

In this review, Birch and Gil summarize the regulation and functions of senescence-associated secretory phenotype (SASP) and highlight the therapeutic potential of SASP modulation as complimentary, or an alternative to, current senolytic approaches.
Keywords: senescence, SASP, inflammation, cancer, aging, disease, senolytics, senomorphics, therapeutics
Abstract
Cellular senescence is a stress response that elicits a permanent cell cycle arrest and triggers profound phenotypic changes such as the production of a bioactive secretome, referred to as the senescence-associated secretory phenotype (SASP). Acute senescence induction protects against cancer and limits fibrosis, but lingering senescent cells drive age-related disorders. Thus, targeting senescent cells to delay aging and limit dysfunction, known as “senotherapy,” is gaining momentum. While drugs that selectively kill senescent cells, termed “senolytics” are a major focus, SASP-centered approaches are emerging as alternatives to target senescence-associated diseases. Here, we summarize the regulation and functions of the SASP and highlight the therapeutic potential of SASP modulation as complimentary or an alternative to current senolytic approaches.
Senescence is a cellular stress response triggered by molecular damage, such as that caused by replicative exhaustion, aberrant oncogene activation (oncogene-induced senescence, OIS), or treatment with chemotherapeutics. Senescent cells arrest stably, produce a complex secretome (known as the senescence-associated secretory phenotype, SASP), and undergo characteristic changes including transcriptional, epigenetic, morphological, and metabolic alterations (Gorgoulis et al. 2019). Senescent cells are characterized by a prolonged and generally irreversible cell cycle arrest. The presence of DNA and other types of macromolecular damage ultimately leads to cessation of proliferation through activation of the p53/p21CIP1 and p16INK4a/RB tumor suppressor pathways (McHugh and Gil 2018). Induction of p21CIP1 and p16INK4a inhibits cyclin-dependent kinases CDK4, CDK6, (and CDK2 in the case of p21CIP1) that are necessary to promote cell cycle progression. This stable arrest explains some of the pathophysiological effects associated with senescence, but many others relate to the ability of senescent cells to influence their microenvironment. Senescent cells can communicate via direct cell-cell contact (Nelson et al. 2012; Hoare et al. 2016), cell fusion (Chuprin et al. 2013), through the formation of cytoplasmic bridges (Biran et al. 2017), by extracellular vesicle (EV) signaling (Takasugi et al. 2017), and through the SASP. Most of the nonautonomous effects of senescent cells have been linked to the SASP (Fig. 1).
Figure 1.
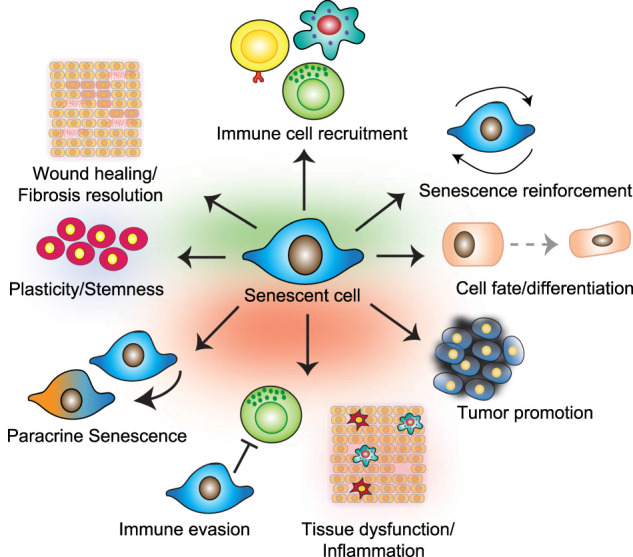
The pleiotropic functions of the SASP. Shown here is a summary of the effects exerted by senescent cells (in the middle) that are mediated by the SASP. The effects above the senescent cell (in green) represent those that are considered beneficial, whereas those at the bottom (in red) reflect some of the detrimental consequences of the SASP.
The pleiotropic effects of the SASP
Cellular senescence might have evolved to induce tissue remodeling during development and in response to damage (Muñoz-Espín and Serrano 2014). In that context, the SASP contributes to recruitment of immune cells that would clear senescent cells. However, it is clear that whatever its primordial function, the SASP can have both beneficial effects and detrimental consequences. The SASP mediates the tumor suppressor functions of senescence. For instance, components of the SASP, such as IL-8, IL-6, plasminogen activator inhibitor 1 (PAI-1), and IGFBP7 reinforce the senescence growth arrest in vitro (Acosta et al. 2008; Kuilman et al. 2008). Moreover, in a fibrosis-associated liver cancer model, the SASP can contribute to an anti-tumor microenvironment by skewing macrophage polarization to a tumor-inhibiting M1 state (Lujambio et al. 2013). TGF-β family members, vascular endothelial growth factor (VEGF), and chemokines such as CCL2 and CCL20 can spread senescence to normal neighboring cells in what is known as paracrine senescence (Acosta et al. 2013). Similarly, ROS signaling through gap junctions induces bystander senescence in vitro and, potentially, in vivo (Nelson et al. 2012). While the role of paracrine senescence in cancer is yet undefined, it has been found to contribute to liver dysfunction upon acetaminophen overdose (Bird et al. 2018). It is possible that paracrine senescence could amplify the anti-tumor response triggered during OIS. Indeed, a key function of the SASP is to signal to different immune cells, including natural killer (NK) cells, macrophages, and T cells. Immune-mediated clearance of senescent cells suppresses tumor initiation (Kang et al. 2011), contributes to tumor regression (Xue et al. 2007), and is essential during development (Muñoz-Espín et al. 2013; Storer et al. 2013). The SASP is also behind many other benefits associated with acute senescence (Muñoz-Espín and Serrano 2014). Senescent fibroblasts contribute to wound healing (Demaria et al. 2014), and the SASP of senescent hepatic stellate cells (HSCs) participates in fibrotic scar degradation and restores tissue homeostasis in liver fibrosis (Lujambio et al. 2013). In response to tissue damage, the SASP can also promote cellular reprogramming in neighboring cells (Mosteiro et al. 2016) while reinforcing plasticity and stemness (Ritschka et al. 2017). On the other hand, coculture systems and xenograft models have shown that the SASP of senescent fibroblasts promotes the tumorigenesis of precancerous epithelial cells (Krtolica at al. 2001). The SASP can also induce epithelial-to-mesenchymal transition (EMT) and increase tumor vascularization, suggesting that it has mostly protumorigenic properties (Coppé et al. 2010). This idea has been cemented using more sophisticated cancer models. For example, the SASP of HSCs promoted hepatocellular carcinoma (HCC) in obese mice treated with carcinogens (Yoshimoto et al. 2013). Similarly, pediatric craniopharyngiomas depend on the SASP of a cluster of senescent stem cells expressing oncogenic β-catenin (González-Meljem et al. 2017). Reconciling the differences in the tumor-suppressing and protumorigenic properties of the SASP is not straightforward, particularly given its context-dependent effects. Developments in transgenic mouse models that allow the detection and elimination of senescent cells has aided in causally determining the role of the SASP in tumorigenesis, but analogous mouse models allowing for regulated modulation of the SASP are required to better understand its effects.
