

Abstract

Photoexcited dihydronicotinamides like NADH and analogues have been found to generate alkyl radicals upon reductive decarboxylation of redox-active esters without auxiliary photocatalysts. This principle allowed aliphatic photocoupling between redox-active carboxylate derivatives and electron-poor olefins, displaying surprising water and air-tolerance and unusually high coupling rates in dilute conditions. The orthogonality of the reaction in the presence of other carboxylic acids and its utility in the functionalization of DNA is presented, notably using visible light in combination with NADH, the ubiquitous reductant of life.
Introduction
Visible light is a prime stimulus to control the conformation of chemical bonds,1 or their cleavage.1a,2 The phototriggered formation of chemical bonds can enable frontier research in medicine and biology,3 but their development is still a challenge in comparison to thermal click reactions4 due to the slower rate and the need for UV-light and/or photocatalysts.5 On one hand, photo-cross-linking methods still rely on unstable precursors like azirines or cyclopropanones.5a,6 On the other hand, recent C–C coupling reactions using photobiocatalytic systems have shown great promise but these are still limited to activated substrates with auxiliary photosensitizers and electron donors.7 As such, developments in self-sensitized, phototriggered, and fast C–C photocoupling between simple functionalities are still highly sought after (Scheme 1A).8,9 Aliphatic linkages are particularly attractive due to their small size, robustness, and flexibility, which maximize the chances to obtain functional and metabolically stable conjugates.8
Scheme 1. Approach Towards Aliphatic Photo-Coupling with Native NADH Bio-Photoreductant.

NAD, Nicotinamide Adenine Dinucleotide; PET, Photoinduced Electron Transfer; 5 – R = Ph, R’= H.
Decarboxylative radical addition reactions (Scheme 1A) have recently emerged as prime tools to create aliphatic ligations in biomolecules.8,9 These methods take advantage of the abundance of carboxylic acids8,10 and the various technologies developed with Michael acceptors.1b,11 Despite their success, radical addition reactions are slow (6–12 h) and require additional catalysts, inorganic reducing suspensions, and/or additives that are not native to biological systems.8 The abundance of endogenous carboxylic acids in biomolecules or biomatrices poses a selectivity challenge for carboxylic acid substrates (1), due to their similar oxidation potentials.8c−8e In contrast, the N-hydroxyphthalimide (NHPI) esters (2) can be orthogonally activated in the presence of other carboxylates via single-electron reduction.8a,8b,8f−8l Recent methods based on desymmetrization8n and late-stage carbene transfer12 illustrate the potential of redox-active esters to be introduced through strategies unavailable to the parent carboxylic acids.
During our synthetic studies with redox-active carbenes,12 we recognized that the coupling of redox-active esters and Michael acceptors8a,8b,8f−8l could significantly expand its capabilities with a suitable biocompatible reductant (Scheme 1B). The reduced nicotinamide adenine dinucleotide (NADH) would be ideal because it is a native component of biological systems.
The redox potential of NADH and its analogs (Eox{5} = 0.57 V vs Ag/Ag+) is insufficient to activate redox-active esters (Ered{2} ∼ – 1.1 ± 0.1 V vs Ag/Ag+).13 These dihydronicotinamides are potent single-electron reductants in the excited state (Eox*{5} = −2.60 V vs Ag/Ag+),14,15 but their short lifetimes in solution (τ{5*} ∼ 0.7 ns)16 have limited their application as autonomous photoreductants.14,17,18 At the onset of our work, these reagents required additional (photo)catalysts8f−8l,18,19 or enzymes20 under rigorously anhydrous and degassed conditions to drive reductive couplings. We reasoned that the short-lived excited states of these systems would have a minimal impact in photoinitiated reactions21 and would avoid side-reactions in the presence of dioxygen derived from triplet-sensitization. The transient generation of the powerful photoreductant 5* would effectively circumvent the incompatibility with oxygen and moisture of other ground state super electron donors.22 Importantly, the expected byproducts of the reaction would be biocompatible: the cofactor NAD+ (or analogues thereof), CO2, and phthalimide (LD50{rat oral} > 5 g/kg).23
Results and Discussion
Toward this end, the reaction of the NADH model BNAH (5) with the redox-active ester 2a and the acrylate acceptor 3a was studied under blue light illumination (λ = 450 nm) without photocatalysts or additives (Scheme 2).8f−8l To our delight, the desired decarboxylative coupling product 4a was obtained in high yield using DMSO as solvent (entry 1). The reaction was found to be surprisingly fast, reaching 66% yield after 5 min of illumination (entry 2). Given the importance of maximizing the reaction rate for its implementation at higher dilution,3,4,4c−4i we explored related photoreductants. It was found that the dihydronicotinamide moiety is essential for high activity (entry 3) as well as the appropriate substitution at the heterocyclic nitrogen (entries 4,5). In line with seminal studies by Overman8h and recent work by Shang,24 the dihydropyridine 9 was found to promote the reaction, but it was slower and less efficient than the more biocompatible dihydronicotinamides (entry 6).25 Interestingly, the N-butyl dihydronicotinamide BuNAH (10), which is the closest structural homologue to NADH among the photoreductants explored, was optimal both in terms of yield and rate (entry 7). This result can be rationalized by the slightly more reductive character of BuNAH (10)26 than the N-benzyl- and N-aryl-dihydronicotinamides 5,8. Moreover, the performance of BuNAH (10) is only marginally affected by concentrations as low as 1 mM (entry 8). The system tolerates water as cosolvent (50% v/v; entry 9) and air atmosphere (entry 10). These are unique features that contrast with sensitive ground-state organic reductants22a,4b,4c,24,25 and other photocatalyzed reactions.8f,8h,18,19c,19d Interestingly, BuNAH (10) can be prepared in multigram amounts, stored indefinitely as a solid, and handled for more than a week as a DMSO stock solution (see SI), thus enabling microdosing in high-throughput studies.
Scheme 2. Discovery of the Photo-Coupling Promoted by BuNAH (10) and NADH (11).

Determined by 1H NMR using 1,1,2,2- tetrachloroethane as internal standard.
3 equiv used.
10 equiv used.
These results led us to explore the performance of NADH (11) due to its relevance as the native reductant in biological systems. The photophysics of NADH (11) have additional challenges due to its shorter excited state lifetime (τ{NADH} ∼ 0.4 ns) and the interaction between its dihydronicotinamide and adenine moieties.27 To our delight, the commercial NADH disodium salt (11) promoted the coupling reaction in a dilute mixture of water and DMSO (1–10 mM; entries 11,12). Unlike that of BuNAH (10), it was found that the use of NADH (11) required inert atmosphere and larger amounts for optimal results, probably due to its higher sensitivity and/or less favorable photophysic properties.
