

Abstract
Effective public response to a pandemic relies upon accurate measurement of the extent and dynamics of an outbreak. Viral genome sequencing has emerged as a powerful approach to link seemingly unrelated cases, and large-scale sequencing surveillance can inform on critical epidemiological parameters. Here, we report the analysis of 864 SARS-CoV-2 sequences from cases in the New York City metropolitan area during the COVID-19 outbreak in spring 2020. The majority of cases had no recent travel history or known exposure, and genetically linked cases were spread throughout the region. Comparison to global viral sequences showed that early transmission was most linked to cases from Europe. Our data are consistent with numerous seeds from multiple sources and a prolonged period of unrecognized community spreading. This work highlights the complementary role of genomic surveillance in addition to traditional epidemiological indicators.
In December 2019, the novel pneumonia COVID-19 emerged in the city of Wuhan, in Hubei Province, China. Shotgun metagenomics rapidly identified the new pathogen as SARS-CoV-2, a betacoronavirus related to the etiological agent of the 2002 SARS outbreak (SARS-CoV), and of possible bat origin (Andersen et al. 2020; Zhou et al. 2020). Building on infrastructure from past outbreaks (Carroll et al. 2015; Park et al. 2015), genomic epidemiology has been applied to track the worldwide spread of SARS-CoV-2 using mutations in viral genomes to link otherwise unrelated infections (Grubaugh et al. 2019; Zhang and Holmes 2020). Collaborative development of targeted sequencing protocols (Quick et al. 2017; Tyson et al. 2020), open sharing of sequences through the Global Initiative on Sharing All Influenza Data (GISAID) repository (Shu and McCauley 2017), and rapid analysis and visualization of viral phylogenies using Nextstrain (Hadfield et al. 2018) have provided unprecedented and timely insights into the spread of the pandemic. Notably, community transmission was identified using surveillance sequencing in the Seattle area in time to implement preventative measures (Bedford et al. 2020; Worobey et al. 2020).
The New York City metropolitan region rapidly became an epicenter of the pandemic following the identification of the first community-acquired case on March 3, 2020 (a resident of New Rochelle in nearby Westchester County who worked in Manhattan). As of May 10, 2020, New York State had 337,055 cases: the highest in the United States and 8% of the worldwide total. Fully 55% of New York State cases lay within the five boroughs of New York City (185,357 cases), followed by the Nassau and Suffolk counties to the east on Long Island (75,248 cases) (NYS Department of Health 2020). The outlying boroughs and suburban counties reported markedly higher infection rates than Manhattan. The outbreak overlaps with the catchment area of the NYU Langone Health (NYULH) hospital system, including hospitals on the east side of Manhattan (Tisch/Kimmel), Brooklyn (formerly Lutheran Hospital), and Nassau County (Winthrop). Because even early COVID-19 cases presented mostly without travel history to countries with existing outbreaks, determining the extent of asymptomatic community spread and transmission paths became paramount. In parallel with increased clinical capacity for diagnostic PCR-based testing, we sought to trace the origin of NYULH-treated COVID-19 cases using phylogenetic analysis to compare them to previously deposited COVID-19 viral sequences. We further aimed to develop an approach to integrate sequencing as a complementary epidemiological indicator of outbreak trajectory.
Results
To assess the spread of SARS-CoV-2 within the NYULH COVID-19 inpatient and outpatient population, we deployed and optimized a viral sequencing, quality control (QC), and analysis pipeline by repurposing existing genomics infrastructure. Samples from unique individuals were selected for sequencing from those confirmed positive between March 12 and May 10, 2020. During this period, positive tests within the NYULH system mirrored those of New York City and nearby counties (Supplemental Fig. S1; Petrilli et al. 2020). Illumina RNA-seq libraries were generated using a ribodepletion strategy starting from total RNA from nasopharyngeal swabs. Hybridization capture with custom biotinylated baits targeting the full SARS-CoV-2 sequence was used to enrich RNA-seq libraries before sequencing (Methods; Supplemental Fig. S2). Of 1107 libraries generated and sequenced, fully 78% yielded a sequence passing QC (see Methods). Pass rates were lower for samples with qRT-PCR Ct values greater than 30 (Supplemental Fig. S3A,B). We observed that high-quality sequences could be generated directly from shotgun libraries for qPCR Ct values less than 30, thereby simplifying pooling and logistical constraints by skipping the capture step. Up to 23 samples were multiplexed in a single capture pool (Supplemental Fig. S3C,D). Samples with similar Ct values were grouped to minimize the range of target cDNA representation across a single capture pool (Supplemental Fig. S3E,F). Our pipeline was verified using a positive control synthetic RNA spiked in to total human RNA, as well as PCR negative and no-sample controls (Supplemental Table S1). This resulted in 864 sequences passing QC, comprising 10% of COVID-19-positive cases in NYULH over that time period (Supplemental Fig. S1; Supplemental Table S2).
