

Abstract
Background:
Ischemia reperfusion injury (IRI) is associated with programmed cell death that promotes inflammation and organ dysfunction. Necroptosis is mediated by members of receptor interacting protein kinases (RIPK1/3). Inhibition of RIPK1/3 provides a pro-survival benefit in kidney IRI. Caspase-8 initiate apoptosis and contributes to IRI. We studied whether inhibiting both RIPK3 and caspase-8 would provide an additional benefit in kidney IRI.
Methods:
A clamp was applied to the left kidney pedicle for 45 minutes followed by right kidney nephrectomy. Kidney and serum from wild type, RIPK3−/−, and RIPK3−/− Caspase-8−/− double knockout (DKO) mice were collected post-IRI for assessment of injury. Tubular epithelial cells (TEC) isolated from wild type, RIPK3−/−, and DKO mice were treated with IFN-γ and IL-1β to induce apoptotic death.
Results:
kidney IRI of DKO mice did not show improvement over RIPK3−/− mice. We have found that DKO triggered ‘intrinsic’ apoptosis in TEC in response to IL-1β and IFN-γ. Upregulation of the B-cell lymphoma 2 (Bcl-2)-associated death promoter (BAD), the Bcl-2-homologous antagonist killer (BAK) and Bcl-2-associated X protein (BAX) and enhanced activation of caspase-3 and 9 were found in DKO TEC. TEC infected with Murine cytomegalovirus (MCMV) that encodes multiple cell death inhibitors resist to death.
Conclusion:
We show that the deletion of both RIPK3 and caspase-8 does not provide additive benefit in IRI or TEC death and may enhance injury by upregulation of intrinsic apoptosis. This suggests blocking multiple death pathways may be required for the prevention of kidney IRI clinically.
Keywords: necroptosis, apoptosis, caspase-8, IRI, kidney, RIPK3
Introduction
Tubular epithelial cells (TEC) are particularly susceptible to damage from ischemia reperfusion injury (IRI). Recent studies have shown that TEC undergo various forms of programmed cell death during inflammatory injury, namely apoptosis, necroptosis, ferroptosis, pyroptosis, autophagy and other1–3. Unfortunately, there are no specific therapeutic strategies or treatments to prevent TEC death.
Apoptosis has long been considered as a major contributor in IRI. In IRI, TEC apoptosis has multiple triggers, including but not limited to NK cell engagement4, or ‘fratricide’ related to TEC co-expression of both Fas and FasL5. TEC apoptosis does not show plasma membrane rupture and dying cells are efficiently removed by circulating phagocytes as well as by resident TEC utilizing Kidney Injury molecule-1(KIM-1), resulting in minimal inflammation6, 7. Necroptosis represents one form of programmed necrosis that has recently been identified and implicated in many injury models. It is dependent on members of receptor interacting protein kinases (RIPK1/3) but regulated by caspase-8 through cleavage and inactivation of RIPK12, 3, 8. RIPK1 and 3 interact with each other to activate downstream mixed lineage kinase domain-like (MLKL) and eventual cell membrane rupture and necroptosis9.
Studies by our group and others have shown that blocking necroptosis through pharmacological inhibition of RIPK110 or genetic elimination of RIPK3 attenuates kidney IRI11, 12. Caspase-8 inhibition also benefits survival in renal IRI13. Thus, these studies would suggest that as kidney injury involves both apoptosis and necroptosis, blocking both forms of death might provide added benefit. However, previous studies blocking caspase-8 apoptosis and necroptosis have produced contradictory results for IRI12, 14. Clearly there are complex interactions between members of programmed cell death pathways and previous studies have not examined the potential effect of blocking caspase-8 apoptosis and necroptosis. In addition, the kidney has high metabolic demands and requires a large number of mitochondria to provide the energy in order to eliminate waste products and regulate ion balances. However, mitochondria also contain multiple molecules that participate in apoptosis and thus may play a dominant role in the ‘intrinsic cell death’ pathway during kidney IRI. In this study, loss of both RIPK3 and caspase-8 did not reduce kidney IRI as expected and indeed may have worsened IRI. Loss of both RIPK3 necroptosis and caspase-8 mediated extrinsic apoptosis lead to enhanced mitochondrial based intrinsic apoptosis. Hence, blocking extrinsic and intrinsic apoptosis, and necroptosis may be required in clinical strategies to effectively prevent kidney IRI. We tested whether murine cytomegalovirus (MCMV) known to encode for inhibitors of death pathways, could alter these programmed cell death pathways in TEC.15
Materials and Methods
Animals
C57BL/6 (B6, H-2b; Jackson Laboratories, Bar Harbor, ME), B6-RIPK3−/− (H-2b; generously provided by Genentech, San Francisco, CA) were used. RIPK3−/− mice are phenotypically normal and show normal kidney function and breeding16. RIPK3−/− mice were bred with RIPK3−/−Caspase-8+/− mice for at least 10 generations before interbreeding RIPK3−/−Caspase-8+/− mice to generate RIPK3−/−Caspase-8−/− (DKO) mice8. Genotyping of RIPK3−/− and DKO was performed as previous described16, 17. All male mice were under 8 weeks old for this study. All experimental procedures were approved by the Western University Animal Care Committee.
TEC culture
TEC from mice were isolated from mouse kidney as described and used in experiments after 4–6 days of culture11. The identity of TEC was confirmed using anti-CD13, anti-CD26, and anti-E-cadherin (Abcam)4. TEC were grown on 0.1% collagen coated plates in complete K1 culture medium (Invitrogen. CA), supplemented with 5% bovine calf serum, hormone mix (5 μg/ml of insulin, 1.25 ng/ml of prostaglandin E1, 34 pg/ml of triodothyronine, 5 μg/ml of transferrin, 1.73 ng/ml of sodium selenite and 18 ng/ml of hydrocortisone), 25 ng/ml of EGF, 100U/ml penicillin and 100 ug/ml streptomycin (Invitrogen, CA).
