that most of the proteins which possess a unique Phospho‐FFAT motif that we tested (namely STARD3, MIGA2, PTPIP51, Kv2.1, and Kv2.2) are anchored to membranes by a transmembrane domain, and thus, regulation must occur on the second determinant of the bridge, i.e., the targeting to the ER, mediated by VAP‐A/VAP‐B/MOSPD2 interaction.
Our search for Phospho‐FFAT motifs was restricted to human proteins, but it can be extended to other species. For instance, yeast cells have two VAPs named Scs2 and Scs22 which were shown to be involved in MCS formation (Murphy & Levine, 2016); it would be interesting to know if Phospho‐FFAT motifs exist and are functional in other species. Besides, it has been shown that viruses and parasites can hijack resources from the host cell by promoting the formation of MCSs. For instance, the obligate intracellular bacterium Chlamydia trachomatis resides in a membrane‐bound compartment which contacts the ER; the pathogen‐containing vacuole bears a Chlamydia protein named IncV which interacts with VAP proteins to promote the formation of contacts with the ER (Stanhope et al, 2017). IncV possesses both conventional and non‐conventional FFAT motifs (threonine in the 4th position) which are equally involved in the interaction with VAP. Norovirus NS2 protein also uses a non‐conventional Phospho‐FFAT motif: NYMTPPE (McCune et al, 2017).
The characterization of the 3D structure of VAP and MOSPD2 bound to conventional and Phospho‐FFATs reveals unique features and specific modes of binding. Regarding the type of FFAT, the main difference lies in the geometry, as phosphoserine is longer than aspartic acid, which modifies several contacts at the FFAT‐MSP interface. Comparing MOSPD2 and VAP‐A, these domains are similar overall: The hydrophobic pocket and the charged residues binding the 2nd (F/Y) and 4th (D/E or pS) residue of the FFAT motif, respectively, are conserved. However, a striking difference lies into their ability to discriminate Phospho‐FFATs with a proline at position 5. However, an even more striking difference is found in MOSPD2 which forms a second hydrophobic pocket accommodating an aromatic residue (F 214) of the Phospho‐FFAT of STARD3. This pocket most probably binds hydrophobic residues present in position +9 of some FFAT motifs and might account for the stronger affinity of FFATs with this feature for MOSPD2 compared to VAP‐A/B.
VAP‐A and VAP‐B are more closely related to each other (82% identity; 93% similarity) than they are to MOSPD2 (~ 30% of identity and ~ 50% of similarity between VAPs and MOSPD2). Our finding that some FFAT‐containing proteins interact with VAP‐A/B but not with MOSPD2 raises the possibility that the repertoires of proteins bound by these receptors are not identical; some FFATs likely bind the three receptors, while others may have a better affinity or a selectivity for one or the other. This is expected because MOSPD2 appeared more recently than VAP during evolution, and is only found in metazoan. Comparative proteomics studies will be required to make an inventory of the partner repertoire of these three proteins.
Overall, this study shows that the formation of MCSs by VAP‐A/VAP‐B/MOSPD2 can be switched on and off by phosphorylation of some of their partners. This mechanism makes contact formation controllable through signaling. Such a regulation by phosphorylation of Phospho‐FFATs could originate through direct signaling in response to extracellular or intracellular signals, or indirectly via transcription. The kinases and phosphatases regulating this process, as yet unknown, will most probably be different depending on their primary target, i.e., the partner of VAP‐A/VAP‐B/MOSPD2. The identification of kinases and phosphatases switching on and off Phospho‐FFATs is the next step to understand how these regulatory processes are integrated in the cell.
Materials and Methods
Cloning and constructs
The GFP‐VAP‐A, GFP‐VAP‐B, GFP‐MOSPD2, Flag‐tagged STARD3, STARD3 FA/YA, and GST‐tagged STARD3 expression vectors were previously described (Alpy et al, 2005, 2013; Di Mattia et al, 2018). STARD3 S209A, STARD3 S209D, STARD3 S209D/P210A, and STARD3 Conv‐FFAT were constructed by site‐directed mutagenesis using the following primers: STARD3 S209A: 5ʹ‐GACAG TTCTA TGCAC CCCCA GAATC CTTTG C‐3ʹ and 5ʹ‐GATTC TGGGG GTGCA TAGAA CTGTC CCTCG G‐3ʹ; STARD3 S209D: 5ʹ‐GACAG TTCTA TGATC CCCCA GAATC CTTTG C‐3ʹ and 5ʹ‐GATTC TGGGG GATCA TAGAA CTGTC CCTCG G‐3ʹ; STARD3 S209D/P210A: 5ʹ‐TTCTA TGATG CCCCA GAATC CTTTG CAGGG TCTGA CAAT‐3ʹ and 5ʹ‐TTCTG GGGCA TCATA GAACT GTCCC TCGGA CAGAG CACC‐3ʹ; STARD3 Conv‐FFAT: 5ʹ‐CCGGT GCTCT GGACG ATGAA GAGTT CTTTG ATGCC TCAGA ATCCT TTGCA GGGTC TGACA ATG‐3ʹ and 5ʹ‐AAAGG ATTCT GAGGC ATCAA AGAAC TCTTC ATCGT CCAGA GCACC GGAGA ACAGC AGGGG TCC‐3ʹ. The cDNA encoding STARD3 insensitive to siRNAs was obtained by gene synthesis (GenScript); silent mutations were introduced in the four sequences targeted by STARD3 siRNAs (SMARTpool ON‐TARGETplus L‐017665‐00; Horizon Discovery).
Plasmids allowing the inducible expression of STARD3 were constructed in the lentiviral vector pLVX‐TRE3G (Clontech) using the following primers: 5ʹ‐TCCGG GCCCG CGGCC GCCAC CATGA GCAAG CTGCC CAGGG AGCTG‐3ʹ and 5ʹ‐CTACC CGGTA GAATT TCACG CCCGG GCCCC CAGCT CGCT‐3ʹ with the SLiCe method (Okegawa & Motohashi, 2015).
Plasmids encoding the MSP domain of VAP‐A (8–212), VAP‐B (1–210), and MOSPD2 (282–490) for expression as C‐terminal His6‐tag proteins in E. coli were previously described (Mesmin et al, 2013; Wilhelm et al, 2017; Di Mattia et al, 2018). VAP‐A K50L, VAP‐B K43L, and MOSPD2 K363L were constructed by site‐directed mutagenesis using the following primers: VAP‐A K50L: 5ʹ‐AGAAA AGTGT GTTTC CTAGT GAAGA CTACA GCACC TCGCC GGTAC TGT‐3ʹ and 5ʹ‐GCGAG GTGCT GTAGT CTTCA CTAGG AAACA CACTT TTCTA TCCGA TGG‐3ʹ; VAP‐B K43L: 5ʹ‐CGAAA TGTGT GTTTT CTAGT GAAGA CTACA GCACC ACGTA GG‐3ʹ and 5ʹ‐TGCTG TAGTC TTCAC TAGAA AACAC ACATT TCGGT CTGTC GG‐3ʹ; MOSPD2 K363L: 5ʹ‐GGAGC TGTTG TTCTC ACCAG AAATG CCACT ATATT TTTAG TTA‐3ʹ and 5ʹ‐TAACT AAAAA TATAG TGGCA TTTCT GGTGA GAACA ACAGC TCC‐3ʹ. For protein crystallization, a plasmid encoding MOSPD2 (315–445) fused to an N‐terminal His6‐tag followed by a thrombin cleavage site was generated.
A plasmid encoding STARD3 (196–445) with a cysteine substitution (L196C) to link the soluble part of STARD3 to MPB‐PE containing liposomes, and fused with an N‐terminal GST tag for purification, was generated. The protein encoded by this construct (without GST) is hereafter referred to as cSTD3. To obtain the pS209 cSTD3 encoding construct, the S209 codon was replaced by an amber codon (TAG) by site‐directed mutagenesis using the following primers 5ʹ‐GGACA GTTCT ATTAG CCCCC AGAAT CCTTT GCAGG G‐3ʹ and 5ʹ‐GGATT CTGGG GGCTA ATAGA ACTGT CCCTC GGACA G‐3ʹ. All constructs were verified by DNA sequencing (Eurofins).