In some instances, the SASP can also have immunosuppressive functions (Di Mitri et al. 2014; Eggert et al. 2016). For example, the SASP produced by preneoplastic hepatocytes recruits immature myeloid cells that inhibit NK cells, therefore contributing to HCC progression (Eggert et al. 2016). Chronic inflammation, often referred to as inflammaging (Franceschi and Campisi 2014), underlies many age-related pathologies. Eliminating senescent cells reduces the levels of proinflammatory cytokines in old mice (Baker et al. 2011, 2016), suggesting that the SASP may also contribute to inflammaging. Interestingly, transplanting small numbers of senescent cells causes physical dysfunction and is sufficient to increase systemic inflammation (Xu et al. 2018). The SASP may also contribute to disease by disrupting tissue homeostasis. The SASP can either suppress differentiation (of preadipocytes) or promote it (of keratinocytes), resulting in alterations such as the lipodystrophy and skin phenotypes common in old mice (Wiley et al. 2016). The SASP can also explain chemotherapy side effects, such as bone marrow suppression, cardiac dysfunction, blood clotting, or cancer relapse (Demaria et al. 2017; Wiley et al. 2019). Why the SASP exerts such pleiotropic and often opposing effects is difficult to disentangle, and much remains to be deciphered. However, the senescence inducer, cell type undergoing senescence, stage of senescence, and context of the tissue microenvironment all likely play roles in shaping the outcome.
The dynamic and heterogeneous composition of the SASP
While cellular context may explain the pleiotropic functionality of the SASP, its heterogeneous composition likely also plays an important role in accounting for its paradoxical effects. Early reports of the SASP documented a plethora of factors secreted from senescent cells, including proinflammatory and immune-modulatory cytokines and chemokines such as IL-6, IL-8, CCL2, and CXCL1, growth modulators including amphiregulin (AREG), IGFBPs, and prosurvival molecules such as the neurotrophic factor GDNF (Coppé et al. 2008). The true extent of the SASP is now better understood; it is also comprised of hundreds of protein and non-protein signaling molecules, including proteases, hemostatic factors, ceramides, bradykinins, extracellular matrix (ECM) components, and damage-associated molecular patterns (DAMPs) (Davalos et al. 2013; Wiley et al. 2019; Basisty et al. 2020). The cell type undergoing senescence and how senescence is triggered, determines SASP composition. For example, dysfunctional mitochondria cause a distinct senescence response, termed mitochondrial dysfunction-associated senescence (MiDAS), which lacks the IL-1-dependent proinflammatory arm of the SASP (Wiley et al. 2016). A recent analysis identified a core of SASP components common across different senescence inducers and in distinct cell types, some of which overlapped with aging markers observed in human plasma such as growth differentiation factor 15 (GDF15), stanniocalcin 1, and serine protease inhibitors (SERPINs) (Basisty et al. 2020). SASP strength and composition is also regulated temporally (Hernandez-Segura et al. 2017). In OIS, fluctuations in NOTCH1 levels switch an early TGF-β-rich immunosuppressive secretome to a proinflammatory SASP (Hoare et al. 2016). Moreover, in very late replicative, stress, and OIS cells, the SASP becomes characterized by the expression of type-I interferons (IFN-I) (De Cecco et al. 2019). Thus, the dynamic and complex nature of the SASP helps explain, at least in part, the diverse biological functions associated with senescence.
Regulation of the SASP
Despite the complexity of the SASP and its pleiotropic nature, we are beginning to understand the mechanisms underlying its regulation. While the events that activate the SASP are still poorly understood, they are typically connected to the DNA damage response (DDR) and converge to induce a transcriptional program required for SASP induction (Fig. 2).
Figure 2.

Regulation of the senescence-associated secretory phenotype (SASP). Scheme showing the different factors contributing to SASP induction. Transcription factors are shown in yellow. Intracellular signaling components are shown in orange. Sensors and receptors and ligands are shown in red. (DSB) Double-strand breaks, (CCF) chromatin cytoplasmic foci.
SASP induction
Senescent cells have co-opted components of the innate immune machinery to sense macromolecular damage and activate the SASP. For example, RIG-I, a sensor for cytoplasmic RNA, mediates senescence-associated inflammation (Liu et al. 2011). Another key mediator explaining SASP induction is the inflammasome (Acosta et al. 2013). Inflammasomes are groups of pattern recognition receptors (PRR) capable of recognizing different DAMPs and activating the IL-1 inflammatory cascade or inducing pyroptosis (Schroder and Tschopp 2010). The NLRP3 inflammasome is at least partially responsible for inflammasome activity during senescence (Acosta et al. 2013). In OIS, toll-like receptor 2 (TLR2) priming by acute phase serum amyloids A1 and A2 triggers inflammasome activation (Hari et al. 2019). Other DAMPs, including high-mobility group box 1 (HMGB1) can also activate TLR signaling to induce the SASP (Davalos et al. 2013). Cytosolic DNA is a key signal inducing the SASP. Sources of cytosolic DNA in senescent cells include retrotransposable elements like LINE-1 (L1) that become transcriptionally derepressed in late senescence (De Cecco et al. 2019), mitochondrial DNA, or cytosolic chromatin fragments (CCFs) (Ivanov et al. 2013). CCFs originate from the blebbing of the nuclear membrane and are linked to autophagy-mediated degradation of lamin B1 (Dou et al. 2017). Dysfunctional mitochondria (Correia-Melo et al. 2016) and a persistent DDR (Rodier et al. 2009) both promote the SASP, in part by increasing CCF formation (Vizioli et al. 2020). Cytosolic DNA is sensed by the cyclic GMP-AMP synthase (cGAS), which then produces cyclic GMP-AMP (cGAMP), activating the stimulator of interferon genes (STING). The cGAS/STING pathway is considered a key regulator of SASP induction (Dou et al. 2017; Glück et al. 2017; Yang et al. 2017).
