

Abstract

Over the past several decades, macrocyclic compounds have emerged as increasingly significant therapeutic candidates in drug discovery. Their pharmacological activity hinges on their rotationally restricted three-dimensional orientation, resulting in a unique conformational preorganization and a high enthalpic gain as a consequence of high-affinity macrocycle–protein binding interactions. Synthetic access to macrocyclic drug candidates is therefore crucial. From a synthetic point of view, the efficiency of macrocyclization events commonly suffers from entropic penalties as well as undesired intermolecular couplings (oligomerization). Although over the past several decades ring-closing metathesis, macrolactonization, or macrolactamization have become strategies of choice, the toolbox of organic synthesis provides a great number of versatile transformations beyond the aforementioned. This Outlook focuses on a selection of examples employing what we term unconventional macrocyclizations toward the synthesis of natural products or analogues.
Short abstract
Unconventional retrosynthetic disconnections enable novel approaches to the synthesis of macrocyclic natural products. Several examples spanning the spectrum of organic chemistry are presented.
1. Introduction
Macrocycles are defined as molecules bearing a cyclic framework consisting of usually 12 or more atoms. Macrocyclic compounds comprise a broad spectrum of different ring sizes and varying degrees of molecular complexity. From porphyrin derivatives (e.g., heme or vitamin B12) to cyclic oligopeptides (e.g., cyclosporin A), Nature expresses an enormous variety of complex macrocyclic motifs which have inspired the scientific community, particularly in drug discovery.1−5 In fact, macrocyclic natural products and their analogues have emerged as powerful candidates for therapy against otherwise “undruggable” targets, as a result of their constrained three-dimensional architecture which allows preorganization (entropic penalty) and high-affinity substrate–protein binding (enthalpic benefit).6−8 Synthetic access to macrocycles is therefore critical, and organic synthesis provides versatile and powerful tools to satisfy this need.9 Regardless of the cyclization technique of choice, macrocyclizations are usually of low efficacy as they pay a large entropic penalty due to the reduction of the degrees of freedom and are plagued by undesired oligomerizations. The latter is usually addressed by employing high dilution—diminishing the probabilities of intermolecular couplings—which inevitably, however, decreases the rate and scalability of the process. Among the numerous macrocyclic natural product syntheses over the past decades, the crucial ring closing was most commonly achieved via late-stage ring-closing metathesis,10−12 macrolactonization,13,14 or macrolactamization strategies (Scheme 1a).15−17 The broad array of methods provided for these transformations in the literature set them as textbook approaches. This does not, however, detract from the fact that different retrosynthetic disconnections are possible. The plethora of synthetic approaches toward diazonamide A (1) illustrate this point (Scheme 1b).18 Diazonamide A is a potent antimitotic agent isolated in 1991 from the colonial marine ascidian Diazona angulate(19) and has since attracted the interest of the synthetic community. In 2001, Harran disclosed the total synthesis of the originally proposed structure (not shown herein) for 1, discovering that it was incorrectly assigned, and thereafter elucidating the actual one (Scheme 1b).20,21 Notably, Vedejs and Nicolaou had also reported their efforts toward the originally proposed diazonamide A structure,22,23 with the latter—after the structural revision by Harran—later accomplishing the first total synthesis of the natural product itself.24,25 While ring A of bis-macrocyclic diazonamide A (1) was constructed via a typical macrolactamization, the second macrocycle was delivered by a Witkop-type aryl–aryl coupling (inspired by Harran’s initial report on the original structure). In 2003, Harran subsequently published the total synthesis of the natural product, employing the same Witkop cyclization protocol and an iodine(III)-mediated oxidative cycloaddition, affording the fused dihydrofuran with significant diastereoselectivity.26 Moreover, in the second total synthesis of 1, Nicolaou established the B ring through an unconventional heteropinacol coupling.27,28 More recently, MacMillan published his approach toward 1, based on a macroaldolization for the closure of ring A (setting the stage for one of the two oxazole syntheses) and a Suzuki–Miyaura aryl–aryl coupling for the second ring.29 The interest of several additional groups was sparked by the challenging structure of diazonamide A, and various formal syntheses have been reported.
Scheme 1. (a) Selected Examples of Common Macrocyclizations in Natural Product Synthesis and (b) Employment of Various Ring-Closing Techniques for the Construction of the Bis-Macrocyclic Natural Product Diazonamide A.

In 2007, Magnus developed a powerful O-aryl to C-aryl migration technique, furnishing the C10 quaternary center with high diastereoselectivity.30 An elegant coupling was reported by Sammakia for the construction of the ring A of the natural product via a diastereoselective oxindole arylation,31 and in 2016, the formal synthesis of diazonamide A was disclosed by Moody, featuring a macrolactamization for the closure of ring A.32
The pool of synthetic approaches toward 1 showcases the power of organic synthesis in overcoming complex challenges as well as the large variety of tools for the construction of macrocyclic compounds. This Outlook summarizes selected examples of synthetic endeavors that utilize unconventional macrocyclizations en route to natural products bearing large rings and aims at providing readers with a different perspective of these synthetic challenges.
2. Palladium-Mediated Cross-Couplings
Palladium-catalyzed cross-coupling reactions are a predominant tool for the construction of carbon–carbon bonds.33 Since the initial reports in the late 1960s by Tsuji34 and Heck,35−38 followed by the pioneering works of Negishi39,40 and Suzuki and Miyaura,41 Pd-mediated transformations have been extensively employed in the synthesis of natural products and derivatives, as well as valuable materials. Apart from these early examples, other protocols such as the Tsuji–Trost, Sonogashira, and Stille reactions have enriched the family of palladium-catalyzed cross-couplings.42−45 Unsurprisingly, a number of macrocyclization approaches rely on Pd-catalyzed cross-coupling reactions.46 This section is dedicated to classifying and summarizing a selection of these advances.
2.1. Stille Coupling
The term Stille coupling is used to describe the C–C bond-forming reaction between organostannanes and halides or pseudohalides under Pd(0)-catalysis. The coupling was developed by and named after J. K. Stille,47,48 even though the first reports were disclosed by the research group of M. Kosugi in 1977.49,50 Despite the toxicity of tin reagents, the Stille coupling remains one of the most common tools utilized in the total synthesis of natural products.45,51
The first example of total synthesis of a macrocyclic natural product employing a palladium-catalyzed coupling of vinyl stannanes and vinyl triflates was reported by Hegedus and Stille in 1990 on the synthesis of the diterpene jatrophone.52 In 2010, Kalesse disclosed the first total synthesis of the polyene chivosazole F,53 and in the same year Bach reported the synthesis of thiopeptides amythiamicin C and D.54 A more recent report by Liu described the total synthesis of the natural depsipeptide nannocystin Ax,55 two years after the reported isolation.
One of the most fascinating applications of the Stille coupling is found in Nicolaou’s 1993 total synthesis of rapamycin (4), employing a “stitching” technique to construct the (E,E,E)-trienic moiety (Scheme 2).56 More precisely, the linear precursor 2 was treated with trans-vinylenedistannane 3 in the presence of Hünig’s base and Pd(CH3CN)2Cl2, presumably forming intermediate 4-intvia an intermolecular Stille coupling. Subsequently, a second Stille coupling takes place intramolecularly, forming a 29-membered ring and delivering the enantiomerically pure immunosuppressive natural product in 28% yield.
Scheme 2. Nicolaou’s “Stitching” Technique en Route to the First Total Synthesis of Rapamycin.

Five years later, Smith reported the first enantioselective total synthesis of macrolactin A (7), a potent antiviral polyene macrolide (Scheme 3a).57 Herein, treatment of a highly diluted solution of vinyl stannane 5 (0.7 mM) with Pd2dba3 and DIPEA in NMP formed the macrocycle 6, which was converted to the natural product after deprotection of the alcohols in 29% yield over two steps.
Scheme 3. Intramolecular Stille Couplings in the Syntheses of (a) Macrolactin A and (b) Sarain A.

In 2006, Overman published the first total synthesis of the bis-macrocyclic alkaloid sarain A (10) (Scheme 3b).58 The key macrocyclization event proceeded via an intramolecular Stille coupling of 8—the first macrocycle having been formed using ring-closing metathesis—in the presence of catalytic Pd(PPh3)4 and an excess of lithium chloride, affording 9. Eventually, the enantiomerically pure alkaloid 10 was obtained after three additional steps.
2.2. Heck Coupling
The Heck reaction has undeniably won a place as a textbook methodology enabling carbon–carbon bond formation between an olefin and a (pseudo)halide.59 In addition to the enhanced atom-economy of Heck couplings when compared to the rest of the family of Pd-catalyzed transformations, a notable advantage is the regioselectivity during the ring-closing process.
In 2009, Maier published the formal synthesis of palmerolide A,60 by constructing an advanced intermediate synthesized two years earlier by Nicolaou and Chen en route to the natural product.61 In this article, Maier highlighted the fact that an intramolecular Heck coupling for the regioselective construction of palmerolide A’s 20-membered ring was superior to a Stille approach, which delivered a mixture of olefin isomers. More recently, Reddy reported the total syntheses of solomonamides A and B from a common macrocyclic precursor constructed by a crucial Heck coupling.62,63
The same year, Menche disclosed the total synthesis of the potent antibiotic macrolide etnangien (13) (Scheme 4).64 The key Heck macrocyclization took place after treating cis-vinyl iodide 11 with Pd(OAc)2, tetrabutylammonium chloride, and potassium carbonate. The coupling proceeded successfully with high stereoselectivity (E/Z > 20:1), and the precursor 12 was isolated in 70% yield. Notably, when similar conditions were applied by the authors in an intermolecular context during the synthesis of archazolid A (not shown),65 a 6:1 mixture of E/Z isomers was obtained, underlining the significance of conformational restrictions for the stereoselectivity of Heck coupling reactions. Finally, etnangien (13) was obtained after seven further steps.
Scheme 4. Heck Coupling toward the Synthesis of Etnangien by Menche.

A more recent example of a Heck macrocyclization is the first total synthesis of biselyngbyolide B (16) by Goswami (Scheme 5a).66 The 18-membered polyene macrocycle was constructed from the linear precursor 14, which underwent cyclization under Pd(OAc)2-catalysis affording 15. Despite the presence of potentially competitive olefins in proximity, the Heck cyclization proceeded regio- and stereoselectively in 58% yield. Deprotection of the allylic alcohol quantitatively delivered the natural product 16.
Scheme 5. Stereoselective Macrocyclizations in the Syntheses of (a) Biselyngbyolide B and (b) Isoplagiochin D.
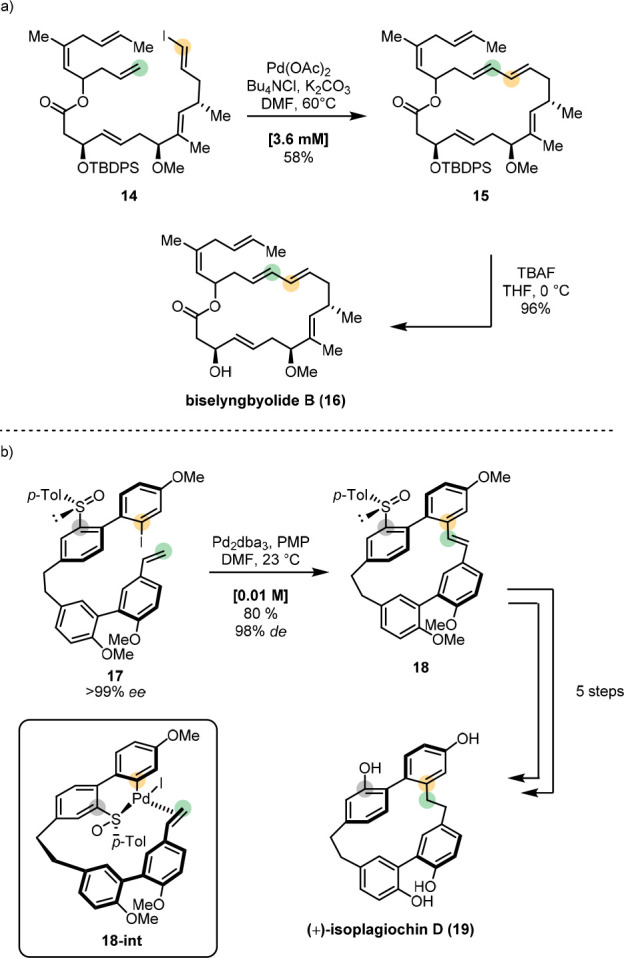
In 2018, Choppin and Speicher established the first enantioselective total synthesis of the cyclophane isoplagiochin D (19) (Scheme 5b).67 Unquestionably, the high rigidity of such compounds provides a great synthetic challenge. The intriguing synthesis of 19 was carried out utilizing a Heck macrocyclization.
Treatment of enantiopure 17 with Pd2dba3 delivered the strained macrocycle 18 in 80% yield with excellent atropo- and diastereoselectivity, selectivities that had not been achieved in a previously reported attempt by the same group.68 The key element of this impressive transformation is the chiral sulfinyl auxiliary which enables a configurational lock, presumably via intermediate 18-int, and dictates the atroposelectivity during the event. The traceless nature of the sulfinyl moiety is implemented with its replacement by a hydroxyl group en route to enantioenriched 19.
2.3. Suzuki–Miyaura Coupling
Organoboron species can be coupled with electrophilic (pseudo)halides under palladium catalysis in the presence of an activating base. This reaction, known as the Suzuki–Miyaura coupling, is one of the most widely employed methods for the synthesis of C(sp2)–C(sp2) and, more recently, C(sp2)–C(sp3) bonds. It has also found broad application in the total syntheses of macrocyclic natural products.69−73
Halcomb’s synthetic strategy toward phomactin A (22) involved an alkyl borane Suzuki–Miyaura coupling to achieve the crucial macrocyclization step (Scheme 6a).74 In particular, stereo- and regioselective hydroboration of the terminal olefin of 20 afforded the corresponding alkyl borane species which was treated with palladium(II) under Johnson’s conditions.75 The strained 12-membered macrocycle 21 was isolated in 37% yield (retaining the sensitive dihydrofuran), and further deprotection afforded the enantiomerically pure natural product 22.
Scheme 6. Suzuki–Miyaura Couplings en Route to the Syntheses of (a) Phomactin A and (b) Isocomplestatin.

