

Abstract
The GGGGCC hexanucleotide expansion in C9orf72 (C9) is the most frequent known cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), yet a clear understanding of how C9 fits into the broader context of ALS/FTD pathology has remained lacking. The repetitive RNA derived from the C9 repeat is known to sequester hnRNPH, a splicing regulator, into insoluble aggregates, resulting in aberrant alternative splicing. Furthermore, hnRNPH insolubility and altered splicing of a robust set of targets have been observed to correlate in C9 and sporadic ALS/FTD patients alike, suggesting that changes along this axis are a core feature of disease pathogenesis. Here, we characterize previously uncategorized RNA splicing defects involving widespread intron retention affecting almost 2000 transcripts in C9ALS/FTD brains exhibiting a high amount of sequestered, insoluble hnRNPH. These intron retention events appear not to alter overall expression levels of the affected transcripts but rather the protein-coding regions. These retained introns affect transcripts in multiple cellular pathways predicted to be involved in C9 as well as sporadic ALS/FTD etiology, including the proteasomal and autophagy systems. The retained intron pre-mRNAs display a number of characteristics, including enrichment of hnRNPH-bound splicing enhancer motifs and a propensity for G-quadruplex (G-Q) formation, linking the defective splicing directly to high amounts of sequestered hnRNPH. Together, our results reveal previously undetected splicing defects in high insoluble hnRNPH-associated C9ALS brains, suggesting a feedback between effective RNA-binding protein dosage and protein quality control in C9, and perhaps all, ALS/FTD.
C9orf72 (C9) is a critical gene at the intersection of the devastating neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Expansion of the variable length hexanucleotide repeat contained within the first intron of C9 is the most common known genetic lesion in both familial and sporadic forms of ALS (DeJesus-Hernandez et al. 2011; Renton et al. 2011), although a large majority of ALS cases have no known mutation. Moreover, C9 repeat expansion exhibits the perplexing behavior of causing FTD in addition to or instead of ALS, differing even among family members with the same parental allele. It is widely believed that the ability to explain and eventually treat the ALS/FTD disease spectrum as a whole will stem from a precise mechanistic understanding of how this polymorphic mutation sets off a cascade ending in highly specific neuronal degeneration (Cook and Petrucelli 2019). To this end, studies seeking to understand the basic molecular properties of the C9 repeats, and to find similarities between C9ALS/FTD and sporadic ALS/FTD, have highlighted an important role for pre-mRNA splicing and its dysregulation (Taylor et al. 2016; Conlon et al. 2018; Nussbacher et al. 2019). RNA splicing is a highly complex process that is known to distinguish neuronal subtypes (Furlanis et al. 2019), thereby suggesting a potential mechanism for the differing cellular vulnerabilities to a ubiquitously expressed somatic mutation.
In the broad interest of identifying a central mechanism of ALS/FTD through consideration of all genetic causes, defects in pre-mRNA processing, resulting from alterations in the activity of RNA-binding proteins (RBPs) (Conlon and Manley 2017), and inadequate protein degradation mechanisms (Blokhuis et al. 2013), through the autophagy pathway (Ramesh and Pandey 2017), have come to the forefront (Ito et al. 2017). Moreover, it has been speculated that the two processes synergize, potentially reinforcing one another in a vicious cycle (Ito et al. 2017). For instance, defects in autophagy and related protein clearance pathways may lead to an overabundance of phase-separated condensates of RBP-rich ribonucleoprotein granules, which may mature into fibril-like RNP aggregates. Another idea is that mislocalization and/or aggregation of RBPs due to genetic mutation may put strain on the protein clearance pathways of the cell. In either scenario, altered or diminished RBP function would lead to mRNA expression and processing changes, for instance through aberrant alternative pre-mRNA splicing. Evidence has been put forth suggesting that detectable splicing changes may preferentially affect transcripts encoding RBPs, reinforcing their intrinsic ability to aggregate and thereby perpetuating this endless cycle (Conlon et al. 2018; Deshaies et al. 2018). Consistent with this theme, splicing defects occurring within transcripts that encode components of the protein degradation machinery may compound the RBP aggregation that is upstream of splicing changes. While this is an interesting idea, no evidence to suggest that such genes are particularly susceptible to altered gene expression programs has been presented.
We initiated this study with the aim of extending previous results that documented genome-wide changes in pre-mRNA splicing in ALS/FTD patient brains. Specifically, we detected a direct correlation between the extent of insolubility of the splicing regulatory RBP HNRNPH (also known as hnRNPH), and other RBPs, and the degree of missplicing, in both C9 and sporadic samples (Conlon et al. 2018). Here, we wished to focus on whether this correlation was maintained with an independent, more stringent method of AS analysis in C9 patient brains, to determine whether specific types of AS events such as intron retention were especially prevalent, and to investigate whether misspliced transcripts reflect specific biological pathways.
Results
We previously characterized extensive pre-mRNA missplicing events in C9 ALS/FTD patient brains and found that the extent of dysregulated splicing correlated with the levels of sequestered, insoluble hnRNPH (Conlon et al. 2016, 2018). To investigate these aberrant splicing patterns further, we first compared the global alternative splicing (AS) profiles in postmortem cerebellum samples from normal controls and from C9 ALS/FTD patients (Table 1). Two subcategories of C9 samples were compared to the controls based on the level of hnRNPH aggregation, as defined by Conlon et al. (2018), in order to stratify sporadic ALS/FTD: those with high insoluble hnRNPH (above 80%; C9high) and those with relatively low levels of insoluble hnRNPH (below 60%; C9low) (Table 1). We used the software tool Junction Usage Model (JUM) (Wang and Rio 2018) for differential AS analysis. JUM provides two important advantages compared to other software tools in that it does not rely on any prior knowledge of transcriptome or splicing event annotations and is thus suitable for tissues like the brain where previously unknown splicing events are prevalent, and it also performs stringent intron retention analysis with very low false positive rates (Wang and Rio 2018).
