

Abstract
Congenital heart disease (CHD), the leading birth defect worldwide, has a largely unknown etiology, likely to result from complex interactions between genetic and environmental factors during heart development, at a time when the heart adapts to diverse physiological and pathophysiological conditions. Crucial among these is the regulation of cardiomyocyte development and postnatal maturation, governed by dynamic changes in DNA methylation. Previous work from our laboratory has shown that exposure to the environmental toxicant tetrachlorodibenzo-p-dioxin (TCDD) disrupts several molecular networks responsible for heart development and function. To test the hypothesis that the disruption caused by TCDD in the heart results from changes in DNA methylation and gene expression patterns of cardiomyocytes, we established a stable mouse embryonic stem cell line expressing a puromycin resistance selectable marker under control of the cardiomyocyte-specific Nkx2-5 promoter. Differentiation of these cells in the presence of puromycin induces the expression of a large suite of cardiomyocyte-specific markers. To assess the consequences of TCDD treatment on gene expression and DNA methylation in these cardiomyocytes, we subjected them to transcriptome and methylome analyses in the presence of TCDD. Unlike control cardiomyocytes maintained in vehicle, the TCDD-treated cardiomyocytes showed extensive gene expression changes, with a significant correlation between differential RNA expression and DNA methylation in 111 genes, many of which are key elements of pathways that regulate cardiovascular development and function. Our findings provide an important clue toward the elucidation of the complex interactions between genetic and epigenetic mechanisms after developmental TCDD exposure that may contribute to CHD.
Keywords: DNA methylation, cardiomyocytes, TCDD, aryl hydrocarbon receptor, epigenetics
Congenital heart disease (CHD) is the most prevalent congenital abnormality worldwide contributing 25%–30% of all birth abnormalities (Armstrong and Bischoff, 2004). Despite emerging research linking heart development and disease, a comprehensive understanding of the etiology and biological mechanisms surrounding CHD pathogenesis remains unclear. Mutations in homeobox transcription factor genes that govern the early events of heart development have been associated with CHD and adult cardiovascular disease in humans and mice (Elliott et al., 2003; Jay et al., 2004; McElhinney et al., 2003; Pashmforoush et al., 2004; Schott et al., 1998; Tanaka et al., 1999). However, epidemiological evidence indicates that <15% of CHD cases are associated with Mendelian inheritance (van der Bom et al., 2011), suggesting a multifactorial etiology arising from complex interactions of genetic, epigenetic, and environmental factors during heart development (Hinton, 2013; Lage et al., 2012; Vecoli et al., 2014). The Developmental Origins of Health and Disease states that the perinatal environment enduringly shapes the structure, function, and metabolism of the adult organism (Barker, 2007). Therefore, exposure to environmental insults during fetal and neonatal life and their interactions with epigenetic and genetic developmental factors may present crucial contributors to CHD pathogenesis.
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor and a member of the basic-region-helix-loop-helix PER/ARNT/SIM (BHLH-PAS) superfamily of transcription factors (Puga, 2011). Members of this family function as environmental sensors (Furness et al., 2007), regulating various signaling pathways related to development, detoxification, immune response regulation, and homeostasis (Kewley et al., 2004). In the context of development, AHR expression is detectable as early as embryonic day (E) E7.5 in the mesoderm of early mouse embryos and after 3 days of differentiation of mouse embryonic stem (ES) cells (Wang et al., 2013). Both in vitro (Jones and Kennedy, 2009; Wang et al., 2013) and in vivo (Abbott et al., 1999; Aragon et al., 2008; Carreira et al., 2015b; Carro et al., 2013; Hofsteen et al., 2013; Puga, 2011; Yoshioka et al., 2011) experimental models have demonstrated that xenobiotic environmental ligands of AHR have adverse effects on the cardiovascular system, regulated through a complex network of endogenous AHR signaling pathways.
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the prototypical and most potent AHR ligand (Schmidt and Bradfield, 1996). Previous work in fish (Plavicki et al., 2013), birds (Walker and Catron, 2000), and mammals (Kopf and Walker, 2009) has shown that the young are more sensitive to TCDD than the adult, and that developmental exposure results in adverse outcomes in adult life. Exposure to TCDD in utero between mouse E7.5 and E11.5 has been shown to disrupt endogenous AHR functions and signaling pathways involved in cardiogenesis and cardiac and mitochondrial functions as early as E13.5 (Carreira et al., 2015b). In addition, activation of the expression of Ahr gene targets by TCDD during ES cell differentiation significantly impairs cardiomyocyte differentiation and perturbs networks responsible for cardiovascular homeostasis (Wang et al., 2013). NKX2-5 signaling networks, important in cardiomyocyte maturation (Rajala et al., 2011), are among the pathways affected by TCDD (Carreira et al., 2015b; Wang et al., 2013). These studies build on previous epidemiological evidence of the association of in utero TCDD exposure of mothers living near waste incinerators with higher incidence of cardiac malformations and lethal CHD (Dummer et al., 2003; Kuehl and Loffredo, 2006). Indeed, the developmental toxicity of TCDD is of concern to humans because pregnant women may transfer a fraction of their dioxin body burden to the fetus during pregnancy and to the infant via breastfeeding (Schecter et al., 2001).
The heart is the first organ to develop during embryogenesis and must adapt to diverse physiological and pathophysiological needs to facilitate organ growth and optimal contractile function (Kruger et al., 2006; Siedner et al., 2003; Taegtmeyer et al., 2010). During early development, cardiomyocytes arise from cardiac progenitors and mature with limited cell division after birth (Bergmann et al., 2009; Senyo et al., 2013). In order to respond to extracellular challenges and because of their limited regenerative capacity, several key gene expression programs tightly orchestrate specific developmental processes related to the maturation of cardiomyocytes and cardiac function. Among these, epigenetic modifications, including microRNAs, chromatin and histone modifications, and DNA methylation, have been implicated as modulators of cardiac gene expression in development and disease (Anand et al., 2013; Grueter et al., 2012; Hon et al., 2013; Wamstad et al., 2012; Zhang et al., 2002).
DNA methylation is a well-characterized regulator of gene expression that occurs at the 5′ cytosine of CpG dinucleotides, and governs the development of specific tissues, maintenance of cell and tissue identity, and genome stability (Bird, 2002; Jones, 2012). Although the majority of the genome is depleted of CpGs, the dinucleotides are mostly found in CpG islands that make up 70% of annotated mammalian promoters (Deaton and Bird, 2011). During cell division, DNA methylation patterns are maintained by DNA methyltransferase 1 (DNMT1), whereas de novo DNA methylation is mediated by DNMT3A and DNMT3B (Reik et al., 2001). Removal of DNA methylation involves oxidation of 5-methyl-cytosine, governed by the 10–11 translocation enzymes TET1-3 (Branco et al., 2012). Most recently, DNA methylation was found to be highly dynamic during cardiomyocyte maturation, with a characteristic demethylation wave occurring among enhancers and gene bodies of cardiomyocyte-specific genes (Gilsbach et al., 2014).
Most studies investigating the effect of TCDD on DNA methylation have focused primarily on adverse outcomes on biological processes such as spermatogenesis (Pilsner et al., 2017; Siddeek et al., 2018) and cancer (Jenkins et al., 2007) but few have addressed cardiovascular outcomes. Here, we tested the hypothesis that exposure of cardiomyocytes to TCDD alters DNA methylation and gene expression patterns critical for cardiomyocyte maturation.