The cohort of 864 sequenced cases included a range of ages (Fig. 1A). Cases originated throughout the NYULH system, which comprises hospitals in the New York City boroughs of Manhattan and Brooklyn, as well as Nassau County, a suburb to the east of the city on Long Island (Fig. 1B). Sixty-six percent of cases resided within New York City; 86%, within New York State (Fig. 1C). Analysis of residential ZIP codes showed that cases reflected the hospital catchment area within the New York metropolitan region (Fig. 1D). Our data set included few cases from Westchester County to the north of the city, where the earliest detected regional outbreak was concentrated, as it is outside of the NYULH catchment area.
Figure 1.

Demographic parameters of sequenced SARS-CoV-2 cases in the NYULH system. Cases are broken down as follows: (A) Age and sex; (B) collecting hospital; (C) residential location, grouped by borough and outlying counties; “Other” includes counties with few cases. (D) Localization of case residences within the New York City region. The color scale indicates number of cases per ZIP code. Collecting hospitals are labeled with rounded boxes. (E) Potential exposure status, categorized by occupation as healthcare worker, travel history, and contact with a COVID-19-positive individual. The pie chart depicts the geographical destination of the potential travel-related exposures. (F) Potential exposure status by collection date.
We compiled a database for 820 of these cases from electronic medical records, including potential exposures from health care worker status, travel history, and close contact with a COVID-19 individual (Methods). We found no recorded potential exposures for 43% of cases (Fig. 1E). Multiple potential exposures were less common: 113 cases were both health care workers and noted a COVID-19 contact, and three health care workers had travel history. Travel history was present for only 17 cases (2%), and all of these cases but one were collected in March (Fig. 1F). Of the 14 cases in which travel destination information was available, nine destinations were within the United States, four were in Europe, and one was in South Asia. This assessment relies upon clinical notes during a period in which clinical capacity was stretched, thus likely underestimates potential exposures. Conversely, the potential exposure may have been coincidental given the uncontrolled community spread at the time.
We inferred a maximum likelihood phylogeny to assess relatedness among cases (Fig. 2). Coloring cases by county of residence within the New York region showed identical or related viral sequences present across multiple counties from the onset of our sampling (Fig. 2). We detected 890 nucleotide and 547 amino acid mutations across all cases (Supplemental Fig. S4). Mutation of D614G in the spike protein, which has been suggested to affect transmission or virulence (Zhang et al. 2020), was present in >95% of sequences. Functional analysis will be required to determine whether functional changes can be ascribed to any of these mutations and what role mutations might play in shaping the ongoing pandemic.
Figure 2.

Phylogenetic relationship of regional viral sequences. Maximum likelihood phylogeny inferred from 864 cases. Nodes with bootstrap support values above 75 are colored. Inner rings indicate groups of clade-defining mutations. Outer ring indicates county of residence. Scale bar, nucleotide substitutions per site.
We then assessed the relatedness of our cases to 5004 sequences from across the world from the GISAID EpiCov repository (Supplemental Fig. S5; Supplemental Table S3). A maximum likelihood tree showed that cases from the New York region showed broader diversity than that initially reported in Seattle (Bedford et al. 2020), the only other U.S. region with a comparable number of viral sequences (Supplemental Fig. S6). To investigate the timing of introductions to New York City, we inferred a rooted timescaled phylogeny (Fig. 3A; Supplemental Fig. S7A). Analysis of our cases within this phylogeny identified 109 genotypes introduced to the northeast United States (Fig. 3B; Supplemental Table S4). Identification of source nodes ancestral to at least one sequence from outside the northeast United States in addition to these transmission chains placed most introductions broadly in late February and early March, slightly earlier than the first detected transmissions within New York City (Fig. 3C; Supplemental Fig. S7B). The timing of these introductions did not differ substantially under alternative nucleotide substitution models or rates (Supplemental Fig. S7C). The number of samples in each transmission chain varied widely, and two early transmission chains each comprised over 300 cases. Only a minority of transmission chains included samples from Asia, whereas samples from Europe and the rest of the United States were well represented (Fig. 3D).
