

Abstract
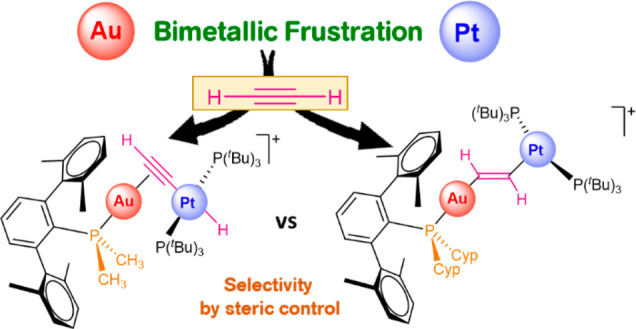
Introducing transition metals into frustrated Lewis pair systems has attracted considerable attention in recent years. Here we report a selection of three metal-only frustrated systems based on Au(I)/Pt(0) combinations and their reactivity toward alkynes. We have inspected the activation of acetylene and phenylacetylene. The gold(I) fragments are stabilized by three bulky phosphines bearing terphenyl groups. We have observed that subtle modifications on the substituents of these ligands proved critical in controlling the regioselectivity of acetylene activation and the product distribution resulting from C(sp)–H cleavage of phenylacetylene. A mechanistic picture based on experimental observations and computational analysis is provided. As a result of the cooperative action of the two metals of the frustrated pairs, several uncommon heterobimetallic structures have been characterized.
Introduction
Frustrated Lewis pairs (FLPs) emerged more than a decade ago as a paradigmatic example of chemical cooperativity, permitting bond activation and catalysis in the absence of transition metals.1 However, the incorporation of the latter as core components of FLP systems has also attracted considerable attention in the last few years.2,3 In fact, the concept of frustration may be applied to a wide variety of transition-metal-mediated transformations that were described considerably earlier than the emergence of FLP chemistry and have so far been associated with the broader concept of chemical cooperativity. From an awareness of this sometimes diffuse frontier between “traditional” transition-metal cooperative chemistry and metallic frustration, the introduction of the latter concept remains useful as a driving force for the discovery of novel transformations. That being said, introducing transition metals into frustrated designs largely increases the amount and structural diversity of Lewis acid/base combinations available. In addition, it provides an array of elementary reactions accessible for transition metals that is foreseen to extend the catalytic usefulness of main-group FLPs beyond their current status. However, a fundamental knowledge on transition-metal FLPs regarding mechanistic aspects, solution dynamics, acid–base interactions, and selectivity effects is rather underexplored in comparison to main-group FLPs, despite the fact that this information is vital for expeditious catalyst development.
In this regard, the Wass group demonstrated that subtle ligand modifications have a strong effect on the ability of zirconocene-based FLPs toward dihydrogen splitting.4 This type of system was also examined to clarify the nature of Lewis acid–base interactions by DOSY NMR spectroscopy.5 Going beyond monometallic systems, our group focused on gaining a fundamental knowledge of FLPs in which the two Lewis components are based on transition metals. Thus, we recently reported the first example of its kind by combining Au(I) and Pt(0) species as the acidic and basic sites, respectively.6 To achieve frustration, we targeted sterically hindered phosphine ligands for both gold and platinum monometallic complexes. Our experimental/computational investigations regarding the heterolytic splitting of dihydrogen mediated by these pairs led us to propose a genuine bimetallic FLP-type pathway7 analogous to the models assumed for main-group counterparts.8 Moreover, we could analyze the strong influence that Au···Pt interactions have on the activation capacity of the bimetallic pairs, as well as the solution dynamic equilibria between the metal-only Lewis pairs and the individual monometallic fragments. This is a particularly important aspect in the field of FLPs that has been widely investigated for metal-free systems, where the term “thermally induced FLPs”9 was coined to refer those pairs in which the Lewis adduct is the resting state. Despite this fact, many of these pairs exhibit a rich FLP reactivity10 and in some cases catalytic performance superior to that of their fully frustrated counterparts.11
In this study, we extend our fundamental knowledge on transition-metal-only FLPs (TMOFLPs) by exploring regioselectivity effects derived from ligand modification during the activation of alkynes, also model substrates widely investigated in the field of frustrated systems.1,12 We have focused on the effects derived from varying the degree of frustration, for which we have used terphenylphosphine ligands PMe2ArXyl2 (a), PMe2ArDipp2 (b), and PCyp2ArXyl2 (c) (Cyp = cyclopentyl), with different steric profiles, to stabilize electrophilic gold fragments 1 (Figure 1). These acidic complexes combined with the basic Pt(0) compound [Pt(PtBu3)2] (2) promote the cooperative activation of terminal alkynes. Our studies demonstrate the potential of using transition-metal Lewis acids (i.e., [PR3AuI]+ fragments) to control the selectivity in the activation of small molecules by tuning the steric properties of the ancillary ligands. This is particularly appealing in view of the challenging available protocols to synthesize the typically used acidic boranes and their scant stability toward moisture.13
Figure 1.