Fibroblast culture and murine cytomegalovirus propagation
NIH 3T3 fibroblasts were grown in 4.5g/mL glucose DMEM mixed with 10% fetal bovine serum, 2mM L-glutamine (Invitrogen, CA). Stocks of wild type K181 BAC MCMV strain were generously provided by Dr. Edward Mocarski (Emory University, USA)18. K181 BAC strain was grown and propagated in NIH 3T3 cells and crude stocks for in vitro experiments were prepared as described18.
Kidney IRI
The right kidney was removed, and then a renal clamp was applied to the left kidney pedicle. It was then removed after 45 minutes, keeping the mouse at 32°C4, 13. Kidneys were collected at 48h post- IRI after being flushed with saline. Serum creatinine levels were tested with an automated CX5 clinic analyzer (Beckman, Fullteron, CA).
RNA isolation and real-time polymerase chain reaction
Total RNA extraction from cultured TEC were performed with Trizol (Invitrogen, USA). cDNA was generated using Superscript II (Invitrogen). Real time PCR was performed using SYBR QPCR kit (Bio-Rad, USA). β-actin was used as the endogenous control. The normalized delta threshold cycle (Ct) value was calculated. Primers include: β-actin: 5’-CTGTGCTATGTTGCTCTA-3’ and 5’-AGGA TTCCATACCCAAGA-3’, BAX: 5’-TTTGCTACAGGGTTTCAT-3’ and 5’-GTCCAGT TCATCTCCAAT-3’, BAK: 5’-CATGAATCCACTGATACCA-3’ and 5’-GTCACTTG TCACCTGAAT-3’, BAD: 5’-CGATGAGTTTGAG GGTTC-3’ and 5’-CTTTGTCGCATCTGTGTT-3’.
Western blot
TEC from B6, RIPK3−/− and DKO mice were cultures to confluence. Protein was isolated using RIPA cell lysis buffer (Sigma, USA). Equal volumes of lysates were loaded for gel electrophoresis. Protein was transferred to a nitrocellulose membrane (BioRad, USA). Blots were incubated with polyclonal rabbit anti-BAD, anti-BAK, and anti-BAX (Abcam, Cambridge, MA, USA.), or mouse anti-β-actin (Santa Cruz Biotech. USA). Protein was visualized using horseradish peroxidase (HRP)-linked anti-rabbit IgG (Sigma-Aldrich) and chemiluminescent HRP substrate (EMD-Millipore, USA). Protein was semi-quantitated by densitometry (Alphaview; ProteinSimple, Santa Clara, CA).
Cell death assays
IL-1β and IFN-γ in combination has been shown to induce BAX-dependent intrinsic apoptosis in other cell types19. We found that 4 ng/mL of IL-1β and 120 ng/mL of IFN-γ most effectively decreased viability in wild type TEC. To induce apoptotic cell death, TEC were grown to confluent monolayers and treated with recombinant murine IFN-γ and IL-1β (R&D Systems, USA) in serum-free media. BAX-inhibiting peptide V5 (BIP; Sigma, Canada) was added 1 hour before cytokine treatment. After 24 hours, TEC were incubated with 12mM MTT (Life Technologies, Canada) for 4 hours before absorbance was measured at 490 nm. Untreated TEC were set as full viability.
Caspase-9 and caspase-3 activities
TECs were grown to confluent monolayers and treated with IL-1β and IFN- γ for 24 h. Caspase-Glo-9 reagent (Caspase-Glo-9; Promega, USA) was added directly to the TEC cultures. Luminescence emission was detected after 1 hour using a VictorX Light (PerkinElmer). Cleaved caspase-3 activity was measured using CellPlayer™ Kinetic Caspase-3/7 Apoptosis Assay Reagent (Essen Bioscience, USA). Incucyte ZOOM (Essen Bioscience) live cell imager was used to scan for the caspase-3 activity over 24 hours.
Histology and Immunochemistry
Kidney sections were stored in 5% formalin (Sigma) and fixed in paraffin before being stained with hematoxylin and eosin (H&E). The slides were scored for acute tubular necrosis (ATN) by a pathologist blinded to experiment settings (0: no change, 1: <25% area change, 2: 25–50% area change, 3: 50–75% area change, 4: 75% area change) using a semi-quantitative method as described4.
Statistical analysis
For parametric data, One way- and Two way- Analysis of Variance (ANOVA) plus Tukey post hoc tests were used to compare multiple groups. Student’s t-test was used for unpaired values. Data are presented as mean ± SEM using p<0.05 for significance.
Results
DKO mice do not show additional benefits over RIPK3−/− mice post renal IRI
We investigated the potential benefit of inhibition of both RIPK3 and caspase-8 by subjecting wild-type (B6), RIPK3−/−, and DKO mice to renal IRI. Compared to wild-type mice, RIPK3−/− mice showed improved kidney function as shown by lower serum creatinine levels post-IRI (51±20 vs. 118±24 μmol/l, Figure 1A). In contrast, DKO mice did not show improvement in injury compared to RIPK3−/− mice, as serum creatinine levels were modestly slightly increased (82±38 vs.51±20 μmol/l, Figure 1A). Histological assessment of ATN showed a similar result, as wild-type mice showed higher injury scores compared to both RIPK3−/− (2.5±0.1 vs. 1.5±0.2, Figure 1B&C) and DKO mice (2.5±0.1 vs.1.6±0.1). DKO mice showed no improvement over RIPK3−/− mice (1.6±0.1 vs. 1.5±0.2). Hence, the absence of both RIPK3 and caspase-8 does not provide additional benefit compared to RIPK3−/− alone to kidney IRI.