Protein expression and purification
For recombinant MSP domains of VAP‐A, VAP‐B, and MOSPD2 (282–490), proteins were expressed in E. coli BL21 DE3 strain at 20°C for 16 h upon induction with 1 mM IPTG (at an optical density OD600nm = 0.5). Cells were suspended in lysis buffer [50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM imidazole, protease inhibitor tablets (cOmplete, Roche)]. Cells were lysed by a Cell Disruptor TS SERIES (Constant Systems Ltd), and the lysate was first centrifuged at 3,500 g for 15 min, then at 50,000 g for 45 min, and filtered through a 0.22‐µm membrane. Purification was performed on an ÄKTA Start chromatography system (GE Healthcare Life Sciences) using HisTrap HP 1 ml columns. Proteins were eluted with Elution buffer [20 mM sodium phosphate, 250 mM imidazole, pH 7.4], and further purified by gel filtration (HiLoad 16/60 Superdex 200; GE) in GF Buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl). Proteins were concentrated with an Amicon Ultra‐15 10 kDa centrifugal filter unit (Merck). Protein concentration was determined by UV‐spectroscopy.
For the recombinant MSP domain of MOSPD2 [315–445], purification was performed with NTA‐Ni2+ agarose beads (HIS‐Select Nickel Affinity Gel, P6611, Sigma). The protein was first bound onto the column. After one washing step with lysis buffer, the His6‐tag was removed to allow the release of MOSPD2 (315–445) by incubating the beads in digestion buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM imidazole, 25U thrombin, CaCl2 50 µM) during 16 h at 4°C, under constant agitation. After incubation, thrombin was inactivated by adding PMSF (200 mM). The protein was recovered in the supernatant after centrifugation, and the beads were washed three times with lysis buffer. The fractions were pooled. The protein was then purified by gel filtration (in GF buffer containing 1 mM TCEP), concentrated, and quantified as described above.
GST‐pS209 cSTD3 was expressed in the E. coli C321 ΔA strain [gift from George Church; Addgene # 48998 (Lajoie et al, 2013)] transformed with the phosphoserine orthogonal translation system SepOTSλ, a plasmid encoding a phosphoserine‐accepting tRNA, a phosphoserine aminoacyl‐tRNA synthetase to load the phosphoserine onto a UAG‐decoding Phosphoserine‐tRNA, and a modified elongation factor Tu (EF‐Sep) to deliver phosphoserine–tRNA to the ribosome. These materials were gifts from Jesse Rinehart [Addgene # 68292; # 68306 (Pirman et al, 2015)]. Bacteria were cultivated in LB medium containing ampicillin (100 µg/ml), kanamycin (25 µg/ml), 0.08 % glucose, and 2 mM 0‐phospho‐l‐serine (P0878, Sigma), at 30°C for 20 h upon induction with 1 mM IPTG (at an optical density OD600nm = 0.7). All purification steps were conducted in TN1 buffer (50 mM Tris, pH 7.4, 150 mM NaCl) containing 2 mM DTT. TN1 buffer was supplemented with 1 mM PMSF, 10 µM bestatin, 10 µM pepstatin, 1 mM orthovanadate, 50 mM sodium fluoride, and protease inhibitor tablets (cOmplete, Roche) for the first purification steps. Cells were lysed by a Cell Disruptor TS SERIES, and the lysate was centrifuged at 186,000 g for 1 h. Then, the supernatant was applied to Glutathione Sepharose 4B beads (17‐0756; GE Healthcare). After three washing steps with TN1 buffer containing 2 mM DTT, the beads were incubated with thrombin at 4°C for 16 h to cleave the GST fusion, and allow the release of pS209 cSTD3. The protein was recovered in the supernatant after centrifugation, and the beads were washed three times with TN1 buffer. The fractions were pooled and concentrated. Stock concentration was estimated with a BCA assay. The day of the experiment, in order to attach pS209 cSTD3 covalently to MPB‐PE‐containing liposomes, 100 µl from stock protein was applied onto an illustra NAP‐5 column (GE Healthcare) and eluted with freshly degassed TN1 buffer according to manufacturer’s indications to remove DTT. The concentration of the eluted protein was determined by UV‐spectroscopy.
GST‐cSTD3 was expressed in E. coli BL21 at 37°C for 3 h upon induction with 1 mM IPTG (at an optical density OD600nm = 0.6). All purification steps were conducted in TN1 buffer containing 2 mM DTT. TN1 buffer was supplemented with 1 mM PMSF, 10 µM bestatin, 10 µM pepstatin, and protease inhibitor tablets (cOmplete, Roche) for the first purification steps. All the following steps of the purification are the same as GST‐pS209 cSTD3, in particular the application onto illustra NAP‐5 column for the experiments.
Cell culture, transfection, and infection
HeLa cells [American Type Culture Collection (ATCC) CCL‐2] were maintained in DMEM with 5% fetal calf serum (FCS) and 40 µg/ml gentamicin. 293T cells (ATCC CRL‐3216) were maintained in DMEM with 10% FCS, penicillin 100 UI/ml, and streptomycin 100 µg/ml. HCC1954 cells (ATCC CRL‐2338) were maintained in RPMI w/o HEPES with 10% FCS and 40 µg/ml gentamicin.
Cells were transfected using X‐tremeGENE 9 DNA Transfection Reagent (Roche). To generate retroviral particles, pQCXIP vectors were co‐transfected with pCL‐Ampho vector (Imgenex) into 293T retroviral packaging cell line. Retroviral infections were used to generate HeLa/Ctrl, HeLa/GFP‐VAP‐A, HeLa/GFP‐VAP‐B, and HeLa/GFP‐MOSPD2 cell lines. The HeLa/Ctrl cell line was obtained using the empty pQCXIP plasmid. For lentiviral infection, pLVX‐TRE3G vectors encoding STARD3 (wild‐type and mutants) or pLVX‐Tet3G vector encoding the transactivator were co‐transfected with three packaging plasmids pLP1, pLP2, and pLP/VSVG (Invitrogen) into the 293T cell line. Viral particles supplemented with 10 µg/ml polybrene and 20 mM HEPES were then incubated with HeLa cells. Selection was performed using 0.5 µg/ml puromycin or 800 µg/ml G418. siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Control siRNA (D‐001810‐10) and STARD3‐targeting siRNAs (L‐017665‐00) were SMARTpool ON‐TARGETplus obtained from Horizon Discovery.
Antibody production and purification
The anti‐phospho‐STARD3‐pS209 antibody (rabbit polyclonal antibody #3144) was raised against the synthetic peptide CDGQFYpSPPESEA. The peptide was coupled to ovalbumin by its N‐terminal cysteine residue, and injected into rabbit. The immunoreactive serum was purified using a two‐step protocol: First, the serum was loaded on an affinity chromatography column bearing the non‐phosphorylated synthetic peptide (CDGQFYSPPESEA), and the unbound fraction was collected; second, this latter fraction was purified on an affinity chromatography column bearing the phosphorylated synthetic peptide, and the bound fraction was collected. In these experiments, peptides were coupled to SulfoLink Coupling Gel (Thermo Scientific Pierce) using conditions recommended by the manufacturer.
Peptide synthesis
Peptides were synthesized on an Applied Biosystems 433A peptide synthesizer using standard Fmoc chemistry, and purified by reverse phase HPLC using a preparative scale column (Phenomenex: Kinetex EVO C18, 100 A, 5 µM, 250 × 21.2 mm). Molecular weight and purity of the peptides were confirmed by mass spectrometry.