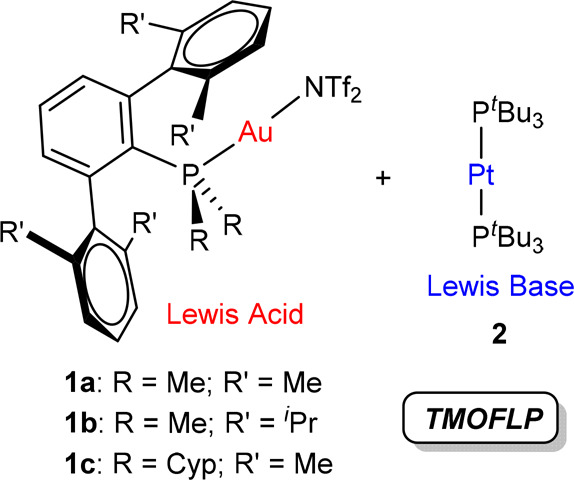
Au(I)/Pt(0) transition-metal-only frustrated Lewis pair (TMOFLP) combinations used in this work (Cyp = cyclopentyl; NTf2– = N(SO2CF3)2– = triflimide).
Results and Discussion
To investigate regioselectivity effects during alkyne activation, we first examined the reactivity of the three Au/Pt bimetallic pairs depicted in Figure 1 toward acetylene, a reaction that we had previously reported with the 1b:2 combination.6a When a dichloromethane or benzene solution of the latter pair is exposed to acetylene (0.5 bar, 25 °C), a rapid color change from bright yellow to intense orange takes place. Multinuclear NMR spectroscopic analysis revealed the formation of a clean mixture of two structurally different isomers, namely a bridging σ,π-acetylide (3b) and a rather unusual heterobimetallic vinylene (−CH=CH−) (4b), which are produced in a 4:1 ratio (Scheme 1). These metallic species are highly reminiscent of the organic products derived from the reactivity of traditional phosphine/borane FLPs with alkynes, where the prevalence of one or the other isomer typically depends on the basicity of the phosphine.14
Scheme 1. Regioselectivity in the Activation of Acetylene by TMOFLPs 1:2.

We now tested the analogous reactivity using the gold precursors [(PMe2ArXyl2)Au(NTf2)] (1a) and [(PCyp2ArXyl2)Au(NTf2)] (1c) in our search for regioselectivity effects, while the basicity of the metallic base was kept unaltered (2). Moreover, the acidities of the gold precursors 1a–c barely differ from one another;15 thus, any anticipated outcomes mostly build on steric grounds. In fact, we found a drastic change in product distribution from the less hindered system (1a, PMe2ArXyl2) to the more congested one (1c, PCyp2ArXyl2), as determined by NMR spectroscopy. While the former yields around 95% of the bridging Au/Pt acetylide 3a and only a residual amount of the vinylene (4a, <5%), the more hindered pair comprising the [(PCyp2ArXyl2)Au]+ fragment (1c) fully reversed the selectivity toward the exclusive formation of the corresponding vinylene 4c (Scheme 1). Attempts to isolate 3c by the reaction of independently prepared compounds [(PCyp2ArXyl2)Au(C≡CH)] (5c) and [Pt(PtBu3)2H][NTf2]16 (6) proved unsuccessful and resulted in intractable mixtures.