In 2005, Snapper and Hoveyda reported the total synthesis and structural revision of isocomplestatin (24) (Scheme 6b),76 an atropisomer of the natural product complestatin, originally reported as isocomplestatin after its isolation in 2001. In this regard, advanced precursor 23 (the macrocycle of which was accessed via a Chan–Evans–Lam coupling similar to the chloropeptin I synthesis by the same research group—see Section 9.2)77 was treated with PdCl2(dppf) and an excess of potassium carbonate, delivering the product of aryl–aryl coupling for the construction of the second macrocycle of isocomplestatin 24, which was eventually obtained after hydrolysis of the carboxylic acid.
More recently, Maulide accomplished the total synthesis of the potent immunosuppressive natural products FR252921 (27), FR252922 (28), and FR256523 (29), embedding a Suzuki–Miyaura coupling protocol in a carefully designed domino sequence (Scheme 7).78 Specifically, initial macrocyclization was achieved via a Suzuki–Miyaura coupling of 25, containing a vinylboronic ester and a remotely attached 1,2-disubstituted cis-chlorocyclobutene carboxylic ester. The 17-membered, fused macrobicycle immediately underwent in situ, thermal 4π-electrocyclic ring opening of intermediate 26-int, providing the enlarged 19-membered trienic macrocycle 26 in high yields. Conrotatory electrocyclic ring-opening ensured the (E,E,E)-triene configuration, which had been shown by computation and prior reports not to be thermodynamically preferred.79,80 Final TBAF-deprotection allowed the isolation of the enantiomerically pure natural products.
Scheme 7. Domino Suzuki–Miyaura/4π-Electrocyclic Ring Opening Developed by Maulide for the Syntheses of FR252921, FR252922, and FR256523.

2.4. Tsuji–Trost Coupling
The coupling reaction of allylic electrophiles and nucleophiles under palladium(0)-catalysis is well-known as the Tsuji–Trost reaction.34,42 The scope of electrophiles ranges from allylic halides to allylic epoxides and even allylic cyclopropanes, but it is dominated by allylic acetates and carbonates. That, in combination with the large variety of potential nucleophilic partners, renders the Tsuji–Trost reaction a highly versatile tool. It is well established that the reaction proceeds via a π-allylpalladium(II) complex which provides the potential construction of two regioisomeric species, depending on the site of the attack of the approaching nucleophile. The enantioselective Tsuji–Trost coupling usually delivers the adducts with high regio- and stereoselectivity.81−83 This selectivity tool has been exploited in various macrocyclization strategies in total synthesis.46
In 1997, Yamamoto reported the stereoselective total synthesis of humulene (32) (Scheme 8a).84 Therein, the sodium salt of β-ketoester 30 was treated with Pd(PPh3)4, affording the 11-membered ketone 31 in 45% yield and with excellent stereo- and regioselectivity. The first total synthesis of the antibiotic roseophilin (35) was disclosed by Fürstner, six years after its structural elucidation (Scheme 8b).85 Macrocyclization of the allylic epoxide 33 under Pd(0)-catalysis proceeded smoothly, and the 13-membered allylic alcohol 34 was obtained in high yield. The natural product 35 was obtained in 11 further steps. The first total synthesis and structural revision of macquarimicin A (38) were disclosed by Tadano in 2003 (Scheme 8c).86 In this report, carbonate 36 was successfully cyclized to the 17-membered ketone 37 after being slowly added to a dilute mixture of a catalytic amount of Pd(PPh3)4 and DPPE in THF. Subsequent elaboration of this ketone through nine more steps afforded the target molecule 38 as a single enantiomer.
Scheme 8. Tsuji–Trost Macrocyclizations in the Syntheses of (a) Humulene by Yamamoto, (b) Roseophilin by Fürstner, and (c) Macquarimicin A by Tadano.

2.5. Sonogashira Coupling
The Sonogashira reaction enables the coupling between terminal alkynes and vinyl or aryl halides in the presence of palladium and copper(I) cocatalysts and an amine base. It is considered one of the most efficient methods for the construction of substituted alkynes and has found application in numerous natural product and drug syntheses.44 Nevertheless, examples of macrocyclization protocols employing Sonogashira coupling en route to natural product total syntheses are rather rare, presumably due to the increased rigidity of an alkynyl-containing ring (sp hybridization forces four consecutive atoms into a linear arrangement), as well as the minute number of natural product cycloalkynes.
In 1990, Schreiber serendipitously discovered a route to access complex scaffolds that share structural characteristics with enediyne natural products such as dynemicin A (41) (Scheme 9a), known for its cytotoxic properties. Schreiber reported the treatment of 39 with Pd(PPh3)4 and CuI, aiming at the formation of the 15-membered ring (cf. 40-int) fused to the 1,2-dihydroquinoline.87 Instead, the formation of a transannulation product 40, likely the product of domino Sonogashira cross-coupling/intramolecular Diels–Alder (IMDA) cycloaddition, was reported. Although surprising, as depicted in Scheme 9a, the 1,3-diene moiety of 40-int adopts the required s-cis conformation and is locked (by the enediyne moiety) in proximity to the electron-poor dienophile. IMDA cycloadditions, including further attempts toward advanced intermediate 40 will be discussed later (Section 5).
Scheme 9. (a) Cascade Sonogashira Macrocyclization and Transannular Diels–Alder in the Synthesis of the Dynemicin A Core by Schreiber and (b) Hirama’s Development of a Kedarcidin Chromophore Fragment.

Highlighting the need for a suitable approach to enediyne natural products, Lear and Hirama successfully constructed diyne 44, a framework of the unstable and structurally strained kedarcidin chromophore (45) (Scheme 9b).88 The crucial macrocyclization succeeded via an intramolecular Sonogashira coupling of iodide 42, when treated with Pd2dba3·CHCl3 and copper(I) iodide in the presence of Hünig’s base. This efficient and reproducible coupling delivered the cyclic product 43 in 88–90% yield. Eventually, the target molecule 44 was accessed with just three subsequent operations.
2.6. Other Palladium-Mediated Couplings
The great utility of Pd-catalyzed cross-coupling reactions for the construction of unique macrocyclic scaffolds has been highlighted thus far. The reign of palladium-mediated macrocyclizations, however, expands beyond that.46
In 2008, Trost developed an efficient route for the atom-economical total synthesis of bryostatin 16 (48) (Scheme 10a),89 a natural product commonly considered a synthetic precursor to the bryostatin family. The crucial macrocyclization was accomplished when diyne 46 was treated with palladium(II) acetate and tris(2,6-dimethoxyphenyl)phosphine (TDMPP) in toluene. Impressively, the 22-membered macrocycle 47 was obtained in 56% yield, while other attempts employing several solvent systems or metal/ligand ratios proved unsuccessful. From a mechanistic point of view, insertion of palladium into the carbon–hydrogen bond of the terminal alkyne allows the following regioselective carbometalation of the internal alkyne which, eventually, delivers the new enyne moiety and the macrocycle 47. Finally, the target molecule 48 is accessed in three subsequent operations.
Scheme 10. (a) Palladium-Catalyzed Macrocyclization Developed by Trost en Route to Bryostatin 16 and (b) Boger’s Palladium-Mediated Transannular Larock Macrocyclization in the Synthesis of Streptide.

The first total synthesis of streptide (51) was reported by Boger in 2019, brilliantly utilizing a Larock-indole-synthesis strategy for the ring-closing event (Scheme 10b).90 Here, linear peptide 49 underwent a palladium(0)-mediated indole annulation, delivering the 20-membered macrocycle 50 in 60% yield en route to the natural product 51. While this type of indole synthesis usually takes place in the presence of catalytic amounts of palladium, it is postulated that either the linear precursor or the cyclized product traps the catalyst thereby necessitating the use of stoichiometric amounts of palladium.
3. Olefinations
Since the 1953 discovery that the reaction of phosphonium ylides and carbonyl compounds results in olefination by G. Wittig, the eponymous reaction has emerged as one of the most powerful tools for the construction of carbon–carbon double bonds.91−93 In addition, a series of newer protocols and modifications has been developed, allowing versatile approaches for the synthesis of double bonds with high chemo- and E/Z-stereoselectivity. Olefinations have thus found application in various total syntheses of natural products. It should also be stressed that, although olefin metathesis appears to dominate in the synthesis of macrocyclic olefins, most of the alkenes engaging in these metatheses are, ironically, constructed by an olefination. This section briefly summarizes a selection of macrocyclization events via Wittig and Horner–Wadsworth–Emmons reactions in the total synthesis of natural products.
3.1. Wittig Olefination
The Wittig reaction remains a staple reaction for alkene synthesis whereby the stereochemistry of the alkene product depends on the nature of the phosphonium ylide. While Wittig olefination is present in many synthetic protocols, the employment in macrocyclizations is rather rare, and two selected examples are discussed herein.
In 1993, Nógrádi reported the total synthesis and structural revision of garuganin III (54) (Scheme 11a).94 In the pursuit of an efficient synthetic strategy, macrocyclization proved quite challenging. Ring-closing approaches such as an Ullmann-type diaryl ether synthesis, aldol condensation, or intramolecular Wurtz-type coupling proved ineffective. Ultimately, Wittig olefination provided an alternative approach for the macrocyclization of 52, and the isoxazole served as a masked 1,3-dicarbonyl dedicated to the vinylogous ester of garuganin III. In the event, the strained macrocycle 53 was formed in good yield upon treatment of 52 with potassium tert-butoxide and was elaborated to the target natural product 54 in three further steps. A similar isoxazole-based strategy was published by Nógrádi the same year in the synthesis of garugamblin-1 utilizing a Wurtz-type macrocyclization, as will be discussed later (Section 7.3).
Scheme 11. (a) Wittig Macrocyclization by Nógrádi toward Garuganin III and (b) Double Wittig Olefination for the Syntheses of Trienomycins A and F by Smith.

A remarkable strategy for the first enantioselective syntheses of trienomycins A (58) and F (59) was established by Smith in 1996 (Scheme 11b).95,96 The compelling ring-closing event hinged on a novel double Wittig olefination of dialdehyde 55. To this effect, excess NaHMDS was slowly added to a dilute solution of bis phosphonium chloride salt 56 and 55, initiating the domino inter- and intramolecular olefinations and successfully delivering the (E,E,E)-trienic ring in 21% yield. Gratifyingly, the resulting macrocycle 57 was a common precursor of both 58 and 59, the syntheses of which were realized in seven further steps.
3.2. Horner–Wadsworth–Emmons Olefination
Olefin synthesis through the coupling of phosphonate-stabilized carbanions with carbonyl compounds is known as the Horner–Wadsworth–Emmons (HWE) olefination.97−99 Advantages of the HWE reaction reside on the increased reactivity of phosphonate anions compared to phosphonium ylides and on the easy removal of the water-soluble, phosphonate salt byproducts. Additionally, HWE olefination is often the reaction of choice when high E-selectivity is required. This selectivity can be reversed under certain modification protocols (e.g., Still–Gennari, Corey–Kwiatkowski, or Ando modifications).100−103 It is no surprise, therefore, that HWE reactions occupy a privileged position in the synthesis of natural products, being employed in various transformations, including macrocyclizations.
In 2003, Kang reported the enantioselective total synthesis of lasonolide A (62) (Scheme 12).104 An intramolecular HWE olefination proved to be the method of choice for the construction of this highly strained 20-membered polyene from phosphonate 60. The macrocyclization delivered the (E,E)-dienoate macrolactone 61 in 71% yield and provided access to the product of interest (62) after three subsequent operations.
Scheme 12. Ring-Closing Horner–Wadsworth–Emmons Olefination for in the Synthesis of Lasonolide A by Kang.

The enantioselective total synthesis of dactylolide (65) was accomplished by Keck in 2005 (Scheme 13a),105 four years after the macrolide’s isolation. Therein, the crucial ring closure was achieved in 60% yield, when phosphonate 63 was treated with NaHMDS. The resulting 18-membered, enantioenriched macrolactone 64 was subjected to two further operations en route to the natural product (65). Another example of an HWE reaction employed in macrocyclization can be found in the 2007 synthesis of archazolid A (67) by Menche (Scheme 13b).65 Therein, the α-methyl phosphonate 66 was treated with sodium hydride, furnishing the E-olefin with excellent stereoselectivity. Subsequently, diastereoselective reduction of the unsaturated ketone under Corey–Itsuno conditions106−109 followed by deprotection afforded the target natural product in 26% yield over three steps.
Scheme 13. HWE Macrocyclizations in the Syntheses of (a) Dactylolide by Keck and (b) Archazolid A by Menche.