MATERIALS AND METHODS
Culture and preparation of differentiated cardiomyocytes from mouse embryonic stem (ES) cells
Undifferentiated C57BL/6N-C2 ES cells (Gertsenstein et al., 2010) were maintained in ES medium containing high-glucose Dulbecco’s minimal essential medium (DMEM; Gibcol Carlsbad, CA) supplemented with 15% ES cell qualified serum (Knockout Serum Replacement; Gibco), 2 mM glutamine, 1% nonessential amino acids, 1% sodium pyruvate, 100 U/ml penicillin, 100 µg/ml streptomycin and 0.1 mM β-mercaptoethanol, and 1000 U/ml ESGRO leukemia inhibitory factor (LIF; Bioscience Research Reagents, Temecula, California). Cells were seeded on 0.1% gelatin-coated plates, incubated at 37°C (95% humidity, 5% CO2) and passaged every second or third day. The establishment and maintenance of the G418-resistant Nkx2-5-ES cell line containing the pNkx2-5PuroIRES2eGFP plasmid were performed using a modified version of the protocols for obtaining cardiomyocytes from mouse ES cells described by Yuasa et al. (2005) and Wang et al. (2012). Briefly, cells were transfected by the pNkx2-5PuroIRES2eGFP plasmid bearing as well a G418-resistance marker regulated by the CMV promoter, seeded on 0.1% gelatin-coated plates and ES medium, and allowed to attach for 24 h before adding 600 µg/ml G418. The transfected cells were selected for 3–5 passages to insure all cells not resistant to G418 were removed. The media was then replaced with ES medium without G418. After 24 h, cells were trypsinized and seeded into wells of gelatin-coated 6-well plates with 5 × 104 cells in ES medium. After 24 h, ES medium was replaced with differentiation medium containing LIF-free DMEM supplemented with 15% non-ES qualified fetal bovine serum and subjected to differentiation treatment sequentially as follows: 150 ng/ml Noggin for 1 day, 5 ng/ml BMP4 for the next 2 days, and then 3 µg/ml puromycin for 4 days. Thereafter, Nkx2-5-positive cells were selected by treatment with 3 µg/ml puromycin every 2–3 days and examined by microscopy until day 14 for the presence of beating cardiomyocytes.
Treatment
Nkx2-5-positive cells were treated for 24, 72, and 96 h with TCDD at a concentration of 5 nM, a concentration in the range of doses commonly used for tissue culture studies of the high-affinity AHR of C57BL/6 mice and established in our previous work with cardiomyocytes differentiated from mouse ES cells to be effective in AHR activation while causing no overt cytotoxicity up to a 10-nM dose (Wang et al., 2010). In addition, our choice of dose is relevant to our previous work investigating the disruption of endogenous AHR functions in vivo (Carreira et al., 2015a; Wang et al., 2010). Given that cardiomyocytes lose regenerative capacity and are effectively withdrawn from the cell cycle after birth (Sim et al., 2015), we believe that timepoints representing early treatment (24 h) and late treatment (72 and 96 h) would be most reflective of the changes being measured. TCDD was dissolved in DMSO (vehicle), which never exceeded 0.1% of the volume of medium in either treatment or vehicle control. Differentiated Nkx2-5-positive cells treated with vehicle for 24 h served as controls because our work has previously demonstrated the stability of the cardiomyocyte phenotype up to 14 days after differentiation (Wang et al., 2013). A population of undifferentiated, untreated parental ES cells was cultured and sampled in parallel to serve as the point-of-departure base line.
Total RNA isolation, reverse transcription, and real-time reverse transcription polymerase chain reaction (RT-qPCR)
Total RNA for RT-qPCR was extracted with the RNeasy Mini Kit (Qiagen, Valencia, California) according to the manufacturer’s specifications. Total RNA for RNA-seq was extracted using TRIzol Reagent (Life Technologies, Carlsbad, California) according to the manufacturer’s instructions. To prepare for RT-qPCR, first-strand complementary DNAs (cDNAs) were synthesized from 10 μg of total RNA in a volume of 15 μl containing 1× reverse transcriptase buffer, 7 mM random hexamers primer, 0.5 mM dNTP mix, 10 mM dithiothreitol, 5 mM magnesium chloride, 20 U RNase inhibitor (RNasin; Promega, Madison, Wisconsin), and 100 U SuperScript III reverse transcriptase (Invitrogen). Samples were denatured and annealed to the primer for 10 min at 70°C and reverse transcribed for 3 h at 42°C. Before amplification, the reverse transcriptase was inactivated by heating to 70°C for 15 min; RNA was hydrolyzed by incubation with 0.05 N sodium hydroxide at 70°C for 10 min and neutralized with 0.05 N hydrochloric acid (HCl); and the cDNA was precipitated with ethanol. The resulting cDNA products were dissolved in a final volume of 200 μl, and a 2-μl aliquot was used as template for subsequent quantification by real-time PCR amplification. PCR reactions were conducted in duplicate or triplicate in a total volume of 25 μl containing SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, New York) and 0.1 μM of each primer. Primer specifics for the genes tested (Ahr, Cyp1a1 [cytochrome P450 1A1], Acta2 [actin alpha 2, smooth muscle], Gata4 [GATA-binding protein 4], Kdr [kinase insert domain protein receptor], Mef2c [myocyte enhancer factor 2C], Myh6 [myosin, heavy polypeptide 6, cardiac muscle, alpha], Myh7 [myosin, heavy polypeptide 7, cardiac muscle, beta], Nkx2.5 [NK2 homeobox 5], Shox2 [short stature homeobox 2], Tnnt2 [troponin T2, cardiac type], Cd31 [cluster of differentiation 31], Hand2 [heart and neural crest derivatives expressed 2], and T [Brachyury]) are shown in Supplementary File 1a, and can also be found in our previous work (Wang et al., 2010, 2013).
Amplification was performed in an ABI 7500 real-time PCR system (Applied Biosystems); the reaction was heated to 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing elongation at 60°C for 60 s. Detection of the fluorescent product was carried out during the 72°C extension period, and emission data were quantified using threshold cycle (Ct) values. Ct values for all genes analyzed were determined in biological duplicates or triplicates, and means were determined from the average Ct values for each biological duplicate. All means were then normalized to values for Gapdh mRNA. The relative gene expression level to ES cells (ΔΔCt) was calculated as sample ΔCt (Ct of gene—Ct of Gapdh) relative to ES cell ΔCt (sample ΔCt—ES cell ΔCt). PCR product specificity from each primer pair was confirmed using melting curve analysis and subsequent polyacrylamide gel electrophoresis.
Immunofluorescence
To confirm the identity of Nkx2-5-positive cells, cells were seeded on 10-mm glass coverslips and fixed with 4% paraformaldehyde in PBS for 20 min. Coverslips were then stained with Hoechst solution. We examined the cells and captured the images using a Zeiss Axio microscope (Carl Zeiss Microscopy, Thornwood, New York). At least 5 fields were evaluated for each treatment group.
RNA-seq
Directional RNA-seq was performed by the Genomics, Epigenomics and Sequencing Core (GESC) at the University of Cincinnati. The RNA quality was determined by Bioanalyzer (Agilent, Santa Clara, California). To isolate the polyA RNA, NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs, Ipswich, Massachusetts) was used with a total of 1 μg of good quality total RNA as input. The NEBNext Ultra II Directional RNA Library Prep Kit (New England BioLabs) was used for library preparation to generate dUTP-based single-stranded library. The library was indexed and amplified under a PCR cycle number of 8. After library Bioanalyzer QC analysis and quantification via qPCR using NEBNext Library Quant Kit (New England BioLabs) combined with QuantStudio 5 Real-Time PCR System (Thermo Fisher, Waltham, Massachusetts), individually indexed libraries were proportionally pooled and sequenced using Illumina Hiseq sequencer (Illumina, San Diego, California). Under the sequencing setting of single read 1 × 51 bp, about 25 million pass filter reads per sample were generated.
DNA isolation and methyl-seq
DNA was extracted after each treatment timepoint using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Methyl-seq was performed by Genomics, Epigenomics and Sequencing Core at the University of Cincinnati. SureSelect Methyl-Seq Target Enrichment System for mouse (Agilent, Santa Clara, California) was used to prepare the sequencing library. First, a total of 1 μg high-quality genomic DNA quantified by Qubit assay (Thermo Fisher, Waltham, Massachusetts) was sheared by Covaris S2 focused-ultrasonicator (Covaris, Woburn, Massachusetts) to about 200 bp under the recommended settings, and validated by 2100 Bioanalyzer (Agilent). Next, the DNA fragments were end prepared and ligated to the methylated adaptor. The size of the ligated libraries was then validated by Bioanalyzer, followed by hybridization with biotin-labeled RNA-baits to capture the regions where methylation is known to be differentially regulated and impact gene regulation. After hybridization, libraries were captured with streptavidin beads and bisulfite modified with EZ DNA Methylation-Gold kit (Zymo, Irvine, California), and enriched with 8 cycles of PCR. These individually amplified libraries were labeled with unique indices by 6 cycles of PCR. After purification and size selection of the indexed libraries using AMPure XP beads (Beckman Coulter, Indianapolis, Indiana), the quality and quantity of the libraries were assessed by Bioanalyzer High Sensitivity DNA assay. To quantify the library concentration for the clustering generation, the libraries were qPCR analyzed by NEBNext Library Quant Kit (New England BioLabs, Ipswich, Massachusetts) using QuantStudio 5 Real-Time PCR System (Thermo Fisher, Waltham, Massachusetts). The sequencing was performed using Illumina Nextseq 550 sequencer under the sequencing setting of PE 2 × 150 bp to generate ∼60 M pass filter reads per sample.
