

Abstract
Approximately 50% of infants with biliary atresia (BA) undergoing Kasai portoenterostomy show survival with native liver (SNL) at age 2 years. Predictors of disease progression after age 2 years are unknown, despite estimates of 20%‐30% undergoing liver transplant (LT) between age 2 and 18 years. We sought to address this knowledge gap by developing prognostic models in participants of the multicenter prospective National Institutes of Health‐supported Childhood Liver Disease Research Network. We extracted 14 clinical and biochemical variables at age 2 years to develop two models for future outcomes: 1) LT or death (LTD) and 2) first sentinel event (SE), either new onset ascites, hepatopulmonary syndrome (HPS), or gastrointestinal (GI) bleed. A total of 240 participants, enrolled between 2004 and 2017, were followed until a median age of 5.1 years (range, 2.0‐13.3 years). Of these participants, 38 underwent LT (n = 37) or death (n = 1); cumulative incidence, 23.7% (95% confidence interval [CI], 16.2%‐32.0%). Twenty‐seven experienced either new‐onset ascites (n = 13), HPS (n = 1), or GI bleed (n = 14). One participant had ascites and GI bleed concurrently; cumulative incidence, 21.5% (95% CI, 14.2%‐29.8%) by age 10 years. The Cox proportional hazard model predicted risk of LTD, using total bilirubin, albumin, platelet count, and history of either ascites or cholangitis (BA LTD model), with a C‐index of 0.88 (range, 0.86‐0.89). A cause‐specific hazard competing risk model predicted SE using platelet count and gamma glutamyltransferase levels (BA SE model) with a C‐index of 0.81 (range, 0.80‐0.84). Internal model validity was assessed using Harrell’s C‐index with cross‐validation. Conclusion: Stratification using these models identified risk of poor outcomes in patients with BA SNL after age 2 years. The models may identify those who would benefit from enhanced clinical surveillance and prioritization in clinical trials.
Abbreviations
- ALT
alanine aminotransferase
- APRI
aspartate aminotransferase to platelet ratio index
- AST
aspartate aminotransferase
- BA
biliary atresia
- BASIC
Biliary Atresia Study in Infants and Children
- ChiLDReN
Childhood Liver Disease Research Network
- CI
confidence interval
- GGT
gamma‐glutamyltransferase
- GI
gastrointestinal
- HPS
hepatopulmonary syndrome
- HR
hazard ratio
- INR
international normalized ratio
- IQR
interquartile range
- KPE
Kasai portoenterostomy
- LT
liver transplant
- LTD
liver transplant or death
- PROBE
A Prospective Database of Infants with Cholestasis
- SE
sentinel event
- SNL
survival with native liver
- TB
total bilirubin
Biliary atresia (BA) is a congenital, idiopathic, obliterative, neonatal cholangiopathy that rapidly leads to end‐stage liver disease if untreated. There is no medical treatment for BA. Even after surgical intervention with Kasai portoenterostomy (KPE), where the fibrotic biliary remnant is excised and drainage of the biliary tree attempted with Roux‐en‐Y, almost 50% of patients with BA will undergo liver transplant (LT) before 2 years of age.( 1 , 2 , 3 )
Efforts at avoiding early LT have targeted early identification and diagnosis, timing of KPE before 45 days of age, and centralization of surgery at experienced centers.( 4 , 5 , 6 , 7 ) Interventions immediately following KPE have been attempted but without significant impact on outcome.( 8 , 9 )
A “draining KPE” with improved bilirubin and survival with native liver (SNL) status at age 2 years is considered a successful outcome.( 10 , 11 ) However, progression of disease and liver‐related sequelae persist. Cross‐sectional studies of 5‐ to 20‐year outcomes after KPE reveal that patients avoiding early LT still experience complications of chronic liver disease, including cholangitis, gastrointestinal (GI) bleeding, and ascites during childhood and early adulthood.( 5 , 12 , 13 , 14 ) Reductions in SNL to as low as 18%‐26% in the 20‐year follow‐up after KPE have been reported.( 15 , 16 ) Factors that predict progression of liver disease in patients with BA SNL remain unknown.
To address this knowledge gap, we used data from participants with BA enrolled in the National Institutes of Health‐funded multicenter Childhood Liver Disease Research Network (ChiLDReN) between 2004 and 2017. The aim of this study was to develop prognostic models of outcomes in BA after age 2 years using readily available biochemical and clinical parameters.
Patients and Methods
Study Population
Two prospective registries of infants and children with BA are included in ChiLDReN: A Prospective Database of Infants with Cholestasis (PROBE; clinicaltrials.gov NCT00061828) and the Biliary Atresia Study in Infants and Children (BASIC; clinicaltrials.gov NCT00345553). PROBE has enrolled infants ≤180 days of age with neonatal cholestasis since June 1, 2004, as described.( 14 , 17 ) Baseline data and biospecimens are collected at the time of KPE; follow‐up visits occur periodically until 18 months of age and then annually from age 2 to 20 years, unless LT, death, or loss to follow‐up occur. Since May 1, 2006, BASIC has enrolled participants with BA who are over 6 months of age, with BA confirmed after review of biopsy, radiographic, and surgical reports, as described.( 14 ) Baseline data are collected at enrollment and annually until age 20 years, LT, or loss to follow‐up.
Institutional Review Board approval was obtained from each ChiLDReN site and the Data Coordinating Center; parents or legal guardians of infants provided written informed consent. The current study analyzed data from participants with BA with KPE in PROBE and BASIC with enrollment at or before age 2 years, with SNL at age 2, and follow‐up data collected thereafter.
Outcome Variables
We analyzed two time‐to‐event outcomes: (1) time from age 2 years to LT or death (LTD) and (2) time from age 2 years to a composite outcome of a sentinel event (SE), defined as the first report of ascites, GI bleeding, or hepatopulmonary syndrome (HPS), whichever happened first.
