

Abstract
The mammalian epididymis not only plays a fundamental role in the maturation of spermatozoa, but also provides protection against various stressors. The foremost among these is the threat posed by oxidative stress, which arises from an imbalance in reactive oxygen species and can elicit damage to cellular lipids, proteins, and nucleic acids. In mice, the risk of oxidative damage to spermatozoa is mitigated through the expression and secretion of glutathione peroxidase 5 (GPX5) as a major luminal scavenger in the proximal caput epididymidal segment. Accordingly, the loss of GPX5-mediated protection leads to impaired DNA integrity in the spermatozoa of aged Gpx5-/- mice. To explore the underlying mechanism, we have conducted transcriptomic analysis of caput epididymidal epithelial cells from aged (13 months old) Gpx5-/- mice. This analysis revealed the dysregulation of several thousand epididymal mRNA transcripts, including the downregulation of a subgroup of piRNA pathway genes, in aged Gpx5-/- mice. In agreement with these findings, we also observed the loss of piRNAs, which potentially bind to the P-element-induced wimpy testis (PIWI)-like proteins PIWIL1 and PIWIL2. The absence of these piRNAs was correlated with the elevated mRNA levels of their putative gene targets in the caput epididymidis of Gpx5-/- mice. Importantly, the oxidative stress response genes tend to have more targeting piRNAs, and many of them were among the top increased genes upon the loss of GPX5. Taken together, our findings suggest the existence of a previously uncharacterized somatic piRNA pathway in the mammalian epididymis and its possible involvement in the aging and oxidative stress-mediated responses.
Keywords: epididymis, GPX5, oxidative stress, piRNA, PIWI-interacting RNA, small noncoding RNA
INTRODUCTION
The mammalian epididymis plays important roles not only in the functional maturation of spermatozoa, but also in their storage and protection. The latter is particularly important because mature spermatozoa lack protective enzymatic activities and are thus placed at heightened vulnerability to various stressors. Among the potential stressors that spermatozoa may encounter, oxidative damage is recognized as one of the major threats, with the potential to elicit damage to the key elements of sperm structure (e.g., plasma membrane, proteome, and nucleus) and function.1 Counteracting the damaging effects of oxidative stress is an extensive array of antioxidant strategies in the mammalian epididymis.2 Among these is the glutathione peroxidase 5 (Gpx5) gene that is both highly and selectively expressed under androgenic control within the proximal caput segment of the mouse epididymis. The corresponding GPX5 protein is secreted into the luminal compartment where it protects transiting spermatozoa from the damaging effects of hydrogen peroxide (H2O2).3 In addition, GPX5 indirectly contributes to the maintenance of the optimal level of H2O2-mediated disulfide bridging events during sperm epididymal maturation by regulating the luminal H2O2 concentration. Such regulation is essential for correct sperm nucleus condensation that protects the paternal genome.4,5 Genome condensation ensures sperm DNA integrity, which, in turn, determines completion of the developmental program and the health of offspring.6
In our previous work, we generated a conventional Gpx5 knockout (KO) mouse model (Gpx5-/-), in order to analyze the contribution of the GPX5 protein to posttesticular sperm maturation.3 Using the Gpx5-/- model, we observed nuclear oxidative alterations in not only the spermatozoa, but also the epididymal tissue of aging animals.3,4,5 In comparison to the mating outcomes of wild-type (WT) animals of the same age, the mating of GPX5-deficient aged males (i.e., males over 13 months of age) with WT females of proven fertility resulted in a higher incidence of miscarriages, abnormal development, and an increase in perinatal mortality.3 While these studies highlight the importance of GPX5 enzymatic activity, we have yet to resolve the mechanistic basis by which the loss of this protein leads to such pronounced changes in epididymal physiology. Therefore, in order to document the molecular consequence of the removal of GPX5 from the epididymis, and to identify the mechanisms underlying sperm nuclear oxidative alterations in aged mice, transcriptomic and small RNA sequencing (RNA-seq) analyses were performed on the epididymides of 13-month-old WT and Gpx5-/- animals. This approach identified a potentially somatic cell-specific P-element-induced wimpy testis (PIWI)-interacting RNA (piRNA) pathway that operates in the caput epididymidis of aged mice and the possible involvement of this pathway in mediating the antioxidative responses of the tissue within this specific epididymal segment.
MATERIALS AND METHODS
Animal care and sample preparation
The WT (strain C57BL/6) and Gpx5-/- mice used in this study were generated as previously reported.3 All protocols in this study were conducted after obtaining approval from the Institutional Animal Care Committee of Shanghai Institute of Biochemistry and Cell Biology (Permit number: SYXK2007-0017). Tissue samples were prepared from seven 13-month-old WT mice and seven 13-month-old Gpx5-/- mice, which were sacrificed by cervical dislocation. The epididymides were removed and freed of connective and adipose tissues before being divided into caput, corpus, and cauda segments. The testes and corpus epididymidis were directly processed for RNA extraction, whereas the caput and cauda epididymidis were transferred to a 1.0-ml droplet of phosphate-buffered saline (PBS) within a small glass dish. The spermatozoa were removed via repeated puncturing of the epididymal tissue with a 26G needle and incubation at 37°C for 15 min. After dispersion of the spermatozoa, caput and cauda tissues were collected for RNA extraction. It is important to note that because of the scarcity of samples from aged Gpx5-/- mice, the spermatozoa of the caput and cauda epididymidis were collected for additional experimental analyses not reported here. As a consequence, the preparation procedures for the caput and cauda epididymidis differed from that of the corpus epididymidis, which may have resulted in a potential biasing of the comparisons between the three epididymal segments.
RNA isolation, library construction, sequencing, and data analysis
Owing to the scarcity of samples from aged Gpx5-/- mice, we elected to pool biological samples to generate a single replicate for transcriptomic and small RNA (sRNA) sequencing experiments. For each sample, total RNA isolation was performed with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The content of each total RNA extraction was determined via measurement on a Nanodrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and the quality was assessed in a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA integrity numbers (RIN) for these samples ranged from 7.2 to 9.1 (Supplementary Figure 1 (1.1MB, tif) ). For transcriptome sequencing, the cDNA library was prepared from full-length mRNA transcripts by using reverse transcriptase with terminal transferase activity. After cDNA synthesis, 2.7 μg of cDNA per sample was sonicated to an expected size of 200–400 nucleotides and was used for library preparation with the Nextera DNA Sample Prep Kit according to the manufacturer's protocol (Illumina, San Diego, CA, USA). Each library was sequenced by Illumina HiSeq 2000, and 100 base pair (bp) single-ended reads were mapped to mousegenome (mm9) by the TopHat2 (version 2.1.0; https://ccb.jhu.edu/software/tophat/index.shtml) short-read mapper.7 For each mapped transcript, differentially expressed genes were putatively identified from the “generalized fold change (GFOLD)” program (version 1.1.4; https://zhanglab.tongji.edu.cn/softwares/GFOLD/index.html). The GFOLD algorithm overcomes the shortcomings of P value and fold change calculated by other algorithms, to provide a more reliable gene ranking score when only one single biological replicate is used,8 as was the case for each sample assessed in this study. A sequencing library was also constructed to identify changes in the global sRNA population between WT and Gpx5-/- samples, with each library constructed using the TruSeq Small RNA Sample Preparation kit according to the manufacturer's protocol (Illumina). In brief, 5' and 3' RNA adapters were ligated to the sRNA fraction of 1.0-μg unmodified, high-quality total RNA. Next, a reverse transcription reaction was performed to generate cDNA libraries, and post library generation, PCR was used to amplify and add unique index sequences to each library. The amplified libraries were separated on a polyacrylamide gel via electrophoresis, and the gel region corresponding to the desired fragment size (18–40 nt) was excised, purified, and sequenced by Illumina HiSeq 2000 sequencer. After sequencing, low-quality reads, the unwanted adaptor sequences, and other contaminants were filtered out and removed. The remaining clean reads were next mapped to the mouse mm9 genome by using Bowtie (version 0.12.7)9 with the default parameters except of “-v 2”, which allows no more than two mismatches during read mapping. Annotation of the sRNA libraries was performed via library alignment to GenBank (http://www.ncbi.nlm.nih.gov/genbank/), mirBase (http://mirbase.org/),10 Rfam (http://rfam.sanger.ac.uk/),11 and piRNABank (http://pirnabank.ibab.ac.in/).12 The piRNA sequence data of pachytene spermatocytes (GSM610966), round spermatids (GSM610967), and elongated spermatids (GSM992826) were obtained from previously published studies.13,14
Reverse transcriptase polymerase chain reaction-based quantification of gene expression and small RNA abundance
For the assessment of gene expression and the determination of sRNA abundance, a reverse transcription (RT) step was performed before quantitative PCR (qPCR). To quantify the changes in the expression of genes of interest, random primers were used to prime the reverse transcription of cDNAs from mRNA templates. For sRNA abundance determination, a stem-loop RT primer specific to each sRNA of interest was hybridized to the sRNA molecule for the generation of a sRNA-specific cDNA, according to a previous study.15 Specifically, 1.0 μg of each total RNA extract was denatured and mixed with 62.5 μmol l−1 of each dNTP, 50 nmol l−1 of the stem-loop primer, and 1 unit of M-MLV reverse transcriptase (Promega, Madison, WI, USA) at 16°C for 30 min, 42°C for 1 h, 16°C for 10 min, and then incubated on ice. qPCR was performed by the SYBR Green assay (TOYOBO, Osaka, Japan) with a Rotor-Gene 3000 thermal cycler (Corbett Life Science, Sydney, Australia). In brief, SYBR Green I Master Mix was mixed with 0.5 μmol l−1 forward and reverse primers and 5 μl of the RT template to give a total volume of 20 μl. The samples were incubated at 95°C for 1 min, followed by 40 cycles of 95°C for 10 s, 60°C for 15 s, and 72°C for 20 s. Data were normalized to the internal control gene Beta-Actin (Atcb)for calculation of 2−ΔΔCT values. The average 2−ΔΔCT values of a gene or a piRNA from the WT, or caput, or 13-month-old groups were used as the divisor to determine the relative expression levels. All primers used in this study are listed in Supplementary Table 1 (396.4KB, pdf) ). Notably, all RT-qPCR validation experiments were performed on nonpooled tissues isolated from three individual mice (i.e., n = 3 biological replicates).
