

SUMMARY
Cyclin D1 encodes the regulatory subunit of a holoenzyme that phosphorylates RB and functions as a collaborative nuclear oncogene. The serine threonine kinase Akt plays a pivotal role in the control of cellular metabolism, survival, and mitogenic signaling. Herein, Akt1-mediated phosphorylation of downstream substrates in the mammary gland is reduced by cyclin D1 genetic deletion and is induced by mammary-gland-targeted cyclin D1 overexpression. Cyclin D1 is associated with Akt1 and augments the rate of onset and maximal cellular Akt1 activity induced by mitogens. Cyclin D1 is identified in a cytoplasmic-membrane-associated pool, and cytoplasmic-membrane-localized cyclin D1—but not nuclear-localized cyclin D1—recapitulates Akt1 transcriptional function. These studies identify a novel extranuclear function of cyclin D1 to enhance proliferative functions via augmenting Akt1 phosphorylation at Ser473.
Graphical Abstract
In Brief
Chen et al. show that the rate of onset and maximal cellular Akt1 activity induced by mitogens was augmented by cyclin D1. Cyclin D1 bound and phosphorylated Akt1 at Ser473. These studies identify a novel extranuclear function of cyclin D1 to enhance proliferative functions via augmenting Akt1 phosphorylation at Ser473.
INTRODUCTION
The cell survival oncoprotein Akt, also known as protein kinase B, conveys distinct pathophysiological processes promoting cellular survival, proliferation, growth, and migration (Hers et al., 2011). Akt is frequently hyperactivated in human cancers. In mammalian cells, oncogenic stimuli and growth factors induce Akt kinase activity to promote anti-apoptotic signaling. Three separate genes encode the major isoforms of Akt (Akt1/PKB, Akt2/PKB, and Akt3/PKB). The phosphatase that negatively regulates Akt, the tumor suppressor gene PTEN, is frequently deleted or mutated in human cancer, resulting in constitutive activation of Akt1 kinase.
Akt activation occurs through a complex, multistep, phosphorylation-dependent mechanism that is incompletely understood (Manning and Cantley, 2007). Akt is phosphorylated in response to growth factor signaling by phosphoinositide-dependent kinase (PDK1) in the activation loop (Thr308) (Dibble and Cantley, 2015) and by mammalian target of rapamycin complex 2 (mTORC2) in the C-terminal hydrophobic motif (Ser473) (Sarbassov et al., 2005). Reflecting the diverse cellular contexts in which AKT plays a role, Akt S473 phosphorylation is enhanced by many signaling pathways, including IKKα (Dan et al., 2016); the serine threonine kinase integrin-linked kinase (ILK) (McDonald et al., 2008); members of the phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) family, including DNA-dependent protein kinase catalytic subunit (DNA-PKc) (Bozulic et al., 2008), mitogen-activated protein kinase-activated protein kinase 2, (p38) (Kim et al., 2008), protein kinase CbII (PKCbII) (Kawakami et al., 2004); ataxia-telangiectasia mutant; and ataxia-telangiectasia and Rad3 related (Halaby et al., 2008). BSD-domain-containing signal transducer and Akt interactor (BSTA) also promote phosphorylation of Akt1 at Ser473 (Yao et al., 2013). The priming phosphorylation by Akt1-pS477/pT479 is mediated by Cdk2/cyclin A, mTORC2, or DNA-dependent protein kinase (DNA-PK) under cell-cycle progression, growth factor stimulation, or DNA-damaging conditions, respectively (Liu et al., 2014). Akt activity fluctuates across the cell cycle (Liu et al., 2014). Under cell-cycle conditions, the cyclin A/cdk2 priming phosphorylation becomes more important and is, therefore, relatively mTORC2/Rictor independent (Liu et al., 2014). These distinct signaling cascades play differing roles under different physiological perturbations. For example, inactivation of mTORC2 by depleting Rictor led to a more dramatic reduction of Akt1-pS477/pT479 in response to insulin than under synchronized cell-cycle conditions. Furthermore, the cyclin A/cdk2-mediated priming of S477/479 was considered to be important in cellular transformation (Liu et al., 2014).
Downstream substrates of Akt1 coordinating the pro-survival functions include tuberous sclerosis (TSC), gene 2, which disrupts the TSC1/TSC2 complex, thereby derepressing mTOR (mammalian target of rapamycin). Additional Akt substrates include BAD, MDM2, Huntington, Arfaptin2, and Forkhead Ligand 1 (FKHR-L1) (Datta et al., 1999; Hay, 2005). The pro-proliferative and pro-survival functions of Akt1 involve caspase-9, BAD, IKβ kinase α, and a GSK3β/cyclin D1 pathway. Akt hyperactivation contributes to human cancer correlating with poor prognosis and therapy resistance (Manning and Cantley, 2007; Yuan and Cantley, 2008), and genetic deletion demonstrated that Akt1 is required for ErbB2-induced breast cancer progression and tumor metastases in vivo (Ju et al., 2007). Maximal activation of Akt requires phosphorylation on the carboxy-terminal site S473 by mTORC2 (Hresko and Mueckler, 2005; Dibble and Cantley, 2015). Activity of mTORC2 is determined, in part, by the abundance of its components, including mTOR, regulatory associated protein of mTOR (Raptor), mammalian lethal with SEC13 protein 8 (mLST8), 40-kDa proline-rich Akt substrate (PRAS40), DEP-domain-containing mTOR interacting protein (DEPTOR), and Sin1 (Sabatini, 2017; Dibble and Cantley, 2015). Rictor and Sin1 are specific components for the mTORC2 kinase complex.
Downstream targets of Akt promoting cellular proliferation include cyclin D1 (Albanese et al., 2003), the regulatory subunit of the holoenzyme that phosphorylates and inactivates the RB and NRF1 proteins (Sherr, 1993; Wang et al., 2006). Phosphorylation of RB promotes DNA synthesis, whereas phosphorylation of NRF1 restrains mitochondrial metabolism. The promotion of DNA synthesis and reduction in mitochondrial metabolism reflects a triage function of cyclin D1 by which metabolic substrates enhance the Warburg effect (Sakamaki et al., 2006) as frequently observed in cancer (Ward and Thompson, 2012; Finley et al., 2013). In genetic deletion studies, the phenotype of the cyclin D1−/− (Sicinski et al., 1995) and Akt1−/− mice (Chen et al., 2001; LaRocca et al., 2011) revealed several common features, including defective mammary gland development. In addition to a kinase function, cyclin D1 conveys distinct noncanonical functions (Pestell, 2013), including a nuclear function promoting transcription factor activity, often in the context of local chromatin, wherein distinct enzyme complexes are recruited to target transcription factor binding sites (Bienvenu et al., 2010; Fu et al., 2005; Casimiro et al., 2012), as reviewed in (Casimiro et al., 2014).
Evidence for an extranuclear function of cyclin D1 includes the location of cyclin D1 and related cell-cycle proteins in the cytoplasmic membrane (Fusté et al., 2016), the association of cyclin D1 with cytoplasmic membrane proteins (Zhong et al., 2010; Meng et al., 2011; Fusté et al., 2016), and evidence for an extranuclear function of cyclin D1 in estrogen (estradiol, or E2) signaling (Li et al., 2014). Cyclin D1 has been shown to bind cytoplasmic-membrane-associated protein PACSIN 2 (protein kinase C [PKC] and casein kinase substrate in neurons protein 2) (Meng et al., 2011) and Filamin A (Zhong et al., 2010) and co-localizes in the cell membrane with paxillin (Fusté et al., 2016). Furthermore, cyclin D1 promotes cellular migration, in part through interacting with Rho GTPase (Li et al., 2006b). Cyclin D1 is required for both estrogen- and androgen-dependent gene expression and function in vivo (Ju et al., 2014; Li et al., 2014), including the estrogen-dependent attenuation of the DNA damage response. In prior studies, estrogen dendrimers that were functionally excluded from the nucleus determined cyclin D1-dependent estrogen signaling (Li et al., 2014), suggesting that the estrogen-dependent attenuation of the DNA damage response involves an extranuclear pool of cyclin D1 (Di Sante et al., 2017). In this study, we identified a novel extranuclear function of cyclin D1 to enhance proliferative functions via augmenting phosphorylation of Akt1 at Ser473 and established that cytoplasmic localized cyclin D1, but not nuclear localized cyclin D1, recapitulates Akt1 transcriptional signal transduction function.
RESULTS
Cyclin D1 Augments Akt Activity In Vivo
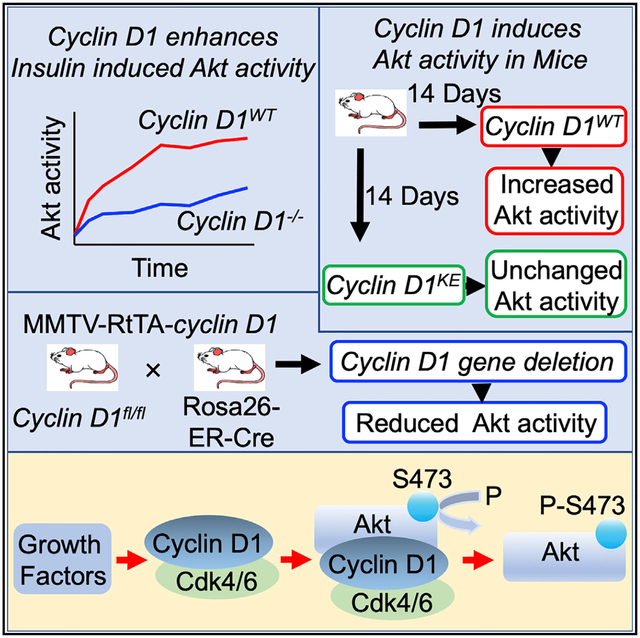
In recent studies, transient expression of a cyclin D1 cDNA at physiological levels was sufficient to promote proliferative signaling and gene expression associated with metabolism, ribosomal biogenesis, and mitogenesis (Casimiro et al., 2012). In order to determine at a higher level of resolution the mechanisms by which cyclin D1 expression induced mitogenic signaling, we examined proliferative kinase activity in the mammary gland of transgenic mice in which either the cyclin D1 cDNA (Casimiro et al., 2012) or a point mutant of the cyclin D1 cDNA K112E (cyclin D1KE) (Casimiro et al., 2015) was intercrossed with MMTV-RtTA mice to ensure doxycycline-inducible expression of cyclin D1 in the mammary gland of mice (Figure 1A). The cyclin D1 cDNA K112E (cyclin D1KE) generates a mutation previously shown to convey reduced ability to phosphorylate RB (Casimiro et al., 2015; Baker et al., 2005). Animals were treated for 14 days, and analysis of Akt1 and Akt1 Ser473 was conducted by immunohistochemistry (IHC) using an Akt1 isoform-specific antibody (Figure 1; Figure S1). The presence of the cyclin D1 transgene was confirmed by IHC to the amino-terminal FLAG epitope of the cyclin D1 cDNA in both the nucleus and cytoplasm (Figure 1B). Induction of the cyclin D1 transgene was associated with increased phosphorylation of Akt1 Ser473, which was seen in both the nucleus and cytoplasm, without a change in Akt1 abundance. Consistent with cyclin D1-mediated activation of Akt1, several downstream targets of Akt were measured in the mammary epithelial cell of the mammary gland in the bi-transgenic mice after induction of the cyclin D1 transgene by doxycycline (TSC2 Ser939, FKHR Ser319, and BAD Ser136) (Figures S1B–S1E).
Figure 1. Cyclin D1 Augments Akt1 Activity In Vivo.
(A) Transgenic mice expressing a doxycycline-inducible cyclin D1WT and cyclin D1KE mutant cDNA targeted to the mammary gland by MMTV-RtTA were treated with doxycycline for 14 days and immunohistochemical analysis was conducted for phosphorylated targets of Akt1.
(B–D) Representative immunohistochemistry (IHC) results are shown, and quantitative data are indicated as mean ± SEM at the right of the panels. IHC was conducted for (B) cyclin D1 cDNA (FLAG), (C) phospho-Akt1 Ser473, and (D) Akt1.
(E) The cyclin D1fl/fl mice were intercrossed with ROSA26-ER-Cre, and the mice treated with tamoxifen for 5 days. Immunohistochemical staining of the mammary epithelium was conducted.
(F–H) Immunohistochemical staining of the mammary gland for cyclin D1 and Akt1 or to downstream Akt signaling substrates.
The data are indicated as semiquantitative data, as mean ± SEM for the relative abundance of proteins.
In order to determine whether endogenous cyclin D1 maintains Akt1 activity in the mammary gland in vivo, cyclin D1fl/fl mice were intercrossed with ROSA26-ER-Cre mice and analyzed after 5 days of tamoxifen treatment used to induce Cre expression and to thereby delete cyclin D1 gene expression (Figure 1E). Consistent with the induction of Akt1 activity upon transgenic expression of cyclin D1, Cre-mediated deletion of the cyclin D1 gene reduced cyclin D1 protein abundance (Figure 1F) and reduced Akt1 activity, as evidenced by reduced phosphorylation of Akt1 (pAkt Ser473) and its substrates (pTSC2 Ser939 and pFKHR Ser319), without altering the abundance of the substrates (Akt1 and TSC2) (Figures S2B–S2D). As recent studies had demonstrated that cyclin A2 can induce Akt1 activity (Liu et al., 2014), we examined cyclin A2 levels in the mammary gland of mice in which cyclin D1 was either overexpressed or reduced in abundance. Cyclin A2 levels were unchanged by cyclin D1 expression, whereas Akt1 Ser473 phosphorylation was increased (Figures S3A and S3B). Induction of the cyclin D1KE cDNA did not induce pAkt1 Ser473 (Figures S3C and S3D). In the cyclin D1−/− mammary gland, cyclin A2 levels were unchanged, although pAkt Ser473 levels were reduced (Figures S3E–S3H).