RNA- and methyl-seq analysis
We performed differential gene expression analysis for the untreated parental ES cells, control cardiomyocytes treated with vehicle for 24 h, and cardiomyocytes treated with TCDD for 24, 72, and 96 h. We performed statistical analysis to identify differentially expressed genes comparing each group to cardiomyocytes treated with vehicle for 24 h using the quasi-likelihood F-test of read counts as implemented in the Bioconductor edgeR package (Robinson et al., 2010). All genes with fdr values < 0.05 were used for further analyses.
CpG methylated and unmethylated counts were summarized and annotated using relevant Bioconductor packages (Genomic Features, circlize, edgeR, and org. Mm.eg.db) for managing analysis of DNA methylation data. For these analyses, the difference in mean methylation ratio, β, between control cardiomyocytes and each of the other groups was used to characterize the direction of methylation change. Among differentially expressed genes, CpG sites adjacent to these genes with the most significant methylation changes were used for our final analyses, yielding 240 genes in total. To assess the correlation between differential methylation and gene expression, the Pearson’s correlation coefficient test was performed for each gene.
To understand the functional and biological significance of our findings, all genes with significant correlation between differential methylation and gene expression were analyzed using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, http://www.ingenuity.com; last accessed September 28, 2020).
Statistical analysis
All statistical analyses were performed using R version 4.0.0. Significant changes in gene expression for each group compared with control treated with vehicle for 24 h were measured using the quasi-likelihood F test. This test reflects the uncertainty in estimating the dispersion for each gene and provides more robust and reliable error rate control when the number of replicates is small (Strimmer, 2003). Differential methylation was assessed using the methylKit R package. Significant changes in gene expression and methylation at a CpG site were selected based on a false-discovery rate-adjusted p-value <.05, adjusting for multiple comparisons A significant correlation between differential gene expression and differential methylation was determined if the log10 p-value > 1.301.
RESULTS
Differentiated and Selected pNkx2-5PuroIRES2eGFP Cells Faithfully Express a Suite of Cardiomyocyte Markers
We have previously shown that the AHR/TCDD axis disrupts the expression of homeobox transcription factors that control key developmental cardiac events in cardiomyocytes (Wang et al., 2013). If dynamic DNA methylation plays a role in the establishment of cardiomyocyte maturation patterns, it is reasonable to expect that TCDD treatment may also affect these patterns. To that end, we aimed to characterize the role of DNA methylation in gene expression changes resulting from TCDD treatment of selected cardiomyocytes differentiated from murine ES cells. Because less than 30%–40% of all heart cells are cardiomyocytes (Wang et al., 2010), to ensure that we tracked only cardiomyocytes, we established the pNkx2-5PuroIRES2eGFP cell line, a stable ES cell line that expresses the selection markers puromycin resistance and eGFP under the control of the promoter for Nkx2-5, a well-characterized marker for cardiomyocytes (Serpooshan et al., 2017) that is also among the known homeobox transcription factors affected by TCDD (Wang et al., 2013). After differentiation of the cells and selection with puromycin, cardiomyocyte characteristics were confirmed in this cell line through expression of cardiomyocyte markers Nkx2-5, Myh6, Myh7, Tnnt2, Brachyury, Kdr, Hand2, and Gata4 via qPCR (Figure 1A), and of eGFP via immunofluorescence (Figure 1B). As expected for cardiomyocytes, Ahr was also expressed, while the AHR-responsive gene, Cyp1a1, was relatively lower in expression compared with that found in ES cells. In addition, Acta2 was expressed at relatively low levels compared with other cardiomyocyte markers, as expected because this gene was previously shown to be disproportionately expressed in ventricular-like cardiomyocytes compared with the rest of the population (Potta et al., 2010). Unexpectedly, Mef2c and Shox2 were expressed at lower, but approximately similar levels to that found in ES cells. Because these genes are also involved in cardiac morphogenesis and the development of the sinoatrial node (Dal-Pra et al., 2019; Scavone et al., 2013), it is possible that the expression of these genes in our findings are reflective of relatively early maturation of these cardiomyocytes. Finally, Cd31 was relatively lower in expression compared with that found in ES cells, as expected because ES cells negative for Cd31 expression have the greatest potential for cardiomyogenic differentiation (Pfister et al., 2005).
Figure 1.

Cells differentiated from mouse ES cells containing the plasmid pNkx2-5ProIRES2eGFP and selected for puromycin resistance express mesodermal markers characteristic of cardiomyocytes. RT-qPCR performed against Ahr (aryl hydrocarbon receptor), Cyp1a1 (cytochrome P450 1A1), Acta2 (actin alpha 2, smooth muscle), Gata4 (GATA-binding protein 4), Kdr (kinase insert domain protein receptor), Mef2c (myocyte enhancer factor 2C), Myh6 (myosin, heavy polypeptide 6, cardiac muscle, alpha), Myh7 (myosin, heavy polypeptide 7, cardiac muscle, beta), Nkx2.5 (NK2 homeobox 5), Shox2 (short stature homeobox 2), Tnnt2 (troponin T2, cardiac type), Cd31 (cluster of differentiation 31), Hand2 (heart and neural crest derivatives expressed 2), and T (Brachyury) (A). Emission data were quantified using threshold cycle (Ct) values. Ct values for all genes analyzed were determined in biological duplicates or triplicates, and means were determined from the average Ct values for each biological duplicate. All means were normalized to Gapdh. The relative gene expression level to embryonic stem (ES) cells (ΔΔCt) was calculated as sample ΔCt (Ct of gene – Ct of Gapdh) relative to ES cell ΔCt (sample ΔCt – ES cell ΔCt). Immunofluorescence confirming eGFP driven by Nkx2-5 (B–D). Scale bar represents 100 µm at 40×.
TCDD Treatment Disrupts Cardiomyocyte Gene Expression and DNA Methylation Patterns
Cardiac progenitors give rise to cardiomyocytes that mature with a limited number of cell divisions after birth (Bergmann et al., 2009; Senyo et al., 2013). Because of this limited regenerative capacity, their response to extracellular challenges is tightly orchestrated by key gene expression programs that control developmental processes related to cardiomyocyte maturation and cardiac function. Primary among these are dynamic changes in DNA methylation that regulate cardiomyocyte development and postnatal maturation (Wang et al., 2013). To test the hypothesis that the disruption caused by TCDD in cardiomyocytes results from changes in DNA methylation and gene expression patterns, we used global gene expression and DNA methylation profiling at different times of treatment to characterize the effect of TCDD treatment on gene expression and DNA methylation in the Nkx2-5-positive pNkx2-5PuroIRES2eGFP cardiomyocytes. To analyze gene expression and DNA methylation changes across time, we compared the control Nkx2-5-positive cells treated with vehicle for 24 h with untreated ES cells as well as Nkx2-5-positive cells treated with 5 nM TCDD for 24, 72, and 96 h. Among all comparisons with vehicle control Nkx2-5-positive cells, we found 4821 genes with significant differences in gene expression (Supplementary File 1b). Differential gene expression by sample for each group is shown in Supplementary File 1c. In addition, significant DNA methylation differences were found in ES cells, as well as in Nkx2-5-positive cells treated with TCDD for 72 and 96 h (fdr < 0.05). Specifically, in ES cells, there were 2619 CpG sites with significant differences in DNA methylation compared with control Nkx2-5-positive cells (Supplementary File 2a). In Nkx2-5-positive cells treated with TCDD, there were 573 CpG sites with significant differences after 72 h (Supplementary File 2 b) and 3026 CpG sites with significant differences after 96 h (Supplementary File 2c). There were no significant DNA methylation differences in the comparison of Nkx2-5-positive cells treated for 24 h with vehicle or with TCDD.
