

Abstract
Abstract Of the multiple and diverse homeostatic events that involve the lung vascular endothelium, participation in preserving vascular integrity and therefore organ function is paramount. We were the first to show that the lipid growth factor and angiogenic factor, sphingosine-1-phosphate, is a critical agonist involved in regulation of human lung vascular barrier function (Garcia et al. J Clin Invest, 2011). Utilizing both in vitro models and preclinical murine, rat, and canine models of acute and chronic inflammatory lung injury, we have shown that S1Ps, as well as multiple S1P analogues such as FTY720 and ftysiponate, serve as protective agents limiting the disruption of the vascular EC monolayer in the pulmonary microcirculation and attenuate parenchymal accumulation of inflammatory cells and high protein containing extravasated fluid, thereby reducing interstitial and alveolar edema. The vasculo-protective mechanism of these therapeutic effects occurs via ligation of specific G-protein-coupled receptors and an intricate interplay of S1P with other factors (such as MAPKS, ROCKs, Rho, Rac1) with rearrangement of the endothelial cytoskeleton to form strong cortical actin rings in the cell periphery and enhanced cell-to-cell and cell-to-matrix tethering dynamics. This cascade leads to reinforcement of focal adhesions and paracellular junctional complexes via cadherin, paxillin, catenins, and zona occludens. S1P through its interaction with Rac and Rho influences the cytoskeletal rearrangement indicated in the later stages of angiogenesis as a stabilizing force, preventing excessive vascular permeability. These properties translate into a therapeutic potential for acute and chronic inflammatory lung injuries. S1P has potential for providing a paradigm shift in the approach to disruption of critical endothelial gatekeeper function, loss of lung vascular integrity, and increased vascular permeability, defining features of acute lung injury (ALI), and may prove to exhibit an intrinsically protective role in the pulmonary vasculature ameliorating agonist- or sepsis-induced pulmonary injury and vascular leakage.
Keywords: Endothelial cells, S1P, Rac, Rho, Cytoskeleton
1. Introduction
The lung vascular endothelium is a critical participant in multiple homeostatic events that preserve the lung’s physiologic function. These range from maintenance of vascular tone, blood coagulation, inflammation, angiogenesis, cell homeostasis, and the maintenance of lung fluid balance. For example, the lung endothelium is essential to adequate gas transfer and tissue oxygenation but serves as a primary cellular target in the profound physiologic derangement that accompanies acute and chronic inflammatory lung injuries. Via disruption of critical endothelial gatekeeper function, the loss of lung vascular integrity results in parenchymal accumulation of leukocytes and extravascular lung water and increased vascular permeability, defining features of acute lung injury (ALI) that contribute to the increased morbidity and mortality of this devastating syndrome. Consequently, there is substantial interest in the development and utilization of clinically effective agents that either interfere with or prevent lung vascular endothelial cell (EC) barrier dysfunction, restore EC barrier integrity, reduce alveolar flooding, and improve respiratory mechanics (Schuchardt et al. 2011; Snider et al. 2010; Belvitch and Dudek 2012; Bazzoni and Dejana 2004; Jacobson and Garcia 2007; Komarova et al. 2007).
The vascular endothelium is composed of a single layer of ECs and an underlying layer of extracellular matrix and serves as a semipermeable barrier, regulating exchange of gases, water, and solutes across it (Donati and Bruni 2006; Belvitch and Dudek 2012; Bazzoni and Dejana 2004). The transport of fluids and macromolecules across the endothelium is regulated by three main processes. Capillary beds contain fenestrae that are small diaphragm covered pores in the endothelium allowing the passive diffusion based transport of the particles based on the size and charge selective characteristics of the particle. In contrast to diffusion, transcytosis is an active process that involves the fusion of endocytic vesicles with the luminal endothelial membrane in response to ligation with cell surface glycoproteins. The third process, and the most important in pathological conditions, is the paracellular pathway, which involves the transit of particles through the gaps between ECs (McVerry and Garcia 2004). The pulmonary vascular and microvascular network is extremely extensive and is composed of non-fenestrated type of endothelium and is the major hub of fluid and solute exchange. Prolonged disruption of this homeostatic exchange is an indication of inflammatory pathological conditions such as acute lung injury and sepsis (Snider et al. 2010; Donati and Bruni 2006; Belvitch and Dudek 2012; Bazzoni and Dejana 2004; English et al. 2001).
The class of cell membrane components termed the sphingolipids (Schuchardt et al. 2011), aptly named in 1884 after the mysterious Sphinx of Greek mythology, is recognized as integral components of cellular function and modulators of EC function. This chapter focuses on the role of sphingolipids as critical integral participants in numerous vascular physiological and biological responses (Hla and Brinkmann 2011; Snider et al. 2010) with a particular focus on lung vascular barrier regulation (Lucke and Levkau 2010). The cumulative effects of these actions are the reduction of barrier permeability resulting in decreased fluid collection, diminished inflammatory cell migration, and vascular cellular disruption, ultimately leading to enhancement of barrier integrity. We outline the molecular mechanisms of both homeostatic and pathobiologic lung vascular barrier regulation and barrier restoration processes that occur in response to the bioactive sphingolipid growth factor, sphingosine-1-phosphate (S1P), and ligation of the family of S1P G-protein-coupled receptors and the translation of this information into novel barrier-modulatory therapeutic strategies (Lucke and Levkau 2010; Donati and Bruni 2006; Belvitch and Dudek 2012; Bazzoni and Dejana 2004; Bode et al. 2010; Boguslawski et al. 2002; Rosen et al. 2007).
2. Sphingosine-1-Phosphate Biosynthesis in Lung Endothelium
As noted elsewhere in this book, over 300 different sphingolipids have been identified, each having a unique head group and a common long chain sphingoid base backbone of ceramide. Sphingosine-1-phosphate (S1P) is a sphingolipid resulting from the phosphorylation of sphingosine, a product of sphingomyelinase catabolism of sphingomyelin, catalyzed by sphingosine kinase (SphK) (Fig. 1). The catabolism of ceramide also may lead to the production of S1P (Schuchardt et al. 2011; Lucke and Levkau 2010). Various enzymes are involved in this process of S1P genesis and may take one of two routes: de novo generation or via a salvage pathway. The endoplasmic reticulum is the main site for the de novo pathway and involves various intermediates such as serine, palmitoyl coenzyme A, and fatty acids, leading to the formation of ceramide. The remaining steps ultimately leading to S1P production occur in the Golgi apparatus and involve the phosphorylation of sphingosine by SphK which exists in two isoforms (SphK1, SphK2) exhibiting a similar structure but distinct catalytic properties, cellular distribution, and expression traits (Schuchardt et al. 2011; Lucke and Levkau 2010; Waeber et al. 2004). The salvage pathway involves the recycling of sphingolipids and ceramide by catabolic actions of glucocerebrosidases and sphingomyelinases and the action of ceramidases that breakdown ceramides to sphingosine and finally the formation of S1P, which then exerts its influence through the S1P receptors (Schuchardt et al. 2011; Snider et al. 2010). All cells in the body have the ability to produce S1P during the course of sphingomyelin metabolism; however, erythrocytes and ECs are very prominent source of S1P present in plasma (Fig. 2). The transfer of the S1P from the intracellular environment of its genesis to the extracellular environment of its action is facilitated by ATP-binding cassette (ABC)-type transporters, although additional mechanisms must exist as experiments in mice lacking these transporters showed unchanged levels of S1P in the plasma. The concentration of S1P in the plasma ranges between 200 and 1,000 nM, primarily bound to HDL but also to albumin, LDL, and VLDL. Even though the total quantity of S1P in the plasma is greater than the concentration of its receptors, the biologically active available fraction of S1P is only about 1–2 %. This indicates that the major portion of the S1P in the plasma is present as a buffered form or in a condensed form, explaining the enormous capacity of the HDL to transport S1P in the plasma, buffer it, and to carry away excess S1P produced at sites of inflammation. This relationship is also involved in many HDL–S1P biological activities such as NO-induced vasodilatation, antioxidant, anti-apoptotic, angiogenic, and anti-inflammatory responses. A likely explanation is that the HDL transports S1P such that only a limited portion of its cargo is exposed to S1P receptors, potentially intricate process involving HDL engagement with its own receptors before S1P docking with S1P receptors. This mechanism is supported by the observation that experimental exogenous loading of HDL with S1P enhances inhibition of apoptosis in EC induced by oxidized LDLs (Schuchardt et al. 2011; Lucke and Levkau 2010; Hla and Brinkmann 2011; Donati and Bruni 2006; Zhao et al. 2007; Berdyshev et al. 2011; Venkataraman et al. 2006; Yang et al. 1999; Yatomi et al. 1995; Waeber et al. 2004; Boguslawski et al. 2002; Saba and Hla 2004).
Fig. 1.
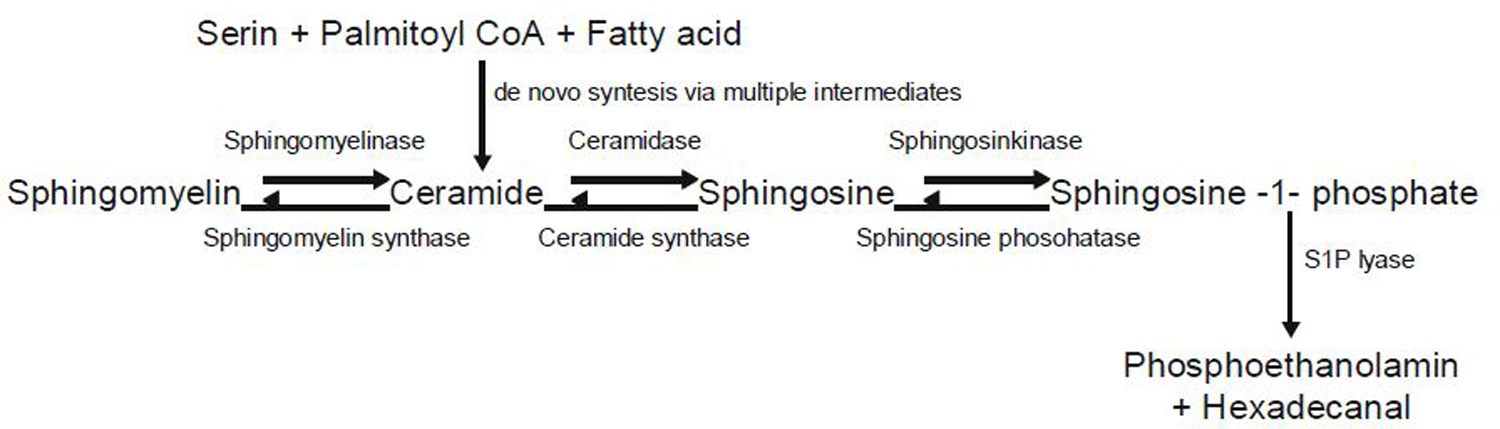
Ceramide is either formed de novo from serine, palmitoyl coA, and fatty acid or via breakdown of membrane sphingomyelin. Ceramide is further converted to sphingosine, which can be phosphorylated to generate S1P. Degradation of S1P could be reversible by dephosphorylation or irreversible by S1P lyase. S1P, sphingosine-1-phosphate [Modified from Schuchardt et al. (2011)]
Fig. 2.
