

Abstract
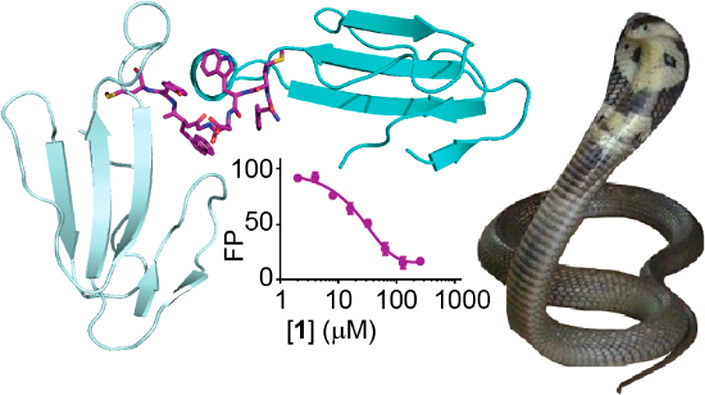
Venomous snakebites cause >100 000 deaths every year, in many cases via potent depression of human neuromuscular signaling by snake α-neurotoxins. Emergency therapy still relies on antibody-based antivenom, hampered by poor access, frequent adverse reactions, and cumbersome production/purification. Combining high-throughput discovery and subsequent structure–function characterization, we present simple peptides that bind α-cobratoxin (α-Cbtx) and prevent its inhibition of nicotinic acetylcholine receptors (nAChRs) as a lead for the development of alternative antivenoms. Candidate peptides were identified by phage display and deep sequencing, and hits were characterized by electrophysiological recordings, leading to an 8-mer peptide that prevented α-Cbtx inhibition of nAChRs. We also solved the peptide:α-Cbtx cocrystal structure, revealing that the peptide, although of unique primary sequence, binds to α-Cbtx by mimicking structural features of the nAChR binding pocket. This demonstrates the potential of small peptides to neutralize lethal snake toxins in vitro, establishing a potential route to simple, synthetic, low-cost antivenoms.
Introduction
Snakebite envenoming kills or maims hundreds of thousands of people every year,1,2 and loss of livestock is also emerging as a substantial problem.3 Treatment requires rapid transport to a medical facility and intravenous administration of antivenom, a mix of immunoglobulin G antibodies isolated from, e.g., horse or sheep immunized with snake venom.1 As the only effective treatment, antivenoms save numerous lives but have the disadvantages of time-consuming and expensive production, poor access for those in need, and alarming numbers of adverse reactions.4−6 There is therefore an urgent need for novel treatments and new strategies, reflected in growing attention from the World Health Organization.7,8
In the case of elapid snakes, such as cobras and kraits, three-finger α-neurotoxins such as α-cobratoxin (α-Cbtx) and α-bungarotoxin (α-Bgtx) are the most prominent and lethal venom components.9−11 α-Cbtx and α-Bgtx potently inhibit postsynaptic nicotinic acetylcholine receptors (nAChRs), which depresses neuromuscular signaling, causing paralysis and loss of respiration.12 The effectiveness of conventional antivenoms is limited by immunogenicity, abundance of α-neurotoxin antibodies in the immunized animal at the time of production,13 and which type of snake inflicts the bite. Newer antibody-based strategies have emerged, with a focus on smaller, less immunogenic, and venom-specific antivenoms. Smaller camelid antibody fragments raised against cobra venom were shown to neutralize the effects of α-Cbtx in mice and may allow for greater tissue penetration than conventional antivenoms.14 Phage display of a library of human-derived antibody single-chain variable fragments against α-Bgtx from krait venom led to one fragment that neutralized toxin injected into mice.15 In the future, combinations of such lead entities could enable development of safe and effective treatments, as reflected in the recent application of cocktails of human monoclonal antibodies targeting mamba toxins.16 These showed promising neutralization of dendrotoxins in mice, although some improvement is needed in neutralizing α-neurotoxins.
As an alternative to antibody-based strategies, we sought to discover short peptide-based neutralizers of α-Cbtx by screening phage display peptide libraries for α-Cbtx binders. Hits were then tested for neutralization of α-Cbtx inhibition of nicotinic nAChRs using electrophysiology. This led to the identification of an effective 8-mer peptide, for which we solved the peptide:α-Cbtx X-ray cocrystal structure, revealing the detailed binding mode. The identification and characterization of short peptide neutralizers of α-Cbtx could lead to the development of targeted, fully synthetic, next-generation antivenoms for snakebite envenoming.
Results
High-Throughput Identification of Peptide Binders of α-Cbtx
Seeking novel binders of α-Cbtx, we screened naïve phage display libraries containing ≥1 × 109 disulfide-constrained 7-mers and linear 7-mers, 12-mers, 16-mers, or 20-mers against immobilized α-Cbtx from Naja kaouthia (monocled cobra). Isolation and sequencing of the most prevalent phages after five rounds of biopanning identified two 16-mers, peptides 1 and 2 (Table 1). Additionally, deep sequencing of phages from all rounds identified six additional prevalent peptides, 3–8, that were also considered further (Table 1; see Experimental Section).
Table 1. Phage Display Hits, Derivatives Thereof, and Their Prevention of α-Cbtx Inhibition of nAChRs.
| peptide | sequence | fold prevention of α-Cbtx inhibitiona | affinityb | identified by |
|---|---|---|---|---|
| no peptide (α-cbtx alone) | 1.0 | |||
| 1 | Ac-HAKTYMWDGWYMPTSH-NH2 | 7.4 | 2 ± 2 μM | phage displayc |
| 2 | Ac-HVNTYMWDGRYMRTSH-NH2 | 1.0 | phage displayc | |
| 3 | H-NWAPAQHGQKWYMQDYTSLE-NH2 | 3.8 | phage displayd | |
| 4 | H-TSNTTPWQTSWELMYAQQNY-NH2 | 1.6 | phage displayd | |
| 5 | H-KPHTYINDHNFIQYDNQQWL-NH2 | not soluble | phage displayd | |
| 6 | H-YVTHPWIDWWDNEPYM-NH2 | not soluble | phage displayd | |
| 7 | H-GHHHRVYMTDVTEHFRWMGK-NH2 | 0.6 | phage displayd | |
| 8 | H-GHHHRVYMTDVTEHFRWWVN-NH2 | 0.5 | phage displayd | |
| 9 | TAMRA-HAKTYMWDGWYMPTSH-NH2 | not tested | 52 ± 3 μM | optimization |
| 10 | Ac-TYMWDGWYMPT-NH2 | 4.5 | optimization | |
| 11 | Ac-YMWDGWYM-NH2 | 6.9 | optimization | |
| 12 | [PEG]6-YMWDGWYM-NH2e | 6.2 | 26 ± 4 μM | optimization |
| 13 | [PEG]6-YMWDGW-OHe | 0.3 | optimization | |
| 14 | [PEG]6-YMWDGW-NH2e | 0.5 | optimization | |
| 15 | H-WDGWYM-[PEG]6e | 0.3 | optimization | |
| 16 | Ac-WDGWYM-[PEG]6e | 0.3 | optimization | |
| 17 | [PEG]6-YMWEGWYM-OHe | 4.8 | optimization | |
| 18 | [PEG]6-YM(Nal)DGWYM-OHe,f | 2.8 | optimization |
α-Cbtx (40 nM) inhibited ACh (100 μM) gated currents through nAChRs to 8 ± 2% their control level, which is here normalized to 1.0. In the presence of, e.g., peptide 1 (100 μM), α-Cbtx (40 nM) only inhibited ACh (100 μM) gated currents to 60 ± 18% of control, here normalized to 7.4-fold prevention of inhibition.