As mentioned above, this dramatic shift in regioselectivity seems to be dominated by steric effects, which contrasts with prior strategies to modulate alkyne activation by FLPs that mostly rely on phosphine basicity. More importantly, it evinces the potential of FLP systems that incorporate transition-metal Lewis acids to easily tune the selectivity during bond activation processes and, as such, in subsequent catalytic applications that incorporate those activation events. As was pointed out earlier, this could be seen as a key advantage in comparison to traditional FLP designs that usually involve fluorinated boranes, since accessing these moieties already entails substantial synthetic challenges and limitations, not to mention their limited stability toward moisture and air.13 In stark contrast, the preparation of terphenylphosphines PR2Ar′ is straightforward and highly versatile,17 while the resulting gold precursors 1 are readily obtained in high yields and exhibit stability toward water or under moderate oxidizing conditions.18
The nature of the new heterobimetallic compounds 3a and 4c was ascertained by a comparison of their 1H and 31P{1H} NMR signals with those derived from their analogous species based on PMe2ArDipp2: that is, 3b and 4b, respectively.6a The heterobimetallic nature of compounds 4 is evinced by the 195Pt satellites that flank the 31P{1H} resonances associated with terphenylphosphines, which appear at 2.1 (4b, 4JPPt = 282 Hz) and 51.7 (4c, 4JPPt = 277 Hz) ppm. The bridging vinylene (−CH=CH−) moiety displays a distinctive pair of 1H NMR signals in the region between 4.0 and 4.5 ppm that reveal scalar coupling to the 195Pt center in the range 120–200 Hz (see the Experimental Section for details). By analogy, we attribute a 31P{1H} NMR resonance at 3.67 ppm to the minor species (ca. 5%) in the PMe2ArXyl2 system (4a), with an identical 1H NMR pattern comprised of signals at 4.54 and 4.37 ppm, though their corresponding 195Pt satellites could not be observed due to the low concentration of isomer 4a. Corresponding 13C NMR resonances for compounds 4b,c emerge at ca. 155 (1JCH ≈ 175 Hz) and 115 (1JCH ≈ 190 Hz) ppm, respectively, supporting the proposed formulation and the sp2 hybridization of the carbon atoms.
The molecular structure of compounds 3a and 4c was further corroborated by X-ray diffraction studies (Figure 2). The presence of the σ,π-acetylide or vinylene linker distorts the linearity around the platinum center: P−Pt−P angles of around 165°, shifted from the ideal 180° due to the steric pressure exerted by the bulky gold fragments. The Pt1–C1 (2.016(6) Å) and Au1–C1 (2.311(5) Å) bond distances in 3a appear slightly shortened in comparison to 3b (dPt1–C1 = 2.044(7) Å; dAu1–C1 = 2.360(7) Å). The average C=C bond distance of the vinylene linkers in the two crystallographically independent molecules of 4c accounts for 1.278(13) Å, comparable to those of 4b (1.287(11) Å). These and other geometric features are similar to those of previously reported related species.19
Figure 2.