LTDs are recorded on data collection forms at the time of the event. SE data are collected at protocolized follow‐up visits. Study definitions of SEs are standardized within the PROBE and BASIC protocols. The presence of ascites was determined by either physical exam or patient receipt of diuretics. HPS required documentation of hypoxia plus evidence of intrapulmonary shunting by bubble contrast echocardiography with agitated saline.( 18 ) GI bleeding was defined as hematemesis, hematochezia, or melena with endoscopic documentation of actively bleeding esophageal or gastric varices or visualization of esophageal varices in the absence of any other identifiable cause of hemorrhage.
Candidate Predictors
Candidate predictors analyzed for the first outcome of LTD included 14 variables: platelet count (103/mm3), total bilirubin (TB, mg/dL), aspartate aminotransferase (AST, U/L), alanine aminotransferase (ALT, U/L), prothrombin time international normalized ratio (INR), albumin (g/dL), gamma‐glutamyltransferase (GGT, U/L), AST to platelet ratio index (APRI), height failure, weight failure, and history of ascites, cholangitis, GI bleed, and splenomegaly.
Baseline laboratory and anthropologic measurement values were obtained from the closest visit within 6 months before or after 2 years of age. Medical history variables were reported before age 2 years. Height and weight failure were defined as age‐ and sex‐adjusted z score <−2, calculated using SAS macros provided by the Centers for Disease Control and Prevention (https://www.cdc.gov/nccdphp/dnpao/growthcharts/resources/sas.htm). Splenomegaly was defined as palpable >2 cm below the costal margin. Diagnosis of cholangitis required presence of fever of >38°C without other obvious clinical source of infection as well as a combination of clinical findings. Clinical findings included new onset of acholic stools; right upper quadrant pain or tenderness; both elevation of direct bilirubin by 25% and at least >1 mg/dL above previous baseline; rise in 2 or more of AST, ALT, and alkaline phosphatase; GGT to 1.5 times the upper limit of normal or >25% above baseline values if previously elevated; and clinical and biochemical improvement after treatment with antibiotics. History of ascites before 2 years of age was defined as described above and limited to reports after 6 months of age.
For developing the prognostic model of SE, the list of candidate predictors included all the above predictors except for history of GI bleed or ascites.
Statistical Analysis
We used Cox proportional hazard models and cause‐specific hazard competing risk models for modeling the risk of LTD and SE, respectively.( 19 ) In the model for SE, LT and death were treated as competing events for SE. Time 0 (baseline) is 2 years of age for both models. Those participants with an SE before age 2 years were excluded from the SE model development because the focus was on new onset after age 2.
Before modeling, distributions of all predictors were examined for extreme values. We used P‐spline functions to explore the potential nonlinear effect of continuous predictors. Covariates with nonlinear effect were transformed to obtain the best model fit. A random forest survival model and a random forest competing risk model were used to explore potential interactions between candidate predictors.( 20 , 21 ) Twenty‐two multiple imputed data sets were generated to fill in missing covariate values, using a sequence of regression models implemented in IVEWARE, incorporating laboratory measures from the visit closest to 1 year of age.( 22 )
To select the best prediction model, we used a stepwise selection procedure, with entry criteria P < 0.10 and stay criteria P < 0.05 on each of the 22 imputed data sets. The selected variables were ordered according to how frequently they were included in the 22 final models. We then selected our final model based on the combined results of the 22 imputed data sets, using Rubin’s rule and the algorithm described as follows.( 23 ) We added predictors into the model one at a time in the order of frequency obtained above, starting with the variable with the highest frequency. We stopped this process when the newly added variable had a stay P > 0.05.
In addition to calculating the apparent Harrell’s C‐index, cross‐validation assessed internal validity of the prognostic models.( 24 ) For each imputed data set, a 5‐fold cross‐validation was conducted by partitioning the study sample into five equal‐size subsamples. Of these, four were used as the training set and one was used as the test set. Median values of Harrell’s C‐index for the tests set across all imputed data sets were calculated.
Results
Study Population
Between June 2004 and August 2017, 1,151 participants with BA with KPE were enrolled (543 in PROBE; 608 in BASIC) (Fig. 1). Of these, we identified a final cohort of 240 participants with BA (196 from PROBE; 44 from BASIC) enrolled before age 2 years, had an age 2‐year study visit, and had follow‐up after age 2 years. The median age at last observation was 5.1 years (range, 2.0‐13.3 years).
FIG. 1.
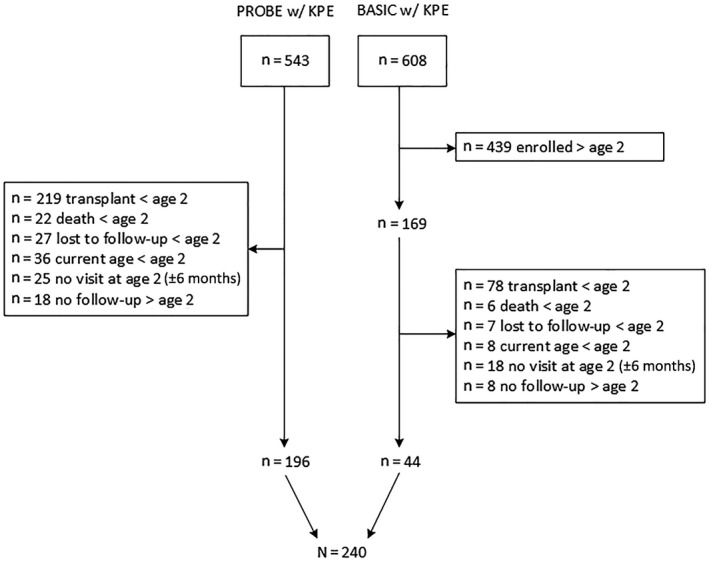
Flow diagram showing identification and inclusion of ChiLDReN participants with BA SNL at age 2 years.