As a form of control for Akt1, we examined the activity of Akt and its substrates in vivo. We examined the mammary gland of mice deleted of the Akt1 gene (Ju et al., 2007). A similar reduction in pTSC2 Ser939, pFKHR Ser319, and pBad Ser136 was observed upon Akt1 gene deletion (Figure S4). We considered the possibility that cyclin D1 may indirectly induce pAkt1 Ser473 via regulating expression of mTORC2 components. The abundance of the mTORC2 components in the MMTV-cyclin D1 mammary tumors were unchanged by prolonged expression of cyclin D1 (Figures S5A and S5B). Acute induction of cyclin D1 for 1 week using the rtTA cyclin D1 transgenics, compared with control mice, demonstrated no significant change in the abundance of the mTORC2 components (Figures S5C and S5D). Cyclin D1−/− 3T3 cells transduced with an expression vector encoding cyclin D1 or a control expression vector showed no change in abundance of mTORC2 components (Figure S5E). Rictor abundance was assessed by IHC in the mammary gland of transgenic mice and showed no alteration with the acute induction of either the wild-type (WT) cyclin D1 (or cyclin D1WT) or point mutation of the lysine residue K112 (cyclin D1KE) transgene (Figure S5F) or upon chronic deletion in cyclin D1+/+ versus cyclin D1−/− mammary epithelium (Figure S5G).
Cyclin D1/CDK Phosphorylation of Akt1
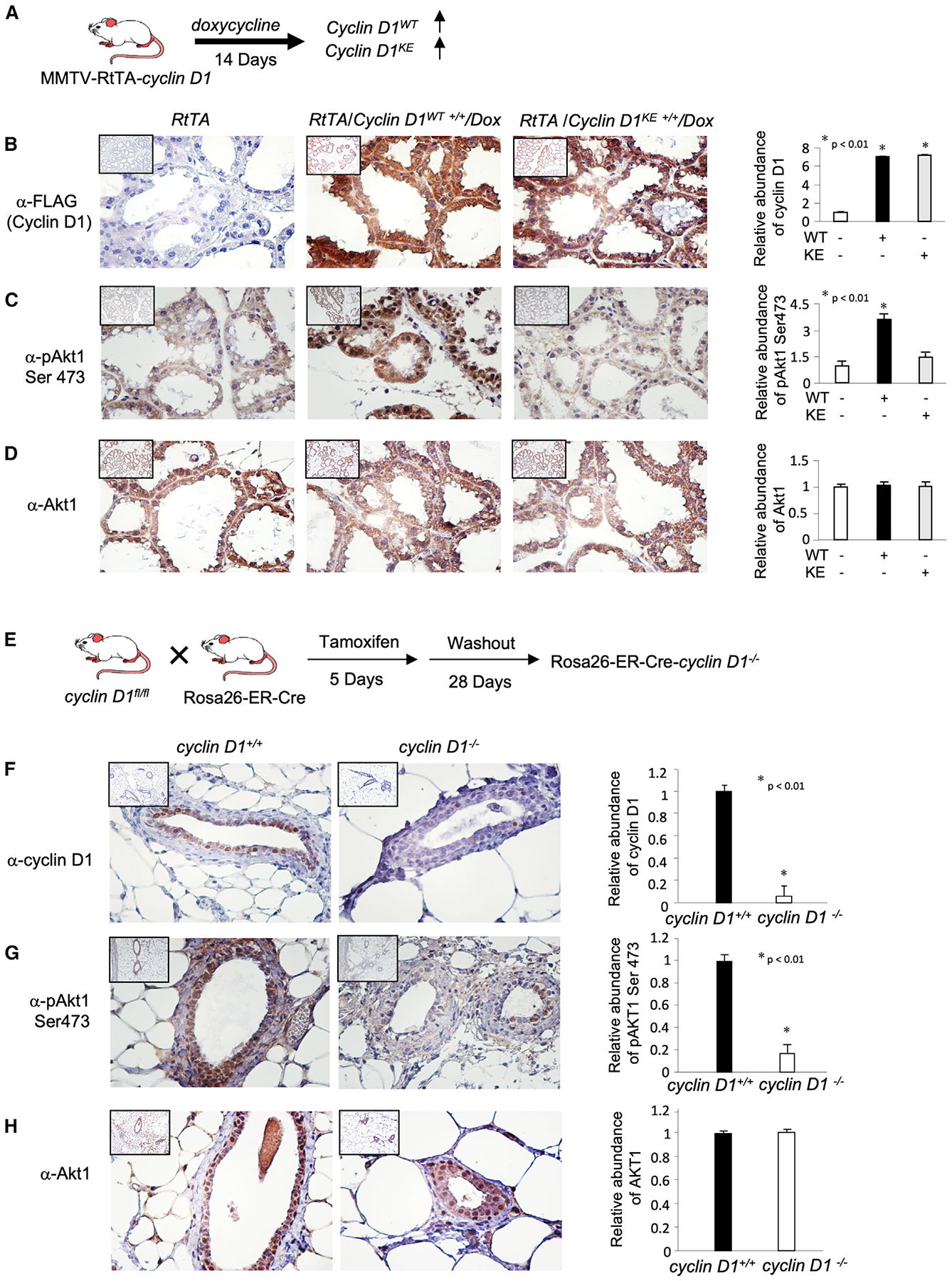
In order to examine a potential role for cyclin D1 in directly phosphorylating Akt1, MCF-7 cells were transduced with an expression vector encoding the amino-terminal FLAG-tagged cyclin D1 expression vector, and western blot was conducted (Figure 2A). The expression of cyclin D1 was identified by the FLAG epitope, and the relative abundance of cellular cyclin D1 was increased ~1.5-fold (exogenous cyclin D1, labeled as “Ex” in Figure 2A). This change in abundance is within the physiological 5-fold change in abundance that occurs in MCF-7 cells upon serum or E2 stimulation or during cell-cycle transition (Li et al., 2014). The relative abundance of Akt1 was not significantly altered. The phosphorylation of Akt1 on Serine 473, which activates Akt (Sarbassov et al., 2005), was enhanced 2.2-fold (Figure 2A). The phosphorylation of the Akt1 substrate TSC2 at Ser392 was increased 6-fold without any change in TSC2. Reintroduction of cyclin D1 into the cyclin D1−/− 3T3 cells increased cyclin D1 levels to that of cyclin D1WT 3T3 cells (Li et al., 2006a), associated with a ~48-fold increase in phosphorylation of Akt at Ser473, without a change in Akt1 abundance (Figure 2B). The phosphorylation of key downstream substrates of Akt1 was increased without a significant change in the abundance of the substrates, including TSC2 (Ser393), FOXO1 (Ser319), and Bad (Ser126) (Figure 2B).
Figure 2. Cyclin D1 Phosphorylates Akt1 at S473.
(A) Western blot analysis of MCF-7 cells transduced with an expression vector encoding cyclin D1 with antibodies as indicated to cyclin D1, Akt1, Akt1(Ser473), and the Akt1 signaling pathway and its substrate TSC2 (Ser939). The comparison of FLAG-tagged cyclin D1 (high molecular weight, labeled “Ex” for exogenous) and endogenous (labeled “End”) cyclin D1 indicates a 2-fold increase in cyclin D1 abundance. The data are representative of n = 3 separate experiments.
(B and C) (B) Cyclin D1−/− cells transduced with a retroviral vector for (B) cyclin D1 or (C) cyclin D1WT and cyclin D1KE with antibodies as indicated.
(D and E) In (D), cyclin D1 immune-precipitation kinase assays were conducted using GST fusion proteins as substrates including pRB, and Akt1. Left panel: proteins on an SDS-PAGE stained with Coomassie. Right panel: γ32 p IP-kinase assay reactions, with relative incorporation into substrates indicated in (E) as mean ± SEM for n = 3 separate experiments.
(F) Representative mass spectrometry spectrum to map the Akt1 Ser473 phosphorylation status in vivo. The liquid chromatography-tandem mass spectrometry (LC-MS/MS) spectrum of the singly phosphorylated doubly charged peptide RPHFPQFpSYSASGTA representing S473 in the modified Akt1 sequence. The neutral loss of phosphate confirms the phosphorylation status, and sites are localized to S10 (S473 full length) in the peptide based on the b-ion series (N-terminal fragments) starting at b7 and the y-ion series starting at y10 that contain a phosphate group.
(G) Cyclin D1 immune-precipitation kinase assays were conducted using GST fusion proteins including Akt1-WT and Akt1 Ser473 mutation. Left panel: proteins on an SDS-PAGE stained with Coomassie. Right panel: γ32P IP-kinase assay reactions.
In order to define further the mechanisms by which cyclin D1 enhanced Akt1 phosphorylation, cyclin D1−/− cells were transduced with expression vectors encoding either cyclin D1WT or cyclin D1KE. Similar levels of cyclin D1WT and cyclin D1KE mutant proteins were identified within the cell by western blot (Figure 2C). Re-expression of cyclin D1 in cyclin D1−/− cells enhanced Akt1 phosphorylation at Ser473. However, cyclin D1KE abrogated the induction of Akt1 phosphorylation (Figure 2C). In order to consider potential mechanism by which cyclin D1 augmented Akt1 activity, we considered the possibility that cyclin D1 directly phosphorylated Akt1. Cyclin D1 immune-precipitation kinase assays were conducted using immune-precipitated cyclin D1 from HEK293T cells overexpressing cyclin D1 as the enzyme source (Ashton et al., 1999) and expressed proteins including a pRB fragment that encodes two CDK phosphorylation sites (Ashton et al., 1999) and full-length immune-purified human Akt1 (Figures 2D and 2E) as substrates. The glutathione S-transferase (GST) fusion protein input was electrophoresed and stained with Coomassie, and the γ32p incorporated product was compared in order to derive relative incorporation of γ32p into the substrates (Figures 2D and 2E). Next, HEK293T cells were transfected with expression vector encoding hemagglutinin (HA)-Akt1 and either vector or FLAG N-amino-terminal tagged cyclin D1WT. HA immunoprecipitation (HA-IP) was conducted on the cells, and mass spectrometry compared the relative abundance in post-translational modification between vector control and cyclin D1-transfected cells (Figure 2F). As Akt1 Ser473 was identified in western blot and by mass spectrometry as a residue phosphorylated in response to cyclin D1 expression, cyclin D1 IP kinase assays were conducted of Akt1 WT versus Akt1 S473A. In contrast with Akt1 WT, point mutation of Akt1 Ser473 reduced γ32p incorporated by −80-fold (Figure 2G). This study demonstrates that RB and Akt1 serve as efficient substrates in cyclin D1 immune complex kinase assays (Figures 2D–2E). Cyclin A2 levels were unchanged by cyclin D1 expression (Figures 2B and 2C).
Extranuclear Interaction of Cyclin D1 with Akt1 via the N Terminus
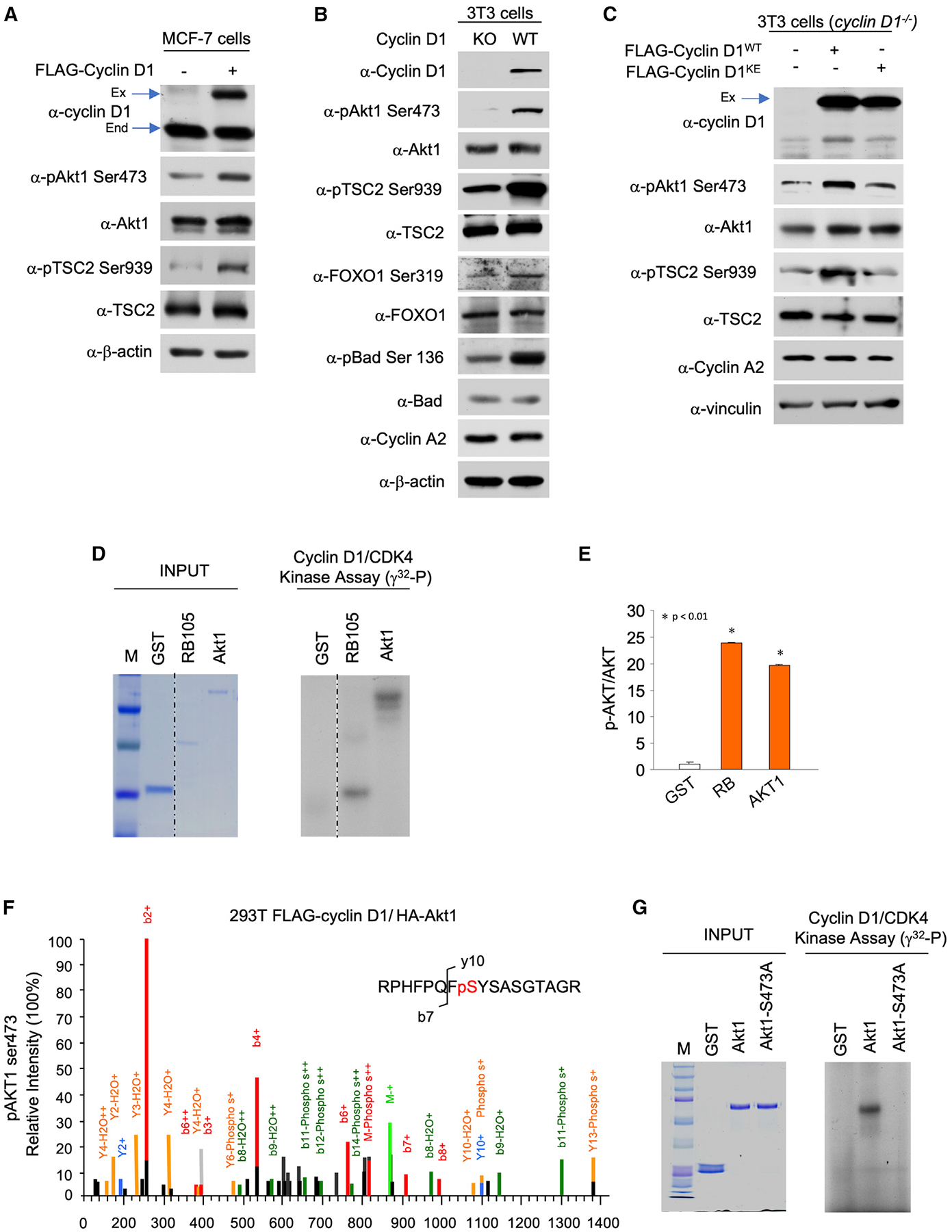
In order to determine whether cyclin D1 resides in proximity to Akt1 within the cell, we conducted proximity ligation assays (PLAs). The PLA is an antibody-based assay that generates a fluorescent signal only when the two target antigens are within 40 nm of each other (Gustafsdottir et al., 2005). We performed PLAs with antibodies recognizing cyclin D1 or Akt1. Figure 3A shows the signal identified when cyclin D1 was introduced into cyclin D1−/− 3T3 cells, whereas background signal only was identified in cells transduced with a control vector (GFP) (Figure 3B) or with control immunoglobulin G (IgG) (Figures S6 and S7). The signal was primarily extranuclear based on DAPI (blue staining) (Figure 3A). The signal for Akt1 and cyclin D1 proximity was maintained with a mutant of cyclin D1 (cyclin D1KE), which is defective in several functions, including kinase activity (Figure 3C). A mutant of a carboxy-terminal cyclin D1 phosphorylation site (cyclin D1T286) that is known to be primarily nuclear in localization (Di Sante et al., 2019; Alt et al., 2000) did not show association with Akt1 (Figure 3D; Figure S8). Deletion of the acidic rich carboxy-terminal motif from cyclin D1 (cyclinΔE) maintained binding to Akt1; however, deletion of the N-terminal residues 1–91 (cyclin D1C6) abrogated Akt1 binding (Figure 3E–3G), and mutants including deletions of this region (e.g., cyclin D1C4 and cyclin D1C5) also failed to bind Akt1 (Figure S8). The murine cyclin D1/ CDK4/Akt1 (mCD1/CDK4/Akt1) model (Figure 3H) was generated as described in the STAR Methods section. The high sequence identity between mCD1/CDK4/Akt1 and the high-resolution X-ray crystal structures used to generate this model provides a high degree of confidence in the model. The model indicates that Akt1 S473 is in proximity of the ATP γ-phosphate for nucleophilic attachment to allow for its phosphorylation.