4. Oxidative Aryl–Aryl Couplings
Biaryl systems are common moieties, abundant in various natural products. In previous sections, we have already met a small series of such macrocyclic natural products, including diazonamide A (1), isocomplestatin (24), and roseophilin (35). Therefore, it is no surprise that many strategies for the total syntheses of natural products rely on aryl–aryl coupling events. This section covers selected examples of such couplings that led to the total syntheses of natural products.
As will be discussed in Section 6, C–H activation has emerged as a powerful logic in synthesis.110−115 Among the many elegant applications of the C–H activation strategy in total synthesis, aryl–aryl macrocyclization events are of present interest.
In 1998, Evans reported a remarkable synthesis of the aglycon of vancomycin (70), an antibiotic with a synthetically challenging core, given its triple atropisomeric architecture (Scheme 14a).116 The first macrocyclization for the synthesis of this complex molecule took place when 68 was treated with VOF3, BF3·Et2O (crucial for prevention of undesired attack by nucleophilic oxygens on the ring), and AgBF4 in a mixture of TFA and dichloromethane at 0 °C, followed by a reductive quench (NaHB(OAc)3 replacing the zinc powder commonly employed in this transformation).117 Interestingly, macrocycle 69 was delivered in 65% yield, possessing the unnatural R-atropisomeric configuration of the biaryl (d.r. > 19:1), which was, nevertheless, isomerized at a later stage to the desired S-configuration. The complex target 70 was achieved through a series of subsequent operations including two SNAr couplings for the construction of the remaining two macrocycles.
Scheme 14. Aryl–Aryl Couplings Developed for the Syntheses of (a) Vancomycin Aglycon by Evans and (b) Arylomycin Core (72) by Baran.

Recently, Romesberg and Baran published a route for the facile and scalable synthesis of arylomycins (e.g., arylomycin A2 (73)), based on a copper-mediated coupling of arenes (Scheme 14b).118 Other strategies, such as macrolactamization or palladium-catalyzed cross-coupling, proved unfruitful in initial efforts. Their interest was then turned to oxidative coupling, and the arylomycin core 72 was successfully obtained upon treatment of 71 with 2 equiv of [Cu(MeCN)4][PF6] and TMEDA.
5. Cycloaddition
Cycloadditions represent an additional method of forming macrocyclic structures through formation of carbon–carbon or carbon–heteroatom bonds. One of the key features of this approach is the fact that, in addition to the macrocyclic structure, another is invariably formed, and further functionalization, rearrangement, or degradation of this fragment is often a mandatory feature.
5.1. Diels–Alder Reaction
The Diels–Alder reaction, arguably the most prominent of the cycloaddition reactions, has been implemented in key macrocyclizations for natural product synthesis.119 In particular, the intramolecular Diels–Alder reaction (IMDA) has proven a powerful synthetic tool for the formation of macrocyclic structures with annulated cyclohexenes and cyclohexadienes. Early examples of the application of IMDA reactions in total synthesis were laid forth by Stork in the syntheses of cytochalasins,120 Yamaguchi in the synthesis of 24-O-methylchlorothricolide,121 and Kishi in the synthesis of pinnatoxin A.122 While elegantly leading to the desired connectivity, these examples all suffered from only moderate levels of diastereoselectivity.
In 2005, Sorensen published a highly diastereoselective intramolecular Diels–Alder approach to macrocyclization, elegantly implemented in the synthesis of abyssomicin C (76) (Scheme 15a).123
Scheme 15. (a) Sorensen’s Diastereoselective Intramolecular Diels–Alder Reaction en Route to Abyssomicin C and (b) Corey’s Enantioselective Syntheses of Palominol and Dolabellatrienone.

Therein, treatment of β-silyloxy ketone 74 with lanthanum(III) triflate in boiling toluene smoothly led to β-elimination and subsequent IMDA of 75-int. This one-pot sequence afforded a 50% yield of the desired 11-membered tricyclic product 75, which was converted to the target molecule abyssomicin C (76) in three further steps. The same year, Corey reported the use of a chiral catalyst (78) in an enantioselective IMDA reaction for the syntheses of palominol (80) and dolabellatrienone (81) (Scheme 15b).124 The success of this approach hinged on three key interactions between the catalyst and the substrate (77), as can be seen in the endo-transition state 79-TS: while a Lewis acid–Lewis base interaction between the boron and the aldehyde promoted the cycloaddition by increasing the dienophilicity of the enal, a formyl hydrogen bond to the oxazaborolidine and π–π stacking between a phenyl group and the dienophile ensured facial selectivity. The substituted bicyclo[9.4.0]pentadecatriene framework 79 (formed with an enantiomeric excess of 90%) was further elaborated to 80 and 81 in six and seven steps, respectively. The relative ease with which this transformation proceeds is also highlighted by the comparatively high concentrations used for the reaction.
A delicate Diels–Alder macrocyclization/retro-hetero-Diels–Alder strategy was elegantly applied in Baran’s 2006 synthesis of the alkaloid haouamine A (83) (Scheme 16a),125 a sequence similarly applied in Beaudry’s 2013 synthesis of cavicularin.126
Scheme 16. (a) Baran’s Diels–Alder/Retro-Diels–Alder Approach for the Synthesis of Haouamine A’s Bent Aromatic Ring and (b) Hetero-Diels–Alder Reaction Enabling Nicolaou’s Synthesis of Sporolide B.

The crucial [4 + 2] cycloaddition between a pyrone and a distal alkyne present in 82 led to the formation of ester-bridged cyclohexadiene 83-int, which underwent cycloreversion in situ, extruding carbon dioxide and delivering the natural product 83. Notably, the formation of the desired bent aromatic ring was facilitated by this type of approach, involving a nonaromatic intermediate to introduce the desired conformation. Employing a hetero-Diels–Alder reaction, Nicolaou was able to construct a macrocyclic precursor for the synthesis of sporolide B (86) (Scheme 16b).127 Proceeding via the transition state 85-TS, the o-quinone 84 was transformed to 85 with remarkable regio- as well as facial selectivity.
A further example of a Diels–Alder reaction in the synthesis of the macrocyclic framework of a natural product was described in Schreiber’s 1993 synthesis of tri-O-methyl dynemicin A methyl ester (88) (Scheme 17).128,129 Therein, the seco acid 87 was initially treated with trichlorobenzoyl chloride (Yamaguchi reagent) and PyBOP to effect macrolactonization and the formation of 40-int (see also Scheme 9a). This intermediate was ideally poised for direct intramolecular Diels–Alder reaction, contracting the original 15-membered ring to the desired 10-membered enediyne motif (40).
Scheme 17. Macrolactonization/Diels–Alder Cyclization Sequence to Form the Core of a Dynemicin A Derivative.

5.2. 1,3-Dipolar Cycloaddition
Another mode of cycloaddition that has been employed in the synthesis of natural products is the 1,3-dipolar cycloaddition. In Evans’ 1999 report on different synthetic approaches toward (+)-miyakolide (91), macrocyclization is effected by two alternative pathways.130 While the classical approach involves a macrolactonization reaction (not shown), the authors also investigated the (3 + 2) cycloaddition between an alkyne and a nitrile oxide which is generated in situ (90-int) from the corresponding N-oxide 89 (Scheme 18). This reaction leads to the formation of an isoxazole (90) that is readily cleaved to the corresponding β-dicarbonyl. A similar strategy was employed in the syntheses of 11-hydroxycurvularins by Lee,131 wherein an alkene-nitrile oxide cycloaddition ultimately affords a diastereomeric mixture of β-hydroxy ketones.
Scheme 18. Evans’ Synthesis of Miyakolide via 1,3-Dipolar Cycloaddition.

5.3. oxa-[3 + 3] Cycloaddition
An unusual oxa-[3 + 3] cycloaddition was envisioned by Hsung to form the macrocyclic phomactin A core (93) (Scheme 19).132 Therein, the silyl enol ether 92 was engaged in a formal cycloaddition with an iminium ion formed in situ from the appended enal. While the original approach, starting from the diketone,133 provided the desired regioisomer 93 as the minor product, switching to the silyl enol ether was able to improve regioselectivity. Interestingly, the undesired regioisomer was isolated as an inseparable mixture of atropisomers (94 and 95), while no such atropisomerism was observed for 93.
Scheme 19. Hsung’s oxa-[3 + 3] Cycloaddition to Build the Phomactin A Core.

6. C–H Activation
The activation of C–H bonds by transition metal catalysis has found widespread application in organic synthesis.110,111 The potential for this type of carbon–carbon and carbon–heteroatom bond formation has also been exploited in the synthesis of macrocyclic natural products. An elegant and highly enantioselective example of rhodium-catalyzed C–H activation for the synthesis of the core macrocyclic structure of cylindrocyclophanes (97) was reported by Davies in 2018 (Scheme 20).134 Remarkably, the choice of catalyst enables the highly selective functionalization of the most accessible unactivated methylene group, which is favored even over benzylic CH2 moieties present in the substrate.
Scheme 20. Enantioselective, Rh-Catalyzed Synthesis of the Cylindrocyclophane Core by Davies.

7. Radical Processes
The generation of radical species allows swift carbon–carbon bond formation in contexts that are often orthogonal to, and not as susceptible to the negative effects of steric hindrance as, classical polar reactivity. It is therefore only logical that radical processes have been heavily employed in the formation of large rings.135 Examples of the wide range of radical-based named reactions utilized for macrocyclization in natural product synthesis shall be presented in this section.
7.1. Pinacol Coupling
The treatment of carbonyls with single-electron reductants to form ketyl radical anions, followed by the radical coupling of two of these intermediates, is referred to as pinacol coupling.136 In 1989, McMurry reported the synthesis of crassin (100), which relied on the treatment of keto aldehyde 98 with titanium(III) chloride and zinc–copper couple to form diol 99 (Scheme 21a).137 While this major product was shown to possess the undesired stereochemical configurations at both alcohols, 2-fold hydroxyl inversion of this intermediate of the classical McMurry reaction138 enabled the formation of 100 in just four additional steps. Employing samarium(II) iodide as the single-electron reductant in the synthesis of isoedunol (103) and β-araneosene (104), Corey was able to synthesize 102 as a single diastereomer, starting from diketone 101 (Scheme 21b).124 While the diastereoselectivity itself is remarkable, two-step inversion of the secondary alcohol was necessary to effect the desired ring expansion through pinacol rearrangement (upon treatment with methanesulfonyl chloride, not shown). One and three further steps, respectively, were required to furnish 103 and 104.
Scheme 21. (a) Incomplete McMurry Reaction Affording Crassin-Intermediate 99, (b) SmI2-Induced Pinacol Coupling, as Applied by Corey, and (c) Nicolaou’s Heteropinacol-Macrocyclization en Route to Diazonamide A.

Samarium(II) iodide was also employed in Nicolaou’s 2003 synthesis of diazonamide A (1) (Scheme 21c), in which a heteropinacol coupling between an aldehyde and an oxime (105) led to the desired macrocyclization.23,27 Proceeding alongside N–O bond cleavage and followed immediately by a peptide coupling with Fmoc-protected l-valine to afford 106, this reaction directly set the stage for the formation of the target molecule’s second oxazole. As exemplified by Waldmann’s use of [V2Cl3·(THF)6][Zn2Cl6] in the synthesis of (9S, 18R)-cyclamenol A, other single-electron reductants can also be employed to effect pinacol-macrocyclization (not shown).139
7.2. Witkop-Type Cyclization
As mentioned in the Introduction, many strategies have been followed for the synthesis of diazonamide A (1). An approach chosen both by Harran and by Nicolaou involves variants of the Witkop cyclization, classically proceeding via a photoinduced electron transfer from the excited state of an indole chromophore.140 Apart from the Witkop reaction, Harran’s second attempt toward diazonamide A, the first having been thwarted by original misassignment of the structure,20,21 featured a further unusual macrocyclization (Scheme 22a).26 Oxidation of phenol 107 led to intermediate formation of a phenoxenium ion which engaged in an oxidative cycloaddition with the remote indole, forming indoline 108. The Witkop cyclization itself was initiated by treatment of the advanced intermediate 109 with lithium hydroxide and photoinduced electron transfer between the electron-rich indole and the comparatively electron-deficient bromoarene, as depicted in a simplified form (truncated structures 110-int1 and 110-int2, Scheme 22a). After mesolytic elimination of the bromide, radical–radical coupling afforded 110, which was elaborated to diazonamide A (1) in seven further steps. Notably, the presence of the phenolate—albeit ultimately superfluous—promoted the electron transfer to a large extent. This becomes particularly apparent when comparing this transformation to Nicolaou’s process (Scheme 22b).24,25 Having constructed the Western macrocycle of 111 through a macrolactamization, a very similar Witkop cyclization, in the absence of an additional electron donor, proceeded to afford 112 in only 30% yield (compared to Harran’s 72%). Despite this comparably low yield, the Witkop approach proved far superior to classical, stannane-based radical-cyclization conditions.
Scheme 22. Witkop-Cyclization Approaches to Diazonamide A by (a) Harran and (b) Nicolaou.