Figure 3.

Timescaled phylogeny showing global sequence context. (A) Colored edges highlight transmission chains. Black squares indicate source nodes; dots, detected presence in the northeast United States. (B) Schematic of approach to infer introductions and transmission chains. (C,D) Transmission chains in the New York City region ordered by inferred divergence date from source. (C) Dates estimated for source transmission (orange) and earliest detected local transmission (purple) inferred from sequenced cases; lines represent 90% confidence intervals. Point size corresponds to the number of strains under source and all transmission chains. (D) Representation of global regions in each source transmission. Bar at top shows overall representation of regions in the phylogeny.
To assess the ongoing trajectory of the outbreak, we applied phylodynamic analysis to estimate viral effective population size from a subsample of sequences (Methods) (Pybus and Rambaut 2009). Under moderate assumptions, effective population size will be proportional to epidemic prevalence, and growth rates of effective population size will correspond to epidemic growth (Volz et al. 2013). This analysis identified a period of rapid growth, followed by return nearly to the start point (Fig. 4A,B). We estimate that the peak effective population size occurred on March 29 (95% CI: March 19–April 5). The growth rate decreased steadily after March 1 and was negative with high confidence by mid-April (Fig. 4C), consistent with the epidemic curve of confirmed infections in the New York City region (Supplemental Fig. S1A).
Figure 4.
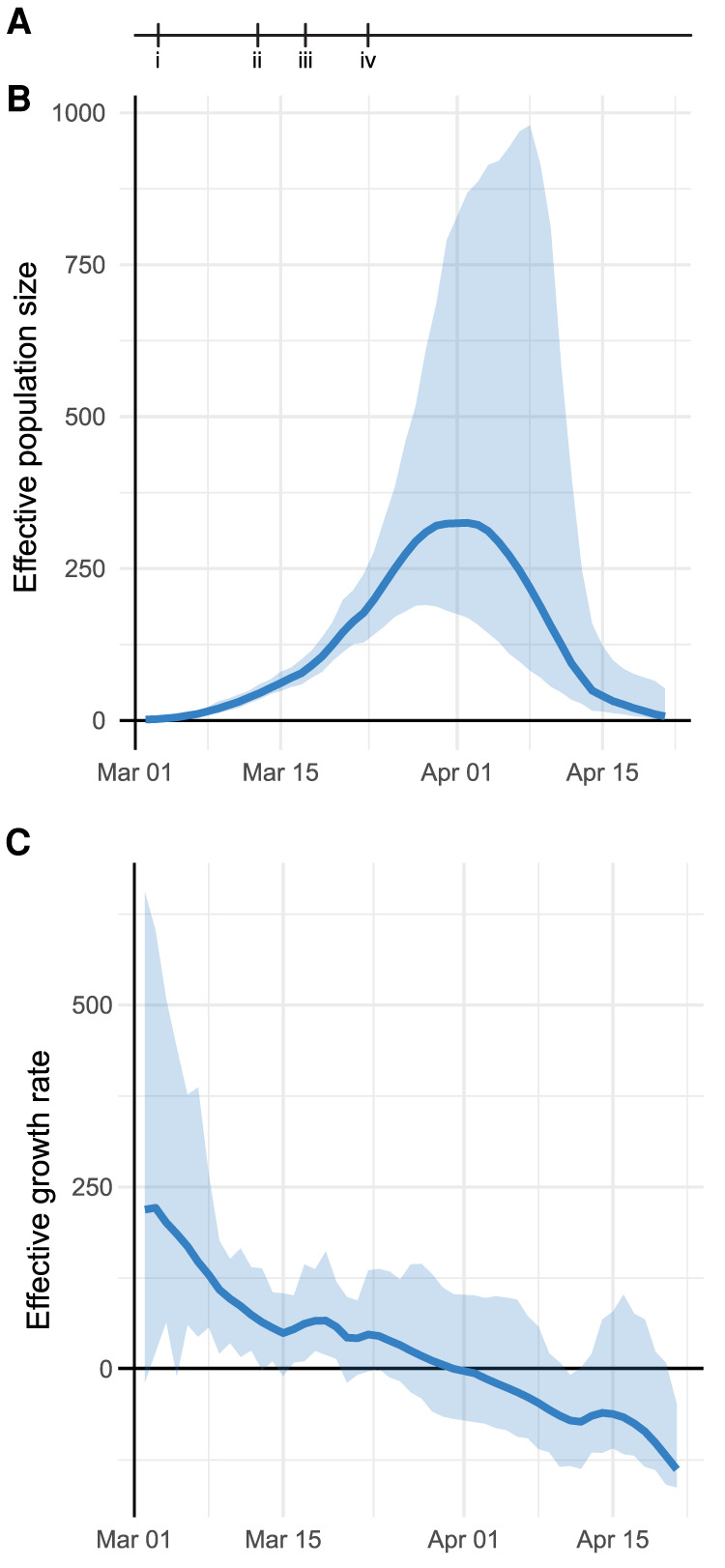
Phylodynamic analysis of outbreak trajectory. (A) Timeline of New York City outbreak, highlighting (i) announcement of first community-acquired case (March 3); (ii) ban on gatherings exceeding 500 people (March 12); (iii) closure of schools, restaurants, and bars, and other venues (March 16); (iv) closure of nonessential businesses (March 22). (B,C) Outbreak trajectory estimated from genetic data showing effective population size relative to March 1 (B) and growth rate of effective population size (C; units of 1/yr). Shaded regions represent 95% credible interval.