Table 1.
Patient sample information
| Sample | RNA-seq ID | Sex | Clinical diagnosis | Age at death | Disease duration | % Insoluble hnRNPH | Group |
|---|---|---|---|---|---|---|---|
| C91 | CGND HRA 00714 | F | ALS | 64 | 1–1.5 yr | 87.2 | C9high |
| C92 | CGND HRA 00728 | F | ALS | 64 | 0.5 yr | 56 | C9low |
| C93 | CGND HRA 00715 | F | ALS | 58 | 1–1.5 yr | 94.7 | C9high |
| C94 | CGND HRA 00730 | M | ALS-FTD | 60 | 0.5–1 yr | 41.3 | C9low |
| C95 | CGND HRA 00729 | F | ALS | 65 | 7–7.5 yr | 89.2 | C9high |
| C96 | CGND HRA 00716 | M | ALS | 73 | 4.5 yr | 37.2 | C9low |
| C98 | CGND HRA 00726 | M | ALS | 59 | 1 yr | 81.8 | C9high |
| C99 | CGND HRA 00718 | M | ALS | 70 | <2 yr | 15.8 | C9low |
| C910 | CGND HRA 00453 | F | FTD | 75 | 9 yr | 95.3 | C9high |
| C911 | CGND HRA 00719 | M | ALS-FTD | 73 | 2 yr | 89.5 | C9high |
| C912 | CGND HRA 00720 | F | ALS | 57 | 1 yr | 33.2 | C9low |
| C913 | CGND HRA 00713 | F | ALS-FTD | 68 | 6 yr | 27 | C9low |
| C916 | CGND HRA 00707 | F | ALS-FTD | 68 | 6 yr | 40.3 | C9low |
| non6 | CGND HRA 00206 | F | Non-neurological control | 54 | n/a | n/a | Control |
| non9 | CGND HRA 00721 | F | Non-neurological control | 90+ | n/a | n/a | Control |
| non10 | CGND HRA 00077 | F | Non-neurological control | 90+ | n/a | n/a | Control |
| non11 | CGND HRA 00197 | F | Non-neurological control | 70 | n/a | n/a | Control |
| non12 | CGND HRA 00196 | F | Non-neurological control | 52 | n/a | n/a | Control |
| non13 | CGND HRA 00753 | M | Non-neurological control | 89 | n/a | n/a | Control |
We first analyzed splicing patterns in RNA samples isolated from C9high brains and from controls. A total of 4681 significantly differentially spliced AS events (q-value ≤ 0.1; ΔΨ ≥ 10%) were identified by JUM (Fig. 1A, upper panel). Seventy-five percent of these events were intron retention (IR) events, indicating a more general deficiency in splicing in these patients, beyond only changes in AS (Fig. 1A, upper panel). In contrast, we found that there were no significant differences in splicing between the C9low brain samples and controls (Fig. 1A, lower panel), consistent with our previous results (Conlon et al. 2016, 2018) and indicating that widespread intron splicing deficiency is highly correlated with the extent of hnRNPH sequestration. We note that this significant elevation in IR in C9high samples was not observed in our previous study (Conlon et al. 2018), which used the AS analysis tool LeafCutter (Li et al. 2018) with the same samples. Although LeafCutter is also annotation free, it does not detect IR (Li et al. 2018).
Figure 1.

Widespread, elevated intron retention is observed in C9high patient brain samples. (A) Number of significantly differentially spliced AS events in six AS categories comparing C9high patient samples with normal controls and C9low patient samples with normal controls, respectively. (B) Volcano plot showing the magnitude and direction of changes in intron retention for a total of 3177 changed intron retention events between C9high patient samples and normal controls. X-axis shows the difference of intron-retained isoform levels between normal controls and C9high patient samples. (C) Volcano plot showing the magnitude and direction of changes in intron retention between C9low patient samples and normal controls. (D,E) Sashimi plot and genome browser shots for two significantly elevated intron retention events in two gene transcripts, HNRNPH1 and PSMA3. Exon coverage from RNA-seq data is shown in three normal control samples (blue) and three C9high samples (red); arcs represent splice junctions identified from the RNA-seq data and the number of uniquely mapped RNA-seq reads mapped to the junctions are shown across the arc; human annotation (hg38) of the transcripts is shown at the bottom. The black arrow indicates the direction of the promoter. The dotted lines indicate the region of the transcript that is enlarged to highlight the retained intron region.
We next characterized the altered IR events in more detail. Among the 3177 significantly changed IR events, we found that 2986 introns (94%) were more retained in C9high patients (Fig. 1B,D,E). Among them, 621 introns displayed elevated IR levels of more than 25% in C9high samples compared to normal controls. Both the direction and magnitude of change were, in general, consistent across all C9high patient samples (Fig. 2A). We next investigated how these elevated retained introns affected accumulation of the transcripts containing them. Previous reports suggested that IR can lead to decreased expression of the affected gene through nonsense-mediated decay (NMD), due to a premature stop codon introduced within the retained intron (Ge and Porse 2014; Jacob and Smith 2017). Among the 1944 genes with significantly elevated retained introns in C9high brains, only 11 (0.6%) displayed significant decreases (q-value ≤ 0.1; fold change ≥ 1) in transcript levels, and the magnitude of down-regulation was relatively small, mostly around one- to twofold (Fig. 2B). This strongly suggests that the prevalent elevated IR in C9high brains does not affect the overall expression levels of the relevant genes.