7.3. Wurtz-Type Cyclization
In 1993, Nógrádi disclosed the synthesis of garugamblin-1 (115) using a Wurtz-type macrocyclization.141 Herein, treatment of dibromide 113 with the radical anion generated from elemental sodium and tetraphenylethene led to sequential single-electron reduction of one of the benzylic halides, followed by substitution of the formed sodium organyl on the remaining bromoalkene (Scheme 23). Simultaneous reductive cleavage of the isoxazole moiety afforded the vinylogous amide 114, which was hydrolyzed and methylated to obtain a mixture of the target compound 115 and its methylation and double bond regioisomers.
Scheme 23. Wurtz-Type Coupling Enabled Nógrádi’s Synthesis of Garugamblin-1.

7.4. Giese Addition
Reversible addition of a thiyl radical to a vinyl cyclopropane moiety (116) initiated Feldman’s 1993 synthesis of the brefeldin A and C core (Scheme 24).142 After formation of intermediate 121, radical opening of the cyclopropane, followed by Giese addition of the resultant radical to the enone, led to the formation of the α-keto radical 122. Subsequent transannular addition of this radical to the allyl sulfide moiety effected intramolecular allylic homolytic elimination to afford a mixture of four diastereomeric products, the main components of which were 117 (displaying the brefeldin stereochemistry) and 118.
Scheme 24. Temporary Addition of a Thiyl Radical Promotes a Giese Macrocyclization, Forming the Brefeldin A and C Core Structure.

8. Addition to Carbonyls
Carbonyls are arguably the prototypical reactive functional group in organic synthesis. Carbonyl addition as a tool for the formation of macrocyclic structures will be summarized in this section, highlighting well-known examples in the formation of large rings during natural product synthesis.
8.1. Aldol Addition
The aldol reaction, probably the most well-known reaction not named after its discoverers, Alexander Borodin and Charles-Adolphe Wurtz, is one of the best-developed transformations in organic chemistry. As such, it is not surprising that it has found application in the formation of macrocyclic natural products. As aldol reactions can be run under both basic and acidic conditions, a large variety of protocols has been developed.
An example of a classical base-promoted aldol reaction was presented in Danishefsky’s 1996 synthesis of epothilone A (125) (Scheme 25a).143
Scheme 25. (a) Base- and (b) Lewis-Acid-Promoted Macroaldolization Reaction for the Syntheses of Epothilone A and Diazonamide A.

Formation of the potassium enolate of acetate 123 at low temperatures led to the formation of 124 after addition to the nonenolizable aldehyde. The authors noted that protonation of the intermediate potassium aldolate at low temperatures favored formation of the undesired diastereomer, while protonation at 0 °C allowed for the exclusive formation of the desired isomer (on analytical scale; preparative scale was less selective, yet still showed a preference of 6:1 for 124 over its isomer). For the synthesis of epothilone B, differing from A only by one methyl group at C-12 (see 123), the authors reported a considerably lower diastereoselectivity of 2.1:1 (not shown).144
Lewis-acid activation is also often employed to promote enolization and thereby aldol addition. Examples of such an approach to macrocycle formation in natural product synthesis include the Danishefsky synthesis of rapamycin (not shown)145 and MacMillan’s contribution to the variety of diazonamide A syntheses, published in 2011 (Scheme 25b).29 Herein, the macroaldolization was triggered by the addition of magnesium bromide and triethylamine to α-amino thioester 126. Trimethylsilyl chloride proved a necessary additive for this reaction, trapping the magnesium alkoxide, and thereby preventing retro-aldol addition. The product 127 was formed in high yield and as a single, ultimately inconsequential, diastereomer.
Magnus’ 1997 synthesis of a protected derivative of calicheamicinone (131) was also achieved with the help of a macroaldolization (Scheme 26).146 The conjugate addition of an aluminium thiophenolate to cyclohexenone 128 generated an enolate, which added to the cobalt-protected ynal moiety to provide the aldol product 129 under highly concentrated conditions. Further transformations led to 130, which proved resistant to deprotection and further elaboration to the target compound 131.
Scheme 26. Synthesis of an Advanced Intermediate of Calicheamicinone by Means of a Thiophenolate-Promoted Macroaldolization.

8.2. Reformatsky Reaction
The classical Reformatsky reaction describes the condensation of aldehydes or ketones with α-halo esters or amides using metallic zinc.147 While therein oxidative addition of zinc into the carbon–halogen bond, and subsequent metallotropic equilibration to the zinc enolate, represent the first steps, the use of other reducing agents can initiate the same reaction by different mechanisms. As shown by the 2014 synthesis of cebulactam A1 (134) by Yang (Scheme 27),148 reduction of an α-bromo amide (132) by samarium(II) iodide forms a samarium enolate that can add to a tethered α-chiral aldehyde. While product 133 was obtained as a pair of diastereomers on the newly formed chiral centers, subsequent oxidation to a 1,3-dicarbonyl moiety allowed access to a single, desired isomer due to in situ epimerization.
Scheme 27. Use of a Samarium-Mediated Reformatsky Reaction in the Synthesis of Cebulactam A1.

8.3. Hosomi–Sakurai Reaction
The nucleophilic addition of an allylsilane to an activated carbonyl moiety under (Lewis) acid catalysis is referred to as the Hosomi–Sakurai reaction.149 Mechanistically, the nucleophilic addition of an allylsilane can be considered a silyl-variation of the Prins reaction in which a simple alkene adds to an activated carbonyl.150 The Hosomi–Sakurai reaction, however, relies on the β-stabilization of positive charge by silicon, thereby ensuring increased reactivity and providing greater control over product distribution than its conceptual relative. As such, the Hosomi–Sakurai reaction is most often used as a method for allylation, especially in cases where the allyl moiety exhibits increased steric hindrance (cf. reverse prenylation).149
Owing to the increased reactivity of allylsilanes and the facile control of product formation, the Hosomi–Sakurai reaction has also been employed in total synthesis (Scheme 28). The synthesis of bryostatin 1 (137), published by Wender in its entirety in 2017,151−153 showcases the macrocyclization of an allylsilane onto an enal (135) (Scheme 28a). Herein, the aldehyde is activated by pyridinium p-toluenesulfonate in combination with trimethyl orthoformate and the in situ TES-deprotected alcohol, forming an oxocarbenium ion which is readily attacked by the nucleophilic alkene, now in close proximity. Thus, in one step, tetrahydropyran 136 is formed in excellent yield, allowing access to bryostatin 1 in just four additional steps. A Sakurai-dimerization/macrocyclization strategy was followed in Rychnovsky’s 2011 synthesis of the cyanolide A aglycon (140) (Scheme 28b).154 The activation of acetal 138 initially led to dimerization via the intermolecular Hosomi–Sakurai reaction, once again employing an adjacent alcohol to form a tetrahydropyran. Subsequent macrocyclization, also initiated by acetal-activation in the same step, afforded 139, which was elaborated to the target molecule in just two further operations.
Scheme 28. Hosomi–Sakurai Reactions Led to the Desired Macrocyclizations in the Syntheses of (a) Bryostatin 1 by Wender and (b) the Cyanolide A Aglycon by Rychnovsky.

8.4. Barbier Reaction
While Grignard reagents are generally prepared in a separate flask before addition to an electrophile, the in situ generation of an organometallic reagent from an alkyl halide in the presence of a carbonyl derivative is termed the Barbier reaction. Therefore, by definition, macrocyclizations cannot be performed using Grignard reagents. Barbier-type reactions, however, have been employed in macrocyclic natural product synthesis. In 2011, Romo disclosed the synthesis of gymnodimine (143) and its C-4 epimer, relying on macrocyclic ring closure of iodoalkane 141 (Scheme 29a).155 Treatment of 141 with an excess of tert-butyllithium at ambient temperature—this comparatively high temperature being critical to achieve conformational equilibria favoring macrocyclization, thereby preventing unselective additions and quenching pathways—led to halogen-metal exchange and subsequent addition to the tosyl-protected lactam, providing 142 in varying, but good, yields. A further Barbier-type protocol, involving the in situ generation of a carbanionic species and subsequent addition onto a carbonyl, was presented in Danishefsky’s synthesis of calicheamicinone (131) (Scheme 29b).156,157 Herein, as in several other syntheses of enediyne natural products,158,159 deprotonation of a terminal alkyne (144) triggered cyclization onto an aldehyde, forming propargylic alcohol 145 with high diastereoselectivity.
Scheme 29. (a) Romo’s Room-Temperature Barbier Reaction en Route to Gymnodimine and (b) Danishefsky’s Acetylide Addition for the Synthesis of Calicheamicinone.

8.5. Nozaki–Hiyama–Kishi Reaction
The coupling of halide-substituted sp2- or sp-carbon atoms with aldehydes using chromium salts and catalytic amounts of nickel is named the Nozaki–Hiyama–Kishi (NHK) reaction.160 This transformation has found widespread application in organic synthesis due to its remarkable selectivity for aldehydes over a range of other reactive functional groups, including ketones. In the context of macrocyclic natural product synthesis, the NHK reaction has particularly found application in approaches toward phomactin A (22) (Scheme 30).
Scheme 30. Nozaki–Hiyama–Kishi Approaches toward Phomactin A.

Pattenden’s synthetic strategy,51 similarly evoked by Maleczka,161 involved the treatment of vinyl iodide 146 with chromium(II) and nickel(II) chlorides, to afford the allylic alcohol 147 in moderate yield (Scheme 30a). Following inversion of the secondary alcohol and diastereoselective monoepoxidation, 22 was obtained after ultimate double deprotection concomitant with acetal formation and epoxide opening. In contrast to this, Frontier’s synthesis of the phomactin A core relied on an NHK reaction of the acyclic precursor 148, which gave 149 in good yield (Scheme 30b).162
8.6. Loh-Type α-Allylation
The Loh allylation allows regioselective α- or γ-allylation of aldehydes with allylindium reagents, enabling a switch in selectivity by choice of the solvent system and reaction conditions.163 In 2016, Banwell exploited this protocol’s selectivity in a macrocyclization en route to the resorcylic acid lactones paecilomycin F (152) and cochliomycin C (153) (Scheme 31).164 The treatment of allylic chloride 150 with indium powder in methylene chloride and water led to α-allylation of the remote aldehyde, resulting in macrocyclization and the formation of 151. Notably, initial attempts involving Nozaki–Hiyama–Kishi conditions provided exclusively the regioisomeric γ-allylation product. Subsequent global deprotection afforded paecilomycin F (152), and aromatic chlorination of this material provided cochliomycin C (153).
Scheme 31. Loh-Type Allylation Employed by Banwell in the Syntheses of Paecilomycin F and Cochliomycin C.

9. Miscellaneous Macrocyclizations
9.1. Nickel-Catalyzed Alkyne-Aldehyde Reductive Coupling
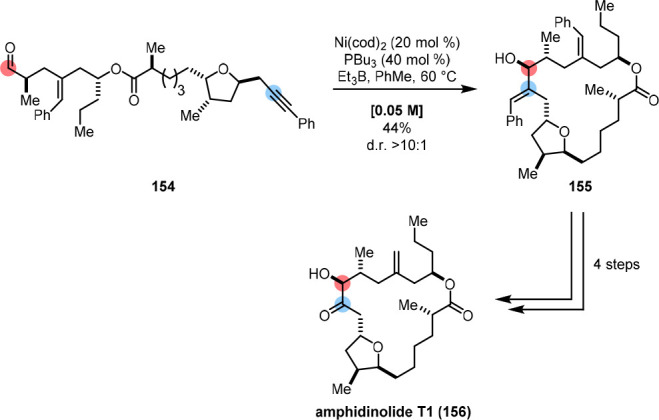
Jamison employed an alkyne-aldehyde reductive coupling on 154 in the syntheses of amphidinolides T1 (156) and T4 (Scheme 32),165 and terpestacin,166 using a nickel catalyst, tributylphosphine and triethylborane.167 Crucially, elevated temperatures had to be employed in order to observe the formation of the desired product 155, and optimal conditions included a comparatively high catalyst loading of 20%.
Scheme 32. Nickel-Catalyzed Alkyne-Aldehyde Reductive Coupling in the Synthesis of Amphidinolide T1 by Jamison.
9.2. Chan–Evans–Lam Coupling
The coupling of an arylboronic ester and a phenol to form a macrocyclic biaryl ether was achieved by Hoveyda in 2003 (Scheme 33).77
Scheme 33. Hoveyda’s Synthesis of Chloropeptin I through a Chan–Evans–Lam Coupling.

Under modified Chan–Evans–Lam conditions,168−170 precursor 157 was first treated with sodium periodate, cleaving the pinacol ester and liberating the free boronic acid, after which coupling was performed using cupric acetate, triethylamine (instead of pyridine), and methanol as an additive. Methanol proved critical for the success of this transformation leading to 158, as yields lower than 20% were generally observed in its absence. The authors suspected methanol to play either of two roles: (i) enabling the in situ formation of the boronic acid dimethyl ester or (ii) increasing the solubility of the copper salt. The total synthesis of chloropeptin I (159) was achieved in 13 further steps, including a Stille coupling to form the second macrocycle.
9.3. Ullmann-Type Coupling
The copper-mediated oxidative coupling of aryl halides for the synthesis of symmetrical biaryls is known as Ullmann coupling and usually proceeds under extreme conditions with elemental copper. However, incorporation of a nucleophile results in a Cu-nucleophile intermediate which adds oxidatively to the aryl-halide bond and eventually delivers a new substituted arene after reductive elimination. This nucleophilic aromatic substitution is also referred to as an Ullmann-type reaction and serves as one of the exceptional strategies for the construction of macrocyclic natural products. A first example was disclosed by Boger, who utilized this reactivity in the enantioselective synthesis of piperazinomycin in 1993.171 More recently, Uchiro reported the synthesis of hirsutellone B (162) (Scheme 34),172 aiming at a more robust synthesis compared to Nicolaou’s,173 who succeeded by a Ramberg–Bäcklund ring contraction (not shown). Uchiro approached the synthetic challenge of the highly strained 13-membered macrocycle by subjecting 160 to copper(I)-catalysis at 160 °C. The macrocyclization successfully afforded aryl ether 161, which was transformed into the natural product 162 in four subsequent operations.
Scheme 34. Ullmann-Type Etherification for the Macrocyclization of Hirsutellone B Developed by Uchiro.