Signaling to express the SASP
A number of factors signal to induce the SASP. STING activation stimulates TBK1 and downstream from TBK1, IRF3, and nuclear factor (NF)-κB become activated to induce IFN-I and inflammatory responses. Persistent DDR activity is required for the induction of several proinflammatory SASP factors, such as IL-6 and IL-8 (Rodier et al. 2009). Activation of p38, a member of the mitogen-activated protein kinase (MAPK) family, promotes the SASP by activating NF-κB (Freund et al. 2011). Metabolic fluctuations, such as altered nicotinamide adenine dinucleotide (NAD+) to NADH ratios, underlie the MiDAS SASP by influencing 5′AMP-activated protein kinase (AMPK)-mediated activation of p53 (Wiley et al. 2016). Conversely, increased nicotinamide phosphoribosyltransferase (NAMPT) activity, which salvages NAD+, promotes a “high” proinflammatory SASP in OIS by enhancing glycolysis and mitochondrial respiration (Nacarelli et al. 2019). All these pathways eventually converge to induce a transcriptional program required for SASP induction.
Transcriptional control of the SASP
The inflammatory SASP is regulated through the activation of two main transcription factors: NF-κB and CCAAT/enhancer binding protein β (C/EBPβ) (Kuilman et al. 2008). Indeed, Loss of both NF-κB and C/EBPβ reduce IL-8 and other chemokine receptor (CXCR)2 ligands affecting growth arrest in OIS (Acosta et al. 2008). Additionally, the NOTCH1-driven inhibition of the later proinflammatory SASP in OIS is mediated by repression of C/EBPβ transcriptional activity (Hoare et al. 2016). Increased stabilization of the transcription factor GATA-binding protein 4 (GATA4) connects the DDR with NF-κB activation to induce the SASP (Kang et al. 2015). The janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway has also been involved in regulating SASP expression (Toso et al. 2014).
Epigenetic regulation of the SASP
During OIS, enhancer landscapes are remodeled with recruitment of the bromodomain and extra-terminal (BET) family protein BRD4 to superenhancer elements adjacent to SASP genes (Tasdemir et al. 2016). Additional chromatin modifications have been implicated in SASP regulation, such as decreased histone H3K9 dimethylation at promoter regions of key SASP factors (Takahashi et al. 2012) and increased expression of the histone variant macroH2A1 (Chen et al. 2015). The histone variant H2AJ that accumulates in senescent cells also participates in SASP induction (Contrepois et al. 2017). Other epigenetic modifiers, such as histone–lysine–N-methyltransferase 2A (MLL1) and HMGB2 also favor SASP production by keeping their genetic loci in an open and active state (Aird et al. 2016; Capell et al. 2016).
Post-transcriptional control of the SASP
The mechanistic target of rapamycin (mTOR) is a key regulator of protein translation in senescent cells. mTOR and lysosomes accumulate at the trans side of the Golgi in a TOR autophagy spatial coupling compartment (TASCC) connecting autophagic processes to mTOR activation, which fuels the SASP in OIS (Narita et al. 2011). mTOR also promotes the translation of IL-1α and NF-κB activation (Laberge et al. 2015). mTOR regulates the translation of MAPKAPK2, a kinase downstream from p38, which phosphorylates and inhibits the RNA-binding protein ZFP36L1 during senescence, stabilizing SASP mRNA transcripts (Herranz et al. 2015). Additionally, the polypyrimidine tract-binding protein 1 (PTBP1) has been shown to regulate alternative splicing of genes involved in intracellular trafficking, such as EXOC7 (Georgilis et al. 2018) to control SASP expression. Overall, our understanding of SASP regulation, although incomplete, provides us with several ways to target it for therapeutic benefit.
Senescence and the SASP contribute to aging and disease
Removal of senescent cells increases healthy life span in murine models (Baker et al. 2011, 2016), but how senescent cells contribute to aging and disease is difficult to discern. This problem is exacerbated as only a small percentage of cells become senescent, even at very old age (Biran et al. 2017). Multiple scenarios explaining senescence-mediated organ dysfunction likely exist (Fig. 3). The progressive accumulation of senescent cells can deplete the organism of functional cells required for tissue repair and regeneration, such as stem and progenitor cells. For example, senescence of neural progenitors (Molofsky et al. 2006) contributes to age-related decline in neurogenesis. Similarly, senescence is responsible for the age-related decline in muscle stem cell regenerative function (Garcia-Prat et al. 2016). These effects are cell intrinsic, but in many settings senescent cells exert their effects in a paracrine fashion. For example, the injection of senescent preadipocytes (representing <1% of cells) causes widespread physical dysfunction in young mice (Xu et al. 2018). Thus, the SASP is a primary mediator of the detrimental effects of senescent cells. Chronic, low-grade inflammation, characterized by elevated levels of circulating cytokines and increased immune infiltration associated with inflammaging, fuels loss of resilience, and increased risk of disease with age (Franceschi and Campisi 2014). Since around 40% of SASP factors are also associated with age in human plasma (Basisty et al. 2020), the SASP likely contributes to inflammaging. Indeed, the clearance of senescent cells reduces levels of secreted inflammatory factors known to drive pathologies (Baker et al. 2011, 2016). Besides systemic effects, specific components secreted by senescent cells might also be responsible for inducing tissue dysfunction locally. For example, hemostatic factors, including SERPINs and PAI-1 may link the SASP with chemotherapy-associated thrombosis (Wiley et al. 2019). Similarly, TGF-β-driven paracrine senescence limits liver regeneration following acute injury with acetaminophen (Bird et al. 2018). Another way in which senescent cells, and the SASP, could contribute to disease is through remodeling of the tissue microenvironment and disruption of tissue homeostasis. This could be mediated by the secretion of ECM components, recruitment of immune cells, or by affecting the fate of other cells in the tissue. This is, for example, the case during fibrosis (Schafer et al. 2017). Finally, the physical build-up of senescent cells could contribute to disease, as is the case with the accumulation of senescent foamy macrophages in atherosclerotic plaques (Childs et al. 2016).