Kd value of 9 determined in fluorescence polarization (FP) saturation experiment (n = 2). Ki values for peptides 1 and 12 determined by inhibition of 9 binding to α-Cbtx in FP competition experiments (n = 6), only measured for two lead peptides.
After five rounds of selection by biopanning.
From deep sequencing of all phages.
PEG, polyethylene glycol.
Nal, 3-(2-naphthyl)-l-alanine.
Characterization of Peptides with YM Motifs That Modulate α-Cbtx Inhibition of Nicotinic Acetylcholine Receptors
We next tested these binders for functional modulation of α-Cbtx activity in electrophysiological assays. We heterologously expressed nicotinic α7-homomeric nAChRs in Xenopus laevis oocytes and measured acetylcholine-gated currents in response to acetylcholine alone, in the presence of α-Cbtx, or in the presence of α-Cbtx preincubated with the identified peptides. As expected, α-Cbtx alone (40 nM) inhibited nAChR responses to ∼10% compared to the control level, and washout of this effect was minimal, with subsequent responses to ACh in the absence of α-Cbtx recovering to only 20% (Figure 1A,B). In the presence of peptide 1 (100 μM), α-Cbtx (40 nM) only inhibited responses to ∼50% of the control level, with responses after washout reaching 70% (Figure 1A,B). Peptide 1 thus appears to prevent the effects of α-Cbtx. Peptide 2 had no effect on α-Cbtx inhibiting nAChRs (Figure 1B). Among the six additional binders, peptide 3 prevented α-Cbtx inhibition of nAChRs comparably to 1 (Figure 1B). However, peptide 4 showed no effect (Figure 1B), and peptides 5 and 6 were poorly soluble and could not be tested. Curiously, peptides 7 and 8enhanced both the onset and the washout of α-Cbtx inhibition of nAChRs (Figure 1B). Thus, for two of the eight α-Cbtx binders identified by phage display, the desired effect of preventing α-Cbtx inhibition of nAChRs could be demonstrated, and the shorter peptide 1 was chosen for the further investigations.
Figure 1.

Peptide 1 prevents α-Cbtx inhibition of nAChRs. (A) Example two electrode voltage clamp recordings of ACh-gated currents in Xenopus laevis oocytes expressing L247V mutant α7 nAChRs and RIC-3 (see Experimental Section, Electrophysiological Assays). In testing peptides, ACh was applied for 10 s in regular conditions (“control”), three times after the application of either α-Cbtx (blue bars) or α-Cbtx with peptide (blue and magenta bars; “App 1–App 3”), and then three more times to observe washout of any effects (“Wash 1–Wash 3”). (B) Mean ± SEM ACh-gated current amplitude, normalized to control (“I/Icontrol”). Each peptide was tested with α-Cbtx on at least three oocytes: n (magenta columns) = 3–4. α-Cbtx alone was tested on the same day on at least two oocytes: n (blue columns) = 2 (peptides 1 and 2) – 4 (all others).
We next established the dose-dependency of the effects of 1 on α-Cbtx in our electrophysiological assay. The initial protection from α-Cbtx inhibition (“IApp3/IControl”) occurred at concentrations of 1 in the range 30–100 μM, whereas apparent enhancement of α-Cbtx washout (“IWash3/IControl”) occurred at concentrations of 1 between 3–30 μM (Figure 2A). To better estimate binding affinity, we turned to a fluorescence polarization (FP) assay, for which we generated peptide 9, a rhodamine dye-labeled derivative of peptide 1 (Figure 2C). From incubating 9 in increasing concentrations of α-Cbtx, we determined the dissociation constant for the α-Cbtx–9 interaction (Kd 52 ± 3 μM, n = 2, Figure 2D). By incubating premixed α-Cbtx (40 μM) and 9 (50 nM) with increasing concentrations of 1, we determined an IC50 value of 29 ± 6 μM for compound 1 (n = 6, Figure 2E), corresponding to a Ki value of 1.9 μM (see Experimental Section, Fluorescence Polarization Assay). Regarding concentrations of 1 that elicited functional effects, this Ki is more consistent with the enhanced washout of α-Cbtx than with the prevention of α-Cbtx inhibition.
Figure 2.
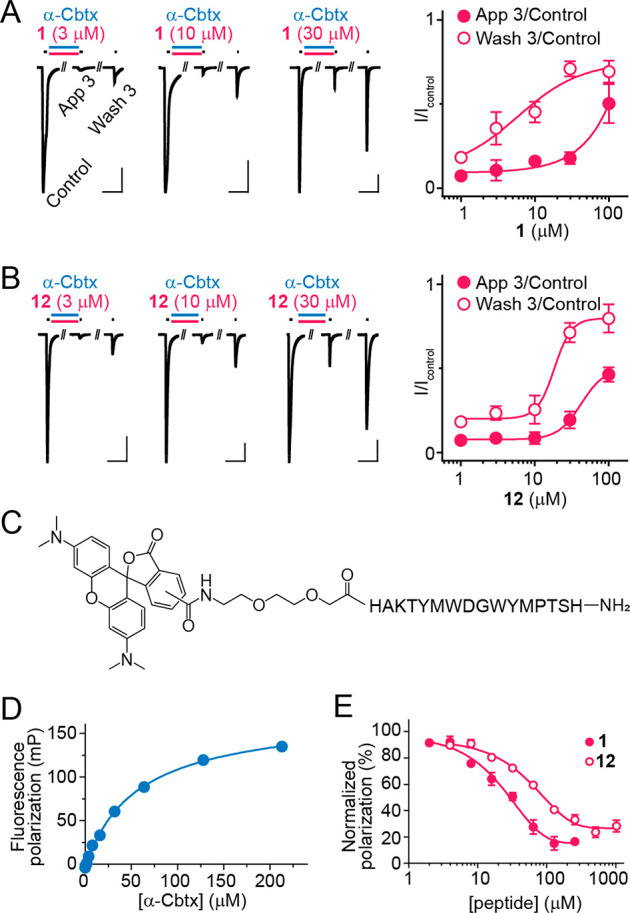
Concentration dependence of peptide activity. (A,B) Electrophysiological experiments as per Figure 1A, but here only initial ACh application (“Control”), the third ACh application after peptide + α-Cbtx (40 nM) (“App 3”), and the third ACh application after washout (“Wash 3”) are shown. Mean ± SEM (n = 3) are shown to the right. (C) Chemical structure of 9. (D) Fluorescence polarization of 9 (50 nM) by increasing concentrations of α-Cbtx. Mean ± SEM, n = 2. (E) Concentration-dependent displacement of 9 (50 nM) from α-Cbtx (40 μM) by compound 1 and 12. Mean ± SEM, n = 6.