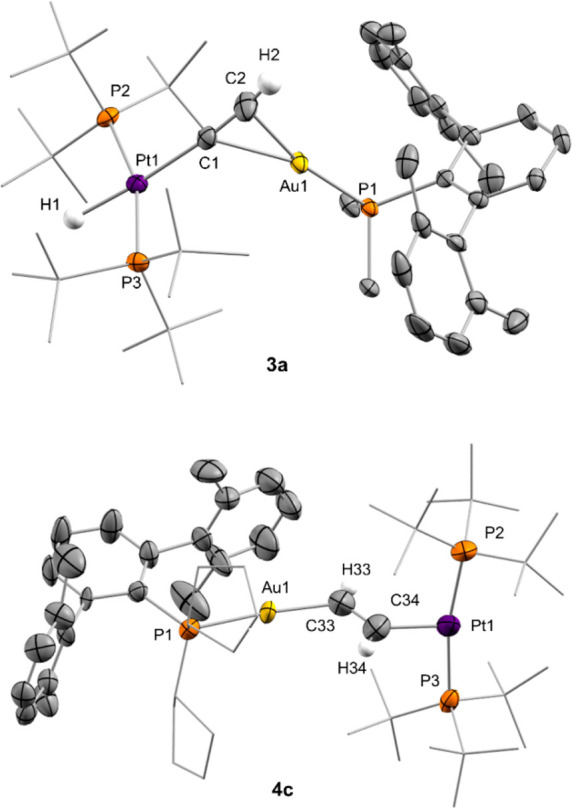
ORTEP diagrams of compounds 3a and 4c. For the sake of clarity most hydrogen atoms and triflimide anions are excluded and some substituents have been represented in wireframe format, while thermal ellipsoids are set at 50% probability.
We had previously observed that the three investigated terphenylphosphines permit control of the equilibrium between complete frustration and bimetallic adduct formation.7 Thus, while a dative Pt→Au bond is immediately formed between [(PMe2ArXyl2)Au(NTf2)] (1a) and [Pt(PtBu3)2] (2), the formation of an identical adduct based on PCyp2ArXyl2 is endergonic and could not be experimentally detected. An intermediate situation is reached for the medium-sized phosphine PMe2ArDipp2, where the prevalence of the monometallic fragments or the bimetallic adduct depends upon experimental conditions. In this context, we have observed that TMOFLPs (1:2) based on the gold precursors [(PMe2ArDipp2)Au(NTf2)] (1b) and [(PCyp2ArXyl2)Au(NTf2)] (1c) are considerably more active toward alkyne activation in comparison to that one built on [(PMe2ArXyl2)Au(NTf2)] (1a). While full conversion toward compounds 3 and 4 was recorded by the time of placing the sample in the NMR probe (<5 min) in the case of using 1b:2 or 1c:2, the analogous transformation essayed with 1a required up to 24 h to reach completion under otherwise identical conditions (C2H2, 0.5 bar, 25 °C, toluene or C6D6). This fact speaks in favor of a genuine FLP mechanism that imposes an energetic demand to overcome the Pt→Au bond cleavage prior to acetylene activation, a requirement that only applies to the less hindered gold precursor 1a. Similar effects have been observed in the gold-catalyzed addition of P-nucleophiles to alkynes.20
In addition, it is key to highlight that the cooperative reactivity depicted in Scheme 1 contrasts with that of the individual Au(I) or Pt(0) fragments (Scheme 2). For instance, [Pt(PtBu3)2] (2) readily catalyzes acetylene polymerization, evinced by the rapid formation of a purple-black solid accompanied by the disappearance of a 1H NMR resonance at 1.34 ppm due to C2H2, while signals due to 2 remained unchanged. At variance, no indication of polyacetylene formation is apparent when gold is also present in solution. In the case of the individual gold compounds 1 there is no sign of chemical transformation in the short term (ca. 30 min), while at longer reaction times the gold triflimide precursors evolve to bridging σ,π-acetylide compounds 7 (Scheme 2A),21 albeit only in moderate yields accompanied by other unidentified gold-containing species.
Scheme 2. (A) Reactivity of Individual Gold (1) and Platinum (2) Compounds toward Acetylene and (B) Reaction of σ,π-Acetylide 7b and T-shaped Platinum Hydride 6.