Figure 3. Association of Akt1 via the Cyclin D1 N-Terminal Residues 1–91.
(A–F) The PLA for endogenous Akt1 with WT cyclin D1 or cyclin D1 mutants is indicated by red dots, and nucleus was stained by DAPI. Cyclin D1 expression plasmids were introduced by transduction of cyclin D1−/− 3T3 cells. 3D reconstruction of the z stack is indicated for representative examples of multiplicate experiments.
(G) Schematic representation of cyclin D1 expression plasmids used.
(H) Model of the murine cyclin D1-CDK4/CDK6-Akt1 (C-terminal peptide) complex. The model indicates cyclin D1 and CDK4/CDK6 in blue and yellow cartoons, respectively. The Akt1 c-terminal peptide bound at the active site of CDK4/CDK6 is indicated in green stick. The ATP and Mg2+ ions are indicated in blue sticks and red spheres, respectively.
Cyclin D1 Augments Akt1 Phosphorylation at the Plasma Membrane
Previously, studies demonstrated the presence of Akt in the cytoplasmic membrane (Zhang et al., 2012) and cyclin D1 was shown to associate with the membrane-associated protein PACSIN 2 (Meng et al., 2011), likely serving to tether cyclin D1 to the cytoplasmic membrane compartment. In cyclin D1 WT 3T3 cells, endogenous cyclin D1 co-localized with pAkt1 Ser473 (Figure 4A) as seen in the Z series. In cyclin D1−/− 3T3 cells, immunohistochemical staining revealed trivial phosphorylation of Akt1 at Ser473 (Figure 4B). Rescue of cyclin D1−/− 3T3 cells with a cyclin D1 expression retrovirus, which resulted in a physiological rescue of cyclin D1 abundance similar to that of cyclin D1WT 3T3 cells (Casimiro et al., 2015), enhanced Akt1 phosphorylation of Akt1 Ser473 at the cytoplasmic membrane, with enrichment at sites of focal contacts (Figure 4C). In contrast, rescue of cyclin D1−/− 3T3 cells with a cyclin D1KE cDNA induced trivial Akt1 Ser473 phosphorylation at the plasma membrane (Figure 4D).
Figure 4. Cyclin D1 Augments Phosphorylation of Akt1 at Ser473.
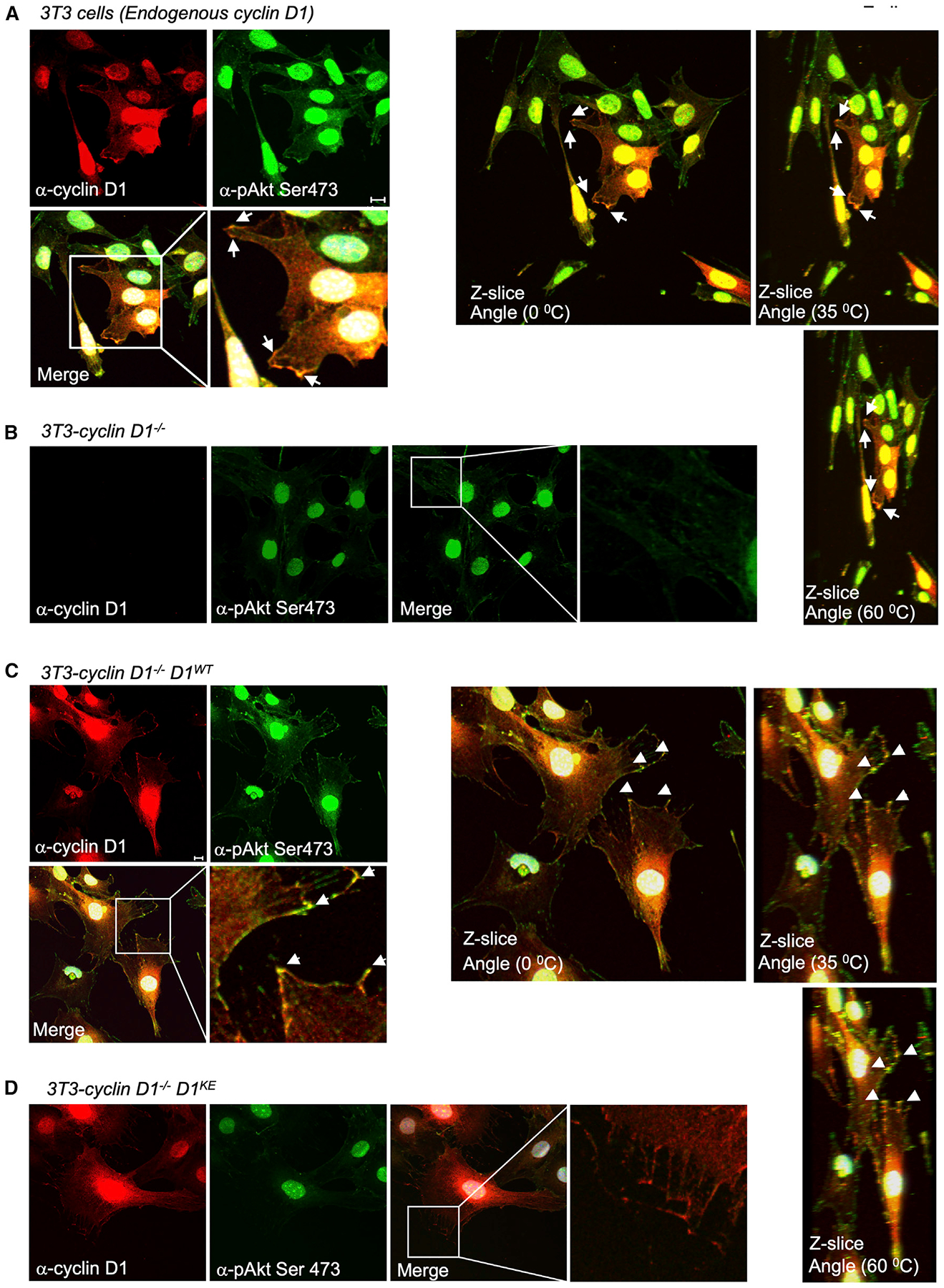
(A–D) Immunohistochemical staining of 3T3 cells (A), cyclin D1−/− 3T3 cells (B), or cyclin D1−/− 3T3 cells transduced with retrovirus encoding either cyclin D1WT (C) or cyclin D1KE (D). Immunohistochemical staining with Z slice reconstruction to show staining with antibody as indicated. Cyclin D1 co-localizes with pAkt1 Ser473 at the cytoplasmic membrane and is enriched at focal contacts.
The Onset and Peak Activity of Akt1 by Mitogens Require Cyclin D1/CDK4/6 Activity
Near-infrared (NIR) dye fluoresces at two different wavelengths (dichromic fluorescence). Recent studies identified a dichromic fluorescent (DCF) dye substrate for cellular Akt1 activity (Shen et al., 2013). The diserine DCF substrate was shown to serve as a specific substrate for Akt1, which can be used to quantitatively assess the enzyme’s activity in real time (Shen et al., 2013). Insulin activation of cellular Akt phosphorylates a single serine residue of the diserine DCF substrate in a time-dependent manner and can be used to assess longitudinally the stimulation and reversibility of Akt1 activity. The dichromic dye LS456 is phosphorylated by Akt1 but not a variety of other kinases (including protein kinase A [PKA], PKC, RSK1, P70S6K, and PI3K) (Shen et al., 2013). The binding of insulin to its cell-surface receptor stimulates PI3K, which then induces the second messenger, phosphotidylinositol-3, 4, 5-triphosphate (PIP3) (Lawlor and Alessi, 2001). PIP3 activates Akt and additional downstream effectors. As LS456 was shown to serve as a specific substrate for Akt1 in response to 150 nM insulin, we examined the kinetics of insulin-mediated activation of LS456 in in cyclin D1−/− mouse embryonic fibroblasts (MEFs) compared with WT MEFs (Figure 5A). Insulin stimulation of Akt1 activity assessed by LS456 was delayed with reduced induction in cyclin D1−/− cells compared with the cyclin D1+/+ cells (Figure 5A). The response to epidermal growth factor (EGF) was also delayed (Figure 5B). Analysis in Cdk4/Cdk6−/− MEFs demonstrated a delayed and reduced induction of insulin- and EGF-mediated activation of LS456 in Cdk4/Cdk6−/− MEFs compared with WT MEFs (Figures 5C and 5D).
Figure 5. Selective Phosphorylation of LS456 by Mitogen Activation Requires Cyclin D1 and CDK4/6.
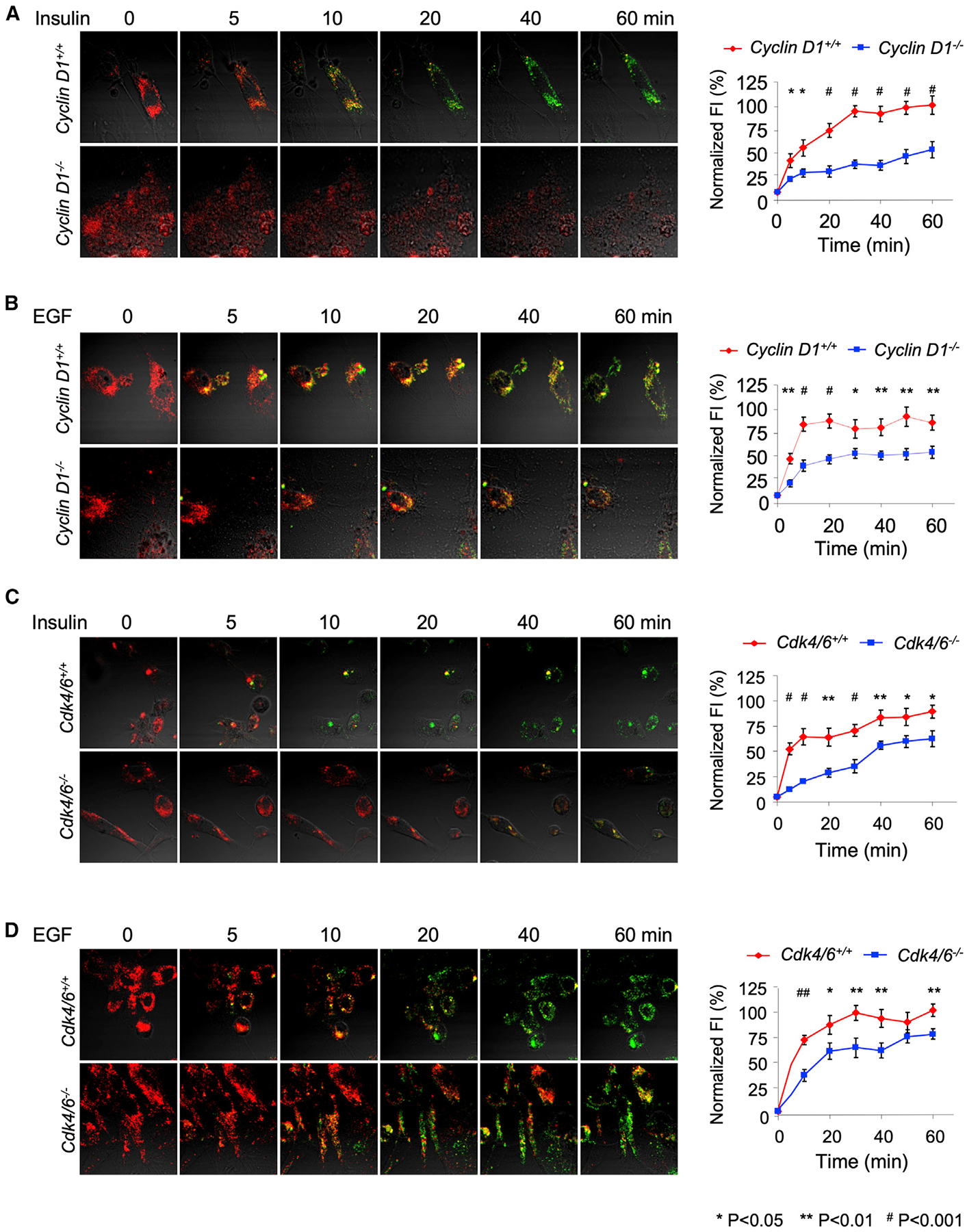
(A and B) The fluorescence intensity of LS456 in response to either (A) insulin (150 nM) or (B) EGF (10 ng/mL) decreased in the 800-nm channel and increased in the 700-nm channel with delayed changes in cyclin D1-deficient MEFs. Data are indicated as mean ± SEM for n = 8 WT and n = 24 for cyclin D1−/− in separate experiments (*p < 0.05; **p < 0.01; #p < 0.001). Quantitative analysis of the FI in WT and cyclin D1-deficient cells. Fluorescent images in cells were superimposed on differential interference contrast images; scale bars, 20 μm.