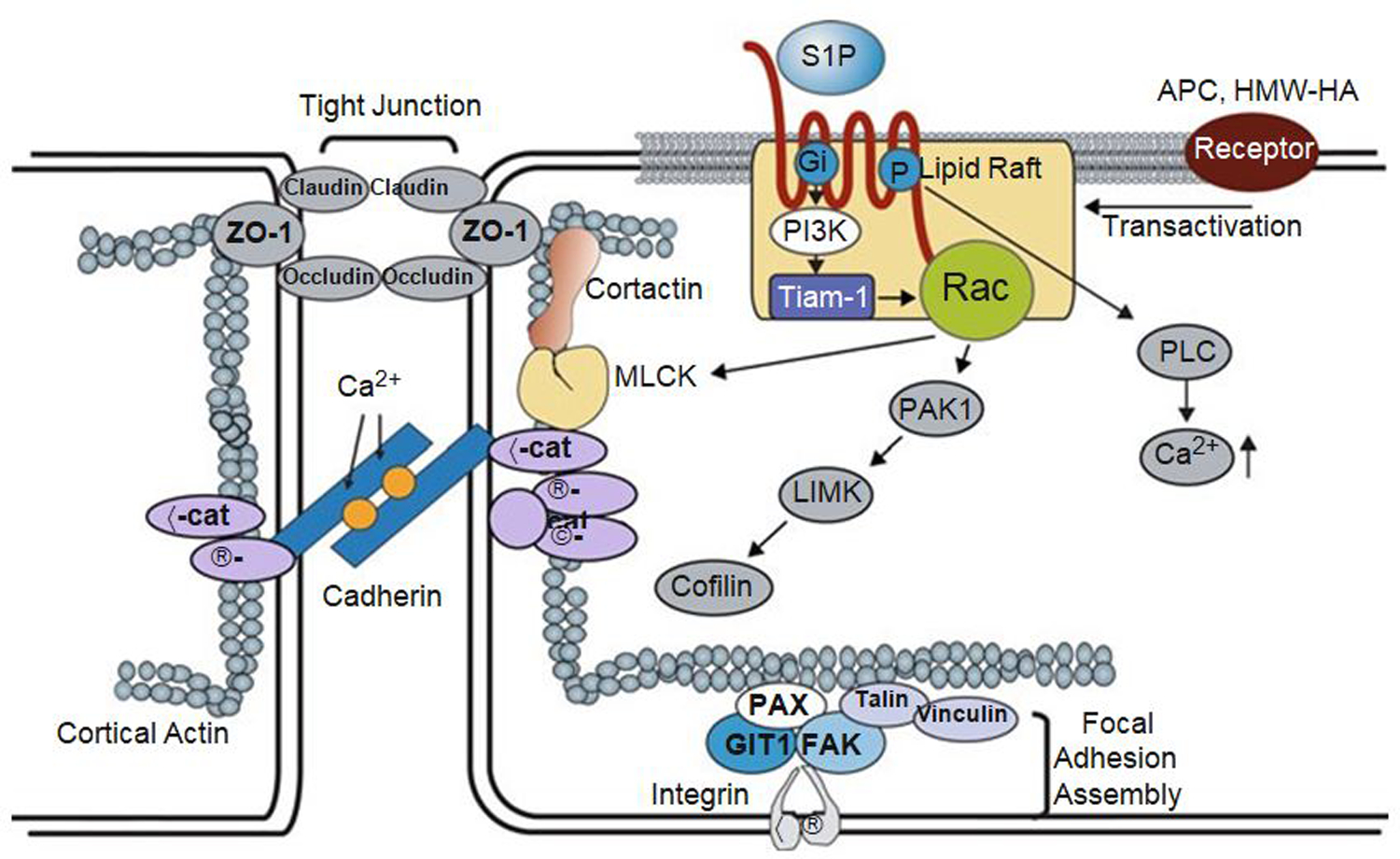
Regulation of vascular permeability by S1P/ SIP1 signaling. Binding of S1P to the SIP1 receptor stimulates the Gi-dependent recruitment of PI3 kinase, Tiam1, and Rac1 to lipid rafts (CEM), which serves to activate Rac1 in a Gi-PI3K-Tiam1-dependent manner. In addition, S1P induces an increase in intracellular Ca2+ concentration via a Gi-PLC pathway with additional activation of Rac1. After the activation of Rac1, S1P induces a series of profound events including adherens junction and tight junction assembly, cytoskeletal reorganization, and formation of focal adhesions that combine to enhance vascular barrier function. Furthermore, the transactivation of S1P1 signaling by other barrier-enhancing agents is recently recognized as a common mechanism for promoting endothelial barrier function. TJ tight junction, AJ adherens junction, S1P sphingosine-1-phosphate, SIP1 sphingosine-1-phosphate receptor 1, PI3K phosphoinositide 3-kinase, Tiam 1 t-lymphoma invasion and metastasis gene 1, Rac1 Rho family of GTPase Rac1, PAK1 p21-activated protein kinase 1, LIMK LIM kinase, PLC phospholipase C, ZO-1 zona occluden protein-1, nmMLCK non-muscle myosin light-chain kinase, VE-Cad vascular endothelial cadherin, a-Cat a-Catenin, β-Cat β-Catenin, Vin vinculin, Pax paxillin, FAK focal adhesion kinase, GIT2 G-protein-coupled receptor kinase interactor-1, ECM extracellular matrix, APC activated protein C, HMW-HA high molecular weight hyaluronan [Modified from Wang and Dudek (2009)]
3. S1P Signal Transduction via Lung Endothelial S1P Receptors and Rho Family GTPases
The neglected status of S1P changed with the discovery and study of the five G-protein-coupled S1P cell surface receptors (S1PR1–S1PR5), formerly known as endothelial differentiation gene or Edg receptors, resulting in the appreciation of S1P as a multifunctional bioactive signaling lipid molecule. Although in mouse embryonic fibroblasts devoid of S1P receptors, S1P continued to mediate growth and survival, the majority of S1P-associated functions are performed through receptor-mediated signaling. The various S1PRs interact mainly with α subunits of G proteins: Gi, Gq, and G 12/13 to initiate signaling associated with S1P (Snider et al. 2010; Garcia et al. 2001; Waeber et al. 2004; Rosenfeldt et al. 2003; Lee et al. 1998; Liu et al. 2000; An et al. 2000; Ohmori et al. 2001). These receptors have prominent effects on the vasculature, promoting endothelial cell mitogenesis, chemotaxis, and angiogenesis. The primary S1P receptors expressed in ECs are S1PR1 and S1PR3, which exhibit distinct coupling to Rho family GTPases (Rho, Rac, and Cdc42), a group of regulatory molecules that channel signals from cell surface receptors to their downstream effectors including actin cytoskeletal components. Rac activity is associated with adherens junctional assembly, cytoskeletal rearrangement, and lamellipodia formation (Lucke and Levkau 2010; Snider et al. 2010; Garcia et al. 2001; Berdyshev et al. 2011; Gosens et al. 2004; Lee et al. 1996, 1998, 1999; Birukova et al. 2004; Rosen and Goetzl 2005; Rosenfeldt et al. 2003; Amano et al. 1996; Ohmori et al. 2001). We demonstrated that S1PR1 is the critical S1P receptor for barrier enhancement (McVerry and Garcia 2004; Sun et al. 2009) mediated by the small GTPase Rac1, inducing a signaling pathway that leads to cytoskeletal rearrangement with increased cortical peripheral actin resulting in increased EC junctional integrity and focal adhesion strength (Garcia et al. 2001; Adyshev et al. 2011; Wang and Dudek 2009; Lee et al. 1996; Singleton et al. 2005) (Fig. 3). Ligation of S1PR1 increases Rac GTPase activity resulting in increased endoplasmic reticulum-derived cytosolic calcium (Worthylake et al. 2001), processes critical to S1P-mediated vascular effects. S1P activates Rac GTPase in pertussis toxin-sensitive fashion to aid in the enhancement of vascular integrity via the construction of adherens junctions and cytoskeletal modifications and lamellipodia formation (Fig. 3). Microinjection of dominative-negative Rac into EC reduces S1P-induced VE-cadherin and β-catenin accumulation at the cell–cell junctions, translocation of cortactin, and the polymerization of cortical actin. Overexpression of activated Rac GTPase institutes changes in the cortical actin similar to those produced by S1P ligation of S1PR1 (Adyshev et al. 2011; Ebnet et al. 2000; Wojciak-Stothard et al. 2006; Corada et al. 1999; Carmeliet et al. 1999; Mitra et al. 2005; Owen et al. 2007; Chae et al. 2004; Waterman-Storer et al. 1999). In addition to lamellipodia, there is increased actin polymerization at the cell periphery (i.e., the cortical actin ring) that occurs with increased force driven by the actin-binding proteins, cortactin, and nmMLCK, which also translocate to this spatially defined region. Like lamellipodia formation, Rac GTPase-dependent increases in cortical actin follow, after exposure to multiple EC barrier-enhancing or maintaining functions, with cortactin directly interacting with nmMLCK, an association which is increased by p60 Src tyrosine phosphorylation of either cortactin or nmMLCK (Parizi et al. 2000). Rac activation is in conjunction with Akt-mediated phosphorylation events known to be involved in EC proliferation and migration (Singleton et al. 2007) and EC barrier enhancement (McVerry and Garcia 2004; Krump-Konvalinkova et al. 2005; Mehta et al. 2005; Arce et al. 2008; Birukova et al. 2004, 2007; Ryu et al. 2002).
Fig. 3.