Peptide Aromatic Side Chains Bind to α-Cbtx Key Residues R33 and D27
Seeking insight into the peptide−α-Cbtx interaction, we turned to structure determination by X-ray crystallography. This required improved peptide solubility, and we therefore designed and tested truncated (peptides 10 and 11) and truncated, polyethylene glycolated (PEG6-ylated, peptides 12–16) derivatives of 1 (Table 1, Supporting Information, Figure S1). The PEG6-ylated 8-mer, peptide 12, exhibited similar potency to 1 in electrophysiological assays, with 12 (100 μM) restoring α-Cbtx (40 nM) inhibited ACh-gated currents from ∼10% to ∼50% (Figure 2B). In the FP assay, 12 outcompeted 9 from α-Cbtx similarly to 1, despite slightly reduced affinity compared to 1 (12Ki = 26 ± 4 μM, n = 6, P < 0.003, Figure 2E). We also observed increased stability of 12 compared to 1 at both room temperature and at 37 °C (Supporting Information, Figure S2). Given the similar functional properties but improved solubility of 12, it was used for X-ray crystallography.
We obtained diffraction-quality cocrystals and solved the X-ray crystal structure of the 12:α-Cbtx complex at 1.9 Å resolution (Supporting Information, Table S1). The asymmetric unit contains four copies of α-Cbtx and two copies of 12 (Figure 3A). One molecule of 12 binds to two toxin molecules (designated α-Cbtx-A and α-Cbtx-B), forming a 1:2 complex (Figure 3A, Supporting Information, Figure S3). The two α-Cbtx-A:12:α-Cbtx-B complexes present in the asymmetric unit are related by noncrystallographic symmetry and highly similar to a root mean square deviation (rmsd) of 0.2 Å. Thus, we observed two different binding modes of 12 with α-Cbtx (Figure 3B,C). Whereas the structure of α-Cbtx-B is very similar to the conformation of α-Cbtx observed in previous crystal structures, 12 binding induces substantial structural rearrangements of loop II and indirectly also of loop III in α-Cbtx-A (Figure 3D,E). The formation of a 1:2 12:α-Cbtx complex was surprising considering that 12 was added in 1.2-fold molar excess in the crystallization set-ups. This raises the possibility that the mechanism of α-Cbtx inhibition by 12 involves inducing toxin dimerization. There is very little contact between the two α-Cbtx molecules themselves in the complex (52 Å2) as determined by computational analysis (PISA),17 suggesting that these interactions do not contribute to the stabilization of the complex. We reasoned that if both motifs A and B were acting independently as inhibitors, they should prevent α-Cbtx inhibition of nAChRs on their own. However, shorter peptides designed to include only binding motif A (peptides 13 and 14) or motif B (peptides 15 and 16) did not have any effect on α-Cbtx activity in electrophysiological assays (Table 1, Supporting Information, Figure S1). We therefore tentatively conclude that the simultaneous binding of 12 to two α-Cbtx molecules leads to a mutual stabilization of both interfaces or that peptide 12 blocks α-Cbtx by linking two α-Cbtx molecules and preventing them from further binding to nAChRs. However, analysis of 12-induced dimerization of α-Cbtx in solution by size exclusion chromatography was not possible due to the high hydrophobicity of the peptide, which led to aggregation on the gel filtration column.
Figure 3.

Structure of the 12:α-Cbtx complex (PDB 6ZFM). (A) Asymmetric unit of the 12:α-Cbtx crystal structure. Each molecule of 12 (magenta) binds two toxin molecules A and B (light and dark cyan, respectively). A second α-Cbtx-A:12:α-Cbtx-B complex in the asymmetric unit is related by noncrystallographic symmetry. (B) Magnified view showing interaction of α-Cbtx loop II (light cyan) with 12 (magenta) in binding mode A. Two alternative conformations for the side chain of 12M2 were modeled. Key interacting amino acid residues shown in stick representation and labeled. Cation−π-stacking interactions and hydrogen bonds are highlighted by dashed and dotted lines, respectively. (C) Binding mode B, with second α-Cbtx molecule (dark cyan), from same structure with interacting residues drawn as in (B). (D) Binding mode A leads to conformational changes in α-Cbtx (light cyan) compared to the structure observed of α-Cbtx from Naja naja siamensis (dark gray, PDB 2CTX, root-mean-square deviation (rmsd) 1,31 Å), Naja naja oxania (light gray, PDB 1NTN, rmsd 1.7 Å), or Naja naja kaouthia in complex with AChBP (green, PDB 1YI5, rmsd 1.47 Å). The greatest displacement is observed in loop II (11 Å) and loop III (4.7 Å). (E) Binding mode B (cyan) resembles more closely previously reported structures of α-Cbtx from Naja naja siamensis (dark gray, rmsd 1,07 Å), Naja naja oxania (light gray, rmsd 0.67 Å), or Naja naja kaouthia in complex with AChBP (green, rmsd 0.87 Å). (F) Interaction of α-Bgtx (blue) loop II with the principal (α subunit) and complementary (δ subunit) faces of the muscle nAChR (pink, PDB 6UWZ). Key interacting residues drawn as in (B).
Although binding modes A and B are distinct, they share common features (Figure 3B,C, Table 2). (i) Two different YM motifs bind at equivalent positions in binding mode A and B, forming a hydrogen bond between 12Y7 or 12Y1 and α-CbtxD27 and interacting with the surface of α-Cbtx at the beginning of loop II. (ii) Using different aromatic residues, modes A and B both create, together with α-CbtxF29, an aromatic pocket for α-CbtxR33 through extensive cation−π stacking interactions of parallel and T-shaped geometry (Figure 3B,C). Thus, the key residue α-CbtxR33 that has been implicated in penetrating the ACh binding pocket of nAChRs18 is tightly blocked by 12 in both binding modes. In binding mode A, a tryptophan residue that precedes the YM motif also interacts with α-CbtxF65 at the C-terminus of α-Cbtx and forms a hydrogen bond to the backbone carbonyl of α-CbtxP7 (Figure 3B). Exchange of this tryptophan for arginine (along with three other substitutions) in peptide 2 led to a loss of the preventive activity of the peptide (Table 1, Figure 1B), underscoring the functional importance of this tryptophan residue.
Table 2. Key Residues of the 12–α-Cbtx Interaction (PDB 6ZFM) in Comparison to the nAChR−α-Bgtx Complex.