Demographic and baseline laboratory/clinical characteristics of the cohort are listed in Table 1. The median age at KPE was 59 days (interquartile range [IQR], 42‐74 days). The median age at enrollment into PROBE and BASIC was 1.8 months (range, 0.5‐5.0 months) and 16.0 months (range, 4.5‐23.9 months), respectively. None of the included participants in BASIC had been referred to a ChiLDReN site for LT evaluation.
Table 1.
Demographics and baseline characteristics of participants with BA SNL at age 2 years
| Variable | BASIC (N = 44) | PROBE (N = 196) | Total (N = 240) | ||||
|---|---|---|---|---|---|---|---|
| N | n (%) or Median (IQR) | N | n (%) or Median (IQR) | N | n (%) or Median (IQR) | ||
| Demographics | |||||||
| Female | 44 | 28 (63.6%) | 196 | 104 (53.1%) | 240 | 132 (55%) | |
| Race | White | 43 | 21 (48.8%) | 195 | 108 (55.4%) | 238 | 129 (54.2%) |
| Black | 43 | 7 (16.3%) | 195 | 29 (14.9%) | 238 | 36 (15.1%) | |
| Other | 43 | 15 (34.9%) | 195 | 58 (29.7%) | 238 | 73 (30.7%) | |
| Hispanic ethnicity | 42 | 5 (11.9%) | 196 | 48 (24.5%) | 238 | 53 (22.3%) | |
| Age at Kasai (days) | 44 | 61 (40, 76) | 196 | 57 (43, 74) | 240 | 59 (42, 74) | |
| Associated anomalies | |||||||
| Asplenia/polysplenia | 37 | 1 (2.7%) | 195 | 8 (4.1%) | 232 | 9 (3.9%) | |
| Cardiovascular anomaly | 37 | 5 (13.5%) | 195 | 30 (15.4%) | 232 | 35 (15.1%) | |
| Gastrointestinal anomaly | 37 | 8 (21.6%) | 195 | 18 (9.2%) | 232 | 26 (11.2%) | |
| Clinical features | |||||||
| History of GI bleed | 44 | 1 (2.3%) | 196 | 10 (5.1%) | 240 | 11 (4.6%) | |
| History of cholangitis | 44 | 23 (52.3%) | 196 | 81 (41.3%) | 240 | 104 (43.3%) | |
| History of ascites | 44 | 8 (18.2%) | 196 | 34 (17.3%) | 240 | 42 (17.5%) | |
| History of splenomegaly | 44 | 21 (47.7%) | 196 | 117 (59.7%) | 240 | 138 (57.5%) | |
| Weight growth failure | 35 | 4 (11.4%) | 178 | 6 (3.4%) | 213 | 10 (4.7%) | |
| Height growth failure | 35 | 0 (0%) | 179 | 13 (7.3%) | 214 | 13 (6.1%) | |
| Antibiotics | 27 | 11 (40.7%) | 194 | 58 (29.9%) | 221 | 69 (31.2%) | |
| Ursodeoxycholic acid | 27 | 17 (63%) | 189 | 123 (65.1%) | 216 | 140 (64.8%) | |
| Laboratory features | |||||||
| TB (mg/dL) | 41 | 0.5 (0.3, 1.3) | 186 | 0.5 (0.3, 0.9) | 227 | 0.5 (0.3, 0.9) | |
| GGT (U/L) | 35 | 127 (50, 239) | 152 | 134 (43, 332) | 187 | 130 (43, 327) | |
| Platelet count (×103/µL) | 38 | 189 (132, 246) | 179 | 206 (134, 280) | 217 | 205 (134, 271) | |
| APRI | 36 | 1.1 (0.7, 2.7) | 176 | 1.2 (0.6, 2.1) | 212 | 1.2 (0.7, 2.2) | |
| AST (U/L) | 40 | 80 (56, 138) | 189 | 88 (56, 159) | 229 | 84 (56, 154) | |
| ALT (U/L) | 40 | 65 (39, 117) | 189 | 85 (45, 163) | 229 | 83 (42, 155) | |
| Albumin (g/dL) | 39 | 4.1 (3.8, 4.4) | 187 | 4.1 (3.8, 4.4) | 226 | 4.1 (3.8, 4.4) | |
| INR | 30 | 1.0 (1.0, 1.2) | 161 | 1.0 (1.0, 1.1) | 191 | 1.0 (1.0, 1.1) | |
Polysplenia/asplenia was documented in 3.9% of cases, cardiac anomaly in 15.1%, and GI anomalies in 11.2%. A history of cholangitis, ascites, and GI bleed was documented in 43.3%, 17.5%, and 4.6%, respectively. Baseline laboratory values were available from 187 to 229 individuals (depending on the missingness of each collected variable) and notable for a median TB of 0.5 mg/dL (IQR, 0.3‐0.9 mg/dL), platelet count of 205 103/mm3 (IQR, 134‐271 103/mm3), GGT of 130 U/L (IQR, 43‐327 U/L), ALT of 83 U/L (IQR, 42‐155 U/L), AST of 84 U/L (IQR, 56‐154 U/L), and APRI of 1.2 (IQR, 0.7‐2.2) (Table 1).
Forty‐eight participants had a reported SE before age 2 years (n = 42 ascites, n = 1 HPS, n = 11 GI bleed, n = 6 with more than one SE) and were excluded from SE modeling, resulting in a total of 192 participants in ChiLDReN used in the SE model. Demographic and baseline characteristics of participants in the SE model compared with those excluded due to a history of SE are listed in Supporting Table S1.