Peptide pull‐down assays
For pull‐down assays of whole cell protein extracts, the affinity resin was prepared by incubating 60 nmol of biotinylated peptide with 15 µl of streptavidin beads (PureProteome Streptavidin Magnetic Beads, Merck) in a total volume of 1 ml pull‐down buffer 1 (PDB1) [50 mM Tris–HCl pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X‐100, protease inhibitor tablet (cOmplete, Roche)] at room temperature for 1 h, under constant agitation. The beads were then washed twice with 1 ml of PDB2 [50 mM Tris–HCl pH 7.4, 500 mM NaCl, 1 mM EDTA, 1% Triton X‐100, protease inhibitor tablets (cOmplete, Roche)], and two times with 1 ml of PDB1. Adherent cells were washed two times with cold PBS (phosphate buffer saline) and lysed in PDB3 [50 mM Tris–HCl pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X‐100, cOmplete protease inhibitor cocktail, protease inhibitor tablets (cOmplete, Roche), PhosSTOP (Roche)]. After a 20 min of incubation on ice, the protein extract was separated from cell debris by centrifugation (10 min; 9,500 g; 4°C). One milligram of proteins was incubated with peptide‐coupled streptavidin beads in a total volume of 1 ml of PDB3 at 4°C for 3 h, under constant agitation. The beads were washed three times with 1 ml of PDB3, and proteins were eluted with Laemmli buffer.
For pull‐down assays of recombinant proteins, the affinity resin was prepared by incubating 20 nmol of biotinylated peptide with 15 µl of streptavidin beads (PureProteome Streptavidin Magnetic Beads, Merck) in 1 ml of PDB4 [50 mM Tris–HCl pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 1 mM DTT, protease inhibitor tablets (cOmplete, Roche)]. The beads were washed three times with 1 ml of PDB4, and a specific binding sites were blocked with BSA (20 µg) at 4°C for 1 h, under constant agitation. The beads were then washed three times with 1 ml of PDB4. Thirty micrograms of recombinant protein with 20 µg of BSA were incubated in a total volume of 1 ml of PDB5 [50 mM Tris–HCl pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.25 mM DTT, protease inhibitor tablets (cOmplete, Roche)] at 4°C for 2 h, under constant agitation. The beads were washed five times with 1 ml of PDB5, and proteins were eluted at room temperature with Laemmli buffer.
GFP‐Trap
GFP‐Trap beads (GFP‐Trap_MA; Chromotek) were washed three times with 1 ml of cold PDB6 buffer [50 mM Tris–HCl pH 7.4, 50 mM NaCl, 1 mM EDTA, 1% Triton X‐100, protease inhibitor tablets (cOmplete, Roche)]. GFP‐VAP‐A (WT or KD/MD mutant), GFP‐VAP‐B, or GFP‐MOSPD2 stably expressing cells were transfected with plasmids expressing wild‐type or mutant STARD3. Two days after transfection, adherent cells were washed two times with cold PBS and lysed in PDB6. The protein extract (500 µg of proteins) was incubated with 20 µl of beads in a total volume of 1 ml at 4°C for 2 h, under constant agitation. The beads were washed three times with 1 ml of PDB6, and proteins were eluted with Laemmli buffer at room temperature.
GST pull‐down
Glutathione Sepharose 4B beads (GE Healthcare) were washed three times with 1 ml of cold PDB7 buffer [50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, protease inhibitor tablets (cOmplete, Roche), PhosSTOP (Roche)]. HeLa cells were transfected with a plasmid expressing the GST‐STARD3 fusion protein. Two days after transfection, adherent cells were washed 2 times with cold TBS1 (248 mM Tris–HCl pH 7.5, 137 mM NaCl, 27 mM KCl) and lysed in PDB7. The protein extract (1.5 mg of proteins) was incubated with 80 µl of beads in a total volume of 1 ml at 4°C for 2 h, under constant agitation. The beads were washed twice with PDB2 containing PhosSTOP, two times with PDB8 [50 mM Tris–HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% Triton X‐100, protease inhibitor tablets (cOmplete, Roche), PhosSTOP (Roche)] and one time with PDB7. Proteins were eluted at room temperature with Laemmli buffer.
STARD3 immunoprecipitation
Fifty microliters of protein A and 50 µl of protein G sepharose (GE Healthcare) was washed three times with 1 ml of cold PDB7 buffer. HeLa or HCC1954 cells were washed two times with cold TBS1 and lysed in PDB7. The protein extract (500 µg) was incubated with 4 µg of rabbit anti‐STARD3 antibody (1611) in a total volume of 1 ml at 4°C for 2 h, under constant agitation. Protein A/G sepharose was added and incubated at 4°C overnight under constant agitation. The beads were washed two times with PDB7, and proteins were eluted at room temperature with Laemmli buffer.
SDS–PAGE, Western blot, CIP treatment, and Coomassie Blue staining
SDS–PAGE and Western blot analysis were performed as previously described (Alpy et al, 2005). For STARD3 protein detection, the samples were not boiled. Note that an additional low molecular weight band is present for GFP‐tagged proteins that are not boiled before loading. The following antibodies were used as follows: rabbit anti‐GFP (1:5,000; GFP‐2A3, Merck), rabbit anti‐FLAG (1:1,000; F7425, Sigma), mouse anti‐VAP‐A (1:1,000; 4C12, Santa Cruz Biotechnology, sc‐293278), rabbit anti‐VAP‐B [1:1,000; kind gift from Dr. L. Dupuis (Kabashi et al, 2013)], mouse anti‐MOSPD2 [1:7; 1MOS‐4E10, (Di Mattia et al, 2018)], mouse anti‐STARD3 [1:1,000; 3G11, (Wilhelm et al, 2017)], rabbit anti‐STARD3 S209 (1:1,000; 3144), mouse anti‐eIF4A3 [1:1,000; 2E5, (Daguenet et al, 2012)], and mouse anti‐actin (1:5,000; ACT‐2D7, Euromedex). For phospho‐specific antibodies, nitrocellulose membranes were blocked and incubated with the primary antibody in TBS2 (20 mM Tris–HCl pH 7.5, 137 mM NaCl) containing 5% BSA and 0.1% Tween 20 (TBSBT). Washes and secondary antibody incubation were performed in TBST (TBS2 buffer containing 0.1% Tween 20).
For calf intestinal alkaline phosphatase (CIP) treatment, the nitrocellulose membrane was blocked in TBS2 buffer containing 5 % BSA and 0.1% Triton X‐100, at room temperature for 45 min. The membrane was then incubated in CIP buffer (50 mM Tris–HCl pH 9, 1 mM MgCl2, 0.1 mM ZnCl2) containing 1 U/ml CIP, at 37°C for 1 h. Primary antibodies were then incubated in TBST at 4°C.
Protein gels were stained with Coomassie blue (PageBlue Protein Staining Solution; Thermo Fisher Scientific) or with fluorescent SYPRO Orange stain (Thermo Fisher Scientific).