We set out to explore the scope of the photocoupling using artificial BuNAH (10; conditions A) or natural NADH (11; conditions B) as photoreductants in aqueous (50% H2O in DMSO) and dilute conditions (1 mM; Scheme 3) most relevant in Chemical Biology. Alternatively, preparative scale reactions can be undertaken at higher concentrations in DMSO (see SI for details). Various Michael acceptors bearing electron-withdrawing groups such as ester (4a,b), amide (4c),11c aldehyde (4d), ketone (4e), nitrile (4f), or sulfone (4g) were accommodated. Among these, acrolein was significantly less efficient as an acceptor, probably due to degradation of the sensitive aliphatic aldehyde product 4c. The maleimide scaffold (4h) that is common in bioconjugation reactions1b,5e,11a,11b was found to be very efficient. In stark contrast, no coupling product was obtained using the dihydropyridine 9,24,25 thus illustrating the superior reactivity of BuNAH (10) or NADH (11) as photoreductants.24,25 High yields and fast reactions also occur across a wide range of redox-active esters. Tertiary sites are coupled efficiently, thus allowing interesting structures to be functionalized, including bicyclic (4i), adamantyl (4j), piperidine (4k), cyclopropane (4l), and more complex scaffolds such as the drug gemfibrozil (4m,n). Secondary radical precursors are equally effective in the reaction (4o,q).
Scheme 3. Scope Study.
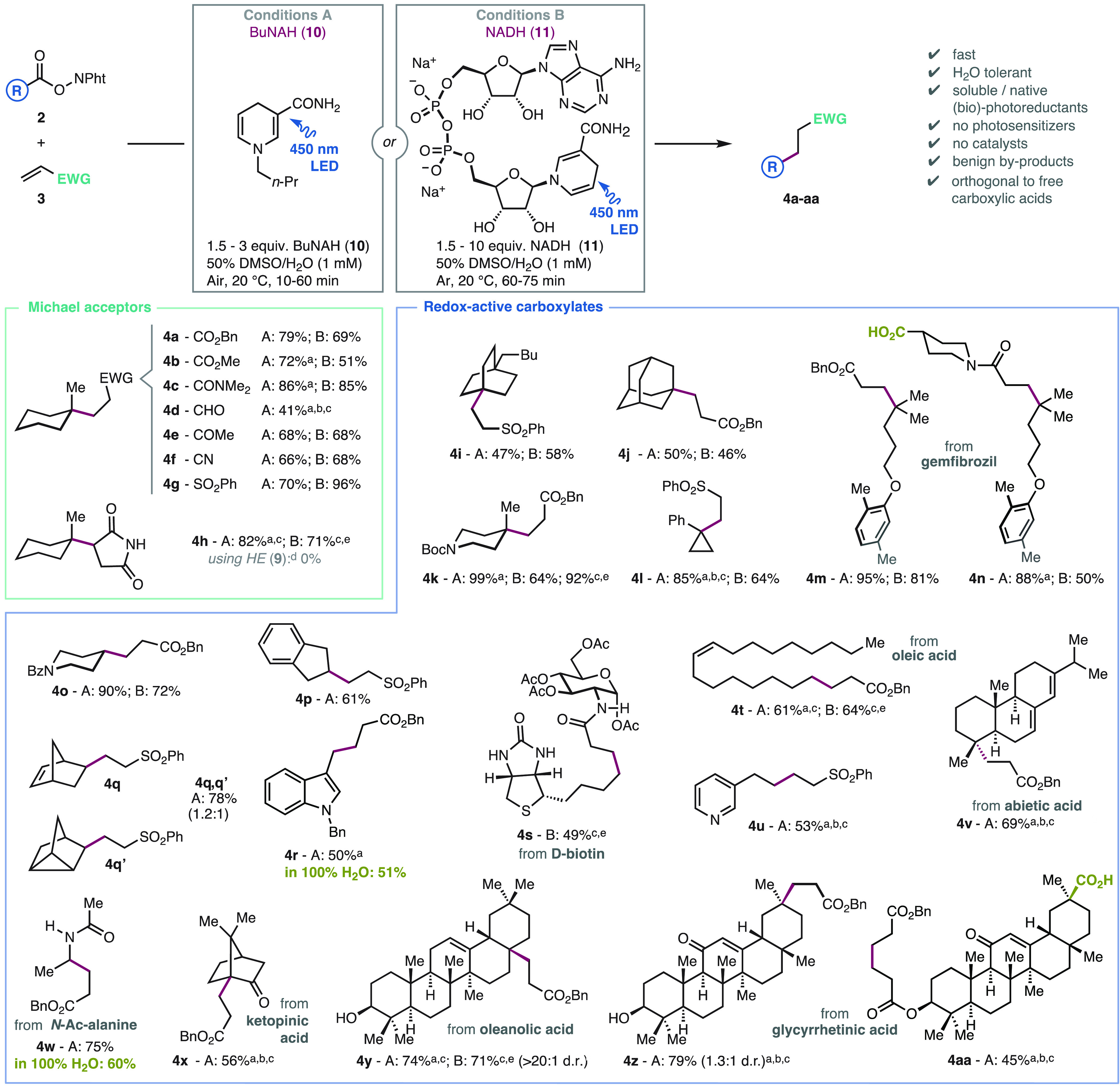
Yields were determined by 1H NMR using an appropriate internal standard; for isolated yields at preparative concentrations, see SI. HE; Hantzsch ester (9).
Ar atmosphere.
100 mM concentration.
DMSO was used as solvent.
Dihydropyridine 9 was used instead of BuNAH (10) for comparison.
20 mM concentration.
Interestingly, the products 4q,q’ demonstrate that the norbornenyl–nortricyclyl radical equilibrium28 can be established before their capture by the Michael acceptor. Primary carboxylate derivatives led to the products (4r–u) featuring robust and flexible alkyl-ligations. These include the cross-coupling of indole (4r), D-biotin (4s), a fatty acid (4t), and pyridine (4u) derivatives. Among those, the biotinylated product 4s displays an easily oxidizable thioether, a polar urea, a secondary amide, and an anomerically activated sugar.29 Moreover, the reaction was proven to be useful in the late-stage functionalization of natural products, including the peptide model derived from alanine (4w), and various densely functionalized terpenes with unprotected ketone, enone, olefin, diene, alcohol, and ester functions (4v,x-aa). The orthogonality between redox-active esters and unprotected carboxylic acids is demonstrated on the products 4n,aa. These substrates would lead to mixtures of products and/or polymers through existing coupling reactions based on oxidative decarboxylation.8c−8e Furthermore, the coupling reactions were complete in 10–75 min. This is substantially faster than previous methods despite the dilute conditions. Particularly sensitive or apolar substrates were understandably less efficient in the standard dilute aqueous media of the reaction (i.e., 4d,s,u,v,y,z,aa). In these cases, coupling efficiencies are enhanced simply by using higher concentration, inert atmosphere, and DMSO as solvent. However, in more favorable substrates, the reaction could operate even in pure water as solvent with similar results (4r,w).