Discussion
Our work documents the genomic epidemiology of the COVID-19 outbreak in the New York City region in the spring of 2020. Analysis of the genetic data suggests that the New York outbreak was seeded by mid-February, largely by way of Europe, which can be placed within the context of reduced travel flows from Asia to the United States, the earlier spread of the pandemic from Asia to Europe, and the low overall prevalence in rest of the United States. Several other reports of the initial stages of the New York City region outbreak have identified early community spread on a similar time frame (Davis et al. 2020; Fauver et al. 2020; Gonzalez-Reiche et al. 2020). Although the low rate of travel history among our cases could reflect incomplete ascertainment of potential exposures, the extent of uncontrolled community spread likely reduces the representation of index travel cases in our data set. Indeed, the ability to track past transmissions is a key advantage of a genetic approach in the face of inadequate testing.
It is important to caution that fine-scale delineation of individual introductions and transmissions through genomic epidemiology is limited by viral mutation rate, incomplete sampling, and incomplete availability of exposure history (Villabona-Arenas et al. 2020). In particular, many early sequences show identical genotypes, which could be consistent with additional transmission events, possibly by way of unsampled regions. Although our estimate of 109 introductions is thus likely to underestimate the total number of introductions, the genomic data are sufficiently informative to outline an unrecognized early spread in February that enabled rapid development of the outbreak in March. Further analysis (Worobey et al. 2020) and sequencing of archival samples will be needed to refine assessments of the initial spread.
Our demonstration of rapid sample processing, deposition, and analysis underscores the potential for genomic epidemiology to provide an independent estimate of disease transmission, as well as its potential to recognize impending resurgence of a regional outbreak. Further surveillance by medical centers, regional public health departments, and national efforts will be needed to monitor genomic epidemiology, pandemic spread, and public responses (Supplemental Fig. S5). Given the logistical, regulatory, and methodological challenges to establishing such surveillance during an outbreak, it is critical to have this infrastructure already in place (Kim et al. 2020) for future waves of COVID-19 or other future pandemics.
Methods
Bioethics statement
The collection of COVID-19 human biospecimens for research has been approved by NYULH institutional review board under S16-00122, Universal Mechanism of Human Bio-Specimen Collection and Storage for Research.
The approved IRB protocol allows for the collection and analysis of clinical, travel, exposure, and demographic data (Osman et al. 2020). Electronic medical records were reviewed to compile a clinical database for 820 cases listing health care worker status, travel history, and close contact with a known COVID-19 case. For cases in which a given exposure was not directly stated in the clinical record, we recorded that field as missing data but included other exposures in our analysis. A summary field of exposure history per case was generated from the presence of a COVID-19 contact, travel history, or health care worker status, in that order.
Sample collection
All samples were collected as part of clinical diagnostics. Nasopharyngeal swabs were collected and placed in 3 mL of viral transport medium (VTM; Copan universal transport medium) following clinical protocols. Samples were transported to the clinical microbiology laboratory at room temperature and tested for SARS-CoV-2 the same day. Remnant samples were stored at −70°C.