Mass spectrometry
Samples were reduced (5 mM TCEP for 30 min at RT) and alkylated (10 mM Iodoacetamide for 30 min at RT in dark). Two types of digestion were performed: (i) digestion with LysC (1:100) during 4 h at 37°C [in Tris 0.1M pH8.5, CaCl2 2 mM] followed by trypsin (1:100) digestion overnight at 37°C. (ii) Digestion with chymotrypsin [in Tris 0.1 M pH 8.5, CaCl2 10 mM] overnight at 25°C. Peptides were then analyzed with a nano‐LC‐MS/MS system (Ultimate nano‐LC and LTQ Orbitrap, Thermo Fisher Scientific). Briefly, peptides were separated on a C18 nano‐column with a 1–30% linear gradient of acetonitrile and analyzed with a TOP20, TOP15, and TOP10 CID or HCD data‐dependent MS method. Peptides were identified with SequestHT algorithm in Proteome Discoverer 2.2 (Thermo Fisher Scientific) using Human SwissProt database (20,368 sequences). Precursor and fragment mass tolerance were set at 7 ppm and 0.6 Da, respectively. Trypsin or chymotrypsin was set as enzyme, and up to two missed cleavages were allowed. Oxidation (M) and phosphorylation (S) were set as variable modifications, and carbamidomethylation (C) as fixed modification. Proteins were identified with a minimum of two unique peptides and were filtered with a 1% FDR (false discovery rate). Probabilities of phosphorylation for all potential phosphorylation sites were determined with PhosphoRS (Taus et al, 2011).
cSTD3 recombinant protein was analyzed by liquid chromatography coupled to a mass spectrometer equipped with a heated electrospray ionization (HESI) probe. HPLC was performed using a Dionex U3000 RSCL Instrument. The injection volume was fixed at 5 µl (Ultimate 3000, Thermo Fisher Scientific). Protein analysis was performed on a 2.1 mm i.d. × 100 mm (3.5 µm, 300 Å) Xbridge Protein BEH C4 column at a flow rate of 250 µl/min. The elution program was based on water (solvent A) and acetonitrile (solvent B) both containing 0.1% formic acid (v/v): 0 min 5% B, 24 min 80% B, 25 min 90% B. The Q‐exactive plus spectrometer completely controlled by the Xcalibur software was operating in electrospray‐positive mode. Typical ESI conditions were as follows: electrospray voltage 4 kV; capillary temperature 320°C, probe temperature 325°C, sheath gas flow 30U and auxiliary gas 10U. The MS scan was acquired in the 500–1,800 m/z range with the resolution set to 140,000. Data analysis was performed with BioPharma Finder, and intact protein spectra were automatically deconvoluted with ReSpect (precursor mass between 23,000 and 33,000 Da and 20 ppm mass tolerance, charge state range between 15 and 45).
Surface plasmon resonance
SPR data were collected on a Biacore T200 instrument (GE Healthcare, Uppsala, Sweden) at 25°C with autosampler rack base cooled at 15°C. Briefly, a CM5 sensor surface was first conditioned with 10 mM HCl, 50 mM NaOH, 0.1% SDS, and 1 M NaCl pulses and extensively washed before attaching reasonable amount of streptavidin (a few hundred RU) using the standard EDC/NHS protocol provided by the manufacturer, followed by immobilization of biotinylated peptides. In order to minimize SPR artifact effects, the levels of immobilized peptides (50 RU) were kept low by injecting a highly diluted peptide solution (1–10 ng/ml) at a high flow rate (90 µl/min). The running buffer for kinetic measurements was 50 mM Tris, 75 mM NaCl, pH 7.0 (filtered through a 0.22 µm membrane), supplemented with 0.005% (v/v) surfactant P20. The control peptide was immobilized on one flow cell of every chip in order to serve as a control for non‐specific binding of the analyte to the matrix and for monitoring changes in solution refractive index. Analytes were simultaneously injected over the four flow cells at 8–12 different monomer concentrations ranging between 0 and 20 µM. At least three concentrations have been duplicated in non‐consecutive cycles to check for signal reproducibility. Injection and post‐injection times were 120 and 180 s, respectively. The flow rate was 50 µl/min. Data were initially processed using the BiaEvaluation 3.2 software (GE Healthcare, Uppsala, Sweden) using “double referencing” (Fournane et al, 2011) in which sensorgrams were corrected for both buffer effects and bulk refractive index changes. The steady‐state binding signal (R eq) was derived by averaging the signals in a 5‐s window at equilibrium. Subsequently, steady‐state analysis using in‐house Python scripts was performed by fitting the average signal R eq as a function of total analyte concentration, assuming a simple 1:1 interaction binding isotherm model. Note that the analyte concentration was considered according to dimers for VAP‐A and VAP‐B, and monomers for MOSPD2. The quality of the fit was assessed by two criteria: (i) the match of the fitted maximum capacity of the surface (R max) with the expected maximum capacity (R theo) inferred from the immobilized level of biotinylated peptide and (ii) the chi‐square parameter which is a statistical measure of how closely the model fits the experimental data. The square root of this parameter should always be below 10% of the globally fitted R max values (BiaEvaluation software user manual, 2005). The values and uncertainties of the R max and KD fitted parameters were estimated with a Monte Carlo approach by reproducing the fit using 1,000 datasets in which noise fluctuations were introduced, and then calculating the mean and the standard deviation of the obtained parameters, respectively.
Lipids
DOPC (1.2‐dioleoyl‐sn‐glycero‐3‐phosphocholine); DNS‐PE (1.2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(5‐dimethylamino‐1‐naphthalenesulfonyl)); NBD‐PE (1.2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(7‐nitro‐2‐1.3‐benzoxadiazol‐4‐yl)); DOGS‐NTA‐Ni2+ (1.2‐dioleoyl‐sn‐glycero‐3‐[(N‐(5‐amino‐1‐carboxypentyl) iminodiacetic acid) succinyl]); 18:1/18:1 MPB‐PE (1.2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[4‐(p‐maleimidophenyl) butyramide]); 18:1/18:1 NBD‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(7‐nitro‐2‐1,3‐benzoxadiazol‐4‐yl)); and 18:1/18:1 Liss Rhod‐PE (1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(lissamine rhodamine B sulfonyl)) were purchased from Avanti Polar Lipids. Dehydroergosterol (DHE) was from Sigma‐Aldrich. The concentration of DHE in stock solution in methanol was determined by UV‐spectroscopy using an extinction coefficient of 13,000/M/cm.
Liposome preparation
Lipids stored in stock solutions in CHCl3 or methanol were mixed at the desired molar ratio. The solvent was removed in a rotary evaporator under vacuum. DOGS‐NTA‐Ni2+ and MPB‐PE lipid films were pre‐warmed to 33°C for 5 min prior to drying. The films were hydrated in TN2 buffer (50 mM Tris pH 7.4, 120 mM NaCl) to obtain a suspension of multilamellar liposomes. The suspension was extruded through a polycarbonate filter of 0.2 µm pore size using a mini‐extruder (Avanti Polar Lipids).
Liposome flotation assay
Each protein (cSTD3 and pS209 cSTD3 at 1.5 µM) was incubated with NBD‐PE containing liposomes (750 µM total lipids) in 150 µl of TN2 buffer at room temperature for 10 min under agitation. The suspension was adjusted to 28 % (w/w) sucrose by mixing 100 µl of a 60% (w/w) sucrose solution in TN2 buffer and overlaid with 200 µl of TN2 buffer containing 24% (w/w) sucrose and 50 µl sucrose‐free TN2 buffer. The sample was centrifuged at 240,000 × g in swing rotor (TLS 55 Beckmann) for 1 h. The bottom (250 µl), middle (150 µl), and top (100 µl) fractions were collected. The bottom and top fractions were analyzed by SDS–PAGE using SYPRO Orange staining and a FUSION FX fluorescence imaging system.
Dynamic light scattering measurements of liposome aggregation
The experiments were performed at 25°C in a Dynapro apparatus (Protein Solutions). LA liposomes (DOPC 97 mol% and MPB‐PE 3 mol%, 50 µM total lipids) in 20 µl of freshly degassed TN3 buffer (50 mM Tris–HCl pH 8, 75 mM NaCl) were added to the quartz cell. A first set of about 12 autocorrelation curves was acquired to measure the size distribution of initial liposome suspension. Then, cSTD3 or pS209 cSTD3 (380 nM final concentration) was added manually and mixed thoroughly. After a 5 min of incubation, LB liposomes (DOPC 90 mol% and DOGS‐NTA‐Ni2+ 10 mol%, 50 µM total lipids) were added followed by the addition of VAP‐AHis6 (700 nM). For all the experiments, the kinetics of aggregation was measured by acquiring one autocorrelation curve every 10 s. At the end of the experiment, a set of 12 autocorrelation functions was acquired. The data were analyzed using two different algorithms provided by the Dynamics v6.1 software (Protein Solutions). During the kinetics, the autocorrelation functions were fitted assuming that the size distribution is a simple Gaussian function. This mode, referred as the monomodal or cumulant algorithm, gives a mean hydrodynamic radius, RH, and the width (or polydispersity). The polydispersity is represented in the kinetics measurements by the shaded area and can reach very large values because of the simultaneous presence of free liposomes and of liposome aggregates of various size. Before and after the aggregation process, the autocorrelation functions were fitted using a more refined algorithm, referred as a regularization algorithm. This algorithm is able to resolve several populations of different sizes, such as free liposomes and liposome aggregates.