| α-Cbtx | 12-A | 12-B | α-Bgtx | nAChR (PDB 6UWZ) |
|---|---|---|---|---|
| D27 | Y7 (H bond) | Y1 (H bond) | D30 | αY190 (H bond) |
| F29 | W6 (π–π) | F32 | γW55/δW57, αW149 (hydrophobic) | |
| R33 | W3, Y7 (cation−π) | Y1, W6 (cation−π) | R36 | Y190α, Y198α (cation−π); γW55/δW57, αW149 (hydrophobic) |
| R36 | D4 (salt bridge) | Y1 (hydrophobic) | V39 | |
| F65 | W6 (hydrophobic) | H68 | backbone (H-bond) |
The Inhibitory Peptide Mimics the α-Cbtx Interaction Site and Binding Mode of the nAChR
The structure of the nAChR:α-Cbtx complex has not been determined, but the structure of the muscle-type (heteromeric α–γ–α–δ−β) nAChR:α-Bgtx complex was recently reported.19 Because α-Cbtx and α-Bgtx share high sequence and structural homology,18 we can use the nAChR:α-Bgtx complex to compare α-Cbtx interactions with the peptide inhibitor to those with its native target (Figure 3F). The nAChR:α-Bgtx interaction is much more extensive (∼1100 Å2) compared to the 12:α-Cbtx complex (∼440 Å2 in mode A and ∼510 Å2 in mode B), but interestingly, the peptide inhibitor 12 partially mimics the binding site of the natural substrate. Most strikingly, 12Y1 (in binding mode B) and 12Y7 (in binding mode A) adopt the same position as nAChRαY190, which forms a conserved hydrogen bond with α-CbtxD27 and α-BgtxD30, respectively (Table 2). This interaction is also conserved in the complex of α-Cbtx with the acetylcholine binding protein (AChBP),20 where AChBPY185 binds α-CbtxD27 (Supporting Information, Figure S4A). The similarity between 12 and nAChR is also apparent in the pocket for α-CbtxR33/α-BgtxR36 and α-CbtxF29/α-BgtxF32 in binding mode A, which is formed by aromatic side chains 12W3/12Y7 in 12-A and nAChRαY190/nAChRαY198 in the nAChR. In contrast, 12Y1/12W6 in 12-B are also engaged in cation−π interactions with α-CbtxR33, but the orientations of side chains differ from nAChR:α-Bgtx. The aromatic pocket for α-BgtxR36 is closed by ynAChRγW55 /nAChRγδW57 and nAChRαW149 through hydrophobic interaction, which lacks a counterpart in the 12:α-Cbtx complex. It is also interesting to note that aspartate nAChRαD200 superposes with 12D4 of binding mode A. nAChRαD200 does not play a role in α-Bgtx binding to the receptor, but the substitution of α-BgtxV39 to α-CbtxR36 likely leads to the formation of a salt bridge upon binding of α-Cbtx to the nAChR. Thus, 12D4 would further imitate the target binding site. Taken together, the structural comparison reveals that the phage display selection provided a peptide that blocks the binding site of α-Cbtx by imitating the cognate interface through an unrelated sequence.
Discussion
The urgent need for alternatives to conventional antivenom for snakebite has given rise to a handful of novel approaches, including camelid- and human-derived antibodies that target particular venom components,14−16,21 nanoparticles to adsorb plasma proteins,22 and small molecules or peptides to inhibit key components of venom.23,24 Smaller peptides are likely cheaper than traditional antivenom and may lead to more convenient and effective drugs that can be injected subcutaneously.7,25 Consequently, we pursued the idea that short, synthetic peptides inhibiting critical toxin–receptor interactions could serve as lead compounds for the development of novel peptide-based antivenoms. Furthermore, our results suggest that such small peptides are conducive to crystallization and rational design and could be useful for the academic community as pharmacological tools. Starting from some five billion 7-mers, 12-mers, 16-mers, and 20-mers, we arrived at an 8-mer that prevents α-Cbtx from inhibiting nicotinic nAChRs.
Our initial search identified eight peptides that bound α-Cbtx. Despite the absence of primary sequence homology with nAChRs, each peptide contained at least one YM motif. The tyrosine forms a hydrogen bond to α-CbtxD27, and this Y–D bond is conserved in both peptide binding modes that we observed in AChBP:α-Cbtx and nAChR:α-Bgtx structures19,20 and also in a nAChR-like 13-mer binding to α-Bgtx26 (Supporting Information, Figure S4B), suggesting that this tyrosine residue represents an essential feature for α-Cbtx-binding peptides. The methionine occupies a unique binding site at the base of α-Cbtx loop II that might be exploited in future optimizations. Remarkably, the YM motif is flanked by a tryptophan in the two peptides (and derivatives thereof) that had the desired effect of preventing nAChR inhibition by α-Cbtx. The YM sequence in combination with an upstream or downstream tryptophan residue thus represents a novel motif that might serve as a scaffold for designing future peptide inhibitors or peptide-derived small molecule inhibitors of α-Cbtx. Simultaneous binding of our inhibitor to two α-Cbtx molecules seems to be required for functionality. This is evident in the inhibition of α-Cbtx by peptides 1, 10, 11, and 12, which contain both motifs A and B of 12, while peptides 13–16, which contain either motif A or motif B of 12, fail to inhibit α-Cbtx. We suspect that binding of two α-Cbtx molecules results in stabilization of the peptide conformation. Introducing dimerization of α-Cbtx might also be part of the inhibitory mechanism of the identified peptides. Thus, strategies to rigidify the two identified epitopes might be employed for improved activity.
Optimization will also require improved affinity for α-Cbtx, as half-maximal effective concentrations and Ki values for our 8-mer, 16-mer, and 20-mer inhibitors were in the low micromolar range. Much greater affinity for α-Bgtx was seen with previously studied 13-mers,24 possibly due to greater stability through the additional intramolecular hydrogen bonds afforded by the β-hairpin structure of those peptides. Although also identified through phage display, those peptides bear striking similarity to the principal face of nAChRs.24 Our identification of a nAChR-unrelated sequence with a mimicking effect opens up entirely new avenues for future work. It is also noteworthy that, in addition to inhibitors of α-Cbtx, we also identified enhancers of α-Cbtx activity, tentatively suggesting that toxin activity can be manipulated by peptides via distinct mechanisms. The desired improvement of affinity of 1, 12, and similar molecules from the present study may be possible through subtle modifications. In limited structure–activity relationship experiments, we observed that single substitutions of D4 and W3 to glutamic acid and naphthylalanine, respectively, yielded two peptides, 17 and 18, that retained the inhibitory effect on α-Cbtx (Table 1, Supporting Information, Figure S1B). Tolerance of isofunctional substitutions at W3 and D4, which indeed interact with α-CbtxR33 and α-CbtxR36 (Figure 3B), suggests some scope for optimization at these positions.
That PEGylation was also well tolerated could prove useful in future antivenom drug development. Weaknesses of peptide-based drugs include short in vivo half-life/rapid elimination and poor membrane permeability.25 However, these can sometimes be overcome by increasing molecular mass via PEGylation and conjugation to larger peptides or other moieties.27−29 The fact that the YMWDGWYM motif was effective on its own (as 11), PEGylated (as 12), straddled by other residues (in 1), or even as a divergent, longer peptide (as 3) is also promising when considering typical approaches to improve stability and membrane permeation, such as cyclization, stapling, and conjugation.30,31 Our measurements of peptides 1 (free peptide) and 12 (PEGylated) in phosphate-buffered saline show that both are reasonably stable over the course of 1 day, and notably, the purity of 12 was decreased by just 7% after incubation for 9 days at room temperature or 2 days at 37 °C (Supporting Information, Figure S2).
Conclusion
Current therapies for snakebite envenoming are difficult to produce, far too inaccessible for those in need, and often cause adverse reactions.1,32 We speculate that a combination of several peptides targeting the key venom components will provide simpler, more distributable, safer, and subcutaneous- and/or intramuscular-capable alternatives to conventional antivenom, or at least a feasible short-term solution that prevents excess toxin from reaching nAChRs immediately after snakebite. Our report on the identification of such snake toxin-binding peptides, which we term “serpentides”,33 the characterization of their effects on the physiological action of the toxin, and the high-resolution structural characterization of the peptide–toxin interaction should facilitate future endeavors in developing such molecules into therapies.