To determine whether there was any overlap between differentially expressed genes and differentially methylated genes, we first defined genes that were differentially methylated by mapping differentially methylated CpG sites to the transcription start site (TSS) of the nearest gene. Among the 4821 genes that were differentially expressed, 240 were also differentially methylated (Figure 2). In addition, 565 genes that were differentially methylated were not differentially expressed. There were 4581 genes that were differentially expressed, but not differentially methylated (Supplementary File 3a).
Figure 2.
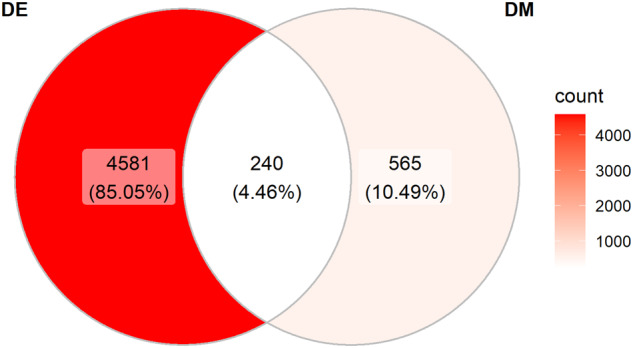
Venn diagram illustrating overlap of differentially expressed and differentially methylated genes. ES cells and Nkx2-5 genes that were differentially methylated were defined by mapping differentially methylated CpG sites to the transcription start site (TSS) of the nearest gene. DE, differentially expressed; DM, differentially methylated.
During early embryogenesis in mammals, DNA demethylation occurs at a genome-wide level from fertilization until the blastocyst stage, when ES cells undergo changes that prepare them for differentiation and reestablishment of DNA methylation patterns in a cell-specific manner (Li et al., 2018). Consequently, we expected that ES cells, derived from preimplantation blastocysts, would be mostly hypomethylated on a global scale relative to differentiated cells. Compared with vehicle control Nkx2-5-positive cells, CpG sites for ES cells were divided into two large clusters of hypomethylation and two smaller clusters of hypermethylation These groups included a total of 240 differentially expressed genes (Supplementary File 3b) that mapped within −550 to + 150 kb from CpG sites with the most significant methylation changes (Figure 3A). Indeed, small levels of methylation are to be expected for control of differentiation programs and the maintenance of epigenetic memory (Cheng et al., 2015). These patterns were accompanied by higher expression levels in adjacent genes for all clusters in ES cells compared with Nkx2-5 positive vehicle control cells. The cluster with the greatest degree of hypomethylation in CpG sites for ES cells showed a high prevalence of promoters occurring within 200 kb upstream of the TSS. Nkx2-5-positive cells treated with TCDD for 24 h showed a modest level of hypermethylation at CpG sites overall, occurring in two clusters, and similarly modest level of hypomethylation in the two other clusters, compared with control (Figure 3B). The majority of CpG-containing promoter regions tended to be hypermethylated compared with control, whereas hypomethylated CpGs tended to occur further away from the adjacent TSS, mostly in intronic and coding regions. The same patterns and clusters occurred after 72 h of TCDD treatment, with overall higher degrees of DNA methylation among the same clusters with hypermethylation compared with 24 h of treatment (Figure 3C). After 96 h of treatment, there was a near-complete loss of the demethylation patterns characteristic of cardiomyocytes, with all clusters becoming significantly hypermethylated compared with control (Figure 3D).
Figure 3.

Comparison of gene expression and methylation changes from RNA-seq and DNA methyl-seq analyses between vehicle-treated Nkx2-5-positive cells and ES cells (A), and Nkx2-5-positive cells treated with TCDD for 24 (B), 72 (C), and 96 (D) h. type, cell type; methylation, mean methylation ratio; direction, direction of methylation change; expression, expression in rpkm; −log10(cor_p), correlation significance; location, genomic location; dist_tss, distance from transcription start site (TSS). Each experimental group had 2 biological replicates. Significance of correlation between DNA methylation and gene expression changes was measured by Pearson’s coefficient test, with threshold of significance at −log10 p ≥ 1.301.
Correlation Between TCDD-Induced Disruption of Gene Expression and Changes in DNA Methylation
We used Pearson’s correlation coefficient analysis to investigate whether there was a correlation between changes in methylation at CpG sites and changes in adjacent gene expression for the 240 targets in which differential DNA methylation and gene expression overlapped. Among all comparisons, there were significant correlations between DNA methylation and expression changes at 111 adjacent genes (Figs. 3A–D; Supplementary File 4a–d). Significant negative correlations between DNA methylation and gene expression occurred mainly among intronic regions, whereas positive correlations occurred among promoter regions up to 1000 kb from the TSS (Supplementary File 4a–d; Figs. 3B–D). In ES cells, close to 100% of all CpG sites in all genomic regions were hypomethylated (Figure 4A). Nearly 65% of these sites were associated with upregulation of adjacent genes. Similar patterns for ES cells also existed among introns. (Figure 4B). Among coding regions, the association between hypomethylation and upregulation for a gene in ES cells was even stronger, encompassing 80% of all genes (Figure 4C). Among promoter regions, hypomethylation of CpG sites in ES cells appeared to be more strongly correlated with downregulation of adjacent genes than among any other genomic region (Figure 4D). Among cardiomyocytes treated with TCDD for 24 h, little correlation between changes in DNA methylation and gene expression was observed among genomic regions (Figs. 4A–D). As time of TCDD treatment was increased to 72 and 96 h, there was progressively higher hypermethylation among CpG sites that tended to correlate with downregulation among introns and coding regions (Figs. 4B and 4C). Among promoters, the progressively higher hypermethylation observed at CpG sites tended to correlate with both up- and down-regulation of genes. Taken together, TCDD seems to affect the genomic region-specific correlation patterns between DNA methylation and gene expression that were found in previous findings in cardiomyocytes in vivo (Gilsbach et al., 2014). Specifically, TCDD appears to cause progressively increasing methylation which affects gene expression in a genomic-region-specific manner.
Figure 4.

Direction of methylation and gene expression changes among the 111 targets in which differential methylation and gene expression are significantly correlated in all genomic regions (A), introns (B), coding regions (C), and promoters (D). Hypo + up, hypomethylated and upregulated gene expression; Hypo + down, hypormethylated and downegulated gene expression; Hyper + up, hypermethylated and upregulated gene expression; Hyper + down, hypermethylated and downregulated gene expression. Each experimental group had 2 biological replicates.
Functional Analyses of Correlated Gene Expression and DNA Methylation Changes in Cardiomyocytes Resulting From TCDD Treatment
DNA methylation has been shown to play an important role in cardiomyocyte maturation and function (Gilsbach et al., 2014). If TCDD treatment results in increased DNA methylation that gives rise to downregulation of genes, it is reasonable to expect that genes important in cardiac function would be affected. To test this hypothesis, we input the RNA-seq data for the 111 genes differentially methylated and expressed into the Ingenuity Knowledge Base (Ingenuity Pathway Analysis http://www.ingenuity.com) to analyze the effects of TCDD treatment on biological function. In ES cells and among all timepoints of TCDD treatment, the most significant change in biological function compared with controls took place in cell signaling and cardiovascular development and function, specifically the aggregation of cells, angiogenesis, vasculogenesis, and development of vasculature. (Figs. 5A–D). Among the genes involved in angiogenesis from our analyses, differential methylation of Kit, Enpp2, and Klf5 has been implicated in various cancers, specifically breast cancer and pulmonary blastoma (Janostiak et al., 2018; Kamalakaran et al., 2011; Liu et al., 2020; Macher-Goeppinger et al., 2011). To further extend these observations, we searched the Ingenuity Knowledge Base for upstream regulatory molecules of the genes involved in these functions and found that Ctnnb1, the gene coding for catenin-β1, an integral part of the canonical WNT signaling pathway (Linask et al., 2005), was predicted to be inhibited at all timepoints of TCDD treatment with a significant p-value (Supplementary File 5a–c). These results are consistent with previous findings in our laboratory that predicted that WNT signaling would be affected by TCDD (Wang et al., 2013). In addition, among the genes that we found to be differentially expressed after TCDD exposure, Kit, involved in angiogenesis and hematopoiesis, and Nrp2, involved in cardiovascular development, are regulated by WNT/CTNNB1 signaling (Supplementary File 5a) and implicated in cancers such as pulmonary blastoma and biliary tract cancer (Macher-Goeppinger et al., 2011; Papadopoulou et al., 2018).