Fusion assay
Experiments were performed in a Shimadzu RF 5301‐PC fluorimeter equipped with a cylindrical quartz cuvette. A suspension (570 µl) of LA liposomes (95 mol% DOPC, 1 mol% NBD‐PE, 1 mol% Rhod‐PE, 3 mol% MPB‐PE, 62.5 µM total lipids final concentration) was incubated with 475 nM pS209 cSTD3 at 37°C under constant stirring in buffer. After 5 min, 30 µl of a suspension of LB liposomes (90 mol% DOPC, 10 mol% DOGS‐NTA‐Ni2+, 62.5 µM total lipids final concentration), pre‐incubated with VAP‐AHis6 (1 µM final concentration), was added. Fusion was measured by recording the NBD‐PE signal at 530 nm (bandwidth 5 nm) upon excitation at 450 nm (bandwidth 5 nm). The percentage of fusion is equal to 100 × ((F − F 0)/(F max − F 0)) where F 0 is the signal measured before the addition of LB liposomes decorated with VAP‐AHis6, and F max is the signal measured after adding Triton X‐100 (1% v/v final concentration). Liposomes and proteins are injected from stock solutions with Hamilton syringes through a guide in the cover of the fluorimeter.
DHE transport assay
Experiments were carried out in a Shimadzu RF 5301‐PC fluorimeter equipped with a cylindrical quartz cuvette. A suspension (570 µl) of LA liposomes (62.5 µM total lipids final concentration) made of DOPC and containing 3 mol% MPB‐PE was incubated with 475 nM cSTD3 or pS209 cSTD3 at 37°C under constant stirring in TN3 buffer. After 5 min, 30 µl of a suspension of LB liposomes (77.5 mol% DOPC, 10 mol% DHE, 2.5 mol% DNS‐PE, 10 mol% DOGS‐NTA‐Ni2+, 62.5 µM total lipids final concentration), pre‐incubated or not with VAP‐AHis6 or VAP‐A (KD/MD)His6 (1 µM final concentration) was added. Lipid transport was measured by recording the DNS‐PE signal at 525 nm (bandwidth 10 nm) upon DHE excitation at 310 nm (bandwidth 1.5 nm). The quantity of DHE transported from LB to LA membrane is expressed in terms of mol% DHE in LB liposomes. It is equal to 10 × ((F − F 0)/(F max − F 0)) where F max is the signal measured in the absence of pS209 cSTD3 upon the addition of LB liposomes and F 0 is the signal measured upon total DHE extraction by 10 mM methyl‐β‐cyclodextrin (Sigma). Liposomes and proteins are injected from stock solutions with Hamilton syringes through a guide in the cover of the fluorimeter.
Immunofluorescence
Cells were grown on glass coverslips, fixed in 4% paraformaldehyde in PBS, and permeabilized with 0.1% Triton X‐100 in PBS. After blocking with 1% bovine serum albumin in PBS (PBS‐BSA), cells were incubated overnight at 4°C with the primary antibody in PBS‐BSA (rabbit anti‐Flag; 1:1,000; F7425, Sigma). Cells were washed twice in PBS and incubated for 30 min with AlexaFluor 555 donkey anti‐rabbit secondary antibodies (Thermo Fisher Scientific). After two washes with PBS, the slides were mounted in ProLong Gold (Invitrogen). Observations were made with a confocal microscope (Leica TCS SP5 inverted, 63×, NA 1.4).
For filipin staining, cells transduced with pLVX‐TRE3G (encoding STARD3 wild‐type and mutants) and pLVX‐Tet3G vectors were treated for 48 h with 100 ng/ml doxycycline. Intracellular cholesterol labeling using filipin was performed as previously described (Wilhelm et al, 2017, 2019). The Fiji software was used to quantify filipin fluorescence intensity (http://fiji.sc/). The macro code is deposited in GitHub: https://github.com/fabienalpy/Fiji_Filipin_Staining_Quantification_Endosome_Mask). Cell contours were manually segmented, and Lamp1 (H4A3; 1:50; Developmental Studies Hybridoma Bank) signal was used to build a segmentation mask corresponding to late endosomes. The mask was then applied to the filipin image, and the mean filipin intensity in endosomes for each individual cell was measured (Appendix Fig S4).
Colocalization analysis
Colocalization was visualized using the colocalization highlighter plug‐in for ImageJ. Pearson correlation coefficient was determined using the Colocalization Threshold plug‐in in Fiji software. The auto‐thresholding was performed using the Costes method (Costes et al, 2004).
Crystallization
The crystallization experiments were carried out by the sitting drop vapor diffusion method at 293 K using a Mosquito Crystal nanolitre dispensing robot (TTP Labtech). A mixture of protein solution (in GF buffer) and reservoir solution was equilibrated against 50 µl of reservoir solution. Several commercially available screens were used, including the JCSG + suite, the Classics suite, the PACT suite, the PEGs suite (Qiagen), Morpheus, BCS (Molecular Dimensions), Wizard Classic 1 & 2 (Rigaku), and the TOP96 (in‐house) (Fazio et al, 2014).
Crystallization of VAP‐A (8–212) in complex with the phosphorylated FFAT of STARD3 (GALpSEGQFYpSPPEpSFAG): The MSP domain of VAP‐A (30 mg/ml) was mixed with the peptide (1:2 molar ratio); 0.2 µl of this solution was mixed with 0.2 µl of reservoir solution. One crystal appeared after 10 weeks in condition 18 of the PEGS suite (25% PEG 2000 MME, 0.1 M HEPES pH 7.5), reaching full size after a further 3 weeks. The crystal was flash‐frozen by direct immersion in liquid nitrogen without further cryo‐protection.
Crystallization of MOSPD2 (282–490) in complex with the conventional FFAT of ORP1 (Biotin‐GAMRSILSEDEFYDALSDSES): The MSP domain of MOSPD2 (25 mg/ml) was mixed with the peptide (1:1.2 molar ratio); 0.15 µl of this solution was mixed with 0.15 µl of reservoir solution. Plates were checked for crystal appearance over 6 months without any result. Twelve months later, a final check was made before discarding the plates, and crystals were found in 10 drops, all containing a PEG/salt mixture. The best crystal was obtained in condition 18 of the TOP96 screen (20% PEG 3350, 0.2 M sodium sulfate). This crystal was transferred to a solution containing 25% PEG 3350, 0.2 M sodium sulfate and 10% glycerol, and flash‐frozen in liquid nitrogen.
Crystallization of MOSPD2 (282–490) in unbound form: The MSP domain of MOSPD2 was prepared at 19 mg/ml and mixed with the Proti‐Ace Kit (Hampton Research) for in situ proteolysis experiments. Alpha‐chymotrypsin, trypsin, elastase, papain, subtilisin, and endoproteinase Glu‐C were prepared as described in the protocol and mixed all together with the protein to create a proteases:sample ratio of 1:190 (w/w). A mix of 0.1 µl protein solution and 0.1 µl reservoir solution was used. One crystal appeared after only 12 h in condition 30 of the JCSG + suite (40% PEG 300, 0.1 M Sodium Phosphate Citrate pH 4.2), reaching full size after 8 days. The crystal was flash‐frozen by direct immersion in liquid nitrogen without further cryo‐protection.