The features of this system in terms of rate, concentration, water tolerance, and solubility of its components made it an ideal candidate for the in vitro alkyl photocoupling on polar biomacromolecules. To benchmark the performances of BuNAH (10) and NADH (11) in this context against comparable decarboxylative coupling methods, we set out to explore coupling reactions on DNA.8b,8d These are important in the synthesis of DNA-encoded libraries (DEL)8b,8d,9e yet challenging due to the complex functionality of the substrates and low scale at which these reactions need to occur. To our delight, DEL headpieces 12a,b were coupled efficiently using either BuNAH (10) or NADH (11) and blue light at 10 nmol-scale to deliver “on-DNA”-functionalized products 13aa-ae,ba-be (Scheme 4). These reactions are generally completed in 1 h with excellent yields despite the micromolar concentrations (100 μM) in buffered media. Importantly, all the components in this system can be handled as dilute solutions, thus facilitating mixing in small reaction volumes (<300 μL). These features are characteristic of this system and can facilitate the future implementation of this reaction in automatic platforms.
Scheme 4. Alkyl Photo-Coupling on DNA.
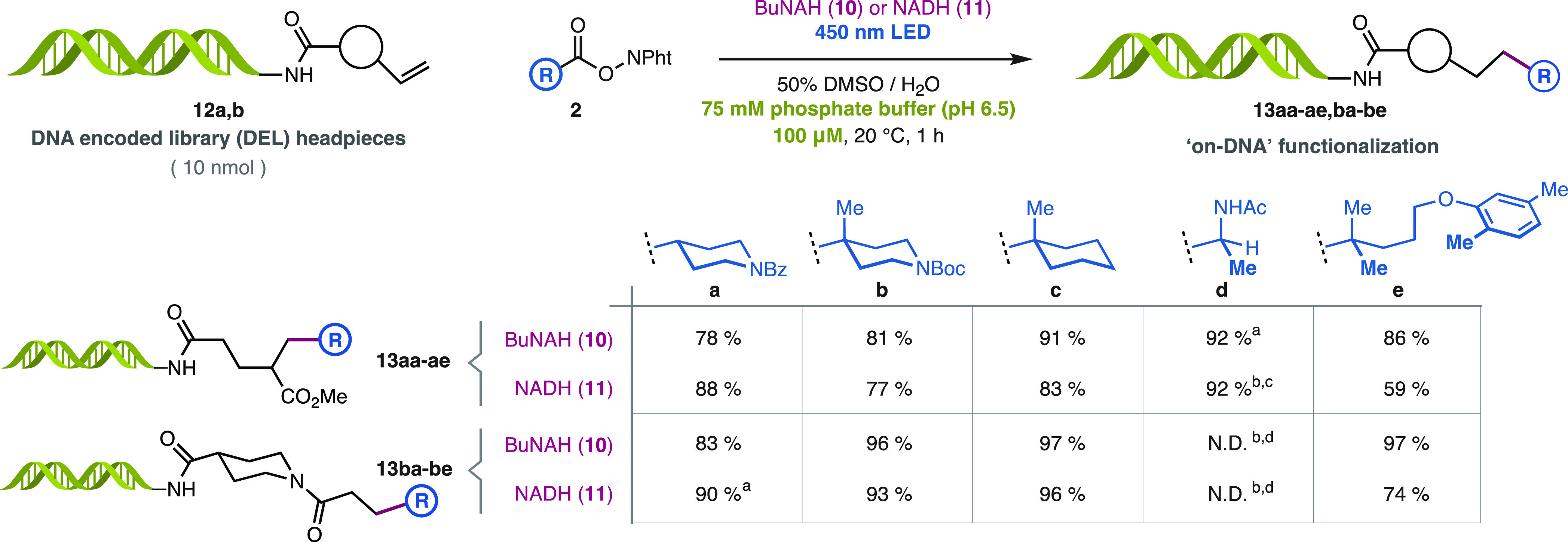
Reaction time 2 h.
Reaction time 4 h.
Buffer pH 5.5.
Coupling product was detected by MS but could not be quantified due to insufficient chromatographic resolution. ND, Not Determined.
The kinetic time-profile of the reaction with BuNAH (10) was obtained using in situ no-D NMR monitoring.30 Nondeuterated DMSO was used to prevent any potential artifacts due to solvent isotopic effects in the propagation of the radical chain. However, it was found that the reaction proceeds similarly in DMSO and DMSO-d6, without any solvent-derived byproducts (see SI). This way it was possible to confirm that the reaction can be completed in 4.3 min at 100 mM concentration (Scheme 5A; gray).8f−8l Moreover, 10-fold (10 mM) and even 100-fold (1 mM) dilutions resulted in a surprising rate acceleration (Scheme 5A; blue traces). The reaction is completed in just 80 s of illumination at 1–10 mM with identical efficiency. To discern the origin of the acceleration, a control experiment was run in the least favorable concentration (100 mM) using a thinner reactor tube (1.25 mm diameter) to minimize the light path on the system. This resulted in significantly faster kinetics (Scheme 5A; red). This result demonstrates that the acceleration observed upon dilution stems from the attenuation of the inner filter effect.31 With the usage of NADH (11) as photoreductant, the reaction is slower (Scheme 5B) but faster than previous decarboxylative coupling reactions.8,24 The reaction is completed in 60 min (>80% in less than 25 min). These results are remarkable considering the dilute conditions (20 mM) in the presence of only 1.5 equiv of the acceptor 3a and NADH (11). Importantly, the system is stable in the absence of light (Scheme 5C). After a long dark period, the system was illuminated obtaining an identical kinetic profile to that of a standard experiment, as evidenced by the time-shifted overlay (Scheme 5D). This demonstrates the absence of static deactivation in the dark, which may be relevant in cases where other equilibria need to be established before the C–C coupling event is phototriggered.3,5,6
Scheme 5. Kinetic Profiling of the Photo-Coupling by No-D 1H NMR.