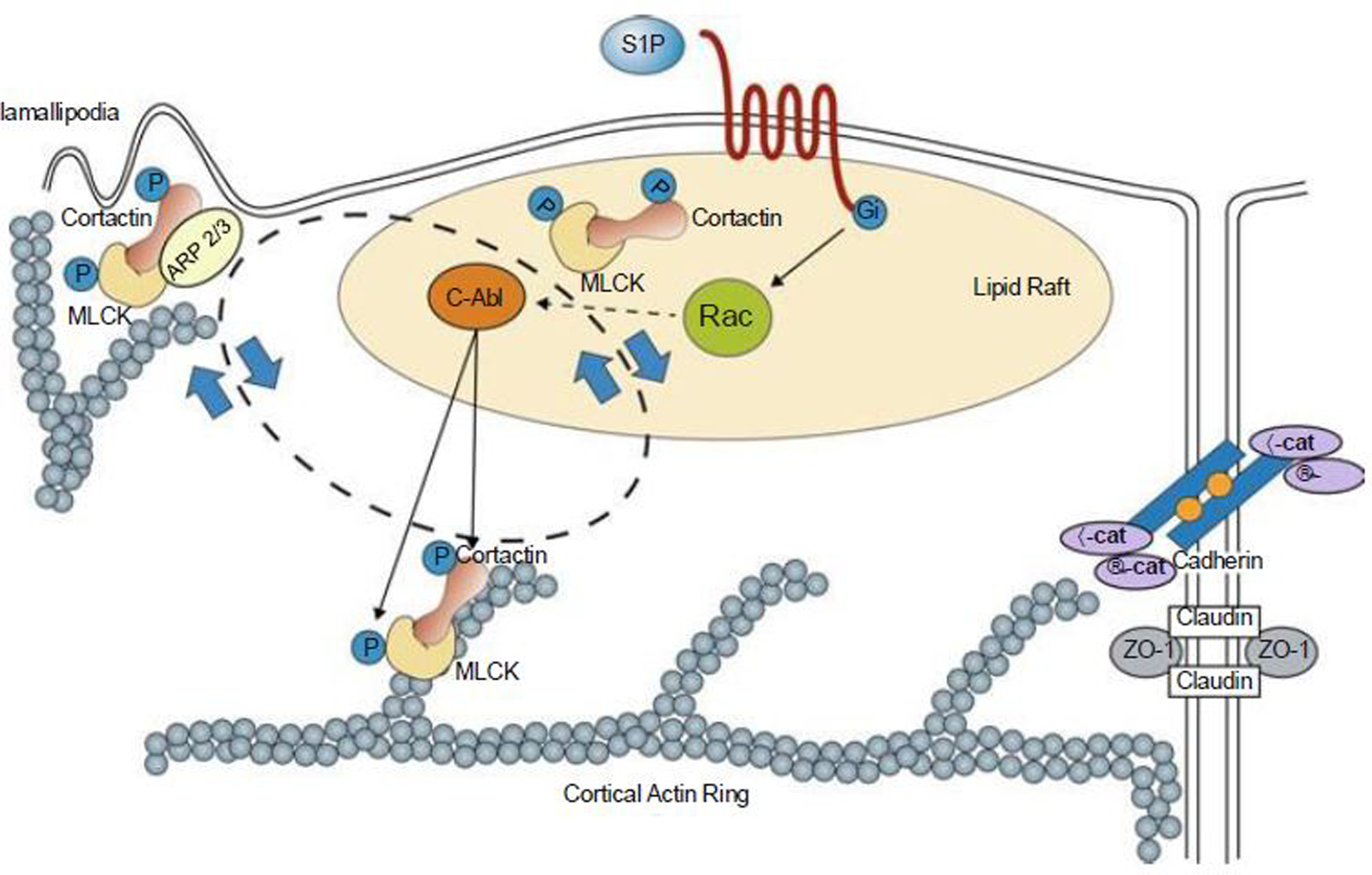
S1P regulates enhanced EC barrier function. Ligation of the S1PR1 Gi protein-coupled receptor by S1P rapidly (within 1–5 min) activates Rac and recruits signaling molecules and cytoskeletal effectors such as c-Abl, cortactin, and nmMLCK to lipid rafts (or CEMs). Tyrosine phosphorylation of these molecules is observed both in lipid rafts and at the EC periphery in association with cortical actin and lamellipodia formation. This activated complex likely interacts with Arp 2/3 machinery to produce lamellipodia protrusion at the cell periphery, which serves to increase overlap between adjacent ECs. The initiation and precise sequence of events responsible for these protein movements are unclear, but within 5 min after S1P stimulation, these proteins are found simultaneously distributed in lipid rafts, cortical actin structures, and peripheral membrane ruffling/lamellipodia (indicated by the bidirectional circle). S1P also induces adherens junction (AJ) and tight junction (TJ) assembly that serve to further strengthen the endothelial barrier. Multiple other signaling and cytoskeletal effector molecules participate in this process as reviewed elsewhere (Wang and Dudek 2009). MLCK non-muscle myosin light-chain kinase, VE-cad vascular endothelial cadherin, ZO-1 zona occluden protein-1 [Modified from Belvitch and Dudek (2012)]
The cortical cytoskeletal rearrangement produced by the S1P involves specific signaling sequences that bind to the p21-associated Ser/Thr kinase (PAK), an important downstream Rac target (Shikata et al. 2003b; Arce et al. 2008), as its binding to Rac results in the phosphorylation and activation of LIM kinase and the subsequent inactivation of the LIM kinase target, cofilin (Shikata et al. 2003b; Arce et al. 2008). Cofilin is an actin-binding protein with actin-severing capabilities leading to actin disassembly, events resulting in EC barrier enhancement. PAK and cofilin allow polymerization–depolymerization cycling to occur and thus facilitate rearrangement of actin from primarily transcytoplasmic to primarily cortical in a spatially distinct organization as a cortical actin cellular ring, findings which are integral to EC barrier function (Schuchardt et al. 2011). Transfection of EC with PAK-1 dominant negative construct significantly reduces S1P-mediated increases in the cortical restructuring, whereas adenoviral-mediated cofilin overexpression prominently attenuates the barrier stabilizing actions of S1P. Rac activation is also critical to the translocation of cortactin, an actin-binding protein that stimulates actin polymerization and stabilizes actin filaments. (Belvitch and Dudek 2012; Shikata et al. 2003b; Singleton et al. 2005; Mitra et al. 2005; Narumiya et al. 1997) And via binding to MLCK localizes to the site of cortical actin polymerization (Belvitch and Dudek 2012; Sun et al. 2009). S1P induced increases in transendothelial electrical resistance (TER) responses, a reflection of barrier integrity, are also reduced when Rac expression is experimentally reduced using siRNA. Of the remaining two members the Rho family of GTPases, Rho has been shown to be involved in the phenotypic modulation and contraction in smooth muscles along with the formation of stress fibers and focal adhesions, whereas Cdc42 has been shown to be a regulator in filopodia formation (Adyshev et al. 2011; Arce et al. 2008; Wojciak-Stothard et al. 2006; Birukova et al. 2004; Wysolmerski and Lagunoff 1990; Ohmori et al. 2001).
4. S1P Recruitment of Signaling Molecules to Lipid Rafts
Lipid rafts are a complex aggregation of various proteins and lipids with sphingolipids and cholesterol embedded within the plasma membrane. Integral components of the lipid rafts include the ganglioside GM1 and caveolin-1 that are directly involved in lipid raft-mediated cell signaling and in S1P-directed vascular barrier regulation as disruption of the liquid-ordered phase of lipid rafts results in inhibition of S1P-mediated barrier enhancement. The dependence of S1P on raft-mediated signaling to maintain and enhance vascular barrier integrity was confirmed through two-dimensional gel electrophoresis (2-DE) immunoblots of phosphotyrosine proteins that demonstrated an attenuation in levels of phosphotyrosine induced by S1P when the membrane raft formation is interrupted. Mass spectrometry identified over 200 proteins in membrane rafts with S1P inducing recruitment of >20 barrier-regulatory phosphotyrosine proteins established such as focal adhesion kinase (FAK), cortactin, p85α phosphatidylinositol 3-kinase (p85α PI3K), myosin light-chain kinase (nmMLCK), filamin A/C, and the non-receptor tyrosine kinase, c-Abl (McVerry et al. 2004). In addition, S1P-induced signaling in human lung EC caveolin-rich microdomains (CEMs) identified additional upstream effectors that contribute to the barrier regulatory properties including Rac, PAK, cofilin, S1P1, S1P3, PI3 kinase catalytic subunits p110α/β, Tiam1 (T-cell lymphoma invasion and metastasis-inducing protein 1), and α-actinin 1/4 to CEMs (Quadri et al. 2003; Vestweber 2008; Venkiteswaran et al. 2002; Parizi et al. 2000; Spiegel and Milstien 2003; Rosenfeldt et al. 2003). Experimental reduction or inhibition of each upstream effector components derails the PI3 kinase-Tiam-Rac1 pathway and impairs S1P-induced cytoskeletal rearrangement imperative for barrier enhancement. Disruption of the CEM using methyl-β-cyclodextrin (MβCD), a cholesterol exhausting agent, hampers the recruitment of S1PR and PI3 kinase p110α/β by S1P to CEMs and negatively impacts EC barrier enhancement. Similarly, reductions in expression of S1PR1, Tiam1, or PI3k all yield the same negative results on barrier dynamics. Finally, advanced studies employing quantitative proteomic analysis (iTRAQ) revealed additional proteins involved with S1P in barrier maintenance such as myristoylated alanine-rich protein kinase C substrate (MARCKS) and MARCKS-related protein (MRP) whose silencing also attenuates S1P-mediated EC barrier enhancement. Endoplasmic reticulum Ca2+ also plays a vital role in the S1P-mediated Rac activation. S1P produces an increase in the intracellular Ca2+ concentration via the Gβ-dependent pathway, which can be blocked by inhibition of Gβ, phospholipase C (PLC), or inositol triphosphate receptors, thus preventing the increase in Ca2+, Rac activation, adherens junction assembly, and ultimately barrier enhancement (Mehta et al. 2005; Singleton et al. 2005; Brinkmann et al. 2002; Argraves et al. 2004; Rosen et al. 2007).
5. S1PR1 Signaling to the Lung Endothelial Cytoskeleton and Restoration of Vascular Integrity
As noted above, we demonstrated that S1PR1 is the critical S1P receptor for barrier enhancement (Berdyshev et al. 2011; Adyshev et al. 2011; Arce et al. 2008) mediated by Rac1 GTPase-mediated signaling pathway, leading to cytoskeletal rearrangement with increased cortical peripheral actin resulting in increased EC junctional integrity and focal adhesion strength (Berdyshev et al. 2011; Adyshev et al. 2011; Arce et al. 2008; Birukova et al. 2004) (Fig. 3). Consistent with the conceptual framework that barrier regulation is intimately linked to the cytoskeleton, changes in the actin cytoskeleton were essential for S1P-mediated barrier enhancement as cytochalasin B, an actin depolymerizing agent, and latrunculin B, which inhibits actin polymerization; each prevent the barrier-enhancing effects of S1P. While increases in MLC phosphorylation within stress fibers are critical to barrier disruption (Fig. 4), MLC phosphorylation is also a key element in S1P-mediated barrier enhancement and occurs in a peripheral distribution within the cortical actin ring (Schuchardt et al. 2011), providing strength to this spatially directed scaffolding force and enhancing cell–cell tethering as we described via atomic force microscopy (Belvitch and Dudek 2012). Immunofluorescence studies demonstrated that overexpressed GFP-nmMLCK distributes along cytoplasmic actin fibers, but rapidly translocates to the cortical regions of the cell after S1P treatment, rapidly catalyzing MLC phosphorylation. In addition, confocal microscopy studies show EC challenged with S1P demonstrates colocalization of nmMLCK with the key actin-binding and EC barrier-regulatory protein, cortactin (Belvitch and Dudek 2012; Garcia et al. 2001; Sammani et al. 2010) (Fig. 4). Cortactin is involved in stimulating actin polymerization (Owen et al. 2007) and cortical actin rearrangement (Belvitch and Dudek 2012), and tyrosine phosphorylation of cortactin is seen after stimuli which cause cytoskeletal rearrangement (Belvitch and Dudek 2012). The C-terminal SH3 region of phosphorylated cortactin directly interacts with nmMLCK at higher rates than non-phosphorylated cortactin, and the interaction of cortactin and nmMLCK decreases cortactin-stimulated actin polymerization (Belvitch and Dudek 2012; Sun et al. 2009; Garcia et al. 2001; Sammani et al. 2010; Shikata et al. 2003b; Singleton et al. 2005) and is essential to S1P barrier protection. A cortactin-blocking peptide (CBP), which competitively blocks the cortactin SH3 site and nmMLCK interaction, did not affect S1P-induced cortactin translocation or cortical actin ring formation, but significantly attenuated S1P-induced barrier enhancement. Immunofluorescence showed that S1P as well as other barrier-enhancing agents (such as HGF) produced rapid translocation of cortactin to the EC periphery, an effect not seen when EC are treated with the barrier-disrupting agent, thrombin (Narumiya et al. 1997). Thus, tyrosine phosphorylation of cortactin is not necessary for peripheral translocation of cortactin after S1P but is necessary for S1P-induced barrier enhancement. p60src is not involved in this pathway, but other tyrosine kinases, such as c-abl, are likely involved (Garcia et al. 2001; Sammani et al. 2010; Shikata et al. 2003b; Singleton et al. 2005). Thus, cortactin, nmMLCK, and the intracellular location of phosporylated MLC are all critically important in the barrier enhancement induced by S1P (Wang and Dudek 2009; Vouret-Craviari et al. 2002; Xu et al. 2007; Vestweber 2008; Venkiteswaran et al. 2002; Narumiya et al. 1997; Parizi et al. 2000; Wysolmerski and Lagunoff 1990; Rosenfeldt et al. 2003; Zondag et al. 1998). Antisense oligonucleotide techniques to create depleted cortactin states result in a significant blunting of EC barrier response to S1P. Similarly studies also showed elevated TER readings in EC following overexpression of wild-type cortactin after administration of S1P. EC with a reduced expression of cortactin, when exposed to S1P, showed AFM elasticity patterns that closely resembled the elasticity patterns of unstimulated EC. It has been observed that in order to achieve peak S1P-induced barrier enhancement phosphorylation of cortactin at three essential tyrosine residues (Tyr421, Tyr466 and Tyr 482) is required. This phosphorylation is mediated through Src. When parallel studies are done in mutant strains of cortactin that lack these tyrosine residues, significantly lower TER patterns are obtained. As with cortactin, S1P-stimulated rapid displacement of nmMLCK to areas of active membrane ruffling acting in concert with the relocated cortactin combined actions essential for the optimal action of S1P. Thus, cortactin and nmMLCK play a critical role in the enhancement of vascular barrier integrity in response to S1P (Donati and Bruni 2006; Sun et al. 2009; Zhang et al. 1997).