(C and D) The fluorescence intensity of LS456 in response to either (C) insulin (150 nM) (n = 8 Cdk4/6 WT and n = 24 Cdk4/6−/− MEFs) or (D) EGF (10 ng/mL) (n = 8 Cdk4/6 WT and n = 8 Cdk4/6−/− MEFs) was determined. The data are presented as mean ± SEM for n = 8 separate experiments (*p < 0.05; **p < 0.01; #p < 0.001).
Akt Ser473 phosphorylation is induced within 15 min of serum release, peaking at 1–3 h (Rosner et al., 2007). In order to determine whether cyclin D1 rescue of cyclin D1−/− cells augmented cell-cycle progression during this time frame, we compared the cyclin D1−/− and cyclin D1−/−cyclin D1WT rescued cells. Fluorescence-activated cell sorting (FACS) analysis demonstrated trivial differences in cell-cycle distribution in the 0- to 4-h time frame, with an augmentation of S phase enrichment (9.9 versus 13.5) occurring at 24 h (Figure S9A). CDK inhibitors have been shown to reduce the RB kinase function of the cyclin D1/CDK4 complex; however, significant additional interactions have been identified (reviewed in Di Sante et al., 2019), warranting an analysis of CDK inhibitor impact on Akt1 phosphorylation at Ser473 and the interaction with Rictor. To this end, we deployed MEFs encoding tamoxifen-inducible Rictor small interfering RNA (siRNA) (Cybulski et al., 2012). Acute reduction in Rictor abundance reduced Akt1 Ser473 compared with Akt1 (Figure S9B, lane 1 versus lane 4). The addition of the two distinct CDK inhibitors palbociclib (8 μm) or abemociclib (8 μm) reduced RB phosphorylation at Ser780 without significant effects on the relative phosphorylation of Akt1 Ser473 (Figure S9B, lanes 1 versus lanes 2 and 3). The reduction on RB phosphorylation by acute siRictor may be due to the previously described reduction in cyclin D1 (Hietakangas and Cohen, 2008). In contrast, after chronic reduction in Rictor (30 days), the addition of a CDK inhibitor—abemaciclib (4–8 μm) or palbociclib (4–8 μm)—reduced Akt1 Ser473 abundance compared with that of Akt1 (Figure S9C, lane 1 versus lane 5). Together, these studies are consistent with the known importance of Rictor to induce Akt1 Ser473 phosphorylation, and the new evidence herein that endogenous cyclin D/CDK4/6 activity augments Akt1 Ser473 phosphorylation, in part dependent upon the relative activity of Rictor in the cells.
A Membrane-Tethered Cyclin D1 Is Sufficient for the Induction of Akt1 Transcriptional Induction
In order to determine whether the onset and peak Akt1 activity induced by insulin required the CDK binding residue of cyclin D1, NIH 3T3 cells, which had been either deleted of the cyclin D1 gene or rescued with either the cyclin D1WT cDNA or a cyclin D1KE, were compared (Figures 6A and 6B). Insulin stimulation of Akt1 activity assessed by LS456 was delayed with reduced induction in cyclin D1−/− cells compared with the cyclin D1WT rescued cells. Cyclin D1−/− NIH 3T3 transduced with a cyclin D1KE was defective in the insulin-mediated induction of Akt1 activity, as assessed by LS456 dichromic fluorescence (Figure 6B).
Figure 6. Induction of Immediate Early Gene Akt Signaling via Membrane-Associated Cyclin D1.
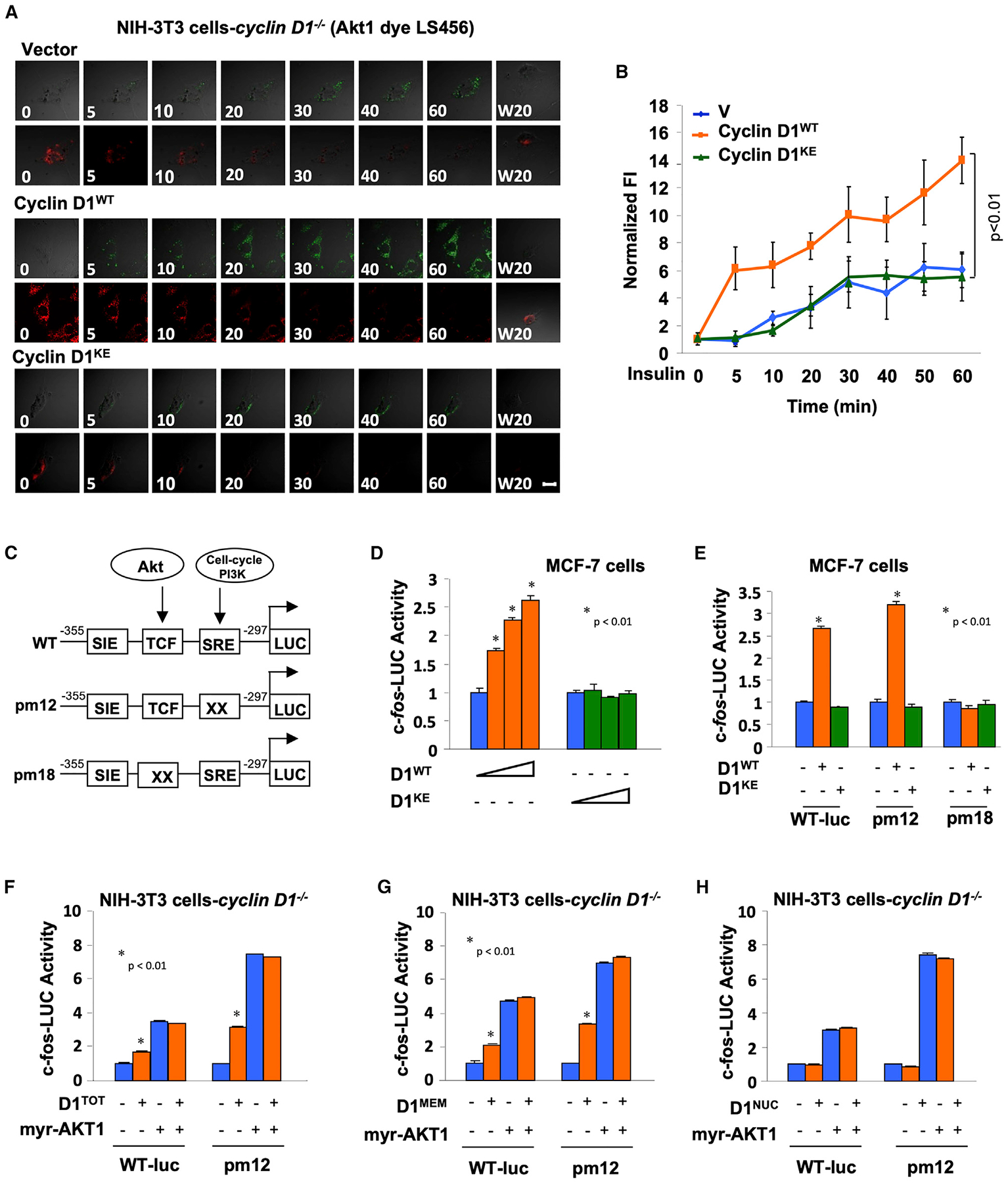
(A) The fluorescent intensity (FI) of LS456 in cyclin D1−/− 3T3 cells transduced with either vector, cyclin D1WT, or cyclin D1KE. The FI of LS456 decreased in the 800-nm channel and increased in the 700-nm channel in response to insulin, indicated as fluorescent images in cells.
(B) Quantitative analysis of the FI in cyclin D1WT versus cyclin D1KE or vector control. Fluorescent images in cells are superimposed on differential interference contrast images. Scale bars, 10 μm. Data are presented as mean ± SEM for n = 8 separate cells.
(C) Schematic representation of c-fos promoter luciferase reporter genes including point mutations of the SRE and TCF site.
(D and E) c-fos-LUC promoter activity in MCF-7 cells (D) transfected with expression vectors encoding either cyclin D1WT or cyclin D1KE or (E) transfected with c-fos-LUC reporter mutants.
(F–H) Cyclin D1−/− 3T3 cells were co-transfected with expression vector encoding c-fos-LUC WT or mutant and activated Akt (myr-Akt1) or (F) cyclin D1, (G) membrane-localized cyclin D1, or (H) nuclear-localized cyclin D1.
The promoter of the immediate early gene c-fos is induced acutely via Akt. The promoter reporter c-fos-LUC is activated by EGF-Akt1 via the ternary complex factor (TCF) site (Figure 6C) (Wang et al., 1998). Co-expression of the cyclin D1WT, but not the CDK-binding defective mutant of cyclin D1, also induced c-fos transcriptional activity assessed using the c-fos promoter linked to a luciferase reporter gene (Figure 6D). A series of point mutations of the TCF site and the serum response element (SRE) of the c-fos promoter were assessed, as prior studies had demonstrated that Akt mediates activity of the c-fos promoter via the TCF site. In MCF7 cells, a point mutation of the TCF site abolished induction of the c-fos promoter, indicating the importance of the TCF site for activation by cyclin D1 (Figure 6E). In contrast, the SRE was identified as the site responding to the serum-induced acute signals that drive cells from Go to G1 via PI3K-Rac/Rho (Wang et al., 1998). Cyclin D1 induced the c-fos-LUC reporter and the SRE mutant reporter (Figure 6F).
We next compared the impact of cyclin D1 when tethered to the cytoplasmic membrane or when localized to the nucleus. An expression vector encoding cyclin D1, cytoplasmic-membrane-tethered cyclin D1, or a constitutively active Akt1 (myr-Akt1) induced the c-fos promoter and the SRE point mutant in cyclin D1−/− cells (Figures 6F and 6G). In contrast, nuclear-targeted cyclin D1 failed to induce c-fos promoter activity (Figure 6H). The myr-Akt1 expression vector induced c-fos-LUC activity ~4- to 6-fold, and membrane-tethered cyclin D1 induced c-fos-LUC activity ~2.5-fold. There was no difference in c-fos-LUC activation by membrane-tethered cyclin D1 versus cyclin D1WT (Figures 6F and 6G). There was no further induction of c-fos-LUC activity by membrane-tethered cyclin D1 in the presence of myr-Akt1. The activity of pm12-LUC is higher than that of WT (c-fos-LUC) in the presence of myr-AKT1. Pm12 has a mutation of the SRE site, which reduces basal level activity maintained by Akt-independent signaling pathways in the cell. pm12-LUC maintains WT activation by Akt1. Therefore, when the activity of pm12 is normalized to 1 for basal level activity, the relative activity of pm12 in the presence of Akt1 is greater. These studies suggest that membrane-tethered cyclin D1, but not nuclear-localized cyclin D1, induces c-fos transcriptional activity.
Cyclin D1 and Akt Promote Common Signaling Pathways in the Mammary Gland In Vivo
In order to determine whether Akt1 and cyclin D1 govern common signaling pathways in vivo, MMTV-ErbB2 transgenic mice were intercrossed with Akt1+/− mice (Figure 7A) and mammary tumor cell lines were derived that were either ErbB2/Akt1+/+ or ErbB2/Akt1−/− (Ju et al., 2007). Microarray analysis of 3 independent lines of each genotype identified expression of 823 genes that were either induced or repressed by endogenous Akt1 (Figure 7B; GEO: GSE138957). The 1,127 genes regulated by cyclin D1 in the mammary gland were derived by transient induction of a cyclin D1 cDNA in the mammary gland under control of the tetracycline-regulated promoter unit (MMTV-RtTA-cyclin D1) (Figures 7A and 7B) (Casimiro et al., 2012). The Akt1-regulated and cyclin D1-regulated genes shared 60 genes when using a <2-fold change in expression as a cutoff (p < 0.05) (Table S1). Approximately 50% of the cyclin D1-regulated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways overlapped with Akt1-regulated pathways (18/38; Figure 7C), including several types of “cancer signaling,” “metabolism in cancer,” “FOXO signaling,” and “meiosis” (Figure 7D) with the relative fold enrichment and gene number given in Figure 7E.
Figure 7. Cyclin D1 and Akt1 Govern Similar Signaling Modules in the Mammary Gland In Vivo.