Figure 1. Loss of RIPK3 and caspase-8 does not provide additional benefits over RIPK3 deficiency alone in renal IRI.
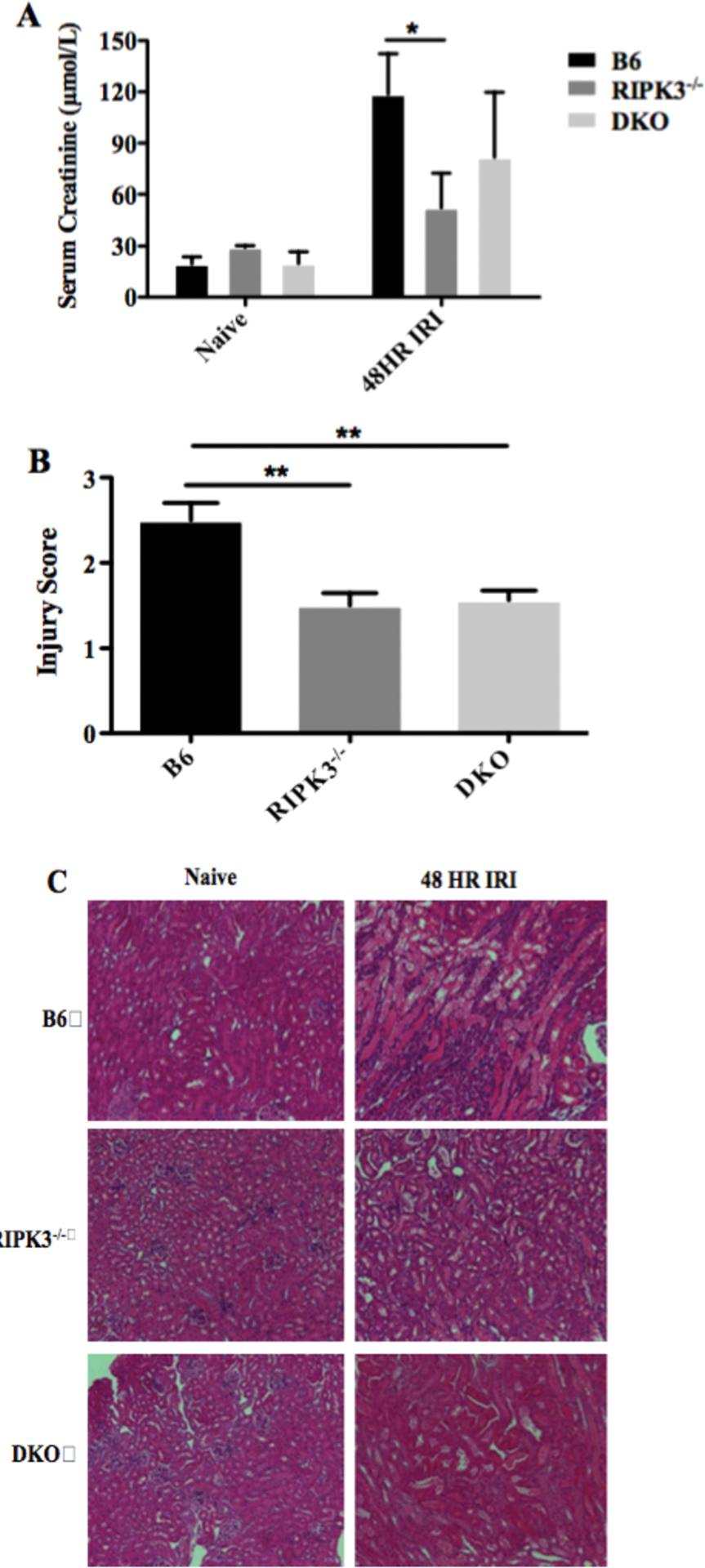
A renal clamp was applied to the left kidney pedicle of wild type B6, RIPK3−/−, and RIPK3−/− Caspase-8−/− mice, and released after 45 minutes and the right kidney was removed. Kidneys and serum were collected at 48 hrs post-IRI to measure acute tubular injury and creatinine. A. Serum creatinine levels were averaged from each group (n=6–9/group, *: p <0.05, Two-Way ANOVA). B. kidneys were used for H&E staining. Slides were scored for acute tubular injury on a scale from 0–4 by pathologist. (**: p <0.01, One-Way-ANOVA). C. Histological pictures are representative of one of kidneys from each group. magnification=200 fold.
Absence of RIPK3 and caspase-8 results in increased basal mRNA levels of pro-apoptotic BCL-2 family members
We assessed mRNA levels of pro-apoptotic BCL-2 family BAX, BAK, and BAD in TEC. Compared to RIPK3−/− TEC, DKO TEC showed a significantly higher basal expression of pro-apoptotic BAX (1.7±0.2 vs. 0.8±0.1, Figure 2A) and BAK (9.4±0.3 vs. 2.4±0.3, Figure 2B). RIPK3−/− TEC also showed higher level of BAK compared to wild type TEC (2.4±0.3 vs. 1±0). BAD mRNA expression was higher in DKO TEC compared to wild type TEC (7.4 ± 2.0 vs. 1 ± 0, Figure 2C). Next, we confirmed our PCR findings by analyzing protein expression of mitochondrial proteins by western blot. Consistent with mRNA results, protein levels of BAD and BAK were all higher in DKO TEC, while BAX was not significantly different each other (Figure 3A–D). Our results suggest enhanced intrinsic apoptosis by DKO TEC may be due to up-regulation of pro-apoptotic BAX, BAK, and BAD.