Risk Factors and a Prognostic Model for LTD
LTD was reported in 38 (37 LT and 1 death) study participants, with a cumulative incidence of 23.7% (95% confidence interval (CI), 16.2%‐32.0%) by 10 years of age (Fig. 2A).
FIG. 2.
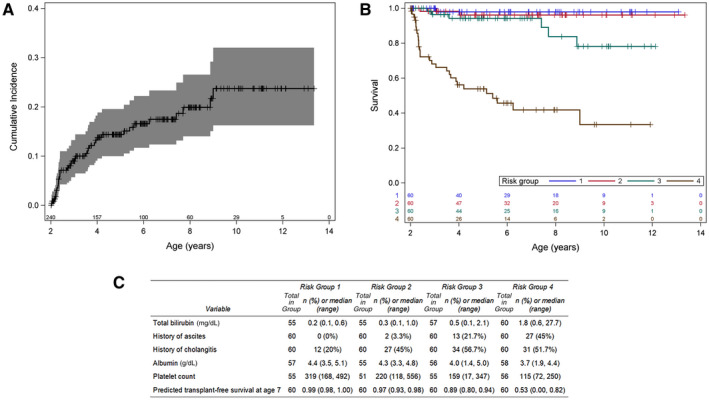
Incidence and risk for the LTD model. (A) Cumulative incidence of LTD among the 240 study participants. (B) Kaplan‐Meier curves for LT‐free survival stratified by quartile of risk score. Stratification of participants shows a high‐risk group (group 4; brown) and a medium‐risk group (group 3; green), with the remaining two quartiles showing a similar lower risk. (C) Risk factor distribution of participants in the analysis by quartiles of risk is provided.
A slowing of the rate of LTD was observed over the study follow‐up by comparing the time to event from ages 2 to 5 years (~15% cumulative incidence by age 5 years) to ages 6‐10 years (additional incidence of 8% by age 10 years). Only 5 participants were followed until age 12 years, and no events were observed thereafter, limiting the ability to predict events beyond age 10 years (29 participants). Exploratory random forest analysis found no significant interactions between candidate predictors.
Clinical variables associated with risk of LTD in univariate analysis (Table 2) were history of GI bleed (hazard ratio [HR], 6.56; 95% CI, 3.00‐14.35), history of ascites (HR, 3.38; 95% CI, 1.75‐6.56), splenomegaly (HR, 6.46; 95% CI, 2.29‐18.20), and height growth failure (HR, 2.91; 95% CI, 1.12‐7.56).
Table 2.
Univariate analysis of laboratory and clinical variables for LTD
| Variable | HR (95% CI) | P Value |
|---|---|---|
| TB (mg/dL), log2 | 2.64 (2.11‐3.29) | <0.001 |
| GGT (U/L), log2 | 1.40 (1.11‐1.76) | 0.004 |
| Platelet count (per 10×103/µL) | 0.88 (0.84‐0.93) | <0.001 |
| APRI, log2 | 1.96 (1.57‐2.44) | <0.001 |
| AST (U/L), log2 | 1.85 (1.43‐2.38) | <0.001 |
| ALT (U/L), log2 | 1.32 (1.04‐1.67) | 0.022 |
| Albumin (g/dL) | 0.21 (0.15‐0.31) | <0.001 |
| INR (per 0.1) | 1.88 (1.55‐2.28) | <0.001 |
| History of GI bleed | 6.56 (3.00‐14.35) | <0.001 |
| History of cholangitis | 1.19 (0.63‐2.26) | 0.587 |
| History of ascites | 3.38 (1.75‐6.56) | <0.001 |
| Splenomegaly | 6.46 (2.29‐18.20) | <0.001 |
| Weight growth failure | 0.50 (0.07‐3.65) | 0.494 |
| Height growth failure | 2.91 (1.12‐7.56) | 0.028 |
All laboratory variables and APRI (Table 2) were significantly associated with risk of LTD (P < 0.05). Of note, doubling the TB (for example, TB rise from 0.5 mg/dL to 1.0 mg/dL) was associated with an HR increase of 2.64 (95% CI, 2.11‐3.29), and doubling the APRI (e.g., 1.2 vs. 0.6) was associated with an HR increase of 1.96 (95% CI, 1.57‐2.44) for the occurrence of LTD.
The stepwise selection approach developed a model (BA LTD) with five variables: TB, platelet count, albumin, and history of ascites or cholangitis (Table 3). The clinical applicability of the BA LTD model is illustrated in the corresponding nomogram (Supporting Fig. S1).
Table 3.
Prognostic model for LTD (n = 240, number of events = 38)
| At age of 5 years: S0 (5) | 94.6% (90.8%, 98.7%) | ||
|---|---|---|---|
| At age of 7 years: S0 (7) | 92.3% (86.9%, 97.9%) | ||
| At age of 10 years: S0 (10) | 86.1% (76.6%, 96.8%) | ||
| Variable | Log (HR) (95% CI) | HR (95% CI) | P Value |
| TB (mg/dL), log2 | 0.70 (0.41, 1.00) | 2.02 (1.51, 2.72) | <.001 |
| History of ascites | 0.74 (0.03, 1.45) | 2.10 (1.03, 4.28) | 0.040 |
| History of cholangitis | 0.90 (0.20, 1.61) | 2.47 (1.22, 5.00) | 0.012 |
| Albumin (g/dL) | −1.11 (−1.84, −0.37) | 0.33 (0.16, 0.69) | 0.003 |
| Platelet count (per 10×103/µL) | −0.07 (−0.13, −0.01) | 0.93 (0.88, 0.99) | 0.014 |
Equation for calculating chance of LTD at age of t years:
1–S0(t)exp(0.7×log2[TB]+0.74×History of Ascites+0.9×History of Cholangitis–1.11×Albumin–0.007×Platelet Count+5.84).