Crystallization of MOSPD2 (315–445) in complex with the phosphorylated FFAT of STARD3 (CLFSGALpSEGQFYpSPPEpSFAG): The MSP domain of MOSPD2 (21 mg/ml) was mixed with the peptide (1:2 molar ratio); 0.2 µl of this solution was mixed with 0.1 µl of reservoir solution. Crystals appeared after 3 weeks in condition 53 of the BCS screen (7.5% PEG 2000, 7.5% PEG 3350, 7.5% PEG 4000, 7.5% PEG 5000 MME, 0.05 M magnesium sulfate, 0.1 M HEPES pH 7.5), reaching full size after a further 2 weeks. Crystals were transferred to the same solution supplemented with 15% glycerol, and flash cooled in liquid nitrogen.
Data collection and structure determination
All data were collected from crystals at 100 K, as detailed below, and processed, integrated, and scaled using XDS (Kabsch, 2010). All molecular replacement was performed using PHASER (McCoy et al, 2007) in the PHENIX (Liebschner et al, 2019) suite. Refinement of the structures was performed using PHENIX and BUSTER (Smart et al, 2012; Bricogne et al, 2019) with iterative model building performed in COOT (Emsley et al, 2010). The quality of the final refined models was assessed using MOLPROBITY (Williams et al, 2018) and PROCHECK (Laskowski et al, 1993). Data collection and refinement statistics are given in Table EV3. Structural figures were prepared using PyMOL (www.pymol.org). Surface coloring was performed with the YRB scheme with all carbon atoms not bound to nitrogen and oxygen atoms in yellow, nitrogen atoms of K and R in blue, oxygen of D and E in red, and all the remaining atoms in white (Hagemans et al, 2015).
For the complex of VAP‐A (8–212: MSP and coiled‐coil domains) with the phosphorylated FFAT motif of STARD3 (200–216), data were collected from two fragments of the same crystal on a PILATUS 6M detector (Dectris) at the ID23‐1 beamline of the ESRF and using the MxCuBE software (Gabadinho et al, 2010). After data processing, the two datasets were scaled together using XSCALE, and anisotropic truncation and correction were performed on the merged dataset using the STARANISO server (Tickle et al, 2018). The crystal diffracted anisotropically to 1.85 Å (3.0 Å in the worst direction) and belonged to the primitive triclinic space group P1, with unit cell dimensions a = 39.1 Å, b = 43.8 Å, c = 83.7 Å, α = 89.46°, β = 92.91°, γ = 105.13°. The structure was solved by molecular a monomer of the MSP domain of rat VAP‐A (Kaiser et al, 2005) (PDB ID: 1Z9O) as a search model. The asymmetric unit contains four copies of the MSP domain (~residues 9–135) and two copies of the peptide, with a corresponding Matthews’ coefficient (Matthews, 1968) of 2.21 Å3/Da and a solvent content of approximatively 44%. The asymmetric unit is too small to contain four copies of the construct crystallized, indicating that the coiled‐coil domain was cleaved in the drop, and is not present as a disordered domain in the crystal.
For the complex of MOSPD2 (282–490) with the conventional FFAT of ORP1 (469–485), data were collected at on an EIGER X 9 M detector (Dectris) at the Proxima 2A beamline of Synchrotron SOLEIL. The crystal diffracted to 2.3 Å and belonged to a primitive hexagonal space group (P6122 or P6522) with unit cell dimensions a = b = 126.9 Å, c = 184.4 Å. The first model from the NMR structure of the MSP domain of MOSPD2 (PDB ID 1WIC) was extracted and truncated to the rigid core, and used as a search model in PHASER with a solution found in P6122. The asymmetric unit contains six copies of the MSP domain (~residues 315–447) and five copies of the peptide, with a corresponding Matthews’ coefficient of 2.16 Å3/Da and a solvent content approximatively 43%. As with VAP‐A, the asymmetric unit is too small to contain six copies of the construct crystallized, implying that the N‐ and C‐terminal extensions had been cleaved in the drop during the long crystallization time.
For unbound MOSPD2 [282‐490], data were collected on our in‐house diffractometer. The diffractometer comprises an FR‐X rotating anode X‐ray generator (Rigaku) fitted with Osmic VariMax HF Arc) Sec confocal optics and an Eiger R 4M detector (Dectris). The crystal diffracted to 1.5 Å and belonged to the primitive orthorhombic space group P212121 with a = 28.1 Å, b = 51.2 Å, c = 78.8 Å. The structure was solved by molecular replacement using a monomer of our MOSPD2/ORP1 complex structure. The asymmetric unit contains one copy of the MSP domain (residues 315‐445) with a corresponding Matthews’ coefficient of 1.97 Å3/Da and a solvent content of approximatively 38%.
For the complex of MOSPD2 (315–445) with the phosphorylated FFAT motif of STARD3 (196–216), data were collected on an EIGER X 9 M detector (Dectris) at the Proxima 2A beamline of Synchrotron SOLEIL. The crystal diffracted to 2.35 Å and belonged to a primitive tetragonal space group (P41212 or P43212) with unit cell dimensions a = b = 87.9 Å, c = 95.1 Å. The structure was solved by molecular replacement in PHASER using our MOSPD2 MSP domain unbound structure as a search model with a solution found in P41212. The asymmetric unit contains two copies of the MOSPD2 MSP domain/STARD3 FFAT motif complex with a corresponding Matthews’ coefficient of 1.97 Å3/Da and a solvent content of approximatively 38%.
In silico identification of Phospho‐FFATs
The algorithm used for Phospho‐FFAT identification is derived from the position weight matrix shown in Table EV4. The scoring system is based on 19 continuous residues: the seven residues forming the core, six residues upstream, and it is extended from the matrix previously described to include the six residues downstream (Slee & Levine, 2019). These residues were allocated a score in the position weight matrix. All residues in all human protein sequences obtained from UniProt were scored by scripts enacted in Python. Thus, for proteins with n residues, n scores were calculated; the two best scores (named SCORE1 and SCORE2) are shown Tables EV1 and EV2. Venn diagrams and sequence logos were constructed using InteractiVenn and Weblogo, respectively (Crooks et al, 2004; Heberle et al, 2015).
Statistical analyses
Statistical analyses were performed using the Mann–Whitney, or the Kruskal–Wallis non‐parametric tests (Prism, GraphPad). In the latter case, all conditions were compared with the Dunn’s multiple comparison test. P‐values < 0.05, < 0.01, and < 0.001 are identified with 1, 2, and 3 asterisks, respectively. ns: P ≥ 0.05. The number of replicates (n) used for calculating statistics is specified in the figure legends. For Pearson correlation coefficient calculations, individual cells from at least three independent experiments were used for calculating statistics.