Figure 2. Absence of RIPK3 and caspase-8 augments mRNA expression of BCL-2 family members in TEC.
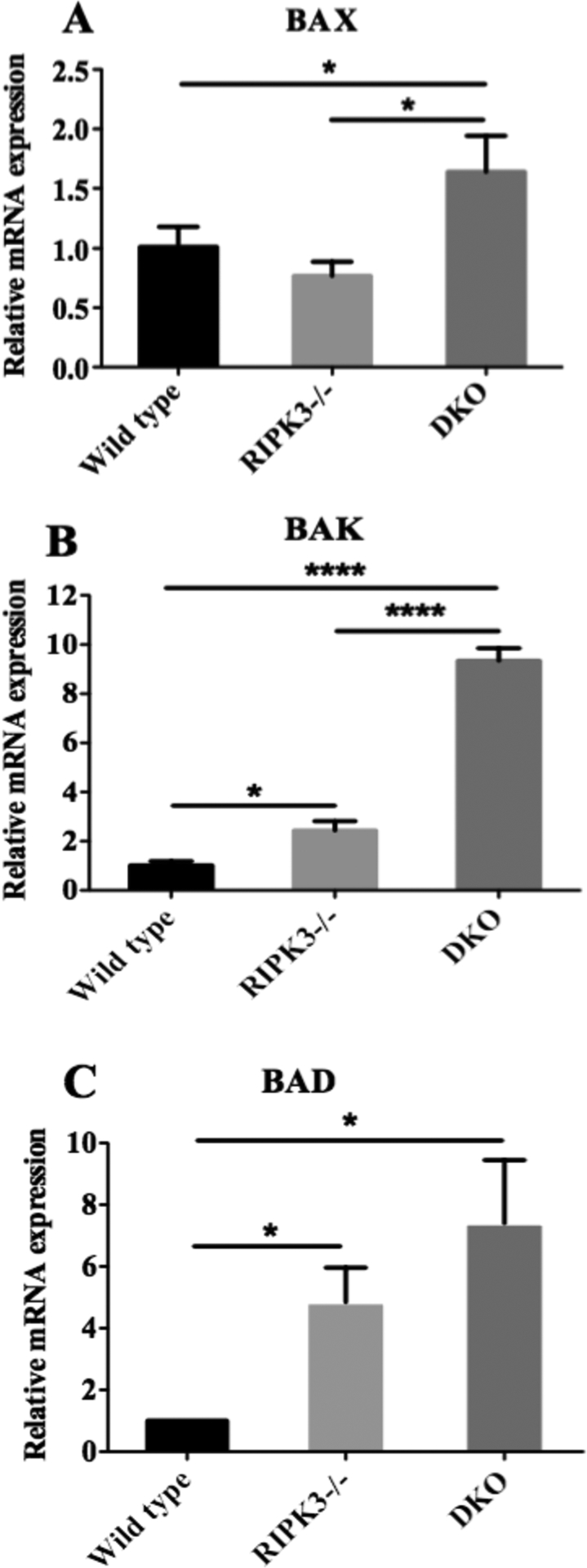
TEC from wild type B6, RIPK3−/−, and RIPK3−/− Caspase-8−/− mice were isolated and cultured for 6 day and RNA were extracted as described in the methods. mRNA levels of pro-apoptotic genes: BAX, BAK and BAD were quantified by real time PCR as described in the methods (A-C). β-actin was used as loading control. Each figure represents mean of triplicates (n=3, *: p < 0.05, **: p<0.01, ****: p<0.0001, One-Way ANOVA). Three independent experiments showed a similar result.
Figure 3. RIPK3 and caspase-8 double knockout augments protein expression of BCL-2 family members in TEC.
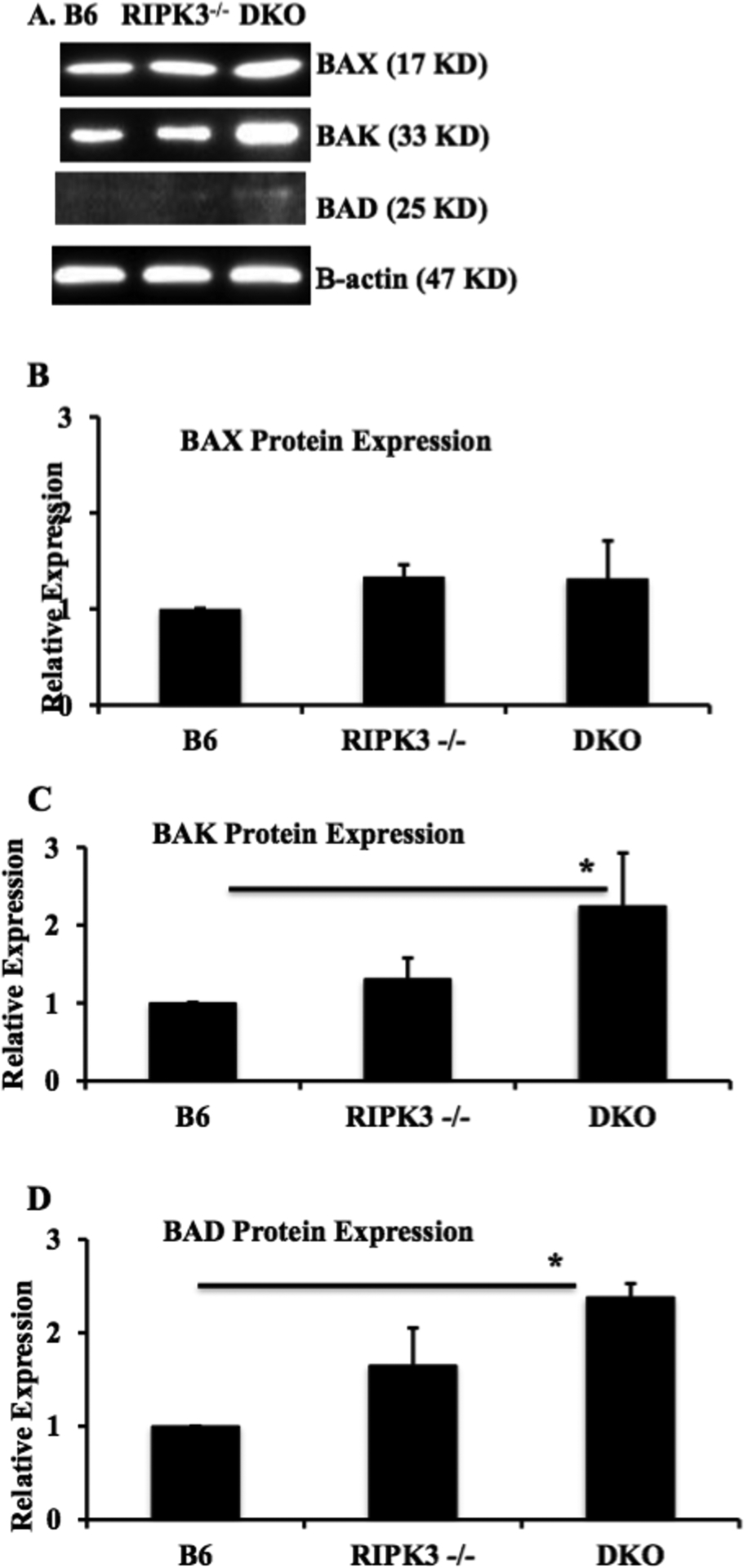
TEC from wild type B6, RIPK3−/−, and RIPK3−/− Caspase-8−/− mice were isolated and cultured followed by proteins extraction as described in the methods. Expression of BAD, BAK and BAX proteins were analyzed by Western blot. Protein was semi-quantitated by densitometry. Data shown as mean ± SEM and representative of 3 different mice in each group. *P ≤ 0.05, One-Way ANOVA.
DKO TEC are sensitized to BAX-dependent death
TEC are capable of undergoing forms of cell death independent of RIPK3 and caspase-8. Since no additive benefits in renal IRI were observed from IRI experiments (Figure 1A–C), and with observed increased levels of BAX, BAK, and BAD (Figure 2&3), we hypothesized that loss of both RIPK3 and caspase-8 may have biased TEC death to intrinsic apoptosis. A reduction in survival was clearly greatest in DKO TEC (17.9±1.8%) as compared to wild type (51.7±2.1%, Figure 4A) and RIPK3−/− (39±5.5%). BAX inhibiting peptide V5 (BIP) increased survival in all TEC (Figure 4A) but DKO TEC showed the greatest response compared to wild type (relative survival restoration: 156±7.9% vs. 62.1±7.9%, Figure 4B) and RIPK3−/− TEC (69.0±7.4%), suggesting that DKO TEC may be more susceptible to intrinsic apoptosis.
Figure 4. Absence of RIPK3 and caspase-8 augments caspase 3 and 9 activities and sensitizes TEC to intrinsic apoptotic death.