9.4. Glaser–Hay Coupling
In order to overcome the inherent need for high dilution in macrocyclization reactions, Collins employed a biphasic solvent system in the formal synthesis of ivorenolide A (165) (Scheme 35).174 The crucial Glaser–Hay coupling of bis-alkyne 163 to afford 164 was performed in a mixture of PEG400 and methanol, ensuring solubilization of the substrate and the catalysts in different media. By limiting substrate–catalyst interaction to the liquid/liquid interface, the effective concentration of the substrate was considerably lowered, allowing the reaction to be performed in continuous flow at roughly 120 times the concentration of similarly successful batch reactions (24 mM compared to 0.2 mM).
Scheme 35. Biphasic Conditions Allow Glaser–Hay Coupling in Continuous Flow at High Concentrations for the Formal Synthesis of Ivorenolide A.

9.5. Corey–Seebach Reaction
While classical nucleophilic substitution reactions have not heavily featured in this article,175 the synthesis of macrocyclic structures through the Corey–Seebach reaction shall be mentioned. In Harrowven’s 2016 synthesis of riccardin C (168), dithiane 166 was deprotonated with n-butyllithium, leading to umpolung of the former aldehyde carbon, and immediately engaged in substitution of the benzylic chloride to form 167 (Scheme 36).176 While only moderately successful in terms of yield, this reaction provided rapid access to the core structure of the target molecule which was synthesized in three additional steps.
Scheme 36. Harrowven’s Corey–Seebach Reaction Enabled the Synthesis of Riccardin C.

