

Abstract
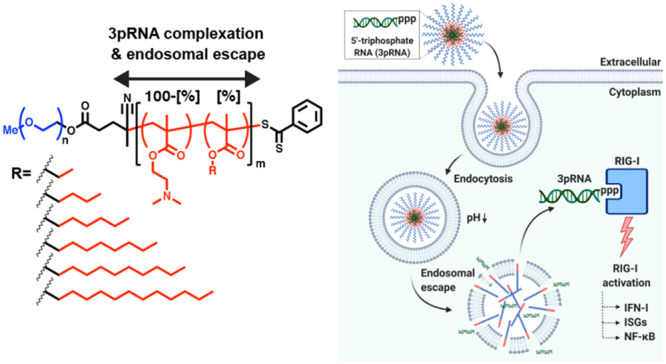
RNA ligands of retinoic acid-inducible gene I (RIG-I) hold significant promise as antiviral agents, vaccine adjuvants, and cancer immunotherapeutics, but their efficacy is hindered by inefficient intracellular delivery to the cytosol where RIG-I is localized. Here, we address this challenge through the synthesis and evaluation of a library of polymeric carriers rationally designed to promote the endosomal escape of 5′-triphosphate RNA (3pRNA) RIG-I agonists. We synthesized a series of PEG-block-(DMAEMA-co-AnMA) polymers, where AnMA is an alkyl methacrylate monomer ranging from n = 2–12 carbons, of variable composition, and examined effects of polymer structure on the intracellular delivery of 3pRNA. Through in vitro screening of 30 polymers, we identified four lead carriers (4–50, 6–40, 8–40, and 10–40, where the first number refers to the alkyl chain length and the second number refers to the percentage of hydrophobic monomer) that packaged 3pRNA into ∼100-nm-diameter particles and significantly enhanced its immunostimulatory activity in multiple cell types. In doing so, these studies also revealed an interplay between alkyl chain length and monomer composition in balancing RNA loading, pH-responsive properties, and endosomal escape, studies that establish new structure–activity relationships for polymeric delivery of 3pRNA and other nucleic acid therapeutics. Importantly, lead carriers enabled intravenous administration of 3pRNA in mice, resulting in increased RIG-I activation as measured by increased levels of IFN-α in serum and elevated expression of Ifnb1 and Cxcl10 in major clearance organs, effects that were dependent on polymer composition. Collectively, these studies have yielded novel polymeric carriers designed and optimized specifically to enhance the delivery and activity of 3pRNA with potential to advance the clinical development of RIG-I agonists.
Short abstract
We designed and structurally optimized polymeric nanocarriers for RNA agonists of RIG-I that overcome critical drug delivery barriers to potently enhance their immunostimulatory activity in mice.
Introduction
Retinoic acid-inducible gene I (RIG-I) is a pattern recognition receptor (PRR) that activates antiviral innate immunity upon recognition of 5′ di- or triphosphorylated double-stranded RNA (2p- or 3pRNA) present in the cytosol.1−3 Activation of RIG-I triggers a multifaceted innate immune response characterized by the expression of type-I interferons (IFN-I) and interferon stimulated genes (ISGs) with a broad spectrum of antiviral effector functions.3,4 In addition to its well-established role as an important sensor of viral pathogen invasion, RIG-I expression in tumor cells has recently been shown to play a vital role in promoting responsiveness to anti-CTLA-4 immune checkpoint blockade, consistent with a strong association between expression of RIG-I, T cell infiltration, and overall survival in melanoma patients.5 Accordingly, RNA ligands of the RIG-I pathway have recently emerged as promising antiviral agents, vaccine adjuvants, and cancer immunotherapeutics.4−10 Notably, RNA RIG-I agonists have recently advanced to clinical trials in immuno-oncology (NCT03065023).
Despite their broad therapeutic potential, RNA RIG-I ligands face multiple barriers to efficacy and clinical translation similar to other classes of nucleic acid therapeutics, including susceptibility to extracellular nuclease degradation, poor intracellular uptake, and, importantly, endo/lysosomal degradation with minimal delivery to the cytosol where RIG-I is localized.11,12 There have been decades of extensive work focused on the development of carriers for siRNA, miRNA, mRNA, and DNA,13,14 including cationic polymers, inorganic materials, and lipid-based nanoparticles, to name only a few. However, there has been minimal investigation into the evaluation and optimization of delivery systems for RIG-I agonists. Instead, the majority of studies either exploring mechanisms or potential applications of RIG-I ligands have utilized commercial in vitro lipid-based transfection agents (e.g., Lipofectamine) or polyethylenimine (PEI),3,7,8,15 which have been widely explored for nucleic acid delivery, but have not been optimized for 2p- or 3pRNA delivery, nor approved for human use. Hence, there is a need to design, evaluate, and optimize new delivery platforms for this emerging class of RNA therapeutics. To date, we are aware of only three previous reports describing new or customized carriers for 3pRNA delivery. Chakravarthy et al. utilized cationic gold nanorods for electrostatic complexation of 3pRNA, and demonstrated the ability of this approach to inhibit viral replication in vitro.16 Huang and co-workers recently described the design of lipid calcium phosphate nanoparticles for 3pRNA delivery, and evaluated immunotherapy responses in a model of pancreatic cancer.17 Finally, our group has recently described the use of cationic endosome-releasing polymer nanoparticles for 3pRNA delivery.18 This carrier has a cationic dimethylaminoethyl methacrylate (DMAEMA) first block for electrostatic complexation of RNA, and an endosome-destabilizing terpolymer block, composed of DMAEMA, butyl methacrylate (BMA), and propylacrylic acid (PAA). In syngeneic mouse models of breast and colon cancer, delivery of 3pRNA using these NPs triggered immunogenic tumor cell death, reduced tumor growth, and improved response to immune checkpoint blockade. Importantly, we also demonstrated the importance of integrating an active endosomal escape mechanism into the carrier for enhancing cytosolic delivery and immunostimulatory activity of 3pRNA.
While this is a promising 3pRNA carrier for applications such as vaccination or intratumoral immunotherapy where localized administration (e.g., intratumoral, intranasal, subcutaneous)18−21 can be leveraged for prophylactic or therapeutic benefit, the cationic DMAEMA corona of NPs is not well-suited for systemic, intravenous administration, which may be desirable for many applications; for example, the treatment of metastatic cancer or antiviral therapy. Additionally, while our previous work implicates endosomal escape as an important design parameter in maximizing the activity of 3pRNA,18 there has not yet been a rigorous investigation into how carrier properties affect 3pRNA activity nor a systematic structural optimization of nanocarriers for this unique class of RNA therapeutic. Therefore, the objective of this work was to develop and optimize polymeric carriers for systemic administration of 3pRNA RIG-I agonists based on elucidation of relationships between polymer structure and 3pRNA activity. To accomplish this, we drew motivation from recent work describing the use of PEG-block-(DMAEMA-co-butyl methacrylate) (PEG-b-DB) for siRNA delivery.22,23 The DB block imparts pH-responsive, endosomolytic activity while also facilitating electrostatic complexation of siRNA into a polymeric nanoparticle with a PEGylated corona. Building upon this design, we synthesized a novel library of 30 mPEG-block-(DMAEMA-co-AnMA[%]) containing 20–60% of AnMA monomers ranging between n = 2–12 carbons (i.e., ranging from ethyl methacrylate (n = 2) to lauryl methacrylate (n = 12)) and investigated the effect of polymer composition on pH-responsive activity and 3pRNA delivery (Figure 1). Interestingly, while varying lipid chain length has been previously explored for several other types of delivery platforms (e.g., lipid nanoparticles, poly(β-amino ester)s)24,25 as a strategy to enhance delivery of associated cargo, the effect of alkyl chain length has not yet been explored for this class of actively endosomolytic carriers, offering an unexplored mechanism for tuning or optimizing polymer properties for 3pRNA delivery. Using this polymer library, we elucidated new structure–property–activity relationships that informed the design of novel carriers for 3pRNA, resulting in identification of several lead candidates for systemic delivery of RIG-I agonists with potential to accelerate their clinical translation and utility.
Figure 1.

Design and optimization of polymeric carriers to enhance the immunostimulatory activity of 3pRNA RIG-I ligands. (a) Library of mPEG-block-[DMAEMA-co-AnMA[%]] polymers composed of 20–60% of alkyl methacrylate monomers (AnMA) containing n = 2–12 carbons was synthesized and screened to identify lead carriers for 3pRNA delivery. (b) Polymers are solubilized at low pH (pH 4) and mixed with 3pRNA to maximize electrostatic interactions followed by exchange into neutral pH buffer to form PEGylated nanoparticles loaded with 3pRNA. (c) Carriers capable of complexing 3pRNA and promoting endosomal escape increase 3pRNA delivery to cytosol, resulting in enhanced activation of RIG-I signaling. Image created with BioRender.com.
Results and Discussion
Balancing Monomer Composition and Alkyl Chain Length Enables Control of pH-Responsive Properties
We synthesized a library of 30 diblock copolymers using a mPEG (10 kDa) functionalized chain transfer agent (CTA) for reversible addition–fragmentation chain-transfer (RAFT) synthesis of a second block comprising DMAEMA and a hydrophobic methacrylate monomer with alkyl side chains ranging from 2 to 12 carbons (AnMA where n = 2–12) at compositions ranging from 20% to 60% (Figure 1a, Scheme 1). mPEG-block-[DMAEMA-co-AnMA[%]] polymers are referred to henceforth by their alkyl chains lengths (n) and hydrophobic monomer compositions ([%]); for example, 4–50 refers to a second block composed of 50% butyl methacrylate (n = 4)). Polymer degree of polymerization, molecular weight, and composition are summarized in Table 1.