The BA LTD risk equation was evaluated by examination of Kaplan‐Meier curves for LTD, stratified by quartile of the estimated risk in the cohort (Fig. 2B,C). Groups 1 to 4 are ranked by lowest to highest risk. Transplant‐free survival is substantially lower at all ages in group 4, reaching just 53% at age 7 years. In group 3, predicted transplant‐free survival deviates from groups 1 and 2 (89% vs ≥97%) at around 7 years of age. The distribution of each of the variables that contribute to the risk of LTD by quartile provides the characteristics of participants in our cohort at the baseline age of 2 years. Approximately half the subjects in groups 2‐4 (having had a history of cholangitis and the median albumin of all groups) were in a normal range. The median platelet count of <150 103/mm3 and a median TB of 1.8 mg/dL were noteworthy in the highest risk group 4. A visual representation of predicted probability of LTD with preselected laboratory values along with the clinical variables in the model is shown in Supporting Fig. S2.
The median of apparent Harrell’s C‐index among the 22 imputed data sets for this model was 0.88 (range, 0.86‐0.89), indicating very good discrimination. Internal validation using the jackknife method on the 22 imputed data sets resulted in a median Harrell’s C‐index of the test sets of 0.87 (range, 0.40‐0.99), indicating a very small magnitude of overfitting to the data set.
Risk Factors and a Prognostic Model for SEs
Of 192 participants, a first SE occurred in 27 participants (14 GI bleed, 13 ascites, and 1 HPS; n = 1 with ascites and GI bleed reported on the same date). The cumulative incidence of first SE was 21.5% (95% CI, 14.2%‐29.8%) by 10 years of age (Fig. 3A). Exploratory analysis using random forest analysis did not reveal significant interactions between candidate predictors. Of these 27 participants, 11 underwent LT during the follow‐up period.
FIG. 3.
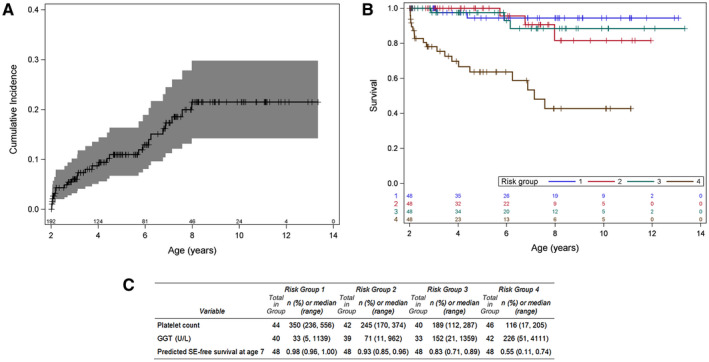
Incidence and risk for the SE model. (A) Cumulative incidence of SE among 192 study participants. (B) Kaplan‐Meier curves for SE‐free survival stratified by quartile of risk score. Stratification of participants shows a high‐risk group (group 4; brown) for the development of an SE. (C) Risk factor distribution of participants in the analysis by quartiles of risk is provided.
The only clinical variable associated with the risk of an SE in the univariate analysis (Table 4) was a history of splenomegaly, with an HR of 2.96 (95% CI, 1.25‐7.01). All laboratory variables, except INR, were associated with risk of SE in the univariate analysis (P < 0.05). Doubling the TB level was associated with an HR of 1.69 (95% CI, 1.30‐2.19). Doubling the APRI level was associated with an HR of 1.88 (95% CI, 1.48‐2.40).
Table 4.
Univariate analysis of laboratory and clinical variables for SE
| Variable | HR (95% CI) | P Value |
|---|---|---|
| TB (mg/dL), log2 | 1.69 (1.30‐2.19) | <0.001 |
| GGT (U/L), log2 | 1.48 (1.17‐1.88) | 0.001 |
| Platelet count (per 10×103/µL) | 0.87 (0.82‐0.92) | <0.001 |
| APRI, log2 | 1.88 (1.48‐2.40) | <0.001 |
| AST (U/L), log2 | 1.62 (1.19‐2.19) | 0.002 |
| ALT (U/L), log2 | 1.35 (1.03‐1.78) | 0.030 |
| Albumin (g/dL) | 0.34 (0.19‐0.59) | <0.001 |
| INR (per 0.1) | 1.25 (0.89‐1.74) | 0.194 |
| History of cholangitis | 0.51 (0.22‐1.21) | 0.129 |
| Splenomegaly | 2.96 (1.25‐7.01) | 0.013 |
| Weight growth failure | 1.24 (0.29‐5.24) | 0.774 |
| Height growth failure | 2.28 (0.68‐7.64) | 0.180 |
The stepwise selection approach developed a BA SE model with two predictors: platelet count and GGT (Table 5). The applicability of the BA SE risk equation was evaluated by examination of the cumulative incidence function curves for SE, stratified by quartile of estimated risk based on the BA SE risk equation (Fig. 3B,C). The first two quartiles showed lower risk of SE in contrast to the higher quartiles, where predicted SE‐free survival at age 7 years is 83% in group 3 and only 55% in group 4. Similar to the BA LTD model and consistent with the known association of thrombocytopenia and portal hypertension, the median platelet count of 116 103/mm3 (range, 17‐205 103/mm3) in group 4 of this model reflects the group most likely to have an SE. Median GGT was >150 U/L in group 3 and median GGT was >200 U/L in group 4. GGT may represent a novel biomarker of those at risk for an SE in combination with the platelet count.
Table 5.
Prognostic model for SE (n = 192, number of events = 27)
| Variable | Log (HR) (95% CI) | HR (95% CI) | P Value |
|---|---|---|---|
| Platelet count (per 10×103/µL) | −0.12 (−0.18, −0.06) | 0.89 (0.84, 0.94) | <0.001 |
| GGT (U/L), log2 | 0.30 (0.04, 0.55) | 1.34 (1.04, 1.73) | 0.023 |
Median of apparent Harrell’s C‐index among the 22 imputed data sets for this model is 0.81 (range, 0.80‐0.84), indicating very good discrimination. Internal validation applying the jackknife method to the 22 imputed data sets resulted in a median Harrell’s C‐index of the test data of 0.85 (range, 0.67‐1.00).