Author contributions
FA, CT, and GD conceived and supervised the project. TDM and AM performed and analyzed most of the biochemistry and cell biology experiments, with the help of CW, CT, and FA. SI and GD performed and analyzed all in vitro membrane tethering/sterol transport experiments. LV performed in vivo cholesterol labeling experiments with the help of TDM and AGM. YN conceived, performed, and analyzed the SPR experiments. PE synthesized the peptides. FR performed and analyzed mass spectrometry experiments. JS and TPL conceived and coded the algorithm identifying Phospho‐FFATs, with the help of AM and FA. PP‐C crystallized the different complexes. AGM collected the X‐ray diffraction data and solved the structures, with the help of JC. FA, CT, GD, AGM, and YN wrote the manuscript and prepared the figures, with the help of TDM and AM. All authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Expanded View and Appendix
DataAppendixFigs
DataAppendixFigs
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We thank the members of the Molecular and Cellular Biology of Breast Cancer team (IGBMC) for helpful advice and discussions. We thank Julie Milanini (IPMC) and the IGBMC cell culture facility (Betty Heller), imaging center (Bertrand Vernay, Elvire Guiot, and Erwan Grandgirard), polyclonal and monoclonal antibody facility (Gilles Duval and Mustapha Oulad‐Abdelghani), proteomics platform (Luc Negroni and Bastien Morlet), and structural biology and genomics platform (Catherine Birck) for their excellent technical assistance. We thank Delphine Debayle for helping us to determine the molecular weight of cSTD3 constructs by mass spectrometry. We wish to thank Luc Dupuis (INSERM U118, Strasbourg, FR) for providing the anti‐VAP‐B antibody. We acknowledge the European Synchrotron Radiation Facility (proposal number MX‐1636) and SOLEIL (proposal number 20170871) for provision of synchrotron radiation facilities, and we would like to thank Andrew McCarthy for assistance in using beamline ID23‐1, and William Shepard and Martin Savko for assistance in using beamline Proxima 2A. T.D.M. received a fellowship from the Fondation pour la Recherche Médicale (https://www.frm.org/). A.M. and L.V. received an allocation from the Ministère de l’Enseignement Supérieur et de la Recherche (France; http://www.enseignementsup-recherche.gouv.fr/). This work was supported by grants from the Agence Nationale de la Recherche ANR (grant ANR‐19‐CE44‐0003; https://anr.fr/), and from the Ligue Contre le Cancer (Conférence de Coordination Interrégionale du Grand Est; https://www.ligue-cancer.net), SEVE Sein et Vie, the Institut National Du Cancer INCA (INCA_9269; www.e-cancer.fr), by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR‐10‐INSB‐05‐01 and INSTRUCT‐ERIC. We also acknowledge funds from the Institut National de Santé et de Recherche Médicale (http://www.inserm.fr/), the Centre National de la Recherche Scientifique (http://www.cnrs.fr/), the Université de Strasbourg (http://www.unistra.fr), and the grant ANR‐10‐LABX‐0030‐INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR‐10‐IDEX‐0002‐02.
The EMBO Journal (2020) 39: e104369.
Contributor Information
Catherine Tomasetto, Email: Catherine-Laure.Tomasetto@igbmc.fr.
Fabien Alpy, Email: Fabien.Alpy@igbmc.fr.
Data availability
The coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 6TQR (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQR; VAP‐A/STARD3 complex), 6TQS (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQS; MOSPD2/ORP1 complex), 6TQT (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQT; MOSPD2 unbound), and 6TQU (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQU; MOSPD2/STARD3 complex).
References
- Alli‐Balogun GO, Levine TP (2019) Regulation of targeting determinants in interorganelle communication. Curr Opin Cell Biol 57: 106–114 [DOI] [PubMed] [Google Scholar]
- Alpy F, Wendling C, Rio M‐C, Tomasetto C (2002) MENTHO, a MLN64 homologue devoid of the START domain. J Biol Chem 277: 50780–50787 [DOI] [PubMed] [Google Scholar]
- Alpy F, Latchumanan VK, Kedinger V, Janoshazi A, Thiele C, Wendling C, Rio M‐C, Tomasetto C (2005) Functional characterization of the MENTAL domain. J Biol Chem 280: 17945–17952 [DOI] [PubMed] [Google Scholar]
- Alpy F, Tomasetto C (2005) Give lipids a START: the StAR‐related lipid transfer (START) domain in mammals. J Cell Sci 118: 2791–2801 [DOI] [PubMed] [Google Scholar]
- Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C, Spiegelhalter C, Kessler P, Mathelin C, Rio M‐C et al (2013) STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J Cell Sci 126: 5500–5512 [DOI] [PubMed] [Google Scholar]
- Amarilio R, Ramachandran S, Sabanay H, Lev S (2005) Differential regulation of endoplasmic reticulum structure through VAP‐Nir protein interaction. J Biol Chem 280: 5934–5944 [DOI] [PubMed] [Google Scholar]
- Balla T, Kim YJ, Alvarez‐Prats A, Pemberton J (2019) Lipid dynamics at contact sites between the endoplasmic reticulum and other organelles. Annu Rev Cell Dev Biol 35: 85–109 [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Rojas R (2006) Retrograde transport from endosomes to the trans‐Golgi network. Nat Rev Mol Cell Biol 7: 568–579 [DOI] [PubMed] [Google Scholar]
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart O, Vonrhein C et al (2019) BUSTER version 2.10.2, Cambridge, UK: Global Phasing Ltd; [Google Scholar]
- Costello JL, Castro IG, Hacker C, Schrader TA, Metz J, Zeuschner D, Azadi AS, Godinho LF, Costina V, Findeisen P et al (2017a) ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J Cell Biol 216: 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JL, Castro IG, Schrader TA, Islinger M, Schrader M (2017b) Peroxisomal ACBD4 interacts with VAPB and promotes ER‐peroxisome associations. Cell Cycle 16: 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S (2004) Automatic and quantitative measurement of protein‐protein colocalization in live cells. Biophys J 86: 3993–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J‐M, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daguenet E, Baguet A, Degot S, Schmidt U, Alpy F, Wendling C, Spiegelhalter C, Kessler P, Rio M‐C, Le Hir H et al (2012) Perispeckles are major assembly sites for the exon junction core complex. Mol Biol Cell 23: 1765–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21: 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mattia T, Wilhelm LP, Ikhlef S, Wendling C, Spehner D, Nominé Y, Giordano F, Mathelin C, Drin G, Tomasetto C et al (2018) Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep 19: e45453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P (2016) Endosome‐ER contacts control actin nucleation and retromer function through VAP‐dependent regulation of PI4P. Cell 166: 408–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio VJ, Peat TS, Newman J (2014) A drunken search in crystallization space. Acta Crystallogr Sect F Struct Biol Commun 70: 1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournane S, Charbonnier S, Chapelle A, Kieffer B, Orfanoudakis G, Travé G, Masson M, Nominé Y (2011) Surface plasmon resonance analysis of the binding of high‐risk mucosal HPV E6 oncoproteins to the PDZ1 domain of the tight junction protein MAGI‐1. J Mol Recognit 24: 511–523 [DOI] [PubMed] [Google Scholar]
- Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D, Tamkun MM (2015) Induction of stable ER–plasma‐membrane junctions by Kv2.1 potassium channels. J Cell Sci 128: 2096–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyre CAC, Rauher PC, Ejsing CS, Klemm RW. (2019) MIGA2 links mitochondria, the ER, and lipid droplets and promotes de novo lipogenesis in adipocytes. Mol Cell 76: 811–825.e14 [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, DiBenedetto JR, West M, Rowland AA, Voeltz GK (2013) Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24: 1030–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuita K, Jee J, Fukada H, Mishima M, Kojima C (2010) Electrostatic interaction between oxysterol‐binding protein and VAMP‐associated protein A revealed by NMR and mutagenesis studies. J Biol Chem 285: 12961–12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabadinho J, Beteva A, Guijarro M, Rey‐Bakaikoa V, Spruce D, Bowler MW, Brockhauser S, Flot D, Gordon EJ, Hall DR et al (2010) MxCuBE: a synchrotron beamline control environment customized for macromolecular crystallography experiments. J Synchrotron Radiat 17: 700–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go et al (in preparation) A proximity biotinylation map of a human cell. Httpscell‐Maporg
- Hagemans D, van Belzen IAEM, Morán Luengo T, Rüdiger SGD (2015) A script to highlight hydrophobicity and charge on protein surfaces. Front Mol Biosci 2: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan J‐L, Mizushima N (2008) FIP200, a ULK‐interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R (2015) InteractiVenn: a web‐based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16: 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hönscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 30: 86–94 [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K et al (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162: 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H et al (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545: 505–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, Leek AN, Solé L, Maverick EE, Levine TP, Tamkun MM (2018) Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci USA 115: E7331–E7340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, El Oussini H, Bercier V, Gros‐Louis F, Valdmanis PN, McDearmid J, Mejier IA, Dion PA, Dupre N, Hollinger D et al (2013) Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum Mol Genet 22: 2350–2360 [DOI] [PubMed] [Google Scholar]