Figure 3.

Mechanism by which senescent cells contribute to disease. Both SASP-independent (top) and SASP-dependent (bottom) effects of senescent cells likely explain how senescence promotes tissue and organ dysfunction. The build-up of senescent cells can deplete stem and progenitor pools and could lead to dysfunctions associated with senescent cell accumulation, such as the case in atherosclerosis. Alternatively, paracrine functions of senescent cells, such as the ability of SASP factors to contribute to chronic systemic inflammation and tissue inflammation and remodeling are also at play. Specific SASP components, such as hemostatic factors including SERPINs and PAI-1, promote platelet activation and increased thrombosis following chemotherapy. Similarly, liver injury, induced by acetaminophen, is driven by macrophage-dependent TGF-β signaling that favors hepatocyte paracrine senescence limiting regeneration.
Senolytics: a wide therapeutic opportunity
Genetic models enabling the specific ablation of senescent cells have made the case to target senescence for therapeutic benefit (Fig. 4A). The most popular strategy has been the identification of drugs that selectively kill senescent cells, known as senolytics. As many senescence triggers also cause apoptosis, senescent cells are primed for death but avoid it by inducing prosurvival pathways. Indeed, enhanced resistance to apoptosis is a widely recognized feature of senescent cells (Gorgoulis et al. 2019). The first senolytic drugs were identified on the hypothesis that senescent cells are more sensitive than their normal counterparts to inhibition of these prosurvival networks. This led to the discovery of dasatinib (D), a multityrosine kinase inhibitor, and quercetin (Q), a flavonol, as senolytics (Zhu et al. 2015). Given in combination, D + Q reduce senescent cell burden in aged, radiation-exposed and progeroid mice, while improving health span parameters, including cardiovascular and physical function (Xu et al. 2018). Similar approaches led to the identification of inhibitors of the B-cell lymphoma 2 (BCL-2) family of anti-apoptotic proteins, and peptides inhibiting FOXO4 as senolytics (Ovadya and Krizhanovsky 2018). D + Q and the BCL-2 inhibitors, ABT-263 (Navitoclax) and ABT-737, are the most frequently used senolytics in vivo, showing promise in improving a wide range of pathologies (van Deursen 2019). Despite the significant success of ABT-263 and ABT-737 in preclinical studies, there are reservations regarding their clinical use. Trials assessing navitoclax as an anticancer agent report neutropenia and thrombocytopenia (Wilson et al. 2010). Senolytic drugs may not need to be administered continuously to produce the benefits associated with senolysis, which could mitigate their side effects. Nevertheless, many labs have set up different efforts to circumvent the detrimental effects of existing senolytics. For example, DT2216, a proteolysis targeting chimera (PROTAC) that targets BCL-XL to the Von Hippel-Lindau (VHL) E3 ligase for degradation, reduced toxicity in platelets, as they express low levels of VHL (He et al. 2020). Multiple groups are actively pursuing the identification of new senolytics. Phenotypic drug screens have pinpointed compounds as varied as heat-shock protein inhibitors (Fuhrmann-Stroissnigg et al. 2017), cardiac glycosides (Guerrero et al. 2019; Triana-Martínez et al. 2019), and p53–MDM2 interaction inhibitors (Jeon et al. 2017) as harboring senolytic activity. A further strategy to develop senolytics is based on exploiting the peculiarities of senescent cells, such as the senescence-associated β-galactosidase (SA-β-Gal) activity. Galacto-oligosaccharide encapsulated nanoparticles (GalNP) containing cytotoxic or senolytic drugs (Muñoz-Espín et al. 2018) and galactose-derived prodrugs display senolytic properties (González-Gualda et al. 2020; Guerrero et al. 2020) and are two examples of this approach. Given their success in preclinical studies, senolytic regimes are the more popular modality of senotherapies and are starting to be tested in clinical trials (Hickson et al. 2019).
Figure 4.

Targeting senescent cells and the SASP in age-related disease. Different strategies to target senescent cells in age-related pathologies have been proposed with the two main approaches being cell death induction via senolytics (A) and modulation of the SASP by interfering with its regulatory pathways (B).
Targeting the SASP as an alternative to senolytic drugs
Consistent with a role for senescence in regeneration, initial studies using the INK-ATTAC and p16-3MR mouse models showed defects in wound healing when senescent cells were eliminated (Baker et al. 2011; Demaria et al. 2014), suggesting that removing senescent cells can lead to unwanted outcomes. Moreover, the recent finding that clearing certain populations of senescent cells, such as liver sinusoid endothelial cells, promotes fibrosis (Grosse et al. 2020) has further strengthened the case for caution and the value of identifying alternatives to senolytics. Since many of the negative effects associated with senescence are driven by the SASP, compounds that modulate the senescence secretome (often referred to as senomorphics) are an alternative to senolytics (Fig. 4B). For example, nucleoside reverse transcriptase inhibitors (NRTIs), such as lamivudine, have been used to suppress cytoplasmic DNA for L1. As a result, lamivudine reduces the late SASP response and ameliorates several phenotypes of aging in old mice, including kidney glomerulosclerosis, skeletal muscle atrophy, and tissue immune infiltrations (De Cecco et al. 2019). Genetic interference with the cGAS-STING pathway results in SASP inhibition and reduces tissue inflammation in vivo (Dou et al. 2017; Glück et al. 2017). Importantly, genetic models have also served to link the cGAS-STING pathway with pathologies such as cancer, fibrosis, neurodegeneration, metabolic, and auto-inflammatory diseases. Given the intricate correlation between cGAS-STING, inflammation, senescence, and disease, several inhibitors of cGAS (Lama et al. 2019) and STING (Haag et al. 2018) have been developed. These are prime candidates to modulate the SASP. Similarly, the NLRP3 inflammasome is a key regulator of SASP induction, widely involved in disease and for which drug development is active (Zahid et al. 2019).