Discussion
The major gap in our understanding of BA SNL disease progression has been a lack of large multicenter prospective studies with sufficient contemporaneous clinical and biochemical data for model development. Using the large North American prospective multicenter ChiLDReN data set, we developed two novel prognostic models of BA in children who have survived to age 2 years with native liver: the BA LTD model for the risk of LTD, using five clinical and laboratory values, and the BA SE model for the risk of developing an SE, using platelet count and serum GGT.
In our study of patients who had SNL at 2 years of age, the cumulative incidence of either LTD or SE was substantial, 23.7% and 21.5%, respectively. Similarly, in a French series, transplant‐free survival rate decreased to about 25% by age 20 years; in an Italian study, it decreased from about 50% at age 2 years to 18% by age 20 years.( 5 , 15 , 16 )
Few predictive models of outcome in BA SNL have been developed. A Japanese series reported factors at 10 years of age associated with 20‐year transplant‐free survival, but small sample size precluded multivariable analysis.( 25 ) Similarly, a retrospective single‐center study reported prognostic markers at age 16 years predictive of LT after age 20 years. Although this may be helpful in guiding surveillance of liver disease progression during transition to adulthood, earlier intervention before adolescence and before liver damage occurs would be more beneficial.( 26 ) The Netherlands registry reported factors associated with LT; however, analysis was limited in its retrospective nature, the small cohort, and use of only very early laboratory values.( 27 )
The biochemical parameters chosen for this analysis are obtained routinely by clinicians. Previous studies of TB as a prognostic marker have focused on outcomes in the few months after KPE.( 17 ) Notably, in our study cohort, the median TB at age 2 years was 0.5 mg/dL. Using TB as a continuous variable revealed that a small increase in TB conferred a substantial contribution to the LTD risk score. Additional variables (decreasing albumin and platelet count and history of ascites and cholangitis) improved the discriminative ability (Harrell’s C‐index, 0.88) to predict LTD over the 10‐year follow‐up. When we examined a subset of our cohort with TB <2 mg/dL at 2 years of age, the median Harrell’s C‐index was 0.79, indicating good discriminability among patients traditionally viewed as having low risk of progression.
Collecting detailed data on intermediate SEs that occur before LT remains one of the primary aims of ChiLDReN. Our study is the first to present cumulative incidence of complications of chronic liver disease occurring in BA SNL in those without prior SE occurrence in infancy. We chose a composite of new onset GI bleed or ascites or HPS, as these complications provide a unique opportunity to model morbidity in BA SNL. Identifying those at risk for an SE may help target future interventions to avoid LT.
Platelet count and GGT were the only two predictors in the selected BA SE model. Platelet count is an established clinical marker of portal hypertension, and “prediction rule” models of varices (APRI, Clinical Prediction Rule, Varices Prediction Rule) in pediatric liver disease consistently include platelet count.( 28 , 29 , 30 ) Conversely, few studies have evaluated GGT as a marker of disease progression in BA. One study of BA SNL showed that GGT >100 at 2 years of age predicted thrombocytopenia at 4‐6 years of age.( 31 ) Similarly, GGT >550 IU/L at 5 months was associated with poor prognosis at 5 years of age, despite clearance of jaundice.( 32 ) In the BA SE model, increasing GGT conferred an increased risk of a first SE and should be a standard test in assessing disease severity in BA SNL at age 2 years.
Both early and late onset cholangitis are described as potentiating fibrosis and progressive liver disease.( 33 , 34 ) A history of cholangitis was a predictor in the BA LTD model, although not for the BA SE model. However, a Japanese cohort describes no significant difference in 20‐year SNL in patients with a history of cholangitis.( 35 ) As the medical management of patients is not protocolized in ChiLDReN, standardized prophylactic antibiotic therapy that could modify the clinical course in BA is lacking, and further evaluation in prospective trials is required.
Our study had several limitations. We were targeting the specific subpopulation of patients with BA who still remain with their native liver at 2 years of age. The moderate number of events could subject the analysis to potential overfitting of the models. Although our internal validation results indicate only a small magnitude of overfitting, future external validations of both the BA LTD and BA SE models are warranted. Variables, such as ascites or splenomegaly, could be made more objective by use of radiographic techniques. Data from the BASIC study include patients after 6 months of age and allow for referral of patients from nonstudy site centers where inclusion criteria in PROBE require that patients undergo KPE at the study site. This difference did not allow for inclusion of some variables in the analysis.
Although we lacked substantial clinical or biochemical information beyond age 10 years, the rate of LTD and SE by age 10 illustrates progression of liver disease. Continued collection of prospective data from the ChiLDReN cohort will help determine rates of LTD or SE after age 10 years. As application of these two models to BA cohorts from international centers occurs, more robust models of BA outcomes beyond age 10 may be developed. Moreover, these models are derived from data obtained from a single time point of a child with BA, i.e., age 2 years. Dynamic prediction models using longitudinal laboratory data accrued beyond 2 years of age may provide better prediction for both outcomes and are warranted for our future work. Recent therapeutics for cholestatic liver diseases are being tested in children and adults, suggesting that in the near future, patients with BA SNL, especially those in the highest risk quartiles, should be considered candidates for clinical trials.( 2 , 36 , 37 , 38 )
Supporting information
Supplementary Material
Acknowledgment
Heather Van Doren, M.F.A., Senior Medical Editor with Arbor Research Collaborative for Health, provided editorial assistance on this manuscript.