Having the previous experimental findings on hand, we were interested in further understanding the cooperative action of TMOFLPs 1:2 to gain a fundamental knowledge of significance for future catalytic applications. As such, we initially wondered about the possible role of acetylide-bridged digold compounds such as 7 as precursors toward complexes 3 and 4. In fact, the reaction between 7b and [Pt(PtBu3)2H][NTf2] (6) immediately yielded the corresponding heterobimetallic σ,π-acetylide compound 3b, where the unsaturated linker is now σ-bonded to the platinum center instead of the gold nucleus. Nevertheless, the complete absence of the vinylene isomer 4b during the latter reaction would require an additional competing route to provide access to this unusual bimetallic motif, which actually is the exclusive isolated isomer for the bulkier PCyp2ArXyl2-based system (Scheme 1). Moreover, the formation of compounds 7 requires several hours to proceed to appreciable conversions, while the activation of acetylene by 1:2 pairs is immediate (<5 min), except for the gold precursor 1a bearing PMe2ArXyl2, which as noted earlier takes around 24 h to convert into 3a in the presence of [Pt(PtBu3)2] (2).
To further investigate the mechanism and the reasons for the drastically different regioselectivity observed, we carried out computational studies (DFT, ωB97XD/6-31G(d,p)+SDD). Focusing on the system based on PMe2ArDipp2, we began searching for initial acetylene activation steps by approaching the acetylene molecule to the individual Au(I) (1b) and Pt(0) (2) fragments, assuming that Au–Pt dissociation is a prerequisite for alkyne activation, as deduced from the reduced reactivity of the pair 1a:2 in comparison to 1b:2 and 1c:2 and previous computational results.7 Interestingly, the formation of 3 and 4 seems to share a common intermediate, namely a Au(I) acetylene adduct of formula [(PR2Ar′)Au(C2H2)]+ (9). The formation of this type of π complex has been previously proposed in the context of alkyne12,14a and alkene22 activation by P/B pairs, but no experimental proofs of their existence have been reported. In an attempt to spectroscopically identify such an intermediate, we recorded the formation of a new gold-containing species by low-temperature NMR (−80 °C) as the major species (ca. 80%) upon exposing a CD2Cl2 solution of 1b to an acetylene atmosphere. This species exhibits a distinctive 1H NMR signal at 3.43 ppm that correlates with a 31P NMR resonance at −0.3 ppm (in comparison to δP −9.9 ppm for 1b) and presents dynamic exchange with free acetylene (δH 2.11 ppm) as seen by EXSY NMR experiments, while other 1H NMR resonances are comparable to those of 1b. This finding constitutes an additional benefit of TMFLPs for mechanistic investigations in frustrated systems, since they provide additional modes of stabilizing otherwise fleeting intermediates.
With the acetylene adduct 9b as the starting point, our calculations indicate that the attack of the platinum compound 2 over 9b leads to either of the two bimetallic isomers 3b and 4b depending on the trajectory followed by 2 while it approaches 9b (Figure 3). Thus, if 2 approaches the acetylene adduct along the Au–C2H2 direction, the corresponding vinylene 4b forms (ΔG⧧ = 23.4 kcal mol–1 from the 1b:2 Lewis pair + acetylene), in a process somehow reminiscent of the gold-mediated nucleophilic attack over activated alkynes (e.g., gold-catalyzed hydroamination).23 In contrast, the alignment of the basic 2 in an orthogonal disposition with respect to the Au–C2H2 bond results in deprotonation of the activated acetylene (15.6 kcal mol–1 for the pair 1b:2) to yield the corresponding Au(I) terminal acetylide [(PMe2ArDipp2)Au(C≡CH)] (5b) and [Pt(PtBu3)2H][NTf2] (6). These two fragments readily rearrange to intermediates [(PR2Ar′)Au(μ-η1:η2-C≡CH)Pt(H)(PtBu3)2]+ (A) that subsequently evolve to compounds 3 by rapid σ,π isomerization of the bridging μ-C≡CR unit. In experimental agreement, the reaction of independently synthesized [(PMe2ArDipp2)Au(C≡CH)] (5b) and [Pt(PtBu3)2H][NTf2] (6) rapidly yields complex 3b. As mentioned above, this does not apply to compound 5c, whose reaction with 6 resulted in a complex mixture of products that include decomposition into elemental gold. Nevertheless, the corresponding [(PCyp2ArXyl2)Au(μ-η2:η1-C≡CH)Pt(H)(PtBu3)2]+ (3c) has not been detected during the bimetallic activation of acetylene. An alternative orthogonal mechanism involving the initial oxidative addition of acetylene over 2, followed by cis–trans isomerization and coupling with 1, was ruled out on the basis of significantly higher overall activation barriers (see Figure S2).