Figure 5.

Biological functions that differ from vehicle-treated Nkx2-5-positive cells in ES cells (A), and after 24 (B), 72 (C), and 96 (D) h of TCDD treatment in Nkx2-5-positive cells. A z-score represents the difference between the mean change in expression for a subset of genes in a category and the population mean in units of standard error. A z-score < 0 represents downregulation and a z-score > 0 represents upregulation of the respective functions. Significance is measured by log10 p-value, representing the probability that a z-score for a given category occurs by chance, with a threshold of log p < −1.301.
DISCUSSION
This study assesses the impact of TCDD on DNA methylation dynamics that are essential for cardiomyocyte maturation. The primary pitfall in interpreting data elucidating epigenetic and regulatory patterns in a heterogenous population of differentiating ES cells is lineage diversity. To circumvent this issue, we adopted a promoter-mediated dominant selection system, previously established for obtaining cardiomyocytes from mouse ES cells by inhibition of bone morphogenetic protein (BMP) signaling (Yuasa et al., 2005). When cultured, this ES cell line derivative showed expression patterns that are consistent with hallmarks of the cardiomyocyte lineage. Distinctive DNA methylation features of cardiomyocytes and ES cells were previously identified using bisulfite sequencing (Gilsbach et al., 2014). For example, a hallmark of maturing cardiomyocytes is a demethylation wave occurring in the coding regions of cardiomyocyte genes, in parallel with de novo DNA methylation, enduringly shaping the epigenome after birth. Consistent with these findings, our data show that vehicle-treated control cardiomyocyte-like Nkx2-5-positive differentiated cells were significantly hypomethylated compared with ES cells. For the first time, our data demonstrate that TCDD treatment in a cardiomyocyte-like Nkx2-5-positive differentiated cell line disrupted global DNA methylation and gene expression patterns. Specifically, as time of TCDD treatment increased to 96 h, cells became progressively more hypermethylated in nearly all regions compared with control, suggesting a strong departure from the typical prevalence of demethylation patterns in cardiomyocytes. In addition, although previous findings have shown prevalent CpG hypomethylation in neonatal and adult cardiomyocytes correlated with increased expression (Gilsbach et al., 2014), our findings suggest that TCDD may impair the demethylation wave found to be associated with gene expression among gene bodies. Althhough the extent to which DNA methylation changes throughout the cell cycle is currently unknown (Vandiver et al., 2015), given that the demethylation wave among enhancers and gene bodies, governed by the oxidation of 5-methyl-cytosine from TET1-3 during DNA replication, is a characteristic of cardiomyocyte maturation (Gilsbach et al., 2014), it may be possible that impaired activity of the enzymes may play a role in TCDD-induced hypermethylation, progressively increasing with every round of DNA replication and loss of TET activity. Further studies that investigate hydroxymethylation changes induced by TET enzymes, targeting some of our top candidate genes for which there was progressive hypermethylation, can address this hypothesis. In addition, because DNA methylation maintenance or loss is time dependent and related to cell proliferation (Vandiver et al., 2015), it seems reasonable to conclude that the lack of TCDD-induced DNA methylation changes after 24 h of treatment may be due to insufficient time for enough cell proliferation to cause these changes, further compounded by the progressive loss of regenerative capacity and withdrawal from the cell cycle by cardiomyocytes (Sim et al., 2015).
Previous studies have suggested that transcription factor activity, together with histone marks such as poised H3K4me1 and active H3K27 could be key factors for active DNA demethylation of enhancer regions (Feldmann et al., 2013; Stadler et al., 2011). Given the established presence of these marks in demethylated enhancers among newborn cardiomyocytes (Gilsbach et al., 2014), it is reasonable to expect that TCDD may also alter these marks when giving rise to hypermethylation. In addition, though the effect of TCDD on DNA methylation during cardiogenesis has not previously been shown, there is emerging evidence for TCDD disrupting DNA methylation patterns important in other developmental processes. Recently, TCDD exposure was shown to cause demethylation at the promoter of the AHR target gene Cyp1a1, facilitated by the recruitment of the 10–11 translocation enzymes TET2 and TET3, involved in the removal of DNA methylation by oxidation of 5-methyl-cytosine (Amenya et al., 2016). Furthermore, TCDD caused hypermethylation of DNA in murine CD8+ T cells and the rat Brca1 promoter, causing adverse responses to influenza infection and increased incidence of breast cancer respectively in adulthood (Aluru et al., 2015; Boule et al., 2015). Finally, during the in vitro differentiation of ES cells, TCDD exposure caused the downregulation of Dnmt3a and Dnmt3b, suggesting that endogenous AHR signaling may play a role in de novo DNA methylation (Wang et al., 2013).
Among the 4821 genes differentially expressed in our findings, only 240 genes were differentially methylated, suggesting that other epigenetic factors may account for the differential expression found in a large fraction of genes. Indeed, several other epigenetic processes such as microRNAs (Grueter et al., 2012) as well as chromatin and histone modifications (Anand et al., 2013; Wamstad et al., 2012; Zhang et al., 2002) have been implicated as modulators of gene expression during cardiac maturation and disease, necessitating the need for future studies to ascertain the extent to which these epigenetic factors play a role in TCDD-induced differential gene expression.
Interestingly, there was also a significant correlation between DNA methylation and gene expression among 111 genes, with a negative correlation mostly among promoters and a positive correlation among coding (genic) regions. Our findings are consistent with previous evidence in maturing cardiomyocytes indicating that genic CpG methylation correlates with gene expression and active histone marks, whereas a negative correlation occurs in promoter and enhancer regions (Gilsbach et al., 2014). Indeed, although the significance of DNA methylation in genic regions is only recently emerging, studies have shown both positive and negative correlations of gene expression and DNA methylation in these regions (Jones, 2012). For example, demethylation of gene bodies downstream of the TSS was identified in highly expressed genes for neurons, hematopoietic stem cells, and other cell lineages (Jeong et al., 2014; Lister et al., 2013; Xie et al., 2013). On the other hand, in human ES cells, mouse oocytes, and B lymphocytes, a positive correlation has been shown between DNA methylation and gene expression (Ball et al., 2009; Kobayashi et al., 2012; Lister et al., 2009).
When we input these 111 genes into the Ingenuity Knowledge Base for pathway analysis, we found that TCDD disrupted cell signaling and cardiovascular development functions including aggregation of cells, angiogenesis, vasculogenesis, and development of vasculature. Emerging studies using in vitro and 3D organoid culture models have demonstrated that cardiomyocytes play a crucial role in the above functions. Exosomes secreted by primary cardiomyocytes were shown to confer protection against oxidative-induced lesion, promote proliferation of endothelial cells, stimulate the formation of capillary structures, and strengthen adhesion complexes and barrier properties (Ribeiro-Rodrigues et al., 2017). Specifically, under ischemic conditions, these exosomes secreted from cardiomyocytes promoted angiogenesis. More recently, a 3D culture model of tissue-like spheroids in co-cultures of cardiomyocytes with endothelial cells released various vascular endothelial growth factor isoforms and fibronectin, and promoted the production of vascular structures and angiogenic sprouting, leading to long-term survival and contractile capacity of the cardiac microtissues (Garzoni et al., 2009). When cardiomyocytes were co-cultured with bone marrow mesenchymal cells in a similar 3D model, isolated cells aggregates formed which led to vasculogenesis. Taken together, our studies and previous findings show that TCDD may adversely impact cardiomyocytes’ function in the maintenance of a healthy myocardial environment, through angiogenesis or vasculogenesis.
These pathways were also implicated when comparing ES cells to untreated Nkx2-5-positive cells. Indeed, because the major role of DNA methylation is in cell type and tissue specificity, it is reasonable to conclude from our results that a potential effect of TCDD treatment is reverting differentiated cells toward an ES-like phenotype, where lineage-specific functions are suppressed in a pluripotent state. Similar to these findings, TCDD-driven AHR activation during the early stages of ES cell differentiation was suggested to maintain a pro-proliferative state and inhibit differentiation by the identification of regulators of pluripotency pathways through IPA analysis such as SOX2, NANOG, KLF4, and OCT4 (Wang et al., 2013). Whether TCDD treatment reverts cardiomyocytes toward an ES-like phenotype remains to be validated in further studies.