Wild type, RIPK3−/−, and DKO TECs were cultured as described in the method and treated with combination of 4 ng/mL of IL-1β and 120 ng/mL of IFN-γ for 24 hours. TEC were also treated with 1 hour pre-incubation of 50 μM BIP. A. Cell viability was measured and quantified using MTT assay analyses. B. Relative cell death restoration after BIP treatment. Cell viability in cytokines-treated TEC without BIP was used as reference. Each figure represents mean of quadruplicates. Four independent experiments showed a similar response (n=4, *: p < 0.05, **: p<0.01, ****: p<0.0001, Two-Way ANOVA). C. TECs were grown to confluent monolayers in triplicates and treated with recombinant murine IL-1β and IFN- γ with or without BIP for 24 hrs. 70 μl of Caspase-Glo-9 (Promega) was added directly to the TEC cultures. Luminescence emission was detected after 1 hour using a VictorX Light (PerkinElmer). Three independent experiments showed a similar result. D. Cleaved caspase-3 activity was measured using CellPlayer™ Kinetic Caspase-3/7 Apoptosis Assay Reagent (Essen Bioscience). Fluorescent intensity (caspase 3 activity) was detected using Incucyte ZOOM (Essen Bioscience) live cell imaging at 24 hours (n=3. *: p < 0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001, Two-Way ANOVA). Three independent experiments showed a similar result.
Loss of RIPK3 and caspase-8 augments caspase 3 and 9-mediated intrinsic apoptosis in TEC
As BAX-mediated intrinsic apoptosis requires caspase-9 and downstream executioner caspase-3, we measured caspase-9 and caspase-3 activity. TEC were exposed to IL-1β+IFN-γ in the absence or presence of 50 μM BIP. Up-regulation of caspase-9 activity was detected in activated wild type (luminescence intensity:13730±601 vs. 6261±467), RIPK3−/− (10670±1881 vs. 3734±292), and DKO TEC (19370±2397 vs. 4119±1236, Figure 4C) compared to control TEC. Of note, caspase-9 activity in activated DKO TEC was higher than RIPK3−/− or wild type TEC. The addition of BIP decreased caspase-9 activity only in DKO TEC (luminescence intensity: 11070±1342 vs. 19370±2397) with no effect in wild type (13710±2308 vs. 13730±601) or RIPK3−/− TEC (11650±2285 vs. 10670±1881).