Absorption spectroscopy revealed that the light absorption of BuNAH (10) is similar to those of other dihydronicotinamides,16 featuring a strong band at 350 nm that extends into the visible region (Scheme 6A, left). In the presence of the redox-active ester 2a, which only absorbs below 350 nm, the absorption increases marginally at 450 nm using concentrations as high as 0.1 M (12% increase; Scheme 6A, right), which may indicate the formation of a donor–acceptor complex (EDA).32,33 Thus, we set out to study the relevance of this possible EDA interaction in the photoactivation of this reaction. Stern–Volmer studies evidenced a linear quenching of the steady-state fluorescence of BuNAH (10; Scheme 6B; blue) with increasing concentrations of the redox-active ester 2a. Nevertheless, the linear decrease in luminescence intensity is not a definitive proof of the mechanism by which this phenomenon occurs.34 Therefore, the fluorescence lifetime of the excited state 10* (τ0(10*) = 1.08 ns) was measured using Time-Correlated Single Photon Counting (TCSPC). This study revealed a decrease in the lifetime of excited BuNAH (10*, Scheme 6B; purple) upon increase of the concentration of redox-active ester 2a. However, the significantly different slopes of the steady-state and lifetime Stern–Volmer plots were not consistent with a conventional dynamic quenching scenario.34 Instead, the data supports the formation of a nonemissive EDA complex 10·2 in equilibrium with the free 10 (Scheme 6B; right). The corresponding equilibrium constant could be estimated through fitting of the steady-state and lifetime data (Keq ∼ 7; see SI).34 Consistently, no additional luminescence bands corresponding to the EDA complex 10·2 could be observed in either excitation or emission spectra (see SI). At this point, it is unclear which of these coexisting dynamic and static interactions between BuNAH (10) and the redox-active ester 2 are most important for the reactivity. However, it is known that the formation of EDA complexes is affected by changes in the substrate, solvent, concentration, and/or temperature.32 The fact that the reaction is not inhibited in dilute conditions disfavors the EDA complex to be critical in the photoactivation of this system.24 In this sense, the direct reduction by photoexcited dihydronicotinamides without engagement in donor–acceptor complexes32,33 has been documented but only in the context of more activated alkyl halide substrates.17b
Scheme 6. Mechanistic Studies.

The expected intermediacy of free-diffusing alkyl radicals was demonstrated by the different ratios of the products 4ab,ab’ that were obtained using the 5-hexenyl radical clock precursor 2ab at different initial concentrations (Scheme 6C). To discern the fate of the radical intermediate that would result from the addition of the alkyl radical into the electron-deficient olefin, we conducted a series of experiments with the dideuterated BuNAH derivative 10-d2 (Scheme 6D). These experiments revealed that hydrogen atom transfer (HAT) from BuNAH (10) is the main process to quench the putative radical addition product.8g,8h,19c−19e Further control experiments confirmed that the solvents (DMSO and H2O) do not exchange with 10-d2 under the reaction conditions and do not have any relevant role in the HAT process (see SI). The involvement of a radical chain mechanism was studied measuring the average quantum yield. This was determined in triplicate at 20–25% conversion of 2a, obtaining a value of 2.9 ± 0.5, which points to the propagation of a radical chain.31
The mechanistic proposal in Scheme 7 comprises the electron–proton–electron transfer manifold that is typical in radical reductions mediated by dihydronicotinamides14,17,35 and our own experiments (Schemes 5 and 6). Photoinduced electron and proton transfer from dihydronicotinamide 10 to the redox-active ester 2 through the dynamic and/or static mechanisms discussed above (Scheme 6B) produces the carbon centered radical 14, a nicotinyl radical 15, phthalimide (16), and CO2. The radical 14 adds to the olefin 3 to produce the radical 17, which after concerted8g or stepwise35 HAT yields the coupling product 4 and the nicotinyl radical 15.8g The latter could reduce the redox-active ester 2 to produce the pyridinium salt 18, CO2, and the alkyl radical 14 that propagates the chain reaction (see Scheme 6C,D).8g The formation of the pyridinium salts 18 derived from BuNAH (10) and NADH (11) and their kinetic correlation with the formation of the product 4 has also been evidenced by in situ NMR monitoring (see SI).
Scheme 7. Proposed Mechanism.

Conclusions
Herein, we report that the dihydronicotinamides BuNAH (10) and NADH (11) promote the photocoupling of redox-active esters and Michael acceptors upon illumination with blue light. These reactions do not require external photocatalysts or additives, have no detectable background reactivity, can run in water, and have an unusually high rate even at low concentration. This system has demonstrated its utility in the functionalization of DNA macromolecules in extremely dilute conditions. The mechanistic experiments demonstrate the multifaceted role of these dihydropyridines as photoinitiators, reductants, and hydrogen-atom donors to drive this fast photocoupling using a minimal homogeneous system. This work introduces NADH (11) as an autonomous photoreductant and opens prospects for new artificial coupling reactions that our group is currently investigating.
Acknowledgments
We are indebted to the personnel of AstraZeneca Gothenburg and the Dept. of Organic Chemistry at Stockholm University for unrestricted support. The authors particularly thank Dr F. Juliá-Hernández for fruitful discussions, and Prof. E. Borbas and S. Kiraev (Uppsala U.) for assistance with early stage fluorescence measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09678.
Experimental procedures, characterization, and other data (PDF)
Author Contributions
$ These authors contributed equally.
Financial support from the Knut and Alice Wallenberg Foundation (KAW2016.0153), and the European Research Council (714737) is gratefully acknowledged.
The authors declare no competing financial interest.
Notes
Raw data for this article can be downloaded from Zenodo DOI: 10.5281/zenodo.4106400.