Bioinformatic analyses
Gene ontology (GO) analyses of up- and down-regulated gene sets were performed using the DAVID Bioinformatics Database 6.8 (http://david.ncifcrf.gov) with the default settings.16
The gene set enrichment analysis (GSEA) was performed by the GSEA software (version 3.0, http://software.broadinstitute.org/gsea/index.jsp)17 with the same parameters as previously reported.18 In brief, a predefined ranked gene list was constructed via the sorting of all genes according to their decreasing GFOLD levels between WT and Gpx5-/- groups. The test gene set of “piRNA pathway genes” was adapted from the gene ontology database, term GO: 0034587 “piRNA metabolic process.” The GSEA calculates the “enrichment score (ES)”, “significance level (P value)”, and “false discovery rate (FDR)”, to determine whether the set of genes was significantly enriched in the top or bottom of the predefined rank gene list.17 The enrichment was considered statistically significant if a P < 0.05 and an FDR of <0.25 were obtained. In practice, 1000 permutations were conducted to evaluate the P value and FDR for the enrichment score of a gene, with the enrichment statistic parameter set to “classic” and permutation type set to the “gene set”.
To create predefined lists of piRNA sequences that bind to different PIWI proteins, including piwi-like RNA-mediated gene silencing 1 (PIWIL1), PIWIL2, and PIWIL4, the previously published datasets GSM1565018 (PIWIL1),19 GSM475280 (PIWIL2),20 and GSM1220989 (PIWIL4)21 were downloaded. The top 5000 most abundant piRNAs from each dataset were selected to build the lists. Next, the piRNA sequences identified here by RNA-seq were assigned to specific PIWIL groups based on sequence identities.
Target prediction for the RNA-seq-identified piRNAs was largely performed as outlined in a previous study,22 but minor modifications were introduced. In brief, the top 300 most highly abundant piRNAs identified in the caput epididymidis of aged WT animals were initially filtered by the criterion of having at least a 10-fold reduction in the corresponding tissue of Gpx5-/- animals. The remaining piRNAs were then subjected to the prediction of putative target genes which harbor piRNA target sequences in their 3' UTR by miRanda software23 (with the parameters, “-strict” and “-quiet”).
The set of “oxidative response genes” was downloaded from the gene ontology database, term GO: 0006979 “response to oxidative stress”.
Statistical analysis
The results were presented as means ± standard deviation (s.d.). A standard Student's t-test was used to compare the differences between the WT and Gpx5-/- samples once the sample data had passed the normality (Shapiro–Wilk) and equal variance tests. For the nonparametric tests, the two-sample Kolmogorov–Smirnov test and the Mann–Whitney–Wilcoxon test were used. For all comparisons, the determined P value was presented in the appropriate figures, and differences were considered statistically significant when P < 0.05.
Data availability
The RNA-seq datasets generated and analyzed during the current study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the accession number of GSE122087.
RESULTS
Analysis of the transcriptomes of the caput epididymidis of aged WT and Gpx5-/- mice
To investigate the molecular consequence of removing GPX5 function from the mouse epididymis, the transcriptomic profiles of WT and Gpx5-/- capita epididymidum of 13-month-old mice were compared (Figure 1a) (full transcriptomes are presented in Supplementary Table 2 (1.6MB, pdf) ). Mice of 13 months of age were selected for transcriptomic profiling as this is the age at which Gpx5-/- mice display their reproductive phenotype.3 Using a GFOLD change threshold of >+1 or <−1, 250 and 3257 up- and down-regulated genes were, respectively, identified in the caput epididymidal tissue of 13-month-old Gpx5-/- mice (Figure 1a). The abundance of the Gpx5 transcript was dramatically reduced in the Gpx5-/- mice (Figure 1a and 1b). In addition, the transcript abundance of three randomly selected genes, including the upregulated Spag11a (a β-defensin-type epididymal protein), and the not significantly changed mitochondrial membrane ATP synthase β-subunit (Atp5b) and zinc finger protein 106 (Zfp106), was confirmed via RT-qPCR to follow the expression trends identified via transcriptomic profiling (Figure 1b).
Figure 1.