Scheme 1. RAFT Synthesis of mPEG-block-[DMAEMA-co-AnMA[%]] Copolymers.
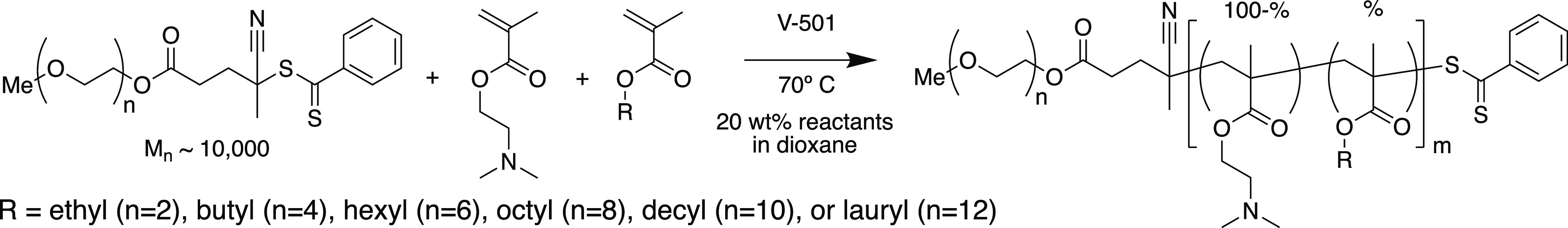
Table 1. Summary of PEG-block-(DMAEMA-co-AnMA) Copolymer Properties.
| n | % | total Mna (kg/mol) | DPb | compositionc (% AnMA) |
|---|---|---|---|---|
| 2 | 20 | 33.2 | 160 | 19.1% |
| 30 | 32.3 | 158 | 28.7% | |
| 40 | 33.5 | 171 | 38.3% | |
| 50 | 33.5 | 176 | 47.5% | |
| 60 | 31.5 | 165 | 56.6% | |
| 4 | 20 | 31.5 | 143 | 19.0% |
| 30 | 32.4 | 150 | 29.6% | |
| 40 | 34.2 | 163 | 40.2% | |
| 50 | 39.1 | 194 | 50.9% | |
| 60 | 34.9 | 170 | 58.1% | |
| 6 | 20 | 35.1 | 161 | 20.6% |
| 30 | 37.1 | 171 | 33.9% | |
| 40 | 36.1 | 163 | 43.6% | |
| 50 | 34.0 | 149 | 51.6% | |
| 60 | 33.0 | 141 | 59.8% | |
| 8 | 20 | 38.6 | 177 | 20.8% |
| 30 | 35.3 | 152 | 32.1% | |
| 40 | 38.7 | 168 | 41.6% | |
| 50 | 38.4 | 162 | 51.6% | |
| 60 | 38.2 | 157 | 60.3% | |
| 10 | 20 | 35.2 | 149 | 23.2% |
| 30 | 39.0 | 163 | 34.6% | |
| 40 | 40.7 | 170 | 37.5% | |
| 50 | 46.9 | 195 | 49.2% | |
| 60 | 44.2 | 176 | 56.6% | |
| 12 | 20 | 34.0 | 141 | 18.4% |
| 30 | 48.1 | 207 | 30.8% | |
| 40 | 37.3 | 141 | 40.1% | |
| 50 | 41.5 | 155 | 49.9% | |
| 60 | 40.0 | 139 | 61.3% |
Number-average molecular weight (Mn) of diblock copolymer calculated by conversion NMR.
Degree of polymerization (DP) of second block calculated by conversion NMR.
Fraction of second block composed of indicated hydrophobic monomer as measured by NMR of purified polymer.
An important and distinguishing feature of this class of carriers is a balance of cationic and hydrophobic content in the second block that can drive the formation of PEGylated micelles at neutral pH, but, upon protonation of amino groups at endo/lysosomal pH values (i.e., pH 5.8–6.2), allows for particle disassembly and the unveiling of membrane-destabilizing segments that can facilitate endosomal escape of associated cargo.22,23,26,27 We therefore first characterized the effect of n and [%] on particle size both at pH 7.4 and at pH 5.8 using dynamic light scattering. First evaluating effects on micelle formation at pH 7.4 (Figure 2a), all polymers with n > 4 and [%] > 40 formed 15–40 nm micelle-sized particles at pH 7.4, and increasing n enabled micellization at lower [%] values; for example, 30% BMA (n = 4) and hexyl methacrylate (n = 6) was insufficient to drive micelle formation, whereas a step increase in particle diameter was observed between 20% and 30% when n > 8. Next, taking the ratio of the particle size at pH 7.4 to pH 5.8 (Figure 2b) we found that, similar to particle formation, pH-responsive disassembly was dictated by an interplay between both n and [%]. Polymers comprising hydrophobic side chain lengths of n = 2 and n = 4 tended to exhibit a pH-dependent size change only at higher hydrophobic monomer compositions between [%] = 50 and [%] = 60, while polymers with hydrophobic side chain lengths of n = 6–10 demonstrated pH-responsiveness at lower hydrophobic monomer compositions between [%] = 30 and 50. Hence, as alkyl chain length is increased, the amount of DMAEMA must also be increased in order to achieve sufficient second block solubility at low pH to allow for a micelle-to-unimer transition. Interestingly, we found that incorporation of >20% lauryl methacrylate (n = 12) eliminated pH-responsive particle disassembly, thereby defining an upper boundary for monomer alkyl chain length beyond which pH-responsive micelle-to-unimer transition does not occur, which was previously unknown for this class of polymers.
Figure 2.

Effect of alkyl methacrylate monomer chain length and composition and on pH-responsive responsive properties of diblock copolymers. (a) Particle diameter (number-average) and polydispersity (PDI) of mPEG-block-[DMAEMA-co-AnMA[%]] composed of alkyl methacrylate monomers of different chain length (n) and composition ([%]) in phosphate buffered saline (PBS) at pH 7.4. Data are mean ± SD with n = 3 experimental replicates. (b) Heat map demonstrating the fold-decrease in particle diameter between pH 7.4 and pH 5.8 for polymers of indicated n and [%]. (c) Fraction of red blood cell hemolysis mediated by polymers (1 μg/mL) at indicated pH values. Data are mean ± SD and representative of two independent experiments, with n = 4 biological replicates. Statistical analysis is provided in Tables S1 and S2 (Supporting Information). (d) Heat map summarizing hemolysis data in (c) at pH 7.4, pH 6.6, and pH 5.8.
To further understand relationships between second block composition and pH-responsive properties, we next evaluated the pH-responsive, membrane-destabilizing activity of all polymers using a well-established erythrocyte lysis assay (Figure 2c,d). While pH-responsive hemolytic activity is not fully predictive of the efficiency of cytosolic drug delivery,28 the assay offers a facile approach to directly investigate the capacity of materials to interact with and lyse biological membranes at different pH values. In general, similar trends were observed between pH-responsive particle disassembly and hemolysis activity. Polymers of insufficient hydrophobicity to self-assemble into micelles (e.g., 2–40, 4–30) also did not possess membrane destabilizing activity, whereas polymers with highly hydrophobic second blocks (e.g., 10–60, 12–60) were unable to disassemble at low pH values to expose membrane-destabilizing segments that facilitate hemolysis. Potential exceptions were 6–30 and 10–30, which formed smaller micelles and thus demonstrated a less pronounced pH-responsive size change, but displayed strong hemolysis activity at pH 5.8, and 2–60, which displayed a size transition, but did not possess membrane destabilizing activity, likely due to insufficiently long alkyl chains for disrupting the lipid bilayer of erythrocytes.
Additionally, both n and [%] had an impact on the pH at which hemolytic activity was observed. This was particularly evident at [%] = 50 where 4–50 begins to demonstrate hemolytic activity at pH 6.6, whereas initiation of activity is shifted to pH 6.2 for carriers with n > 4 (i.e., 6–50, 8–50, 10–50). The ability to more precisely tune the transition pH at which membrane-destabilizing activity manifests may have important implications for optimizing the delivery of 3pRNA and other nucleic acid therapeutics. For example, in an application of cancer therapeutics, avoiding premature particle destabilization in the low pH microenvironment of many types of solid tumors,29 while still achieving endosomal escape of cargo, may merit selection of a carrier that transitions to a membrane-disruptive state at a slightly lower pH value. Together, these studies identify upper and lower boundaries for both composition and alkyl chain length, which act cooperatively to generate a “hot spot” in copolymer composition where pH-responsive, membrane destabilizing activity is observed (Figure 2d) and the potency of which can be tuned via control of both alkyl chain length and copolymer composition.