Supplementary Material
References
- a Ankenbruck N.; Courtney T.; Naro Y.; Deiters A. Optochemical Control of Biological Processes in Cells and Animals. Angew. Chem., Int. Ed. 2018, 57 (11), 2768–2798. 10.1002/anie.201700171. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fehrentz T.; Schönberger M.; Trauner D. Optochemical Genetics. Angew. Chem., Int. Ed. 2011, 50 (51), 12156–12182. 10.1002/anie.201103236. [DOI] [PubMed] [Google Scholar]; c Albert L.; Vázquez O. Photoswitchable Peptides for Spatiotemporal Control of Biological Functions. Chem. Commun. 2019, 55 (69), 10192–10213. 10.1039/C9CC03346G. [DOI] [PubMed] [Google Scholar]; d Szymański W.; Beierle J. M.; Kistemaker H. A. V.; Velema W. A.; Feringa B. L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113 (8), 6114–6178. 10.1021/cr300179f. [DOI] [PubMed] [Google Scholar]; e Heinrich B.; Bouazoune K.; Wojcik M.; Bakowsky U.; Vázquez O. ortho-Fluoroazobenzene Derivatives as DNA Intercalators for Photocontrol of DNA and Nucleosome Binding by Visible Light. Org. Biomol. Chem. 2019, 17 (7), 1827–1833. 10.1039/C8OB02343C. [DOI] [PubMed] [Google Scholar]; f Broichhagen J.; Trauner D. The in vivo Chemistry of Photoswitched Tethered Ligands. Curr. Opin. Chem. Biol. 2014, 21, 121–127. 10.1016/j.cbpa.2014.07.008. [DOI] [PubMed] [Google Scholar]
- a Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113 (1), 119–191. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kolarski D.; Sugiyama A.; Breton G.; Rakers C.; Ono D.; Schulte A.; Tama F.; Itami K.; Szymanski W.; Hirota T.; Feringa B. L. Controlling the Circadian Clock with High Temporal Resolution through Photodosing. J. Am. Chem. Soc. 2019, 141 (40), 15784–15791. 10.1021/jacs.9b05445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4 (8), 952–959. 10.1021/acscentsci.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of nonlight regulated bioorthogonal reactions, see:; a Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Patterson D. M.; Nazarova L. A.; Prescher J. A. Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol. 2014, 9 (3), 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]; c Whiting M.; Muldoon J.; Lin Y.-C.; Silverman S. M.; Lindstrom W.; Olson A. J.; Kolb H. C.; Finn M. G.; Sharpless K. B.; Elder J. H.; Fokin V. V. Inhibitors of HIV-1 Protease by Using In Situ Click Chemistry. Angew. Chem., Int. Ed. 2006, 45 (9), 1435–1439. 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]; d Fang Y.; Zhang H.; Huang Z.; Scinto S. L.; Yang J. C.; Am Ende C. W.; Dmitrenko O.; Johnson D. S.; Fox J. M. Photochemical Syntheses, Transformations, and Bioorthogonal Chemistry of Trans-Cycloheptene and Sila Trans-Cycloheptene Ag(I) complexes. Chem. Sci. 2018, 9 (7), 1953–1963. 10.1039/C7SC04773H. [DOI] [PMC free article] [PubMed] [Google Scholar]; e An P.; Lewandowski T. M.; Erbay T. G.; Liu P.; Lin Q. Sterically Shielded, Stabilized Nitrile Imine for Rapid Bioorthogonal Protein Labeling in Live Cells. J. Am. Chem. Soc. 2018, 140 (14), 4860–4868. 10.1021/jacs.8b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Blackman M. L.; Royzen M.; Fox J. M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc. 2008, 130 (41), 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Patterson D. M.; Nazarova L. A.; Xie B.; Kamber D. N.; Prescher J. A. Functionalized Cyclopropenes As Bioorthogonal Chemical Reporters. J. Am. Chem. Soc. 2012, 134 (45), 18638–18643. 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]; h Sachdeva A.; Wang K.; Elliott T.; Chin J. W. Concerted, Rapid, Quantitative, and Site-Specific Dual Labeling of Proteins. J. Am. Chem. Soc. 2014, 136 (22), 7785–7788. 10.1021/ja4129789. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Laughlin S. T.; Baskin J. M.; Amacher S. L.; Bertozzi C. R. In Vivo Imaging of Membrane-Associated Glycans in Developing Zebrafish. Science 2008, 320 (5876), 664–667. 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of light-regulated bioorthogonal reactions:; a Tasdelen M. A.; Yagci Y. Light-Induced Click Reactions. Angew. Chem., Int. Ed. 2013, 52 (23), 5930–5938. 10.1002/anie.201208741. [DOI] [PubMed] [Google Scholar]; b Chen R. T.; Marchesan S.; Evans R. A.; Styan K. E.; Such G. K.; Postma A.; McLean K. M.; Muir B. W.; Caruso F. Photoinitiated Alkyne–Azide Click and Radical Cross-Linking Reactions for the Patterning of PEG Hydrogels. Biomacromolecules 2012, 13 (3), 889–895. 10.1021/bm201802w. [DOI] [PubMed] [Google Scholar]; c Singh K.; Fennell C. J.; Coutsias E. A.; Latifi R.; Hartson S.; Weaver J. D. Light Harvesting for Rapid and Selective Reactions: Click Chemistry with Strain-Loadable Alkenes. Chem. 2018, 4 (1), 124–137. 10.1016/j.chempr.2017.11.007. [DOI] [Google Scholar]; d Kaur G.; Singh G.; Singh J. Photochemical Tuning of Materials: A Click Chemistry Perspective. Mater. Today Chem. 2018, 8, 56–84. 10.1016/j.mtchem.2018.03.002. [DOI] [Google Scholar]; e Bordoni A. V.; Lombardo M. V.; Wolosiuk A. Photochemical radical thiol–ene click-based methodologies for silica and transition metal oxides materials chemical modification: a mini-review. RSC Adv. 2016, 6 (81), 77410–77426. 10.1039/C6RA10388J. [DOI] [Google Scholar]; f Ramil C. P.; Lin Q. Photoclick chemistry: a fluorogenic light-triggered in vivo ligation reaction. Curr. Opin. Chem. Biol. 2014, 21, 89–95. 10.1016/j.cbpa.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li G.; Liu Y.; Liu Y.; Chen L.; Wu S.; Liu Y.; Li X. Photoaffinity Labeling of Small-Molecule-Binding Proteins by DNA-Templated Chemistry. Angew. Chem., Int. Ed. 2013, 52 (36), 9544–9549. 10.1002/anie.201302161. [DOI] [PubMed] [Google Scholar]; b Herner A.; Lin Q. Photo-Triggered Click Chemistry for Biological Applications. Top. Curr. Chem. 2016, 374 (1), 1. 10.1007/s41061-015-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Choi D. S.; Kuk S. K.; Park C. B. Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem., Int. Ed. 2018, 57 (27), 7958–7985. 10.1002/anie.201710070. [DOI] [PubMed] [Google Scholar]