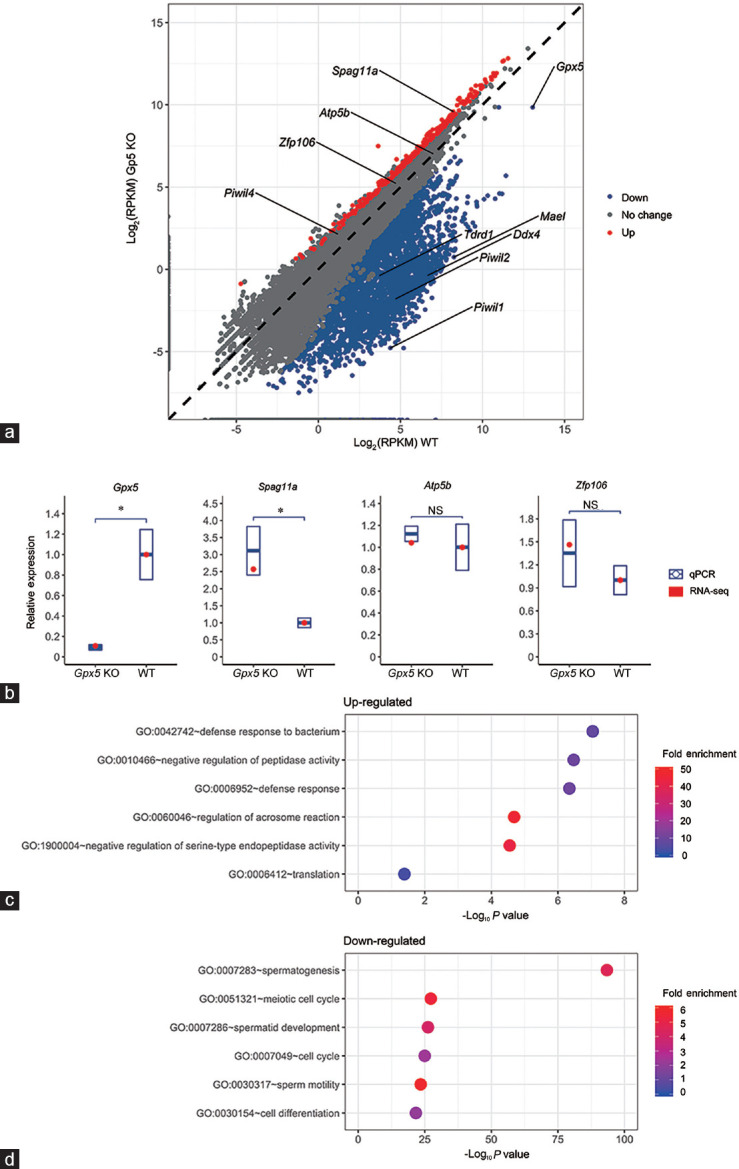
Analysis of the transcriptomes of the caput epididymidis of aged WT and Gpx5-/- mice. (a) Difference in mRNA expression between the WT and Gpx5-/- groups. Each gene is represented by a dot located according to its expression level between the WT and KO groups. (b) RNA-seq data validation by RT-qPCR of Gpx5 and another three randomly chosen genes in the 13-month-old WT and Gpx5-/- mice. Quantitative PCR data are presented as mean ± s.d. from three independent animals. NS: not significant; *P < 0.05; Student's t-test. (c) GO enrichment result for the up-regulated genes in the caput epididymis of Gpx5-/- mice. (d) GO enrichment result for the down-regulated genes in the caput epididymis of Gpx5-/- mice. RPKM: reads per kilobase of transcript per million mapped reads; GO: gene ontology; RT-qPCR: quantitative reverse transcription PCR; Gpx5: glutathione peroxidase 5; s.d.: standard deviation; WT: wild-type; KO: knockout.
To investigate further the putative functions of genes with altered abundance in Gpx5-/- animals, GO analysis was performed on the up- (Figure 1c) and down-regulated (Figure 1d) gene sets, a strategy revealing substantial differences in the genes being positively and negatively influenced by the loss of GPX5. For genes with elevated expression in Gpx5-/- animals, functional enrichment was noted among GO categories related to cellular defense, including “defense response to bacterium” (GO: 0042742) and “defense response” (GO: 0006952). This finding is consistent with the up-regulation of defensin family members in the caput epididymidis of Gpx5-/- animals (Supplementary Table 2 (1.6MB, pdf) ). By contrast, GO analysis of genes with down-regulated expression in Gpx5-/- animals revealed a highly significant functional enrichment of several categories related to sperm development, including “spermatogenesis” (GO: 0007283), “meiotic cell cycle” (GO: 0051321), “spermatid development” (GO: 0007286), and “sperm motility” (GO: 0030317) (Figure 1d).
Alterations in piRNA pathway gene expression in the caput epididymidis of aged Gpx5-/- mice
Prompted by the significant GO enrichment of spermatogenesis-related genes that were down-regulated in the caput epididymidis of aged Gpx5-/- animals (Figure 1d), we performed an additional curation of this subset of genes. Among them, we noticed quite a few that are implicated in the mouse piRNA pathway.24,25,26,27 To confirm this observation, we conducted a GSEA analysis on the set of “piRNA pathway genes” (listed in Supplementary Table 3 (96.1KB, pdf) ). GSEA revealed significantly higher expression of this gene set in the WT caput epididymidis indicated by a positive Normalized Enrichment Score (NES) of 1.88 with P < 0.001 and thus, a concomitant tendency of their repression within the Gpx5-/- animals (Figure 2a). Of the six piRNA pathway genes analyzed, RT-qPCR confirmed significantly reduced Piwil1, Piwil2, Ddx4, Mael, and Tdrd1 transcript abundance in the caput epididymidis of Gpx5-/- animals (Figure 2b). Curiously, however, the level of Piwil4 transcript remained at its approximate WT abundance.
Figure 2.
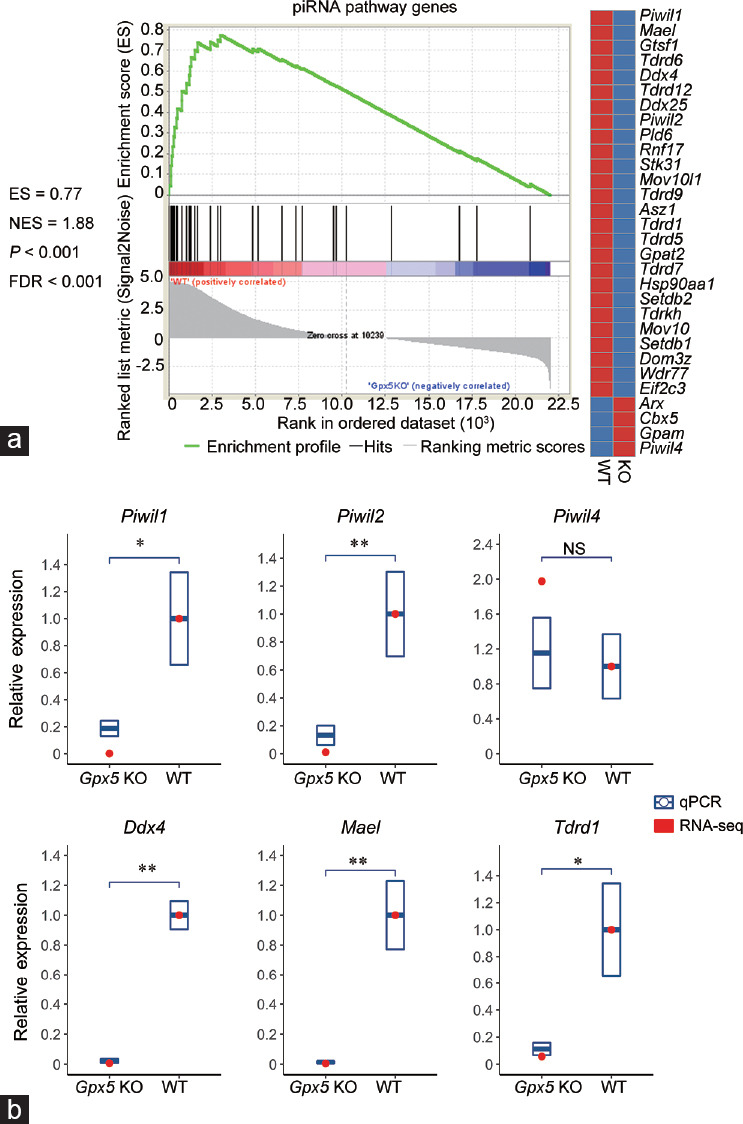
Alteration in the mRNA expression of piRNA pathway genes in the caput epididymidis of aged Gpx5-/- mice. (a) GSEA analysis result of the piRNA pathway genes. A positive ES with a significant P value and FDR indicates higher mRNA levels of piRNA pathway genes in the WT group and the significant downregulation in the Gpx5-/- mice. (b) RNA-seq data validation by RT-qPCR of six known piRNA pathway genes in the 13-month-old WT and Gpx5-/- groups. Quantitative PCR data are presented as mean ± s.d. from three independent animals. NS: not significant; *P < 0.05; **P < 0.01; Student's t-test. GSEA: gene set enrichment analysis; ES: enrichment score; FDR: false discovery rate; RT-qPCR: quantitative reverse transcription PCR; Gpx5: glutathione peroxidase 5; s.d.: standard deviation; WT: wild-type; KO: knockout; NES: normalized enrichment score; piRNA: P-element-induced wimpy testis (PIWI)-interacting RNA.