Elucidation of Property–Activity Relationships Identify Lead Carriers for 3pRNA Delivery
To further understand relationships between pH-responsive polymer properties and 3pRNA delivery, we next evaluated the ability of all carriers to enhance the immunostimulatory activity of 3pRNA. To accomplish this, we first evaluated the efficiency at which each polymer could electrostatically complex dsRNA at N:P (NH3+:PO4–) ratios of 2, 4, 8, 12, 16, and 20 (assuming 50% protonation of DMAEMA) using an RNA intercalating dye (Ribogreen) to evaluate the degree of complexation (Figure 3a). Interestingly, there was an interplay between n and [%] in defining the N:P ratio where complete RNA complexation was achieved. Not surprisingly, carriers with a higher DMAEMA content tended to complex RNA at lower charge ratios; however, this was offset by the incorporation of longer alkyl chains which tended to inhibit complexation at a given composition, potentially owing to an increased steric hindrance between the RNA and cationic DMAEMA residues on the polymer backbone. Nonetheless, an N:P ratio of 20:1 was selected for subsequent studies, as this represented the ratio at which >70% complexation was achieved for all carriers, with the exception of 8–50, 8–60, 10–50, 10–60, 12–50, and 12–60, which all plateaued in their capacity to complex RNA at around ∼40–50% owing to their low charge density and longer, sterically bulky alkyl side chains. Hence, some carriers exhibit pH-responsive hemolytic activity (e.g., 8–50, 10–50) but have a poor capacity for RNA loading. While this limits their utility for delivery of electrostatically complexed nucleic acids, the increased core hydrophobicity of 4–60, 6–50, 8–50, and 10–50 may confer enhanced particle stability and/or offer advantages for delivery of covalently linked or hydrophobized nucleic acid therapeutics (e.g., lipid-modified siRNA).30−32
Figure 3.

Identification of lead polymeric carriers for delivery of 3pRNA RIG-I agonists. (a) Effect of charge ratio (N:P) on degree of electrostatic complexation of dsRNA by polymeric carriers. (b) Representative dose response curve in A549 ISG reporter cells using polymers containing variable amounts of BMA (n = 4) to deliver 3pRNA. (c) Summary of approximated half-maximal effective concentration (EC50) of 3pRNA delivered with polymeric carriers. For graphical representation, a value of 1000 nM is assigned to all carriers for which an EC50 could not be estimated due to insufficient activity in the dose range explored. (d) Heat map depicting the ratio of the minimum estimated EC50 (4–50) to the EC50 of the indicated polymer. All values plotted as mean ± SD.
Using a 20:1 N:P ratio to complex 3pRNA, we next performed a dose response study in A549 ISG reporter cells that express a secreted luciferase (Lucia) under control of an ISG54 minimal promoter in conjunction with five IFN-stimulated response elements. A series of representative dose response curves for n = 4 is shown in Figure 3b with curves for all carriers shown in Figure S1. Based on these data, we estimated an EC50 value for each carrier (Figure 3c,d). A number of carriers were capable of enhancing 3pRNA activity, and as anticipated, there was a statistically significant (P < 0.0001) Spearman correlation between EC50 and hemolytic activity at pH 5.8 (Figure S2). Among polymers that enhanced 3pRNA activity, we identified four carriers with estimated in vitro EC50 values below 5 nM: 4–50, 6–40, 8–40, and 10–40–and more accurately determined EC50 values for these four lead carriers (Figure S3). 4–50 and 6–40 had similar activities with EC50 values of 0.5–0.9 nM, slightly lower than 8–40 and 10–40 which had a virtually identical EC50 of 4 nM. An analogous RNA with a 5′ hydroxyl (OH-RNA; i.e., lacking the triphosphate group) complexed to lead carriers did not stimulate an IFN-I response in A549 ISG reporter cells (Figure S4), confirming that the response is RIG-I dependent. All lead carriers complexed with dsRNA formed nanoparticles (NPs) of approximately 100 nm in diameter by dynamic light scattering (Figure S5) and displayed similar levels of cytotoxicity in A549 cells (Figure S6). We also evaluated the serum stability of lead NP/3pRNA complexes (Figure S7). Though we observed minimal release of RNA from all complexes upon incubation in 10% serum for 6 h as measured by gel electrophoresis (Figure S7a), a loss ∼30–40% in activity for 4–50, 6–40, and 10–40 and ∼60% loss in activity for 8–40 was measured after 2 h in 80% serum (Figure S7b), which further decayed to a complete loss of activity by 12 h, with the exception of 6–40, which maintained ∼20% of initial activity even after 24 h in serum. This may reflect the superior capacity of 6–40 to protect RNA from nuclease degradation and/or hydrolysis of the triphosphate group, and demonstrates that second block composition may also modestly impact particle and/or RNA stability. Additionally, all carriers are able to maintain 40% or greater activity even after 2 h in serum; while there is room for improvement, that several lead carriers can confer significant enhancement in 3pRNA activity even after extended incubation in serum bodes favorably for in vivo applications.
It is notable that while each of the lead carriers identified from bioactivity screens also demonstrate the targeted pH-responsive size change and strong hemolysis at pH 5.8, they represent only a subset of carriers with these desirable physicochemical properties. This supports recent findings that indicate that pH-responsive hemolysis is a prerequisite for this class of carriers to deliver RNA cytosolically, but is not perfectly predictive of endosomal escape and suggests that other carrier properties (e.g., stability, RNA release) also influence delivery efficiency.28 To gain further insight into this, we evaluated the endosomal escape capacity of carriers using a recently described Galectin 8 (Gal8) recruitment assay used to quantify the relative degree of endosomal escape (Figure 4a−c).28 Gal8 is cytosolically dispersed but redistributes to the endosomal membrane upon endosomal disruption, a process that can be imaged with fluorescent microscopy and quantified in real-time in cells expressing a Gal8-YFP fusion protein. Using this assay, we quantified the number of Gal8+ vesicles per cell upon treatment with dsRNA-loaded polymers comprising [%] = 40, 50, and 60 with n = 2–12, a significant subset of the polymer library that also represented a range of hemolytic potency and activity as 3pRNA carriers. Consistent with their capacity to enhance 3pRNA activity, 4–50, 6–40, 8–40, and 10–40 all demonstrated among the highest increases in Gal8 recruitment, whereas carriers comprising ethyl methacrylate (n = 2) or lauryl methacrylate (n = 12) or 60% hydrophobic monomer stimulated lower levels of Gal8 recruitment, also consistent with their relatively inefficient 3pRNA delivery. Interestingly, 8–50, which displayed hemolytic activity but only moderately enhanced 3pRNA delivery, resulted in a significant increase in Gal8 recruitment, potentially reflecting the inefficient RNA packaging capacity of this carrier (e.g., ∼50% RNA complexation at 20:1 N:P). Additionally, one lead carrier, 10–40, demonstrated a nearly 3-fold stronger increase in Gal8 recruitment relative to the other leads (i.e., 6–40, 8–40, 4–50). We speculated that this may reflect less efficient packaging of 3pRNA or an enhanced capacity to promote endosomal escape in the specific cell type used (MDA-MB-231 human breast cancer cells). To further evaluate this, we replaced 3pRNA with an siRNA targeting luciferase and evaluated knockdown in MDA-MB-231 cells constitutively expressing luciferase. This enabled investigation of endosomal escape and RNA delivery efficiency without the potential confounding effects of RIG-I activation on cell health/viability or changes in endocytosis or vesicular trafficking. Luciferase knockdown was consistent with the magnitude of Gal8 recruitment, with 10–40 demonstrating a lower EC50 value for luciferase knockdown (1.5 nM) than 4–50, 6–40, and 8–40 which all had an EC50 of approximately 3–4 nM (Figure 4d,e). Taken together, these data suggest that both composition and alkyl chain length are important variables for controlling the efficiency of endosomal escape, and that 10–40 can enhance endosomal escape and cytosolic delivery over the 4–50 carrier, at least in the MDA-MB-231 cell line. Furthermore, the optimal carrier composition may be different for specific cell types.
Figure 4.

Effect of alkyl monomer chain length and composition on endosomal escape. (a) Schematic of Galectin 8 (Gal8) recruitment assay used to investigate endosomal escape of selected carriers. Image created with BioRender.com. (b) Mean number of YFP-Gal8 vesicles observed per cell for indicated polymers loaded with control dsRNA. (c) Representative fluorescent images of cells expressing Gal8-YFP fusion protein upon treatment with 12–60 (low degree of endosomal escape), 8–40 (medium escape), 10–40 (high escape), or untreated (NT). (d) Dose response curves of relative luminescence upon treatment of luciferase-expressing MDA-MB-231 with indicated carrier complexed with luciferase siRNA. (e) Summary of estimated half-maximal effective concentration values (EC50) of the data shown in (e). All values are plotted as mean ± SD **P < 0.01, ****P < 0.0001 by one-way ANOVA with Tukey’s posthoc test.
3pRNA/NP Complexes Enhance RIG-I Signaling in Cancer and Immune Cells
Unlike the toll-like receptors, which are predominantly expressed in subsets of immune cells, RIG-I is ubiquitously expressed in virtually all nucleated cell types.33,34 This has rendered it a more universal agonist for innate immune activation, opening up potential broad applications ranging from cancer immunotherapy to antiviral therapy. Accordingly, we next evaluated the activity of 3pRNA/polymer complexes assembled using lead carriers (i.e., 4–50, 6–40, 8–40, 10–40) to activate ISGs and proinflammatory gene expression in macrophages (RAW264.7), dendritic cells (DC2.4), and three common murine cancer cell lines, 4T1 breast cancer, Lewis lung carcinoma (LLC), and B16.F10 melanoma (Figure 5). In general, all carriers increased 3pRNA activity over vehicle control (PBS) as evidenced by elevated expression of Ifnb1, Cxcl10, an ISG, and Tnf in most cell types, with 8–40 demonstrating the highest activity in RAW264.7 macrophages, DC2.4 cells, B16.F10 melanoma, and LLC cells. Consistent with A549 ISG reporter cell data (Figure S6), a control RNA with a 5′-hydroxyl (OH-RNA) instead of a 5′ triphosphate moiety displayed negligible activity when delivered using a commercial lipid transfection reagent (Lipofectamine2000; Figure S8) or all lead carriers (Figure S9), demonstrating the dependence of RIG-I in mediating the observed response. Interestingly, 6–40 tended to be the most active carrier in 4T1 cells, again highlighting the possibility that modulation of alkyl chain length may provide a mechanism to selectively control the magnitude of endosomal escape in specific cell types. This possibility merits further investigation.