Figure 2.

Prevalent, elevated intron retention in C9high patient samples does not change overall gene expression levels of the affected transcripts. (A) Heat map of the changed intron retention levels for 3177 introns (column), comparison across six normal control samples and six C9high patient samples. Magnitude and direction of changes in intron retention between normal controls and C9 samples are color-coded. (B) Volcano plot showing the overall expression level changes in genes that undergo significantly elevated intron retention. X-axis shows fold changes in gene expression level between normal controls and C9high patient samples. (C) Box plot showing intron length distribution between introns that are more retained in C9high patient samples, introns that are more retained in normal controls, and all introns detected in the brain samples. (D) Box plot showing intron CG content comparison between introns that are more retained in C9high patient samples, introns that are more retained in normal controls, and all introns detected in the brain samples.
We then investigated whether the significantly elevated retained introns in C9high brains possess any unique features compared to other introns in general. We found that the retained introns tend to have a smaller size (median 602 nt vs. 1793 nt for all introns detected in the brain samples, and 819 nt for the introns that are more retained in the normal controls) (Fig. 2C). In addition, these introns have significantly higher G/C content (Fig. 2D), and relatively weaker 5′ and 3′ splice sites (Supplemental Fig. S1B), suggesting that they have higher dependence on accessory splicing factors, such as hnRNPH.
We next compared overall levels of gene expression, independent of retained introns, between C9 patient samples and normal controls. We found only a relatively small number of genes (731) that displayed significant expression level changes between C9high and controls (mostly approximately twofold change) (Supplemental Fig. S2A,C). Similar to the differential AS analysis, there were almost no differences in gene expression profiles between C9low samples and normal controls (Supplemental Fig. S2B). This latter result indicates that the presence of an expanded C9 allele is not sufficient to cause changes in gene expression in the absence of measurable RBP insolubility. Also, the small number and magnitude of changes in the C9high samples are consistent with the idea that hnRNPH and related RBP insolubility (Conlon et al. 2018) affect primarily splicing and not transcription and/or mRNA stability. Extending the comparison, only 24 out of the 631genes showing reduced expression overlapped with the set of 2927 genes that displayed significant differential AS (Supplemental Fig. S2D).
We next analyzed whether genes whose transcripts displayed elevated IR in C9high brains were enriched in specific functional pathways, using Gene Ontology analysis (P-value < 10−3). We noticed that the retained introns are found in transcripts functionally enriched in many ALS-implicated pathways (van Blitterswijk and Landers 2010; Arnold et al. 2013; Blasco et al. 2014; Kim and Taylor 2017; Webster et al. 2017), including the protein aggregation/misfolded protein clearance pathways (Blokhuis et al. 2013; Cirulli et al. 2015; Webster et al. 2017), protein transport control pathways (Kim and Taylor 2017), and pre-mRNA splicing regulatory pathways (Fig. 3A; Conlon et al. 2016). The fact that transcripts encoding proteins involved in pre-mRNA splicing also displayed intron retention raises the possibility that some splicing defects could be secondary to the events that are due to direct sequestration of RBPs such as hnRNPH, as discussed previously (Conlon et al. 2018).
Figure 3.

Transcripts that undergo significantly changed intron retention in C9high patient samples encode proteins functionally enriched in cellular protein quality control pathways. (A) Gene Ontology enrichment analysis of transcripts that undergo significantly changed intron retention in C9high patient samples compared to normal controls. (B) Volcano plots showing the magnitude and direction of changes in intron retention for introns embedded in transcripts that encode proteins functioning in the proteasomal pathway, the autophagy pathway, and the splicing regulatory pathway, respectively.
Both of the two major protein quality control pathways required for cellular homeostasis were affected: the ubiquitin-proteasome pathway (Deng et al. 2011) and the autophagy pathway (Fig. 3A,B, left and middle panels; Dikic 2017). Both of these pathways have been implicated in ALS pathology, because compromised protein quality control can contribute to accumulation of protein aggregates, a hallmark of ALS-diseased neurons (Blokhuis et al. 2013). For the proteasome pathway, elevated IR has the potential to affect almost all categories of the protein subunits that assemble and maintain the function of the proteasome machinery (Fig. 3B; Supplemental Figs. S3–S12; Table 2; Supplemental Table S1). Most of the retained introns are predicted to change either partially or completely the C termini of the encoded proteins (PSMA3, PSMC3, PSME1, for example) (Table 2; Supplemental Table S1; Supplemental Figs. S3–S12). In one extreme case (PSMA4), IR has the potential to produce an entirely distinct protein, because the retained intron, which contains a significant open reading frame (ORF), resides before the authentic ORF (Table 2; Supplemental Table S1). As a result, these C9high-associated IR events can have significant effects on the integrity and function of the proteasome machinery. For example, the C terminus of PSMA3, which would be significantly affected by IR, is known to be an interaction hub for certain intrinsically disordered proteins, functioning to facilitate their degradation (Sánchez-Lanzas and Castaño 2014). IR thus potentially compromises degradation of such proteins.
Table 2.