Targeting transcriptional regulators of the SASP is another option. For instance, the JAK1/2 inhibitor ruxolitinib reduces the proinflammatory SASP, in vitro and in vivo and enhances insulin sensitivity, limits osteoporosis and reduces frailty in ageing mice (Xu et al. 2015a,b; Farr et al. 2017). Inhibiting inflammation is a promising concept to treat the age-associated diseases in which senescence plays a role. However, as an anticancer therapy, the situation is more complex. While the SASP can drive cancer progression, a robust SASP is also necessary to mount immune surveillance against preneoplastic lesions or in response to therapy. In prostate tumors treated with chemotherapy, the JAK2/STAT3 pathway is required to induce an immunosuppressive SASP that leads to increased tumor growth. In this context, JAK inhibitors reprogram the SASP to trigger an antitumor immune response (Toso et al. 2014). This suggests that in some instances, rather than blunting the SASP response, modifying it might be more desirable. Indeed, inhibiting IL1 signaling, or cGAS-STING, interferes with immune surveillance and senescent reinforcement and could contribute to tumor escape (Acosta et al. 2013; Dou et al. 2017). To address this problem, a screen aiming to identify strategies to limit the proinflammatory arm of the SASP, without promoting senescence escape, identified PTBP1. As proof-of-principle, genetic interference with PTBP1 prevented tumor growth caused by the presence of senescent cells in vivo without resulting in increased tumor initiation (Georgilis et al. 2018). Interference with pathways signaling for SASP induction has also been used to limit the SASP, including using drugs targeting epigenetic changes, such as the BRD4 Inhibitor IBET762 (Tasdemir et al. 2016) and the histone deacetylase (HDAC) inhibitor, Trichostatin A (TSA). At low concentrations, TSA behaves as a pan-SASP suppressor, blunting SASP-associated inflammation in vitro and in an acetaminophen-driven liver injury model in vivo (Vizioli et al. 2020). However, at millimolar doses, TSA has been shown to promote senescence and development of the SASP (Pazolli et al. 2012), emphasizing the importance of fine balance when attempting to tweak these processes. Another concern when using drugs that inhibit epigenetic or signaling factors is how much of the observed effect can be attributed to SASP modulation. This observation applies to several other drugs. Glucocorticoid treatment decreases the production of selected components of the SASP, namely, proinflammatory cytokines like IL-6, across different inducers of senescence by inhibiting NF-κB (Laberge et al. 2012). The antidiabetic drug metformin alleviates a number of age-related conditions in experimental animal models and humans and inhibits multiple proinflammatory SASP factors by blocking NF-κB nuclear translocation (Moiseeva et al. 2013). However, these compounds exert other effects, making it difficult to prove causality of SASP inhibition per se. For instance, the mTOR inhibitor rapamycin increases life span and health span (Miller et al. 2011) and acts as a selective SASP suppressor. Rapamycin also limits the tumor-promoting abilities of senescent cells in vivo (Herranz et al. 2015; Laberge et al. 2015). However, given the wide range of actions associated with mTOR inhibition, it is difficult to know what the contribution of SASP inhibition is. Nevertheless, a number of senomorphic compounds are being tested in humans, including metformin and analogs of rapamycin (rapalogs). Metformin has been shown to improve cardiovascular function and prolong life span in diabetic patients, prompting investigation into the ability of metformin to delay aging in nondiabetic individuals (Targeting Aging with Metformin; TAME study) (Kulkarni et al. 2020). Several rapalogs, including everolimus, have also been tested in humans, with mTOR inhibition leading to improved vaccination responses in the elderly, possibly through targeting immunosenescence (Mannick et al. 2014).
Targeting of mitochondrial-derived ROS with MitoQ suppresses the SASP and has been shown to limit skin aging in 3D equivalents (Victorelli et al. 2019; Vizioli et al. 2020), suggesting that indirect effects on the SASP, via improved mitochondrial function, are possible. Consistently, the use of FK866, a specific NAMPT inhibitor, suppresses the proinflammatory SASP and limits pancreatic ductal adenocarcinoma (PDAC) progression (Nacarelli et al. 2019). These findings suggest that mitochondrial targeting could be a viable way to modulate the SASP. Alternatively, targeting specific SASP components (e.g., using anakinra, an IL-1R antagonist or tocilizumab, an antibody against IL-6R) could dampen the SASP, since its persistence relies on self-amplifying feedback loops. Other times, individual SASP components, such as TGF-β, might be pinpointed as disease drivers and targeting them specifically might be appropriate (Bird et al. 2018).
Engaging senescence in cancer treatment
While most senotherapies rely on opposing senescence and its phenotypes, induction of senescence (prosenescence therapies) is being actively investigated for cancer treatment. Antitumor strategies, including radiotherapies and many chemotherapies, cause extensive DNA damage, inducing senescence (Gorgoulis et al. 2019). Some targeted therapies, such as inhibitors of aurora kinase or CDK4/6 also trigger tumor senescence and show promise in treating a number of cancers (Wagner and Gil 2020). While senescence induction in cancer cells is an attractive therapeutic option, care must be taken to limit the detrimental effects associated with the chronic presence of senescent cells. These include increases in local and systemic inflammation and cancer reoccurrence (Demaria et al. 2017). Therapy-induced senescence (TIS) also promotes stem cell and self-renewing features, which can contribute to cancer initiation and aggressiveness (Milanovic et al. 2018). Recent developments in senotherapies have provided additional ways in which senescence and the SASP could be targeted for the treatment of cancer while circumventing potential undesirable effects of senescence induction (Fig. 5).
Figure 5.
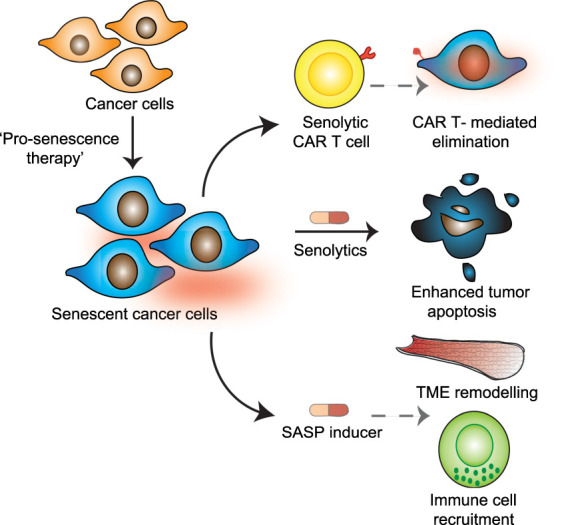
Targeting senescence in cancer. Engaging senescence using “prosenescence therapies” in cancer can be followed up by strategies that then exploit vulnerabilities of senescent cancer cells. Inducing senescent cell death by senolytic treatment or through the development of senolytic CAR T cells that recognize factors expressed on the surface of senescent cancer cells is one approach. An alternative method is to use drugs that potentiate the SASP. Drugs that induce or reinforce the SASP have been found to promote immune cell surveillance and senescent cell clearance. SASP inducers can also remodel the tumor microenvironment, favoring access by immune cells and chemotherapeutic drugs used as part of anticancer treatments. TME, tumor microenvironment.