We next measured downstream pro-caspase-3 activation. The pan-caspase inhibitor Z-vad-fmk was used to block caspase mediated apoptotic cell death. Results for IL-1β+IFN-γ treatment are shown at 24 hours for comparison to no treatment (Figure 4D), and shows the largest increase in induced caspase-3 activity in DKO TEC (luminescence intensity: 62760±3808 vs. 4310±400) as compared to wild type (7636±806 vs. 4503±120), and RIPK3−/− TEC (6605±631 vs. 3207±933). Addition of BIP decreased caspase-3 activity in DKO TEC (8199±1078 vs.62760±3808) but not in wild type (8075±1465vs. 636±806) or RIPK3−/− TEC (8573±1887 vs. 6605±631). Z-vad-fmk was effective in wild type (285±140 vs.7636±806), RIPK3−/− (521±389 vs. 6605±631), and DKO TEC (62760±3808). These data support that the enhanced level of caspase-3 and 9 activities in DKO TEC is related to enhanced intrinsic apoptotic cell death.
MCMV abrogates intrinsic apoptosis in TEC
Murine and human MCMV possess a potent capacity to inhibit multiple cell death pathways simultaneously using endogenous viral inhibitor proteins to ensure their survival15, 20, 21. CMV encode a series of ‘anti-death proteins’ that can inhibit RIPK1/3 (necroptosis), caspase-8 (extrinsic apoptosis), and BAK or BAX (intrinsic apoptosis).22 To test whether MCMV can alter TEC survival and in particular, intrinsic apoptosis, we measured the viability of MCMV-infected TEC following IL-1β+IFN-γ exposure. Wild type and RIPK3 null TEC were unaffected by MCMV in the absence of cytokine stimulation while DKO TEC had somewhat reduced survival (64.0±4.0%, Figure 5A–C). As previously observed, the addition of cytokines had the greatest negative effect on survival in DKO TEC (wild type TEC survival rate: 57.0±9.6%, Figure 5A), RIPK3−/− (39.3±3.77%, Figure 5B) and DKO TEC (29.3±0.3%, Figure 5C). With MCMV infection, there was recovery of survival in wild type (85.0±9.3% vs.57.0±9.6%), RIPK3−/− (87.3±3.53% vs.39.3±3.8%), and DKO TEC survival (53.7±3.93% vs.29.3±0.3%, Figure 5A–C), which approached that of infected TEC without cytokine treatment. These data support that inhibition of multiple pathways of cell death has utility in TEC injury, and that perhaps exploiting this unique capacity of MCMV might be applied to clinical strategies.
Figure 5. MCMV abrogates IL-1β /IFN- γ induced intrinsic apoptosis in TEC.
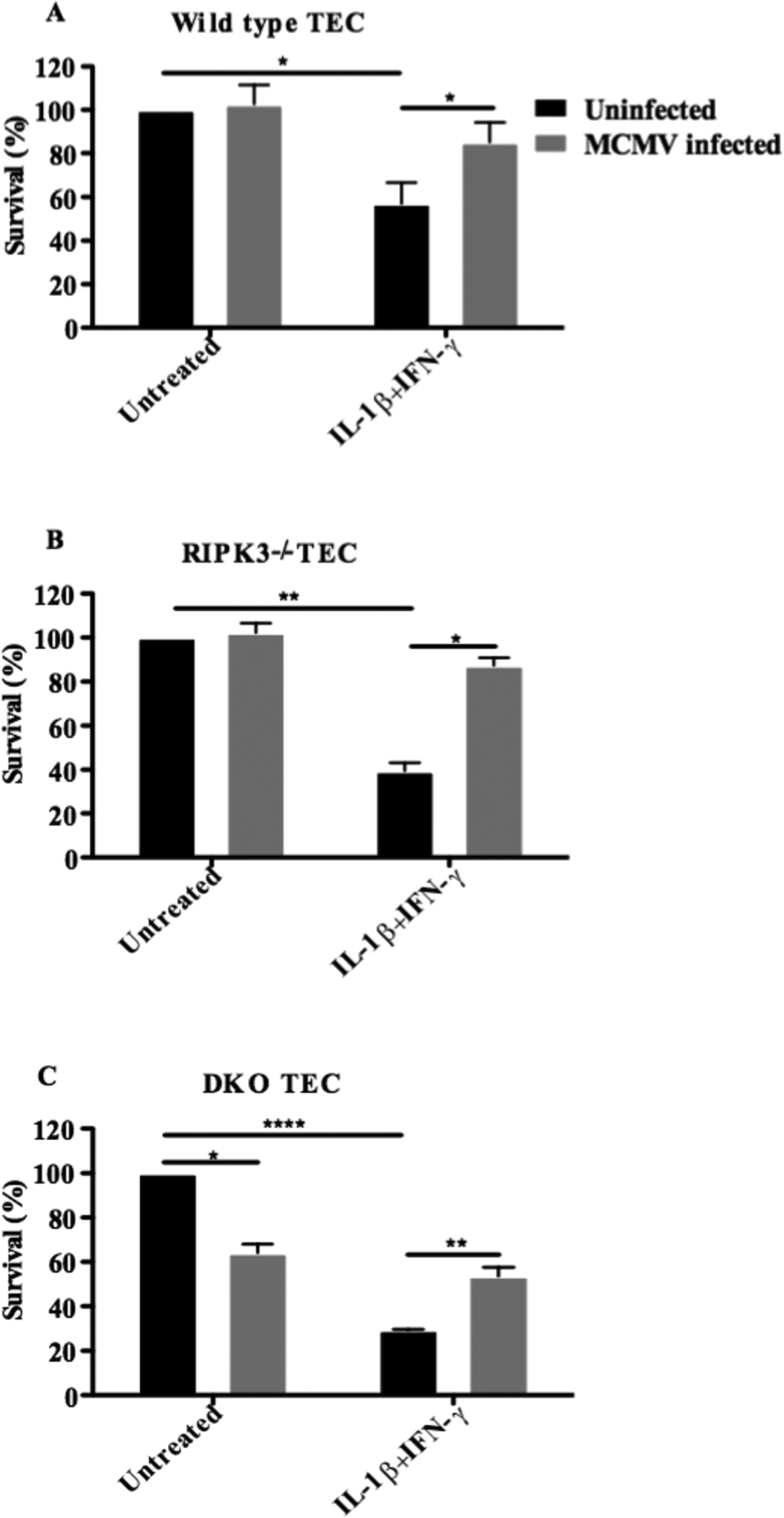
Wild type (A), RIPK3−/− (B) and DKO TEC (C) were infected with wild type K181 BAC MCMV strain respectively. TEC then were treated with combination of 4 ng/ml IL-1β and 120 ng/ml IFN- γ for 24 hours to induce intrinsic apoptosis. Cell viability was measured and quantified using MTT assay analyses. Figures represent mean of triplicates. (n=3, *: p < 0.05, **: p<0.01, ****: p<0.0001, Two-Way ANOVA). Three independent experiments showed a similar result.