Summary of elevated intron retention events in C9high patient samples compared to normal controls that affect transcripts encoding subunits of the proteasome machinery
| Gene | Basic molecular function | # of IR events | Effect from IR |
|---|---|---|---|
| PSMA3 | Proteasome (prosoma, macropain) subunit, alpha type, 3 | 3 | IR 1: C terminus truncation IR 2: C terminus truncation and change IR 3: C terminus change |
| PSMA4 | Proteasome (prosoma, macropain) subunit, alpha type, 4 | 2 | IR 1: Complete coding protein change IR 2: Complete coding protein change |
| PSMA7 | Proteasome (prosoma, macropain) subunit, alpha type, 7 | 1 | IR: C terminus truncation |
| PSMB4 | Proteasome (prosoma, macropain) subunit, beta type, 4 | 2 | IR 1: C terminus truncation and change IR 2: C terminus truncation |
| PSMB10 | Proteasome (prosoma, macropain) subunit, beta type, 10 | 1 | IR: C terminus truncation and change |
| PSMC3 | Proteasome (prosoma, macropain) 26s subunit, atpase type, 3 | 1 | IR: C terminus change |
| PSMC4 | Proteasome (prosoma, macropain) 26s subunit, atpase type, 4 | 1 | IR: C terminus truncation and change |
| PSMC5 | Proteasome (prosoma, macropain) 26s subunit, alpha type, 5 | 2 | IR 1: C terminus truncation and change IR 2: C terminus truncation |
| PSMC6 | Proteasome (prosoma, macropain) 26s subunit, alpha type, 6 | 1 | IR: C terminus truncation and change |
| PSMD3 | Proteasome (prosoma, macropain) 26s subunit, non-atpase, 3 | 1 | IR: C terminus truncation and change |
| PSMD6 | Proteasome (prosoma, macropain) 26s subunit, non-atpase, 6 | 1 | IR: C terminus truncation and change |
| PSMD7 | Proteasome (prosoma, macropain) 26s subunit, non-atpase, 7 | 1 | IR: C terminus truncation and change |
| PSMD11 | Proteasome (prosoma, macropain) 26s subunit, non-atpase, 11 | 2 | IR 1: C terminus truncation and change IR 2: C terminus truncation and change |
| PSMD13 | Proteasome (prosoma, macropain) 26s subunit, non-atpase, 13 | 1 | IR: C terminus truncation |
| PSME1 | Proteasome (prosoma, macropain) activator subunit 1 (pa28 alpha) | 1 | IR: C terminus truncation and change |
| PSME2 | Proteasome (prosoma, macropain) activator subunit 2 (pa28 beta) | 3 | IR 1: C terminus truncation and change IR 2: C terminus truncation and change IR 3: C terminus truncation and change |
| PSME4 | Proteasome (prosoma, macropain) activator subunit 4 | 1 | IR: C terminus truncation and change |
We next wished to gain more insight into the basis for the greatly increased IR in C9high patients. Specifically, is this elevated IR mechanistically linked to the sequestration level of hnRNPH? HnRNPH is known to bind to NGGG(+)N (three or more consecutive guanines) motifs downstream of a 5′ splice site in the intronic region (Caputi and Zahler 2001; Wang and Cambi 2009; Xiao et al. 2009; Uren et al. 2016). These poly(G) motifs typically act as intronic splicing enhancers (ISEs), meaning that when hnRNPH binds to these motifs, it enhances splicing of the intron (Caputi and Zahler 2001; Wang and Cambi 2009; Xiao et al. 2009; Uren et al. 2016). We thus first calculated and compared the enrichment of NGGGN elements in three intron groups: introns that were significantly more retained in C9high patients compared to normal controls (Fig. 4A, red), the much smaller number of introns that were instead significantly more retained in the controls (Fig. 4A, blue), and all introns detected in the patient brain samples (Fig. 4A, gray). We found that introns that were more retained in C9ALS patients were significantly more likely to contain NGGGN elements within 100 nt downstream from the 5′ splice sites compared to total introns (Mann–Whitney U test P-value < 10−16) (Fig. 4A). In contrast, introns that were more retained in the normal controls are deficient in such poly(G) motifs (Mann–Whitney U test P-value < 10−8) (Fig. 4A). Moreover, enrichment for the NGGGN motif was especially prominent within 10 to 40 bp downstream from the 5′ splice site in the C9ALS retained introns (Fig. 4A), matching the previously reported general locations of hnRNPH-associated ISEs in introns (Xiao et al. 2009). These results strongly suggest that the splicing of the C9ALS-retained introns tends to have higher dependency on hnRNPH and consequently will be more vulnerable in C9high ALS samples where the levels of soluble, functional hnRNPH is significantly reduced (Conlon et al. 2016).
Figure 4.

Introns that are significantly more retained in C9high patient samples are enriched in hnRNPH-associated intronic splicing enhancer motif activities. (A) Cumulative distribution function plot showing the fraction of introns with at least one NGGG + N motif at the designated intronic position (bp) downstream from the 5′ splice site (x-axis showing a total of 0–100 bp downstream from the 5′ splice site) for six groups of introns. (B) Cumulative distribution function plot showing the fraction of introns with at least one NGGG + N motif of four and more Gs in the poly(G) tract at the designated intronic position (bp) downstream from the 5′ splice site for six groups of introns. (C) Cumulative distribution function plot showing the fraction of introns with at least four non-overlapping NGGG + N motifs at the designated intronic position (bp) downstream from the 5′ splice site for six groups of introns.
We next examined subcategories of C9ALS-elevated retained introns, in the proteasome, autophagy, and splicing regulatory pathways. We again observed enrichment of the NGGGN motif relative to general introns, with the highest enrichment in the proteasomal pathway (Fig. 4A). This suggests that splicing of transcripts encoding components of the protein quality control pathway tends to be more susceptible to the reduced hnRNPH levels in C9high ALS brains.