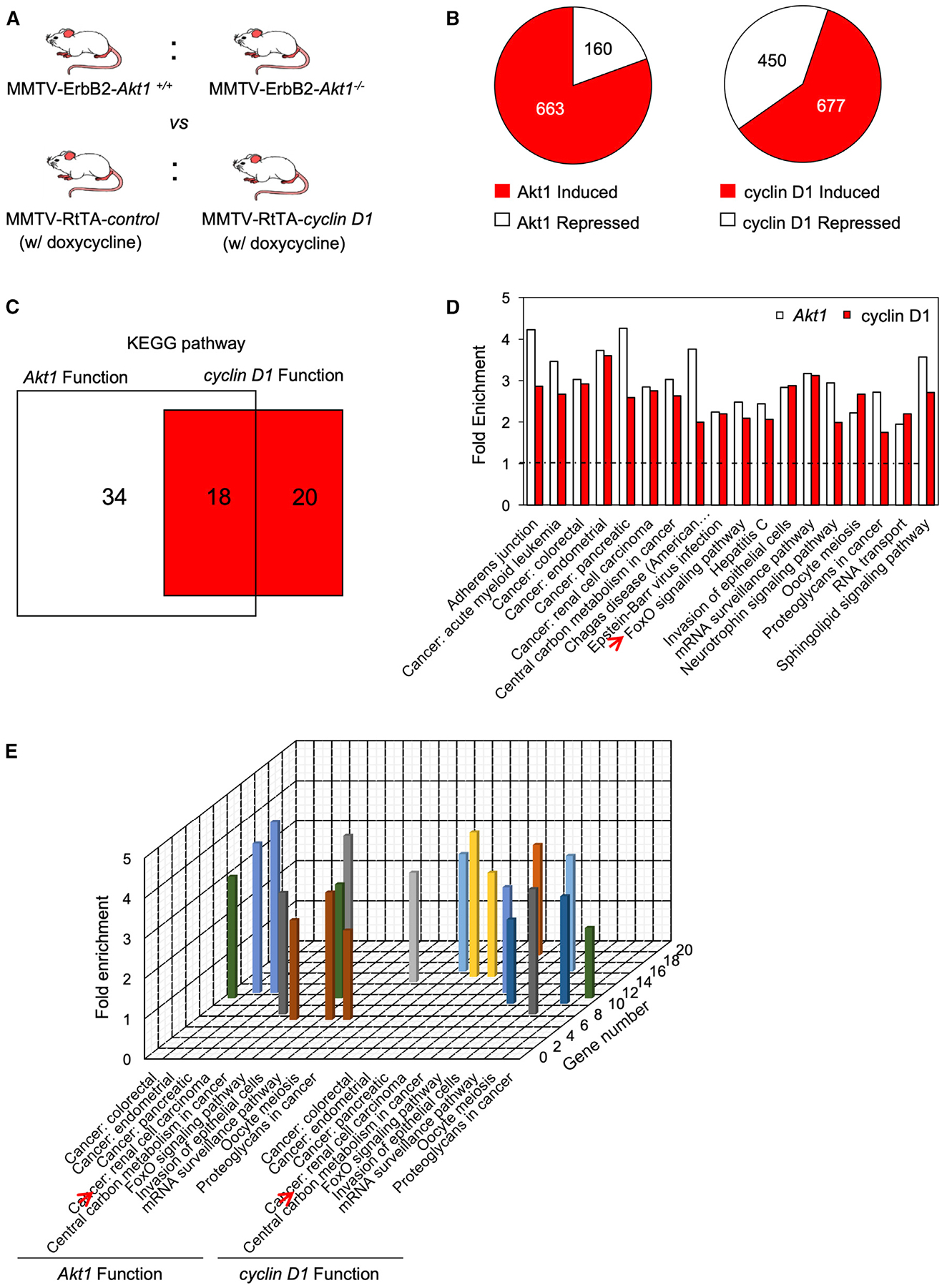
(A) Schematic representation of transgenic mice used for gene expression analysis. MMTV-Akt+/− mice were intercrossed with MMTV-ErbB2 transgenics, and the mammary epithelium was used for a source of mRNA. The cyclin D1-induced state was recapitulated through doxycycline induction of MMTV-RtTA-cyclin D1 transgenics treated for 10 days.
(B) Pie diagrams representing the number of genes either induced or repressed by Akt or cyclin D1.
(C) KEGG pathway analysis used to identify functional pathways regulated by either Akt1 or cyclin D1 and those regulated by both Akt1 and cyclin D1.
(D) The names of pathways identified by KEGG analysis regulated by both cyclin D1 and Akt1 in the mammary gland (migration 1, adherens junction; migration 2, tight junction).
(E) Schematic representation of the relative fold enrichment in gene number of the individual KEGG pathways regulated by both cyclin D1 and Akt1.
Akt1 Gene Expression Signature Correlates with Cyclin D1 Expression in Human Breast Cancer
For luminal A and luminal B breast cancer subtypes, a high Akt1-induced gene expression signature correlated with poor outcome (Figures S9D and S9E). In order to examine the relationship between cyclin D1 expression and gene expression reflecting Akt1 pathway signaling, a superset of 2,254 patient breast cancer gene expression profiles was interrogated (Casimiro et al., 2012). The gene expression signature for Akt1 signaling in the mammary tumors (Figure 7B) was examined for correlation with the abundance of cyclin D1 mRNA in human breast cancer using the Pearson product-moment correlation coefficient. In healthy individuals, the Akt1 gene expression signature and cyclin D1 expression were highly correlated (R = 0.39, p < 10−4) (Figure S10A). Cyclin D1 expression and the Akt1-induced gene expression signature were positively correlated in each genetic subtype of breast cancer, with the highest significance for luminal A (R = 0.34, p < 10−9), luminal B (R = 0.44, p < 10−10), and basal (R = 0.43, p = < 10−17) (Figures S10B–S10F).
DISCUSSION
This study provides evidence for a novel mechanism in which cyclin D1 kinase augments Akt1 activity through phosphorylation of Akt1 at Ser473. In this study, mass spectrometry evidenced that cyclin D1 bound to, and augmented, Akt1 phosphorylation at Ser473. Cyclin D1 restored Akt1 signaling in a kinase-dependent manner, as evidenced by increased phosphorylation of Akt1 and its substrates, shown by western blot, immunofluorescence, and IHC in tissue culture and in transgenic mice. Using cyclin D1−/− cells for reconstitution, we demonstrated that cyclin D1 is sufficient to augment Akt1 signaling, as demonstrated by phosphorylation of Akt1 (Ser473) and downstream substrates of Akt1 (TSC2 Ser939, FOXO1 Ser319, and Bad Ser126). The phosphorylation of Akt1 Ser473 by cyclin D1-associated kinase was observed upon acute cyclin D1 overexpression in mammary epithelium of tissue-specific inducible transgenic mice and was shown to be a function of endogenous cyclin D1 using mammary epithelial cell tissue-specific cyclin D1 knockout. In the mammary epithelium and in tissue culture, cyclin A2 levels were unchanged, evidencing that the mechanism by which cyclin D1 augments Akt1 activity is distinct from cyclin A2-induced phosphorylation at S477/T479 (Liu et al., 2014).
In this study, using PLAs, cyclin D1 co-localized with Akt1 in an extranuclear pool. The interaction between Akt1 and cyclin D1 required the amino-terminal domain of cyclin D1. Restoration of cyclin D1 levels rescued Akt1 phosphorylation at the plasma membrane. Cyclin D1 restored phosphorylation of Akt1 at Ser473. Previous studies identified an association between cyclin D1 and cytoplasmic membrane proteins, including PACSIN 2 (Meng et al., 2011), Filamin A (Zhong et al., 2010), and Paxillin (Fusté et al., 2016); and several components of the cell-cycle control apparatus are located in the cytoplasmic membrane, including cyclin D1 (Nebot-Cegarra and Domenech-Mateu, 1989; Alhaja et al., 2004), p27Kip1, and p16INK4a (Alhaja et al., 2004; Fåhraeus and Lane, 1999). Although the physiological function of cytoplasmic-membrane-associated cell-cycle components was previously not well understood, p16INK4a and CDK6 co-localized in membrane ruffles of spreading cells and functioned upstream of alpha-v-beta3-dependent activation of PKC to regulate matrix-dependent cell migration (Fåhraeus and Lane, 1999). The present study extends our understanding of cyclin D1 function in the extranuclear pool and directly links two hall-marks of cancer: cell-cycle control and Akt activity.
Herein, cyclin D1 enhanced phosphorylation of TSC2 at Ser939. mTOR functions within two distinct complexes (mTORC1 and mTORC2) that couple growth factors to anabolic signaling (Dibble and Cantley, 2015). The TSC complex, which is composed of three subunits—TSC1, TSC2, and TBC1D7—integrates many growth signals that sense the abundance of growth factors glucose, oxygen, and energy (ATP) (Manning and Cantley, 2007; Yuan and Cantley, 2008). TSC2 Ser939 is important in organismal growth, and mutation of the Akt-target sites on TSC2 (corresponding to S939 and T1462) can block Akt-induced growth in flies (Potter et al., 2002). The finding that cyclin D1 augments TSC2 Ser939 phosphorylation, via activation of Akt1, may provide a novel mechanism by which a component of the cell cycle can rapidly cross-couple and thereby fine tune growth sensing and anabolic metabolism.
In this study, cyclin D1 restored the induction of Akt1 signaling to the c-fos promoter. Cyclin D1 was sufficient to reconstitute the Akt1 transcriptional signaling program, and membrane-tethered cyclin D1 was sufficient to reconstitute the 2.5-fold induction of c-fos-LUC seen with the rescue by WT cyclin D1. Importantly, signaling to the c-fos promoter by membrane-tethered cyclin D1 occurred via the TCF site. which is known to be activated by EGF-Akt (Wang et al., 1998). In contrast, the SRF site, which responds to serum-induced signals via Rac/Rho (Wang et al., 1998; Norman et al., 1988), was not involved in activation of c-fos-LUC activity by membrane-tethered cyclin D1. Although the signaling to the SRE and TCF sites is complex, our findings are consistent with a model in which membrane-tethered cyclin D1 augments an Akt signaling pathway. The noncanonical functions of cyclin D1 include the ability to regulate gene transcription via specific target transcription factors. Such activity has been designated as a nuclear transcriptional activity mediated by a pool of cyclin D1 located in the context of chromatin (Bienvenu et al., 2010; Fu et al., 2005; Casimiro et al., 2012, 2014). The present studies extend these findings by demonstrating that, in addition to the nuclear transcriptional activity of cyclin D1 (Pestell, 2013), a membrane-associated cyclin D1 also augments gene transcription via phosphorylation of Akt1 and, thereby, induction of downstream signaling.
Both insulin and EGF signaling were reduced but not abolished in cyclin D1−/− cells, indicating that cyclin D1-dependent and -independent Akt1 signaling occur. As cyclin D1 tethered to the membrane induced Akt1 transcriptional activity, we sought to determine whether increased cyclin D1 expression correlated with increased Akt1 gene signaling in human breast cancer. To test this hypothesis, we generated a surrogate measure of Akt1 genetic activity by creating an Akt1 genetic signature. The Akt1 activity genetic signature was derived by comparing genetic deletion of Akt1 with Akt1 WT mammary gland. In order to interrogate cyclin D1 and Akt1-mediated gene expression, we interrogated a superset of 2,254 breast cancer samples and samples from healthy breast tissue. This study shows that increased cyclin D1 expression correlated with increased Akt1-mediated gene activity. The abundance of cyclin D1 gene expression correlated with the gene signature for Akt1-induced activity in normal breast tissue and in breast cancer samples. Furthermore, cyclin D1 expression and the Akt1-induced gene expression signature were positively correlated in each genetic subtype of breast cancer. Furthermore, genome-wide expression analysis of murine mammary glands from mice with the Akt1 gene deletion and mice with the cyclin D1 gene deletion demonstrated substantial overlap in KEGG functions, including the “cell-cycle” and “insulin signaling” pathways and “mTOR signaling,” consistent with the phenotype of cyclin D1−/− (Sicinski et al., 1995) and Akt1−/− mice (Chen et al., 2001; LaRocca et al., 2011), which share several common features, including defective mammary gland development. Together, these studies are consistent with a model in which cyclin D1 induction correlates with increased Akt1-target gene activity.
This study define a mechanism by which the cell cycle interacts with Akt signaling that is distinct from that in several previous studies. Under cell-cycle conditions, the cyclin A/Cdk2 priming phosphorylation becomes more important and is, therefore, relatively mTORC2/Rictor independent (Liu et al., 2014). We propose that Rictor can function upstream of cyclin D1/CDK4/6 (by priming S477/T479 phosphorylation) and, in certain physiological circumstances, in parallel to mTORC2/Rictor (direct phosphorylation of Ser473). The induction of Akt1 activity by cyclin D1 herein appears to be independent of previously described cell-cycle-related Akt activity, which occurs during the first 15 min to 6 h of the cell cycle (Rosner et al., 2007), preceding the cell-cycle changes of cyclin D1, which occurred at 16 to 24 h, as assessed herein by FACS analysis. The Sin1 component of mTORC2 binds hyper-phosphorylated RB, which inhibits mTORC2-mediated activation of Akt (Zhang et al., 2016). Although our studies did not directly address the role of pRB/Sin1 in regulating Akt1 activity, the mechanisms of the two studies appear to be distinct. Cyclin A2 can induce Akt1 activity (Liu et al., 2014); however, herein, cyclin A2 levels were unchanged by cyclin D1 expression in the mammary gland of mice in which cyclin D1 was either overexpressed or reduced in abundance. Similarly, the cyclin D1 rescue analysis in 3T3 cells conducted herein showed induction of cyclin D1, but not cyclin A, abundance.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard G. Pestell (richard.pestell@bblumberg.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The Microarray data generated during this study are available at The Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo/. The accession code for the cyclin D1-dependent gene signature in mouse mammary gland with transient induction of a cyclin D1 expression under control of the tetracycline-regulated promoter unit is GEO: GSE43216. The accession code for the Akt1-dependent gene signature from the murine ErbB2 mammary tumors is GEO: GSE138957.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MCF-7 and HEK293T cell lines were from the American Type Culture Collection (Manassas, VA). The rictor inducible siRNA MEFs (iRicKO) (Cybulski et al., 2012) were a generous gift from Dr. Michael N. Hall. The cyclin D1+/+ and cyclin D1−/− MEFs and 3T3 cells (Casimiro et al., 2012) and the ErbB2-Akt1+/+ or ErbB2-Akt1−/− (Ju et al., 2007) mammary tumor cell lines were generated from transgenic mouse tumors developed by this laboratory as described previously (Albanese et al., 1999). MCF-7 and HEK293T were recently authenticated by ATCC. The MEFs generated by this laboratory were authenticated by IDEXX Bioresearch. All cell lines were tested for mycoplasma contamination using the ATCC Universal Mycoplasma Detection Kit. All cell lines were cultured at 37°C in 5% CO2 with DMEM medium supplemented with 10% fetal bovine serum, 100 unit/mL penicillin and 100 mg/mL streptomycin.