Among the genes involved in angiogenesis from our analyses, differential methylation of KIT, ENPP2, and KLF5 in humans has been implicated in various cancers, specifically breast cancer and pulmonary blastoma (Janostiak et al., 2018; Kamalakaran et al., 2011; Liu et al., 2020; Macher-Goeppinger et al., 2011). For example, hypermethylation of KIT and ENPP2 was associated with loss of expression and malignancy, and poor prognosis among human tumor samples (Janostiak et al., 2018; Liu et al., 2020) while hypomethylation of KLF5 was associated with poor breast cancer prognosis (Kamalakaran et al., 2011). Together with these studies, our findings suggest that the differential methylation of these genes caused by TCDD during cardiomyocyte maturation may be implicated in key processes such as angiogenesis linked to cardiovascular development and disease.
If DNA methylation patterns that affect faithful maturation and function of cardiomyocytes are affected by TCDD treatment, it can also be expected that other factors that regulate lineage specification may be affected. To that end, we searched the Ingenuity Knowledge Base for upstream regulators of the genes and biological functions affected. We found that CTNNB1, an integral component of the canonical WNT signaling pathway, was predicted to be inhibited and that Kit and Nrp2 were among the target genes in our dataset predicted to be affected in this pathway. Kit has important roles in hematopoiesis, specifically hematopoietic stem cell self-renewal and erythropoiesis (Deshpande et al., 2013), and its hypermethylation was previously associated with malignancy in breast cancer (Janostiak et al., 2018). Although Kit was hypermethylated in our dataset compared with controls, TCDD did not affect Nrp DNA methylation, despite its differential expression, suggesting that other signaling pathways such as CTNNB1 may play a larger role. Nrp plays an important role in cardiovascular development (Ding et al., 2018) and the WNT signaling that regulates it plays a key role in the faithful maturation of cardiomyocytes (Lian et al., 2013; Wang and Dey, 2006). Mutations in any of the interacting molecules, including CTNNB1, or environmental insults affecting these pathways can result in embryonic lethality or fetuses born with severe heart defects (Linask et al., 2005). Consistent with our findings, extensive work has already shown that TCDD disrupts WNT signaling in zebrafish (Hofsteen et al., 2013; Lanham et al., 2012; Mathew et al., 2009) and this finding was more recently implicated in the inhibition of cardiomyocyte differentiation by maintenance of a pro-proliferative state (Wang et al., 2013, 2016). Therefore, disruption of canonical WNT signaling and resulting inhibition of cardiomyocyte function are potentially major consequences of altered DNA methylation dynamics by TCDD that remain to be determined.
In this study, continuous treatment of maturing cardiomyocytes with TCDD for 24, 72, and 96 h was utilized. To date, studies investigating the effect of TCDD on cardiomyocyte maturation have been limited to early developmental phases after early ES cell differentiation (Wang et al., 2013). Indeed, Gilsbach et al. (2014) showed that cardiomyocytes can adapt to pathological stress through characteristic DNA methylation patterns linked to gene regulation and activity. Given these findings, it may be possible that continuous exposure of cardiomyocytes to TCDD may have long-lasting effects on the transcriptome and methylome, and future research can shed new light in this area.
Our results add credence to a growing body of evidence that TCDD inhibits cardiomyocyte maturation and function, in this case specifically by altering DNA methylation patterns that are crucial for this process. Given that DNA methylation patterns have been implicated in cardiac disease (Gilsbach et al., 2014), a phenotype linked to TCDD in our own work in vivo (Carreira et al., 2015a, 2015b), DNA methylation may present another mechanistic layer in the role that TCDD plays in fetal heart and vascular injury related to impaired cardiomyocyte function. We also showed that TCDD caused differential gene expression in cardiomyocytes not attributable to DNA methylation, necessitating examination into further epigenetic mechanisms and signaling pathways that may be at play.
DATA Availability
Code and data for RNA-seq and DNA methyl-seq analyses are available at https://doi.org/10.5281/zenodo.4006612 (degama01 2020). Raw and processed genomic data were submitted to Gene Expression Omnibus (GEO; accession number GSE154599), available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154599.
FUNDING
National Institute of Environmental Health Sciences (R01 ES024744, P3 ES06096).
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary Material
REFERENCES
- Abbott B. D., Schmid J. E., Pitt J. A., Buckalew A. R., Wood C. R., Held G. A., Diliberto J. J. (1999). Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol. Appl. Pharmacol. 155, 62–70. [DOI] [PubMed] [Google Scholar]
- Aluru N., Kuo E., Helfrich L. W., Karchner S. I., Linney E. A., Pais J. E., Franks D. G. (2015). Developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters DNA methyltransferase (DNMT) expression in zebrafish (Danio rerio). Toxicol. Appl. Pharmacol. 284, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenya H. Z., Tohyama C., Ohsako S. (2016). Dioxin induces AHR-dependent robust DNA demethylation of the cyp1a1 promoter via tdg in the mouse liver. Sci. Rep. 6, 34989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P., Brown J. D., Lin C. Y., Qi J., Zhang R., Artero P. C., Alaiti M. A., Bullard J., Alazem K., Margulies K. B., et al. (2013). Bet bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon A. C., Kopf P. G., Campen M. J., Huwe J. K., Walker M. K. (2008). In utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: Effects on fetal and adult cardiac gene expression and adult cardiac and renal morphology. Toxicol. Sci. 101, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong E. J., Bischoff J. (2004). Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 95, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M. P., Li J. B., Gao Y., Lee J. H., LeProust E. M., Park I. H., Xie B., Daley G. Q., Church G. M. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D. J. (2007). The origins of the developmental origins theory. J. Intern. Med. 261, 412–417. [DOI] [PubMed] [Google Scholar]
- Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabe-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H., et al. (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Boule L. A., Burke C. G., Fenton B. M., Thevenet-Morrison K., Jusko T. A., Lawrence B. P. (2015). Developmental activation of the AHR increases effector cd4+ t cells and exacerbates symptoms in autoimmune disease-prone gnaq+/- mice. Toxicol. Sci. 148, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. R., Ficz G., Reik W. (2012). Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 13, 7–13. [DOI] [PubMed] [Google Scholar]
- Carreira V. S., Fan Y., Kurita H., Wang Q., Ko C. I., Naticchioni M., Jiang M., Koch S., Zhang X., Biesiada J., et al. (2015. a). Disruption of Ah receptor signaling during mouse development leads to abnormal cardiac structure and function in the adult. PLoS One 10, e0142440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira V. S., Fan Y., Wang Q., Zhang X., Kurita H., Ko C. I., Naticchioni M., Jiang M., Koch S., Medvedovic M., et al. (2015. b). Ah receptor signaling controls the expression of cardiac development and homeostasis genes. Toxicol. Sci. 147, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro T., Dean K., Ottinger M. A. (2013). Effects of an environmentally relevant polychlorinated biphenyl (PCB) mixture on embryonic survival and cardiac development in the domestic chicken. Environ. Toxicol. Chem. 32, 1325–1331. [DOI] [PubMed] [Google Scholar]
- Cheng Y., Xie N., Jin P., Wang T. (2015). DNA methylation and hydroxymethylation in stem cells. Cell Biochem. Funct. 33, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal-Pra S., Hodgkinson C. P., Dzau V. J. (2019). Induced cardiomyocyte maturation: Cardiac transcription factors are necessary but not sufficient. PLoS One 14, e0223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton A. M., Bird A. (2011). Cpg islands and the regulation of transcription. Genes Dev. 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- degama01. 2020. Degama01/tcddmeth: Third release of tcddmeth repository. Zenodo.