The strength of hnRNPH binding to the NGGG(+)N motifs and the corresponding enhancement of the intron involved is further determined by the length of the G-tract in the poly(G) run motif (Caputi and Zahler 2001; Wang and Cambi 2009; Xiao et al. 2009; Uren et al. 2016). We therefore next examined possible enrichment of longer (four consecutive guanines or more) G tracts in different intron groups (Fig. 4B). In accordance with the results above, we indeed observed significant enrichment of longer G tracts in introns that were more retained in C9ALS patient brain samples (Mann–Whitney U test P-value < 10−10) (Fig. 4B). We also observed that a significantly smaller fraction of introns that were more retained in control brains possess long G tracts in poly(G) motifs (Mann–Whitney U test P-value < 10−18) (Fig. 4B).
It is also possible that hnRNPH protein can bind to several poly(G) motifs in a cooperative manner. As a result, having multiple poly(G) tracts near the 5′ splice site in the intron can lead to the coordination of multiple hnRNPH proteins and a resulting stronger enhancement of splicing of the intron. Moreover, when four or more poly(G) runs are in close proximity, they can fold into RNA G-quadruplexes (G-Qs), and these highly stable structures may preclude binding of general splicing factors, such as U1 snRNP (Tan et al. 2019). Proteins that have high affinity for individual poly(G) motifs, like hnRNPH, may play important roles in blocking structure formation (Guo and Bartel 2016), and their sequestration may lead to especially severe splicing defects in introns that have G-Q potential. We thus also calculated and compared enrichment of multiple (four or more) non-overlapping poly(G) run elements in the vicinity of 5′ splice sites in the different intron groups (Fig. 4C). We again observed enrichment of multiple, non-overlapping poly(G) motifs in introns that were significantly more retained in C9high samples, especially in the protein quality control pathway-associated genes (Mann–Whitney U test P-value < 10−18) (Fig. 4C). While these criteria are indicators of potential structure formation, sequence-based prediction of G-Q existence is quite complicated due to the presence of competing RNA structures (Wang et al. 2019). Thus, while we conclude that the retained introns are more likely to contain multiple poly(G) motifs in the immediate downstream intron, further experimental analysis would be needed to show that G-Q formation occurs at these sites.
Finally, to provide in vivo support for the hnRNPH binding motif analyses described above, we examined whether empirically determined hnRNPH binding events are enriched in affected retained introns in the vicinity of their 5′ splice sites. We profiled hnRNPH binding sites from a previously published hnRNPH CLIP-seq data set that utilized HEK 293T cells (Katz et al. 2010) and compared the enrichment of hnRNPH peaks (B-H corrected P-value < 0.05) in the vicinity of 5′ splice sites in different intron groups (Fig. 5). Overall, a relatively small fraction of the introns detected in patient cerebellum had HEK 293T-derived hnRNPH CLIP peaks. This could be a function of several factors—for instance, the low-sequencing depth and lack of concordance in overall gene expression profiles between human adult cerebellum and HEK 293T cells. Nonetheless, we indeed observed significant enrichment of hnRNPH binding sites within 100 nt downstream from the 5′ splice sites in introns that were more retained in C9ALS patients (Mann–Whitney U test P-value < 10−17) (Fig. 5), while introns that were more retained in the normal controls were deficient in hnRNPH binding sites (Mann–Whitney U test P-value < 10−19) (Fig. 5). Together, these and the above results further suggest that the C9ALS retained introns tend to be more dependent on hnRNPH-mediated regulation and thus more sensitive to the reduction in functional hnRNPH levels in the C9high ALS patient brains.
Figure 5.
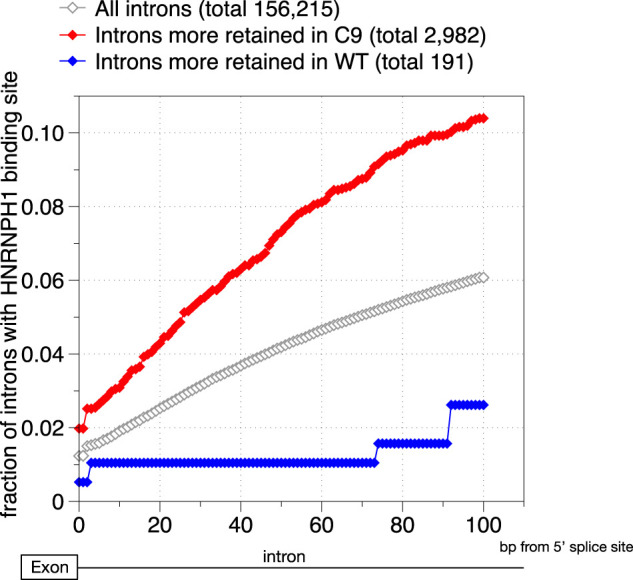
Cumulative distribution function plot showing the fraction of introns with at least one hnRNPH binding site at the designated intronic position (bp) downstream from the 5′ splice site (x-axis showing a total of 0–100 bp downstream from the 5′ splice site) for three groups of introns.