Fig. 4.
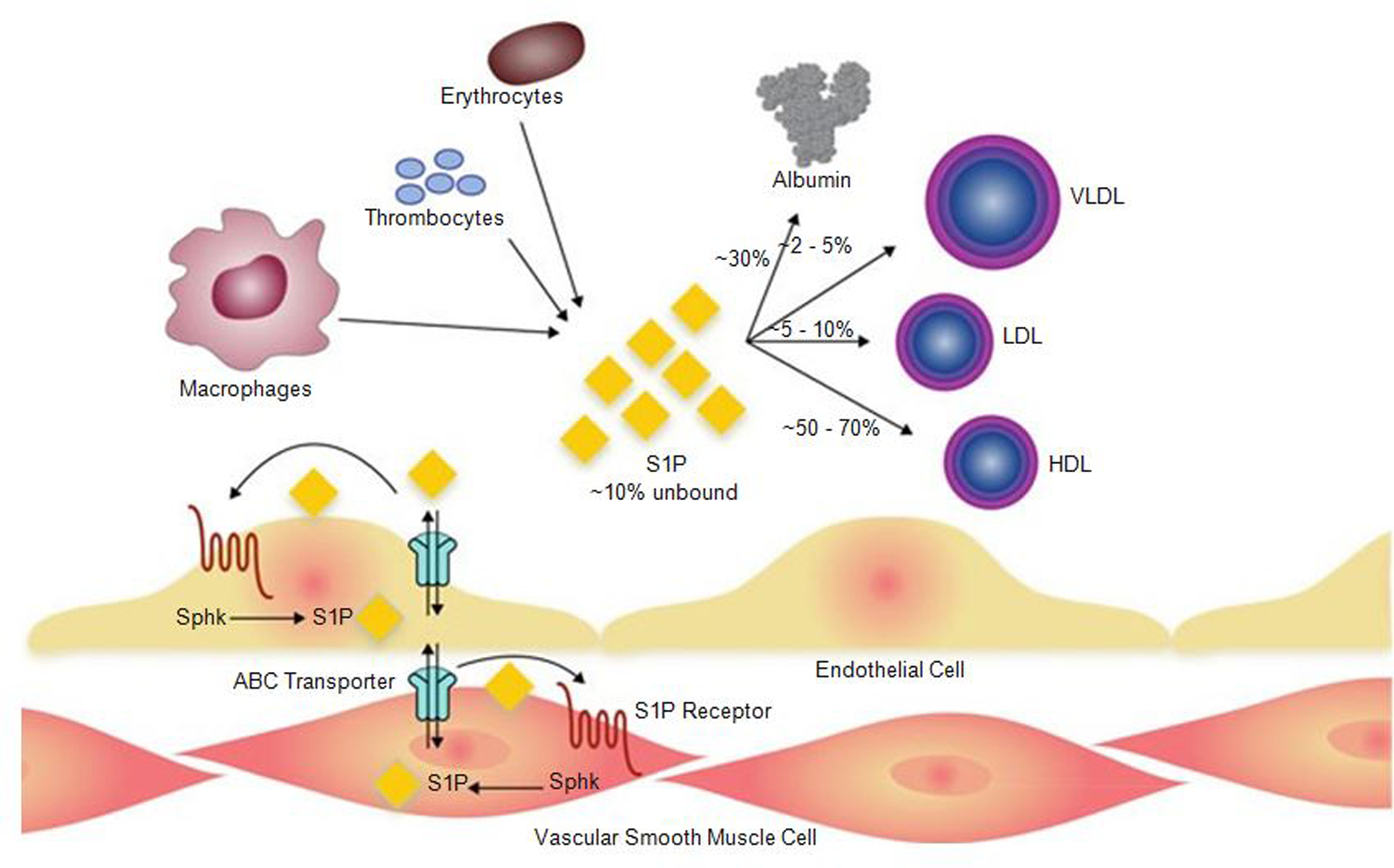
Secretion of S1P by erythrocytes, platelets, macrophages, and endothelium. Once secreted, most of the S1P is uptaken by serum albumin or various serum lipoproteins. Intracellular-produced S1P in ECs or vascular smooth muscle cells could be transported across the membrane by ABC transporters. HDL high-density lipoprotein, LDL low-density lipoprotein, Sphk sphingosine kinase, S1P sphingosine-1-phosphate, VLDL very low-density lipoprotein [Modified from Belvitch and Dudek (2012)]
6. S1P Signaling to Lung Endothelial Adherens Junctions and Focal Contacts
Highly specialized proteins serve as points of contact between the individual ECs and between the ECMs with three main types of junctional points of contact between the ECs: the adherens junctions (AJ or zonula adherens), tight junctions (TJ or zonula occludens), and gap junctions (GJ). AJ and TJ have very few junctional proteins in common and institute intracellular adhesion through the formation of paracellular zipper-like structures along their transmembrane adhesion sites (Bazzoni and Dejana 2004; Furuse et al. 1994; Lee et al. 2006; Ebnet et al. 2000; Mehta et al. 2002, 2005). The major structural protein of AJ in the EC is the vascular endothelial cadherin (VE-cadherin) composed of five extracellular cadherin domains and employing a Ca2-dependent mechanism to mediate homophilic interaction between adjacent ECs. The VE-cadherin cytoplasmic tail is similar to other cadherins and avidly binds to β-catenin or plakoglobin (ɣ-catenin). β-catenin or plakoglobin actively bind to α-catenin, an actin-binding protein, firmly anchoring the AJ to the actin cytoskeleton. α-catenin can also bind α-actinin and vinculin, further adding stability to the AJ complexes. An additional non-actin-binding player that also assists in AJ stability is p120, binding to a membrane proximal domain of VE-cadherin. VE-cadherin is essential in barrier function and normal vascular development as evident from VE-cadherin knockout mice, which do not survive embryonically due to immature vascular development. In mouse model, exposure to anti-VE-cadherin antibodies results in increased pulmonary vascular permeability; similarly, overexpression of VE-cadherin lacking mutant, or the chelation of extracellular Ca2+ by EDTA, also yields the same results along with EC barrier disruption. Severing interaction between β-catenin and VE-cadherin negatively alters the AJ junction formation and its adhesive strength with adjacent cells and the actin cytoskeleton. Studies in human EC show a substantial increase in the levels of VE-cadherin and β-catenin at the cell–cell contact region to enhance AJ assembly and interaction. Both protein complexes are critical to maintaining EC barrier function and signal transduction from within each cell to its surrounding matrix and neighboring cells (Sun et al. 2009; Bazzoni and Dejana 2004; Furuse et al. 1994; Lee et al. 1996; Ebnet et al. 2000; Mehta et al. 2002, 2005). Co-immunoprecipitation studies revealed the increased association of VE-cadherin with FAK and paxillin in S1P-challenged EC with enhancement of VE-cadherin interaction with α-catenin and β-catenin associated with the increased formation of FAK-β-catenin protein complexes. Depletion of β-catenin using specific siRNA resulted in complete loss of S1P effects on VE-cadherin association with FAK and paxillin rearrangement. These results demonstrate that S1P-induced endothelial barrier enhancement involves β-catenin-linked adherens junction/focal adhesion interaction (Hla and Brinkmann 2011; Sun et al. 2009; Bazzoni and Dejana 2004; Furuse et al. 1994; Lee et al. 1996; Quadri and Bhattacharya 2007; Shikata et al. 2003a, b; Venkiteswaran et al. 2002; Corada et al. 1999). Experiments involving human embryonic kidney cell lines show that overexpression of S1P increases the expression values of P and E-cadherin, but not α- and β-cadherin, to promote cell–cell aggregation through a Ca2-based mechanism. Similarly S1P silencing leads to an underexpression of both VE-cadherin and platelet endothelial cell adhesion molecule-1 (PECAM-1). Despite all this evidence regarding the important role of VE-cadherin in assisting in AJ junctional stability, a direct link between VE-cadherin and S1P-mediated barrier regulation is still vague; furthermore, recent studies have shown that VE-cadherin may not be involved in the rapid and immediate effects of S1P on the barrier regulation as shown through TER studies but may play a role in the delayed onset-sustained effects of S1P on barrier enhancement. Thus, the S1P/S1P1 signaling pathways not only plays a crucial role in the translocation of cadherin molecules and AJ assembly but also oversees the expression of integral junctional molecules (Bazzoni and Dejana 2004; Furuse et al. 1994; Gosens et al. 2004; Ebnet et al. 2000; Mehta et al. 2005; Quadri and Bhattacharya 2007; Corada et al. 1999; Carmeliet et al. 1999; Birukova et al. 2007; Narumiya et al. 1997; Vestweber et al. 2009).
The influence of S1P in relation is not limited to just AJ but also includes tight junctions (TJ). The TJ are positioned on the outer leaflets of the lateral membranes between adjacent cells. TJ regulates the movement of solutes across intercellular spaces (barrier function) and the movement of membrane proteins between the apical and basolateral domains of the plasma membrane (fence function). The TJ consists of a complex of proteins including claudins, occludins, and junctional adhesion molecules (JAM). The TJ anchors into the actin cytoskeleton through the interaction and binding of the occludins, claudins, and JAM with the zona occludens proteins (ZO-1, ZO-2, or ZO-3). Following stimulation by S1P, ZO-1 is reassigned to the lamellipodia and to the cell–cell junctions via the S1P1/Gi/Akt/ Rac pathway, whereas the barrier-enhancing actions of S1P are blunted by siRNA-induced downregulation of ZO-1 expression. So like the AJ, the TJ also plays an important role in barrier regulation in association with S1P (Donati and Bruni 2006; Bazzoni and Dejana 2004; Furuse et al. 1994; Gosens et al. 2004; Ebnet et al. 2000; Mehta et al. 2005; Mitra et al. 2005; Narumiya et al. 1997).