Figure 3.
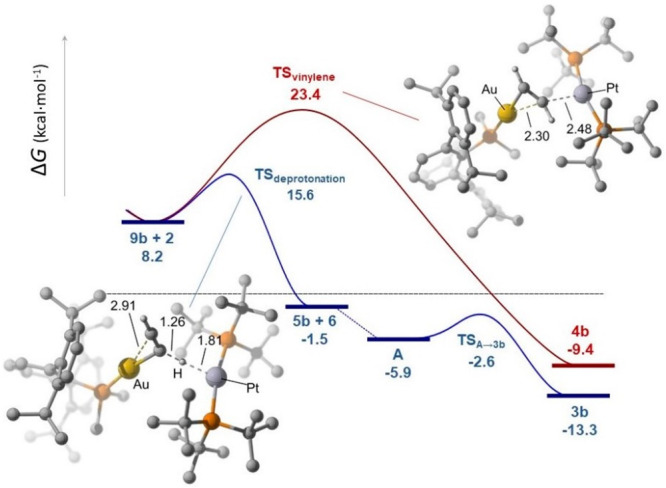
Energy profiles for the formation of the acetylide and vinylene complexes 3b and 4b from the common intermediate 9b and platinum(0) compound 2. The origin of energies is set to the 1b:2 Lewis pair + acetylene.
Information on the activation of the simplest alkyne (C2H2) by FLPs is rather scarce.12a For the sake of completeness and to better compare our results with prior studies on main-group FLPs, we decided to test other more commonly employed triply bonded hydrocarbons. With regard to internal alkynes, all our attempts to access bimetallic vinylenes were unsuccessful. Reactions of 1:2 pairs with diphenylacetylene, 2-butyne, and 1,4-diphenylbutadiyne did not result in the formation of any new species even under more forcing experimental conditions. At variance, addition of phenylacetylene to an equimolar mixture of 1 and 2 provided phosphine-dependent divergent outcomes derived from C(sp)–H bond cleavage (Scheme 3). In parallel with acetylene activation with the more congested pairs based on 1b,c, the reaction of these systems with PhC≡CH was also immediate. In contrast, while acetylene activation took up to 24 h for the nonfrustrated 1a:2 pair, the reaction was complete after around 15 min in the case of phenylacetylene.
Scheme 3. Product Distribution from the Activation of Phenylacetylene by TMOFLP Pairs 1:2.

The appearance of a distinctive low-frequency 1H NMR resonance at around −10 ppm flanked by 195Pt satellites (1JHPt = 608 Hz, 1a:2; 1JHPt = 533 Hz, 1b:2 and 1c:2) prompted us to believe that corresponding heterobimetallic σ,π-acetylide complexes were formed in all cases. However, a more careful analysis revealed unexpected differences in product distribution for the less bulky terphenylphosphine PMe2ArXyl2 in comparison to the more hindered systems (Scheme 3). Extracting the crude reaction mixtures with pentane permitted isolation of the same platinum-containing compound 10 for the pairs 1b:2 and 1c:2, while the less hindered gold fragment 1a did not led to any metallic species soluble in nonpolar hydrocarbon solvents. An infrared band at 2090 cm–1 was recorded for a Pt-hydride ligand in 10, while characteristic 13C{1H} NMR resonances at 118.8 and 117.6 ppm accounted for a σ-bonded acetylide ligand. On the basis of these spectroscopic features and the high solubility, we proposed a molecular formulation for 10 as [Pt(PtBu3)2(H)(C≡CPh)]: that is, the formal oxidative addition of phenylacetylene over Pt(0) compound 2. This assumption was further corroborated by X-ray diffraction analysis (see Figure S3). Nevertheless, it is important to remark that compound 2 does not react with phenylacetylene even after longer reaction times (48 h) or at elevated temperatures (80 °C), which suggests a cooperative action between platinum and gold to account for alkyne C–H activation.