Discussion
Recent advances in the molecular genetics of ALS and FTD have pointed to a convergence of protein clearance pathway dysfunction, RBP aggregation, and RNA processing defects (Ito et al. 2017). However, despite this growing consensus (Ito et al. 2017; Cook and Petrucelli 2019) and the increased availability of transcriptomic data (Prudencio et al. 2015; Conlon et al. 2018), an outstanding challenge that remains is how to identify the most central and meaningful gene expression changes that potentially drive the disease phenotype. A drawback of any transcriptomic analysis involving human patients is that the standard assumption is to compare patients to unaffected controls. In doing this, any meaningful variability between patients can be grossly underestimated and skew the outcome. Furthermore, computational analyses done with different algorithmic methods can lead to vastly different results, as evidenced by the differences we have observed using JUM and LeafCutter (Conlon et al. 2018). In this manuscript, we have carefully considered both of these factors. Through our use of functional stratification of C9ALS/FTD patients by hnRNPH insolubility, and through the use of the intron-aware method JUM (Wang and Rio 2018), we have identified an important prevalence of IR events in proteasomal genes that may have otherwise been overlooked in different analyses (Prudencio et al. 2015; Conlon et al. 2018). Furthermore, our data illustrate how defects in the proteasome can potentially be both cause and consequence of dysregulated splicing programs in C9 ALS/FTD, synergizing with what is known about proteostasis effects in C9 and other forms of ALS and FTD.
Intron retention is a particular form of alternative splicing that has been previously associated with human disease. It has been observed, for example, in some cancers (Dvinge and Bradley 2015) as well as in ALS (Luisier et al. 2018; Humphrey et al. 2020). Perhaps more than other forms of alternative splicing, intron retention has the ability to explain the subtle accumulation of defective transcripts, leading to dysregulated protein expression even in cases where mutations in the relevant genes are strongly selected against. For instance, while mutations affecting proteasomal subunits directly may have the potential to recapitulate some aspects of ALS/FTD, they may be too severe to allow viability or may lead to other deleterious consequences before the onset of ALS/FTD at later stages in life. Indeed, few genes encoding proteasome subunits have been shown to be mutated in disease (Gomes 2013). In contrast, through our comprehensive analysis of IR in stratified C9 patient samples, we have found evidence for pervasive defects in transcripts encoding proteasomal subunits.
Evidence has existed for some time that defects in the ubiquitin-proteasome system (UPS) play a significant role in ALS/FTD pathology (Bendotti et al. 2012). This is not only reflected in mutations in the UBQLN2 gene (Deng et al. 2011) that are found in a fraction of ALS/FTD patients, but evidence for UPS defects has been observed widely in ALS/FTD, including in sporadic cases (Kabashi et al. 2012). However, apart from UBQLN2 mutations, the basis for UPS dysfunction has been largely unknown. Our results that defects in splicing, specifically IR, of transcripts encoding multiple UPS subunits offers an explanation, at least in C9 patients displaying high levels of hnRNPH insolubility. The concept that an individual RBP can regulate a set of functionally coherent transcripts has been demonstrated previously, as in the case of the neuronal RBP NOVA (Ule et al. 2003).
It has recently been shown that poly Gly-Ala dipeptide repeat proteins, which can be produced by repeat-associated non-AUG (RAN) translation of the C9 GGCCCC repeats, can form aggregates that interact with 26S proteasomes and disrupt their activity (Guo et al. 2018). Thus, it may be, in C9 ALS/FTD, affected neurons suffer from a two-pronged attack on the UPS system, reflecting both missplicing and RAN translation. In addition, while our study dealt with C9 samples, our recent work showed that a significant fraction of sporadic ALS/FTD brains display high levels of hnRNPH aggregation and extensive missplicing (Conlon et al. 2018). Thus, it is possible that missplicing, including IR, contributes to the defect in the UPS observed in sporadic as well as C9 ALS/FTD.
Although we have focused heavily on hnRNPH, it should be mentioned that we previously observed levels of insoluble hnRNPH to be highly correlated to the insolubilities of several other ALS/FTD-relevant RBPs, such as TARDBP (also known as TDP-43) and FUS (Conlon et al. 2018). While we have argued that hnRNPH is a driver of such “multi-RBP proteinopathy,” we cannot rule out the possibility that the other proteins that display this highly insoluble behavior contribute to splicing changes and could be used to functionally stratify this patient group in a similar manner. Nonetheless, as we have carefully measured hnRNPH insolubility values across a wide range of patients, it is the most reliable metric we have to describe this relationship. Similar to our previously reported findings on splicing in C9 and sporadic ALS/FTD (Conlon et al. 2018), quantifiable changes in IR were detected only in patients with high hnRNPH insolubility (C9high). As discussed previously (Conlon et al. 2018), it may be that the samples with lower measurable insolubility also undergo similar changes in splicing, including IR, but that due to the smaller magnitude of these changes or their presence in only certain cell populations, they fail to pass stringent filters set to identify significant changes relative to controls. This argument makes further sense in light of the “vicious cycle” we propose is at play here. Given that RBP aggregation leads to these splicing changes and the splicing changes affect the cell's ability to clear them, we anticipate a strong compounding effect, where the initial amount of RBP aggregation is amplified through IR and resultant proteasomal dysfunction. Indeed, this may complement or exacerbate another vicious cycle we proposed previously, in which the extensive missplicing of transcripts encoding splicing regulatory RBPs that occurs in C9high brains leads to altered levels of these proteins, which in turn causes additional missplicing.
In conclusion, our analyses have shed new insights into the complex, interconnected relationship between RNA processing defects and protein-clearance pathways in ALS/FTD, highlighting the ways in which they may mutually reinforce the pathological events that culminate in this devastating disease spectrum.
Methods
Human patient samples
Patient brain samples used in this study are identified according to previously published conventions (Conlon et al. 2018). All human samples were donated for research purposes by next-of-kin. See detailed patient sample information in Table 1.
RNA-seq data
Previously published RNA-seq data (Conlon et al. 2018) were downloaded from the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) accession GSE116622.