- Kaiser SE, Brickner JH, Reilein AR, Fenn TD, Walter P, Brunger AT (2005) Structural basis of FFAT motif‐mediated ER targeting. Structure 13: 1035–1045 [DOI] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K (2015) Assessing and maximizing data quality in macromolecular crystallography. Curr Opin Struct Biol 34: 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M, Kumagai K, Nishijima M, Hanada K (2006) Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP‐associated protein‐interacting FFAT motif of CERT. J Biol Chem 281: 30279–30288 [DOI] [PubMed] [Google Scholar]
- Kirmiz M, Vierra NC, Palacio S, Trimmer JS (2018) Identification of VAPA and VAPB as Kv2 channel‐interacting proteins defining endoplasmic reticulum‐plasma membrane junctions in mammalian brain neurons. J Neurosci 38: 7562–7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai K, Kawano M, Shinkai‐Ouchi F, Nishijima M, Hanada K (2007) Interorganelle trafficking of ceramide is regulated by phosphorylation‐dependent cooperativity between the PH and START domains of CERT. J Biol Chem 282: 17758–17766 [DOI] [PubMed] [Google Scholar]
- Kumagai K, Kawano‐Kawada M, Hanada K (2014) Phosphoregulation of the ceramide transport protein CERT at serine 315 in the interaction with VAMP‐associated protein (VAP) for inter‐organelle trafficking of ceramide in mammalian cells. J Biol Chem 289: 10748–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie MJ, Rovner AJ, Goodman DB, Aerni H‐R, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA et al (2013) Genomically recoded organisms expand biological functions. Science 342: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM (1993) Main‐chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067 [DOI] [PubMed] [Google Scholar]
- Levine T, Loewen C (2006) Inter‐organelle membrane contact sites: through a glass, darkly. Curr Opin Cell Biol 18: 371–378 [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung L‐W, Jain S, McCoy AJ et al (2019) Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr Sect Struct Biol 75: 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, Trimmer JS (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25: 385–397 [DOI] [PubMed] [Google Scholar]
- Loewen CJR, Roy A, Levine TP (2003) A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J 22: 2025–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord SJ, Velle KB, Mullins RD, Fritz‐Laylin LK. (2020) SuperPlots: communicating reproducibility and variability in cell biology. J Cell Biol 219: e202001064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv BF, Yu CF, Chen YY, Lu Y, Guo JH, Song QS, Ma DL, Shi TP, Wang L (2006) Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a novel mitochondria protein with an N‐terminal mitochondrial targeting sequence and induces apoptosis. Apoptosis 11: 1489–1501 [DOI] [PubMed] [Google Scholar]
- Matthews BW (1968) Solvent content of protein crystals. J Mol Biol 33: 491–497 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune BT, Tang W, Lu J, Eaglesham JB, Thorne L, Mayer AE, Condiff E, Nice TJ, Goodfellow I, Krezel AM et al (2017) Noroviruses co‐opt the function of host proteins VAPA and VAPB for replication via a phenylalanine–phenylalanine‐acidic‐tract‐motif mimic in nonstructural viral protein NS1/2. MBio 8: e00668‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas‐Gervais S, Drin G, Antonny B (2013) A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 155: 830–843 [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Polidori J, Jamecna D, Lacas‐Gervais S, Antonny B (2017) Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J 36: 3156–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikitova V, Levine TP (2012) Analysis of the key elements of FFAT‐like motifs identifies new proteins that potentially bind VAP on the ER, including two AKAPs and FAPP2. PLoS One 7: e30455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SE, Levine TP (2016) VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT‐like motifs in the VAPome. Biochim Biophys Acta 1861: 952–961 [DOI] [PubMed] [Google Scholar]
- Okegawa Y, Motohashi K (2015) A simple and ultra‐low cost homemade seamless ligation cloning extract (SLiCE) as an alternative to a commercially available seamless DNA cloning kit. Biochem Biophys Rep 4: 148–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes‐Carter F, Campbell NH, Chavali G, Chen C, del‐Toro N et al (2014) The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42: D358–D363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oughtred R, Stark C, Breitkreutz B‐J, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R et al (2019) The BioGRID interaction database: 2019 update. Nucleic Acids Res 47: D529–D541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H‐S, Hohn MJ, Umehara T, Guo L‐T, Osborne EM, Benner J, Noren CJ, Rinehart J, Söll D (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirman NL, Barber KW, Aerni HR, Ma NJ, Haimovich AD, Rogulina S, Isaacs FJ, Rinehart J (2015) A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat Commun 6: 8130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA, Toulmay A, Balla T (2019) The functional universe of membrane contact sites. Nat Rev Mol Cell Biol 21, 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7–RILP–p150Glued and late endosome positioning. J Cell Biol 185: 1209–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee JA, Levine TP (2019) Systematic prediction of FFAT motifs across eukaryote proteomes identifies nucleolar and eisosome proteins with the predicted capacity to form bridges to the endoplasmic reticulum. Contact 2: 2515256419883136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G (2012) Exploiting structure similarity in refinement: automated NCS and target‐structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68: 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope R, Flora E, Bayne C, Derré I (2017) IncV, a FFAT motif‐containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen‐containing vacuole. Proc Natl Acad Sci USA 114: 12039–12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R, Vos KJD, Paillusson S, Mueller S, Sancho RM, Lau K‐F, Vizcay‐Barrena G, Lin W‐L, Xu Y‐F, Lewis J et al (2014) ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun 5: ncomms4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struck DK, Hoekstra D, Pagano RE (1981) Use of resonance energy transfer to monitor membrane fusion. Biochemistry 20: 4093–4099 [DOI] [PubMed] [Google Scholar]
- Sugiki T, Egawa D, Kumagai K, Kojima C, Fujiwara T, Takeuchi K, Shimada I, Hanada K, Takahashi H (2018) Phosphoinositide binding by the PH domain in ceramide transfer protein (CERT) is inhibited by hyperphosphorylation of an adjacent serine‐repeat motif. J Biol Chem 293: 11206–11217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K (2011) Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10: 5354–5362 [DOI] [PubMed] [Google Scholar]
- Tickle I, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G (2018) STARANISO. Cambridge, UK: Global Phasing Ltd; http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi [Google Scholar]
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott‐Schwartz J (2017) Applying systems‐level spectral imaging and analysis to reveal the organelle interactome. Nature 546: 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrate LM, Lee JE, Prinz WA, Voeltz GK (2015) Form follows function: the importance of endoplasmic reticulum shape. Annu Rev Biochem 84: 791–811 [DOI] [PubMed] [Google Scholar]
- Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard M‐P, Tomasetto C, Drin G, Alpy F (2017) STARD3 mediates endoplasmic reticulum‐to‐endosome cholesterol transport at membrane contact sites. EMBO J 36: 1412–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm LP, Voilquin L, Kobayashi T, Tomasetto C, Alpy F. (2019) Intracellular and plasma membrane cholesterol labeling and quantification using filipin and GFP‐D4 In Intracellular lipid transport: methods and protocols, Drin G. (ed.) pp 137–152. New York, NY: Springer; [DOI] [PubMed] [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB et al (2018) MolProbity: more and better reference data for improved all‐atom structure validation. Protein Sci 27: 293–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Carvalho P, Voeltz GK (2018) Here, there, and everywhere: the importance of ER membrane contact sites. Science 361: eaan5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu X, Bai J, Tian X, Zhao X, Liu W, Duan X, Shang W, Fan H‐Y, Tong C (2016) Mitoguardin regulates mitochondrial fusion through MitoPLD and is required for neuronal homeostasis. Mol. Cell 61: 111–124 [DOI] [PubMed] [Google Scholar]
- Zhao YG, Liu N, Miao G, Chen Y, Zhao H, Zhang H (2018) The ER contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr Biol 28: 1234–1245.e4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Expanded View and Appendix
DataAppendixFigs
DataAppendixFigs
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
The coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes 6TQR (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQR; VAP‐A/STARD3 complex), 6TQS (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQS; MOSPD2/ORP1 complex), 6TQT (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQT; MOSPD2 unbound), and 6TQU (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TQU; MOSPD2/STARD3 complex).