Animal models
The MMTV-RtTA-cyclin D1, MMTV-RtTA-cyclin D1 KE and MMTV-ErbB2-Akt1−/− transgenic mice were previously described (Casimiro et al., 2012). Cyclin D1fl/fl (Choi et al., 2012) and ROSA26-ER-Cre mice (Ventura et al., 2007) were intercrossed to form Cyclin D1fl/fl-ROSA26-ER-Cre bi-transgenic mice and treated with Tamoxifen as previously described (Choi et al., 2012). Comparison was made between the cyclin D1fl/fl-ROSA26-ER-Cre and cyclin D1 wt. ROSA26-ER-Cre (Ventura et al., 2007). All of the mice used were in FVB background, female and 10–12 weeks old. Experimental procedures with transgenic mice were approved by the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University.
METHOD DETAILS
Plasmids
pMSCV-IRES-GFP encoding 3xFlag tagged wild-type cyclin D1 or its series of mutants were generated by inserting the coding region of the human cyclin D1 cDNA or its mutants into the MSCV-IRES-GFP vector at the EcoRI site upstream of the IRES driving expression of GFP (Li et al., 2006b). The inserts were PCR amplified from p3xFLAG-CMV-10 (Sigma) encoding 3xFlag tagged wild-type cyclin D1 or its mutants (Wang et al., 2003). pECFP-mem-cyclin D1 was generated by inserting the cyclin D1 cDNA, which was amplified by PCR from pMSCV-cyclin D1-IRES-GFP, into Nhe1 and Age1 site of the pECFP-mem vector (Clonetech), which encodes a fusion protein consisting of the N-terminal 20 amino acids of neuromodulin, also called GAP-43, and a cyan fluorescent variant of the enhanced green fluorescent protein (ECFP) (Hynes et al., 2004). The neuromodulin fragment contains a signal for posttranslational palmitoylation of cysteines 3 and 4 that targets ECFP to cellular membranes. Expression of ECFP-Mem in mammalian cells results in strong labeling of the plasma membrane (Piljic and Schultz, 2006) and had been used to target proteins including ERα to the plasma membrane (Razandi et al., 2003). Cherry-lacR-NLS-CD1NUC which encodes a nuclear localized form of cyclin D1 was generated by inserting the cyclin D1 cDNA at the COOH-terminus of the Cherry-lacR-NLS vector (Soutoglou and Misteli, 2008) into the KpnI/XmaI sites. The primers used were the following: cyclin D1 forward: cggggtaccgaacaccagctcctgtgct; cyclin D1a reverse: tccccccgggtca gatgtccacgtcccgca; cyclin D1b reverse: tccccccgggtcacccttgggggccttg (Li et al., 2010). The Akt1 retroviral expression plasmids encoding constitutively active Akt1 (myr-Akt1) linked through an internal ribosomal entry site to a GFP fusion protein (pBABE-myrAkt1-IRES-GFP) was generous gift from Dr. Nissim Hey at the University of Illinois at Chicago (Eves et al., 1998). pCMV5-HA-Akt1 was from Dr. Alessi at the University of Dundee, UK (Alessi et al., 1996). pGEX-GST-Akt1 and pGEX-GST-Akt1(S473A) were from Dr. Wenyi Wei from Harvard University at Boston. The c-fos-LUC, serum response element (SRE), or ternary complex factor (TCF) mutants (pm12 and pm18) were gift from Dr. Ron Prywes at Columbia University, New York (Wang et al., 1998). pRSV-β-Gal was a gift from Dr. Frederick Stanley at NYU Langone Medical Center (now available at Addgene, plasmid # 24058). All plasmid DNA constructs were verified by sequencing.
Antibodies
The antibodies used in western blot analysis were cyclin D1 (sc-20044, Santa Cruz), b-actin (sc-47778, Santa Cruz), Akt1 (sc-5298, Santa Cruz), pAkt1 Ser473 (#9018, CST), pAkt1/2/3 Ser473 (sc-7985-R, Santa Cruz), TSC2 (sc-893, Santa Cruz), pTSC2 Ser939 (ab59269, Abcam), FKHR (sc-11350, Santa Cruz), pFKHR Ser319 (sc-101682, Santa Cruz), cyclin A2 (ab16726, Abcam), Bad (sc-8044, Santa Cruz), pBad Ser136 (ab28824, Abcam), Rictor (SC#271081, H11) and phosphorylated RB (S780) (Cell Signaling). Mouse anti-FLAG (M2), mouse anti-vinculin (hVIN-1) antibodies were from Sigma (St. Louis, MO). The antibody used for Immunoprecipitation was HA (sc-805, Santa Cruz). The antibodies used for immunohistochemistry and immunofluorescence were cyclin D1 (sc-20044, Santa Cruz), for Akt Ser473 phosphorylation (pAkt1/2/3 Ser473) (SC-7985-R, Santa Cruz), for pAkt1 Ser473 (#9018, CST), TSC2 (sc-893, Santa Cruz), pTSC2 Ser939 (ab59269, Abcam), pFKHR Ser319 (sc-101682, Santa Cruz), pBad Ser136 (ab28824, Abcam), cyclin A2 (ab16726, Abcam), paxillin (05–417, Millipore-Sigma), Rictor (H278, sc-99004), tyrosine phosphorylated paxillin (44–722G, Thermo Fisher), PACSIN2 (10518–2-AP, Proteintech).
Other reagents
Purified Akt1 used in the cyclin D1 immune-precipitation kinase assays was from OriGene, (AKT1, CAT#TP320257) or GST fusion proteins for pRB, which contains 2 CDK phosphorylation sites (Wang et al., 2006), Akt1 or Akt1-S473A, were generated in this laboratory as previously described (Wang et al., 2006). Insulin was from Millipore-Sigma and epidermal growth factor (EGF) was from R&D.
Immunohistochemistry (IHC)
IHC for tissue was conducted at the Translational Research/Pathology Core Facility of Thomas Jefferson University using a DAKO Autostainer Plus equipment with an enzyme labeled biotin–streptavidin system. The IHC images were acquired with a 10x or 40x objective and semi-quantified with Fiji software. The procedure for semi-quantification involved first opening the image software, then in the heading dropdown menu selecting the option of “image,” then clicking on the menu item “color” then “color deconvolution” with selection of “H&E, DAB.” This procedure splits the original image into H&E and DAB-images. When selected within the DAB-image modality, the threshold was established to cover the target signals and this threshold value was then applied to all samples being compared. Subsequently, under the heading dropdown menu entitled “analyze” was activated, then the menu item entitled “setup measurement” and then “integrated intensity” were selected. Within the threshold-image modality, the heading “measure” in the heading drop down menu and then the heading “analyze” were chosen to obtain the results of integrated intensity. In the H-images setting, the threshold to cover the target nuclear staining was established, then the heading dropdown menu titled “process” was selected followed by activating the operation entitled “binary,” then “fill holes” and then “watershed.” The heading dropdown menu item titled “analyze,” was then used, activating the item titled “analyze particles” and then within the pop-out menu, the target size was defined, “show outlines” and “summary” were selected, and then clicking “ok” to obtain the total cell number. The relative intensity was calculated by dividing the integrated intensity with the total cell number. In order to obtain the ratio of positive cells, the DAB-image was processed as an H-image above to obtain the total number of positive cells. The ratio was calculated by dividing the total positive cell number with the total cell number. Double check if the outline images of the nucleoli matched the staining shown in the original images and if not, therefore it was necessary to count the cell number manually.
Immunofluorescence (IF)
IF staining and confocal microscopy of cultured cells was conducted as described previously (Li et al., 2010, Jiao et al., 2008). The cells that were grown in 4-well-chamber slides (Lab-Tek II, Fisher) were fixed with 10% paraformaldehyde in PBS for 20 min at room temperature. The chambers were removed after fixation. The slides were rinsed with PBS and permeated with 0.05% NP-40 in PBS. The primary antibodies were diluted in PBS containing 5% goat sera, 5% FBS and 0.05% NP-40 and incubated with samples for 1 hour. The secondary antibodies used were Alexa Fluor 488, 568 or 633–conjugated F(ab’)2 fragment of goat anti-mouse immunoglobulin G (IgG; Molecular Probes, 1/500). The samples were visualized on a Zeiss LSM 510 META Confocal Microscope with a 63x objective. The images were processed with Fiji software.
Western blot
Western blot analyses were conducted as described previously (Li et al., 2006b). Whole-cell lysates (60 μg) were electrophoresed in a 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) in a running buffer (25 mM Tris base, 190 mM glycine, 0.1% SDS, pH8.3) and semi-dry transferred to nitrocellulose membrane (Amersham Corp.) in a transfer buffer (48 mM Tris, 39 mM glycine, 0.037% SDS, pH 8.3, 20% methanol). After semi-dry transfer, the membrane was stained with ATX Ponceau S Red staining solution (09189, Fluka) for blotting efficiency. The blotting membrane was blocked for 1 h at room temperature in PBST buffer (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4 and 0.05% Twenn-20) supplemented with 5% (w/v) dry milk or 2% Bovine Serum Albumin (BSA). Following an overnight incubation with primary antibody in PBST buffer containing 0.8% Fish Gelatin (G-7765, Sigma). The membrane was washed 10 min, 3 times with PBST and then incubated with horseradish peroxidase conjugated second antibody (1/2000) for 1 hour and washed again. The target band were visualized by the enhanced chemiluminescence system.
Immunoprecipitation/kinase assays
The protocol is described previously (Ashton et al., 1999) with modification. HEK293T cells transfected with an expression vector encoding 3xFLAG tagged cyclin D1, were grown in 10-cm plates at 37°C in 5% CO2 with DMEM medium supplemented with 10% fetal bovine serum, 100 unit/mL penicillin and 100 mg/mL streptomycin. The cells were washed with 10 mL PBS, and one ml of cell lysis buffer (50 mM HEPES pH7.2, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1% Tween 20, 0.1 mM PMSF (Sigma), 2.5 μg/mL leupeptin (Sigma), 100 μM sodium orthovanadate) was added. Cells were incubated on ice for 20 min, then scraped, transferred to a 1.5 mL Eppendorf tube and then mixed thoroughly by pipetman tips. The cell lysates were cleared by centrifugation at 10000 rpm for five minutes at 4°C. The supernatant was collected and the protein concentration was measured. 100 μg of soluble proteins were incubated overnight at 4°C in a rocker with 2 μg of anti-Flag antibody (M2, F1804, Sigma) previously bound to 20 μL Protein G agarose beads, which were prewashed with cold lysate buffer without inhibitors. The beads were spun down, the supernatant was carefully removed with a pipetman and then washed three times with cold lysis buffer and two times cold assay buffer (50 mM HEPES buffer, pH7.2, 10 mM MgCl2, 5 mM MnCl2 and 1 mM DTT). The beads were incubated in 40 μL of assay buffer supplemented with 10 μM ATP, 5 μCi γ32P-ATP and 2 μg of purified Akt1, Akt1(S473A) or GST-Rb for 30 minutes at 30°C. The samples were denatured by boiling in SDS sample buffer and separated on SDS-PAGE gels. The gel was subsequently stained with Coomassie blue, dried, and exposed to radiographic film.
Identification of Akt1 phosphorylation sites by mass spectrometry
The mass spectrometry was conducted as previously described (Wang et al., 2006). HEK293T cells were transfected with expression vectors encoding HA-Akt1 and either Vector or FLAG N-amino terminal target cyclin D1WT. HA-IP was conducted of the cells and mass spectrometry conducted, comparing the relative abundance in post-translational modification between vector control and cyclin D1 transfected cells. The Akt1 protein was isolated by HA immunoprecipitation and separated by gel electrophoresis. The gels were stained with Coomassie G250 and the bands were excised and digested with trypsin. Phosphorylated peptides were isolated using affinity purification using TiO2 Nu-tips from Glygen. Briefly, the extracted peptides were loaded on the tip in a buffer containing 300 mg/ml DHB in 80% Acetonitrile, 0.1% TFA, washed once with the loading buffer and once with 80% Acetonitrile, 0.1% TFA and eluted in 0.4M Ammonium Hydroxide. Peptides were immediately acidified with Formic Acid and were analyzed by ESI-MS/MS on a on a Q Exactive Plus mass spectrometer. MS/MS spectra were searched against a custom UniProt human database plus the HA-Akt1 sequence using MaxQuant 1.5.2.8 with Carbamidomethyl as a fixed modification and Oxidation (M), Phospho (ST), Phospho (Y) as variable modifications. False discovery rates for protein, peptide and PTM site are set at 1%.