Profiling of piRNA abundance in the mouse testis and epididymis
The demonstration of repressed piRNA pathway gene expression led us next to determine the sRNA landscape of the testis and the caput, corpus, and cauda segments of the epididymis of 13-month-old mice via a high-throughput sequencing approach. In this analysis, the testis was included as a reference material for an expectedly high level of the piRNA population.13 In the testis, the piRNA class of sRNA was indeed clearly the most abundantly expressed sRNA species (Figure 3a). This was distinct from the sRNA profiles documented for the three segments of the epididymis where the microRNA (miRNA) class of sRNA was the predominantly expressed species (Figure 3a). In addition to the miRNAs, the transfer RNA (tRNA)-derived sRNAs (tsRNAs) were also accumulated in the epididymal samples. Interestingly, this sRNA species almost failed to register in the testicular sample (Figure 3a). Figure 3a and 3b also clearly reveal a distinct sRNA profile for the caput epididymidis, compared with that of the corpus and cauda, with sRNAs of length 28–32 nt representing approximately 16% of the global sRNA population of the proximal segment of the epididymis. This population of sRNAs was of the expected size of the piRNA class (Figure 3b), and, further, was determined to have a sequence composition that perfectly matched those of previously characterized piRNAs. Further comparison of this caput epididymidis-enriched sRNA class revealed that it had the same length distribution as known piRNAs detected in round and elongated spermatids, which are themselves slightly longer than those piRNAs reported in pachytene spermatocytes (Figure 3c). Furthermore, the epididymal piRNAs were also determined to harbor predominantly a uracil (U) residue at their 5' terminal nucleotide (Figure 3d), a sequence composition feature that matched that of the piRNAs detected in the testis (Figure 3d).
Figure 3.

Regional expression of epididymal piRNAs in aged animals. (a) Profiles of regional sRNA constitution in 13-month-old mouse testis and epididymis by RNA-seq. (b) Composition of sRNA categories according to their length distributions in each sample. (c) Length distribution comparison of piRNAs from different testicular sperm cell types and aged caput epididymidis. (d) Composition of the 5' end nucleotide of piRNAs from each sample. (e) RT-qPCR validation of small RNA-seq data for six selected piRNAs. Quantitative PCR data are presented as mean ± s.d. from three independent animals. The “snRNA Frag”, “rRNA Frag”, and “mRNA Frag” refer to the RNA fragments derived from snRNAs, rRNAs, and mRNAs, respectively. RT-qPCR: quantitative reverse transcription PCR; s.d.: standard deviation; piRNA: P-element-induced wimpy testis (PIWI)-interacting RNA; snRNA: small nuclear RNA; sno-sRNA: small nuclear RNA-derived small RNA; mito-sRNA: mitochondrial RNA-derived small RNA; tsRNA: tRNA-derived small RNA.
In order to validate the sequencing data experimentally, an RT-qPCR approach was applied to quantify known piRNA abundance across the testis and caput, corpus, and cauda epididymidal segments. Of the six piRNAs quantified via RT-qPCR (Figure 3e), piRNAs piR-125360, piR-14721, piR-17753, and piR-20751 were abundant in the testicular and caput epididymidal samples. Interestingly, the piRNA piR-22206 was determined by RT-qPCR to be mostly expressed in the caput epididymidal region, whereas piR-1330 was more abundant in all the three epididymal segments than that in the testis (Figure 3e). Nevertheless, these quantifications of piRNA abundance did match the abundance trends profiled by sRNA sequencing.
While these data attest to the accuracy of our sequencing data, they stand in contrast to previous reports in which negligible levels of piRNAs were detected in the epididymal tissue of young WT mice (approximately 2 months of age).28 In view of this apparent dichotomy, we elected to explore whether the expression of the piRNAs detected in the caput epididymidis of 13-month-old mice in this study was also conserved in younger animals. Thus, we repeated our sRNA profiling of caput epididymidis collected from 2-month-old WT mice, revealing striking age-dependent alterations in the global sRNA composition of this tissue (Figure 4a and 4b). The sRNA population of the caput epididymidis of the 2-month-old mice was almost entirely composed of miRNAs. In contrast, the proximal segment of the epididymis of 13-month-old mice also contained an abundance of piRNAs and tsRNAs (Figure 4a). The difference in the sRNA profiles of the caput epididymidis of 2- and 13-month-old animals was further demonstrated by assessment of the size distribution of the sRNA molecules that accumulated in each population. Indeed, in the caput epididymidis of 2-month-old animals, the 22-nt size class was clearly the most predominant sRNA (Figure 4b); an sRNA length that corresponds to miRNAs. The 22-nt size class of sRNA also represented the majority of the sRNA population of the caput epididymidis of 13-month-old animals. However, in these animals, an additional population of longer sRNA species (i.e., 26–34 nt), corresponding to the sizes predicted for piRNAs and tsRNAs, also accumulated in the proximal segment of the epididymis of aged animals.
Figure 4.

Temporal expression of caput epididymidal piRNAs. (a) Profiles of temporal sRNA constitution in the caput epididymidis by RNA-seq. (b) Composition of sRNA categories according to their length distributions in each sample. (c) RT-qPCR validation of small RNA-seq data for six piRNAs. Quantitative PCR data are presented as mean ± s.d. from three independent animals. NS: not significant; *P < 0.05; **P < 0.01; Student's t-test. The “snRNA Frag”, “rRNA Frag”, and “mRNA Frag” refer to the RNA fragments derived from snRNAs, rRNAs, and mRNAs, respectively. RT-qPCR: quantitative reverse transcription PCR; s.d.: standard deviation; WT: wild-type; KO: knockout; piRNA: P-element-induced wimpy testis (PIWI)-interacting RNA; snRNA: small nuclear RNA; sno-sRNA: small nuclear RNA-derived small RNA; mito-sRNA: mitochondrial RNA-derived small RNA; tsRNA: tRNA-derived small RNA.
RT-qPCR was next used to confirm the differences in the abundance of the piRNA class in the caput epididymidis of young and aged animals. This approach revealed that for the six piRNAs quantified (including piR-1330, piR-14721, piR-17753, piR-20751, piR-22206, and piR-125360), the abundance of each was clearly much greater in aged animals than in the corresponding tissue of young, 2-month-old animals (Figure 4c). Taken together, the results presented in Figures 3 and 4 strongly suggest that the piRNAs are almost exclusively expressed in the proximal caput segment of the mouse epididymis, and, further, that the observed piRNA accumulation is greatly enhanced in aged animals.
The caput epididymidal piRNA population of aged mice is predominantly bound by the PIWI-like proteins PIWIL1 and PIWIL2
Having demonstrated that the accumulation of the piRNA class of sRNA was enriched in the caput epididymidis of aged mice, we next sought to characterize the individual members of this class of sRNA via documenting their potential binding to the PIWI-like proteins PIWIL1, PIWIL2, and PIWIL4. For this purpose, we took advantage of the published datasets that catalog preferential PIWIL1-, PIWIL2-, and PIWIL4-binding piRNAs.19,20,21 In order to maximize the accuracy of this analysis via minimizing the rate of false-positive assignment of PIWIL/piRNA binding, only the 5000 most abundant binding piRNA sequences from each PIWIL protein piRNA-binding dataset were included (Supplementary Table 4 (199.2KB, pdf) ). First, the preferential binding of each “top-5000” piRNA to each PIWIL protein was assessed. Unsurprisingly, the vast majority of piRNA sequences from each of the three top-5000 lists was determined to bind only to the PIWIL protein from which the piRNA sequence was originally sourced; that is, piRNAs sourced from the PIWIL1 dataset were determined to bind only to PIWIL1 (Figure 5a). After establishing these expected interactions, we next mapped our own caput epididymidal piRNA sequences to the PIWIL1, PIWIL2, and PIWIL4 piRNA binding top-5000 lists. When the respective abundance of the total caput epididymidal piRNA read population was considered, it was revealed that the majority of the piRNA population potentially only bound to either PIWIL1 (756 071 reads, 49.4%), PIWIL2 (144 185 reads, 9.4%), or both PIWIL1 and PIWIL2 (447 768 reads, 29.3%) (Figure 5b). In direct contrast, only approximately 5.5% (84 834 reads) of the detected piRNAs were determined to be preferentially bound by PIWIL4 (Figure 5b).