Senolytics in combined therapies against cancer
One way to potentiate anticancer therapies is to pair them with senolytic treatments. This strategy of following up prosenescence treatment with senolytics has been termed a “one–two punch approach” and has been demonstrated in vitro using human cancer cells, including melanoma and lung cancer lines (Wang et al. 2017). Treatment of tumors with chemotherapy can induce vulnerabilities associated with senescence that could be targeted therapeutically. Indeed, one of the first descriptions of the viability of synthetic lethal targeting of senescence to improve cancer treatment was from Dörr et al. (2013). Here, proteotoxic stress accompanying TIS, driven in part by SASP demand, rendered senescent cells vulnerable to the inhibition of glucose processing and autophagy, with pharmacological disruption of these altered metabolic processes leading to tumor regression and improved outcomes in murine lymphoma models (Dörr et al. 2013). Senescence induction by inhibition of DNA replication kinase CDC7 has also been paired with a senolytic that disrupts mTOR signaling, inducing apoptosis in senescent human liver cancer cells and improving survival in mouse models of HCC (Wang et al. 2019). Such “double-hit” strategies are still to be tested in patients. However, preclinical findings of this approach appear promising. While a “gold standard” senolytic, the use of ABT-263 in the clinic is limited due to its reported side effects. Other senolytic compounds, including cardiac glycosides, are widely used clinically and cooperate with prosenescence therapies to kill cancer cells (Guerrero et al. 2019). Moreover, such broad-spectrum senolytics, which target different types of senescent cells that coexist in the tumor microenvironment, could have benefits that go beyond potentiating prosenescence therapies (Guerrero et al. 2019).
Harnessing the immune system to eliminate senescent cells
Recently, a different approach has been described to eliminate tumor cells undergoing senescence. Rather than using senolytics, Amor et al. (2020) engineered chimeric antigen receptor (CAR) T cells recognizing uPAR, a protein expressed by senescent cells, a strategy that mirrors normal senescence immunosurveillance. Importantly, these “senolytic CAR T cells” can be used not just as an anticancer therapy, but to treat other senescence-associated disorders, such as nonalcoholic steatohepatitis (NASH) (Amor et al. 2020). The identification of senescence-specific antigens opens up the possibility of targeting them by other means, such as using bispecific antibodies or antibodies conjugated with toxic ligands.
Potentiating the SASP as an anticancer therapy
An alternative to artificially engineered CAR T cells is to potentiate the clearance of senescent cells by the immune system. This could be achieved using drugs that enhance the SASP. Two recent studies exemplify this approach. Ruscetti et al. (2018) showed that while palbociclib treatment of lung tumors alone induces features of senescence, its combination with the MEK inhibitor Trametinib (T/P) results in a stronger growth arrest and a more potent SASP. Indeed, T/P induces senescence in a KRAS-mutant lung cancer model and activates an immunomodulatory SASP, comprised of TNF-α and intercellular adhesion molecule−1 (ICAM1). This promotes NK cell surveillance and tumor regression and prolongs survival (Ruscetti et al. 2018). The effects of SASP induction seem to be context dependent. In PDAC, treatment with T/P led to SASP-facilitated remodeling of the tumor microenvironment (TME), which exposed therapeutic vulnerabilities. Specifically, VEGF increased blood vessel density, whereas the proinflammatory SASP, namely, IL-6, CCL5, and CXCL1, promoted endothelial cell activation, increasing vessel permeability. This led to improved access of the chemotherapeutic drug gemcitabine and facilitated tumor infiltration by CD8+ T cells, promoting tumor destruction (Ruscetti et al. 2020). While, the infiltrated CD8+ T cells were exhausted, they could be reawakened by checkpoint blockade. In this way, combining either gemcitabine or anti-PD-1 therapy with T/P produces a potent antitumor effect.
Conclusions, challenges, and future directions
Targeting senescent cells offers a myriad of therapeutic opportunities, particularly in the context of age-associated disease and cancer. This knowledge has fueled testing of senolytic drugs as a novel therapeutic paradigm. Although the SASP likely explains many of the detrimental consequences attributed to senescence, direct evidence of its role is often lacking. Transgenic models that allow specific targeting of senescent cells has helped prove causality. The development of ingenious models allowing the manipulation of the SASP will help to determine its precise involvement as a mediator of the effects exerted by senescent cells. This is likely to be complicated given SASP heterogeneity and its context-dependent effects. Clarifying SASP involvement will contribute to placing senomorphics on par with senolytic approaches, increasing the arsenal available to treat senescence-associated diseases.
To design senotherapies targeted to specific diseases, we will need to better understand what cell types undergo senescence, particularly since eliminating certain senescent cell types can be harmful (Grosse et al. 2020). Clarification of the molecular and physiological properties of senescent cells, including their SASP profile, is also needed. The advancement of single-cell technologies will likely further these efforts. Indeed, while a number of SASP regulators have been identified to date, most modulators interrogated thus far impact on the proinflammatory and immune-modulatory SASP, but it is unclear which signaling pathways influence the profibrotic or proangiogenic SASP, for example. It is also important to be able to decipher regulators of the SASP per se rather than those underpinning more generic inflammatory processes. Separating the SASP from inflammatory events distinct from senescence is a current challenge; this will hopefully be helped by the identification of reliable in vivo markers of senescence and models that allow for targeted manipulation of SASP regulatory factors within senescent cells. It is unlikely that complete SASP disruption will ever be beneficial; rather a better appreciation of the different arms of the SASP, and the effects that they exert, is needed. Knowledge of the regulatory processes that are at play in bystander cells, and how bystander responses change depending on tissue, with age, or in particular disease states, will also be of value. This is partly exemplified by the complex, and often varied, responses elicited by the SASP in cancer. A clear understanding of these differences will help inform when and how prosenescence therapies and senotherapies are used. Answers to some of these questions are needed promptly, particularly as induction of senescence in cancer cells is emerging as a therapeutic concept and senolytics are now being trialed in humans, albeit the safety and efficacy of these compounds for human use still remain to be fully determined (Hickson et al. 2019). While the dynamic and ever-evolving consequences of senescence appear increasingly complex, one thing is certain: The therapeutic opportunities can be transformative, provided we harness them correctly.