Experimental Section
Phage Display Screening
Maleic anhydride activated 96-well plates (ThermoFisher Scientific) wells were incubated with 100 μL of 10.0 μM α-cobratoxin (≥99%, from Naja kaouthia, Latoxan) in phosphate buffered saline (PBS) or PBS alone (as control), capped with 200 μL 10% (v/v) ethanolamine, for 1 h at room temperature (RT, ∼21 °C) and then overnight at 4 °C. Subsequently, the wells were washed 3× with 200 μL of 0.05% Tween-20 (PBST) and 2× with 200 μL of PBS. Wells were then blocked by incubation in 100 μL blocking buffer (0.1 M NaHCO3 pH 8.6, 0.02% NaN3, 0.1 μg/mL streptavidin (incidental, as the selection process did not utilize biotinylation), 5 mg/mL bovine serum albumin at RT on a plate shaker for 2 h. Wells were then washed 10× with PBST and 2× with PBS to remove excess blocking agent.
For the first biopanning round, 2.5 μL of both TriCo-16 and TriCo-20 libraries (Creative Biolabs) were combined in 105 μL of blocking buffer and incubated for 30 min at RT. This phage solution (100 μL) was transferred to a blocked but not α-cobratoxin-coated MaxiSorp plate and incubated for 1 h at RT to remove unspecific binding phages. Unbound phage (100 μL) was then transferred to the α-cobratoxin-coated wells and incubated for 1 h at RT. The solution was then removed, and wells were washed 10x PBST and 2× with PBS. Bound phages were eluted by incubating for 10 min with triethylamine (200 μL, 100 mM) followed by neutralization with a solution of Tris-HCl (400 μL, 1 M, pH 7.4). Before each of four subsequent rounds of selection, the eluted phages were amplified in 2xYT medium (8–10 mL) overnight at 37 °C after infecting TG1 Escherichia coli. Amplified phages were precipitated using PEG6000/NaCl and concentrated in PBS (300 μL). Phages (115 μL) mixed with blocking buffer were incubated for 1 h in a MaxiSorp well before the next round of panning. The specific binder/control ratios (based on numbers of plaque forming units) were 24, 203, 755, and 175 for rounds two through five, indicating that specific α-Cbtx binders had been amplified (except in the final round).
Twenty-four single plaques were picked and amplified overnight, centrifuged for 10 min at 8000 g, and 125 μL supernatant was added to α-Cbtx-coated wells for overnight incubation. The solution was then removed, and wells washed, blocked, and washed again, and each eluate (100 μL in PBS) was mixed 1:1 with blocking buffer. 100 μL was added to α-Cbtx-coated (or control) wells, incubated for 1 h at RT on a plate shaker, and then washed 10× with 100 μL PBST and 2× with PBS. Bound phages were then incubated in 100 μL of a solution of monoclonal horseradish peroxidase (HRP) conjugated anti-M13 phage antibody (GE Healthcare) diluted 1:100 in blocking buffer, for 1 h on a plate shaker, washed again (10× with PBST, 2× with PBS), and each well was incubated in 100 μL HRP substrate (ortho-phenylenediamine (OPD, Dako, 2 mg in 3 mL water) with 5 μL H2O2) for ∼30 min. Reactions were stopped by the addition of 100 μL of 0.5 M H2SO4, and absorbance (490 nm) was measured (VersaMax Tunable microplate reader, Molecular Devices). Empty wells were used as blank controls for subtraction of signal. The 12 clones that gave the highest responses in the ELISA were selected for further processing. They were amplified overnight and PEG6000/NaCl precipitated. ssDNA was isolated and Sanger sequenced (Eurofins Genomics). Eleven encoded the same peptide, 1, and the remaining encoded 2, which is 75% identical to 1 (Table 1).
Deep Sequencing of Phage Libraries and Selection of an Additional Six Peptides for Testing
Amplified phage eluates from biopanning rounds one, two, three, and five were used for deep sequencing of variable regions along with rounds two, three, four and five for the control selections. The libraries after those rounds were PCR amplified with barcoded primers (based on the above phage libraries and according to suppliers’ instructions), gel purified, and sequencing adapters were ligated onto the DNA using the Ion Plus fragment library kit (ThermoFisher Scientific). Finally, the DNA was ligated to Ion Sphere Particles, subjected to emulsion PCR according to the Ion OneTouch 200 protocol. The Ion Sphere Particles were loaded onto an Ion 318 chip and sequenced in an Ion Torrent Personal Genome Machine system. The phage sequences were extracted, split according to barcodes and converted to peptide reads. For unknown reasons, the sequencing failed for the fifth biopanning round against α-Cbtx as well as the second biopanning round for the control. The number of peptides allocated to these samples were <400 compared to >100 000 for the remaining six samples. Accordingly, these samples were disregarded in further analyses. A total of 1,469,811 sequences were obtained representing an average of 224,969 per sample (range 110 503–499 660). The overall quality of the sequencing run was relatively low: if the quality (Phred) score cutoff was increased from 15 to 20, the approved number of sequences dropped to 674,989. Regarding the phage selection process, the number of unique peptides dropped with each subsequent round. Specifically, from 4938 to 410 for round one to three for the toxin-selected samples and similarly from 413 to 108 from rounds three to five in the control samples.
The most frequent peptide sequence identified in deep sequencing of the third selection round was identical to peptide 1, a known binder from phage display/ELISA, suggesting that frequently occurring sequences in this round could be potential binders. Accordingly, three additional peptides (peptides 7, 3, and 5 in Table 1) ranking second through fourth in frequency in round three were selected for further studies. Another peptide, 8, was included because it ranked seventh in round three and was consistently prevalent in the later rounds. The most frequent peptide in the first selection round, 6, was also selected. Finally, a peptide ranking sixth in the third round, 4, was included because it was highly similar to 8 differing only in 3 C-terminal amino acids out of the 20-mer peptide.
Electrophysiological Assays
Stage V/VI Xenopus laevis oocytes were prepared as described previously34 and under license 2014-15-0201-00031 from the Danish Veterinary and Food Administration. Oocytes were injected with a volume of 40 nL containing 4 ng of cRNA for α7 nicotinic acetylcholine receptor (rat) carrying the L247V mutation to reduce desensitization35 and 4 ng of cRNA for the chaperone RIC-3 (human), which enhances acetylcholine receptor function.36 cRNAs were prepared and oocytes were maintained as described previously.34 One to 3 days after injecting, two electrode voltage clamp recordings were made as follows: A single oocyte was transferred to a custom recording chamber37 and continuously perfused with bath solution (in mM; 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4, with NaOH). The oocyte was impaled with borosillicate glass micropipettes containing 3 M KCl (resistance ∼0.5–1.5 MΩ), membrane voltage was clamped at −40 mV, and current recorded with OC-725C amplifier (Warner Instruments), Digidata 1550 digitizer, and pClamp 10 Software (Molecular Devices) at 1 kHz with 200 Hz filtering. Replicate experiments were generally performed on four individual oocytes (see Quantification and Statistical Analysis below). Solution exchange was performed with a ValveBank 8 perfusion system (AutoMate Scientific). ACh and α-Cbtx were prepared from stocks dissolved in water (100 mM and 10 μM, respectively, stored at −20 °C). Peptide (1–18) stocks of 10 mM were prepared in bath solution and typically used within 1 week (if not used in 1 day, these were stored at −20 °C). Peptides were mixed with α-Cbtx 20 min to 1.5 h before application to oocytes. Current amplitude was measured in Clampfit 10 (pClamp, Molecular Devices) and current amplitude was plotted and analyzed in GraphPad Prism 7.