Clinical testing
All initial detections of COVID-19 cases were performed as part of the clinical care. Clinical testing was performed using the following three FDA emergency use authorization (EUA)–approved COVID-19 PCR-based tests:
NYULH-validated PCR test using the U.S. CDC primer design, targeting three regions of the virus nucleocapsid (N) gene, and an internal control primer targeting the human RNase P (RP) gene (https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html) with PCR performed on an ABI7500 Dx system. The limit of detection is 10,000 copies per milliliter.
The Roche Cobas 6800 RT-PCR platform targeting the Orf1/a and E sequences, per the manufacturer's instructions. The limit of detection is 180 copies per milliliter.
The Cepheid Xpert Xpress RT-PCR platform targeting the N2 and E viral sequences, per the manufacturer's instructions. The limit of detection is 250 copies per milliliter.
RNA extraction
RNA extraction was performed using two platforms for parallel sample processing:
By using the Maxwell RSC instrument (Promega AS4500), total RNA was extracted from 300 µL of VTM with the buccal swab DNA kit (Promega AS1640). The following modifications were introduced to extract total RNA as opposed to total nucleic acids: Samples were incubated for 30 min at 65°C for Proteinase K digestion and virus deactivation, and DNase I (Promega) was added to the reagents cartridge to remove genomic DNA during nucleic acids extraction. Total RNA was eluted in 50 µL of nuclease-free water.
By using the KingFisher flex system (Thermo Fisher Scientific), RNA was extracted from heat-inactivated nasopharyngeal swab samples in batches of 96 samples, following the manufacturer's instructions and the MagMax mirVana total RNA isolation kit (Thermo Fisher Scientific A27828). Briefly, 250 µL of nasopharyngeal swab collection was lysed in lysis buffer and β-mercaptoethanol and subsequently bound to magnetic beads and loaded into the KingFisher flex instrument. A DNase I treatment step was performed as part of the instrument protocol, and RNA samples were eluted in 50 µL of elution buffer and immediately stored at −80°C.
Library preparation and sequencing
lllumina sequencing libraries were prepared from 10 µL of total RNA. Two ribodepletion methods for cDNA RNA-seq library preps were used:
KAPA RNA HyperPrep kit with RiboErase (HMR; Roche Kapa KK8561). We followed the manufacturer's protocol with the following modifications: For the adapter ligation step, we prepared a plate of barcoded adapters (IDT) at a concentration of 500 nM and performed 15 cycles of PCR amplification of the final library.
Nugen trio with human rRNA depletion (Tecan Genomics 0606-96), including DNase I treatment, cDNA synthesis, single primer isothermal amplification (SPIA), enzymatic fragmentation, library construction, final PCR amplification (12–16 cycles), and an AnyDeplete step to remove host rRNA transcripts. An automated protocol was implemented on a Biomek FXP liquid handler integrated with a Biometra TRobot 96-well thermal cycler (Beckman Coulter).
Purified libraries were quantified using qPCR (Kapa Biosystems KK4824). Library size distribution was checked using an Agilent TapeStation 2200.
Libraries presumed more suitable for capture (generally, qPCR Ct value greater than 30) were enriched for SARS-CoV-2 genomic sequences using custom biotinylated DNA probe pools either from Twist Biosciences or from Integrated DNA Technologies:
For capture using the IDT xGen COVID capture panel (Integrated DNA Technologies 10006764), we followed the manufacturer's protocol. Briefly, hybridization of 500 ng–1 µg of combined library DNA with 4 µL of xGen Lockdown probes was performed for 4–16 h at 65°C, followed by PCR amplification for six to 10 cycles.
For capture using the Twist Bioscience custom panel (Twist Design ID: TE-95888003, generously shared by the Seattle Flu Study), we followed the manufacturer's protocol using the Twist hybridization and wash kit (Twist Biosciences 101025). Hybridization of 1–2 µg combined library DNA was performed for 16–20 h at 70°C. Postcapture PCR amplification cycles ranged from 12 to 14 cycles.
In general, we pooled samples with similar Ct values and accounted for variations in parent library concentration, multiplexing up to 23 libraries per reaction. Positive and negative control samples are described in Supplemental Table S1.
Samples were sequenced as paired-end 100- or 150-cycle reads on the NextSeq 500 or NovaSeq 6000 (using SP or S1 flow cells). All flow cells were loaded such that indexing barcode sequences for multiplexed samples differed by ≥3 bp.