7. S1P Signaling to Lung Endothelial Focal Adhesions
Focal adhesions (FA) are a specific set of cellular sites that help anchor cells to the underlying ECM and play an essential role in the maintenance of the endothelial monolayer by adhering the cells to their underlying substrate and providing bidirectional signaling between the ECM and the EC cytoskeleton. Focal adhesions bridge the intracellular and extracellular space and are composed of extracellular matrix proteins, transmembrane proteins, and cytoplasmic focal adhesion plaques (Snider et al. 2010; Ebnet et al. 2000). Focal adhesions facilitate communication between the actin cytoskeleton and the extracellular space. FA is an amalgamation of integrin proteins, actin-binding structural proteins such as vinculin, talin, and α-actinin, adaptor proteins such as paxillin, and focal adhesion kinases (FAK). The integrins exist as multiple α- and β-glycoprotein chains that non-covalently link in parallel arrays to form different heterodimers. These different dimers then specify the ECM-binding target such as collagen, laminin, or fibronectin. FAK is a highly conserved cytoplasmic tyrosine kinase that is involved in the engagement of integrins and the assembly of FA through the breakdown of numerous downstream signals. S1P initiates a series of rapid signaling events in the EC membrane that are realized by downstream effectors into cytoskeletal changes and variations in barrier function. FAK also participates in these events at multiple levels and, with other effector molecules, is recruited to lipid rafts or CEMs and undergo phosphorylation and activation. These CEM signaling platforms are integral for S1P-mediated barrier regulation. S1P promotes FAK phosphorylation at specific sites (Y576), leading to FA disruption and relocation toward the periphery of the cell. S1P also mediates a temporary association between G-protein-coupled receptor kinase-interacting protein 1 (GITI1) and paxillin and induces relocation of the GIT2-paxillin complex toward the cortical ring of the cell (Mehta and Malik 2006; Peng et al. 2004; Ebnet et al. 2000; Quadri and Bhattacharya 2007; Narumiya et al. 1997; Dudek et al. 2007; Yuan et al. 2005; Birukova et al. 2004; Wysolmerski and Lagunoff 1990). In contrast to S1P, thrombin induces FAK phosphorylation at specific sites (Y397, Y576, Y925), resulting in translocation of FA proteins to stress fiber ends and EC barrier disruption. Inhibition of Src with Src-specific inhibitor PP2 abolishes S1P-induced FAK phosphorylation and migration of FA proteins that is potentially unique for specific signaling pathways (Mehta et al. 2005; Shikata et al. 2003b; Singleton et al. 2005; Mitra et al. 2005; Spiegel and Milstien 2003; Miura et al. 2000; Rosenfeldt et al. 2001; Mehta and Malik 2006).
8. Transactivation of S1PR1 in Lung Endothelial Signal Transduction
Activated protein C (APC) has the ability to induce cytoskeletal changes that resemble those produced by S1P. APC itself is an anti-inflammatory protein, and its recombinant human version is used in the clinical settings of severe sepsis. APC quickly increases the phosphorylation of endothelial MLC upon the attenuation of endothelial protein C receptor (EPCR) and induces robust actin-phospho-MLC, restructuring at the periphery of the cell and at the same time reducing the formation of central stress fibers. S1P1 is also phosphorylated by APC on the threonine residue 236 through an EPCR and PI3-kinase/AKT-dependent pathway to mediate Rac1-dependent cytoskeletal changes. On the other hand, ligation of S1P1 results in a blunting of the APC-mediated barrier enhancements, especially against thrombin. All this points toward the importance of APC and EPCR in the transactivation of S1P1-mediated signaling in barrier integrity enhancement (Tani et al. 2007; Xu et al. 2007; Zhang et al. 1997; Wang et al. 2009).
Our earlier studies were the first to link the angiogenic properties of S1P and S1P-receptor ligation to vascular barrier regulation and demonstrated that physiologic doses of S1P induce EC activation, marked cytoskeletal rearrangement, and stabilization of lung EC barrier function in vitro (Belvitch and Dudek 2012; Camp et al. 2009). This novel function for S1P was of particular relevance to clinical medicine as thrombocytopenia is well known to be associated with increased vascular leak (Peng et al. 2004; Shea et al. 2010), and while the mechanism of this effect was unknown, we demonstrated that activated platelets are an important source of S1P and directly enhance barrier function via S1P1 ligation (Arce et al. 2008). Platelets contain significant levels of sphingosine kinase but reduced levels of sphingosine lyase, thereby serving as enriched sources for the barrier-promoting S1P (Arce et al. 2008). Prior to the last decade, permeability-reducing strategies primarily consisted of cAMP augmentation, producing only modest barrier enhancement (Peng et al. 2004; Arce et al. 2008; Shea et al. 2010). More recently, a number of barrier-promoting agents have been identified that share common signal transduction mechanisms that are distinct from cAMP signals and target the endothelial actin cytoskeleton to facilitate barrier-restorative processes. We have conceptualized a paradigm whereby barrier recovery after edemagenic agonists involves development of a cortical actin ring to anchor cellular junctions and a carefully choreographed (but poorly understood) gap-closing process via formation of Rac GTPase-dependent lamellipodial protrusions into the paracellular space between activated ECs. Within these lamellipodia, signals are transduced to actin-binding proteins (nmMLCK and cortactin) and phosphorylated MLCs in spatial-specific cellular locations. Lamellipodia also require formation of focal adhesions (regulated by the cytoskeleton) critical to establishment of linkage of the actin cytoskeleton to target effectors that restore cell–cell adhesion and cell–matrix adhesion (Tani et al. 2007; Wojciak-Stothard et al. 2006; Xu et al. 2007; Birukova et al. 2004; Zhang et al. 1997; Wang et al. 2009; Hla 2004; Kihara and Igarashi 2008; Bode et al. 2010; Boguslawski et al. 2002; Fyrst and Saba 2010; Futerman and Riezman 2005; English et al. 2001). Another molecule that plays a role in the transactivation of S1P1 signaling is the glycosaminoglycan (GAG) called hyaluronan (HA). HA is composed of a high molecular weight HA (HMW-HA) and a low molecular weight HA (LMW-HA). HMW-HA has the ability to induce S1P1 phosphorylation via AKT to enhance barrier integrity. However, LMW-HA promotes Src and ROCK 1/2-mediated phosphorylation of S1P3 to cause barrier disruption. Blocking of S1P1 or S1P3 mutes the actions of HMW-HA and LMW-HA, respectively, on the endothelium in terms of barrier stability. Keeping the negative effects of S1P3 on the endothelium in mind, a novel approach toward enhancing barrier protection would be the blocking of S1P3. One such protein understudy is methylnaltrexone (MNTX), which is mu-opioid receptor antagonist that inhibits the actions of S1P3, and provides barrier protection against thrombin and LPS (Donati and Bruni 2006; McVerry and Garcia 2004; Tani et al. 2007; Xu et al. 2007; Owen et al. 2007; Singleton et al. 2006, 2007; Zhang et al. 1997; Wang et al. 2009; Hla 2004; Kihara and Igarashi 2008; Waterman-Storer et al. 1999).
9. S1PR3 Signaling to the Lung Endothelial Cytoskeleton and Loss of Vascular Integrity
Interestingly, S1P at elevated concentrations (>5 μM) results in S1PR3-dependent RhoA-mediated signaling and increased barrier permeability (Adyshev et al. 2011; Gosens et al. 2004; Narumiya et al. 1997; Birukova et al. 2004). A “conversation” elicited by ligation of G-protein-coupled receptors takes place between transmembrane components (such as large and small GTPases) and cytoskeletal proteins in membrane domains such as caveolin-enriched lipid rafts and Rac GTPase-dependent lamellipodia. These pathways induce EC cytoskeletal rearrangement resulting in enhanced junctional linkages between ECs as well as increased linkage of the cytoskeleton with the underlying extracellular matrix. These events provide the conceptual underpinning for the molecular targeting of these permeability-reducing therapeutic strategies. Barrier-restorative agonists (detailed in reference) evoke a carefully choreographed resolution of inflammation-mediated paracellular gaps by promoting the formation of lamellipodia, which protrude into the paracellular space driven by actin polymerizations with focal contacts, which reseal the gaps between activated ECs (Fig. 3). These lamellipodia contain a variety of actin-binding proteins such as the Ca2+/calmodulin-dependent non-muscle myosin light-chain kinase (nmMLCK), which triggers myosin ATPase activity, actin polymerization, and EC tension development. Increases in vascular permeability must ultimately be attributed to loss or disruption of endothelial intercellular junctions, in combination with a breakdown of the tethering forces characteristic of cell–cell or cell–matrix interactions, which result in paracellular vascular leakage (Zhang et al. 1997; Jaillard et al. 2005; Bode et al. 2010; Boguslawski et al. 2002; Fyrst and Saba 2010; Futerman and Riezman 2005).
Disruption of pulmonary barrier integrity and edema are cardinal characteristics of pulmonary pathologies. Airway administration of S1P induces the pulmonary vasculature leak phenomenon by disruption of epithelial tight junctions that is mediated through the S1P3 receptors. The disruption of tight junctions is accompanied by the development of gaps in the paracellular spaces and a disintegration of cytoplasmic plaques associated with tight junctions along with other integral membrane organizer proteins. This effect can be experimentally compounded by the addition of the pro-inflammatory cytokine TNF. The integral role of S1P3 receptors can be gauged in S1P3-null mice as they are resistant to S1P-induced vascular leakage, indicating a probable protective role of S1P3 antagonism. This role of S1P3 is in sharp contrast to that of S1P1 receptors that enhance the stability of vascular endothelial barrier. Interestingly, IV infusion of S1P inhibits pulmonary leakage in the pulmonary endothelium, when it is exposed to thrombin or LPS. This is again quite the opposite, as the effect of S1P when introduced via IT injection on pulmonary epithelium is to induce vascular leakage leading to acute pulmonary edema. Immunoreactivity studies in mice (WT and S1P3-null mice) have shown the presence of S1P3 receptors on all pulmonary epithelial surfaces and not on the pulmonary endothelium. When these mice were exposed to S1P, disruption was mainly seen in the epithelial tight junctions, indicating that S1P-induced, and S1P3-mediated, vascular disruption was epithelial in nature. In this complex interplay, Rho signaling has been assumed to play a very integral role in the molecular regulation of tight junctional integrity. S1P3 fosters actin cytoskeleton rearrangement by the activation of Gα12 and Gα13 via Rho signaling. Rho activity has been linked to the regulation of the cellular tight junctions as well as seen in the overexpression of constitutively active RhoA which results in a disarray of the structured tight junction morphology. This effect is mediated through ROCK1 (Rho-associated, coiled-coil containing protein kinase 1) which oversees the formation of stress fibers by maintaining the active state of myosin light chains by inactivating myosin light-chain phosphatase (MLCP), and it also stimulates LIM kinases to exert their effect on cofilin, by inhibiting it, ultimately resulting in a reorganization of actin cytoskeleton (Gon et al. 2005; Gosens et al. 2004; Birukova et al. 2004; Worthylake et al. 2001; Wang et al. 2009; Hla 2004; Fyrst and Saba 2010; Ohashi et al. 2000; Amano et al. 1996).