Proximity ligation assay
The PLA was performed using Duolink reagents (Invitrogen) according to the manufacturer’s instructions as previously described (Ivanschitz et al., 2015). Formalin-fixed 3T3 Ccnd1 −/− rescue wt (WT), 3T3 Ccnd1 -/ -rescue GFP (GFP), 3T3 Ccnd1 -/ -rescue KE (KE), 3T3 Ccnd1 -/ -rescue C6 (C6), 3T3 Ccnd1 -/ -rescue ΔE (ΔE) and 3T3 Ccnd1 -/ -rescue 286 (286) cells were incubated with blocking solution (Sigma, DUO82007) for 30 minutes at 37°C and then incubated overnight at 4°C with anti-Akt1 (Santa Cruz biotechnology 1:50, sc-5298) and anti-Cyclin D1 (Santa Cruz biotechnology 1:50, sc-753) in antibody diluent (Sigma, DUO82008). For positive control, 3T3ccnd1−/− rescue WT and 3T3ccnd1−/−rescue GFP cells were incubated with anti-Akt1 (Santa Cruz biotechnology 1:50, sc-5298) and anti-phospho-Akt1 S473 (Cell signaling, 1:50, cat # 9018). For negative control, 3T3 ccnd1−/− rescue WT and 3T3 ccnd1−/−rescue GFP cells were incubated with IgG. After incubation with primary antibody, 3T3 ccnd1 −/− rescue wt and 3T3 ccnd1 −/−rescue GFP cells were incubated with secondary antibody conjugated with oligonucleotides (PLA probe PLUS and PLA probe MINUS) for 60 minutes at 37°C. After this step, the samples were incubated with ligation solution for 30 minutes at 37°C and then incubated with amplification solution for 100 minutes at 37°C. The samples were analyzed using a fluorescence microscope (Zeiss Axiovert 200M).
Generating the model of the cyclin D1, CDK4/CDK6, Akt1 complex
The murine cyclin D1 and CDK4/6 3D models were generated using the software Protein Homology/analogY Recognition Engine V 2.0 (Phyre 2), which produces a model of the protein of interest based on sequence alignment to known structures (Kelley et al., 2015). The 3D structure of murine cyclin D1 was modeled of the X-ray crystal structure of human CD1 (PDB ID: 2W9Z), which has 95% sequence identity to the mouse gene. The murine CDK4/CDK6 structure was modeled of the crystal structure of the human CDK4/CDK6 (PDB ID: 1BLX) to which the murine gene has 69% sequence identity. The presence of X-ray crystal structures with significantly high sequence identity with the murine genes has allowed PHYRE to generate these models with 100% confidence. Subsequently the murine cyclin D1-CDK4/6 assembly was generated using the crystal structure of the human cyclin D1-CDK4/CDK6 complex (PDB ID: 2W9Z). In the absence of a structure of CDK4/CDK6 bound to the Akt1 C-terminal activation peptide, we used the crystal structures of CDK2-cyclin A (PDB ID: 1QMZ) and Akt1-GDC-0068 (PDB ID: 4EKK) protein peptide complexes to model the C-terminal Akt1 peptide at the active site of the murine CDK4/CDK6 model. The model was further refined by applying geometry minimization in Phenix (Afonine et al., 2010). The figures were generated in Pymol (Schrodinger, 2015).
Live cell Akt activity monitoring
Cyclin D1 wt and cyclin D1−/− 3T3 cells and MEFs (Albanese et al., 1999) were maintained at 37°C and 5% CO2 in DMEM medium supplemented with 10% fetal bovine serum, 100 unit/mL penicillin, and 100 mg/mL streptomycin. The cells (105 cell/well) were cultured in glass bottom dishes overnight and transfected with 5 mM of each LS456, using GeneJuice transfection reagent (Novagen, Madison, WI) in the dish for 18 h at 37°C, according to the manufacturer’s instructions. The transfected cells were treated with 150 nM insulin in Tris-buffered saline and imaged at 30°C using an FV1000 confocal microscope with UPLanApo/IR 60X/1.20W objective lens (Olympus, Center Valley, PA). The treated cells were incubated with 150 nM of insulin. Imaging was conducted at different time points. The mean fluorescence intensity (n58) (Ex/Em = 633/670–730 nm; Ex/Em = 785/805–830 nm) in the dishes was determined with FV1000 software. All of the fluorescence intensity changes at each time point were normalized to the positive control (0 min). The image of LS542 (Figures 5 and 6) were acquired with LI-COR Pearl Imager (Lincoln, NE) at Ex/Em of 685/705 nm and 785/810 nm channels. Live cell imaging studies were conducted at 30°C, which is the default room temperature for the imaging platform of the microscope. All images within the same time series were recorded from the same area of the slides. However, as the live cells moved during the imaging sessions, eight representative cells were used to quantify the fluorescence using Olympus FV1000 software. The data shown in Figures 5 and 6B were obtained from a duplicate of 4 different experiments.
Luciferase reporter assays (LUC assay)
MCF-7 or cyclin D1−/− 3T3 cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin. One day before transfection, the cells were seeded in 24-well plates. MCF-7 cells were co-transfected with 200 ng c-fos wt or mutant promoter reporters, 0–400 ng wild-type cyclin D1 or cyclin D1 mutant expression plasmids, and 200 ng transfection efficiency reporter plasmid pRSV-β-gal using Lipofectamine 2000 (Invitrogen) following the manufacturer’s manual. 48 hours post transfection, LUC assays were performed as previously described (Li et al., 2006b). Briefly, the cells were extracted by 100 μL extract buffer (X-buffer, GME buffer (25 mM Gly-gly, 15 mM MgSO4, 4 mM EGTA) supplement with 1 mM DTT, 1% Triton X-100). The cell lysates were divided to two parts: 90 μL was transferred to the assay tube for LUC assay and 10 μL was transferred to 96-well plate for β-gal assay. For LUC assay, 300 μL ATP-mix (GME buffer supplement with 1/6 (v/v) of 100 mM potassium phosphate buffer (pH 7.4), 1 mM DTT and 1 mM ATP) was added in LUC assay tube and the luciferase activity were read by an automatic tube luminometer (Autolumat LB953, EG&G Berthold) with LUC-buffer (GME buffer supplemented with 1 mM DTT and 0.2 mM Luciferin). For β-gal assay, 100 μL PM2 buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MCl2 and 50 mM Mercap-toethanol) and 20 μL ONPG solution were added to each well, which contained 10 μL of the cell lysates, of 96-well plate. Incubation was conducted at 37°C for 1 hour and the optical density read at 420 nm on an ELISA reader. Normalized luciferase activity was calculated by luciferase activity dividing with b-galactosidase reporter activity. The -fold effect was determined by comparison to the empty expression vector cassette.
Comparison of gene expression from Akt1 deficient or cyclin D1 overexpressing mice
Comparison was made of Akt activity from Akt WT or Akt1-deficient (Akt1−/−) cells. The Akt1−/− and Akt1+/+ (WT) cells used in this study were cultured and transfected as previously described (Ju et al., 2007). Briefly, mammary tumor epithelial cells derived from MMTV-ErbB2 transgenic mice tumors were cultured in F-12 medium (Sigma, St. Louis, MO) with EGF (10 ng/mL), hydrocortisone (1 mg/mL), penicillin (100 units/mL), streptomycin (100 mg/mL), and gentamycin (50 mg/mL) and supplemented with 10% FBS. Total RNA isolated from either ErbB2-Akt1+/+ or ErbB2-Akt1−/− mammary tumor cell lines (Ju et al., 2007), or mammary epithelium from doxycycline inducible cyclin D1 transgenic mice (Casimiro et al., 2012, 2015) was treated with Trizol (Sakamaki et al., 2006).
RNA samples were treated with RQ1 DNase I (Promega Inc, Madison, WI) to remove contaminating DNA from RNA preparations followed by re-purification using RNeasy Mini Kit (QIAGEN, Valencia, CA). DNA-free RNA was subjected to reverse transcription reactions, performed using SuperScript III reverse transcriptase kit (Invitrogen, Carlsbad, CA). Affymetrix Expression Console 1.1 or the R statistic console with the limma package was used to compute Robust Multichip Average (RMA) expression values for the Mouse Gene 1.0 ST microarrays and Mouse 430A 2.0 microarrays. The core set of probe-set clusters was used with annotation version na30. The cyclin D1 dataset was imported into MATLAB version R2010b (The Mathworks), and 1-way ANOVA was used to evaluate the significance of differential expression between biological conditions. The -fold change cutoff was > 1.25 and the p value was < 0.05. Akt1 microarray analysis was performed using GeneSpring. Arrays were normalized using robust multi-array analysis, the fold change cutoff of 2 and p value of < 0.05 were applied as a statistical criterion for differentially expressed genes.
Microarray Dataset
A breast cancer microarray dataset that was previously compiled from the public repositories Gene Expression Omnibus (Casimiro et al., 2012) (https://www.ncbi.nlm.nih.gov/geo/, GSE1456, GSE6532, GSE7390, GSE9195, GSE12093) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) was used to evaluate Akt pathway and CCND1 transcript level expression in the context of clinical samples (Casimiro et al., 2012) and displayed using Zebra Plots (Chaves et al., 2012).
A breast cancer microarray dataset used to evaluate the Akt1 pathway and CCND1 transcript level expression in the context of clinical samples was available from the public repositories Gene Expression Omnibus (Casimiro et al., 2012), and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/, E-TABM-158).
Statistical Analysis
Unless specified, 3 to 4 independent experiments were included in each statistical analysis. All statistical analysis unless otherwise specified was conducted with the Student t test in Microsoft Excel. Data was expressed as mean ± SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| cyclin D1 | Santa Cruz | sc-20044; RRID: AB_627346 |
| Akt1 | Santa Cruz | sc-5298; RRID: AB_626658 |
| pAkt1 Ser473 | Cell Signaling | #9018; RRID: AB_2629283 |
| pAkt1/2/3 Ser473 | Santa Cruz | sc-7985-R; RRID: AB_667741 |
| TSC2 | Santa Cruz | sc-893; RRID: AB_632569 |
| pTSC2 Ser939 | Abcam | ab59269; RRID: AB_2210004 |
| FKHR | Santa Cruz | sc-11350; RRID: AB_640607 |
| pFKHR Ser319 | Santa Cruz | sc-101682; RRID: AB_2294254 |
| Bad | Santa Cruz | sc-8044; RRID: AB_626717 |
| pBad Ser136 | Abcam | ab28824; RRID: AB_725616 |
| Rictor | Santa Cruz | sc-271081; RRID: AB_10611167 |
| Rictor | Santa Cruz | sc-99004; RRID: AB_2269610 |
| pRb Ser780 | Cell signaling | #9307; RRID: AB_330015 |
| Flag (M2) | Millipore-Sigma | F1804; RRID: AB_262044 |
| HA | Santa Cruz | sc-805; RRID: AB_631618 |
| cyclin A2 | Abcam | ab16726; RRID: AB_302478 |
| PACSIN 2 | Proteintech | 10518–2-AP; RRID: AB_2161854 |
| Paxillin (5H11) | Millipore-Sigma (Upstate) | 05–417; RRID: AB_309724 |
| pPaxillin Tyr118 | Thermo Fisher | 44–722G; RRID: AB_2533733 |
| β-actin | Santa Cruz | sc-47778; RRID: AB_2714189 |
| Bacterial and Virus Strains | ||
| JM109 Competent Cells | Promega | L2001 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AKT1 (NM_005163) Human Recombinant Protein | Origene | TP320257 |
| Insulin Human Recombinant Protein | Millipore-Sigma | I2643 |
| Recombinant human EGF protein, CF | R&G | 236-EG |
| Critical Commercial Assays | ||
| Duolink In Situ Red Starter Kit Mouse/Rabbit | Millipore-Sigma | DUO92101 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| SuperScript™ III First-Strand Synthesis System | Thermo Fisher | 18080051 |
| Deposited Data | ||
| cyclin D1-dependent gene signature in mouse mammary gland | GEO database | GEO: GSE43216 |
| Akt1-dependent gene signature from the murine ErbB2 mammary tumor | GEO database | GEO: GSE138957 |
| Experimental Models: Cell Lines | ||
| MCF-7 | ATCC | HTB-22 |
| HEK293T | ATCC | CRL-3216 |
| Cyclin D1 wt and ko MEFs/3T3s | Casimiro et al., 2012 | N/A |
| ErbB2-Akt wt and ko mammary tumor cells | Ju et al., 2007 | N/A |
| Inducible rictor shRNA MEFs (iRicKO) | Cybulski et al., 2012 | N/A |
| Experimental Models: Organisms/Strains | ||
| MMTV-Rtta-control, cyclin D1-wt and cyclin D1-KE mice/FVB | Casimiro et al., 2012, 2015 | N/A |
| MMTV-ErbB2-Akt1 wt and ko mice/FVB | Ju et al., 2007 | N/A |
| Flox-cyclin D1/Rosa26-CreERT2 mice/FVB | Choi et al., 2012 | N/A |
| Recombinant DNA | ||
| p3xFlag-CMV-10 encoding cyclin D1 and its mutants | Wang et al., 2003 | N/A |
| pMSCV -IRES-GFP encoding cyclin D1 and its mutants | Li et al., 2006b | N/A |
| ECFP-Mem-Cyclin D1 plasmid | This paper | N/A |
| Cherry-lacR-NLS-CD1NUC Plasmid | Li et al., 2010 | N/A |
| pBABE-myrAkt1-IRES-GFP | Eves et al., 1998 | N/A |
| pCMV5-HA-Akt1 | Alessi et al., 1996 | N/A |
| pGEX-GST-Akt1 and pGEX-GST-Akt1(S473A) | Liu et al., 2014 | N/A |
| c-fos-LUC, SRE orTCF mutant (pm12 or pm18) | Wang et al., 1998 | N/A |
| pRSV-β-Gal | Addgene | #24058 |
| Software and Algorithms | ||
| Fiji (ImageJ) | ImageJ | https://imagej.net/Fiji |
Highlights.