Supported by the National Institute of Diabetes, Digestive, and Kidney Diseases U01 grants (DK062445 to Mt. Sinai School of Medicine, DK062497 to Cincinnati Children’s Hospital Medical Center, DK062470 to Children’s Healthcare of Atlanta, DK062481 to The Children’s Hospital of Philadelphia, DK062456 to University of Michigan, DK084536 to Riley Hospital for Children, DK084575 to Seattle Children’s Hospital, DK062500 to University of California San Francisco [UCSF] Children’s Hospital, DK062466 to Children’s Hospital of Pittsburgh of the University of Pittsburgh Medical Center [UPMC], DK062453 to Children’s Hospital Colorado, DK084538 to Children’s Hospital Los Angeles, DK062436 to Ann & Robert H Lurie Children’s Hospital of Chicago, DK103149 to Texas Children’s Hospital, DK103135 to The Hospital for Sick Children, DK103140 to University of Utah), the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Awards (UL1 TR002535 to University of Colorado, UL1 TR001872 to UCSF Children’s Hospital, UL1 TR001857 to Children’s Hospital of Pittsburgh of UPMC, UL1 TR001878 to The Children’s Hospital of Philadelphia, UL1 TR000423 and UL1 RR025014 to Seattle Children’s Hospital, UL1TR002378 to Children’s Healthcare of Atlanta, UL1TR00130 to Children’s Hospital of Los Angeles).
Potential conflict of interest: Dr. Karpen consults for Albireo, Mirum, and LogicBio. Dr. Kamath and Dr. Loomes consult for and received grants from Albireo and Mirum. Dr. Harpavat is on a Data Monitoring Committee for Syneos Health. Dr. Mack and Dr. Ng consult for Albireo. Dr. Molleston received grants from Albireo, AbbVie, Gilead, and Mirum. Dr. Murray consults for Albireo and Gilead. Dr. Rosenthal consults for and received grants from AbbVie, Albireo, Gilead, Mirum, and Retrophin; he consults for Audentes and Dicerna and received grants from Arrowhead and Merck. Dr. Sokol consults for Albireo, Mirum, and Retrophin. The other authors have nothing to report.
References
- 1. Barshes NR, Lee TC, Balkrishnan R, Karpen SJ, Carter BA, Goss JA. Orthotopic liver transplantation for biliary atresia: the US experience. Liver Transpl 2005;11:1193‐1200. [DOI] [PubMed] [Google Scholar]
- 2. Bezerra JA, Wells RG, Mack C, Karpen S, Hoofnagle JH, Doo E, et al. Biliary atresia: clinical and research challenges for the twenty‐first century. Hepatology 2018;68:1163‐1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sundaram S, Mack C, Feldman AG, Sokol RJ. Biliary atresia: indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl 2017;23:96‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harpavat S, Ramraj R, Finegold D, Brandt ML, Hertel PM, Fallon SC, et al. Newborn direct or conjugated bilirubin measurements as a potential screen for biliary atresia. J Pediatr Gastroenterol Nutr 2016;62:799‐803. [DOI] [PubMed] [Google Scholar]
- 5. Chardot C, Buet C, Serinet M, Golmard J, Lachaux A, Roquelaure B, et al. Improving outcomes of biliary atresia: French national series 1986‐2009. J Hepatology 2013;58:1209‐1217. [DOI] [PubMed] [Google Scholar]
- 6. Serinet M, Broue P, Jacquemin E, Lachaux A, Sarles J, Gottrand F, et al. Management of patients with biliary atresia in France: results of a decentralized policy 1986‐2002. Hepatology 2006;44:75‐84. [DOI] [PubMed] [Google Scholar]
- 7. Davenport M, Ville DE, de Goyet J, Stringer M, Mieli‐Vergani G, Kelly D, et al. Seamless management of biliary atresia in England and Wales. Lancet 2004;363:1354‐1357. [DOI] [PubMed] [Google Scholar]
- 8. Bezerra JA, Spino C, Magee JC, Shneider BL, Rosenthal P, Wang KS, et al.; Childhood Liver Disease Research and Education Network (ChiLDREN) . Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA 2014;311:1750‐1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mack CL, Spino C, Alonso EM, Bezerra JA, Moore J, Goodhue C, et al.; The ChiLDReN Network . A phase I/IIa trial of intravenous immunoglobulin following portoenterostomy in biliary atresia. J Pediatr Gastroenterol Nutr 2019;68:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shneider BL, Brown MB, Haber B, Whitington PF, Schwarz K, Squires R, et al.; Biliary Atresia Research Consortium . A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr 2006;148:467‐474. [DOI] [PubMed] [Google Scholar]
- 11. Sokol RJ, Shepherd R, Superina R, Bezerra Jeronimo SM, Robuck PR, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 2007;46:566‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lykavieris P, Chardot C, Sokhn M, Gauthier F, Valayer J, Bernard O. Outcome in adulthood of biliary atresia: a study of 63 patients who survived for over 20 years with their native liver. Hepatology 2005;41:366‐371. [DOI] [PubMed] [Google Scholar]
- 13. Bijl EJ, Bharwani KD, Houwen RH, de Man RA. The long‐term outcome of the Kasai operation in patients with biliary atresia: a systematic review. Neth J Med 2013;71:170‐173. [PubMed] [Google Scholar]
- 14. Ng VL, Haber BH, Magee JC, Miethke A, Murray KF, Michail S, et al.; Childhood Liver Disease Research and Education Network (CHiLDREN) . Medical status of 219 children with biliary atresia surviving long‐term with their native livers: results from a North American multicenter consortium. J Pediatr 2014;165:539‐546.e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parolini F, Boroni G, Milianti S, Tonegatti L, Armellini A, Magne MG, et al. Biliary atresia: 20‐40‐year follow‐up with native liver in an Italian centre. J Pediatr Surg 2019;54:1440‐1444. [DOI] [PubMed] [Google Scholar]