Figure 5.

Evaluation of lead 3pRNA carriers in different cell types. Expression of (a) Ifnb1, (b) Cxcl10, and (c) Tnf in B16.F10 melanoma cells, Lewis lung carcinoma (LLC) cells, 4T1 breast cancer cells, RAW264.7 macrophages, DC2.4 dendritic cells, and primary murine bone marrow derived dendritic cells (BMDCs) 6 h after treatment with indicated carrier complexed to 3pRNA. (d) Flow cytometric quantification of the median fluorescent intensity (MFI) of MHC-II and CD86 on BMDCs treated with indicated carrier complexed to 3pRNA for 18 h. Vehicle (veh) control is PBS. All values plotted as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA with Tukey posthoc test; for (a) and (b) asterisks directly above bars are with respect to vehicle control and asterisks above brackets are between carriers.
To validate carrier activity in a primary, immunologically relevant cell type, we also evaluated 3pRNA-dependent gene expression in bone marrow derived dendritic cells (BMDCs). Here, all of the carriers demonstrated nearly equivalent capacity to increase Ifnb1 and Cxcl10 expression. Consistent with immunostimulation via the RIG-I pathway, this also triggered elevated cell surface levels of the DC activation and maturation markers MHC-II and CD86 (Figure 5d). These data demonstrated the activity of lead carriers in primary murine cells and motivated further exploration of 3pRNA delivery in vivo.
Systemic Administration of 3pRNA/NPs Complexes Stimulates RIG-I Activation in Mice
We next evaluated the ability of 3pRNA-loaded NPs comprising lead carriers complexed to 3pRNA to activate RIG-I in vivo utilizing an intravenous administration route. In these studies, we utilized a recently described 3p-modifed stem-loop RNA (SLR) ligand for RIG-I.3,8,35 The use of a stem-loop structure instead of a two-piece duplex increases the thermodynamic stability of 3pRNA,36 a key determinant of RIG-I binding affinity, while also presenting a single duplex terminus to ensure binding to RIG-I in a structurally defined orientation that has been characterized crystallographically.3,35 Additionally, a hairpin structure generates only a single blunt end and thereby reduces susceptibility to degradation by serum exonucleases.3,37 Accordingly, SLRs have been previously leveraged pharmacologically for potent and specific RIG-I activation in mice using jetPEI as a carrier.3,8
Healthy, wild-type C57BL/6 mice were administered 3pRNA/NP complexes assembled using 4–50, 6–40, 8–40, and 10–40 at a dose corresponding to 12.5 μg of 3pRNA (∼0.625 mg/kg RNA). Five hours after injection, blood was collected for quantification of serum IFN-α levels, and major clearance organs (liver, lung, kidney, and spleen) were harvested for gene expression analysis of Ifnb1 and Cxcl10 (Figure 6). While delivery of 3pRNA with all four carriers tended to increase serum IFN-α levels relative to vehicle (PBS) treated mice, only 4–50 and 6–40, the carriers with the lowest in vitro EC50 values (Figure 3c), resulted in a statistically significant (P < 0.01) increase in IFN-α levels. We also confirmed that responses were mediated by 3pRNA using 4–50 to deliver an analogous RNA hairpin lacking the 5′ triphosphate group (OH-RNA) (Figure S10).
Figure 6.

In vivo evaluation of lead 3pRNA carriers. (a) Schematic of in vivo analysis of 3pRNA carrier activity in mice. Image created with BioRender.com. (b) Serum levels of IFN-α following intravenous administration of 3pRNA using indicated carrier. qRT-PCR analysis of (c) Ifnb1 and (d) Cxcl10 expression in liver, spleen, lungs, and kidney of mice following intravenous administration of 3pRNA using indicated carrier. Vehicle (veh) is PBS. All values plotted as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs vehicle by one-way ANOVA with Dunnett’s multiple comparison test.
In evaluating ISG expression in clearance organs, there was a similar tendency of carriers to enhance Ifnb1 and Cxcl10 over vehicle to similar extent, reflecting their superior capacity for 3pRNA delivery. Considering their similar particle size and a common 10 kDa PEG corona, we did not expect dramatic differences in their relative capacity of lead carriers to activate RIG-I in tissues. Nonetheless, some carriers appeared to have a modestly increased activity relative to others at some organ sites; for example, 6–40 and 8–40 tended to enhance Ifnb1 and Cxcl10 expression to a greater extent in the spleen, whereas 4–50 tended to confer the greatest activity in the liver and lung. This may reflect differences in the pharmacokinetics or biodistribution of NP/3pRNA complexes that could be attributed to variations in relative complex stability. Alternatively, this may support in vitro findings suggesting that some carriers may have a higher capacity to promote endosomal escape of RNA cargo in specific cell populations. For example, the increased activity of 8–40 in the spleen, which is rich in myeloid cell populations capable of clearing circulating NPs, may reflect the ability of this carrier to preferentially potentiate 3pRNA activity in macrophage and dendritic cell lines. Further investigation into the effect of second block composition on the pharmacokinetic, pharmacodynamic, and biodistribution properties of 3pRNA is necessary to address these possibilities.
Collectively, these investigations have resulted in the identification of a small family of polymeric carriers designed to enhance the immunostimulatory activity of 3pRNA RIG-I agonists delivered via an intravenous route. Specifically, of the 30 mPEG-block-(DMAEMA-co-AnMA) carriers screened, those composed of 50% BMA (4–50), and 40% hexyl methacrylate (6–40), octyl methacrylate (8–40), or decyl methacrylate (10–40) were found to most efficiently enhance 3pRNA activity. Notably, all of these carriers conferred greater activity (i.e., lower EC50) in A549 ISG reporter cells than our previously reported cationic 3pRNA carrier platform (EC50 ∼ 10 nM)18 while also displaying a PEG corona that enabled intravenous administration.
As the objective of this work was to investigate relationships between polymer composition and 3pRNA activity in order to select lead carriers for in vivo use, the potential to adapt and leverage lead carriers for specific applications, including cancer immunotherapy, vaccines, and antiviral therapy, still remains to be explored. Notably, intratumoral administration of 3pRNA complexed to a PEI-based delivery vehicle (RGT100/PEI) has recently advanced to clinical trials (NCT03065023) as an in situ vaccination strategy.34 However, for the treatment of advanced metastatic disease and/or for patients with inaccessible lesions, systemic administration of 3pRNA and broad activation of RIG-I may be necessary to achieve delivery to relevant cell populations and tissues. Evaluating whether and how systemic delivery of the NP/3pRNA complexes developed herein can enhance tumor immunogenicity and immunotherapy responses will be an important next step and the subject of future investigations. However, systemic delivery of 3pRNA is also likely to result in a transient elevation of systemic cytokine levels that may limit the therapeutic window of 3pRNA/NP complexes. Additional investigation will be required to determine a maximum tolerated dose for systemically administered 3pRNA/NP carriers and to further modulate carrier and/or 3pRNA properties to increase this. For example, we have recently described a new class of 3pRNA pro-ligands based on the concept of a “synthetic overhang”, bulky macromolecules (e.g, PEG) linked to the 3′ end of the complement strand via a cleavable linker that blocks RIG-I recognition of 3pRNA until removed under a specific environmental stimulus (e.g., redox, enzymes).38 Therefore, combining such 3pRNA prodrugs with optimized carriers may provide a strategy to enrich RIG-I activation at specific sites (e.g., tumors) while minimizing systemic inflammation, thereby widening the therapeutic window. Nonetheless, it is notable that other systemically administered nanoparticle-based innate immune agonists with a similar cytokine profile have advanced into patients who experienced only transient flu-like symptoms.39,40 Moreover, the clinical experience with cancer immunotherapies (e.g., CAR T cells) suggests that some level of systemic cytokine response can be well-tolerated and may even be beneficial to outcomes. Additionally, strategies have emerged for managing immunotoxicity, including anti-IL-6 antibodies (tocilizumab) and corticosteroids while maintaining efficacy.41
Tuning of both alkyl chain length and composition may also provide a mechanism for tailoring organ-level RIG-I activation profiles for specific therapeutic applications. For example, the liver and lung are common sites of cancer metastasis and viral infection (e.g., respiratory viruses, hepatitis), diseases for which RIG-I ligands have been explored as therapeutics.4,6,8,42,43 Therefore, carriers that demonstrate an enhanced capacity to trigger RIG-I signaling at these sites (e.g., 4–50) may be particularly good candidates for future exploration in relevant disease models. Likewise, 3pRNA is a promising vaccine adjuvant,9,10 and intravenous administration of vaccines has recently been demonstrated to augment cellular immunity likely due to enhanced uptake by dendritic cells residing in the spleen.39,44,45 We have previously demonstrated that a similar type of endosomolytic nanocarrier, which also enables 3pRNA delivery,18 can be leveraged as a vaccine delivery system for enhancing cellular immunity to protein subunit antigens.19,20 Given their superior immunostimulatory potency, the NP/3pRNA complexes developed here also hold potential as vaccine delivery platforms, investigations that will be pursued in future work. Important to such efforts, the synthetic versatility of RAFT synthesis also allows for incorporation of reactive handles into carriers for covalent linkage of peptides and proteins,20,46 offering a strategy to achieve codelivery of antigen and adjuvant, which has been widely demonstrated to enhance vaccine responses.47 Similarly, by using reactive or functionalized chain transfer agents in polymer synthesis, these carriers are also highly amenable to incorporation of targeting ligands (e.g., antibodies, carbohydrates) to improve cell or organ specificity of RIG-I activation.