- For decarboxylative radical additions with redox-active esters in biomolecules, see:; a Qin T.; Malins L. R.; Edwards J. T.; Merchant R. R.; Novak A. J. E.; Zhong J. Z.; Mills R. B.; Yan M.; Yuan C.; Eastgate M. D.; Baran P. S. Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chem., Int. Ed. 2017, 56 (1), 260–265. 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang J.; Lundberg H.; Asai S.; Martín-Acosta P.; Chen J. S.; Brown S.; Farrell W.; Dushin R. G.; O’Donnell C. J.; Ratnayake A. S.; Richardson P.; Liu Z.; Qin T.; Blackmond D. G.; Baran P. S. Kinetically guided radical-based synthesis of C(sp3)–C(sp3) linkages on DNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (28), E6404–E6410. 10.1073/pnas.1806900115. [DOI] [PMC free article] [PubMed] [Google Scholar]; For decarboxylative radical additions with carboxylic acids in biomolecules, see:; c McCarver S. J.; Qiao J. X.; Carpenter J.; Borzilleri R. M.; Poss M. A.; Eastgate M. D.; Miller M. M.; MacMillan D. W. C. Decarboxylative Peptide Macrocyclization through Photoredox Catalysis. Angew. Chem., Int. Ed. 2017, 56 (3), 728–732. 10.1002/anie.201608207. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kölmel D. K.; Loach R. P.; Knauber T.; Flanagan M. E. Employing Photoredox Catalysis for DNA-Encoded Chemistry: Decarboxylative Alkylation of α-Amino Acids. ChemMedChem 2018, 13 (20), 2159–2165. 10.1002/cmdc.201800492. [DOI] [PubMed] [Google Scholar]; e Bloom S.; Liu C.; Kölmel D. K.; Qiao J. X.; Zhang Y.; Poss M. A.; Ewing W. R.; MacMillan D. W. C. Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials. Nat. Chem. 2018, 10, 205–211. 10.1038/nchem.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]; For photocatalyzed coupling of redox-active esters and Michael acceptors, see:; f Brandhofer T.; Mancheño O. G. Versatile Ru-Photoredox-Catalyzed Functionalization of Dehydro-Amino Acids and Peptides. ChemCatChem 2019, 11 (16), 3797–3801. 10.1002/cctc.201900446. [DOI] [Google Scholar]; g Okada K.; Okamoto K.; Morita N.; Okubo K.; Oda M. Photosensitized decarboxylative Michael addition through N-(acyloxy)phthalimides via an electron-transfer mechanism. J. Am. Chem. Soc. 1991, 113 (24), 9401–9402. 10.1021/ja00024a074. [DOI] [Google Scholar]; h Pratsch G.; Lackner G. L.; Overman L. E. Constructing Quaternary Carbons from N-(Acyloxy)phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis. J. Org. Chem. 2015, 80 (12), 6025–6036. 10.1021/acs.joc.5b00795. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Schnermann M. J.; Overman L. E. A Concise Synthesis of (−)-Aplyviolene Facilitated by a Strategic Tertiary Radical Conjugate Addition. Angew. Chem., Int. Ed. 2012, 51 (38), 9576–9580. 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Schwarz J.; König B. Metal-free, visible-light-mediated, decarboxylative alkylation of biomass-derived compounds. Green Chem. 2016, 18 (17), 4743–4749. 10.1039/C6GC01101B. [DOI] [Google Scholar]; k Müller D. S.; Untiedt N. L.; Dieskau A. P.; Lackner G. L.; Overman L. E. Constructing Quaternary Stereogenic Centers Using Tertiary Organocuprates and Tertiary Radicals. Total Synthesis of trans-Clerodane Natural Products. J. Am. Chem. Soc. 2015, 137 (2), 660–663. 10.1021/ja512527s. [DOI] [PubMed] [Google Scholar]; l Jamison C. R.; Overman L. E. Fragment Coupling with Tertiary Radicals Generated by Visible-Light Photocatalysis. Acc. Chem. Res. 2016, 49 (8), 1578–1586. 10.1021/acs.accounts.6b00284. [DOI] [PubMed] [Google Scholar]; for a related decarboxylative alkynylation, see:; m Yang J.; Zhang J.; Qi L.; Hu C.; Chen Y. Visible-light-induced Chemoselective Reductive Decarboxylative Alkynylation under Biomolecule-compatible Conditions. Chem. Commun. 2015, 51, 5275–5278. 10.1039/C4CC06344A. [DOI] [PubMed] [Google Scholar]; n Chen T. – G.; Barton L. M.; Lin Y.; Tsien J.; Kossler D.; Bastida I.; Asai S.; Bi C.; Chen J. S.; Shan M.; Fang H.; Fang F. G.; Choi H. – W.; Hawkins L.; Qin T.; Baran P. S. Building C(sp3)-rich complexity by combining cycloaddition and C–C cross-coupling reactions. Nature 2018, 560, 350–354. 10.1038/s41586-018-0391-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For other types of photochemical radical couplings in biomolecules, see:; a Aycock R. A.; Pratt C. J.; Jui N. T. Aminoalkyl Radicals as Powerful Intermediates for the Synthesis of Unnatural Amino Acids and Peptides. ACS Catal. 2018, 8 (10), 9115–9119. 10.1021/acscatal.8b03031. [DOI] [Google Scholar]; b Kölmel D. K.; Meng J.; Tsai M.-H.; Que J.; Loach R. P.; Knauber T.; Wan J.; Flanagan M. E. On-DNA Decarboxylative Arylation: Merging Photoredox with Nickel Catalysis in Water. ACS Comb. Sci. 2019, 21 (8), 588–597. 10.1021/acscombsci.9b00076. [DOI] [PubMed] [Google Scholar]; c Garreau M.; Le Vaillant F.; Waser J. C-Terminal Bioconjugation of Peptides through Photoredox Catalyzed Decarboxylative Alkynylation. Angew. Chem., Int. Ed. 2019, 58 (24), 8182–8186. 10.1002/anie.201901922. [DOI] [PubMed] [Google Scholar]; d Bottecchia C.; Noël T. Photocatalytic Modification of Amino Acids, Peptides, and Proteins. Chem. - Eur. J. 2019, 25 (1), 26–42. 10.1002/chem.201803074. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Phelan J. P.; Lang S. B.; Sim J.; Berritt S.; Peat A. J.; Billings K.; Fan L.; Molander G. A. Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc. 2019, 141 (8), 3723–3732. 10.1021/jacs.9b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bratt E.; Suárez-Pantiga S.; Johansson M. J.; Mendoza A. Mechanism and regioselectivity of the anionic oxidative rearrangement of 1,3-diketones towards all-carbon quaternary carboxylates. Chem. Commun. 2019, 55 (60), 8844–8847. 10.1039/C9CC03331A. [DOI] [PubMed] [Google Scholar]; b Colas K.; dos Santos A. C. V. D.; Mendoza A. i-Pr2NMgCl·LiCl Enables the Synthesis of Ketones by Direct Addition of Grignard Reagents to Carboxylate Anions. Org. Lett. 2019, 21 (19), 7908–7913. 10.1021/acs.orglett.9b02899. [DOI] [PubMed] [Google Scholar]