Sequence read processing
Reads were demultiplexed with Illumina bcl2fastq v2.20, requiring a perfect match to indexing barcode sequences. All RNA-seq and Capture-seq data were processed using a uniform mapping pipeline. Illumina sequencing adapters were trimmed with Trimmomatic v0.39 (Bolger et al. 2014). Reads were aligned using BWA v0.7.17 (Li and Durbin 2009) to a custom index containing human genome reference (GRCh38/hg38), including unscaffolded contigs and alternate references plus the reference SARS-CoV-2 genome (NC_045512.2, wuhCor1). Presumed PCR duplicates were marked using SAMBLASTER v0.1.24 (Faust and Hall 2014). Only sequences with >23,000 bp of sequence with ≥20× coverage depth were analyzed, resulting in 864 final sequences (Supplemental Table S2). Variants were called across all samples using BCFtools v1.9 (Li et al. 2009):
bcftools mpileup ‐‐redo-BAQ ‐‐adjust-MQ 50 ‐‐gap-frac 0.05 ‐‐max-depth 10000 ‐‐max-idepth 200000 ‐‐output-type u |
bcftools call ‐‐ploidy 1 ‐‐keep-alts ‐‐multiallelic-caller -f GQ
Raw pileups were filtered using
bcftools norm ‐‐check-ref w ‐‐output-type u |
bcftools filter -i “INFO/DP>=10 & QUAL>=10 & GQ>=99 & FORMAT/DP>=10” ‐‐SnpGap 3 ‐‐IndelGap 10 ‐‐set-GTs. ‐‐output-type u |
bcftools view -i ‘GT=“alt”‘ ‐‐trim-alt-alleles
Viral sequences were generated by applying VCF files to the reference sequence using `bcftools consensus` with -m to mask sites below 20× with Ns, and -m N to mask sites of ambiguous genotypes with N.
Geoplotting
The regional case heat map was generated using R v3.6.2 (R Core Team 2020), using the packages ggplot2 v3.3.0 (Wickham 2016) for plotting and sf v0.8 for geospatial data manipulation. Maps were generated based on the 2018 ZIP code tabulated area geographical boundaries obtained from the U.S. Census Bureau (United States Census Bureau 2018).
Phylogenetic analysis
Sequences for non-NYULH cases were downloaded from GISAID EpiCov on June 14, 2020, and filtered to sequences collected on or before May 10, 2020. Sequences from non-human hosts, annotated by Nextstrain as duplicate individuals or highly divergent, with fewer than 27,000 nonambiguous nucleotides or with improperly formatted dates or location were excluded. Sequences from outside New York State were subsampled to a maximum of 20 samples per admin division (United States) or country (outside United States) per month, prioritizing sequences most similar to the focal set of 864 NYULH samples. This priority was penalized if many non-US samples were most similar to the same U.S. sample, and mutations were weighted 333× more heavily than masked sites. Global sequences were then combined with the sequences from this study.
Sequences were analyzed using the augur v7.0.2 pipeline (Hadfield et al. 2018). Sequences were aligned along with the reference genome using MAFFT v7.453 (Katoh and Standley 2013), and the resulting alignment was masked to remove 100 bp from the beginning, 50 from the end, and uninformative point mutations (positions 11083, 13402, 21575, 24389, 24390).
Maximum likelihood phylogenetic reconstruction was performed with IQ-TREE v1.6.12 (Nguyen et al. 2015) using a GTR substitution model and the -czb option. Support values were generated with the ultrafast bootstrapping option with 1000 replicates. This tree was used to tabulate nucleotide and amino acid changes specific to lineages and cases; gaps with respect to the reference were reported as deletions. TreeTime v0.7.4 (Sagulenko et al. 2018) was used to generate a timetree rooted at the reference sequence, using the ‐‐keep-polytomies option, and under a strict mutational clock under a skyline coalescent prior with a rate of 8 × 10−4 mutations per site per year and a standard deviation of 4 × 10−4.
For each NYULH case, the inferred earliest New York City transmission was identified as the most ancestral node or tip with >70% of sequences originating in the Northeast (defined as the states of New York, Connecticut, New Jersey, Pennsylvania) on the timescaled phylogeny using the ape (Paradis and Schliep 2019) and phangorn (Schliep 2011) R packages. The transmission source was identified as the first ancestral node defined by a unique mutation and ancestral to a sequence originating outside the Northeast. Transmissions with identical source nodes were grouped to yield transmission chains. Trees were plotted with the tidygraph and ggraph R packages.