Discussion
Cell death that occurs with IRI is central in regulating the inflammatory processes that further contribute to injury in a ‘self-amplification’ cycle in which DAMP from necrotic cells induce further necrotic cell death1, 2. In the present study, we have shown that loss of both RIPK3-mediated necroptosis and caspase-8 mediated extrinsic apoptosis did not provide an additive or synergistic benefit in IRI over RIPK3 deficiency alone. This appears related to enhanced mitochondrial based BAX-dependent caspase-9 mediated intrinsic apoptosis. These data suggest that blocking both extrinsic and intrinsic apoptosis as well as necroptosis, may be required to abrogate rather than attenuate renal IRI as we and others have shown in blocking individual pathways1, 2, 10, 11.
DR members (TNFR-1, Fas, DR4 or 5) interacting with their respective ligands are known to induce both apoptosis and necroptosis in parenchymal cells, including TEC1, 2, 8–11. Apoptosis has long been considered the prototypic form of programmed cell death, and it has been extensively studied in diverse models in oncology, developmental biology, and transplantation. Caspase-8 silencing or inhibition can protect TEC against TNF-α induced apoptosis in vitro and renal IRI in vivo23, 24. However while silencing caspase-8 and reducing apoptosis has been shown to be beneficial in renal IRI, it decreased kidney allograft survival by promoting necroptosis and inflammation11. These findings emphasize the potential for harm from strategies that do not consider the inter-relation of cell death pathways.
Necrosis was thought to be an unregulated form of cell death. However, under certain conditions in which caspase-8 is inhibited, TNF superfamily receptors including TNFR1 and Fas can induce a programmed form of necrosis which has now been termed necroptosis8. Necroptosis leads to the rupture of plasma membranes and the release of cytoplasmic contents which provoke inflammation2, 9. Necroptosis is dependent on RIPK1 and RIPK3. Inhibition of RIPK1 produced a significant benefit in kidney function and reduced ATN in a renal IRI model10–12. Similarly, inflammation and rejection were attenuated but not eliminated in RIPK3−/− kidney transplants11 and heart transplants25, suggesting that other forms of cell death including extrinsic and intrinsic apoptosis, likely remain operational in long-term transplants. Targeting of both apoptosis and necroptosis in kidneys may be thus expected to have an additive benefit. A previous study showed that caspase-8/RIPK3 DKO mice and RIPK3−/− mice had equivalent protection in a kidney IRI model12. However, a recent study showed that DKO mice having a 100% survival rate compared with approximately 90% survival of RIPK3−/− mice in lethal kidney IRI14. Although these diverging results may be accounted for by differences in models used, it remains unclear why inhibition of several distinct pathways of cell death did not improve IRI overall in a nonlethal model. In our study, RIPK3 and caspase-8 deficiency did not provide additional protection as compared to RIPK3 deficiency alone in a non-lethal kidney IRI model. ATN by blinded scoring of sections was similar in RIPK3−/− mice and DKO mice post-IRI (Figure 1). These results suggested that a compensatory mechanism for TEC death exists in response to the inflammatory stress of IRI, evident only when necroptosis and extrinsic apoptosis are blocked. Indeed, using TEC in vitro as surrogates of parenchymal injury in kidneys, the removal of caspase-8 and RIPK3 resulted in augmentation of intrinsic apoptosis and increased caspase-9 and caspase-3 activation (Figure 4). Furthermore, death could be inhibited by the BAX inhibitor BIP in support of activation of intrinsic apoptosis pathways (Figure 4). This may be a unique response of TEC as a previous study showed that thymocytes from DKO mice did not show different responses compared to wild type or RIPK3−/− thymocytes in response to intrinsic apoptosis stimuli8. Thus, DKO TEC may be predisposed to enhanced intrinsic apoptosis and up-regulation of pro-apoptotic BAX, BAK, and BAD mRNA compared to wild type or RIPK3−/− TEC (Figure 2&3) might account for differences between TEC and thymocytes. In our study, the protein expression of BAK and BAD were higher in DKO TEC. However, BAX protein level is not significantly higher in DKO compared with other TEC. In our study, anti-BAX detected size 17 kD protein, not 21 kD as the manufacture suggested. The reason for this difference is unknown. We postulate but can not prove degradation of BAX protein in this study.