- a Renault K.; Fredy J. W.; Renard P.-Y.; Sabot C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjugate Chem. 2018, 29 (8), 2497–2513. 10.1021/acs.bioconjchem.8b00252. [DOI] [PubMed] [Google Scholar]; b Spicer C. D.; Pashuck E. T.; Stevens M. M. Achieving Controlled Biomolecule–Biomaterial Conjugation. Chem. Rev. 2018, 118 (16), 7702–7743. 10.1021/acs.chemrev.8b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee Y.-J.; Wu B.; Raymond J. E.; Zeng Y.; Fang X.; Wooley K. L.; Liu W. R. A Genetically Encoded Acrylamide Functionality. ACS Chem. Biol. 2013, 8 (8), 1664–1670. 10.1021/cb400267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yu Z.; Mendoza A. Enantioselective Assembly of Congested Cyclopropanes using Redox-Active Aryldiazoacetates. ACS Catal. 2019, 9 (9), 7870–7875. 10.1021/acscatal.9b02615. [DOI] [Google Scholar]; b Montesinos-Magraner M.; Costantini M.; Ramírez-Contreras R.; Muratore M. E.; Johansson M. J.; Mendoza A. General Cyclopropane Assembly by Enantioselective Transfer of a Redox-Active Carbene to Aliphatic Olefins. Angew. Chem., Int. Ed. 2019, 58 (18), 5930–5935. 10.1002/anie.201814123. [DOI] [PubMed] [Google Scholar]
- a Horner L.; Jordan M. Studien zum Vorgang der Wasserstoffübertragung, 53. Beitrag zur Kenntnis der elektroreduktiven Spaltung von Hydroxylaminderivaten. Justus Liebigs Ann. Chem. 1978, 1978 (9), 1518–1525. 10.1002/jlac.197819780916. [DOI] [Google Scholar]; b Li H.; Breen C. P.; Seo H.; Jamison T. F.; Fang Y.-Q.; Bio M. M. Ni-Catalyzed Electrochemical Decarboxylative C–C Couplings in Batch and Continuous Flow. Org. Lett. 2018, 20 (5), 1338–1341. 10.1021/acs.orglett.8b00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzumi S.; Inada O.; Suenobu T. Mechanisms of Electron-Transfer Oxidation of NADH Analogues and Chemiluminescence. Detection of the Keto and Enol Radical Cations. J. Am. Chem. Soc. 2003, 125 (16), 4808–4816. 10.1021/ja029623y. [DOI] [PubMed] [Google Scholar]
- For photocatalysis based on NAD+ excitation, using UV light and sacrificial electron donors, see:; Kim J.; Lee S. H.; Tieves F.; Paul C. E.; Hollmann F.; Park C. B. Nicotinamide adenine dinucleotide as a photocatalyst. Sci. Adv. 2019, 5 (7), eaax0501. 10.1126/sciadv.aax0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens F. M.; Verhoeven J. W.; Gase R. A.; Pandit U. K.; de Boer T. J. On the question of one-electron transfer in the mechanism of reduction by NADH-models. Tetrahedron 1978, 34 (4), 443–446. 10.1016/0040-4020(78)80030-1. [DOI] [Google Scholar]
- For seminal work on the photochemistry of NADH analogues, see:; a Fukuzumi S.; Mochizuki S.; Tanaka T. Photoreduction of phenacyl halides by NADH analogues. Origins of different mechanistic pathways. J. Chem. Soc., Perkin Trans. 2 1989, 2 (10), 1583–1589. 10.1039/p29890001583. [DOI] [Google Scholar]; b Fukuzumi S.; Hironaka K.; Tanaka T. Photoreduction of alkyl halides by an NADH model compound. An electron-transfer chain mechanism. J. Am. Chem. Soc. 1983, 105 (14), 4722–4727. 10.1021/ja00352a034. [DOI] [Google Scholar]; c Fukuzumi S.; Koumitsu S.; Hironaka K.; Tanaka T. Energetic comparison between photoinduced electron-transfer reactions from NADH model compounds to organic and inorganic oxidants and hydride-transfer reactions from NADH model compounds to p-benzoquinone derivatives. J. Am. Chem. Soc. 1987, 109 (2), 305–316. 10.1021/ja00236a003. [DOI] [Google Scholar]
- For a related radical fragmentation of photoexcited 4-alkyl-dihydropyridines, see:; a van Leeuwen T.; Buzzetti L.; Perego L. A.; Melchiorre P. A Redox-Active Nickel Complex that Acts as an Electron Mediator in Photochemical Giese Reactions. Angew. Chem., Int. Ed. 2019, 58 (15), 4953–4957. 10.1002/anie.201814497. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Buzzetti L.; Prieto A.; Roy S. R.; Melchiorre P. Radical-based C-C Bond-Forming Processes Enabled by the Photoexcitation of 4-Alkyl-1,4-dihydropyridines. Angew. Chem., Int. Ed. 2017, 56 (47), 15039–15043. 10.1002/anie.201709571. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bonet A. G.; Tellis J. C.; Matsui J. K.; Vara B. A.; Molander G. A. 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis. ACS Catal. 2016, 6, 8004–8008. 10.1021/acscatal.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related photocatalyzed reactions of redox-active esters, see:; a Okada K.; Okamoto K.; Oda M. A new and practical method of decarboxylation: photosensitized decarboxylation of N-acyloxyphthalimides via electron-transfer mechanism. J. Am. Chem. Soc. 1988, 110 (26), 8736–8738. 10.1021/ja00234a047. [DOI] [Google Scholar]; b Okada K.; Okubo K.; Morita N.; Oda M. Reductive decarboxylation of N-(acyloxy)phthalimides via redox-initiated radical chain mechanism. Tetrahedron Lett. 1992, 33 (48), 7377–7380. 10.1016/S0040-4039(00)60192-2. [DOI] [Google Scholar]; c Pac C.; Ihama M.; Yasuda M.; Miyauchi Y.; Sakurai H. Tris(2,2’-bipyridine)ruthenium(2+)-mediated photoreduction of olefins with 1-benzyl-1,4-dihydronicotinamide: a mechanistic probe for electron-transfer reactions of NAD(P)H-model compounds. J. Am. Chem. Soc. 1981, 103 (21), 6495–6497. 10.1021/ja00411a040. [DOI] [Google Scholar]; d Lackner G. L.; Quasdorf K. W.; Overman L. E. Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of tert-Alkyl N-Phthalimidoyl Oxalates. J. Am. Chem. Soc. 2013, 135 (41), 15342–15345. 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]; For radical couplings using nonredox-ester phthalimide derivatives, see:; e Ito Y.; Kimura A.; Osawa T.; Hari Y. Photoredox-Catalyzed Deformylative 1,4-Addition of 2′-Deoxy-5′-O-phthalimidonucleosides for Synthesis of 5′-Carba Analogs of Nucleoside 5′-Phosphates. J. Org. Chem. 2018, 83 (18), 10701–10708. 10.1021/acs.joc.8b00637. [DOI] [PubMed] [Google Scholar]; f Deng Y.; Nguyen M. D.; Zou Y.; Houk K. N.; Smith A. B. Generation of Dithianyl and Dioxolanyl Radicals Using Photoredox Catalysis: Application in the Total Synthesis of the Danshenspiroketallactones via Radical Relay Chemistry. Org. Lett. 2019, 21 (6), 1708–1712. 10.1021/acs.orglett.9b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhang J.; Li Y.; Zhang F.; Hu C.; Chen Y. Generation of Alkoxyl Radicals by Photoredox Catalysis Enables Selective C(sp3)–H Functionalization under Mild Reaction Conditions. Angew. Chem., Int. Ed. 2016, 55 (5), 1872–1875. 10.1002/anie.201510014. [DOI] [PubMed] [Google Scholar]