Figure 5.
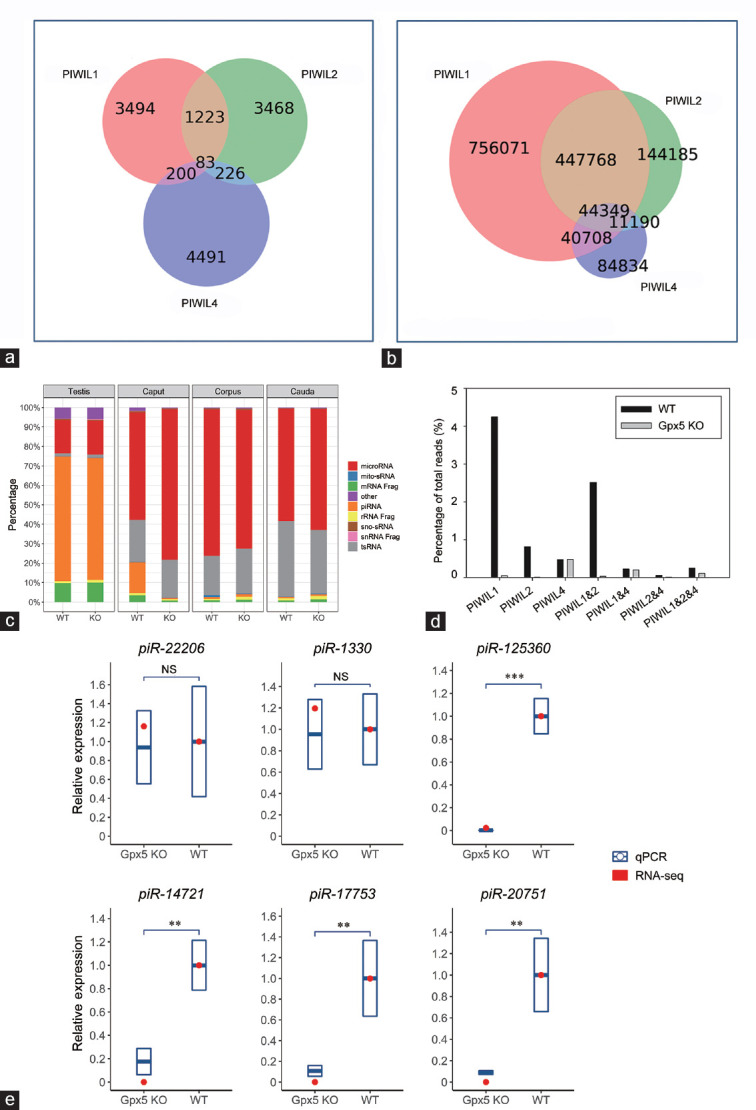
The piRNAs that potentially bind to PIWIL1 and PIWIL2, but not PIWIL4, were absent in the Gpx5-/- caput epididymidis. (a) The known PIWIL1-, PIWIL2-, and PIWIL4-binding piRNA data were, respectively, downloaded from previously published datasets GSM1565018, GSM475280, and GSM1220989. The top 5000 most abundant piRNAs in each dataset were selected to build a predefined list. The Venn diagram shows how these piRNAs overlap each other. (b) Potential interactions of the total piRNA reads of the 13-month-old caput epididymidis. (c) Profile of the sRNA constitution in the testis and different epididymal regions of WT and Gpx5-/- groups by RNA-seq. (d) Alteration of piRNAs that bind to different PIWI proteins in the 13-month-old Gpx5-/- caput epididymidis. (e) RT-qPCR validation of small RNA-seq data for six selected piRNAs in 13-month-old WT and Gpx5-/- mice. Quantitative PCR data are presented as means ± s.d. from three independent animals. NS: not significant; **P < 0.01; ***P < 0.001; Student's t-test. The “snRNA Frag”, “rRNA Frag”, and “mRNA Frag” refer to the RNA fragments derived from snRNAs, rRNAs, and mRNAs, respectively. RT-qPCR: quantitative reverse transcription PCR; PIWI: P-element-induced wimpy testis; PIWIL1: piwi-like RNA-mediated gene silencing 1; PIWIL2: piwi-like RNA-mediated gene silencing 2; PIWIL4: piwi-like RNA-mediated gene silencing 4; s.d.: standard deviation; piRNA: PIWI-interacting RNA; snRNA: small nuclear RNA; sno-sRNA: small nuclear RNA-derived small RNA; mito-sRNA: mitochondrial RNA-derived small RNA; tsRNA: tRNA-derived small RNA.
The expression of PIWIL1- and PIWIL2-binding piRNAs is dramatically decreased in the caput epididymidis of aged Gpx5-/- mice
From the marked reduction in the expression of several piRNA pathway genes in the caput epididymidis of aged Gpx5-/- mice (Figure 2b), we hypothesized that piRNA production in this segment of the epididymis was likely to be defective in the absence of GPX5 function. Therefore, high-throughput sequencing was used to profile the sRNA landscape of the caput, corpus, and cauda epididymidis, as well as the testes of aged Gpx5-/- animals. The testes of WT and Gpx5-/- animals were included in this analysis to demonstrate that loss of GPX5 activity is of little biological relevance in this organ. As expected, the sRNA profiles of the testes of WT and Gpx5-/- animals were almost identical, with the piRNA and miRNA classes representing the majority of the global sRNA population of this tissue (Figure 5c). A similar result was observed for the corpus and cauda segments of the epididymis, in which the loss of GPX5 was revealed to have only a very modest influence on the sRNA landscapes of these two epididymal segments. More specifically, loss of GPX5 activity appeared to influence primarily tsRNA accumulation in both segments, with tsRNA abundance being increased very slightly in the corpus epididymidis and marginally reduced in the cauda epididymidis (Figure 5c). By comparison, Figure 5c reveals the almost complete loss (approximately 96% reduction) of accumulation of the piRNA class of sRNA in the caput epididymidis of aged Gpx5-/- mice.
From our previous demonstration that (1) Piwil1 and Piwil2 expression is reduced in the caput epididymidis of aged Gpx5-/- animals in the absence of any change in Piwil4 transcript abundance and (2) PIWIL1 and PIWIL2 putatively bind the vast majority of piRNAs that accumulate in the caput epididymidis, it was not surprising to document that most of the piRNAs that potentially associate with PIWIL1 (98.8%), PIWIL2 (98.6%), or both (98.7%) were lost in the proximal segment of the epididymis (Figure 5d). Together, these PIWIL1- and PIWIL2-binding piRNAs largely accounted for the observed approximately 96% loss from the total piRNA populations, given that the piRNAs that potentially interact with PIWIL4 largely remained unchanged (Figure 5d). Furthermore, RT-qPCR experiments confirmed the RNA-seq data in illustrating that, despite the dramatic decline of piRNAs in the aged Gpx5-/- caput epididymidis, the expression of the two most abundant remaining piRNAs, piR-22206 (a potential PIWIL4-binding piRNA) and piR-1330 (which potentially binds to all the three PIWI proteins), was not significantly altered (Figure 5e). In contrast, the expression of four piRNAs that can potentially be bound by either PIWIL1 or PIWIL2 (including piR-125360, piR-14721, piR-17753, and piR-20751) was confirmed as being significantly attenuated in the caput epididymidis of aged Gpx5-/- animals (Figure 5e). Taken together, the experimental data presented in Figure 5 clearly show that loss of GPX5 from the caput epididymidis of aged mice results in the selective loss of the piRNAs that are preferentially bound by PIWIL-like proteins, PIWIL1 and PIWIL2.