Cyclin D1 enhances Akt1 activities both in vitro and in vivo
Cyclin D1 bound to and phosphorylated Akt1 at Ser473
Cyclin D1 was required for growth factor induced Akt1 activity
Correlation of cyclin D1 and Akt1 gene signature with breast cancer patient outcomes
ACKNOWLEDGMENTS
This work was supported in part by NIH R01CA132115 and the Breast Cancer Research Program (Breakthrough Award W81XWH1810605) (to R.G.P.), NIH R01CA201312-01 (to E.S.), and a Wistar Cancer Center support grant (P30CA10815, NIH) (to E.S. and R.G.P.). This work was partially funded by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship (to G.D.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108151.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afonine PV, Mustyakimov M, Grosse-Kunstleve RW, Moriarty NW, Langan P, and Adams PD (2010). Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 66, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese C, D’Amico M, Reutens AT, Fu M, Watanabe G, Lee RJ, Kitsis RN, Henglein B, Avantaggiati M, Somasundaram K, et al. (1999). Activation of the cyclin D1 gene by the E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. J. Biol. Chem 274, 34186–34195. [DOI] [PubMed] [Google Scholar]
- Albanese C, Wu K, D’Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze’ev A, et al. (2003). IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol. Biol. Cell 14, 585–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, and Hemmings BA (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alhaja E, Adan J, Pagan R, Mitjans F, Cascalló M, Rodríguez M, Noé V, Ciudad CJ, Mazo A, Vilaró S, and Piulats J (2004). Anti-migratory and anti-angiogenic effect of p16: a novel localization at membrane ruffles and lamellipodia in endothelial cells. Angiogenesis 7, 323–333. [DOI] [PubMed] [Google Scholar]
- Alt JR, Cleveland JL, Hannink M, and Diehl JA (2000). Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev 14, 3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton AW, Watanabe G, Albanese C, Harrington EO, Ware JA, and Pestell RG (1999). Protein kinase Cdelta inhibition of S-phase transition in capillary endothelial cells involves the cyclin-dependent kinase inhibitor p27(Kip1). J. Biol. Chem 274, 20805–20811. [DOI] [PubMed] [Google Scholar]
- Baker GL, Landis MW, and Hinds PW (2005). Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 4, 330–338. [PubMed] [Google Scholar]
- Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, et al. (2010). Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature 463, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozulic L, Surucu B, Hynx D, and Hemmings BA (2008). PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 30, 203–213. [DOI] [PubMed] [Google Scholar]
- Casimiro MC, Crosariol M, Loro E, Ertel A, Yu Z, Dampier W, Saria EA, Papanikolaou A, Stanek TJ, Li Z, et al. (2012). ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. J. Clin. Invest 122, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro MC, Velasco-Velázquez M, Aguirre-Alvarado C, and Pestell RG (2014). Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin. Investig. Drugs 23, 295–304. [DOI] [PubMed] [Google Scholar]
- Casimiro MC, Di Sante G, Crosariol M, Loro E, Dampier W, Ertel A, Yu Z, Saria EA, Papanikolaou A, Li Z, et al. (2015). Kinase-independent role of cyclin D1 in chromosomal instability and mammary tumorigenesis. Oncotarget 6, 8525–8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves KCB, Peron JPS, Chammas R, Turaça LT, Pesquero JB, Braga MS, Foguer K, Schor N, and Bellini MH (2012). Endostatin gene therapy stimulates upregulation of ICAM-1 and VCAM-1 in a metastatic renal cell carcinoma model. Cancer Gene Ther 19, 558–565. [DOI] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al. (2001). Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 15, 2203–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, and Sicinski P (2012). The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski N, Zinzalla V, and Hall MN (2012). Inducible raptor and rictor knockout mouse embryonic fibroblasts. Methods Mol. Biol 821, 267–278. [DOI] [PubMed] [Google Scholar]
- Dan HC, Antonia RJ, and Baldwin AS (2016). PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget 7, 21064–21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, and Greenberg ME (1999). Cellular survival: a play in three Akts. Genes Dev 13, 2905–2927. [DOI] [PubMed] [Google Scholar]
- Di Sante G, Di Rocco A, Pupo C, Casimiro MC, and Pestell RG (2017). Hormone-induced DNA damage response and repair mediated by cyclin D1 in breast and prostate cancer. Oncotarget 8, 81803–81812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sante G, Pagé J, Jiao X, Nawab O, Cristofanilli M, Skordalakes E, and Pestell RG (2019). Recent advances with cyclin-dependent kinase inhibitors: therapeutic agents for breast cancer and their role in immuno-oncology. Expert Rev. Anticancer Ther 19, 569–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, and Cantley LC (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eves EM, Xiong W, Bellacosa A, Kennedy SG, Tsichlis PN, Rosner MR, and Hay N (1998). Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol. Cell. Biol 18, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fåhraeus R, and Lane DP (1999). The p16(INK4a) tumour suppressor protein inhibits alphavbeta3 integrin-mediated cell spreading on vitronectin by blocking PKC-dependent localization of alphavbeta3 to focal contacts. EMBO J 18, 2106–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LWS, Zhang J, Ye J, Ward PS, and Thompson CB (2013). SnapShot: cancer metabolism pathways. Cell Metab 17, 466–466.e2. [DOI] [PubMed] [Google Scholar]
- Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X, Li Z, Yao TP, and Pestell RG (2005). Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J. Biol. Chem 280, 16934–16941. [DOI] [PubMed] [Google Scholar]
- Fusté NP, Fernández-Hernández R, Cemeli T, Mirantes C, Pedraza N, Rafel M, Torres-Rosell J, Colomina N, Ferrezuelo F, Dolcet X, and Garí E (2016). Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat. Commun 7, 11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsdottir SM, Schallmeiner E, Fredriksson S, Gullberg M, Söder-berg O, Jarvius M, Jarvius J, Howell M, and Landegren U (2005). Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem 345, 2–9. [DOI] [PubMed] [Google Scholar]
- Halaby MJ, Hibma JC, He J, and Yang DQ (2008). ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell. Signal 20, 1555–1563. [DOI] [PubMed] [Google Scholar]
- Hay N (2005). The Akt-mTOR tango and its relevance to cancer. Cancer Cell 8, 179–183. [DOI] [PubMed] [Google Scholar]
- Hers I, Vincent EE, and Tavaré JM (2011). Akt signalling in health and disease. Cell. Signal 23, 1515–1527. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, and Cohen SM (2008). TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer 8, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko RC, and Mueckler M (2005). mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem 280, 40406–40416. [DOI] [PubMed] [Google Scholar]
- Hynes TR, Mervine SM, Yost EA, Sabo JL, and Berlot CH (2004). Live cell imaging of Gs and the beta2-adrenergic receptor demonstrates that both alphas and beta1gamma7 internalize upon stimulation and exhibit similar trafficking patterns that differ from that of the beta2-adrenergic receptor. J. Biol. Chem 279, 44101–44112. [DOI] [PubMed] [Google Scholar]
- Ivanschitz L, Takahashi Y, Jollivet F, Ayrault O, Le Bras M, and de Thé H (2015). PML IV/ARF interaction enhances p53 SUMO-1 conjugation, activation, and senescence. Proc. Natl. Acad. Sci. USA 112, 14278–14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Katiyar S, Liu M, Mueller SC, Lisanti MP, Li A, Pestell TG, Wu K, Ju X, Li Z, et al. (2008). Disruption of c-Jun reduces cellular migration and invasion through inhibition of c-Src and hyperactivation of ROCK II kinase. Mol. Biol. Cell 19, 1378–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, et al. (2007). Akt1 governs breast cancer progression in vivo. Proc. Natl. Acad. Sci. USA 104, 7438–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X, Casimiro MC, Gormley M, Meng H, Jiao X, Katiyar S, Crosariol M, Chen K, Wang M, Quong AA, et al. (2014). Identification of a cyclin D1 network in prostate cancer that antagonizes epithelial-mesenchymal restraint. Cancer Res 74, 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Nishimoto H, Kitaura J, Maeda-Yamamoto M, Kato RM, Littman DR, Leitges M, Rawlings DJ, and Kawakami T (2004). Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J. Biol. Chem 279, 47720–47725. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Byun JY, Yun CH, Park IC, Lee KH, and Lee SJ (2008). c-Src-p38 mitogen-activated protein kinase signaling is required for Akt activation in response to ionizing radiation. Mol. Cancer Res 6, 1872–1880. [DOI] [PubMed] [Google Scholar]
- LaRocca J, Pietruska J, and Hixon M (2011). Akt1 is essential for postnatal mammary gland development, function, and the expression of Btn1a1. PLoS ONE 6, e24432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MA, and Alessi DR (2001). PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci 114, 2903–2910. [DOI] [PubMed] [Google Scholar]
- Li Z, Jiao X, Wang C, Ju X, Lu Y, Yuan L, Lisanti MP, Katiyar S, and Pestell RG (2006a). Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res 66, 9986–9994. [DOI] [PubMed] [Google Scholar]
- Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, Dye C, Yang J, Dai M, Ju X, et al. (2006b). Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol. Cell. Biol 26, 4240–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, Wang M, Soutoglou E, Knudsen ES, and Pestell RG (2010). Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res 70, 8802–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen K, Jiao X, Wang C, Willmarth NE, Casimiro MC, Li W, Ju X, Kim SH, Lisanti MP, et al. (2014). Cyclin D1 integrates estrogen-mediated DNA damage repair signaling. Cancer Res 74, 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, and Cantley LC (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, Wederell ED, Bally MB, Foster LJ, and Dedhar S (2008). Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res 68, 1618–1624. [DOI] [PubMed] [Google Scholar]
- Meng H, Tian L, Zhou J, Li Z, Jiao X, Li WW, Plomann M, Xu Z, Lisanti MP, Wang C, and Pestell RG (2011). PACSIN 2 represses cellular migration through direct association with cyclin D1 but not its alternate splice form cyclin D1b. Cell Cycle 10, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebot-Cegarra J, and Domenech-Mateu JM (1989). Association of trache-oesophageal anomalies with visceral and parietal malformations in a human embryo (Carnegie stage 21). Teratology 39, 11–17. [DOI] [PubMed] [Google Scholar]
- Norman C, Runswick M, Pollock R, and Treisman R (1988). Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55, 989–1003. [DOI] [PubMed] [Google Scholar]
- Pestell RG (2013). New roles of cyclin D1. Am. J. Pathol 183, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piljic A, and Schultz C (2006). Annexin A4 self-association modulates general membrane protein mobility in living cells. Mol. Biol. Cell 17, 3318–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, and Xu T (2002). Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol 4, 658–665. [DOI] [PubMed] [Google Scholar]
- Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, and Levin ER (2003). Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol. Cell. Biol 23, 1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner M, Hanneder M, Freilinger A, and Hengstschläger M (2007). Nuclear/cytoplasmic localization of Akt activity in the cell cycle. Amino Acids 32, 341–345. [DOI] [PubMed] [Google Scholar]
- Sabatini DM (2017). Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 114, 11818–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, et al. (2006). Cyclin D1 determines mitochondrial function in vivo. Mol. Cell. Biol 26, 5449–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Schrodinger L (2015). The PyMOL molecular graphics system, version 1.8 (New York, NY: Schrodinger LLC; ). [Google Scholar]
- Shen D, Bai M, Tang R, Xu B, Ju X, Pestell RG, and Achilefu S (2013). Dual fluorescent molecular substrates selectively report the activation, sustainability and reversibility of cellular PKB/Akt activity. Sci. Rep 3, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ (1993). Mammalian G1 cyclins. Cell 73, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, and Weinberg RA (1995). Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82, 621–630. [DOI] [PubMed] [Google Scholar]
- Soutoglou E, and Misteli T (2008). Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320, 1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, and Jacks T (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- Wang Y, Falasca M, Schlessinger J, Malstrom S, Tsichlis P, Settleman J, Hu W, Lim B, and Prywes R (1998). Activation of the c-fos serum response element by phosphatidyl inositol 3-kinase and rho pathways in HeLa cells. Cell Growth Differ 9, 513–522. [PubMed] [Google Scholar]
- Wang C, Pattabiraman N, Zhou JN, Fu M, Sakamaki T, Albanese C, Li Z, Wu K, Hulit J, Neumeister P, et al. (2003). Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol. Cell. Biol 23, 6159–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, and Pestell RG (2006). Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc. Natl. Acad. Sci. USA 103, 11567–11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB (2012). Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Suraokar M, Darnay BG, Hollier BG, Shaiken TE, Asano T, Chen CH, Chang BH, Lu Y, Mills GB, et al. (2013). BSTA promotes mTORC2-mediated phosphorylation of Akt1 to suppress expression of FoxC2 and stimulate adipocyte differentiation. Sci. Signal 6, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, and Cantley LC (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX, and ten Dijke P (2012). USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat. Cell Biol 14, 717–726. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu K, Liu P, Geng Y, Wang B, Gan W, Guo J, Wu F, Chin YR, Berrios C, et al. (2016). Inhibition of Rb Phosphorylation Leads to mTORC2-Mediated Activation of Akt. Mol. Cell 62, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Yeow WS, Zou C, Wassell R, Wang C, Pestell RG, Quong JN, and Quong AA (2010). Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res 70, 2105–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Microarray data generated during this study are available at The Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo/. The accession code for the cyclin D1-dependent gene signature in mouse mammary gland with transient induction of a cyclin D1 expression under control of the tetracycline-regulated promoter unit is GEO: GSE43216. The accession code for the Akt1-dependent gene signature from the murine ErbB2 mammary tumors is GEO: GSE138957.