- Deshpande S., Bosbach B., Yozgat Y., Park C. Y., Moore M. A., Besmer P. (2013). Kit receptor gain-of-function in hematopoiesis enhances stem cell self-renewal and promotes progenitor cell expansion. Stem Cells 31, 1683–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Zhou J., Wang S., Li Y., Mi Y., Gao S., Xu Y., Chen Y., Yan J. (2018). Anti-neuropilin-1 monoclonal antibody suppresses the migration and invasion of human gastric cancer cells via akt dephosphorylation. Exp. Ther. Med. 16, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer T. J., Dickinson H. O., Parker L. (2003). Adverse pregnancy outcomes around incinerators and crematoriums in Cumbria, north west England, 1956–93. J. Epidemiol. Community Health 57, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott D. A., Kirk E. P., Yeoh T., Chandar S., McKenzie F., Taylor P., Grossfeld P., Fatkin D., Jones O., Hayes P., et al. (2003). Cardiac homeobox gene nkx2-5 mutations and congenital heart disease: Associations with atrial septal defect and hypoplastic left heart syndrome. J. Am. Coll Cardiol. 41, 2072–2076. [DOI] [PubMed] [Google Scholar]
- Feldmann A., Ivanek R., Murr R., Gaidatzis D., Burger L., Schübeler D. (2013). Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 9, e1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness S. G., Lees M. J., Whitelaw M. L. (2007). The dioxin (aryl hydrocarbon) receptor as a model for adaptive responses of BHLH/PAS transcription factors. FEBS Lett. 581, 3616–3625. [DOI] [PubMed] [Google Scholar]
- Garzoni L. R., Rossi M. I., de Barros A. P., Guarani V., Keramidas M., Balottin L. B., Adesse D., Takiya C. M., Manso P. P., Otazú I. B., et al. (2009). Dissecting coronary angiogenesis: 3d co-culture of cardiomyocytes with endothelial or mesenchymal cells. Exp. Cell Res. 315, 3406–3418. [DOI] [PubMed] [Google Scholar]
- Gertsenstein M., Nutter L. M., Reid T., Pereira M., Stanford W. L., Rossant J., Nagy A. (2010). Efficient generation of germ line transmitting chimeras from c57bl/6n es cells by aggregation with outbred host embryos. PLoS One 5, e11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach R., Preissl S., Gruning B. A., Schnick T., Burger L., Benes V., Wurch A., Bonisch U., Gunther S., Backofen R., et al. (2014). Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 5, 5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter C. E., van Rooij E., Johnson B. A., DeLeon S. M., Sutherland L. B., Qi X., Gautron L., Elmquist J. K., Bassel-Duby R., Olson E. N. (2012). A cardiac microRNA governs systemic energy homeostasis by regulation of med13. Cell 149, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton R. B. (2013). Genetic and environmental factors contributing to cardiovascular malformation: A unified approach to risk. J. Am. Heart Assoc. 2, e000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofsteen P., Mehta V., Kim M. S., Peterson R. E., Heideman W. (2013). Tcdd inhibits heart regeneration in adult zebrafish. Toxicol. Sci. 132, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G. C., Rajagopal N., Shen Y., McCleary D. F., Yue F., Dang M. D., Ren B. (2013). Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 45, 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janostiak R., Vyas M., Cicek A. F., Wajapeyee N., Harigopal M. (2018). Loss of c-kit expression in breast cancer correlates with malignant transformation of breast epithelium and is mediated by kit gene promoter DNA hypermethylation. Exp. Mol. Pathol. 105, 41–49. [DOI] [PubMed] [Google Scholar]
- Jay P. Y., Harris B. S., Maguire C. T., Buerger A., Wakimoto H., Tanaka M., Kupershmidt S., Roden D. M., Schultheiss T. M., O’Brien T. X., et al. (2004). Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Invest. 113, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins S., Rowell C., Wang J., Lamartiniere C. A. (2007). Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod. Toxicol. 23, 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M., Sun D., Luo M., Huang Y., Challen G. A., Rodriguez B., Zhang X., Chavez L., Wang H., Hannah R., et al. (2014). Large conserved domains of low DNA methylation maintained by dnmt3a. Nat. Genet. 46, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. A. (2012). Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. [DOI] [PubMed] [Google Scholar]
- Jones S. P., Kennedy S. W. (2009). Chicken embryo cardiomyocyte cultures – A new approach for studying effects of halogenated aromatic hydrocarbons in the avian heart. Toxicol. Sci. 109, 66–74. [DOI] [PubMed] [Google Scholar]
- Kamalakaran S., Varadan V., Giercksky Russnes H. E., Levy D., Kendall J., Janevski A., Riggs M., Banerjee N., Synnestvedt M., Schlichting E., et al. (2011). DNA methylation patterns in luminal breast cancers differ from non-luminal subtypes and can identify relapse risk independent of other clinical variables. Mol. Oncol. 5, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kewley R. J., Whitelaw M. L., Chapman-Smith A. (2004). The mammalian basic helix-loop-helix/pas family of transcriptional regulators. Int. J. Biochem. Cell Biol. 36, 189–204. [DOI] [PubMed] [Google Scholar]
- Kobayashi H., Sakurai T., Imai M., Takahashi N., Fukuda A., Yayoi O., Sato S., Nakabayashi K., Hata K., Sotomaru Y., et al. (2012). Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 8, e1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf P. G., Walker M. K. (2009). Overview of developmental heart defects by dioxins, PCBS, and pesticides. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 27, 276–285. [DOI] [PubMed] [Google Scholar]
- Kruger M., Kohl T., Linke W. A. (2006). Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin-i isoform switching are not critically triggered by birth. Am. J. Physiol. Heart Circ. Physiol. 291, H496–506. [DOI] [PubMed] [Google Scholar]
- Kuehl K. S., Loffredo C. A. (2006). A cluster of hypoplastic left heart malformation in baltimore, maryland. Pediatr. Cardiol. 27, 25–31. [DOI] [PubMed] [Google Scholar]
- Lage K., Greenway S. C., Rosenfeld J. A., Wakimoto H., Gorham J. M., Segre A. V., Roberts A. E., Smoot L. B., Pu W. T., Pereira A. C., et al. (2012). Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc. Natl. Acad. Sci. U.S.A. 109, 14035–14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanham K. A., Peterson R. E., Heideman W. (2012). Sensitivity to dioxin decreases as zebrafish mature. Toxicol. Sci. 127, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Fan Y., Li G., Xu X., Duan J., Li R., Kang X., Ma X., Chen X., Ke Y., et al. (2018). DNA methylation reprogramming of functional elements during mammalian embryonic development. Cell Discov. 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X., Zhang J., Azarin S. M., Zhu K., Hazeltine L. B., Bao X., Hsiao C., Kamp T. J., Palecek S. P. (2013). Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating WNT/beta-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linask K. K., Manisastry S., Han M. (2005). Cross talk between cell-cell and cell-matrix adhesion signaling pathways during heart organogenesis: Implications for cardiac birth defects. Microsc. Microanal. 11, 200–208. [DOI] [PubMed] [Google Scholar]
- Lister R., Mukamel E. A., Nery J. R., Urich M., Puddifoot C. A., Johnson N. D., Lucero J., Huang Y., Dwork A. J., Schultz M. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., Nery J. R., Lee L., Ye Z., Ngo Q. M., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Peng Y., Wang J. (2020). Integrative analysis of DNA methylation and gene expression profiles identified potential breast cancer-specific diagnostic markers. Biosci. Rep. 40, 10.1042/BSR20201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macher-Goeppinger S., Penzel R., Roth W., Dienemann H., Thomas M., Schnabel P. A., Schirmacher P., Bläker H. (2011). Expression and mutation analysis of egfr, c-kit, and β-catenin in pulmonary blastoma. J. Clin. Pathol. 64, 349–353. [DOI] [PubMed] [Google Scholar]
- Mathew L. K., Sengupta S., Franzosa J. A., Perry J., La Du J., Andreasen E. A., Tanguay R. L. (2009). Comparative expression profiling reveals an essential role for raldh2 in epimorphic regeneration. J. Biol. Chem. 284, 33642–33653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinney D. B., Geiger E., Blinder J., Benson D. W., Goldmuntz E. (2003). Nkx2.5 mutations in patients with congenital heart disease. J. Am. Coll. Cardiol. 42, 1650–1655. [DOI] [PubMed] [Google Scholar]
- Papadopoulou K., Murray S., Manousou K., Tikas I., Dervenis C., Sgouros J., Rontogianni D., Lakis S., Bobos M., Poulios C., et al. (2018). Genotyping and mRNA profiling reveal actionable molecular targets in biliary tract cancers. Am. J. Cancer Res. 8, 2–15. [PMC free article] [PubMed] [Google Scholar]