The piRNAs lost in the caput epididymidis of aged Gpx5-/- mice are correlated with the increase in mRNA levels of their target oxidative stress response genes
It has previously been shown that, in addition to their well-established roles in repressing the activity of mobile DNA elements such as transposons in the testis, piRNAs also play a role in the posttranscriptional regulation of protein-coding gene expression via mediating mRNA degradation.22,29,30 As the caput epididymidal piRNAs appeared to accumulate only in aged animals and were dramatically reduced following Gpx5 knockout, we sought to determine whether there was any relationship between the change in piRNA abundance and protein-coding gene expression in this tissue. Initially, the 300 most highly abundant piRNAs (accounting for approximately 29% of the entire piRNA population) present in the caput epididymidis of 13-month-old WT animals were selected for this analysis. Of these, 292 (97.3%) were determined to have a greater than 10-fold reduction in abundance in the caput epididymidis of Gpx5-/- animals. From the miRanda software, potential target mRNAs for these 292 piRNAs were predicted and global transcriptomic data were interrogated to evaluate their relative levels of expression in the caput epididymidis of aged WT animals. This strategy revealed an inverse correlation between mRNA abundance and the number of piRNA target sites harbored by the 3'UTR of the mRNA (Figure 6a).
Figure 6.

Expression and function analysis of the potential target genes of epididymal piRNAs. (a) The levels of mRNAs in aged WT caput epididymidis are inversely correlated with the numbers of potential binding piRNAs to their 3'UTRs. The X-axis represents the number of potential binding piRNAs. The Y-axis shows the average levels of mRNAs (RPKM) with the same number of binding piRNAs. (b) Comparison of the cumulative abundance (log10) among the three groups of mRNAs with >50 targeting piRNAs (red line), 1–50 targeting piRNAs (green line), and no apparent targeting piRNAs (blue line). The number of mRNAs in each group is indicated, and P and D values determined by Kolmogorov–Smirnov (KS) test are shown. (c) GO enrichment result for the 3830 genes (Group 1) that are targeted by >50 piRNAs. (d) The number of targeting piRNAs to the set of “oxidative stress response genes” and other genes. The significance of the difference was determined from the Mann–Whitney–Wilcoxon test. (e) Rank-ordered depiction of the GFOLD scores for each gene. The 3830 genes (Group 1) that are targeted by >50 piRNAs are depicted in red. RPKM: reads per kilobase of transcript per million mapped reads; GFOLD: generalized fold change; GO: gene ontology; piRNA: P-element-induced wimpy testis (PIWI)-interacting RNA.
We then subdivided the mRNAs of each gene into three groups on the basis of the number of targeting piRNAs (Supplementary Table 5 (722.4KB, pdf) ) as follows: Group 1: 3830 mRNA transcripts targeted by >50 piRNAs; Group 2: 14 718 mRNA transcripts targeted by 1–50 piRNAs; and Group 3: 3472 mRNA transcripts targeted by no piRNA. As revealed in Figure 6b, compared with the Group 3 transcripts which are potentially targeted by no piRNA, Group 1 (D [value of the discrepancy calculated by the Kolmogorov–Smirnov test] = 0.322; P < 2.2 × 10−16) and Group 2 (D = 0.190; P < 2.2 × 10−16) transcripts had statistically significant inclinations toward positive GFOLD values, which indicate an increase in the Gpx5-/- mice. The statistically significant difference (D = 0.133; P < 2.2 × 10−16) between the Group 1 and Group 2 transcripts further implicates the synergistic downregulation effects of piRNAs.
In view of these correlative data, we sought to explore further the biological functions of these piRNAs through their target genes. We first performed GO analysis on the Group 1 genes that are targeted by >50 piRNAs. Three genes were found to have the top enriched functions such as “transcription” (GO: 0006351), “protein phosphorylation” (GO: 0006468), and “covalent chromatin modification” (GO: 0016569) (Figure 6c). These enriched GO terms implicate an extensive involvement of this group of piRNAs in the gene expression regulation at different levels.
In addition, we hoped to determine whether the caput epididymidal piRNAs also directly take part in the oxidative stress response process via the targeting of genes associated with this pathway, given their dramatic loss in the Gpx5-/- mice. We first surveyed the “oxidative stress response genes” and revealed that they were subject to significantly more targeting piRNAs than other genes (Figure 6d). Only seven of the oxidative stress response genes (approximately 2%) had no targeting piRNAs, whereas 62 (approximately 20%) of them had more than fifty targeting piRNAs. Accordingly, from the rank-ordered plot, these oxidative stress response genes were indeed among the most highly elevated genes upon the loss of GPX5 (Figure 6e).
Together, these results identify the expression of a novel piRNA network in the caput epididymidis of aged mice and implicate this network in the negative regulation of a large number of mRNAs, including those of the oxidative stress response genes. Thus, the absence of piRNAs in the caput epididymidis of Gpx5-/- animals could, at least in part, promote the increased mRNA levels of the oxidative stress response genes that may compensate for the loss of GPX5 expression in that segment.
DISCUSSION
PIWI proteins are a class of highly conserved RNA-binding proteins belonging to the subfamily of Argonaute proteins. They bind to piRNAs, which are a diverse population of around 24–31 nt small noncoding RNAs, and form a conserved piRNA-induced silencing complex that is known to protect the germline against the harmful expression of transposable elements (TEs).31 Both piRNAs and PIWI proteins were previously believed to be predominantly restricted to the germline, where they fulfil important functions in germ cell development, suppression of transposons, and maintenance of genomic integrity against DNA damage.31,32,33 By comparison, relatively little is known regarding the presence and roles of piRNAs in mammalian somatic cells. Nevertheless, continued advances in sequencing technology have underpinned the recent discoveries of piRNA expression in somatic tissues as diverse as the brain,34,35 gastrointestinal tract,36 and epididymis,28,37,38,39 indicating that functional piRNA pathways are not restricted to germ cells.40 Despite progress regarding our understanding of somatic piRNAs, the lack of suitable models and functional readouts related to the change in somatic piRNA expression remains a major obstacle hampering studies of their patterns of expression and functional significances.
In this study, we confirmed the existence of piRNAs in the mouse epididymis, adding to the previous reports of piRNAs in the epididymis of primates, i.e., rhesus macaque38 and humans.37 Epididymal piRNAs from different species have a similar length distribution and typical 1U bias features to those of other somatic piRNAs from brain, stomach, and small intestine.34,36 Unlike the majority of homogeneous tissues, it is well documented that the segmental patterns of expression of both proteins and sRNAs in the epididymis form an integral part of the regulatory network responsible for region-specific functions of this organ.18,41,42,43 Similarly, we found that mouse epididymal piRNAs were also expressed in a spatiotemporal pattern and enriched only in the caput epididymidis and not the more distal regions of the tissue, i.e., the corpus and the cauda, of aged mice. Our observation of increased piRNA expression in caput epididymidis in aged animals is similar to the recent findings in Drosophila heads,44 suggesting their involvement in aging-related events, such as the accumulation of oxidative stress, which may elicit DNA damage and impair genome integrity.
As mentioned in the “Materials and Methods” section, we removed the spermatozoa from the caput and cauda epididymidis by repeated tissue puncture with a 26G needle and incubation at 37°C for 15 min. A limitation of our study is that we have not implemented quality control to ensure the complete separation of spermatozoa from tissue, and thus we cannot rule out the possibility that a portion of the RNAs we identified by RNA-seq are attributed to residual sperm cells. Arguing against this possibility, however, is evidence from previous studies and our ongoing research that has shown that caput spermatozoa in fact harbor very few piRNA and PIWI molecules,28,45 owing to their degradation by the anaphase-promoting complex/cyclosome (APC/C)-26S proteasome pathway in the late testicular spermatids.46 Therefore, the contribution of contaminating spermatozoa to our piRNA profiling is likely to be minimal. The absence of GPX5, a luminal antioxidant with caput-restricted expression, has previously been shown to elicit a state of stress, leading to nuclear oxidative alterations in spermatozoa and epididymal tissue, and to a higher incidence of miscarriages, abnormal development, and perinatal mortality following the crossing of aged Gpx5-/- males with WT females.3 In this study, our transcriptomic analyses provide additional information regarding the profile of altered gene expression within the caput epididymidal tissue of WT and Gpx5-/- animals. The numerous interesting observations that we have not explored directly here include the up-regulation of genes that encode proteins, exerting a negative influence on the activity of peptidases and regulating the sperm acrosome reaction. One such example is the Kazal-type serine protease inhibitor serine protease inhibitor Kazal-type 13 (Spink13), which has been implicated in both of the above-mentioned categories. Indeed, previous work has demonstrated that SPINK13 can bind to maturing spermatozoa and thereafter influence male fertility via regulation of the acrosome reaction.47 Thus, dysregulation of Spink13 expression may, at least in part, explain the fertility defects encountered in the Gpx5-/- mice.