RNA-seq data mapping for JUM
RNA-seq reads were mapped to the human (hg38) genomes using STAR (Dobin et al. 2013). A two-pass mapping mode was applied for read alignment, which has been shown to greatly improve splice junction quantification. The detailed mapping commands and procedures are listed in the JUM manual: https://github.com/qqwang-berkeley/JUM/wiki/0.b.-Input-files (Wang and Rio 2018). Then, only uniquely mapped reads were kept in the output for downstream JUM analysis.
Running JUM on the RNA-seq samples
JUM (version 2.0.2) was run on the RNA-seq samples. Each patient sample in normal control, C9high, and C9low categories is treated as a biological replicate when comparing the overall alternative splicing profile across different conditions. JUM commands are run as detailed in the JUM manual: https://github.com/qqwang-berkeley/JUM/wiki/3.1.-Manual-running-JUM-(v2.0.2-and-up) (Wang and Rio 2018). Statistical cutoff of q value ≤ 0.1, ΔΨ or ΔPSI ≥ 10% is applied to the differential alternative splicing analyses described in this study. A detailed summary of differentially alternatively spliced events between normal controls and C9high samples in six AS categories (cassette exon, A5SS, A3SS, IR, mutually exclusive exons, and composite) are in Supplemental Data S1–S6.
Intron property analysis
GC content in introns was calculated using the nuc option in BEDTools (https://bedtools.readthedocs.io/en/latest/) (Quinlan 2014). The strength of 5′ splice sites and 3′ splice sites of the introns were calculated using MaxEntScan (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) (Yeo et al. 2004). The SeqKit was used to count occurrence of poly(G) motifs in introns and their locations (https://bioinf.shenwei.me/seqkit/) (Shen et al. 2016).
Visualization
All RNA-seq track data and junction reads were visualized using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) and the Sashimi plots tool (Katz et al. 2015). Visualized tracks were further organized using ImageJ (Schneider et al. 2012). All box plots in this paper were plotted using BoxPlotR (Spitzer et al. 2014). Volcano plots in this paper were plotted using R (R Core Team 2019) and ggplot2. Heat maps in this paper were plotted using R (R Core Team 2019) and heatmap.2.
Gene Ontology analysis
Gene Ontology analyses were performed using GOrilla (http://cbl-gorilla.cs.technion.ac.il/) (Eden et al. 2009). A list of transcripts expressed in the normal control brains at greater than 10 reads was used as a background data set.
Differential gene expression analysis between C9ALS and normal control patient brain samples
Differential gene expression analysis between C9ALS patient samples and the normal control samples was performed using DESeq2 (Love et al. 2014). The read to gene count was calculated using the multicopv option in BEDTools (Quinlan 2014). After that, DESeq2 was run on the RNA-seq samples as described in the manual: http://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html.
Detailed summaries of differentially expressed genes between normal controls and C9high samples, as well as between normal controls and C9low samples, are listed in Supplemental Data S7 and S8, respectively.
Analysis of hnRNPH CLIP-seq data
HnRNPH CLIP-seq data sets were downloaded from Katz et al. (2010) (GSE23694). Processing of the data sets, as well as hnRNPH binding peak calling, were done using the CTK software tool (Shah et al. 2017), following the protocol as described in https://zhanglab.c2b2.columbia.edu/index.php/CTK_Documentation.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Dr. Neil Shneider for access to the human patient samples that we have used in this and other related publications (Conlon et al. 2016, 2018; Wang et al. 2019). We thank Jianchao Gao for technical assistance. This work was supported by National Institutes of Health (NIH) grant R35 GM118136 to J.L.M. and NIH grants from the National Institute of General Medical Sciences (R35 GM118121) and from the National Human Genome Research Institute (R21 HG010238) to D.C.R. Q.W. was supported by the Arnold O. Beckman Postdoctoral Fellowship.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.265298.120.
References
- Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. 2013. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci 110: E736–E745. 10.1073/pnas.1222809110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A, De Biasi S. 2012. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol 97: 101–126. 10.1016/j.pneurobio.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Blasco H, Mavel S, Corcia P, Gordon PH. 2014. The glutamate hypothesis in ALS: pathophysiology and drug development. Curr Med Chem 21: 3551–3575. 10.2174/0929867321666140916120118 [DOI] [PubMed] [Google Scholar]
- Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. 2013. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol 125: 777–794. 10.1007/s00401-013-1125-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi M, Zahler AM. 2001. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J Biol Chem 276: 43850–43859. 10.1074/jbc.M102861200 [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347: 1436–1441. 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Manley JL. 2017. RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev 31: 1509–1528. 10.1101/gad.304055.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL. 2016. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNPH to disrupt splicing in ALS brains. eLife 5: e17820 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Fagegaltier D, Agius P, Davis-Porada J, Gregory J, Hubbard I, Kang K, Kim D, New York Genome Center ALS Consortium, Phatnani H, et al. 2018. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. eLife 7: e37754 10.7554/eLife.37754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Petrucelli L. 2019. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101: 1057–1069. 10.1016/j.neuron.2019.02.032 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, et al. 2011. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477: 211–215. 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies JE, Shkreta L, Moszczynski AJ, Sidibé H, Semmler S, Fouillen A, Bennett ER, Bekenstein U, Destroismaisons L, Toutant J, et al. 2018. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 141: 1320–1333. 10.1093/brain/awy062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I. 2017. Proteasomal and autophagic degradation systems. Annu Rev Biochem 86: 193–224. 10.1146/annurev-biochem-061516-044908 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Bradley RK. 2015. Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7: 45 10.1186/s13073-015-0168-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlanis E, Traunmüller L, Fucile G, Scheiffele P. 2019. Landscape of ribosome-engaged transcript isoforms reveals extensive neuronal-cell-class-specific alternative splicing programs. Nat Neurosci 22: 1709–1717. 10.1038/s41593-019-0465-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Porse BT. 2014. The functional consequences of intron retention: alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 36: 236–243. 10.1002/bies.201300156 [DOI] [PubMed] [Google Scholar]