Peptides
All peptides used for subsequent assays, except 9, were purchased from Schafer-N (Copenhagen, Denmark) or GenScript, ≥95% purity (Supporting Information, Figures S5–S22). The 5(6)-carboxytetramethylrhodamine (TAMRA)-labeled peptide 9 was synthesized using an automated peptide synthesizer and standard Fmoc (fluorenylmethoxycarbonyl)-based solid-phase peptide synthesis (SPPS) on Rink amide TentaGel resin (0.23 mmol/g) at a 0.02 mmol scale. Fmoc deprotections were performed in two steps: (1) piperidine in DMF (2:3, v/v) for 3 min and (2) piperidine in DMF (1:4, v/v) for 12 min. Deprotection steps were followed by washing with DMF (2 × 45 s), CH2Cl2 (1 × 45 s), and DMF (2 × 45 s). Coupling steps were performed as double couplings with Fmoc-Xaa-OH (5.00 equiv to the resin loading), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronoium hexafluorophosphate (HBTU) (4.90 equiv), and i-Pr2NEt in NMP (10.0 equiv, 2.0 M) in DMF (final concentration = 0.2 M for Fmoc-Xaa-OH) for 40 min for each coupling. Glycine was incorporated as N-α-Fmoc-N-α-(2,4-dimethoxybenzyl)-glycine (Fmoc-(Dmb)Gly-OH). TAMRA-acid (1.50 equiv) was coupled manually using 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronoium hexafluorphosphate (HATU) (1.50 equiv) and i-Pr2NEt (3.00 equiv) in DMF (final concentration = 0.06 M) for 16 h. Global deprotection and cleavage was conducted in the cleavage cocktail (TFA–i-Pr3SiH–water, 95:2.5:2.5, v/v/v) at room temperature (RT) for 2 h, followed by TFA evaporation and ether precipitation. The crude peptide was purified as single isomer by preparative reverse-phase high performance liquid chromatography (RP-HPLC) on a C8 Phenomenex Luna column (5 μm, 100 Å, 250 mm × 20 mm) using an Agilent 1260 LC system. Fractions were analyzed by matrix assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOF MS), and pure fractions were pooled and lyophilized. Purity was determined using an Agilent 1100 system equipped with C18 Phenomenex Luna column (2.6 μm, 100 Å, 150 mm × 4.60 mm). Analytical HPLC purity 95% (λ = 215 nm; Figure S13). UPLC-MS m/z calcd for C124H157N27O30S22+ [M + 2H]2+: 1284.55, found 1284.38; C124H158N27O30S23+ [M + 3H]3+: 856.70, found 856.82; C124H159N27O30S24+ [M + 4H]4+: 642.78, found 642.91 (Supporting Information, Figure S13).
Fluorescence Polarization (FP) Assays
Protein concentrations and DMSO stock concentrations of unlabeled peptides were determined by UV absorbance (NanoDrop OneC, ThermoFisher Scientific) at 280 nm. The DMSO stock of TAMRA ligand (9) was determined by weight of the corresponding TFA salt (MW + 5 × TFA = 3138 g/mol). Binding affinities were determined in a 384-well plate format (Corning Life Science) using a Safire 2 plate reader (Tecan). The instrument G-factor was calibrated to give an initial millipolarization at 20 (excitation at 530 nm; emission at 580 nm) and the instrumental Z-factor was adjusted to maximum fluorescence. All measurements were conducted in phosphate-buffered saline (PBS, pH = 7.4) at 25 °C. In a FP saturation assay, 50 nM of 9 was mixed with increasing concentrations (0.125–213 μM) of α-Cbtx. The resulting polarization was plotted as a function of the protein concentration and fitted to a one-site binding model GraphPad Prism 7.0 software to yield the Kd at 50% saturation. The binding experiment was performed as a single experiment with two replicates. FP competition experiments were conducted by mixing a preformed α-Cbtx:9 complex at fixed concentration (40 μM/50 nM) with varying unlabeled peptide concentrations ranging from 0.2–1024 μM. The polarization (mP) was plotted as a function of peptide concentration and fitted to a sigmoidal dose response curve using GraphPad Prism 7 software. The Ki values were calculated according to the equations by Nikolovska-Coleska et al.38 The Ki values were determined form three individual experiments performed with two replicates.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
P, protein (α-Cbtx); L, ligand (TAMRA-peptide 9); I, inhibitor (peptide 1 or 12); Kd, dissociation constant of the protein–ligand complex; [P]T, total protein concentration; [L]T, total ligand concentration; [P]0, unbound protein concentration at 0% inhibition; [L]0, unbound ligand concentration at 0% inhibition; [PL]0, protein–ligand complex concentration at 0% inhibition; [P]50, unbound protein concentration at 50% inhibition; [L]50, unbound ligand concentration at 50% inhibition; [PL]50, protein–ligand complex concentration at 50% inhibition; [I]50, unbound inhibitor concentration at 50% inhibition; IC50, half maximal inhibitory concentration; Ki, dissociation constant of the protein–inhibitor complex.
Crystallization and Structure Determination
α-Cbtx (lyophilized powder, ≥99%, Latoxan) and 12 (≥95%, GenScript) were solubilized in 20 mM HEPES, pH 7.2, 100 mM NaCl at concentrations of 14.4 mg mL–1 (1.87 mM) and 8.2 mg mL–1 (5.5 mM), respectively, and mixed at 1:1.2 molar ratio. Crystallization trays were set up with a semiautomated pipetting robot (Gryphon, Art Robbins Instruments). Best crystals grew in 0.1 M carboxylic acids (sodium formate/ammonium acetate/sodium citrate/sodium potassium tartrate/sodium oxamate), 0.1 M HEPES/MOPS, pH 7.5, 30.0% v/v PEG 500 MME/PEG 20000 (Morpheus Crystallization Screen, Molecular Dimensions) and were directly flash frozen in liquid nitrogen. Data sets were collected at beamline MX14.2, BESSY Berlin39 and beamline P13, EMBL Hamburg, respectively. Diffraction data was processed with XDSAPP40,41 and initial phases were obtained by molecular replacement using phenix.phaser42 with the neurotoxin-1 from Naja naja oxiana (PDB 1NTN,43 as a search model. A molecular model for the peptide portion of 12 could be built unambiguously into the remaining difference density. The final model was obtained through iterative cycles of model building in Coot44 and refinement with refmac/phenix.refine42,45 and is deposited at the protein database (PDB 6ZFM).
Quantification and Statistical Analysis
In Phage display screening, sequencing identified only two peptide binders of α-Cbtx, and both were characterized further. In Deep sequencing of phage libraries and selection of an additional six peptides for testing, several more potential peptide binders were identified, but only six were selected for further characterization based on their amino acid sequence identity with proven binders from Phage Display Screening.
Data from Electrophysiological assays are mean ± SEM (n = 2–4 oocytes in Figure 1B; n = 3 oocytes in Figure 2A,B; and n = 2–5 in Supporting Information, Figure S1B, as indicated in figure legends). We have chosen to show SEM error bars for the few groups where n = 2, as this shows the range of these data. To establish that, e.g., peptide 1 prevented α-Cbtx from inhibiting acetylcholine-gated currents through acetylcholine receptors, we compared the mean current amplitude (relative to control) in the presence of α-Cbtx to the mean current amplitude (relative to control) in the presence of both α-Cbtx and peptide 1, using a two-tailed unpaired t test (GraphPad Prism).