- 16. Fanna M, Masson G, Capito C, Girard M, Guerin F, Hermeziu B, et al. Management of biliary atresia in France 1986 to 2015: long‐term results. J Pediatr Gastroenterol Nutr 2019;69:416‐424. [DOI] [PubMed] [Google Scholar]
- 17. Shneider BL, Magee JC, Karpen SJ, Rand EB, Narkewicz MR, Bass LM, et al.;Childhood Liver Disease Research Network (ChiLDReN) . Total serum bilirubin within 3 months of hepatoportoenterostomy predicts short‐term outcomes in biliary atresia. J Pediatr 2016;170:211‐217.e211‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Machicao VI, Fallon MB. Hepatopulmonary syndrome. Semin Respir Crit Care Med 2012;33:11‐16. [DOI] [PubMed] [Google Scholar]
- 19. Dignam JJ, Zhang Q, Kocherginsky M. The use and interpretation of competing risks regression models. Clin Cancer Res 2012;18:2301‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishwaran H, Kogalur UB, Blackstone EH, Lauer MS. Random survival forests. Ann Appl Stat 2008;2:841‐860. [Google Scholar]
- 21. Ishwaran H, Gerds TA, Kogalur UB, Moore RD, Gange SJ, Lau BM. Random survival forests for competing risks. Biostatistics 2014;15:757‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raghunathan TE, Lepkowski JM, Van Hoewyk J, Solenberger P. A multivariate technique for multiply imputing missing values using a sequence of regression models. Surv Methodol 2001;27:85‐95. [Google Scholar]
- 23. Rubin DB. Multiple Imputation for Nonresponse in Surveys. New York, NY: John Wiley and Sons; 1987. [Google Scholar]
- 24. Harrell FE Jr. Regression Modeling Strategies With Applications to Linear Models, Logistic and Ordinal Regression and Survival Analysis. New York, NY: Springer Science+Business Media; 2001. [Google Scholar]
- 25. Nio M, Wada M, Sasaki H, Tanaka H, Okamura A. Risk factors affecting late‐presenting liver failure in adult patients with biliary atresia. J Pediatr Surg 2012;47:2179‐2183. [DOI] [PubMed] [Google Scholar]
- 26. Jain V, Burford C, Alexander EC, Sutton H, Dhawan A, Johsi D, et al. Prognostic markers in adolescence in patients requiring liver transplantation for biliary atresia in adulthood. J Hepatology 2019;71:71‐77. [DOI] [PubMed] [Google Scholar]
- 27. Witt M, van Wessel DBE, de Kleine RHJ, Bruggink JLM, Hulscher JBF, Verkade HJ; NeSBAR (Netherlands Study group on Biliary Atresia Registry) . Prognosis of biliary atresia after 2‐year survival with native lIver: a nationwide cohort. J Pediatr Gastroenterol Nutr 2018;67:689‐694. [DOI] [PubMed] [Google Scholar]
- 28. Isted A, Grammatikopoulos T, Davenport M. Prediction of esophageal varices in biliary atresia:derivation of the "varices prediction rule", a novel noninvasive predictor. J Pediatr Surg 2015;50:1734‐1738. [DOI] [PubMed] [Google Scholar]
- 29. Gana JC, Turner D, Mieli‐Vergani G, Davenport M, Miloh T, Avitzur Y, et al. A clinical prediction rule and platelet count predict esophageal varices in children. Gastroenterology 2011;141:2009‐2016. [DOI] [PubMed] [Google Scholar]
- 30. van Wessel DBE, Witt M, Bax N, Verkade HJ, Scheenstra R, De Kleine RH, et al. Variceal bleeds in patients with biliary atresia. Eur J Pediatr Surg 2018;28:439‐444. [DOI] [PubMed] [Google Scholar]
- 31. Freeman AJ, Ng VL, Harpavat S, Hrycko A, Apted Z, Bulut P, et al. Level of gamma‐glutamyltrasferase in 2‐year‐old children with biliary atresia associates with progression of portal hypertension. Clin Gastroenterol Hepatol 2017;15:1133‐1135. [DOI] [PubMed] [Google Scholar]
- 32. Ihn K, Ho IG, Chang EY, Han SJ. Correlation between gamma‐glutamyl transpeptidase activity and outcomes after Kasai portoenterostomy for biliary atresia. J Pediatr Surg 2018;53:461‐467. [DOI] [PubMed] [Google Scholar]
- 33. Altman RP, Lilly JR, Greenfield J, Weinberg A, van Leeuwen K, Flanigan L. A multivariable risk factor analysis of the portoenterostomy (Kasai) procedure for biliary atresia; twenty‐five years of experience from two centers. Ann Surg 1997;226:348‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nio M, Sano N, Ishii T, Sasaki H, Hayashi Y, Ohi R. Cholangitis as a late complication in long‐term survivors after surgery for biliary atresia. J Pediatr Surg 2004;39:1797‐1799. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki H, Tanaka H, Wada M, Kazama T, Nakamura M, Kudo H, et al. Analysis of the prognostic factors of long‐term native liver survival in survivors of biliary atresia. Pediatr Surg Int 2016;32:839‐843. [DOI] [PubMed] [Google Scholar]
- 36. Trauner M, Fuchs CD, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology 2017;65:1393‐1404. [DOI] [PubMed] [Google Scholar]
- 37. Feldman AG, Sokol RJ. Neonatal cholestasis: Emerging molecular diagnostics and potential novel therapeutics. Nature Rev Gastroenterol Hepatol 2019;16:346‐360. [DOI] [PubMed] [Google Scholar]
- 38. Karpen SJ. Novel bile acid therapies for liver disease. Gastroenterol Hepatol (N Y) 2018;14:117‐119. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material