Previous work exploring similar endosomolytic carriers has utilized butyl methacrylate (n = 4) as the hydrophobic monomer, varying the composition of BMA and DMAEMA or other protonizable amine-containing monomers to optimize delivery efficiency.22,27,48 Here, by examining the effect of alkyl chain length we explored a new parameter space with potential implications for carrier design that extend beyond optimization of 3pRNA activity. First, our data suggest that modulation of alkyl chain length may provide a mechanism to optimize endosomal escape in specific cell populations, which may have different endosomal membrane compositions and physicochemical properties.49 Similar strategies have been employed to enhance cell or tissue tropism of lipid nanoparticles.50,51 This intriguing possibility that second block composition can be tuned to selectively enrich delivery to specific cell populations merits further investigation. Additionally, carriers containing longer alkyl chains may have a lower critical micelle concentration, and therefore a higher degree of stability, and/or allow for codelivery of hydrophobic or amphiphilic cargo (e.g., chemotherapeutics); for example, 6–40 and 10–40 demonstrate comparable in vitro and in vivo activity, but the >1.5× number of methylene groups in 10–40 may offer advantages for such codelivery applications. Similarly, several carriers demonstrated promising hemolysis and/or Gal8 recruitment, but were ineffective 3pRNA carriers, likely owing to inefficient oligo complexation (e.g., 8–50). However, such carriers may prove advantageous for delivery of covalently linked or amphiphilic oligonucleotides.30,31 Finally, our investigations here were focused on enhancing the delivery of 3pRNA, an emerging therapeutic for which very few carrier technologies have been evaluated, let alone optimized. Nonetheless, the carrier library described here also offers promise for enhancing and/or tuning the delivery of a diversity of other nucleic acid therapeutics, including siRNA, mRNA, DNA, and other immunostimulatory nucleic acid molecules. Indeed, an initial evaluation demonstrates the ability of lead carriers to enhance cytosolic delivery of siRNA which may be further augmented through selection of alkyl monomer in the endosomolytic block. The utility of these carriers to enhance delivery of other nucleic acid therapeutics, and the potential to leverage alkyl chain length to potentiate and/or tune activity in specific cell types, merits future investigation.
Conclusion
Critical drug delivery barriers, including poor cellular uptake, endo/lysosomal degradation, and inefficient cytosolic delivery hinder the activity and future development of RNA RIG-I agonists as vaccine adjuvants, antiviral agents, and cancer immunotherapeutics. To address this challenge, we synthesized a novel library of mPEG-block-(DMAEMA-co-AnMA) polymers, where AnMA is an alkyl methacrylate monomer ranging from n = 2–12 carbons in length and DMAEMA is a pH-sensor that also enables electrostatic complexation of nucleic acids. Through in vitro screening of 30 polymers, we identified four lead carriers that significantly enhance the immunostimulatory potency of 5′-triphospate RNA RIG-I ligands in vivo and in vitro: 4–50, 6–40, 8–40, and 10–40, where the first number refers to the alkyl chain length and the second number refers to the percentage of hydrophobic monomer. In doing so, we also established new structure–activity relationships between monomer composition, alkyl chain length, pH-responsive properties, and endosomal escape that inform the design of carriers for 3pRNA and other nucleic acid therapeutics. Importantly, these lead carriers, which packaged 3pRNA into ∼100-nm-diameter particles, enabled intravenous administration of 3pRNA, resulting in increased RIG-I activation as measured by increased levels of serum IFN-α and expression of Ifnb1 and Cxcl10 in major clearance organs, effects that were partially dependent on second block composition. Collectively, this work resulted in the development of the first polymeric carrier systems that were designed and optimized specifically to enhance the delivery of RIG-I ligands and, therefore, offer high potential for increasing the immunostimulatory activity and utility of this emerging class of RNA therapeutic.
Materials and Methods
RAFT Polymerization of PEG-block-(DMAEMA-co-AnMA)
For reversible addition–fragmentation chain transfer (RAFT) polymerizations, the following reagents were used: Poly(ethylene glycol) 4-cyano-4-(phenylcarbonothioylthio)pentanoate (Mn = 10 000 Da, Sigma-Aldrich) was used as the chain transfer agent (CTA), N,N-dimethylaminoethyl methacrylate (DMAEMA, Sigma-Aldrich) and variable hydrophobic side chain length methacrylates (AnMA), including ethyl methacrylate (EMA, Sigma-Aldrich), butyl methacrylate (BMA, Sigma-Aldrich), hexyl methacrylate (HMA, Sigma-Aldrich), octyl methacrylate (OMA, Polysciences), decyl methacrylate (DeMA, Polysciences), and lauryl methacrylate (LMA, Sigma-Aldrich) were used as monomers, 4,4′-Azobis(4-cyanovaleric acid) (V-501, Wako Chemicals) was used as a free-radical initiator, and 60:40 mixture of 1,4-dioxane (Sigma-Aldrich) and dimethylformamide (Sigma-Aldrich) was used as the solvent. Briefly, inhibitor was removed from monomers using gravity filtration through aluminum oxide (Sigma-Aldrich) packed columns. Initiator, CTA, and monomers were mixed into solvent at a ratio of 0.2 I0:1 CTA0:300 M0. Monomers and CTA were 20 wt % of the final solution, and monomers were combined in a ratio of 0:100, 20:80, 30:70, 40:60, 50:50, or 60:40 AnMA:DMAEMA. The mixture was polymerized under a nitrogen atmosphere for 20 h at 70 °C. The resultant diblock copolymers were diluted in acetone and isolated using dialysis (3 kDa MWCO, Thermo) three times against acetone with a final dialysis against molecular grade water (HyClone). After polymer isolation, the purified polymer solution was frozen and lyophilized. Degree of polymerization, monomer conversion, and polymer composition were determined using 1H NMR (CDCl3) spectroscopy (Figure S11) using end group analysis as described elsewhere.22,52
Synthesis of 5′-Triphosphate RNA
Y3, (seq: 5′-ppp-AACAAUUGCACUGAUAAUGAAUUCC-3′), Y6 (seq: 5′-ppp-CGUUAAUCGCGUAUAAUACGCCUAU-3′), and SLR20 (seq: 5′-ppp-GGAUCGAUCGAUCGAUCGGCUUCGGCCGAUCGAUCGAUCGAUCC-3′) were synthesized as previously described.8 5′-Hydroxyl control RNAs for Y3, Y6, and SLR20, as well as the complement strands for Y3 (seq: 5′-GGAAUUCAUUAUCAGUGCAAUUGUU-3′) and Y6 (seq: 5′-AUAGGCGUAUUAUACGCGAUUAACG-3′) were purchased from Integrated DNA technologies (IDT) and resuspended in RNase free water. In the cases of Y3 and Y6, to generate double-stranded RNA, equimolar amounts of top strand with 5′-triphosphorylated or 5′-hydroxyl top strand and the respective complement strand were suspended in 0.3 M NaCl, transferred to a 0.25 mL PCR tube and annealed using a thermocycler by setting the temperature to 90 °C and slowly cooling to 35 °C over 1 h. The resulting duplexes were diluted to 100 μM RNA in RNase free water, and 2% agarose gel electrophoresis was used to confirm hybridization. In the case of SLR20, hairpin formation was obtained using the same method but without a complement strand.
Formulation of NP/3pRNA Complexes
Lyophilized copolymers were dissolved into ethanol at 50 mg/mL and stored at 4 °C. This stock was further diluted to 3.33 mg/mL in citric acid buffer (pH 4, 100 mM) and rapidly mixed with 3pRNA or OH-RNA at charge ratios (N:P) between 20:1 and 1:1. After incubating at room temperature for 30 min, 1.24× volume phosphate buffer (pH 8, 100 mM) was added and mixed rapidly to form nanoparticles (NPs). After 15 min, the solution was further diluted into 1× PBS (pH 7.4, Gibco) before use. The second block DMAEMA content is estimated to have 50% protonation for the purposes of determining N:P ratios. A charge ratio of 20:1 was selected for all in vitro cell culture studies. The same approach was used to prepare formulations for in vivo studies, except that they were suspended at 10 mg/mL in citric acid buffer instead of 3.33 mg/mL, phosphate buffer was added at a ratio of 1:1.26, a charge ratio of 15:1 was used, and formulations were filtered using a 0.22 μm syringe filter (Pall corporation). 3pRNA complexation efficiency was determined by quantifying free 3pRNA after NP/3pRNA formulation with the fluorescence-based RiboGreen reagent (Invitrogen) according to the manufacturer’s instructions.