10. S1P and Lung Angiogenesis
The interface between EC barrier regulation and angiogenesis is an exciting area of vascular biology. New blood vessel formation, or angiogenesis, is a complex process involving EC activation, migration, maturation, and remodeling. These events may occur in a variety of contexts including during normal development and growth, in response to wound healing, or as part of the pathogenesis of a number of cancers and autoimmune diseases (Birukova et al. 2004; Boguslawski et al. 2002). Our initial studies determined that S1P is the most potent EC chemotactic agent present in serum and is ultimately involved in angiogenesis and vascular hemostasis through its ability to evoke various cell-specific responses (Singleton et al. 2007; Birukova et al. 2004; Bode et al. 2010; Boguslawski et al. 2002). In the setting of coagulation, S1P is abundantly released from platelets and, via its pleiotropic effects, potentially contributes to new blood vessel formation. This is evidenced by in vivo studies that establish S1P as remarkably effective in avian chorioallantoic membranes, in Matrigel-implanted plugs in mice (McVerry et al. 2004; Bazzoni and Dejana 2004; Corada et al. 1999; Argraves et al. 2004; Liu et al. 2000; English et al. 2001) and in the avascular mouse cornea. In contrast to VEGF-induced increases in EC permeability, we were the first to report that another angiogenic factor (S1P) can also produce EC barrier restoration and enhancement. S1P strongly enhances TER across human EC monolayers and significantly attenuates thrombin-induced barrier disruption while rapidly restoring barrier integrity in the isolated perfused murine lung (McVerry and Garcia 2004; Peng et al. 2004; Sammani et al. 2010; Mathew et al. 2011; McVerry et al. 2004). A single intravenous dose of S1P, given 1 h after intratracheal endotoxin administration, produced highly significant reductions in multiple indices of inflammatory lung injury, including vascular leak, as demonstrated in both murine (Peng et al. 2004; Sammani et al. 2010; Mathew et al. 2011; McVerry et al. 2004; English et al. 2001) and canine models of ALI (Quadri et al. 2003; Paik et al. 2004; Liu et al. 2001). Furthermore, S1P is a major serum component released by platelets and represents a key mechanism by which platelets nurture the microcirculation and preserve vascular integrity (McVerry and Garcia 2004; Peng et al. 2004; Sammani et al. 2010; Mathew et al. 2011; McVerry et al. 2004; Singleton et al. 2007; Shea et al. 2010; Sanna et al. 2006; English et al. 2001).
11. In Vivo Effects of S1P in Preclinical Models of Human Lung Disease
One of the main pathophysiological mechanisms involved in the genesis of various vascular disease conditions is endothelial dysfunction. Conditions ranging from atherosclerosis, hypertension, pulmonary hypertension, and cerebrovascular disease have a basis in endothelial barrier imbalance and dysregulation. Devastating inflammatory conditions such as acute lung injury (ALI) are characterized by significant and prolonged vascular permeability. Experimentally the intratracheal administration of lipopolysaccharide (LPS) has been used to mimic the clinical presentation of ALI in murine lung models. LPS induces all the responses associated with ALI including inflammatory lung injury, thickening of the alveolar wall, neutrophilic migration into the lung interstitium, and alveolar space. However, after appropriate spacing, the delivery of S1P prominently reduces the inflammatory landscape and attenuates neutrophilic migration into the LPS-exposed lung parenchyma. Similar effects have also been achieved usinga S1P analogue FTY720 in reducing inflammatory changes associated with LPS administration. Experiments on both murine and canine models have yielded promising results into the therapeutic potential of S1P in countering the devastating effects of barrier dysfunction-related pathologies of the vasculature. However, given studies implicating gene expression in the modulation of ALI, the exact manner in which S1P imparts protection may involve the reduction of inflammatory and/or innate immunity gene expression (Donati and Bruni 2006; McVerry and Garcia 2004; Sammani et al. 2010; Mathew et al. 2011; McVerry et al. 2004; Komarova et al. 2007; Mansoor and Melendez 2008; Chae et al. 2004; Brinkmann et al. 2002).
In addition to the abundant in vitro data describing the EC barrier-enhancing effect of S1P, the potential utility of S1P in restoring lung water balance in patients with inflammatory injury was underscored in studies involving small and large animal models of ALI in which S1P provided dramatic attenuation of LPS-mediated lung inflammation and permeability. Mice treated with S1P had significantly less histologic evidence of inflammatory changes/lung injury, with decreased neutrophil alveolitis on BAL and decreased lung myeloperoxidase (MPO) activity. Interestingly, mice treated with S1P after intratracheal LPS also showed an attenuated renal inflammatory response compared to control, measured by tissue MPO activity and Evans blue dye extravasation as a measure of capillary leak. S1P also protected against intrabronchial LPS-induced ALI and concomitant VILI in a canine model, with decreased shunt fraction, decreased BAL protein, decreased extravascular lung water, and improved oxygenation (Kihara and Igarashi 2008). Use of a large animal canine model allowed investigation of regional lung changes in ALI and the effect of S1P on these changes. Computed tomography (CT) scans of animals subjected to LPS/VILI found that animals treated with S1P had dramatic improvement in alveolar air content (with decreased edema) in all lung regions (Kihara and Igarashi 2008). Additional in vivo studies found that S1P protects against ventilator-induced lung injury (VILI) in a murine model as assessed by Evans blue dye extravasation (Sammani et al. 2010; Mathew et al. 2011; McVerry et al. 2004; Mansoor and Melendez 2008; Kihara and Igarashi 2008; Chae et al. 2004; Brinkmann et al. 2002; Rogers et al. 1989).
As a result, we are excited about the potential utility of FTY720, an unphosphorylated S1P analogue and a derivative of the natural immunosuppressant myriocin that has been recently described to cause peripheral lymphopenia by inhibiting cellular egress from lymphoid tissues. We demonstrated FTY to induce delayed endothelial barrier enhancement through a Gi-coupled receptor and to protect against murine inflammatory lung injury. Thus, targeting S1PR1 activation, either directly or via S1PR1 transactivation by agonists such as activated protein C and high molecular weight hyaluronan (both robustly barrier protective), or antagonism of S1PR3 as with methylnaltrexone appears to be promising strategies for attenuating the vascular leak associated with ALI (Peng et al. 2004; Sammani et al. 2010; McVerry et al. 2004; Camp et al. 2009; Yuan et al. 2005; Chae et al. 2004; Paik et al. 2004; Wu 2005; Waeber et al. 2004; Shea et al. 2010). MLC phosphorylation is also a key element in S1P-mediated barrier enhancement and occurs in a peripheral distribution within the cortical actin ring (Belvitch and Dudek 2012; Camp et al. 2009), providing strength to this spatially directed scaffolding force and enhancing cell–cell tethering as we described via atomic force microscopy. Immunofluorescence studies have also demonstrated this via the overexpression of Green Fluorescent Protein–nmMLCK at sites of barrier remodelling. (Birukova et al. 2004; Boguslawski et al. 2002). These observations serve to highlight the importance of the cellular location of cytoskeletal proteins in small and large animal models of ALI in which S1P provided dramatic attenuation of LPS-mediated lung inflammation and permeability. Mice treated with S1P had significantly less histologic evidence of inflammatory changes/lung injury, with decreased neutrophil alveolitis on bronchoalveolar lavage (BAL) and decreased lung myeloperoxidase (MPO) activity (Peng et al. 2004; Mathew et al. 2011; McVerry et al. 2004; Camp et al. 2009; Jacobson and Garcia 2007; Birukova et al. 2004, 2007; Dudek et al. 2007; Brinkmann et al. 2002). Similarly, mice treated with S1P after intratracheal LPS also showed an attenuated renal inflammatory response compared to control, measured by tissue MPO activity and Evan’s blue dye extravasation as a measure of capillary leak. S1P also protected against intrabronchial LPS-induced ALI and concomitant VILI in a canine model, with decreased shunt fraction, decreased BAL protein, decreased extravascular lung water, and improved oxygenation. Use of a large animal canine model allowed investigation of regional lung changes in ALI and the effect of S1P on these changes. Computed tomography scans of animals subjected to LPS/VILI found that animals treated with S1P had dramatic improvement in alveolar air content (with decreased edema) in all lung regions. Additional in vivo studies found that S1P protects against VILI in a murine model as assessed by Evan’s blue dye extravasation (Peng et al. 2004; Mathew et al. 2011; McVerry et al. 2004; Camp et al. 2009; Chae et al. 2004; Paik et al. 2004; Wu 2005; Waeber et al. 2004; Shea et al. 2010; Rosen and Goetzl 2005; Rosenfeldt et al. 2003).
We have also evaluated a potential role for S1P in ameliorating lung ischemia– reperfusion (IR) injury, a common sequelae of lung transplantation, which is characterized by alveolar damage, edema, and inflammation in donor lungs and is a significant cause of transplant failure. Utilizing a rat model of IR injury (pulmonary artery ligation and reperfusion), we determined that rats pretreated with S1P exhibited reduced lung vascular permeability and inflammation compared to controls. Lung myeloperoxidase activity, an index of parenchymal leukocyte infiltration, and levels of IL-6, IL-1b, and IL-2 were also attenuated in S1P-treated animals exposed to IR injury. Together, these findings suggest that S1P may serve as an effective permeability-reducing agent in diverse conditions that share an element of lung inflammatory burden (Peng et al. 2004; Sammani et al. 2010; Mathew et al. 2011; McVerry et al. 2004; Camp et al. 2009; Chae et al. 2004; Paik et al. 2004; Wu 2005; Waeber et al. 2004; Shea et al. 2010; Sanna et al. 2006). Clinically significant radiation-induced lung injury (RILI) is a common toxicity in patients administered thoracic radiotherapy. Although the molecular etiology is poorly understood, we previously characterized a murine model of RILI in which alterations in lung barrier integrity surfaced as a potentially important pathobiologic event and genome-wide lung gene mRNA levels identified dysregulation of sphingolipid metabolic pathway genes. We hypothesized that sphingolipid signaling components serve as modulators and novel therapeutic targets of RILI. Sphingolipid involvement in murine RILI was confirmed by radiation-induced increases in lung expression of sphingosine kinase (SphK) isoforms 1 and 2 and increases in the ratio of ceramide to sphingosine-1-phosphate (S1P) and dihydro-S1P (DHS1P) levels in plasma, bronchoalveolar lavage fluid, and lung tissue. Mice with a targeted deletion of SphK1 (SphK1(−/−)) or with reduced expression of S1P receptors (S1PR1(+/−), S1PR2(−/−), and S1PR3(−/−)) exhibited marked RILI susceptibility. Finally, studies of 3 potent vascular barrier-protective S1P analogues, FTY720, (S)-FTY720-phosphonate (fTyS), and SEW-2871, identified significant RILI attenuation and radiation-induced gene dysregulation by the phosphonate analogue, fTyS (0.1 and 1 mg/kg i.p., 2× per week) and to a lesser degree by SEW-2871 (1 mg/kg i.p., 2× per week), compared with those in controls. These results support the targeting of S1P signaling as a novel therapeutic strategy in RILI (Peng et al. 2004; Mathew et al. 2011; McVerry et al. 2004).