- Pashmforoush M., Lu J. T., Chen H., Amand T. S., Kondo R., Pradervand S., Evans S. M., Clark B., Feramisco J. R., Giles W., et al. (2004). Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117, 373–386. [DOI] [PubMed] [Google Scholar]
- Pfister O., Mouquet F., Jain M., Summer R., Helmes M., Fine A., Colucci W. S., Liao R. (2005). Cd31- but not cd31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ. Res. 97, 52–61. [DOI] [PubMed] [Google Scholar]
- Pilsner J. R., Parker M., Sergeyev O., Suvorov A. (2017). Spermatogenesis disruption by dioxins: Epigenetic reprograming and windows of susceptibility. Reprod. Toxicol. 69, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plavicki J., Hofsteen P., Peterson R. E., Heideman W. (2013). Dioxin inhibits zebrafish epicardium and proepicardium development. Toxicol. Sci. 131, 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potta S. P., Liang H., Winkler J., Doss M. X., Chen S., Wagh V., Pfannkuche K., Hescheler J., Sachinidis A. (2010). Isolation and functional characterization of alpha-smooth muscle actin expressing cardiomyocytes from embryonic stem cells. Cell. Physiol. Biochem. 25, 595–604. [DOI] [PubMed] [Google Scholar]
- Puga A. (2011). Perspectives on the potential involvement of the Ah receptor-dioxin axis in cardiovascular disease. Toxicol. Sci. 120, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala K., Pekkanen-Mattila M., Fau-Aalto-Setälä K., Aalto-Setälä K. (2011). Cardiac differentiation of pluripotent stem cells. Stem Cells Int 2011, 383709. [DOI] [PMC free article] [PubMed]
- Reik W., Dean W., Walter J. (2001). Epigenetic reprogramming in mammalian development. Science 293, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Ribeiro-Rodrigues T. M., Laundos T. L., Pereira-Carvalho R., Batista-Almeida D., Pereira R., Coelho-Santos V., Silva A. P., Fernandes R., Zuzarte M., Enguita F. J., et al. (2017). Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc. Res. 113, 1338–1350. [DOI] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J., Smyth G. K. (2010). Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavone A., Capilupo D., Mazzocchi N., Crespi A., Zoia S., Campostrini G., Bucchi A., Milanesi R., Baruscotti M., Benedetti S., et al. (2013). Embryonic stem cell-derived cd166+ precursors develop into fully functional sinoatrial-like cells. Circ. Res. 113, 389–398. [DOI] [PubMed] [Google Scholar]
- Schecter A., Cramer P., Boggess K., Stanley J., Papke O., Olson J., Silver A., Schmitz M. (2001). Intake of dioxins and related compounds from food in the U.S. population. J. Toxicol. Environ. Health A 63, 1–18. [DOI] [PubMed] [Google Scholar]
- Schmidt J. V., Bradfield C. A. (1996). Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 12, 55–89. [DOI] [PubMed] [Google Scholar]
- Schott J. J., Benson D. W., Basson C. T., Pease W., Silberbach G. M., Moak J. P., Maron B. J., Seidman C. E., Seidman J. G. (1998). Congenital heart disease caused by mutations in the transcription factor nkx2-5. Science 281, 108–111. [DOI] [PubMed] [Google Scholar]
- Senyo S. E., Steinhauser M. L., Pizzimenti C. L., Yang V. K., Cai L., Wang M., Wu T. D., Guerquin-Kern J. L., Lechene C. P., Lee R. T. (2013). Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpooshan V., Liu Y. H., Buikema J. W., Galdos F. X., Chirikian O., Paige S., Venkatraman S., Kumar A., Rawnsley D. R., Huang X., et al. (2017). Nkx2.5+ cardiomyoblasts contribute to cardiomyogenesis in the neonatal heart. Sci. Rep. 7, 12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddeek B., Mauduit C., Simeoni U., Benahmed M. (2018). Sperm epigenome as a marker of environmental exposure and lifestyle, at the origin of diseases inheritance. Mutat. Res. 778, 38–44. [DOI] [PubMed] [Google Scholar]
- Siedner S., Kruger M., Schroeter M., Metzler D., Roell W., Fleischmann B. K., Hescheler J., Pfitzer G., Stehle R. (2003). Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J. Physiol. 548, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim C. B., Ziemann M., Kaspi A., Harikrishnan K. N., Ooi J., Khurana I., Chang L., Hudson J. E., El-Osta A., Porrello E. R. (2015). Dynamic changes in the cardiac methylome during postnatal development. Faseb. J. 29, 1329–1343. [DOI] [PubMed] [Google Scholar]
- Stadler M. B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., van Nimwegen E., Wirbelauer C., Oakeley E. J., Gaidatzis D., et al. (2011). DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495. [DOI] [PubMed] [Google Scholar]
- Strimmer K. (2003). Modeling gene expression measurement error: A quasi-likelihood approach. BMC Bioinformatics 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taegtmeyer H., Sen S., Vela D. (2010). Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 1188, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Chen Z., Bartunkova S., Yamasaki N., Izumo S. (1999). The cardiac homeobox gene csx/nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 126, 1269–1280. [DOI] [PubMed] [Google Scholar]
- van der Bom T., Zomer A. C., Zwinderman A. H., Meijboom F. J., Bouma B. J., Mulder B. J. (2011). The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 8, 50–60. [DOI] [PubMed] [Google Scholar]
- Vandiver A. R., Idrizi A., Rizzardi L., Feinberg A. P., Hansen K. D. (2015). DNA methylation is stable during replication and cell cycle arrest. Sci. Rep. 5, 17911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecoli C., Pulignani S., Foffa I., Andreassi M. G. (2014). Congenital heart disease: The crossroads of genetics, epigenetics and environment. Curr. Genomics 15, 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M. K., Catron T. F. (2000). Characterization of cardiotoxicity induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related chemicals during early chick embryo development. Toxicol. Appl. Pharmacol. 167, 210–221. [DOI] [PubMed] [Google Scholar]
- Wamstad J. A., Alexander J. M., Truty R. M., Shrikumar A., Li F., Eilertson K. E., Ding H., Wylie J. N., Pico A. R., Capra J. A., et al. (2012). Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151, 206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Dey S. K. (2006). Roadmap to embryo implantation: Clues from mouse models. Nat Rev Genet 7, 185–199. [DOI] [PubMed] [Google Scholar]
- Wang J., Chen L., Ko C. I., Zhang L., Puga A., Xia Y. (2012). Distinct signaling properties of mitogen-activated protein kinase kinases 4 (MKK4) and 7 (MKK7) in embryonic stem cell (ESC) differentiation. J. Biol. Chem. 287, 2787–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Chen J., Ko C. I., Fan Y., Carreira V., Chen Y., Xia Y., Medvedovic M., Puga A. (2013). Disruption of aryl hydrocarbon receptor homeostatic levels during embryonic stem cell differentiation alters expression of homeobox transcription factors that control cardiomyogenesis. Environ. Health Perspect. 121, 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Kurita H., Carreira V., Ko C. I., Fan Y., Zhang X., Biesiada J., Medvedovic M., Puga A. (2016). Ah receptor activation by dioxin disrupts activin, bmp, and wnt signals during the early differentiation of mouse embryonic stem cells and inhibits cardiomyocyte functions. Toxicol. Sci. 149, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Fan Y., Puga A. (2010). Dioxin exposure disrupts the differentiation of mouse embryonic stem cells into cardiomyocytes. Toxicol. Sci. 115, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W., Schultz M. D., Lister R., Hou Z., Rajagopal N., Ray P., Whitaker J. W., Tian S., Hawkins R. D., Leung D., et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka W., Peterson R. E., Tohyama C. (2011). Molecular targets that link dioxin exposure to toxicity phenotypes. J. Steroid. Biochem. Mol. Biol. 127, 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa S., Itabashi Y., Koshimizu U., Tanaka T., Sugimura K., Kinoshita M., Hattori F., Fukami S., Shimazaki T., Ogawa S., et al. (2005). Transient inhibition of bmp signaling by noggin induces cardiomyocyte differentiation of mouse embryonic stem cells. Nat. Biotechnol. 23, 607–611. [DOI] [PubMed] [Google Scholar]
- Zhang C. L., McKinsey T. A., Chang S., Antos C. L., Hill J. A., Olson E. N. (2002). Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code and data for RNA-seq and DNA methyl-seq analyses are available at https://doi.org/10.5281/zenodo.4006612 (degama01 2020). Raw and processed genomic data were submitted to Gene Expression Omnibus (GEO; accession number GSE154599), available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154599.