- Emmanuel M. A.; Greenberg N. R.; Oblinsky D. G.; Hyster T. K. Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540, 414–417. 10.1038/nature20569. [DOI] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55 (1), 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- a Tsurugi H.; Mashima K. Salt-Free Reduction of Transition Metal Complexes by Bis(trimethylsilyl)cyclohexadiene, -dihydropyrazine, and −4,4′-bipyridinylidene Derivatives. Acc. Chem. Res. 2019, 52 (3), 769–779. 10.1021/acs.accounts.8b00638. [DOI] [PubMed] [Google Scholar]; b Tsurugi H.; Mashima K. A New Protocol to Generate Catalytically Active Species of Group 4–6 Metals by Organosilicon-Based Salt-Free Reductants. Chem. - Eur. J. 2019, 25 (4), 913–919. 10.1002/chem.201803181. [DOI] [PubMed] [Google Scholar]; c Broggi J.; Terme T.; Vanelle P. Organic Electron Donors as Powerful Single-Electron Reducing Agents in Organic Synthesis. Angew. Chem., Int. Ed. 2014, 53 (2), 384–413. 10.1002/anie.201209060. [DOI] [PubMed] [Google Scholar]; d Murphy J. A.; Khan T. A.; Zhou S.-z.; Thomson D. W.; Mahesh M. Highly Efficient Reduction of Unactivated Aryl and Alkyl Iodides by a Ground-State Neutral Organic Electron Donor. Angew. Chem., Int. Ed. 2005, 44 (9), 1356–1360. 10.1002/anie.200462038. [DOI] [PubMed] [Google Scholar]; e Murphy J. A.; Garnier J.; Park S. R.; Schoenebeck F.; Zhou S.-z.; Turner A. T. Super-Electron Donors: Bis-pyridinylidene Formation by Base Treatment of Pyridinium Salts. Org. Lett. 2008, 10 (6), 1227–1230. 10.1021/ol800134g. [DOI] [PubMed] [Google Scholar]
- Lorz P. M.; Towae F. K.; Enke W.; Jäckh R.; Bhargava N.; Hillesheim W., Phthalic Acid and Derivatives. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: New York, 2007. [Google Scholar]
- Zheng C.; Wang G.-Z.; Shang R. Catalyst-free Decarboxylation and Decarboxylative Giese Additions of Alkyl Carboxylates through Photoactivation of Electron Donor-Acceptor Complex. Adv. Synth. Catal. 2019, 361 (19), 4500–4505. 10.1002/adsc.201900803. [DOI] [Google Scholar]
- During our scope studies, it has been independently disclosed24 similar reactions promoted by the dihydropyridine 9, albeit only under strictly deoxygenated and anhydrous conditions, and with lower efficiency and rate. The dihydronicotinamide BNAH (5) was declared unsuitable for this reaction,24 in clear contrast with our results (Scheme 2, A; entry 5).
- a Brewster M. E.; Simay A.; Czako K.; Winwood D.; Farag H.; Bodor N. Reactivity of biologically important reduced pyridines. IV. Effect of substitution on ferricyanide-mediated oxidation rates of various 1,4-dihydropyridines. J. Org. Chem. 1989, 54 (15), 3721–3726. 10.1021/jo00276a039. [DOI] [Google Scholar]; b Paul C. E.; Gargiulo S.; Opperman D. J.; Lavandera I.; Gotor-Fernández V.; Gotor V.; Taglieber A.; Arends I. W. C. E.; Hollmann F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C=C Bioreduction Using Enoate Reductases. Org. Lett. 2013, 15 (1), 180–183. 10.1021/ol303240a. [DOI] [PubMed] [Google Scholar]
- Scott T. G.; Spencer R. D.; Leonard N. J.; Weber G. Synthetic spectroscopic models related to coenzymes and base pairs. V. Emission properties of NADH. Studies of fluorescence lifetimes and quantum efficiencies of NADH, AcPyADH, [reduced acetylpyridineadenine dinucleotide] and simplified synthetic models. J. Am. Chem. Soc. 1970, 92 (3), 687–695. 10.1021/ja00706a043. [DOI] [Google Scholar]
- a Alnajjar M. S.; Kuivila H. G. Free-radical and anion intermediates in the reactions of 5-halo-2-norbornenes and 3-halonortricyclenes with (trimethyltin)sodium. J. Org. Chem. 1981, 46 (6), 1053–1057. 10.1021/jo00319a002. [DOI] [Google Scholar]; b Walborsky H. M.; Topolski M. The surface nature of Grignard reagent formation. J. Am. Chem. Soc. 1992, 114 (9), 3455–3459. 10.1021/ja00035a044. [DOI] [Google Scholar]; c Wong P. C.; Griller D. A kinetic EPR study of the norbornenyl-nortricyclyl radical rearrangement. J. Org. Chem. 1981, 46 (11), 2327–2329. 10.1021/jo00324a023. [DOI] [Google Scholar]
- We have also detected the successful coupling of the fully unprotected sugar, but only trace amounts could be isolated.
- Hoye T. R.; Eklov B. M.; Ryba T. D.; Voloshin M.; Yao L. J. No-D NMR (No-Deuterium Proton NMR) Spectroscopy: A Simple Yet Powerful Method for Analyzing Reaction and Reagent Solutions. Org. Lett. 2004, 6 (6), 953–956. 10.1021/ol049979+. [DOI] [PubMed] [Google Scholar]
- Buzzetti L.; Crisenza G. E. M.; Melchiorre P. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019, 58 (12), 3730–3747. 10.1002/anie.201809984. [DOI] [PubMed] [Google Scholar]
- a Rosokha S. V.; Kochi J. K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41 (5), 641–653. 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]; b Lima C. G. S.; Lima T. M.; Duarte M.; Jurberg I. D.; Paixao M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6 (3), 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]; c Foster R. Electron donor-acceptor complexes. J. Phys. Chem. 1980, 84 (17), 2135–2141. 10.1021/j100454a006. [DOI] [Google Scholar]
- Cao Z.-Y.; Ghosh T.; Melchiorre P. Enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion-based EDA complex. Nat. Commun. 2018, 9 (1), 3274. 10.1038/s41467-018-05375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lakowicz J. R.Principles of Fluorescence Spectroscopy, Plenum: New York, 1983, ch. 9;. [Google Scholar]; b Fraiji L. K.; Hayes D. M.; Werner T. C. Static and Dynamic Fluorescence Quenching Experiments for the Physical Chemistry Laboratory J. J. Chem. Educ. 1992, 69, 424–428. 10.1021/ed069p424. [DOI] [Google Scholar]
- Martens F. M.; Verhoeven J. W.; Varma C. A. G. O.; Bergwerf P. Photo-oxidation of 1,4-dihydropyridines by various electron acceptors: a laser flash photolysis study. J. Photochem. 1983, 22 (2), 99–113. 10.1016/0047-2670(83)80032-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.