Interestingly, TEC infected with MCMV showed near-complete resistance to intrinsic apoptosis (Figure 5). Human and mouse CMV encode a series of ‘anti-death proteins’ that can inhibit RIPK1/3, caspase-8, and BAK or BAX.22 MCMV’s ability to inhibit necroptosis, extrinsic as well as intrinsic apoptosis has been documented in other cell types, but our study shows for the first time that MCMV exerts its cell death-inhibiting abilities in TEC. However, MCMV has cytotoxic effects on TEC, so exploiting this capacity of MCMV in any therapeutic strategy might involve using only the endogenous viral proteins specific to these 3 pathways15, 21.
Programmed cell death including apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy have been shown to contribute acute kidney injury1, 26–28. How these are selectively triggered and related to organ injury remains unclear. Apoptosis or autophagy may inhibit necroptosis, whereas, necroptosis may have a reciprocal relationship with other forms of death including apoptosis, pyroptosis and ferroptosis25–27. It is reasonable to consider that RIPK3 and caspase-8 deletion could have an impact on additional cell death pathways apart from intrinsic apoptosis as demonstrated in this study. As we seek to translate insights of cell death into clinical scenarios, it is increasingly apparent how critical it is to understand how different cell death program interact each other during organ injury.
In conclusion, we show for the first time that lack of benefit from deletion of both RIPK3 and caspase-8 is related due to enhanced intrinsic apoptosis. These data highlight the complex biology of programmed cell death in kidney IRI and suggests blocking multiple death pathways may be required for the prevention of kidney IRI clinically.
Acknowledgements:
We thank Dr. Francis Chan for RIPK3−/− mice, Dr. Razqallah Hakem for Caspase-8−/− mice. This work was supported by Canadian Institutes of Health Research (CIHR, MOP-111180, MOP-115048, AMJ, ZXZ). This work is also supported by Kidney Foundation of Canada (KFOC160020,AMJ and ZXZ). This work is part of the Canadian National Transplant Research Program (CNTRP) and was supported by the CIHR and partners (TFU-127880).
Footnotes
Disclosure: All the authors have declared no conflicts of interest.
References
- 1.Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013; 13: 2797–804. [DOI] [PubMed] [Google Scholar]
- 2.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017; 18: 127–36. [DOI] [PubMed] [Google Scholar]
- 3.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010; 11: 700–14. [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZX, Wang S, Huang X, Min WP, Sun H, Liu W, et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol. 2008; 181: 7489–98. [DOI] [PubMed] [Google Scholar]
- 5.Du C, Guan Q, Yin Z, Masterson M, Zhong R, Jevnikar AM. Renal tubular epithelial cell apoptosis by Fas-FasL-dependent self-injury can augment renal allograft injury. Transplantation proceedings. 2003; 35: 2481–2. [DOI] [PubMed] [Google Scholar]
- 6.Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest. 2015; 125: 1620–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ismail OZ, Zhang X, Wei J, Haig A, Denker BM, Suri RS, et al. Kidney injury molecule-1 protects against Galpha12 activation and tissue damage in renal ischemia-reperfusion injury. Am J Pathol. 2015; 185: 1207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011; 471: 363–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green DR. Pseudokiller, qu’est-ce que c’est? Immunity. 2013; 39: 421–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012; 81: 751–61. [DOI] [PubMed] [Google Scholar]
- 11.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013; 13: 2805–18. [DOI] [PubMed] [Google Scholar]
- 12.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2013; 110: 12024–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du C, Wang S, Diao H, Guan Q, Zhong R, Jevnikar AM. Increasing resistance of tubular epithelial cells to apoptosis by shRNA therapy ameliorates renal ischemia-reperfusion injury. Am J Transplant. 2006; 6: 2256–67. [DOI] [PubMed] [Google Scholar]
- 14.Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cam M, Handke W, Picard-Maureau M, Brune W. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 2010; 17: 655–65. [DOI] [PubMed] [Google Scholar]
- 16.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004; 24: 1464–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011; 471: 368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormick AL, Meiering CD, Smith GB, Mocarski ES. Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J Virol. 2005; 79: 12205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Storling J, et al. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes. 2009; 58: 1807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010; 7: 302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nature reviews Immunology. 2012; 12: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Handke W, Krause E, Brune W. Live or let die: manipulation of cellular suicide programs by murine cytomegalovirus. Med Microbiol Immunol. 2012; 201: 475–86. [DOI] [PubMed] [Google Scholar]
- 23.Du C, Guan Q, Diao H, Yin Z, Jevnikar AM. Nitric oxide induces apoptosis in renal tubular epithelial cells through activation of caspase-8. Am J Physiol Renal Physiol. 2006; 290: F1044–F54. [DOI] [PubMed] [Google Scholar]
- 24.Du C, Guan Q, Yin Z, Zhong R, Jevnikar AM. IL-2-mediated apoptosis of kidney tubular epithelial cells is regulated by the caspase-8 inhibitor c-FLIP. Kidney Int. 2005; 67: 1397–409. [DOI] [PubMed] [Google Scholar]
- 25.Pavlosky A, Lau A, Su Y, Lian D, Huang X, Yin Z, et al. RIPK3-Mediated Necroptosis Regulates Cardiac Allograft Rejection. Am J Transplant. 2014; 14: 1778–90. [DOI] [PubMed] [Google Scholar]
- 26.Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014; 14: 759–67. [DOI] [PubMed] [Google Scholar]
- 27.Linkermann A Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 2016; 89: 46–57. [DOI] [PubMed] [Google Scholar]
- 28.Mulay SR, Linkermann A, Anders HJ. Necroinflammation in Kidney Disease. J Am Soc Nephrol. 2016; 27: 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]