Phylodynamic analysis
To minimize ascertainment and sampling bias, analysis was performed on a subset of sequenced cases residing in New York City and the outlying Westchester, Nassau, and Suffolk counties and excluded outpatients and known health care workers. Sequence data were aligned to reference (accession NC_045512.2) and ends trimmed using MAFFT 7.450 (Katoh and Standley 2013). A maximum likelihood tree was estimated using IQ-TREE 1.6.1 using a HKY substitution model (Nguyen et al. 2015). A further 20 phylogenies were derived by randomly resolving polytomies and enforcing a small minimum branch length of 7 × 10−6 substitutions per site using the ape R package (Paradis and Schliep 2019). Rooted timescaled phylogenies were estimated using the treedater R package version 0.5.1 (Volz and Frost 2017) and a strict molecular clock. The skygrowth R package version 0.3.1 (Volz and Didelot 2018) was used to estimate effective population size through time with an exponential prior for the smoothing parameter with rate 10−4. The final estimates were generated by averaging results over the 20 estimated timetrees. A script for reproducing these results is available at GitHub (https://gist.github.com/emvolz/d58cce01c3310a01df09faf615b77070).
Software availability
Code used in sequencing data processing is available at GitHub (https://github.com/mauranolab/mapping/tree/master/dnase) and as Supplemental Code.
Data access
All raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA650245; sequencing reads have been filtered to remove the host genome. Sequences have been deposited into the GISAID repository immediately upon QC with virus name “NYUMC”.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank the New York University Langone Health (NYULH) clinicians and laboratory personnel involved in the care and testing of the patients in this study. We thank all the laboratories who have contributed sequences to GISAID (Supplemental Table S3). We thank Lea Starita and the Seattle Flu Study for technical assistance and sharing their bait design. This work was partially funded by National Institutes of Health grants R35GM119703 (M.T.M.), P50CA016087 (I.O. and G.J.), and P30CA016087 (I.O., P.C., and A.H.); UM1AI148574 (M.J.M.), the NYULH Office for Science and Research; and MR/R015600/1 from the Medical Research Council Centre for Global Infectious Disease Analysis, School of Public Health, Imperial College London (M.R-C. and E.M.V.).
Author contributions: M.T.M., M.J.M., P.C., M.S., and A.H. conceived and supervised the study. L.B., G.S., X.F., C.A-G., K.V.A., M.B., A.S., M.E.C., M.J.K., B.B., T.G., A.L., J.P., T.V., L.H.L., A.R., V.R., M.I.S., G.J., I.O., M.A-R., M.J.M., P.C., and M.S. collected clinical samples and data. S.R., P.Z., D.D., G.W., M.S.H., P.M., Y.Z., and A.H. generated sequencing data. C.M., J. Cadley, E.G., and J. Chen contributed informatics tools. M.T.M., A.M.R., N.A.V., M.S.H., M.R-C., L.G., R.O., R.L., E.H., E.M.V., and A.H. performed the data analysis. M.T.M., E.M.V., M.S., and A.H. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.266676.120.
Freely available online through the Genome Research Open Access option.
References
- Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. 2020. The proximal origin of SARS-CoV-2. Nat Med 26: 450–452. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford T, Greninger AL, Roychoudhury P, Starita LM, Famulare M, Huang M-L, Nalla A, Pepper G, Reinhardt A, Xie H, et al. 2020. Cryptic transmission of SARS-CoV-2 in Washington state. Science 370: 571–575. 10.1126/science.abc0523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MW, Matthews DA, Hiscox JA, Elmore MJ, Pollakis G, Rambaut A, Hewson R, García-Dorival I, Bore JA, Koundouno R, et al. 2015. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 524: 97–101. 10.1038/nature14594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JT, Chinazzi M, Perra N, Mu K, Pastore y Piontti A, Ajelli M, Dean NE, Gioannini C, Litvinova M, Merler S, et al. 2020. Estimating the establishment of local transmission and the cryptic phase of the COVID-19 pandemic in the USA. medRxiv 10.1101/2020.07.06.20140285 [DOI]
- Faust GG, Hall IM. 2014. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30: 2503–2505. 10.1093/bioinformatics/btu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG, Vogels CBF, Brito AF, Alpert T, Muyombwe A, et al. 2020. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell 181: 990–996.e5. 10.1016/j.cell.2020.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, Ciferri B, Alshammary H, Obla A, Fabre S, Kleiner G, Polanco J, Khan Z, et al. 2020. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 369: 297–301. 10.1126/science.abc1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh ND, Ladner JT, Lemey P, Pybus OG, Rambaut A, Holmes EC, Andersen KG. 2019. Tracking virus outbreaks in the twenty-first century. Nat Microbiol 4: 10–19. 10.1038/s41564-018-0296-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, Sagulenko P, Bedford T, Neher RA. 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34: 4121–4123. 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AE, Brandstetter E, Graham C, Heimonen J, Osterbind A, McCulloch DJ, Han PD, Starita LM, Nickerson DA, Van de Loo MM, et al. 2020. Seattle Flu study—swab and send: study protocol for at-home surveillance methods to estimate the burden of respiratory pathogens on a city-wide scale. medRxiv 10.1101/2020.03.04.20031211 [DOI]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, Haeseler von A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32: 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYS Department of Health. 2020. New York State statewide COVID-19 testing. New York State Department of Health, Albany, NY. https://health.data.ny.gov/Health/New-York-State-Statewide-COVID-19-Testing/xdss-u53e [accessed June 18, 2020].