Competing interest statement
J.G. has acted as a consultant for Unity Biotechnology, Geras Bio, and Merck KGaA; owns equity in Unity Biotechnology and Geras Bio; and is a named inventor in an MRC patent related to senolytic therapies.
Acknowledgments
Core support from Medical Research College (MC_U120085810) funds research in J.G.’s laboratory.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.343129.120.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. 2013. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978–990. 10.1038/ncb2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, Le L, Alicea G, Yang TL, Johnson FB, et al. 2016. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol 215: 325–334. 10.1083/jcb.201608026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, et al. 2020. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583: 127–132. 10.1038/s41586-020-2403-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. 10.1038/nature10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. 2016. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530: 184–189. 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Sharma V, Ferrucci L, et al. 2020. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18: e3000599 10.1371/journal.pbio.3000599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, Krizhanovsky V. 2017. Quantitative identification of senescent cells in aging and disease. Aging Cell 16: 661–671. 10.1111/acel.12592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TG, Müller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, Lu WY, Jamieson T, Govaere O, Campbell AD, et al. 2018. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med 10: eaan1230 10.1126/scitranslmed.aan1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, et al. 2016. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev 30: 321–336. 10.1101/gad.271882.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. 2015. Macroh2a1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol Cell 59: 719–731. 10.1016/j.molcel.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. 2016. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354: 472–477. 10.1126/science.aaf6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. 2013. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 27: 2356–2366. 10.1101/gad.227512.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrepois K, Coudereau C, Benayoun BA, Schuler N, Roux PF, Bischof O, Courbeyrette R, Carvalho C, Thuret JY, Ma Z, et al. 2017. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat Commun 8: 14995 10.1038/ncomms14995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: e301 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118. 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, et al. 2016. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35: 724–742. 10.15252/embj.201592862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J. 2013. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201: 613–629. 10.1083/jcb.201206006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. 2019. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566: 73–78. 10.1038/s41586-018-0784-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dollé ME, et al. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31: 722–733. 10.1016/j.devcel.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. 2017. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7: 165–176. 10.1158/2159-8290.CD-16-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, D'Antuono R, Montani E, Garcia-Escudero R, Guccini I, et al. 2014. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 515: 134–137. 10.1038/nature13638 [DOI] [PubMed] [Google Scholar]
- Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. 2013. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501: 421–425. 10.1038/nature12437 [DOI] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. 2017. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406. 10.1038/nature24050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, et al. 2016. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30: 533–547. 10.1016/j.ccell.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, et al. 2017. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23: 1072–1079. 10.1038/nm.4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69: S4–S9. 10.1093/gerona/glu057 [DOI] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. 2011. p38MAPK is a novel DNA damage response independent regulator of the senescence-associated secretory phenotype. EMBO J 30: 1536–1548. 10.1038/emboj.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, Grassi D, Gregg SQ, Stripay JL, Dorronsoro A, et al. 2017. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun 8: 422 10.1038/s41467-017-00314-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. 2016. Autophagy maintains stemness by preventing senescence. Nature 529: 37–42. 10.1038/nature16187 [DOI] [PubMed] [Google Scholar]
- Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, Leote AC, D'Artista L, Gallage S, Seehawer M, et al. 2018. PTBP1-mediated alternative splicing regulates the inflammatory secretome and the Pro-tumorigenic effects of senescent cells. Cancer Cell 34: 85–102.e9. 10.1016/j.ccell.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. 2017. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070. 10.1038/ncb3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Meljem JM, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, Kaushal G, Virasami A, Panousopoulos L, Mousavy-Gharavy SN, et al. 2017. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 8: 1819 10.1038/s41467-017-01992-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Gualda E, Pàez-Ribes M, Lozano-Torres B, Macias D, Wilson JR, González-López C, Ou HL, Mirón-Barroso S, Zhang Z, Lérida-Viso A, et al. 2020. Galacto-conjugation of navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19: e13142 10.1111/acel.13142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, et al. 2019. Cellular senescence: defining a path forward. Cell 179: 813–827. 10.1016/j.cell.2019.10.005 [DOI] [PubMed] [Google Scholar]
- Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, Bulavin DV. 2020. Defined p16(high) senescent cell types are indispensable for mouse healthspan. Cell Metab 32: 87–99.e6. 10.1016/j.cmet.2020.05.002. [DOI] [PubMed] [Google Scholar]
- Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, Wolter K, Pombo J, Irvine EE, Innes AJ, et al. 2019. Cardiac glycosides are broad-spectrum senolytics. Nat Metab 1: 1074–1088. 10.1038/s42255-019-0122-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, Guiho R, Herranz N, Uren A, Withers DJ, Martínez-Barbera JP, Tietze LF, Gil J. 2020. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 19: e13133 10.1111/acel.13133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, et al. 2018. Targeting STING with covalent small molecule inhibitors. Nature 559: 269–273. 10.1038/s41586-018-0287-8 [DOI] [PubMed] [Google Scholar]
- Hari P, Millar FR, Tarrats N, Birch J, Quintanilla A, Rink CJ, Fernández-Duran I, Muir M, Finch AJ, Brunton VG, et al. 2019. The innate immune sensor toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci Adv 5: eaaw0254 10.1126/sciadv.aaw0254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang X, Chang J, Kim HN, Zhang P, Wang Y, Khan S, Liu X, Zhang X, Lv D, et al. 2020. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat Commun 11: 1996 10.1038/s41467-020-15838-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. 2017. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27: 2652–2660.e4. 10.1016/j.cub.2017.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al. 2015. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17: 1205–1217. 10.1038/ncb3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, et al. 2019. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47: 446–456. 10.1016/j.ebiom.2019.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama R, et al. 2016. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 18: 979–992. 10.1038/ncb3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. 2013. Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202: 129–143. 10.1083/jcb.201212110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, et al. 2017. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23: 775–781. 10.1038/nm.4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551. 10.1038/nature10599 [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. 2015. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: aaa5612 10.1126/science.aaa5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. 2001. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci 98: 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019–1031. [DOI] [PubMed] [Google Scholar]