Judging from their sequence identities, the majority of piRNAs in the caput epididymidis of aged mice were identified as putative PIWIL1- and PIWIL2-binding piRNAs. Under the stress-associated condition in the absence of GPX5, a luminal antioxidant with restricted expression in the caput epididymidis, the mRNA expression of Piwil1 and Piwil2, together with several other known piRNA pathway genes, was dramatically down-regulated in the caput epididymidis of aged mice. In agreement with this observation, piRNAs putatively binding to these two PIWI proteins were dramatically reduced in the caput epididymidis of the Gpx5-/- animals. The segment-specific loss of piRNA was strongly aligned with the highly regionalized expression of the Gpx5 gene, and accumulation of the GPX5 protein, in the caput epididymidis of aged WT mice.3
PIWIL4 accounted for a smaller fraction of the piRNAs in the caput epididymidis of aged mice. In contrast to the observation that both Piwil1 and Piwil2 transcripts and their potentially binding piRNAs were drastically reduced in Gpx5-/- mice, the Piwil4 transcript and potential PIWIL4-binding piRNAs were almost unaffected. These differential responses suggest functional diversity of the PIWI proteins expressed in the epididymis, as occurs in testicular germ cells.31,48 Unlike the other two PIWI proteins, testicular PIWIL4 function is restricted to a narrow developmental window during primordial germ cell reprogramming and seems not to be required for the subsequent postnatal germline development and function.49 From the current evidence, it appears unlikely that PIWIL4 is involved in the oxidative stress response, or compensates the loss of PIWIL1 and PIWIL2 in the caput epididymidis of aged mice.
On the basis of the theory that piRNAs recognize and bind to target mRNAs like miRNAs, and act as modulators at the posttranscriptional level,22,30 we provided different levels of correlation analysis to show that in the Gpx5-/- genetic background, genes that are predicted to be piRNA targets were indeed up-regulated in response to the loss of piRNAs. More importantly, the set of “oxidative stress response genes” was more prone to piRNA regulation, and their mRNA levels tended to be elevated with the loss of piRNAs in the Gpx5-/- group. These results provide insight into the regulation of the oxidative stress response genes by epididymal piRNAs, as has also been proposed in several other studies.44,50 Furthermore, those genes targeted by >50 piRNAs had significant function enrichments in “transcription”, “protein phosphorylation”, “covalent chromatin modification”, and “intracellular signal transduction”, which suggest that in addition to the oxidative stress response genes, the caput epididymidal piRNAs also tend to target important genes encoding transcription factors, kinases, epigenetic regulators, and signaling molecules and pose indirect but more profound effects.
Given that fewer piRNAs were detected in the caput epididymidis of young WT mice (2 months of age), the accumulation of piRNAs in the caput epididymidis appears to be age dependent. At present, we have no evidence indicating that the GPX5 protein is directly involved in the biogenesis of these caput epididymidal piRNAs. Rather, we anticipate that it is the antioxidant stress response of the caput epididymidis that triggers the up-regulation and accumulation of these piRNAs. We, therefore, propose that the function of these piRNAs is, in part, to regulate the expression of antioxidant genes in the caput epididymidal epithelium and that the loss of GPX5 dysregulates the piRNA pathway within these cells.
Taken together, our current results shed light on an age-dependent group of piRNAs associated with oxidative stress, and a possible function of the piRNAs existing in the mammalian epididymis, that merit further investigation. This study also suggests that the Gpx5-/- mouse is a suitable model for future somatic piRNA research.
AUTHOR CONTRIBUTIONS
CC, LY, YCZ, and JRD conceived, designed, and organized the whole study. CC and LY performed the experiments and analyzed the data. JHB, YFR, ZTL, MH, SSX, WBM, MJN, ZMN, and YLG contributed to the experiments. LTG, AK, AC, and QL contributed to the data collection and preprocessing. CDC, MFL, YCZ, JRD, and YLZ analyzed and organized the results. CC and LY prepared the figures. CC, SS, YZ, ALE, BN, ZLF, JRD, and YLZ wrote and revised the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
Agilent 2100 results and RIN values of all sequencing samples in this study.
Supplementary Table 1 (396.4KB, pdf) : Primers used in this study
Supplementary Table 2 (1.6MB, pdf) : Transcriptome of 13M WT and Gpx5 KO caput epididymis
Supplementary Table 3 (96.1KB, pdf) : Expression of mouse piRNA pathway genes
Supplementary Table 4 (199.2KB, pdf) : Predefined list of piRNAs binding to each PIWI protein
Supplementary Table 5 (722.4KB, pdf) : Potential target piRNAs to each gene
ACKNOWLEDGMENTS
This research was supported by the National Basic Research Program of China (Grant No. 2014CB943103) and National Natural Science Foundation of China (Grant No. 31471104, No. 31671203, No. 31301225, No. 31301226, No. 31701119, and No. 31571192) and was partly realized under the frame of the France-China scientific exchange programs “Xu Guangqi” and “Cai Yuanpei” of the “Partenariat Hubert Curien” attributed to JRD and YLZ. The authors thank Prof. Winnie Wai Chi Shum, Prof. Xiaodong Sun, Ms. Aihua Liu, Dr. Chaobao Zhang, Dr. Zhen Lin, Dr. Xueting Luo, and the Bio-Med Big Data Center, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, and Chinese Academy of Sciences for their kind support.