- Osman I, Cotzia P, Moran U, Donnelly D, Arguelles-Grande C, Mendoza S, Moreira A. 2020. The urgency of utilizing COVID-19 biospecimens for research in the heart of the global pandemic. J Transl Med 18: 219 10.1186/s12967-020-02388-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35: 526–528. 10.1093/bioinformatics/bty633 [DOI] [PubMed] [Google Scholar]
- Park DJ, Dudas G, Wohl S, Goba A, Whitmer SLM, Andersen KG, Sealfon RS, Ladner JT, Kugelman JR, Matranga CB, et al. 2015. Ebola virus epidemiology, transmission, and evolution during seven months in Sierra Leone. Cell 161: 1516–1526. 10.1016/j.cell.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrilli CM, Jones SA, Yang J, Rajagopalan H, O'Donnell L, Chernyak Y, Tobin KA, Cerfolio RJ, Francois F, Horwitz LI. 2020. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 369: m1966 10.1136/bmj.m1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pybus OG, Rambaut A. 2009. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 10: 540–550. 10.1038/nrg2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, et al. 2017. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 12: 1261–1276. 10.1038/nprot.2017.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Sagulenko P, Puller V, Neher RA. 2018. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol 4: 741 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics 27: 592–593. 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, McCauley J. 2017. GISAID: Global initiative on sharing all influenza data—from vision to reality. Euro Surveill 22: 30494 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JR, James P, Stoddart D, Sparks N, Wickenhagen A, Hall G, Choi JH, Lapointe H, Kamelian K, Smith AD, et al. 2020. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 10.1101/2020.09.04.283077 [DOI] [Google Scholar]
- United States Census Bureau. 2018. Cartographic Boundary Files. United States Census Bureau, Washington, DC. https://www.census.gov/geographies/mapping-files/time-series/geo/carto-boundary-file.html [accessed April 15, 2020].
- Villabona-Arenas CJ, Hanage WP, Tully DC. 2020. Phylogenetic interpretation during outbreaks requires caution. Nat Microbiol 5: 876–877. 10.1038/s41564-020-0738-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz EM, Didelot X. 2018. Modeling the growth and decline of pathogen effective population size provides insight into epidemic dynamics and drivers of antimicrobial resistance. Syst Biol 67: 719–728. 10.1093/sysbio/syy007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz EM, Frost SDW. 2017. Scalable relaxed clock phylogenetic dating. Virus Evol 3: vex025 10.1093/ve/vex025 [DOI] [Google Scholar]
- Volz EM, Koelle K, Bedford T. 2013. Viral phylodynamics. PLoS Comput Biol 9: e1002947 10.1371/journal.pcbi.1002947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. Ggplot2: elegant graphics for data analysis. Springer-Verlag, New York: http://ggplot2.org. [Google Scholar]
- Worobey M, Pekar J, Larsen BB, Nelson MI, Hill V, Joy JB, Rambaut A, Suchard MA, Wertheim JO, Lemey P. 2020. The emergence of SARS-CoV-2 in Europe and North America. Science 370: 564–570. 10.1126/science.abc8169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-Z, Holmes EC. 2020. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 181: 223–227. 10.1016/j.cell.2020.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jackson CB, Mou H, Ojha A, Rangarajan ES, Izard T, Farzan M, Choe H. 2020. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 10.1101/2020.06.12.148726 [DOI]
- Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, et al. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.