In FP assays, the binding saturation with 9 and α-Cbtx was performed as a single experiment with two replicates. For competition with 1 and 12, Ki values were determined from three individual experiments performed with two replicates. Data are given as mean ± SEM Ki values were calculated according to Nikolovska-Coleska et al.38 and compared using a two-tailed unpaired t test (GraphPad Prism).
Acknowledgments
This work was supported by Carlsberg Foundation grant 2013_01_0333 and Lundbeck Foundation grant R289-2018-2074 to C.A.O., Lundbeck Foundation Fellowship R139-2012-12390 to S.A.P., and the Tech Transfer Office, University of Copenhagen (to B.L.). We thank Morten Jørgensen, Brian Bentzen, and Jonas Caspersen, at Terrariet, Vissenbjerg, Denmark, for Naja kaouthia venom used in preliminary exploratory studies.
Glossary
Abbreviations Used
- AChBP
acetylcholine binding protein
- nAChRs
acetylcholine receptors
- PEG
polyethelene glycol
- TAMRA
5(6)-carboxytetramethylrhodamine
- α-Bgtx
α bungarotoxin
- α-Cbtx
α cobratoxin
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01202.
Effects of additional peptides on α-cobratoxin inhibition of nAChRs; chemical stability of peptides 1 and 12; architecture of the 12:α-Cbtx complex observed in the crystal structure; structurally homologous α-neurotoxin binding interactions with other proteins; HPLC chromatograms and MS spectra; PDB file 6ZFM (12:α-Cbtx X-ray cocrystal structure) (PDF)
Accession Codes
Coordinates and structure factors for the 12:α-Cbtx X-ray cocrystal structure have been deposited in the RCSB Protein Data Bank with accession number PBD 6ZFM. Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
◇ A.H.L.: Department of Biotechnology and Biomedicine, Building 224 Søltofts Plads, Technical University of Denmark, DK-2800 Kongens Lyngby, Denmark
Author Contributions
B.L. conceived the study; T.L., B.H.G., A.C., M.D., K.D., A.H.L., C.A.O., D.K., S.A.P. and B.L. designed experiments; B.L. and B.H.G. performed peptide synthesis; B.L. and J.J. performed phage display; B.H.G. and M.M.-M. performed fluorescence polarization assays; T.L. performed electrophysiology; A.C. performed deep sequencing; S. Kiontke, S. Kattelmann, and D.K. performed X-ray crystallography; T.L., B.H.G., A.H.L., K.D., D.K., and B.L. analyzed the data. M.D., A.H.L., C.A.O., D.K., S.A.P. and B.L. provided supervision. T.L., D.K., and B.L. wrote the manuscript with input from all other authors.
The authors declare the following competing financial interest(s): Brian Lohse is owner of Serpentides and therefore declares a commercial interest in the development of peptide-based antivenoms.
Supporting Information Figure S2 was misformatted in the version published on November 4, 2020 and was correctly restored on November 5, 2020.
Supplementary Material
References
- Gutiérrez J. M.; Calvete J. J.; Habib A. G.; Harrison R. A.; Williams D. J.; Warrell D. A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17063. 10.1038/nrdp.2017.63. [DOI] [PubMed] [Google Scholar]
- Mohapatra B.; Warrell D. A.; Suraweera W.; Bhatia P.; Dhingra N.; Jotkar R. M.; Rodriguez P. S.; Mishra K.; Whitaker R.; Jha P. Million Death Study, C., Snakebite mortality in India: a nationally representative mortality survey. PLoS Neglected Trop. Dis. 2011, 5 (4), e1018 10.1371/journal.pntd.0001018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon I.; Finat M.; Herrera M.; Nickerson A.; Grace D.; Schütte S.; Babo Martins S.; Ruiz de Castañeda R. Snakebite in domestic animals: first global scoping review. Preventive Veterinary Medicine 2019, 170, 104729. 10.1016/j.prevetmed.2019.104729. [DOI] [PubMed] [Google Scholar]
- de Silva H. A.; Ryan N. M.; de Silva H. J. Adverse reactions to snake antivenom, and their prevention and treatment. Br. J. Clin. Pharmacol. 2016, 81 (3), 446–452. 10.1111/bcp.12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León G.; Vargas M.; Segura Á.; Herrera M.; Villalta M.; Sánchez A.; Solano G.; Gómez A.; Sánchez M.; Estrada R.; Gutiérrez J. M. Current technology for the industrial manufacture of snake antivenoms. Toxicon 2018, 151, 63–73. 10.1016/j.toxicon.2018.06.084. [DOI] [PubMed] [Google Scholar]
- Snake bite—the neglected tropical disease. Lancet, 2015, 386 ( (9999), ) 1110. 10.1016/S0140-6736(15)00247-0 [DOI] [PubMed] [Google Scholar]
- Laustsen A. H.; Engmark M.; Milbo C.; Johannesen J.; Lomonte B.; Gutiérrez J. M.; Lohse B. From fangs to pharmacology: The future of snakebite envenoming therapy. Curr. Pharm. Des. 2016, 22 (34), 5270–5293. 10.2174/1381612822666160623073438. [DOI] [PubMed] [Google Scholar]
- Williams H. F.; Layfield H. J.; Vallance T.; Patel K.; Bicknell A. B.; Trim S. A.; Vaiyapuri S. The urgent need to develop novel strategies for the diagnosis and treatment of snakebites. Toxins 2019, 11 (6), 363. 10.3390/toxins11060363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modahl C. M.; Mukherjee A. K.; Mackessy S. P. An analysis of venom ontogeny and prey-specific toxicity in the Monocled Cobra (Naja kaouthia). Toxicon 2016, 119, 8–20. 10.1016/j.toxicon.2016.04.049. [DOI] [PubMed] [Google Scholar]
- Suryamohan K.; Krishnankutty S. P.; Guillory J.; Jevit M.; Schröder M. S.; Wu M.; Kuriakose B.; Mathew O. K.; Perumal R. C.; Koludarov I.; Goldstein L. D.; Senger K.; Dixon M. D.; Velayutham D.; Vargas D.; Chaudhuri S.; Muraleedharan M.; Goel R.; Chen Y.-J. J.; Ratan A.; Liu P.; Faherty B.; de la Rosa G.; Shibata H.; Baca M.; Sagolla M.; Ziai J.; Wright G. A.; Vucic D.; Mohan S.; Antony A.; Stinson J.; Kirkpatrick D. S.; Hannoush R. N.; Durinck S.; Modrusan Z.; Stawiski E. W.; Wiley K.; Raudsepp T.; Kini R. M.; Zachariah A.; Seshagiri S. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat. Genet. 2020, 52 (1), 106–117. 10.1038/s41588-019-0559-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. Y.; Tan C. H.; Tan N. H. Venom and purified toxins of the spectacled cobra (Naja naja) from Pakistan: insights into toxicity and antivenom neutralization. Am. J. Trop. Med. Hyg. 2016, 94 (6), 1392–1399. 10.4269/ajtmh.15-0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber C. M.; Isbister G. K.; Hodgson W. C. Alpha neurotoxins. Toxicon 2013, 66, 47–58. 10.1016/j.toxicon.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Gutiérrez J. M.; Solano G.; Pla D.; Herrera M.; Segura Á.; Villalta M.; Vargas M.; Sanz L.; Lomonte B.; Calvete J. J.; León G. Assessing the preclinical efficacy of antivenoms: from the lethality neutralization assay to antivenomics. Toxicon 2013, 69, 168–179. 10.1016/j.toxicon.2012.11.016. [DOI] [PubMed] [Google Scholar]