- Gomes AV. 2013. Genetics of proteasome diseases. Scientifica (Cairo) 2013: 637629 10.1155/2013/637629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Bartel DP. 2016. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353: aaf5371 10.1126/science.aaf5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Lehmer C, Martínez-Sánchez A, Rudack T, Beck F, Hartmann H, Pérez-Berlanga M, Frottin F, Hipp MS, Hartl FU, et al. 2018. In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell 172: 696–705.e12. 10.1016/j.cell.2017.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J, Birsa N, Milioto C, McLaughlin M, Ule AM, Robaldo D, Eberle AB, Kräuchi R, Bentham M, Brown AL, et al. 2020. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res 48: 6889–6905. 10.1093/nar/gkaa410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Hatano M, Suzuki N. 2017. RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci Transl Med 9: eaah5436 10.1126/scitranslmed.aah5436 [DOI] [PubMed] [Google Scholar]
- Jacob AG, Smith CWJ. 2017. Intron retention as a component of regulated gene expression programs. Hum Genet 136: 1043–1057. 10.1007/s00439-017-1791-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Agar JN, Strong MJ, Durham HD. 2012. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13: 367–371. 10.3109/17482968.2012.686511 [DOI] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015. 10.1038/nmeth.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Silterra J, Schwartz S, Wong B, Thorvaldsdóttir H, Robinson JT, Mesirov JP, Airoldi EM, Burge CB. 2015. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 31: 2400–2402. 10.1093/bioinformatics/btv034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Taylor JP. 2017. Lost in transportation: Nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96: 285–297. 10.1016/j.neuron.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, Pritchard JK. 2018. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50: 151–158. 10.1038/s41588-017-0004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisier R, Tyzack GE, Hall CE, Mitchell JS, Devine H, Taha DM, Malik B, Meyer I, Greensmith L, Newcombe J, et al. 2018. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat Commun 9: 2010 10.1038/s41467-018-04373-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbacher JK, Tabet R, Yeo GW, Lagier-Tourenne C. 2019. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 102: 294–320. 10.1016/j.neuron.2019.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza-Johnston AE, Desaro P, et al. 2015. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci 18: 1175–1182. 10.1038/nn.4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR. 2014. BEDTools: the Swiss-Army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11.12.1–11.12.34. 10.1002/0471250953.bi1112s47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N, Pandey UB. 2017. Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front Mol Neurosci 10: 263 10.3389/fnmol.2017.00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Lanzas R, Castaño JG. 2014. Proteins directly interacting with mammalian 20S proteasomal subunits and ubiquitin-independent proteasomal degradation. Biomolecules 4: 1140–1154. 10.3390/biom4041140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, Qian Y, Weyn-Vanhentenryck SM, Zhang C. 2017. CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33: 566–567. 10.1093/bioinformatics/btw653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Le S, Li Y, Hu F. 2016. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11: e0163962 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M, Wildenhain J, Rappsilber J, Tyers M. 2014. BoxPlotR: a web tool for generation of box plots. Nat Methods 11: 121–122. 10.1038/nmeth.2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Yang L, Ong AAL, Shi J, Zhong Z, Lye ML, Liu S, Lisowiec-Wachnicka J, Kierzek R, Roca X, et al. 2019. A disease-causing intronic point mutation C19G alters tau exon 10 splicing via RNA secondary structure rearrangement. Biochemistry 58: 1565–1578. 10.1021/acs.biochem.9b00001 [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr., Cleveland DW. 2016. Decoding ALS: from genes to mechanism. Nature 539: 197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215. 10.1126/science.1090095 [DOI] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, de Araujo PR, Vogel C, Qiao M, Burns SC, Smith AD, Penalva LO. 2016. High-throughput analyses of hnRNPH1 dissects its multi-functional aspect. RNA Biol 13: 400–411. 10.1080/15476286.2015.1138030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Landers JE. 2010. RNA processing pathways in amyotrophic lateral sclerosis. Neurogenetics 11: 275–290. 10.1007/s10048-010-0239-4 [DOI] [PubMed] [Google Scholar]
- Wang E, Cambi F. 2009. Heterogeneous nuclear ribonucleoproteins H and F regulate the proteolipid protein/DM20 ratio by recruiting U1 small nuclear ribonucleoprotein through a complex array of G runs. J Biol Chem 284: 11194–11204. 10.1074/jbc.M809373200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rio DC. 2018. JUM is a computational method for comprehensive annotation-free analysis of alternative pre-mRNA splicing patterns. Proc Natl Acad Sci 115: E8181–E8190. 10.1073/pnas.1806018115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Goodrich KJ, Conlon EG, Gao J, Erbse AH, Manley JL, Cech TR. 2019. C9orf72 and triplet repeat disorder RNAs: G-quadruplex formation, binding to PRC2 and implications for disease mechanisms. RNA 25: 935–947. 10.1261/rna.071191.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Shaw PJ, De Vos KJ. 2017. Protein homeostasis in amyotrophic lateral sclerosis: therapeutic opportunities? Front Mol Neurosci 10: 123 10.3389/fnmol.2017.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Wang Z, Jang M, Nutiu R, Wang ET, Burge CB. 2009. Splice site strength–dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol 16: 1094–1100. 10.1038/nsmb.1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, Burge CB. 2004. Variation in alternative splicing across human tissues. Genome Biol 5: R74 10.1186/gb-2004-5-10-r74 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.