- Kulkarni AS, Gubbi S, Barzilai N. 2020. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab 32: 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Zhou L, Sarantos MR, Rodier F, Freund A, de Keizer PL, Liu S, Demaria M, Cong YS, Kapahi P, et al. 2012. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11: 569–578. 10.1111/j.1474-9726.2012.00818.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061. 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, Gogakos T, Steinberg JI, Miller M, Ramos-Espiritu L, et al. 2019. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun 10: 2261 10.1038/s41467-019-08620-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Wu S, Ren H, Gu J. 2011. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat Cell Biol 13: 254–262. 10.1038/ncb2167 [DOI] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153: 449–460. 10.1016/j.cell.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, et al. 2014. mTOR inhibition improves immune function in the elderly. Sci Transl Med 6: 268ra179 10.1126/scitranslmed.3009892 [DOI] [PubMed] [Google Scholar]
- McHugh D, Gil J. 2018. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol 217: 65–77. 10.1083/jcb.201708092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Yu Y, Schmitt CA. 2018. The senescence-stemness alliance–A cancer-hijacked regeneration principle. Trends Cell Biol 28: 1049–1061. 10.1016/j.tcb.2018.09.001 [DOI] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, et al. 2011. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 66A: 191–201. 10.1093/gerona/glq178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. 2013. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 12: 489–498. 10.1111/acel.12075 [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. 2006. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443: 448–452. 10.1038/nature05091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, FernandezMarcos PJ, Muñoz-Martin M, Blanco-Aparicio C, Pastor J, et al. 2016. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354: aaf4445 10.1126/science.aaf4445 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Serrano M. 2014. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482–496. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. 2013. Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118. 10.1016/j.cell.2013.10.019 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, Paez-Ribes M, Llanos S, Chaib S, Muñoz-Martín M, Ucero AC, et al. 2018. A versatile drug delivery system targeting senescent cells. EMBO Mol Med 10: e9355 10.15252/emmm.201809355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, Aird KM, Iwasaki O, Kossenkov AV, Schultz D, et al. 2019. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 21: 397–407. 10.1038/s41556-019-0287-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, et al. 2011. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332: 966–970. 10.1126/science.1205407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. 2012. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11: 345–349. 10.1111/j.1474-9726.2012.00795.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadya Y, Krizhanovsky V. 2018. Strategies targeting cellular senescence. J Clin Invest 128: 1247–1254. 10.1172/JCI95149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. 2012. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res 72: 2251–2261. 10.1158/0008-5472.CAN-11-3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. 2017. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31: 172–183. 10.1101/gad.290635.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. 2009. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11: 973–979. 10.1038/ncb1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Feucht J, et al. 2018. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362: 1416–1422. 10.1126/science.aas9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti M, Morris J, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho YJ, Fennell M, et al. 2020. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181: 424–441.e21. 10.1016/j.cell.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. 2017. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532 10.1038/ncomms14532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140: 821–832. 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. 2013. Senescence Is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130. 10.1016/j.cell.2013.10.041 [DOI] [PubMed] [Google Scholar]
- Takahashi A, Imai Y, Yamakoshi K, Kuninaka S, Ohtani N, Yoshimoto S, Hori S, Tachibana M, Anderton E, Takeuchi T, et al. 2012. DNA damage signaling triggers degradation of histone methyltransferases through APC/CCdh1 in senescent cells. Mol Cell 45: 123–131. 10.1016/j.molcel.2011.10.018 [DOI] [PubMed] [Google Scholar]
- Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. 2017. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun 8: 15729 10.1038/ncomms15728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, et al. 2016. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov 6: 612–629. 10.1158/2159-8290.CD-16-0217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, Pinton S, Zhang J, Kalathur M, Civenni G, et al. 2014. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep 9: 75–89. 10.1016/j.celrep.2014.08.044 [DOI] [PubMed] [Google Scholar]
- Triana-Martínez F, Picallos-Rabina P, Da Silva-Álvarez S, Pietrocola F, Llanos S, Rodilla V, Soprano E, Pedrosa P, Ferreirós A, Barradas M, et al. 2019. Identification and characterization of cardiac glycosides as senolytic compounds. Nat Commun 10: 4731 10.1038/s41467-019-12888-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM. 2019. Senolytic therapies for healthy longevity. Science 364: 636–637. 10.1126/science.aaw1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorelli S, Lagnado A, Halim J, Moore W, Talbot D, Barrett K, Chapman J, Birch J, Ogrodnik M, Meves A, et al. 2019. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J 38: e101982 10.15252/embj.2019101982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, Perez-Garcia A, Kiourtis C, Dasgupta N, Lei X, et al. 2020. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev 34: 428–445. 10.1101/gad.331272.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner V, Gil J. 2020. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 39: 5165–5176. 10.1038/s41388-020-1354-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Leite de Oliveira R, Wang C, Fernandes Neto JM, Mainardi S, Evers B, Lieftink C, Morris B, Jochems F, Willemsen L, et al. 2017. High-throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Rep 21: 773–783. 10.1016/j.celrep.2017.09.085 [DOI] [PubMed] [Google Scholar]
- Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, de Oliveira RL, Morris B, Gadiot J, Wang W, et al. 2019. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574: 268–272. 10.1038/s41586-019-1607-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, et al. 2016. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23: 303–314. 10.1016/j.cmet.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Liu S, Limbad C, Zawadzka AM, Beck J, Demaria M, Artwood R, Alimirah F, Lopez-Dominguez JA, Kuehnemann C, et al. 2019. SILAC analysis reveals increased secretion of hemostasis-related factors by senescent cells. Cell Rep 28: 3329–3337.e5. 10.1016/j.celrep.2019.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, et al. 2010. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 11: 1149–1159. 10.1016/S1470-2045(10)70261-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N, et al. 2015a. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4: e12997 10.7554/eLife.12997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, et al. 2015b. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci 112: E6301–E6310. 10.1073/pnas.1515386112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, et al. 2018. Senolytics improve physical function and increase lifespan in old age. Nat Med 24: 1246–1256. 10.1038/s41591-018-0092-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660. 10.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. 2017. cGAS is essential for cellular senescence. Proc Natl Acad Sci 114: E4612–E4620. 10.1073/pnas.1705499114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101. 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- Zahid A, Li B, Kombe AJK, Jin T, Tao J. 2019. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol 10: 2538 10.3389/fimmu.2019.02538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. 2015. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14: 644–658. 10.1111/acel.12344 [DOI] [PMC free article] [PubMed] [Google Scholar]