Dynamic Light Scattering
Series polymers were diluted
from ethanol stocks to 1 mg/mL in either lysosomal pH range (pH 5.8)
or physiological pH range (pH 7.4) PBS. For each series polymer, NP
particle size distribution and polydispersity index (PDI) was analyzed
via dynamic light scattering (Malvern Zetasizer Nano ZS). Fold pH
responsive size change was determined using the following expression: 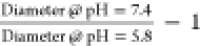 .
.
Erythrocyte Lysis Assay
The ability of polymers to disrupt lipid bilayer membranes at different pH values was performed as previously described.52 Briefly, whole blood from de-identified patients was acquired from the Vanderbilt Technologies for Advanced Genomics (VANTAGE) core. Blood was centrifuged to pellet erythrocytes, plasma was aspirated, erythrocytes were resuspended in pH 7.4 PBS (Gibco), and washed three times. After the final wash, erythrocytes were resuspended in pH 7.4, 7.0, 6.6, 6.2, or 5.8 PBS (150 nM). Polymers were mixed with suspended erythrocytes to a concentration of 1 μg/mL in a 96-well V-bottom plate. The plates were incubated for 1 h at 37 °C and centrifuged to pellet intact erythrocytes, and the supernatant was transferred to a 96-well flat-bottom plate. Membrane disruption was quantified through hemoglobin leakage, which can be measured using absorbance spectroscopy at 575 nm.
Cell Lines
The human lung carcinoma IRF and NF-κB reporter cell line A549-Dual (Invivogen), murine Lewis lung carcinoma (LLC) cell line (ATCC), and the murine macrophage cell line RAW 264.7 (ATCC) were cultured in DMEM (Gibco) supplemented with 2 mM l-glutamine, 4.5 g/L d-glucose, 10% heat inactivated fetal bovine serum (HI FBS, Gibco), and 100 U/mL penicillin/100 μg/mL streptomycin (Gibco). The murine breast cancer cell line 4T1 (ATCC) and the murine melanoma cell line B16–F10 (ATCC) were cultured in RPMI 1640 (Gibco) supplemented with 2 mM l-glutamine, 10% fetal bovine serum (FBS, Gibco), and 100 U/mL penicillin/100 μg/mL streptomycin (Gibco). The murine dendritic cell line DC2.4 was kindly provided by K. Rock (University of Massachusetts Medical School) and cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (HI FBS; Gibco), 2 mM l-glutamine, 100 U/mL penicillin/100 μg/mL streptomycin (Gibco), 50 μM 2-mercaptoethanol (Gibco), 1 × nonessential amino acids (Cellgro), and 10 mM HEPES (Invitrogen). Luciferase-expressing MDA-MB-231 cells53 were cultured in DMEM growth media supplemented with 4.5 g L–1d-glucose, 2 mM l-glutamine, 10% heat-inactivated FBS, and 1% penicillin and streptomycin. All cell types were grown at 37 °C in 5% CO2.
Bone Marrow Derived Dendritic Cell Isolation and Culture
BMDCs were obtained from 6- to 8-week-old C57BL/6 mice. Mouse tibias and femurs were removed and flushed with cold PBS through a 70-μm-wide cell strainer. The cells were pelleted by centrifugation for 5 min at 450 × g and resuspended in RPMI 1640 medium supplemented with 10% HI FBS, 2 mM l-glutamine, 0.4 mM sodium pyruvate, 50 μM 2-mercapthoethanol, and 20 ng/mL mGM-CSF. Then, the cells were seeded in 100 × 20 mm nontreated cell culture dishes in 10 mL of conditioned medium at a density of 9 × 106 cells per dish, and they were incubated for 8 days at 37 °C in 5% CO2. Fresh medium was added on days 4 and 7.
In Vitro Evaluation of 3pRNA Activity
To estimate the half-maximal response concentration (EC50) of each formulation, RNA dose sweeps between 0.05 and 50 nM final RNA concentration were performed in A549-Dual reporter cells (Invivogen). Cells were suspended at 50 000 cells/mL in media, plated at 200 μL in 96-well plates, and allowed to adhere overnight for reporter cell activity. Polymer/RNA complexes were formulated as detailed above and cells were treated with formulations containing 3pRNA, control OH-RNA, or PBS (vehicle) for 24 h. Luminescent reporter assays were performed using QUANTI-Luc (Invivogen) following the manufacturer’s instructions. Luminescence was quantified using a Synergy H1 microplate reader (BioTek, Winooski, VT). All measurements were normalized after baselining to the average value of the PBS-treated negative control group. Values for EC50 were extrapolated from dose–response curve fits using GraphPad Prism software. EC50 ratios shown in Figure 3d were determined using the following formula: EC50min/EC50sample. Cell viability was determined using CellTiter-Glo (Promega) according to manufacturer’s instructions.
For qRT-PCR analyses, 50 000 cells were plated in 12-well plates and allowed to adhere overnight. After 24 h, the media was replaced and cells treated with the indicated formulation at an RNA dose of 20 nM or vehicle control. After 6 h, cells were washed and 700 μL of RLT lysis buffer (Qiagen) was added to each well. Lysates were stored at −80 °C until use. mRNA was extracted from cell lysates using an RNA isolation kit (RNeasy mini kit, Qiagen). cDNA was synthesized for each sample using a cDNA synthesis kit (iScript, Bio-Rad) and analyzed using qRT-PCR using Taqman kits (Thermo Fischer) and a CFX real-time PCR detection system (Bio-Rad) following the manufacturer’s instructions. Taqman probes for mouse Ifnb1 (Mm00439552_s1), Cxcl10 (Mm00445235_m1), Tnf (Mm00443258_m1), and Hmbs (Mm01143545_m1) were purchased from ThermoFisher. Fold change was calculated using the ΔΔCt method.
For flow cytometry studies, BMDCs were plated at 100 000 cell/well in 24 well plates and treated with indicated formulation at an RNA dose of 20 nM for 18 h. Following treatment, cells were washed with PBS and then mechanically detached from plates using cell scrapers. 0.25% Cells were washed 3× in FACS buffer (0.5% BSA in PBS) and stained with labeled antibodies for CD11c (Clone N418 – FTIC, Tonbo), MHC-II (I-A/I-E, Clone M5/114.14.2 – APC/Cy7, Biolegend), and CD86 (Clone GL-1 – PE/Cy7, BioLegend) in FACS buffer following the manufacturer’s protocols. DAPI (Sigma) staining was used to discriminate live from dead cells. Samples were kept on ice and analyzed using a CellSteam flow cytometer. All flow cytometry data were analyzed using FlowJo ver. 10 (Tree Star Inc.).
In Vitro Evaluation of NP/RNA Stability
To evaluate complex stability in serum by agarose gel electrophoresis, lead carriers (4–50, 6–40, 8–40, and 10–40) were formulated as described above with OH-RNA labeled with AlexaFluor 647 (Integrated DNA Technologies) at an N:P ratio of 20:1. Formulated fluorescent OH-RNA/NPs were then supplemented with 10% additional PBS or to a final concentration of 10% fetal bovine serum (FBS; Gibco) and incubated at 37 °C for 0, 2, or 6 h prior to analysis by agarose gel (4%) electrophoresis (1 μg RNA per lane). For the PBS group and t = 0 time point, the NPs and the respective diluent were warmed to 37 °C and mixed immediately before loading. The gel was subsequently imaged on a LICOR Odyssey imaging system. To evaluate the effect of serum incubation on complex activity, lead carriers were formulated with 3pRNA, diluted in 80% FBS for 0, 2, 6, 12, 18, or 24 h, and activity evaluated in A549-Dual reporter cells (Invivogen) as described above at a concentration of 2 nM RNA. Data are plotted as a percentage of luminescence at t = 0 h after subtracting background signal from vehicle (PBS) treated controls.
Luciferase Knockdown Study
Luciferase expressing MDA-MB-231 (MDA-MB-231-Luc) cells were plated in black clear-bottom 96 well plates at a density of 2000 cells per well. The following day, the plated cells were treated with luciferase siRNA as well as negative control siRNA (DS NC1, Integrated DNA Technologies) complexed with each polymer tested at an N:P of 20:1 at concentrations ranging from 0.39 nM to 50 nM. After treating the cells for 24 h, the media was replaced with media containing 150 μg mL–1d-luciferin, and bioluminescence was measured with an IVIS Lumina III imaging system.
Gal8 Recruitment Assay
Gal8 recruitment assays were performed as previously described with minor modifications.28 Gal8-MDA-MB-231 cells were plated in a half area 96 well plate (Corning 4580) at a confluency of ∼40% and allowed to adhere overnight. Media was replaced with 70 μL imaging Media (FluoroBrite DMEM + 25 mM HEPES + 10% FBS + Pen/Strep+ Nucblue Live ReadyProbes nuclear stain (ThermoFisher Scientific)) and treatments were added in 30 μL Opti-MEM (Gibco) for a total well volume of 100 μL and final treatment concentration of 50 nM RNA, formulated with indicated polymers as described above. Imaging began 30 min after treatment. Cells were housed in humid CO2 incubator at 37 °C when not being imaged. The cells were imaged with a 20× objective in an ImageXpress Micro XLS Widefield High-Content Analysis System. Four images were taken per well. Images were analyzed using MetaXpress software Transfluor Application module to quantify the integrated YFP intensity per vesicle and fluorescent nuclei per image. Using the tabulate feature of JMP Statistical Software the total vesicle intensity and number of nuclei were summed together to create one data point per well. Gal8 intensity was then normalized to the cell number in each well. Statistics and graphing were then performed treating each well as an independent replicate.