- Richard G.; Meyers A. J.; McLean M. D.; Arbabi-Ghahroudi M.; MacKenzie R.; Hall J. C. In vivo neutralization of α-cobratoxin with high-affinity llama single-domain antibodies (VHHs) and a VHH-Fc antibody. PLoS One 2013, 8 (7), e69495 10.1371/journal.pone.0069495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkeaw K.; Sakolvaree Y.; Srimanote P.; Tongtawe P.; Maneewatch S.; Sookrung N.; Tungtrongchitr A.; Tapchaisri P.; Kurazono H.; Chaicumpa W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J. Proteomics 2009, 72 (2), 270–282. 10.1016/j.jprot.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Laustsen A. H.; Karatt-Vellatt A.; Masters E. W.; Arias A. S.; Pus U.; Knudsen C.; Oscoz S.; Slavny P.; Griffiths D. T.; Luther A. M.; Leah R. A.; Lindholm M.; Lomonte B.; Gutiérrez J. M.; McCafferty J. In vivo neutralization of dendrotoxin-mediated neurotoxicity of black mamba venom by oligoclonal human IgG antibodies. Nat. Commun. 2018, 9, 3928. 10.1038/s41467-018-06086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372 (3), 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Nirthanan S.; Gwee M. C. E. Three-finger alpha-neurotoxins and the nicotinic acetylcholine receptor, forty years on. J. Pharmacol. Sci. 2004, 94 (1), 1–17. 10.1254/jphs.94.1. [DOI] [PubMed] [Google Scholar]
- Rahman M. M.; Teng J.; Worrell B. T.; Noviello C. M.; Lee M.; Karlin A.; Stowell M. H. B.; Hibbs R. E. Structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins. Neuron 2020, 106 (6), 952–962. 10.1016/j.neuron.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y.; Talley T. T.; Hansen S. B.; Taylor P.; Marchot P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. EMBO J. 2005, 24 (8), 1512–1522. 10.1038/sj.emboj.7600620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgilas P. B.; Neves-Ferreira A. G.; Domont G. B.; Perales J. PO41, a snake venom metalloproteinase inhibitor isolated from Philander opossum serum. Toxicon 2003, 42 (6), 621–628. 10.1016/j.toxicon.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Karain B. D.; Lee M. K. H.; Hayes W. K. C60 fullerenes as a novel treatment for poisoning and envenomation: a proof-of-concept study for snakebite. J. Nanosci. Nanotechnol. 2016, 16 (7), 7764–7771. 10.1166/jnn.2016.12851. [DOI] [Google Scholar]
- Lewin M.; Samuel S.; Merkel J.; Bickler P. Varespladib (LY315920) appears to be a potent, broad-spectrum, inhibitor of snake venom phospholipase A2 and a possible pre-referral treatment for envenomation. Toxins 2016, 8 (9), 248. 10.3390/toxins8090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherf T.; Kasher R.; Balass M.; Fridkin M.; Fuchs S.; Katchalski-Katzir E. A β-hairpin structure in a 13-mer peptide that binds α-bungarotoxin with high affinity and neutralizes its toxicity. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (12), 6629–6634. 10.1073/pnas.111164298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discovery Today 2015, 20 (1), 122–128. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Harel M.; Kasher R.; Nicolas A.; Guss J. M.; Balass M.; Fridkin M.; Smit A. B.; Brejc K.; Sixma T. K.; Katchalski-Katzir E.; Sussman J. L.; Fuchs S. The binding site of acetylcholine receptor as visualized in the X-ray structure of a complex between α-bungarotoxin and a mimotope peptide. Neuron 2001, 32 (2), 265–275. 10.1016/S0896-6273(01)00461-5. [DOI] [PubMed] [Google Scholar]
- Chandy K. G.; Norton R. S. Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Curr. Opin. Chem. Biol. 2017, 38, 97–107. 10.1016/j.cbpa.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Guidotti G.; Brambilla L.; Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol. Sci. 2017, 38 (4), 406–424. 10.1016/j.tips.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Strohl W. R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 2015, 29, 215–239. 10.1007/s40259-015-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17 (1), 134–143. 10.1208/s12248-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalafatovic D.; Giralt E. Cell-penetrating peptides: design strategies beyond primary structure and amphipathicity. Molecules 2017, 22 (11), 1929. 10.3390/molecules22111929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longbottom J.; Shearer F. M.; Devine M.; Alcoba G.; Chappuis F.; Weiss D. J.; Ray S. E.; Ray N.; Warrell D. A.; Ruiz de Castañeda R.; Williams D. J.; Hay S. I.; Pigott D. M. Vulnerability to snakebite envenoming: a global mapping of hotspots. Lancet 2018, 392 (10148), 673–684. 10.1016/S0140-6736(18)31224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse B. Synthetic antivenoms for snakebites: a slithering road!. Biochemist 2019, 41 (6), 6–9. 10.1042/BIO04106006. [DOI] [Google Scholar]
- Lynagh T.; Komnatnyy V. V.; Pless S. A. Unique contributions of an arginine side chain to ligand recognition in a glutamate-gated chloride channel. J. Biol. Chem. 2017, 292 (9), 3940–3946. 10.1074/jbc.M116.772939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revah F.; Bertrand D.; Galzi J. L.; Devillers-Thiéry A.; Mulle C.; Hussy N.; Bertrand S.; Ballivet M.; Changeux J. P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 1991, 353 (6347), 846–849. 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Halevi S.; Yassin L.; Eshel M.; Sala F.; Sala S.; Criado M.; Treinin M. Conservation within the RIC-3 Gene Family: effectors of mammalian nicotinic acetylcholine receptor expression. J. Biol. Chem. 2003, 278 (36), 34411–34417. 10.1074/jbc.M300170200. [DOI] [PubMed] [Google Scholar]
- Dahan D. S.; Dibas M. I.; Petersson E. J.; Auyeung V. C.; Chanda B.; Bezanilla F.; Dougherty D. A.; Lester H. A. A fluorophore attached to nicotinic acetylcholine receptor βM2 detects productive binding of agonist to the αδ site. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (27), 10195–10200. 10.1073/pnas.0301885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolovska-Coleska Z.; Wang R.; Fang X.; Pan H.; Tomita Y.; Li P.; Roller P. P.; Krajewski K.; Saito N. G.; Stuckey J. A.; Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004, 332 (2), 261–273. 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- Mueller U.; Darowski N.; Fuchs M. R.; Forster R.; Hellmig M.; Paithankar K. S.; Puhringer S.; Steffien M.; Zocher G.; Weiss M. S. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 2012, 19 (3), 442–449. 10.1107/S0909049512006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug M.; Weiss M. S.; Heinemann U.; Mueller U. XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45 (3), 568–572. 10.1107/S0021889812011715. [DOI] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickitenko A. V.; Michailov A. M.; Betzel C.; Wilson K. S. Three-dimensional structure of neurotoxin-1 from Naja naja oxiana venom at 1.9 Å resolution. FEBS Lett. 1993, 320 (2), 111–117. 10.1016/0014-5793(93)80073-4. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (12 Part 1), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53 (3), 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.