Summary and Outlook
Macrocyclic natural products and their nature-inspired analogues have sparked the interest of the synthetic and medicinal community as a result of their high potential as pharmaceuticals. This appears to derive from their specific three-dimensional architecture which allows a preorganization profile ensuring high-affinity binding interactions with protein targets. Nature possesses countless macrocyclic scaffolds with high bioactive potency that could serve as candidates in drug discovery. Therefore, the development of robust methodologies and approaches for the synthesis of these macrocyclic natural products and their non-natural analogues is a necessity.
This Outlook aimed to gaze beyond the common go-to-transformations when it comes to macrocyclization—namely, olefin metathesis, macrolactonization, or lactamization—and provide examples of what we termed unconventional approaches (e.g., Pd-mediated couplings, olefinations, aryl–aryl couplings, cycloadditions, C–H activation, radical couplings, addition to carbonyls, and others) for the synthesis of macrocyclic cores en route to natural products or analogues. In this regard, we also highlighted the multidirectional approaches of various research groups toward the total synthesis of diazonamide A (1), a potent antimitotic agent bearing two synthetically challenging macrocycles. While a large variety of approaches enabling highly flexible strategies for the formation of almost any kind of macrocycle has been established, several challenges remain. We hope that this overview of unconventional macrocyclizations can serve as a useful resource that enhances the future development of novel strategies toward the synthesis of macrocyclic, bioactive substances.
Acknowledgments
Generous support by the FWF (Fellowship J 4202-N28 to D.K..; P 32607 to N.M.) and the ERC (CoG VINCAT 682002 to N.M.) is acknowledged. We are also grateful to the University of Vienna for continued support of our research programs.
Author Contributions
§ I.S. and D.K. contributed equally.
The authors declare no competing financial interest.
References
- Newman D. J.; Cragg G. M.. Bioactive Macrocycles from Nature. In Macrocycles in Drug Discovery; Royal Society of Chemistry, 2015; pp 1–36. [Google Scholar]
- Mallinson J.; Collins I. Macrocycles in New Drug Discovery. Future Med. Chem. 2012, 4, 1409–1438. 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- Marsault E.; Peterson M. L. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. The Exploration of Macrocycles for Drug Discovery - an Underexploited Structural Class. Nat. Rev. Drug Discovery 2008, 7, 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- Ermert P. Design, Properties and Recent Application of Macrocycles in Medicinal Chemistry. Chimia 2017, 71, 678–702. 10.2533/chimia.2017.678. [DOI] [PubMed] [Google Scholar]
- Yudin A. K. Macrocycles: Lessons from the Distant Past, Recent Developments, and Future Directions. Chem. Sci. 2015, 6, 30–49. 10.1039/C4SC03089C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí-Centelles V.; Pandey M. D.; Burguete M. I.; Luis S. V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. 10.1021/acs.chemrev.5b00056. [DOI] [PubMed] [Google Scholar]
- Blankenstein J.; Zhu J. Conformation-Directed Macrocyclization Reactions. Eur. J. Org. Chem. 2005, 2005, 1949–1964. 10.1002/ejoc.200400751. [DOI] [Google Scholar]
- Zheng K.; Hong R. Stereoconfining Macrocyclizations in the Total Synthesis of Natural Products. Nat. Prod. Rep. 2019, 36, 1546–1575. 10.1039/C8NP00094H. [DOI] [PubMed] [Google Scholar]
- Grubbs R. H.; Miller S. J.; Fu G. C. Ring-Closing Metathesis and Related Processes in Organic Synthesis. Acc. Chem. Res. 1995, 28, 446–452. 10.1021/ar00059a002. [DOI] [Google Scholar]
- Gradillas A.; Pérez-Castells J. Macrocyclization by Ring-Closing Metathesis in the Total Synthesis of Natural Products: Reaction Conditions and Limitations. Angew. Chem., Int. Ed. 2006, 45, 6086–6101. 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]
- Hoveyda A. H.; Zhugralin A. R. The Remarkable Metal-Catalysed Olefin Metathesis Reaction. Nature 2007, 450, 243–251. 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- Parenty A.; Moreau X.; Niel G.; Campagne J. M. Update 1 of: Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2013, 113, PR1–PR40. 10.1021/cr300129n. [DOI] [PubMed] [Google Scholar]
- Li Y.; Yin X.; Dai M. Catalytic Macrolactonizations for Natural Product Synthesis. Nat. Prod. Rep. 2017, 34, 1185–1192. 10.1039/C7NP00038C. [DOI] [PubMed] [Google Scholar]
- Davies J. S. The Cyclization of Peptides and Depsipeptides. J. Pept. Sci. 2003, 9, 471–501. 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- Montalbetti C. A. G. N.; Falque V. Amide Bond Formation and Peptide Coupling. Tetrahedron 2005, 61, 10827–10852. 10.1016/j.tet.2005.08.031. [DOI] [Google Scholar]
- Santandrea J.; Bédard A.-C.; de Léséleuc M.; Raymond M.; Collins S. K.. Alternative Strategies for the Construction of Macrocycles. In Practical Medicinal Chemistry with Macrocycles: Design, Synthesis, and Case Studies; John Wiley & Sons, Inc., 2017; pp 307–337. [Google Scholar]
- Lachia M.; Moody C. J. The Synthetic Challenge of Diazonamide A, a Macrocyclic Indole Bis-Oxazole Marine Natural Product. Nat. Prod. Rep. 2008, 25, 227–253. 10.1039/b705663j. [DOI] [PubMed] [Google Scholar]
- Lindquist N.; Fenical W.; Duyne G. D.; Van; Clardy J. Isolation and Structure Determination of Diazonamides A and B, Unusual Cytotoxic Metabolites from the Marine Ascidian Diazona Chinensis. J. Am. Chem. Soc. 1991, 113, 2303–2304. 10.1021/ja00006a060. [DOI] [Google Scholar]
- Li J.; Jeong S.; Esser L.; Harran P. G. Total Synthesis of Nominal Diazonamides– Part 1: Convergent Preparation of the Structure Proposed for (−)-Diazonamide A. Angew. Chem., Int. Ed. 2001, 40, 4765–4769. . [DOI] [PubMed] [Google Scholar]
- Li J.; Burgett A. W. G.; Esser L.; Amezcua C.; Harran P. G. Total Synthesis of Nominal Diazonamides– Part 2: On the True Structure and Origin of Natural Isolates. Angew. Chem., Int. Ed. 2001, 40, 4770–4773. . [DOI] [PubMed] [Google Scholar]
- Vedejs E.; Barda D. A. Progress toward Synthesis of Diazonamide A. Preparation of a 3-(Oxazol-5-Yl)-4-Trifluoromethyl-Sulfonyloxyindole and Its Use in Biaryl Coupling Reactions. Org. Lett. 2000, 2, 1033–1035. 10.1021/ol005548p. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Huang X.; Giuseppone N.; Rao P. B.; Bella M.; Reddy M. V.; Snyder S. A. Construction of the Complete Aromatic Core of Diazonamide A by a Novel Hetero Pinacol Macrocyclization Cascade Reaction. Angew. Chem., Int. Ed. 2001, 40, 4705–4709. . [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Bella M.; Chen D. Y.-K.; Huang X.; Ling T.; Snyder S. A. Total Synthesis of Diazonamide A. Angew. Chem., Int. Ed. 2002, 41, 3495–3499. . [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Chen D. Y. K.; Huang X.; Ling T.; Bella M.; Snyder S. A. Chemistry and Biology of Diazonamide A: First Total Synthesis and Confirmation of the True Structure. J. Am. Chem. Soc. 2004, 126, 12888–12896. 10.1021/ja040092i. [DOI] [PubMed] [Google Scholar]
- Burgett A. W. G.; Li Q.; Wei Q.; Harran P. G. A Concise and Flexible Total Synthesis of (−)-Diazonamide A. Angew. Chem., Int. Ed. 2003, 42, 4961–4966. 10.1002/anie.200352577. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Rao P. B.; Hao J.; Reddy M. V.; Rassias G.; Huang X.; Chen D. Y.-K.; Snyder S. A. The Second Total Synthesis of Diazonamide A. Angew. Chem., Int. Ed. 2003, 42, 1753–1758. 10.1002/anie.200351112. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Hao J.; Reddy M. V.; Rao P. B.; Rassias G.; Snyder S. A.; Huang X.; Chen D. Y. K.; Brenzovich W. E.; Giuseppone N.; Giannakakou P.; O’Brate A. Chemistry and Biology of Diazonamide A: Second Total Synthesis and Biological Investigations. J. Am. Chem. Soc. 2004, 126, 12897–12906. 10.1021/ja040093a. [DOI] [PubMed] [Google Scholar]
- Knowles R. R.; Carpenter J.; Blakey S. B.; Kayano A.; Mangion I. K.; Sinz C. J.; MacMillan D. W. C. Total Synthesis of Diazonamide A. Chem. Sci. 2011, 2, 308–311. 10.1039/C0SC00577K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C. M.; Goldberg F. W.; Magnus P.; Russell C. J.; Tumbull R.; Lynch V. An Expedient Formal Total Synthesis of (−)-Diazonamide A via a Powerful, Stereoselective O-Aryl to C-Aryl Migration to Form the C10 Quaternary Center. J. Am. Chem. Soc. 2007, 129, 12320–12327. 10.1021/ja0744448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai C.-K.; Sommons M. F.; Sammakia T. A Concise Formal Synthesis of Diazonamide a by the Stereoselective Construction of the C10 Quaternary Center. Angew. Chem., Int. Ed. 2010, 49, 2397–2400. 10.1002/anie.200906318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David N.; Pasceri R.; Kitson R. R. A.; Pradal A.; Moody C. J. Formal Total Synthesis of Diazonamide A by Indole Oxidative Rearrangement. Chem. - Eur. J. 2016, 22, 10867–10876. 10.1002/chem.201601605. [DOI] [PubMed] [Google Scholar]
- Tsuji J.Palladium in Organic Synthesis; Springer-Verlag Berlin and Heidelberg GmbH & Co. KG, 2010. [Google Scholar]
- Tsuji J.; Takahashi H.; Morikawa M. Organic Syntheses by Beans of Noble Metal Compounds XVII. Reaction of π-Allylpalladium Chloride with Nucleophiles. Tetrahedron Lett. 1965, 6, 4387–4388. 10.1016/S0040-4039(00)71674-1. [DOI] [Google Scholar]
- Heck R. F. Arylation, Methylation, and Carboxyalkylation of Olefins by Group VIII Metal Derivatives. J. Am. Chem. Soc. 1968, 90, 5518–5526. 10.1021/ja01022a034. [DOI] [Google Scholar]
- Heck R. F. The Arylation of Allylic Alcohols with Organopalladium Compounds. A New Synthesis of 3-Aryl Aldehydes and Ketones. J. Am. Chem. Soc. 1968, 90, 5526–5531. 10.1021/ja01022a035. [DOI] [Google Scholar]
- Heck R. F. Aromatic Haloethylation with Palladium and Copper Halides. J. Am. Chem. Soc. 1968, 90, 5538–5542. 10.1021/ja01022a038. [DOI] [Google Scholar]
- Heck R. F. The Addition of Alkyl- and Arylpalladium Chlorides to Conjugated Dienes. J. Am. Chem. Soc. 1968, 90, 5542–5546. 10.1021/ja01022a039. [DOI] [Google Scholar]
- Baba S.; Negishi E. A Novel Stereospecific Alkenyl-Alkenyl Cross-Coupling by a Palladium- or Nickel-Catalyzed Reaction of Alkenylalanes with Alkenyl Halides. J. Am. Chem. Soc. 1976, 98, 6729–6731. 10.1021/ja00437a067. [DOI] [Google Scholar]
- Negishi E.; Baba S. Novel Stereoselective Alkenyl-Aryl Coupling via Nickel-Catalysed Reaction of Alkenylalanes with Aryl Halides. J. Chem. Soc., Chem. Commun. 1976, 596b–597b. 10.1039/C3976000596B. [DOI] [Google Scholar]
- Miyaura N.; Suzuki A. Stereoselective Synthesis of Arylated (E)-Alkenes by the Reaction of Alk-1 -Enylboranes with Aryl Halides in the Presence of Palladium Catalyst. J. Chem. Soc., Chem. Commun. 1979, 866–867. 10.1039/c39790000866. [DOI] [Google Scholar]
- Trost B. M.; Fullerton T. J. New Synthetic Reactions. Allylic Alkylation. J. Am. Chem. Soc. 1973, 95, 292–294. 10.1021/ja00782a080. [DOI] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2943. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- Chinchilla R.; Nájera C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- Cordovilla C.; Bartolomé C.; Martínez-Ilarduya J. M.; Espinet P. The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053. 10.1021/acscatal.5b00448. [DOI] [Google Scholar]
- Ronson T. O.; Unsworth W. P.; Fairlamb I. J. S.. Palladium-Catalyzed Synthesis of Macrocycles. In Practical Medicinal Chemistry with Macrocycles: Design, Synthesis, and Case Studies; John Wiley & Sons, Inc., 2017; pp 281–305. [Google Scholar]
- Milstein D.; Stille J. K. A General, Selective, and Facile Method for Ketone Synthesis from Acid Chlorides and Organotin Compounds Catalyzed by Palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. 10.1021/ja00479a077. [DOI] [Google Scholar]
- Stille J. K. The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles. Angew. Chem., Int. Ed. Engl. 1986, 25, 508–524. 10.1002/anie.198605081. [DOI] [Google Scholar]
- Kosugi M.; Sasazawa K.; Shimisu Y.; Migita T. Reactions of Allyltin Compounds III. Allylation of Aromatic Halides with Allyltributyltin in the Presence of Tetrakis(Triphenylphosphine)Palladium(0). Chem. Lett. 1977, 6, 301–302. 10.1246/cl.1977.301. [DOI] [Google Scholar]
- Kosugi M.; Shimizu Y.; Migita T. Alkylation, Arylation, and Vinylation of Acyl Chlorides by Means of Organotin Compounds in the Presence of Catalytic Amounts of Tetrakis(Triphenylphosphine)Palladium(0). Chem. Lett. 1977, 6, 1423–1424. 10.1246/cl.1977.1423. [DOI] [Google Scholar]
- Pattenden G.; Sinclair D. J. The Intramolecular Stille Reaction in Some Target Natural Product Syntheses. J. Organomet. Chem. 2002, 653, 261–268. 10.1016/S0022-328X(02)01169-5. [DOI] [Google Scholar]
- Gyorkos A. C.; Stille J. K.; Hegedus L. S. Total Synthesis of (±)-Epi-Jatrophone and (±)-Jatrophone Using Palladium-Catalyzed Carbonylative Coupling of Vinyl Triflates with Vinylstannanes as the Macrocycle-Forming Step. J. Am. Chem. Soc. 1990, 112, 8465–8472. 10.1021/ja00179a035. [DOI] [Google Scholar]
- Brodmann T.; Janssen D.; Kalesse M. Total Synthesis of Chivosazole F. J. Am. Chem. Soc. 2010, 132, 13610–13611. 10.1021/ja107290s. [DOI] [PubMed] [Google Scholar]
- Ammer C.; Bach T. Total Syntheses of the Thiopeptides Amythiamicin C and D. Chem. - Eur. J. 2010, 16, 14083–14093. 10.1002/chem.201002144. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-H.; Liu R.; Liu B. Total Synthesis of Nannocystin Ax. Chem. Commun. 2017, 53, 5549–5552. 10.1039/C7CC00469A. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Chakraborty T. K.; Piscopio A. D.; Minowa N.; Bertinato P. Total Synthesis of Rapamycin. J. Am. Chem. Soc. 1993, 115, 4419–4420. 10.1021/ja00063a093. [DOI] [Google Scholar]