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Aitken RJ, Gibb Z, Baker MA, Drevet J, Gharagozloo P. Causes and consequences of oxidative stress in spermatozoa. Reprod Fertil Dev. 2016;28:1–10. doi: 10.1071/RD15325. [DOI] [PubMed] [Google Scholar]
- 2.Vernet P, Aitken RJ, Drevet JR. Antioxidant strategies in the epididymis. Mol Cell Endocrinol. 2004;216:31–9. doi: 10.1016/j.mce.2003.10.069. [DOI] [PubMed] [Google Scholar]
- 3.Chabory E, Damon C, Lenoir A, Kauselmann G, Kern H, et al. Epididymis seleno-independent glutathione peroxidase 5 maintains sperm DNA integrity in mice. J Clin Invest. 2009;119:2074–85. doi: 10.1172/JCI38940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noblanc A, Damon-Soubeyrand C, Karrich B, Henry-Berger J, Cadet R, et al. DNA oxidative damage in mammalian spermatozoa: where and why is the male nucleus affected? Free Radic Biol Med. 2013;65:719–23. doi: 10.1016/j.freeradbiomed.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 5.Kocer A, Henry-Berger J, Noblanc A, Champroux A, Pogorelcnik R, et al. Oxidative DNA damage in mouse sperm chromosomes: size matters. Free Radic Biol Med. 2015;89:993–1002. doi: 10.1016/j.freeradbiomed.2015.10.419. [DOI] [PubMed] [Google Scholar]
- 6.Zini A, Libman J. Sperm DNA damage: clinical significance in the era of assisted reproduction. CMAJ. 2006;175:495–500. doi: 10.1503/cmaj.060218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng J, Meyer CA, Wang Q, Liu JS, Shirley Liu X, et al. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics. 2012;28:2782–8. doi: 10.1093/bioinformatics/bts515. [DOI] [PubMed] [Google Scholar]
- 9.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–7. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sai Lakshmi S, Agrawal S. piRNABank: a web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008;36:D173–7. doi: 10.1093/nar/gkm696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gan H, Lin X, Zhang Z, Zhang W, Liao S, et al. piRNA profiling during specific stages of mouse spermatogenesis. RNA. 2011;17:1191–203. doi: 10.1261/rna.2648411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng H, Shi J, Zhang Y, Zhang H, Liao S, et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012;22:1609–12. doi: 10.1038/cr.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu C, Yu L, Wu B, Ma L, Gou LT, et al. A sequence of 28S rRNA-derived small RNAs is enriched in mature sperm and various somatic tissues and possibly associates with inflammation. J Mol Cell Biol. 2017;9:256–9. doi: 10.1093/jmcb/mjx016. [DOI] [PubMed] [Google Scholar]
- 16.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu C, Zheng G, Hu S, Zhang J, Xie S, et al. Epididymal region-specific miRNA expression and DNA methylation and their roles in controlling gene expression in rats. PLoS One. 2015;10:e0124450. doi: 10.1371/journal.pone.0124450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goh WS, Falciatori I, Tam OH, Burgess R, Meikar O, et al. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev. 2015;29:1032–44. doi: 10.1101/gad.260455.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robine N, Lau NC, Balla S, Jin Z, Okamura K, et al. A broadly conserved pathway generates 3'UTR-directed primary piRNAs. Curr Biol. 2009;19:2066–76. doi: 10.1016/j.cub.2009.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pandey RR, Tokuzawa Y, Yang Z, Hayashi E, Ichisaka T, et al. Tudor domain containing 12 (TDRD12) is essential for secondary PIWI interacting RNA biogenesis in mice. Proc Natl Acad Sci U S A. 2013;110:16492–7. doi: 10.1073/pnas.1316316110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gou LT, Dai P, Yang JH, Xue Y, Hu YP, et al. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 2014;24:680–700. doi: 10.1038/cr.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John B, Enright AJ, Aravin A, Tuschl T, Sander C, et al. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwasaki YW, Siomi MC, Siomi H. PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem. 2015;84:405–33. doi: 10.1146/annurev-biochem-060614-034258. [DOI] [PubMed] [Google Scholar]
- 25.Ku HY, Lin H. PIWI proteins and their interactors in piRNA biogenesis, germline development and gene expression. Natl Sci Rev. 2014;1:205–18. doi: 10.1093/nsr/nwu014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet. 2019;20:89–108. doi: 10.1038/s41576-018-0073-3. [DOI] [PubMed] [Google Scholar]
- 27.Ishizu H, Siomi H, Siomi MC. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012;26:2361–73. doi: 10.1101/gad.203786.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutcheon K, McLaughlin EA, Stanger SJ, Bernstein IR, Dun MD, et al. Analysis of the small non-protein-coding RNA profile of mouse spermatozoa reveals specific enrichment of piRNAs within mature spermatozoa. RNA Biol. 2017;14:1–15. doi: 10.1080/15476286.2017.1356569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiuchi T, Koga H, Kawamoto M, Shoji K, Sakai H, et al. A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature. 2014;509:633–6. doi: 10.1038/nature13315. [DOI] [PubMed] [Google Scholar]
- 30.Rouget C, Papin C, Boureux A, Meunier AC, Franco B, et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature. 2010;467:1128–32. doi: 10.1038/nature09465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 2011;12:246–58. doi: 10.1038/nrm3089. [DOI] [PubMed] [Google Scholar]
- 32.Zheng K, Wang PJ. Blockade of pachytene piRNA biogenesis reveals a novel requirement for maintaining post-meiotic germline genome integrity. PLoS Genet. 2012;8:e1003038. doi: 10.1371/journal.pgen.1003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2008;135:3–9. doi: 10.1242/dev.006486. [DOI] [PubMed] [Google Scholar]
- 34.Lee EJ, Banerjee S, Zhou H, Jammalamadaka A, Arcila M, et al. Identification of piRNAs in the central nervous system. RNA. 2011;17:1090–9. doi: 10.1261/rna.2565011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosheh Y, Seridi L, Ryu T, Takahashi H, Orlando V, et al. Characterization of piRNAs across postnatal development in mouse brain. Sci Rep. 2016;6:25039. doi: 10.1038/srep25039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ortogero N, Schuster AS, Oliver DK, Riordan CR, Hong AS, et al. A novel class of somatic small RNAs similar to germ cell pachytene PIWI-interacting small RNAs. J Biol Chem. 2014;289:32824–34. doi: 10.1074/jbc.M114.613232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Wang HY, Wan FC, Liu FJ, Liu J, et al. Deep sequencing analysis of small non-coding RNAs reveals the diversity of microRNAs and piRNAs in the human epididymis. Gene. 2012;497:330–5. doi: 10.1016/j.gene.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 38.Yan Z, Hu HY, Jiang X, Maierhofer V, Neb E, et al. Widespread expression of piRNA-like molecules in somatic tissues. Nucleic Acids Res. 2011;39:6596–607. doi: 10.1093/nar/gkr298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu C, Zhang YL, Yu L, Sharma S, Fei ZL, et al. Epididymal small non-coding RNA studies: progress over the past decade. Andrology. 2019;7:681–9. doi: 10.1111/andr.12639. [DOI] [PubMed] [Google Scholar]
- 40.Ross RJ, Weiner MM, Lin H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature. 2014;505:353–9. doi: 10.1038/nature12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belleannee C, Thimon V, Sullivan R. Region-specific gene expression in the epididymis. Cell Tissue Res. 2012;349:717–31. doi: 10.1007/s00441-012-1381-0. [DOI] [PubMed] [Google Scholar]
- 42.Cornwall GA. New insights into epididymal biology and function. Hum Reprod Update. 2009;15:213–27. doi: 10.1093/humupd/dmn055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnston DS, Jelinsky SA, Bang HJ, DiCandeloro P, Wilson E, et al. The mouse epididymal transcriptome: transcriptional profiling of segmental gene expression in the epididymis. Biol Reprod. 2005;73:404–13. doi: 10.1095/biolreprod.105.039719. [DOI] [PubMed] [Google Scholar]
- 44.Kuintzle RC, Chow ES, Westby TN, Gvakharia BO, Giebultowicz JM, et al. Circadian deep sequencing reveals stress-response genes that adopt robust rhythmic expression during aging. Nat Commun. 2017;8:14529. doi: 10.1038/ncomms14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuster A, Tang C, Xie Y, Ortogero N, Yuan S, et al. SpermBase: a database for sperm-borne RNA contents. Biol Reprod. 2016;95:99. doi: 10.1095/biolreprod.116.142190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao S, Gou LT, Zhang M, Zu LD, Hua MM, et al. piRNA-triggered MIWI ubiquitination and removal by APC/C in late spermatogenesis. Dev Cell. 2013;24:13–25. doi: 10.1016/j.devcel.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Ma L, Yu H, Ni Z, Hu S, Ma W, et al. Spink13, an epididymis-specific gene of the Kazal-type serine protease inhibitor (SPINK) family, is essential for the acrosomal integrity and male fertility. J Biol Chem. 2013;288:10154–65. doi: 10.1074/jbc.M112.445866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Annu Rev Cell Dev Biol. 2009;25:355–76. doi: 10.1146/annurev.cellbio.24.110707.175327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bao J, Zhang Y, Schuster AS, Ortogero N, Nilsson EE, et al. Conditional inactivation of Miwi2 reveals that MIWI2 is only essential for prospermatogonial development in mice. Cell Death Differ. 2014;21:783–96. doi: 10.1038/cdd.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwon C, Tak H, Rho M, Chang HR, Kim YH, et al. Detection of PIWI and piRNAs in the mitochondria of mammalian cancer cells. Biochem Biophys Res Commun. 2014;446:218–23. doi: 10.1016/j.bbrc.2014.02.112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Agilent 2100 results and RIN values of all sequencing samples in this study.
Data Availability Statement
The RNA-seq datasets generated and analyzed during the current study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the accession number of GSE122087.