S1P-induced Rac activation, and cytoskeletal rearrangement produce increased linkage of actin to VE-cadherin and β-catenin, both important AJ components, as well as S1P-induced phosphorylation of focal adhesion-related proteins paxillin and focal adhesion kinase (FAK), with translocation of these proteins to the EC periphery, further implicating S1P-induced cell–cell adhesive changes as part of the mechanism of S1P-induced barrier enhancement (Quadri et al. 2003; Worthylake et al. 2001; Zhang et al. 1997; Spiegel and Milstien 2003; Kihara and Igarashi 2008). Despite profound attractiveness as a therapeutic agent targeting the endothelium in high permeability states, S1P has several attributes which limit its potential utility as a permeability-reducing strategy. With an affinity for ligation of the S1P3 receptor (S1PR3), intratracheal S1P has been implicated as a cause of pulmonary edema via endothelial/epithelial barrier disruption. S1P also causes bradycardia via ligation of cardiac S1P3 receptors. These findings generated increased interest in FTY720, a derivative of the natural immunosuppressant myriocin and a recently described immunosuppressive agent that causes peripheral lymphopenia by inhibiting cellular egress from lymphoid tissues. FTY720 is structurally similar (but not identical) to S1P and is phosphorylated by sphingosine kinase to FTY720-phosphate, which is an agonist at S1P receptors. This characteristic prompted investigation of the effect of FTY720 on EC barrier function. FTY720 did not have superior efficacy compared to mycophenolate mofetil in preventing renal transplant rejection, but it is in Phase III clinical trials as an immunosuppressant in multiple sclerosis patients. The clinical availability of FTY720 makes it attractive as a potential mediator of EC barrier function in patients with ALI. An in vivo study demonstrated that intraperitoneal FTY720 protected against intratracheal LPS in a murine model of ALI, as measured by Evans blue dye extravasation. Like S1P, FTY720 causes increased TER measurements in pulmonary EC, an effect which is abolished by pertussis toxin (implicating Gi-coupled receptor activation), and requires the generation of signaling components within membrane lipid rafts. Interestingly, however, the mechanism of FTY720-induced EC barrier enhancement diverges from the mechanism described for S1P in several ways including the delayed kinetics of the rise in TER compared to S1P. Decreased expression of the S1P1 receptor prevented S1P-induced increase in TER but did not affect FTY720-induced TER increases. Unlike S1P, FTY720 did not result in threonine phosphorylation of the S1P1 receptor, nor did inhibition of PI3 kinase prevent FTY720-induced EC barrier enhancement. Furthermore, FTY720 did not cause the increased intracellular calcium, the MLC phosphorylation, or the cytoskeletal rearrangement seen in response to S1P (Waeber et al. 2004). Downregulation of Rac or cortactin using siRNAs attenuated the barrier-enhancing effect of S1P, but not that of FTY720. Although FTY720 is an S1P receptor agonist, its mechanism of barrier enhancement is distinct from that of S1P and does not require the S1P1 receptor (Camp et al. 2009; Dudek et al. 2007; Chae et al. 2004; Paik et al. 2004; Wu 2005; Waeber et al. 2004; Shea et al. 2010; Liu et al. 2001).
12. Summary
Sphingosine-1-phosphate is a biologically active lipid growth factor secreted by erythrocytes, activated platelets, and other cells including endothelium that is integral to numerous vascular biological mechanisms ranging from cell differentiation, proliferation, motility, angiogenesis, and barrier regulation. S1P is the natural ligand for five G-protein-coupled receptors and is an integral intracellular secondary messenger exhibiting an intrinsically protective role in the pulmonary vasculature ameliorating agonist- or sepsis-induced pulmonary injury and vascular leakage. These vasculo-protective mechanisms involve an intricate interplay of S1P with other factors (such as MAPKS, ROCKs, Rho, Rac1) with rearrangement of the endothelial cytoskeleton to form strong cortical actin rings in the cell periphery and enhanced cell-to-cell and cell-to-matrix tethering dynamics. This cascade leads to reinforcement of focal adhesions and paracellular junctional complexes via cadherin, paxillin, catenins, and zona occludens. S1P through its interaction with Rac and Rho influence the cytoskeletal rearrangement indicated in the later stages of angiogenesis as a stabilizing force, preventing excessive vascular permeability. These properties translate into a therapeutic potential for S1P, as shown by its analogue FTY720, as a protective agent limiting the disruption of the vascular EC monolayer in the pulmonary microcirculation that results in accumulation of protein and inflammatory cell-filled fluid in the interstitial and alveolar compartments leading to pulmonary edema and ultimately respiratory failure.
Contributor Information
Taimur Abbasi, Department of Medicine, Institute for Personalized Respiratory Medicine, The University of Illinois at Chicago, Chicago, IL 60612, USA.
Joe G.N. Garcia, Earl Bane Professor of Medicine, Pharmacology & Bioengineering, Chicago, IL 60612, USA
References
- Adyshev DM, Moldobaeva NK, Elangovan VR, Garcia JG, Dudek SM (2011) Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell Signal 23(12):2086–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 271(34):20246–20249 [DOI] [PubMed] [Google Scholar]
- An S, Zheng Y, Bleu T (2000) Sphingosine 1-phosphate-induced cell proliferation, survival, and related signaling events mediated by G protein-coupled receptors Edg3 and Edg5. J Biol Chem 275(1):288–296 [DOI] [PubMed] [Google Scholar]
- Arce FT, Whitlock JL, Birukova AA, Birukov KG, Arnsdorf MF, Lal R, Garcia JG, Dudek SM (2008) Regulation of the micromechanical properties of pulmonary endothelium by S1P and thrombin: role of cortactin. Biophys J 95(2):886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argraves KM, Wilkerson BA, Argraves WS, Fleming PA, Obeid LM, Drake CJ (2004) Sphingosine-1-phosphate signaling promotes critical migratory events in vasculogenesis. J Biol Chem 279(48): 50580–50590 [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Dejana E (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Review. Physiol Rev 84(3):869–901 [DOI] [PubMed] [Google Scholar]
- Belvitch P, Dudek SM (2012) Role of FAK in S1P-regulated endothelial permeability. Microvasc Res 83(1):22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdyshev EV, Gorshkova I, Usatyuk P, Kalari S, Zhao Y, Pyne NJ, Pyne S, Sabbadini RA, Garcia JG, Natarajan V (2011) Intracellular S1P generation is essential for S1P-induced motility of human lung endothelial cells: role of sphingosine kinase 1 and S1P lyase. PLoS One 6(1):e16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Smurova K, Birukov KG, Usatyuk P, Liu F, Kaibuchi K, Ricks-Cord A, Natarajan V, Alieva I, Garcia JG, Verin AD (2004) Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol 201(1):55–70 [DOI] [PubMed] [Google Scholar]
- Birukova AA, Malyukova I, Poroyko V, Birukov KG (2007) Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol 293(1):L199–L211 [DOI] [PubMed] [Google Scholar]
- Bode C, Sensken SC, Peest U, Beutel G, Thol F, Levkau B, Li Z, Bittman R, Huang T, Tölle M, van der Giet M, Gräler MH (2010) Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem 109(6):1232–1243 [DOI] [PubMed] [Google Scholar]
- Boguslawski G, Grogg JR, Welch Z, Ciechanowicz S, Sliva D, Kovala AT, McGlynn P, Brindley DN, Rhoades RA, English D (2002) Migration of vascular smooth muscle cells induced by sphingosine 1-phosphate and related lipids: potential role in the angiogenic response. Exp Cell Res 274(2):264–274 [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277(24):21453–21457 [DOI] [PubMed] [Google Scholar]
- Camp SM, Bittman R, Chiang ET, Moreno-Vinasco L, Mirzapoiazova T, Sammani S, Lu X, Sun C, Harbeck M, Roe M, Natarajan V, Garcia JG, Dudek SM (2009) Synthetic analogs of FTY720 [2-amino-2-(2-[4-octylphenyl]ethyl)-1,3-propanediol] differentially regulate pulmonary vascular permeability in vivo and in vitro. J Pharmacol Exp Ther 331(1):54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98(2): 147–157 [DOI] [PubMed] [Google Scholar]
- Chae SS, Proia RL, Hla T (2004) Constitutive expression of the S1P1 receptor in adult tissues. Prostaglandins Other Lipid Mediat 73(1–2):141–150 [DOI] [PubMed] [Google Scholar]
- Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, Lampugnani MG, Martin-Padura I, Stoppacciaro A, Ruco L, McDonald DM, Ward PA, Dejana E (1999) Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci USA 96(17):9815–9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati C, Bruni P (2006) Sphingosine 1-phosphate regulates cytoskeleton dynamics: implications in its biological response. Biochim Biophys Acta 1758(12):2037–2048 [DOI] [PubMed] [Google Scholar]
- Dudek SM, Camp SM, Chiang ET, Singleton PA, Usatyuk PV, Zhao Y, Natarajan V, Garcia JG (2007) Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell Signal 19(8):1754–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D (2000) Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem 275(36):27979–27988 [DOI] [PubMed] [Google Scholar]
- English D, Garcia JG, Brindley DN (2001) Platelet-released phospholipids link haemostasis and angiogenesis. Review. Cardiovasc Res 49(3):588–599 [DOI] [PubMed] [Google Scholar]
- Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1994) Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol 127(6 Pt 1):1617–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Riezman H (2005) The ins and outs of sphingolipid synthesis. Review. Trends Cell Biol 15(6):312–318 [DOI] [PubMed] [Google Scholar]
- Fyrst H, Saba JD (2010) An update on sphingosine-1-phosphate and other sphingolipid mediators. Review. Nat Chem Biol 6(7):489–497, Erratum in Nat Chem Biol. 2010;6(9):689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108(5):689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gon Y, Wood MR, Kiosses WB, Jo E, Sanna MG, Chun J, Rosen H (2005) S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci USA 102(26):9270–9275 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gosens R, Schaafsma D, Meurs H, Zaagsma J, Nelemans SA (2004) Role of Rho-kinase in maintaining airway smooth muscle contractile phenotype. Eur J Pharmacol 483(1):71–78 [DOI] [PubMed] [Google Scholar]
- Hla T (2004) Physiological and pathological actions of sphingosine 1-phosphate. Review. Semin Cell Dev Biol 15(5):513–520 [DOI] [PubMed] [Google Scholar]
- Hla T, Brinkmann V (2011) Sphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology 76(8 Suppl 3):S3–S8 [DOI] [PubMed] [Google Scholar]
- Jacobson JR, Garcia JG (2007) Novel therapies for microvascular permeability in sepsis. Review. Curr Drug Targets 8(4):509–514 [DOI] [PubMed] [Google Scholar]
- Jaillard C, Harrison S, Stankoff B, Aigrot MS, Calver AR, Duddy G, Walsh FS, Pangalos MN, Arimura N, Kaibuchi K, Zalc B, Lubetzki C (2005) Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci 25(6):1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Igarashi Y (2008) Production and release of sphingosine 1-phosphate and the phosphorylated form of the immunomodulator FTY720. Review. Biochim Biophys Acta 1781(9):496–502 [DOI] [PubMed] [Google Scholar]
- Komarova YA, Mehta D, Malik AB (2007) Dual regulation of endothelial junctional permeability. Review. Sci STKE 412:re8. [DOI] [PubMed] [Google Scholar]
- Krump-Konvalinkova V, Yasuda S, Rubic T, Makarova N, Mages J, Erl W, Vosseler C, Kirkpatrick CJ, Tigyi G, Siess W (2005) Stable knock-down of the sphingosine 1-phosphate receptor S1P1 influences multiple functions of human endothelial cells. Arterioscler Thromb Vasc Biol 25(3):546–52 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Evans M, Hla T (1996) The inducible G protein-coupled receptor edg-1 signals via the G(i)/mitogen-activated protein kinase pathway. J Biol Chem 271(19):11272–11279 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S, Hla T (1998) Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 279(5356):1552–1555 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99(3):301–312 [DOI] [PubMed] [Google Scholar]