- Smith A. B. III; Ott G. R. Total Syntheses of (−)-Macrolactin A, (+)-Macrolactin E, and (−)-Macrolactinic Acid: An Exercise in Stille Cross-Coupling Chemistry. J. Am. Chem. Soc. 1998, 120, 3935–3948. 10.1021/ja980203b. [DOI] [Google Scholar]
- Garg N. K.; Hiebert S.; Overman L. E. Total Synthesis of (−)-Sarain A. Angew. Chem., Int. Ed. 2006, 45, 2912–2915. 10.1002/anie.200600417. [DOI] [PubMed] [Google Scholar]
- Beletskaya I. P.; Cheprakov A. V. The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev. 2000, 100, 3009–3066. 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- Jägel J.; Maier M. E. Formal Total Synthesis of Palmerolide A. Synthesis 2009, 2009 (17), 2881–2892. 10.1055/s-0029-1216921. [DOI] [Google Scholar]
- Nicolaou K. C.; Guduru R.; Sun Y. P.; Banerji B.; Chen D. Y.-K. Total Synthesis of the Originally Proposed and Revised Structures of Palmerolide A. Angew. Chem., Int. Ed. 2007, 46, 5896–5900. 10.1002/anie.200702243. [DOI] [PubMed] [Google Scholar]
- Kashinath K.; Jachak G. R.; Athawale P. R.; Marelli U. K.; Gonnade R. G.; Srinivasa Reddy D. Total Synthesis of the Marine Natural Product Solomonamide B Necessitates Stereochemical Revision. Org. Lett. 2016, 18, 3178–3181. 10.1021/acs.orglett.6b01395. [DOI] [PubMed] [Google Scholar]
- Jachak G. R.; Athawale P. R.; Agarwal H.; Barthwal M. K.; Lauro G.; Bifulco G.; Srinivasa Reddy D. Total Synthesis of the Potent Anti-Inflammatory Natural Product Solomonamide A along with Structural Revision and Biological Activity Evaluation. Org. Biomol. Chem. 2018, 16, 9138–9142. 10.1039/C8OB02713G. [DOI] [PubMed] [Google Scholar]
- Li P.; Li J.; Arikan F.; Ahlbrecht W.; Dieckmann M.; Menche D. Total Synthesis of Etnangien. J. Am. Chem. Soc. 2009, 131, 11678–11679. 10.1021/ja9056163. [DOI] [PubMed] [Google Scholar]
- Menche D.; Hassfeld J.; Li J.; Rudolph S. Total Synthesis of Archazolid A. J. Am. Chem. Soc. 2007, 129, 6100–6101. 10.1021/ja071461o. [DOI] [PubMed] [Google Scholar]
- Das S.; Paul D.; Goswami R. K. Stereoselective Total Synthesis of Bioactive Marine Natural Product Biselyngbyolide B. Org. Lett. 2016, 18, 1908–1911. 10.1021/acs.orglett.6b00713. [DOI] [PubMed] [Google Scholar]
- Meidlinger D.; Marx L.; Bordeianu C.; Choppin S.; Colobert F.; Speicher A. Access to the Enantiopure Axially Chiral Cyclophane Isoplagiochin D through Atropo-Diastereoselective Heck Coupling. Angew. Chem., Int. Ed. 2018, 57, 9160–9164. 10.1002/anie.201803677. [DOI] [PubMed] [Google Scholar]
- Groh M.; Meidlinger D.; Bringmann G.; Speicher A. Atroposelective Heck Macrocyclization: Enantioselective Synthesis of Bis(Bibenzylic) Natural Products. Org. Lett. 2012, 14, 4548–4551. 10.1021/ol302132s. [DOI] [PubMed] [Google Scholar]
- Molander G. A.; Dehmel F. Formal Total Synthesis of Oximidine II via a Suzuki-Type Cross-Coupling Macrocyclization Employing Potassium Organotrifluoroborates. J. Am. Chem. Soc. 2004, 126, 10313–10318. 10.1021/ja047190o. [DOI] [PubMed] [Google Scholar]
- Tortosa M.; Yakelis N. A.; Roush W. R. Total Synthesis of (+)-Superstolide A. J. Org. Chem. 2008, 73, 9657–9667. 10.1021/jo801794s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour J.; Neuville L.; Zhu J. Intramolecular Suzuki-Miyaura Reaction for the Total Synthesis of Signal Peptidase Inhibitors, Arylomycins A2 and B2. Chem. - Eur. J. 2010, 16, 10523–10534. 10.1002/chem.201000924. [DOI] [PubMed] [Google Scholar]
- Dieckmann M.; Kretschmer M.; Li P.; Rudolph S.; Herkommer D.; Menche D. Total Synthesis of Rhizopodin. Angew. Chem., Int. Ed. 2012, 51, 5667–5670. 10.1002/anie.201201946. [DOI] [PubMed] [Google Scholar]
- Liao L.; Zhou J.; Xu Z.; Ye T. Concise Total Synthesis of Nannocystin A. Angew. Chem., Int. Ed. 2016, 55, 13263–13266. 10.1002/anie.201606679. [DOI] [PubMed] [Google Scholar]
- Mohr P. J.; Halcomb R. L. Total Synthesis of (+)-Phomactin A Using a B-Alkyl Suzuki Macrocyclization. J. Am. Chem. Soc. 2003, 125, 1712–1713. 10.1021/ja0296531. [DOI] [PubMed] [Google Scholar]
- Johnson C. R.; Braun M. P. A Two-Step, Three-Component Synthesis of PGE1: Utilization of α-Iodoenones in Pd(0)-Catalyzed Cross-Couplings of Organoboranes. J. Am. Chem. Soc. 1993, 115, 11014–11015. 10.1021/ja00076a079. [DOI] [Google Scholar]
- Shinohara T.; Deng H.; Snapper M. L.; Hoveyda A. H. Isocomplestatin: Total Synthesis and Stereochemical Revision. J. Am. Chem. Soc. 2005, 127, 7334–7336. 10.1021/ja051790l. [DOI] [PubMed] [Google Scholar]
- Deng H.; Jung J. K.; Liu T.; Kuntz K. W.; Snapper M. L.; Hoveyda A. H. Total Synthesis of Anti-HIV Agent Chloropeptin I. J. Am. Chem. Soc. 2003, 125, 9032–9034. 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Coussanes G.; Souris C.; Aillard P.; Kaldre D.; Runggatscher K.; Kubicek S.; Di Mauro G.; Maryasin B.; Maulide N. A Domino 10-Step Total Synthesis of FR252921 and Its Analogues, Complex Macrocyclic Immunosuppressants. J. Am. Chem. Soc. 2019, 141, 13772–13777. 10.1021/jacs.9b07185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amans D.; Bellosta V.; Cossy J. Synthesis of a Promising Immunosuppressant: FR252921. Org. Lett. 2007, 9, 4761–4764. 10.1021/ol702110k. [DOI] [PubMed] [Google Scholar]
- Amans D.; Bellosta V.; Cossy J. Synthesis of Two Bioactive Natural Products: FR252921 and Pseudotrienic Acid B. Chem. - Eur. J. 2009, 15, 3457–3473. 10.1002/chem.200802649. [DOI] [PubMed] [Google Scholar]
- Oliveira M. T.; Luparia M.; Audisio D.; Maulide N. Dual Catalysis Becomes Diastereodivergent. Angew. Chem., Int. Ed. 2013, 52, 13149–13152. 10.1002/anie.201305933. [DOI] [PubMed] [Google Scholar]
- Oliveira M. T.; Audisio D.; Niyomchon S.; Maulide N. Diastereodivergent Processes in Palladium-Catalyzed Allylic Alkylation. ChemCatChem 2013, 5, 1239–1247. 10.1002/cctc.201200644. [DOI] [Google Scholar]
- Misale A.; Niyomchon S.; Maulide N. Cyclobutenes: At a Crossroad between Diastereoselective Syntheses of Dienes and Unique Palladium-Catalyzed Asymmetric Allylic Substitutions. Acc. Chem. Res. 2016, 49, 2444–2458. 10.1021/acs.accounts.6b00375. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y.; Itoh A.; Hashimoto S.; Yamamoto H.; Nozaki H. Total Synthesis of Humulene. A Stereoselective Approach. J. Am. Chem. Soc. 1977, 99, 3864–3867. 10.1021/ja00453a069. [DOI] [Google Scholar]
- Fürstner A.; Weintritt H. Total Synthesis of Roseophilin. J. Am. Chem. Soc. 1998, 120, 2817–2825. 10.1021/ja973846k. [DOI] [Google Scholar]
- Munakata R.; Katakai H.; Ueki T.; Kurosaka J.; Takao K. I.; Tadano K. I. Total Synthesis of (+)-Macquarimicin A. J. Am. Chem. Soc. 2003, 125, 14722–14723. 10.1021/ja038732p. [DOI] [PubMed] [Google Scholar]
- Porco J. A.; Schoenen F. J.; Stout T. J.; Clardy J.; Schreiber S. L. Transannular Diels-Alder Route to Systems Related to Dynemicin A. J. Am. Chem. Soc. 1990, 112, 7410–7411. 10.1021/ja00176a060. [DOI] [Google Scholar]
- Koyama Y.; Lear M. J.; Yoshimura F.; Ohashi I.; Mashimo T.; Hirama M. Efficient Construction of the Kedarcidin Chromophore Ansamacrolide. Org. Lett. 2005, 7, 267–270. 10.1021/ol0477374. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Dong G. Total Synthesis of Bryostatin 16 Using Atom-Economical and Chemoselective Approaches. Nature 2008, 456, 485–488. 10.1038/nature07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isley N. A.; Endo Y.; Wu Z. C.; Covington B. C.; Bushin L. B.; Seyedsayamdost M. R.; Boger D. L. Total Synthesis and Stereochemical Assignment of Streptide. J. Am. Chem. Soc. 2019, 141, 17361–17369. 10.1021/jacs.9b09067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig G.; Geissler G. Zur Reaktionsweise Des Pentaphenyl-Phosphors Und Einiger Derivate. Justus Liebigs Ann. Chem. 1953, 580, 44–57. 10.1002/jlac.19535800107. [DOI] [Google Scholar]
- Wittig G.; Schöllkopf U. Über Triphenyl-Phosphin-Methylene Als Olefinbildende Reagenzien (I. Mitteil.). Chem. Ber. 1954, 87, 1318–1330. 10.1002/cber.19540870919. [DOI] [Google Scholar]
- Wittig G.; Haag W. Über Triphenyl-Phosphinmethylene Als Olefinbildende Reagenzien (II. Mitteil.). Chem. Ber. 1955, 88, 1654–1666. 10.1002/cber.19550881110. [DOI] [Google Scholar]
- Keserü G. M.; Dienes Z.; Nógrádi M.; Kajtár-Peredy M. Synthesis and Revised Structure of Garuganin III. J. Org. Chem. 1993, 58, 6725–6728. 10.1021/jo00076a036. [DOI] [Google Scholar]
- Smith A. B. III; Wood J. L.; Wong W.; Gould A. E.; Rizzo C. J.; Barbosa J.; Komiyama K.; Omura S. (+)-Trienomycins A, B, C, and F and (+)-Mycotrienins I and II: Relative and Absolute Stereochemistry. J. Am. Chem. Soc. 1996, 118, 8308–8315. 10.1021/ja961400i. [DOI] [Google Scholar]
- Smith A. B. III; Barbosa J.; Wong W.; Wood J. L. Total Syntheses of (+)-Trienomycins A and F via a Unified Strategy. J. Am. Chem. Soc. 1996, 118, 8316–8328. 10.1021/ja961401a. [DOI] [Google Scholar]
- Horner L.; Hoffmann H.; Wippel H. G. Phosphororganische Verbindungen, XII. Phosphinoxyde Als Olefinierungsreagenzien. Chem. Ber. 1958, 91, 61–63. 10.1002/cber.19580910113. [DOI] [Google Scholar]
- Horner L.; Hoffmann H.; Wippel H. G.; Klahre G. Phosphororganische Verbindungen, XX. Phosphinoxyde Als Olefinierungsreagenzien. Chem. Ber. 1959, 92, 2499–2505. 10.1002/cber.19590921017. [DOI] [Google Scholar]
- Wadsworth W. S.; Emmons W. D. The Utility of Phosphonate Carbanions in Olefin Synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. 10.1021/ja01468a042. [DOI] [Google Scholar]
- Still W. C.; Gennari C. Direct Synthesis of Z-Unsaturated Esters. A Useful Modification of the Horner-Emmons Olefination. Tetrahedron Lett. 1983, 24, 4405–4408. 10.1016/S0040-4039(00)85909-2. [DOI] [Google Scholar]
- Corey E. J.; Kwiatkowski G. T. A New Synthesis of Olefins from Carbonyl Compounds and Phoshonic Acid Bis Amides. J. Am. Chem. Soc. 1968, 90, 6816–6821. 10.1021/ja01026a045. [DOI] [Google Scholar]
- Ando K. Practical Synthesis of Z-Unsaturated Esters by Using a New Horner-Emmons Reagent, Ethyl Diphenylphosphonoacetate. Tetrahedron Lett. 1995, 36, 4105–4108. 10.1016/0040-4039(95)00726-S. [DOI] [Google Scholar]
- Ando K. Highly Selective Synthesis of Z -Unsaturated Esters by Using New Horner–Emmons Reagents, Ethyl (Diarylphosphono)Acetates. J. Org. Chem. 1997, 62, 1934–1939. 10.1021/jo970057c. [DOI] [PubMed] [Google Scholar]
- Kang S. H.; Kang S. Y.; Kim C. M.; Choi H. W.; Jun H. S.; Lee B. M.; Park C. M.; Jeong J. W. Total Synthesis of Natural (+)-Lasonolide A. Angew. Chem., Int. Ed. 2003, 42, 4779–4782. 10.1002/anie.200352016. [DOI] [PubMed] [Google Scholar]
- Sanchez C. C.; Keck G. E. Total Synthesis of (+)-Dactylolide. Org. Lett. 2005, 7, 3053–3056. 10.1021/ol051040g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A.; Itsuno S.; Nakahama S.; Yamazaki N. Asymmetric Reduction of Aromatic Ketones with Chiral Alkoxy-Amine-Borane Complexes. J. Chem. Soc., Chem. Commun. 1981, 315–317. 10.1039/c39810000315. [DOI] [Google Scholar]
- Itsuno S.; Ito K.; Hirao A.; Nakahama S. Asymmetric Reduction of Aromatic Ketones with the Reagent Prepared from (S)-(−)-2-Amino-3-Methyl-1,1-Diphenylbutan-1-Ol and Borane. J. Chem. Soc., Chem. Commun. 1983, 469–470. 10.1039/C39830000469. [DOI] [Google Scholar]
- Corey E. J.; Bakshi R. K.; Shibata S. Highly Enantioselective Borane Reduction of Ketones Catalyzed by Chiral Oxazaborolidines. Mechanism and Synthetic Implications. J. Am. Chem. Soc. 1987, 109, 5551–5553. 10.1021/ja00252a056. [DOI] [Google Scholar]
- Corey E. J.; Bakshi R. K.; Shibata S.; Chen C. P.; Singh V. K. A Stable and Easily Prepared Catalyst for the Enantioselective Reduction of Ketones: Applications to Multistep Syntheses. J. Am. Chem. Soc. 1987, 109, 7925–7926. 10.1021/ja00259a075. [DOI] [Google Scholar]
- Gutekunst W. R.; Baran P. S. C–H Functionalization Logic in Total Synthesis. Chem. Soc. Rev. 2011, 40, 1976. 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- McMurray L.; O’Hara F.; Gaunt M. J. Recent Developments in Natural Product Synthesis Using Metal-Catalysed C–H Bond Functionalisation. Chem. Soc. Rev. 2011, 40, 1885. 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- Fischer D. F.; Sarpong R. Total Synthesis of (+)-Complanadine A Using an Iridium-Catalyzed Pyridine C–H Functionalization. J. Am. Chem. Soc. 2010, 132, 5926–5927. 10.1021/ja101893b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst W. R.; Baran P. S. Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation. J. Am. Chem. Soc. 2011, 133, 19076–19079. 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C. P.; Maimone T. J. C–H Bond Arylation in the Synthesis of Aryltetralin Lignans: A Short Total Synthesis of Podophyllotoxin. Angew. Chem., Int. Ed. 2014, 53, 3115–3119. 10.1002/anie.201311112. [DOI] [PubMed] [Google Scholar]
- O’ Donovan D. H.; Aillard P.; Berger M.; De La Torre A.; Petkova D.; Knittl-Frank C.; Geerdink D.; Kaiser M.; Maulide N. C–H Activation Enables a Concise Total Synthesis of Quinine and Analogues with Enhanced Antimalarial Activity. Angew. Chem., Int. Ed. 2018, 57, 10737–10741. 10.1002/anie.201804551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D. A.; Katz J. L.; West T. R. Synthesis of Diaryl Ethers through the Copper-Promoted Arylation of Phenols with Arylboronic Acids. An Expedient Synthesis of Thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. 10.1016/S0040-4039(98)00502-4. [DOI] [Google Scholar]
- Evans D. A.; Dinsmore C. J.; Ratz A. M.; Evrard D. A.; Barrow J. C. Synthesis and Conformational Properties of the M(4–6)(5–7) Bicyclic Tetrapeptide Common to the Vancomycin Antibiotics. J. Am. Chem. Soc. 1997, 119, 3417–3418. 10.1021/ja964419u. [DOI] [Google Scholar]