Animal Care and Experimentation
Female C57BL/6J mice (6–8 weeks old) were obtained from The Jackson Laboratory (Bar Harbor, ME). All animals were maintained at the animal facilities of Vanderbilt University under specific pathogen-free conditions. All animal experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC).
In Vivo Evaluation of NP/3pRNA Activity
Eight-week-old female C57Bl/6 mice were intravenously administered 15 μg of 3pRNA or OH-RNA complexed to the indicated polymer at an N:P ratio of 15:1, corresponding to approximately 600 μg polymer. Sterile PBS was used as the diluent and injected alone for vehicle-treated mice. Five hours post-treatment, mice were euthanized by carbon dioxide asphyxiation, and blood and organs (lung, liver, spleen, and kidneys) were collected post mortem. Blood was allowed to clot, was spun down, and serum was collected for quantification of IFN-α using the Lumikine mIFN-α kit (Invivogen) following the manufacturer’s instructions. Organs were stored in RNAlater (Invitrogen) at −80 °C until processing on the TissueLyser LT (Qiagen), following manufacturer’s protocol for downstream qRT-PCR analyses. Following tissue lysis, RNA was purified using the RNeasy mini kit (Qiagen), cDNA was synthesized by the iScript kit (Bio-Rad), and qRT-PCR was performed using Taqman probes (ThermoFisher) on a CFX thermocycler (Bio-Rad) as described above for transcript analysis of cells. Fold change was calculated using the ΔΔCt method.
Statistics
Statistical analyses were performed using GraphPad Prism (ver. 8) software. Significance was determined using one-way ANOVA with Tukey’s multiple comparisons test unless otherwise noted. Values represent experimental means, and error bars represent SD unless otherwise noted. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05.
Safety Considerations
No unexpected or unusually high safety considerations were encountered.
Acknowledgments
We gratefully acknowledge Kenneth Rock for providing DC2.4 cells. We thank the core facilities of the Vanderbilt Institute of Nanoscale Sciences and Engineering (VINSE) for use of dynamic light scattering and the Vanderbilt Technologies for Advanced Genomics (VANTAGE) for providing human blood for hemolysis studies. The Table of Contents graphic was generated using BioRender.com. This research was supported by grants from the National Science Foundation CBET-1554623 (JTW), the National Institutes of Health (NIH) 5R21AI121626 (JTW), the NIH Integrated Training in Engineering and Diabetes Training Grant (T32 DK101003; LEP), the American Cancer Society (Institutional Research Grant IRG-58-009-56), the Vanderbilt University Discovery Grant Program (JTW), Alex’s Lemonade Stand Foundation (SID924), and the Congressionally-Directed Medical Research Program (W81XWH-161-0063, W81XWH-20-1-0624). RBF acknowledges a National Science Foundation Graduate Research Fellowship under grant numbers 1445197 and 1937963, and KAC acknowledges the VINSE National Science Foundation Research Experience for Undergraduates (REU) (DMR 1263182). OF is a Research Scientist and AMP is an Investigator with the Howard Hughes Medical Institute.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00568.
Dose response curves; Spearman correlation; Size distribution; Viability of cells after treatment; Evaluation of stability and activity; Serum levels; NMR spectra; Statistical analysis (PDF)
Author Contributions
● MEJ and KWB contributed equally to this work. The authors contributed as follows: Conceptualization, MEJ and JTW; Methodology, MEJ, KWB, JTW, CLD; Investigation, MEJ, KWB, LEP, CRP, RBF, KAC; Resources, OF, CLD, AMP, JTW; Writing – Original Draft, MEJ, JTW; Writing – Review and Editing, MEJ, KWB, LEP, CRP, RBF, OF, CLD, AMP, JTW; Visualization, MEJ, KWB, LEP, CRP, JTW; Supervision, JTW; Funding Acquisition, JTW.
The authors declare no competing financial interest.
Supplementary Material
References
- Hornung V.; Ellegast J.; Kim S.; Brzózka K.; Jung A.; Kato H.; Poeck H.; Akira S.; Conzelmann K. K.; Schlee M.; et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314 (5801), 994. 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Schwerd T.; Hamm W.; Hellmuth J. C.; Cui S.; Wenzel M.; Hoffmann F. S.; Michallet M.-C.; Besch R.; Hopfner K.-P.; et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (29), 12067. 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan M. M.; Dickey T. H.; Molinari E. S.; Fitzgerald M. E.; Potapova O.; Iwasaki A.; Pyle A. M. A minimal RNA ligand for potent RIG-I activation in living mice. Sci. Adv. 2018, 4 (2), e1701854 10.1126/sciadv.1701854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet M.-L.; Olagnier D.; Xu Z.; Paz S.; Belgnaoui S. M.; Lafferty E. I.; Janelle V.; Arguello M.; Paquet M.; Ghneim K.; et al. Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog. 2013, 9 (4), e1003298 10.1371/journal.ppat.1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidegger S.; Wintges A.; Stritzke F.; Bek S.; Steiger K.; Koenig P. A.; Gottert S.; Engleitner T.; Ollinger R.; Nedelko T. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci. Immunol 2019, 4 (39), eaau8943. 10.1126/sciimmunol.aau8943. [DOI] [PubMed] [Google Scholar]
- Elion D. L.; Jacobson M. E.; Hicks D. J.; Rahman B.; Sanchez V.; Gonzales-Ericsson P. I.; Fedorova O.; Pyle A. M.; Wilson J. T.; Cook R. S. Therapeutically Active RIG-I Agonist Induces Immunogenic Tumor Cell Killing in Breast Cancers. Cancer Res. 2018, 78 (21), 6183. 10.1158/0008-5472.CAN-18-0730. [DOI] [PubMed] [Google Scholar]
- Poeck H.; Besch R.; Maihoefer C.; Renn M.; Tormo D.; Morskaya S. S.; Kirschnek S.; Gaffal E.; Landsberg J.; Hellmuth J.; et al. 5′-triphosphate-siRNA: turning gene silencing and RIG-I activation against melanoma. Nat. Med. 2008, 14 (11), 1256. 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Muthusamy V.; Fedorova O.; Kong Y.; Kim D. J.; Bosenberg M.; Pyle A. M.; Iwasaki A. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J. Exp. Med. 2019, 216 (12), 2854. 10.1084/jem.20190801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidegger S.; Kreppel D.; Bscheider M.; Stritzke F.; Nedelko T.; Wintges A.; Bek S.; Fischer J. C.; Graalmann T.; Kalinke U.; et al. RIG-I activating immunostimulatory RNA boosts the efficacy of anticancer vaccines and synergizes with immune checkpoint blockade. EBioMedicine 2019, 41, 146. 10.1016/j.ebiom.2019.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beljanski V.; Chiang C.; Kirchenbaum G. A.; Olagnier D.; Bloom C. E.; Wong T.; Haddad E. K.; Trautmann L.; Ross T. M.; Hiscott J. Enhanced Influenza Virus-Like Particle Vaccination with a Structurally Optimized RIG-I Agonist as Adjuvant. J. Virol. 2015, 89 (20), 10612. 10.1128/JVI.01526-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanasty R.; Dorkin J. R.; Vegas A.; Anderson D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967. 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- Pack D. W.; Hoffman A. S.; Pun S.; Stayton P. S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discovery 2005, 4 (7), 581. 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- Kauffman K. J.; Webber M. J.; Anderson D. G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Controlled Release 2016, 240, 227. 10.1016/j.jconrel.2015.12.032. [DOI] [PubMed] [Google Scholar]
- Hajj K. A.; Whitehead K. A. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nature Reviews Materials 2017, 2, 17056. 10.1038/natrevmats.2017.56. [DOI] [Google Scholar]
- Ellermeier J.; Wei J.; Duewell P.; Hoves S.; Stieg M. R.; Adunka T.; Noerenberg D.; Anders H. J.; Mayr D.; Poeck H.; et al. Therapeutic Efficacy of Bifunctional siRNA Combining TGF-beta1 Silencing with RIG-I Activation in Pancreatic Cancer. Cancer Res. 2013, 73 (6), 1709. 10.1158/0008-5472.CAN-11-3850. [DOI] [PubMed] [Google Scholar]
- Chakravarthy K. V.; Bonoiu A. C.; Davis W. G.; Ranjan P.; Ding H.; Hu R.; Bowzard J. B.; Bergey E. J.; Katz J. M.; Knight P. R. Gold nanorod delivery of an ssRNA immune activator inhibits pandemic H1N1 influenza viral replication. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (22), 10172. 10.1073/pnas.0914561107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M.; Shen L.; Liu Q.; Goodwin T. J.; Huang L. Nanoparticle Delivery of RIG-I Agonist Enables Effective and Safe Adjuvant Therapy in Pancreatic Cancer. Mol. Ther. 2019, 27 (3), 507. 10.1016/j.ymthe.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M. E.; Wang-Bishop L.; Becker K. W.; Wilson J. T. Delivery of 5′-triphosphate RNA with endosomolytic nanoparticles potently activates RIG-I to improve cancer immunotherapy. Biomater. Sci. 2019, 7 (2), 547. 10.1039/C8BM01064A. [DOI] [PubMed] [Google Scholar]