- Lee JF, Zeng Q, Ozaki H, Wang L, Hand AR, Hla T, Wang E, Lee MJ (2006) Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J Biol Chem 281(39):29190–29200 [DOI] [PubMed] [Google Scholar]
- Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 106(8):951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Verin AD, Wang P, Day R, Wersto RP, Chrest FJ, English DK, Garcia JG (2001) Differential regulation of sphingosine-1-phosphate-and VEGF-induced endothelial cell chemotaxis. Involvement of G(ialpha2)-linked Rho kinase activity. Am J Respir Cell Mol Biol 24(6):711–719 [DOI] [PubMed] [Google Scholar]
- Lucke S, Levkau B (2010) Endothelial functions of sphingosine-1-phosphate. Cell Physiol Biochem 26(1):87–96 [DOI] [PubMed] [Google Scholar]
- Mansoor M, Melendez AJ (2008) Recent trials for FTY720 (fingolimod): a new generation of immunomodulators structurally similar to sphingosine. Rev Recent Clin Trials 3(1):62–69 [DOI] [PubMed] [Google Scholar]
- Mathew B, Jacobson JR, Berdyshev E, Huang Y, Sun X, Zhao Y, Gerhold LM, Siegler J, Evenoski C, Wang T, Zhou T, Zaidi R, Moreno-Vinasco L, Bittman R, Chen CT, LaRiviere PJ, Sammani S, Lussier YA, Dudek SM, Natarajan V, Weichselbaum RR, Garcia JG (2011) Role of sphingolipids in murine radiation-induced lung injury: protection by sphingosine 1-phosphate analogs. FASEB J 25(10):3388–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVerry BJ, Garcia JG (2004) Endothelial cell barrier regulation by sphingosine 1-phosphate. Review. J Cell Biochem 92(6):1075–1085 [DOI] [PubMed] [Google Scholar]
- McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG (2004) Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med 170(9):987–993 [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB (2006) Signaling mechanisms regulating endothelial permeability. Physiol Rev 86(1):279–367, Review [DOI] [PubMed] [Google Scholar]
- Mehta D, Tiruppathi C, Sandoval R, Minshall RD, Holinstat M, Malik AB (2002) Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J Physiol 539(Pt 3):779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB (2005) Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem 280(17):17320–17328 [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Review. Nat Rev Mol Cell Biol 6(1):56–68 [DOI] [PubMed] [Google Scholar]
- Miura Y, Yatomi Y, Rile G, Ohmori T, Satoh K, Ozaki Y (2000) Rho-mediated phosphorylation of focal adhesion kinase and myosin light chain in human endothelial cells stimulated with sphingosine 1-phosphate, a bioactive lysophospholipid released from activated platelets. J Biochem 127(5):909–914 [DOI] [PubMed] [Google Scholar]
- Narumiya S, Ishizaki T, Watanabe N (1997) Rho effectors and reorganization of actin cytoskeleton. Review. FEBS Lett 410(1):68–72 [DOI] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275(5):3577–3582 [DOI] [PubMed] [Google Scholar]
- Ohmori T, Yatomi Y, Okamoto H, Miura Y, Rile G, Satoh K, Ozaki Y (2001) G(i)-mediated Cas tyrosine phosphorylation in vascular endothelial cells stimulated with sphingosine 1-phosphate: possible involvement in cell motility enhancement in cooperation with Rho-mediated pathways. J Biol Chem 276(7):5274–5280 [DOI] [PubMed] [Google Scholar]
- Owen KA, Pixley FJ, Thomas KS, Vicente-Manzanares M, Ray BJ, Horwitz AF, Parsons JT, Beggs HE, Stanley ER, Bouton AH (2007) Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. J Cell Biol 179(6):1275–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, Hla T (2004) Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev 18(19):2392–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parizi M, Howard EW, Tomasek JJ (2000) Regulation of LPA-promoted myofibroblast contraction: role of Rho, myosin light chain kinase, and myosin light chain phosphatase. Exp Cell Res 254(2):210–220 [DOI] [PubMed] [Google Scholar]
- Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG (2004) Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med 169(11):1245–1251 [DOI] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharya J (2007) Resealing of endothelial junctions by focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 292(1):L334–L342 [DOI] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharjee M, Parthasarathi K, Tanita T, Bhattacharya J (2003) Endothelial barrier strengthening by activation of focal adhesion kinase. J Biol Chem 278(15):13342–13349 [DOI] [PubMed] [Google Scholar]
- Rogers DF, Boschetto P, Barnes PJ (1989) Plasma exudation. Correlation between Evans blue dye and radiolabeled albumin in guinea pig airways in vivo. J Pharmacol Methods 21(4):309–315 [DOI] [PubMed] [Google Scholar]
- Rosen H, Goetzl EJ (2005) Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol 5(7):560–570 [DOI] [PubMed] [Google Scholar]
- Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ (2007) Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol 28(3):102–107 [DOI] [PubMed] [Google Scholar]
- Rosenfeldt HM, Hobson JP, Maceyka M, Olivera A, Nava VE, Milstien S, Spiegel S (2001) EDG-1 links the PDGF receptor to Src and focal adhesion kinase activation leading to lamellipodia formation and cell migration. FASEB J 15(14):2649–2659 [DOI] [PubMed] [Google Scholar]
- Rosenfeldt HM, Amrani Y, Watterson KR, Murthy KS, Panettieri RA Jr, Spiegel S (2003) Sphingosine 1-phosphate stimulates contraction of human airway smooth muscle cells. FASEB J 17(13):1789–1799 [DOI] [PubMed] [Google Scholar]
- Ryu Y, Takuwa N, Sugimoto N, Sakurada S, Usui S, Okamoto H, Matsui O, Takuwa Y (2002) Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular Rac activity and cell migration in vascular smooth muscle cells. Circ Res 90(3): 325–332 [DOI] [PubMed] [Google Scholar]
- Saba JD, Hla T (2004) Point-counterpoint of sphingosine 1-phosphate metabolism. Circ Res 94(6): 724–734 [DOI] [PubMed] [Google Scholar]
- Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, Sun X, Nunez L, Jacobson JR, Dudek SM, Natarajan V, Garcia JG (2010) Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol 43(4):394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H (2006) Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol 2(8): 434–441 [DOI] [PubMed] [Google Scholar]
- Schuchardt M, Tölle M, Prüfer J, van der Giet M (2011) Pharmacological relevance and potential of sphingosine 1-phosphate in the vascular system. Br J Pharmacol 163(6):1140–1162. doi: 10.1111/j.1476-5381.2011.01260.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM (2010) Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol 43(6):662–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata Y, Birukov KG, Birukova AA, Verin A, Garcia JG (2003a) Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J 17(15):2240–2249 [DOI] [PubMed] [Google Scholar]
- Shikata Y, Birukov KG, Garcia JG (2003b) S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J Appl Physiol 94(3):1193–1203 [DOI] [PubMed] [Google Scholar]
- Singleton PA, Dudek SM, Chiang ET, Garcia JG (2005) Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J 19(12):1646–1656 [DOI] [PubMed] [Google Scholar]
- Singleton PA, Lingen MW, Fekete MJ, Garcia JG, Moss J (2006) Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: role of receptor transactivation. Microvasc Res 72(1–2):3–11 [DOI] [PubMed] [Google Scholar]
- Singleton PA, Moreno-Vinasco L, Sammani S, Wanderling SL, Moss J, Garcia JG (2007) Attenuation of vascular permeability by methylnaltrexone: role of mOP-R and S1P3 transactivation. Am J Respir Cell Mol Biol 37(2):222–231 [DOI] [PubMed] [Google Scholar]
- Snider AJ, Orr Gandy KA, Obeid LM (2010) Sphingosine kinase: role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 92(6):707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4(5):397–407 [DOI] [PubMed] [Google Scholar]
- Sun X, Shikata Y, Wang L, Ohmori K, Watanabe N, Wada J, Shikata K, Birukov KG, Makino H, Jacobson JR, Dudek SM, Garcia JG (2009) Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res 77(3):304–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M, Ito M, Igarashi Y (2007) Ceramide/sphingosine/sphingosine 1-phosphate metabolism on the cell surface and in the extracellular space. Review. Cell Signal 19(2):229–237 [DOI] [PubMed] [Google Scholar]
- Venkataraman K, Thangada S, Michaud J, Oo ML, Ai Y, Lee YM, Wu M, Parikh NS, Khan F, Proia RL, Hla T (2006) Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J 397(3):461–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkiteswaran K, Xiao K, Summers S, Calkins CC, Vincent PA, Pumiglia K, Kowalczyk AP (2002) Regulation of endothelial barrier function and growth by VE-cadherin, plakoglobin, and beta-catenin. Am J Physiol Cell Physiol 283(3):C811–C821 [DOI] [PubMed] [Google Scholar]
- Vestweber D (2008) VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Review. Arterioscler Thromb Vasc Biol 28(2):223–232 [DOI] [PubMed] [Google Scholar]
- Vestweber D, Winderlich M, Cagna G, Nottebaum AF (2009) Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol 19(1):8–15 [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari V, Bourcier C, Boulter E, van Obberghen-Schilling E (2002) Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J Cell Sci 115(Pt 12):2475–2484 [DOI] [PubMed] [Google Scholar]
- Waeber C, Blondeau N, Salomone S (2004) Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Review. Drug News Perspect 17(6):365–382 [DOI] [PubMed] [Google Scholar]
- Wang L, Dudek SM (2009) Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res 77(1):39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, Surks HK (2009) ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res 104(4):531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED (1999) Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol 1(1): 45–50 [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Tsang LY, Paleolog E, Hall SM, Haworth SG (2006) Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290(6):L1173–L1182 [DOI] [PubMed] [Google Scholar]
- Worthylake RA, Lemoine S, Watson JM, Burridge K (2001) RhoA is required for monocyte tail retraction during transendothelial migration. J Cell Biol 154(1):147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MH (2005) Endothelial focal adhesions and barrier function. Review. J Physiol 569(Pt 2): 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysolmerski RB, Lagunoff D (1990) Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci USA 87(1):16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Waters CL, Hu C, Wysolmerski RB, Vincent PA, Minnear FL (2007) Sphingosine 1-phosphate rapidly increases endothelial barrier function independently of VE-cadherin but requires cell spreading and Rho kinase. Am J Physiol Cell Physiol 293(4):C1309–C1318 [DOI] [PubMed] [Google Scholar]
- Yang L, Yatomi Y, Miura Y, Satoh K, Ozaki Y (1999) Metabolism and functional effects of sphingolipids in blood cells. Br J Haematol 107(2):282–293 [DOI] [PubMed] [Google Scholar]
- Yatomi Y, Ruan F, Hakomori S, Igarashi Y (1995) Sphingosine-1-phosphate: a platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood 86(1):193–202 [PubMed] [Google Scholar]
- Yuan CS, Doshan H, Charney MR, O’connor M, Karrison T, Maleckar SA, Israel RJ, Moss J (2005) Tolerability, gut effects, and pharmacokinetics of methylnaltrexone following repeated intravenous administration in humans. J Clin Pharmacol 45(5):538–546 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Magnusson MK, Mosher DF (1997) Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol Biol Cell 8(8):1415–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Kalari SK, Usatyuk PV, Gorshkova I, He D, Watkins T, Brindley DN, Sun C, Bittman R, Garcia JG, Berdyshev EV, Natarajan V (2007) Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J Biol Chem 282(19):14165–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondag GC, Postma FR, Etten IV, Verlaan I, Moolenaar WH (1998) Sphingosine 1-phosphate signalling through the G-protein-coupled receptor Edg-1. Biochem J 330(Pt 2):605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]