- Peters D. S.; Romesberg F. E.; Baran P. S. Scalable Access to Arylomycins via C-H Functionalization Logic. J. Am. Chem. Soc. 2018, 140, 2072–2075. 10.1021/jacs.8b00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Snyder S. A.; Montagnon T.; Vassilikogiannakis G. The Diels-Alder Reaction in Total Synthesis K. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. . [DOI] [PubMed] [Google Scholar]
- Stork G.; Nakamura E. A Simplified Total Synthesis of Cytochalasins via an Intramolecular Diels-Alder Reaction. J. Am. Chem. Soc. 1983, 105, 5510–5512. 10.1021/ja00354a072. [DOI] [Google Scholar]
- Takeda K.; Ierarashi Y.; Okazaki K.; Yoshii E.; Yamaguchi K. An Intramolecular Diels-Alder Approach to the Synthesis of Chlorothricolide. Synthesis of (±)-24–0-Methylchlorothricolide. J. Org. Chem. 1990, 55, 3431–3434. 10.1021/jo00298a003. [DOI] [Google Scholar]
- McCauley J. A.; Nagasawa K.; Lander P. A.; Mischke S. G.; Semones M. A.; Kishi Y. Total Synthesis of Pinnatoxin A. J. Am. Chem. Soc. 1998, 120, 7647–7648. 10.1021/ja981257o. [DOI] [Google Scholar]
- Zapf C. W.; Harrison B. A.; Drahl C.; Sorensen E. J. A Diels–Alder Macrocyclization Enables an Efficient Asymmetric Synthesis of the Antibacterial Natural Product Abyssomicin C. Angew. Chem., Int. Ed. 2005, 44, 6533–6537. 10.1002/anie.200502119. [DOI] [PubMed] [Google Scholar]
- Kingsbury J. S.; Corey E. J. Enantioselective Total Synthesis of Isoedunol and β-Araneosene Featuring Unconventional Strategy and Methodology. J. Am. Chem. Soc. 2005, 127, 13813–13815. 10.1021/ja055137+. [DOI] [PubMed] [Google Scholar]
- Baran P. S.; Burns N. Z. Total Synthesis of (±)-Haouamine A. J. Am. Chem. Soc. 2006, 128, 3908–3909. 10.1021/ja0602997. [DOI] [PubMed] [Google Scholar]
- Zhao P.; Beaudry C. M. Total Synthesis of (±)-Cavicularin: Control of Pyrone Diels-Alder Regiochemistry Using Isomeric Vinyl Sulfones. Org. Lett. 2013, 15, 402–405. 10.1021/ol303390a. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Tang Y.; Wang J. Total Synthesis of Sporolide B. Angew. Chem., Int. Ed. 2009, 48, 3449–3453. 10.1002/anie.200900264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood J. L.; Porco J. A.; Taunton J.; Lee A. Y.; Clardy J.; Schreiber S. L. Application of the Allylic Diazene Rearrangement: Synthesis of the Enediyne-Bridged Tricyclic Core of Dynemicin A. J. Am. Chem. Soc. 1992, 114, 5898–5900. 10.1021/ja00040a085. [DOI] [Google Scholar]
- Taunton J.; Wood J. L.; Schreiber S. L. Total Syntheses of Di- and Tri-O-Methyl Dynemicin A Methyl Esters. J. Am. Chem. Soc. 1993, 115, 10378–10379. 10.1021/ja00075a071. [DOI] [Google Scholar]
- Evans D. A.; Ripin D. H. B.; Halstead D. P.; Campos K. R. Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide. J. Am. Chem. Soc. 1999, 121, 6816–6826. 10.1021/ja990789h. [DOI] [Google Scholar]
- Choe H.; Pham T. T.; Lee J. Y.; Latif M.; Park H.; Kang Y. K.; Lee J. Remote Stereoinductive Intramolecular Nitrile Oxide Cycloaddition: Asymmetric Total Synthesis and Structure Revision of (−)-11β-Hydroxycurvularin. J. Org. Chem. 2016, 81, 2612–2617. 10.1021/acs.joc.5b02760. [DOI] [PubMed] [Google Scholar]
- Cole K. P.; Hsung R. P. Unique Structural Topology and Reactivities of the ABD Tricycle in Phomactin A. Chem. Commun. 2005, 46, 5784–5786. 10.1039/b511338e. [DOI] [PubMed] [Google Scholar]
- Cole K. P.; Hsung R. P. Intramolecular Formal Oxa-[3 + 3] Cycloaddition Approach to the ABD System of Phomactin A. Org. Lett. 2003, 5, 4843–4846. 10.1021/ol030115i. [DOI] [PubMed] [Google Scholar]
- Liu W.; Ren Z.; Bosse A. T.; Liao K.; Goldstein E. L.; Bacsa J.; Musaev D. G.; Stoltz B. M.; Davies H. M. L. Catalyst-Controlled Selective Functionalization of Unactivated C-H Bonds in the Presence of Electronically Activated C-H Bonds. J. Am. Chem. Soc. 2018, 140, 12247–12255. 10.1021/jacs.8b07534. [DOI] [PubMed] [Google Scholar]
- López-Valdez L. G.; Zuleta-Prada H.; Reyes-Trejo B.; Cuevas-Yañez E. Synthesis of 10-Membered and Larger Rings via Free Radical Methods. Tetrahedron 2018, 74, 1581–1612. 10.1016/j.tet.2018.01.056. [DOI] [Google Scholar]
- Suzuki K.; Tamiya M.. Pinacol Coupling Reactions. In Compr. Org. Synth., 2nd Ed.; Elsevier Ltd., 2014; Vol. 3, pp 580–620. [Google Scholar]
- McMurry J. E.; Dushin R. G. Total Synthesis of (±)-Crassin by Titanium-Induced Pinacol Coupling. J. Am. Chem. Soc. 1989, 111, 8928–8929. 10.1021/ja00206a031. [DOI] [Google Scholar]
- Takeda T.; Tsubouchi A.. The McMurry Coupling and Related Reactions. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, 2013; pp 1–470. [Google Scholar]
- Nazare M.; Waldmann H. Synthesis of the (9S,18R) Diastereomer of Cyclamenol A. Angew. Chem., Int. Ed. 2000, 39, 1125–1128. . [DOI] [PubMed] [Google Scholar]
- Gritsch P. J.; Leitner C.; Pfaffenbach M.; Gaich T. The Witkop Cyclization: A Photoinduced C-H Activation of the Indole System. Angew. Chem., Int. Ed. 2014, 53, 1208–1217. 10.1002/anie.201307391. [DOI] [PubMed] [Google Scholar]
- Vermes B.; Keserû G. M.; Mezey-Vándor G.; Nógrádix M.; Tóth G. The Synthesis of Garugamblin-1. Tetrahedron 1993, 49, 4893–4900. 10.1016/S0040-4020(01)80406-3. [DOI] [Google Scholar]
- Feldman K. S.; Berven H. M.; Romanelli A. L.; Parvez M. Macrocyclization through Radical-Mediated Vinylcyclopropane/Alkene Condensation. Facile Entry into the Brefeldin Ring System. J. Org. Chem. 1993, 58, 6851–6856. 10.1021/jo00076a056. [DOI] [Google Scholar]
- Balog A.; Meng D.; Kamenecka T.; Bertinato P.; Su D.; Sorensen E. J.; Danishefsky S. J. Total Synthesis of (−)-Epothilone A. Angew. Chem., Int. Ed. Engl. 1996, 35, 2801–2803. 10.1002/anie.199628011. [DOI] [Google Scholar]
- Meng D.; Bertinato P.; Balog A.; Su D.-S.; Kamenecka T.; Sorensen E. J.; Danishefsky S. J. Total Syntheses of Epothilones A and B. J. Am. Chem. Soc. 1997, 119, 10073–10092. 10.1021/ja971946k. [DOI] [Google Scholar]
- Hayward C. M.; Yohannes D.; Danishefsky S. J. Total Synthesis of Rapamycin via a Novel Titanium-Mediated Aldol Macrocyclization Reaction. J. Am. Chem. Soc. 1993, 115, 9345–9346. 10.1021/ja00073a083. [DOI] [Google Scholar]
- Magnus P.; Miknis G. F.; Press N. J.; Grandjean D.; Taylor G. M.; Harling J. Synthesis of a Protected (±)-Calicheamicinone Derivative by Sequential Introduction of Functionality into the Bicyclo[7.3.1]Enediyne Core Structure. J. Am. Chem. Soc. 1997, 119, 6739–6748. 10.1021/ja970743t. [DOI] [Google Scholar]
- Baba A.; Yasuda M.; Nishimoto Y.. Zinc Enolates: The Reformatsky and Blaise Reactions; Elsevier Ltd., 2014; Vol. 2. [Google Scholar]
- Yang S.; Xi Y.; Chen J. H.; Yang Z. Asymmetric Total Synthesis of (−)-Cebulactam A1. Org. Chem. Front. 2014, 1, 91–99. 10.1039/c3qo00036b. [DOI] [Google Scholar]
- Lade J. J.; Pardeshi S. D.; Vadagaonkar K. S.; Murugan K.; Chaskar A. C. The Remarkable Journey of Catalysts from Stoichiometric to Catalytic Quantity for Allyltrimethylsilane Inspired Allylation of Acetals, Ketals, Aldehydes and Ketones. RSC Adv. 2017, 7, 8011–8033. 10.1039/C6RA27813B. [DOI] [Google Scholar]
- Han X.; Peh G.; Floreancig P. E. Prins-Type Cyclization Reactions in Natural Product Synthesis. Eur. J. Org. Chem. 2013, 2013, 1193–1208. 10.1002/ejoc.201201557. [DOI] [Google Scholar]
- Wender P. A.; DeChristopher B. A.; Schrier A. J. Efficient Synthetic Access to a New Family of Highly Potent Bryostatin Analogues via a Prins-Driven Macrocyclization Strategy. J. Am. Chem. Soc. 2008, 130, 6658–6659. 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Hardman C. T.; Ho S.; Jeffreys M. S.; Maclaren J. K.; Quiroz R. V.; Ryckbosch S. M.; Shimizu A. J.; Sloane J. L.; Stevens M. C. Scalable Synthesis of Bryostatin 1 and Analogs, Adjuvant Leads against Latent HIV. Science 2017, 358, 218–223. 10.1126/science.aan7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.; Lu J.; Song Z. Recent Efforts to Construct the B-Ring of Bryostatins. Chem. Commun. 2013, 49, 10211–10220. 10.1039/c3cc45947k. [DOI] [PubMed] [Google Scholar]
- Gesinski M. R.; Rychnovsky S. D. Total Synthesis of the Cyanolide A Aglycon. J. Am. Chem. Soc. 2011, 133, 9727–9729. 10.1021/ja204228q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K.; Moussa Z.; Lee C.; Romo D. Total Synthesis of the Spirocyclic Imine Marine Toxin (−)-Gymnodimine and an Unnatural C4-Epimer. J. Am. Chem. Soc. 2011, 133, 19844–19856. 10.1021/ja207385y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseltine J. N.; Cabal M. P.; Mantlo N. B.; Iwasawa N.; Yamashita D. S.; Coleman R. S.; Danishefsky S. J.; Schulte G. K. Total Synthesis of Calicheamicinone: New Arrangements for Actuation of the Reductive Cycloaromatization of Aglycon Congeners. J. Am. Chem. Soc. 1991, 113, 3850–3866. 10.1021/ja00010a030. [DOI] [Google Scholar]
- Hitchcock S. A.; Boyer S. H.; Chu-Moyer M. Y.; Olson S. H.; Danishefsky S. J. A Convergent Total Synthesis of Calicheamicin γI1. Angew. Chem., Int. Ed. Engl. 1994, 33, 858–862. 10.1002/anie.199408581. [DOI] [Google Scholar]
- Myers A. G.; Liang J.; Hammond M.; Harrington P. M.; Wu Y.; Kuo E. Y. Total Synthesis of (+)-Neocarzinostatin Chromophore. J. Am. Chem. Soc. 1998, 120, 5319–5320. 10.1021/ja980588y. [DOI] [Google Scholar]
- Groneberg R. D.; Miyazaki T.; Stylianides N. A.; Schulze T. J.; Stahl W.; Schreiner E. P.; Suzuki T.; Iwabuchi Y.; Smith A. L.; Nicolaou K. C. Total Synthesis of Calicheamicin γI1. 1. Synthesis of the Oligosaccharide Fragment. J. Am. Chem. Soc. 1993, 115, 7593–7611. 10.1021/ja00070a004. [DOI] [Google Scholar]
- Fürstner A. Carbon-Carbon Bond Formations Involving Organochromium(III) Reagents. Chem. Rev. 1999, 99, 991–1045. 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]
- Mi B.; Maleczka R. E. J. A Nozaki-Hiyama-Kishi Ni(II)/Cr(II) Coupling Approach to the Phomactins. Org. Lett. 2001, 3, 1491–1494. 10.1021/ol015807q. [DOI] [PubMed] [Google Scholar]
- Ciesielski J.; Cariou K.; Frontier A. J. A Macrocyclic β-Iodoallenolate Intermediate Is Key: Synthesis of the ABD Core of Phomactin A. Org. Lett. 2012, 14, 4082–4085. 10.1021/ol3017116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K. T.; Chng S. S.; Cheng H. S.; Loh T. P. Development of a Highly α-Regioselective Metal-Mediated Allylation Reaction in Aqueous Media: New Mechanistic Proposal for the Origin of α-Homoallylic Alcohols. J. Am. Chem. Soc. 2003, 125, 2958–2963. 10.1021/ja029276s. [DOI] [PubMed] [Google Scholar]
- Ma X.; Bolte B.; Banwell M. G.; Willis A. C. Total Syntheses of the Resorcylic Acid Lactones Paecilomycin F and Cochliomycin C Using an Intramolecular Loh-Type α-Allylation Reaction for Macrolide Formation. Org. Lett. 2016, 18, 4226–4229. 10.1021/acs.orglett.6b01963. [DOI] [PubMed] [Google Scholar]
- Colby E. A.; Jamison T. F. A Comparative Analysis of the Total Syntheses of the Amphidinolide T Natural Products. Org. Biomol. Chem. 2005, 3, 2675–2684. 10.1039/b507315b. [DOI] [PubMed] [Google Scholar]
- Chan J.; Jamison T. F. Enantioselective Synthesis of (−)-Terpestacin and Structural Revision of Siccanol Using Catalytic Stereoselective Fragment Couplings and Macrocyclizations. J. Am. Chem. Soc. 2004, 126, 10682–10691. 10.1021/ja0470968. [DOI] [PubMed] [Google Scholar]
- Montgomery J. Nickel-Catalyzed Cyclizations, Couplings, and Cycloadditions Involving Three Reactive Components. Acc. Chem. Res. 2000, 33, 467–473. 10.1021/ar990095d. [DOI] [PubMed] [Google Scholar]
- Chan D. M. T.; Monaco K. L.; Wang R.-P.; Winters M. P. New N- and O-Arylations with Phenylboronic Acids and Cupric Acetate. Tetrahedron Lett. 1998, 39, 2933–2936. 10.1016/S0040-4039(98)00503-6. [DOI] [Google Scholar]
- Evans D. A.; Katz J. L.; West T. R. Synthesis of Diaryl Ethers through the Copper-Promoted Arylation of Phenols with Arylboronic Acids. An Expedient Synthesis of Thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. 10.1016/S0040-4039(98)00502-4. [DOI] [Google Scholar]
- Lam P. Y. S.; Clark C. G.; Saubern S.; Adams J.; Winters M. P.; Chan D. M. T.; Combs A. New Aryl/Heteroaryl C—N Bond Cross-Coupling Reactions via Arylboronic Acid/Cupric Acetate Arylation. Tetrahedron Lett. 1998, 39, 2941–2944. 10.1016/S0040-4039(98)00504-8. [DOI] [Google Scholar]
- Boger D. L.; Zhou J. Total Synthesis of (+)-Piperazinomycin. J. Am. Chem. Soc. 1993, 115, 11426–11433. 10.1021/ja00077a047. [DOI] [Google Scholar]
- Uchiro H.; Kato R.; Arai Y.; Hasegawa M.; Kobayakawa Y. Total Synthesis of Hirsutellone B via Ullmann-Type Direct 13-Membered Macrocyclization. Org. Lett. 2011, 13, 6268–6271. 10.1021/ol202748e. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Sarlah D.; Wu T. R.; Zhan W. Total Synthesis of Hirsutellone B. Angew. Chem., Int. Ed. 2009, 48, 6870–6874. 10.1002/anie.200903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Léséleuc M.; Godin É.; Parisien-Collette S.; Lévesque A.; Collins S. K. Catalytic Macrocyclization Strategies Using Continuous Flow: Formal Total Synthesis of Ivorenolide A. J. Org. Chem. 2016, 81, 6750–6756. 10.1021/acs.joc.6b01500. [DOI] [PubMed] [Google Scholar]
- Kuroda Y.; Nicacio K. J.; da Silva I. A. Jr; Leger P. R.; Chang S.; Gubiani J. R.; Deflon V. M.; Nagashima N.; Rode A.; Blackford K.; Ferreira A. G.; Sette L. D.; Williams D. E.; Andersen R. J.; Jancar S.; Berlinck R. G. S.; Sarpong R. Isolation, Synthesis and Bioactivity Studies of Phomactin Terpenoids. Nat. Chem. 2018, 10, 938–945. 10.1038/s41557-018-0084-x. [DOI] [PubMed] [Google Scholar]
- Almalki F. A.; Harrowven D. C. A Corey–Seebach Macrocyclisation Strategy for the Synthesis of Riccardin C and an Unnatural Macrocyclic Bis(Bibenzyl) Analogue. Eur. J. Org. Chem. 2016, 2016, 5738–5746. 10.1002/ejoc.201601179. [DOI] [Google Scholar]