- Wilson J. T.; Keller S.; Manganiello M. J.; Cheng C.; Lee C.-C.; Opara C.; Convertine A.; Stayton P. S. pH-Responsive Nanoparticle Vaccines for Dual-Delivery of Antigens and Immunostimulatory Oligonucleotides. ACS Nano 2013, 7 (5), 3912. 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight F. C.; Gilchuk P.; Kumar A.; Becker K. W.; Sevimli S.; Jacobson M. E.; Suryadevara N.; Wang-Bishop L.; Boyd K. L.; Crowe J. E.; et al. Mucosal Immunization with a pH-Responsive Nanoparticle Vaccine Induces Protective CD8+ Lung-Resident Memory T Cells. ACS Nano 2019, 13 (10), 10939. 10.1021/acsnano.9b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland K. M.; Sevimli S.; Kilchrist K. V.; Duvall C. L.; Cook R. S.; Wilson J. T. Microparticle Depots for Controlled and Sustained Release of Endosomolytic Nanoparticles. Cell. Mol. Bioeng. 2019, 12 (5), 429. 10.1007/s12195-019-00571-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. E.; Kintzing J. R.; Hanna A.; Shannon J. M.; Gupta M. K.; Duvall C. L. Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity In Vivo. ACS Nano 2013, 7, 8870. 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werfel T. A.; Jackson M. A.; Kavanaugh T. E.; Kirkbride K. C.; Miteva M.; Giorgio T. D.; Duvall C. Combinatorial optimization of PEG architecture and hydrophobic content improves ternary siRNA polyplex stability, pharmacokinetics, and potency in vivo. J. Controlled Release 2017, 255, 12. 10.1016/j.jconrel.2017.03.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltoukhy A. A.; Chen D.; Alabi C. A.; Langer R.; Anderson D. G. Degradable terpolymers with alkyl side chains demonstrate enhanced gene delivery potency and nanoparticle stability. Adv. Mater. 2013, 25 (10), 1487. 10.1002/adma.201204346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; Zumbuehl A.; Goldberg M.; Leshchiner E. S.; Busini V.; Hossain N.; Bacallado S. A.; Nguyen D. N.; Fuller J.; Alvarez R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26 (5), 561. 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. T.; Postma A.; Keller S.; Convertine A. J.; Moad G.; Rizzardo E.; Meagher L.; Chiefari J.; Stayton P. S. Enhancement of MHC-I Antigen Presentation via Architectural Control of pH-Responsive, Endosomolytic Polymer Nanoparticles. AAPS J. 2015, 17 (2), 358. 10.1208/s12248-014-9697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganiello M. J.; Cheng C.; Convertine A. J.; Bryers J. D.; Stayton P. S. Diblock copolymers with tunable pH transitions for gene delivery. Biomaterials 2012, 33 (7), 2301. 10.1016/j.biomaterials.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilchrist K. V.; Dimobi S. C.; Jackson M. A.; Evans B. C.; Werfel T. A.; Dailing E. A.; Bedingfield S. K.; Kelly I. B.; Duvall C. L. Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS Nano 2019, 13 (2), 1136. 10.1021/acsnano.8b05482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y.; Ozawa S.; Miyamoto C.; Maehata Y.; Suzuki A.; Maeda T.; Baba Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13 (1), 89. 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarett S. M.; Werfel T. A.; Chandra I.; Jackson M. A.; Kavanaugh T. E.; Hattaway M. E.; Giorgio T. D.; Duvall C. L. Hydrophobic interactions between polymeric carrier and palmitic acid-conjugated siRNA improve PEGylated polyplex stability and enhance in vivo pharmacokinetics and tumor gene silencing. Biomaterials 2016, 97, 122. 10.1016/j.biomaterials.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundy B. B.; Convertine A.; Miteva M.; Stayton P. S. Neutral polymeric micelles for RNA delivery. Bioconjugate Chem. 2013, 24 (3), 398. 10.1021/bc300486k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M. F.; Khvorova A. Improving siRNA Delivery In Vivo Through Lipid Conjugation. Nucleic Acid Ther. 2018, 28 (3), 128. 10.1089/nat.2018.0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M. Master sensors of pathogenic RNA - RIG-I like receptors. Immunobiology 2013, 218 (11), 1322. 10.1016/j.imbio.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boorn J. G.; Hartmann G. Turning Tumors into Vaccines: Co-opting the Innate Immune System. Immunity 2013, 39 (1), 27. 10.1016/j.immuni.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Kohlway A.; Luo D.; Rawling D. C.; Ding S. C.; Pyle A. M. Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep. 2013, 14 (9), 772. 10.1038/embor.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier S. M.; Kierzek R.; Jaeger J. A.; Sugimoto N.; Caruthers M. H.; Neilson T.; Turner D. H. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl. Acad. Sci. U. S. A. 1986, 83 (24), 9373. 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder P. S.; DeVine R. J.; Dagle J. M.; Walder J. A. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3′ exonuclease in plasma. Antisense Res. Dev. 1991, 1 (2), 141. 10.1089/ard.1991.1.141. [DOI] [PubMed] [Google Scholar]
- Palmer C. R.; Jacobson M. E.; Fedorova O.; Pyle A. M.; Wilson J. T. Environmentally Triggerable Retinoic Acid-Inducible Gene I Agonists Using Synthetic Polymer Overhangs. Bioconjugate Chem. 2018, 29 (3), 742. 10.1021/acs.bioconjchem.7b00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz L. M.; Diken M.; Haas H.; Kreiter S.; Loquai C.; Reuter K. C.; Meng M.; Fritz D.; Vascotto F.; Hefesha H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534 (7607), 396. 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- Sahin U.; Oehm P.; Derhovanessian E.; Jabulowsky R. A.; Vormehr M.; Gold M.; Maurus D.; Schwarck-Kokarakis D.; Kuhn A. N.; Omokoko T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585 (7823), 107. 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- Neelapu S. S.; Tummala S.; Kebriaei P.; Wierda W.; Gutierrez C.; Locke F. L.; Komanduri K. V.; Lin Y.; Jain N.; Daver N.; et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15 (1), 47. 10.1038/nrclinonc.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q.; Hou Z.; Yin C.; Zhang C.; Zhang J. 5′-triphosphate siRNA targeting HBx elicits a potent anti-HBV immune response in pAAV-HBV transfected mice. Antiviral Res. 2019, 161, 36. 10.1016/j.antiviral.2018.11.006. [DOI] [PubMed] [Google Scholar]
- Coch C.; Stumpel J. P.; Lilien-Waldau V.; Wohlleber D.; Kummerer B. M.; Bekeredjian-Ding I.; Kochs G.; Garbi N.; Herberhold S.; Schuberth-Wagner C.; et al. RIG-I Activation Protects and Rescues from Lethal Influenza Virus Infection and Bacterial Superinfection. Mol. Ther. 2017, 25 (9), 2093. 10.1016/j.ymthe.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn G. M.; Sedlik C.; Baharom F.; Zhu Y.; Ramirez-Valdez R. A.; Coble V. L.; Tobin K.; Nichols S. R.; Itzkowitz Y.; Zaidi N.; et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol. 2020, 38 (3), 320. 10.1038/s41587-019-0390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah P. A.; Zeppa J. J.; Maiello P.; Hackney J. A.; Wadsworth M. H. 2nd; Hughes T. K.; Pokkali S.; Swanson P. A. 2nd; Grant N. L.; Rodgers M. A.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577 (7788), 95. 10.1038/s41586-019-1817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevimli S.; Knight F. C.; Gilchuk P.; Joyce S.; Wilson J. T. Fatty Acid-Mimetic Micelles for Dual Delivery of Antigens and Imidazoquinoline Adjuvants. ACS Biomater. Sci. Eng. 2017, 3 (2), 179. 10.1021/acsbiomaterials.6b00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine D. J.; Swartz M. A.; Szeto G. L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12 (11), 978. 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertine A. J.; Benoit D. S.; Duvall C. L.; Hoffman A. S.; Stayton P. S. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J. Controlled Release 2009, 133 (3), 221. 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9 (2), 112. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Wei T.; Farbiak L.; Johnson L. T.; Dilliard S. A.; Siegwart D. J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15 (4), 313. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sago C. D.; Lokugamage M. P.; Paunovska K.; Vanover D. A.; Monaco C. M.; Shah N. N.; Gamboa Castro M.; Anderson S. E.; Rudoltz T. G.; Lando G. N.; et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (42), E9944. 10.1073/pnas.1811276115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shae D.; Becker K. W.; Christov P.; Yun D. S.; Lytton-Jean A. K. R.; Sevimli S.; Ascano M.; Kelley M.; Johnson D. B.; Balko J. M.; et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol. 2019, 14 (3), 269. 10.1038/s41565-018-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. A.; Bedingfield S. K.; Yu F.; Stokan M. E.; Miles R. E.; Curvino E. J.; Hoogenboezem E. N.; Bonami R. H.; Patel S. S.; Kendall P. L.; et al. Dual carrier-cargo hydrophobization and charge ratio optimization improve the systemic circulation and safety of zwitterionic nano-polyplexes. Biomaterials 2019, 192, 245. 10.1016/j.biomaterials.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.