The only detectable gold-containing species in these reactions were assigned to the corresponding bridging σ,π-acetylide digold complexes 11. Those where characterized by 31P{1H} NMR signals at 0.4 (11b) and 53.9 (11c), shifted to higher frequencies with respect to their precursors 1 (cf. −11.9 for 1b and 48.8 ppm for 1c), in agreement with other related examples.24 Also similar to those results, the presence of a single 31P resonance for each compound suggests rapid exchange of the σ,π coordination in solution.18 However, the process is frozen in the solid state. An ORTEP representation of the molecular structure of 11b is shown in Figure 4a. The σ,π coordination of the acetylide is reflected by a nonsymmetric arrangement characterized by bond distances of 2.021(10) and 2.209(9) Å for the Au2–C1 and Au1–C1 bond distances, respectively. The Au1 center is also connected to C2 by a slightly longer bond length of 2.310(9) Å. The presence of an aurophilic interaction can be inferred from a Au1–Au2 distance of 3.366(1) Å, which is faintly elongated in comparison to its related acetylide analogue [Au2{μ-C≡CH}] (dAu1–Au2 = 3.31 Å), likely as a result of the higher steric pressure exerted by phenylacetylide. Digold complexes 11b,c were accompanied by equimolar amounts of [Pt(PtBu3)2(H)]+ (6) that could not be washed out with pentane, as evinced by a broad low-frequency 1H NMR resonance at 1.16 ppm (1JPPt = 6.5 Hz).
Figure 4.
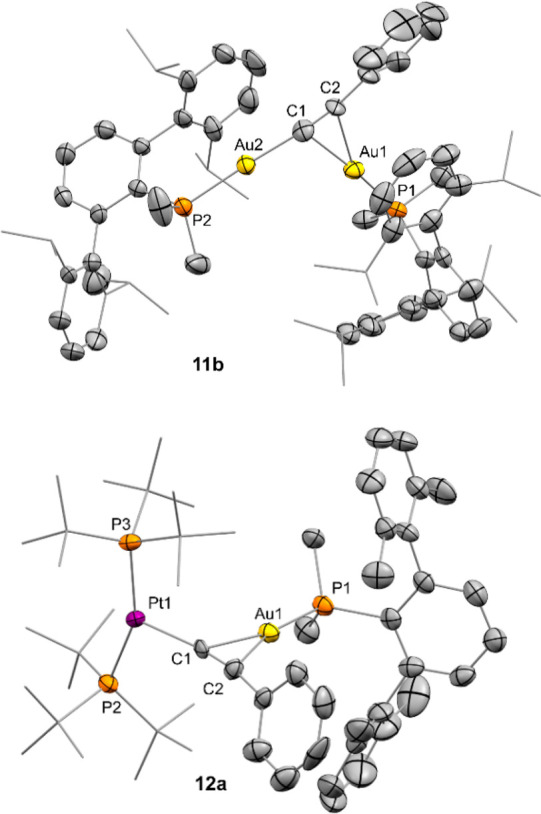
ORTEP diagrams of compounds 11b and 12a. For the sake of clarity hydrogen atoms, solvent molecules, and triflimide anions are excluded and some substituents have been represented in wireframe format, while thermal ellipsoids are set at 50% probability. The hydride ligand bound to platinum in 12a could not be located in the Fourier electron density map.
As noted earlier, the reaction of the less hindered 1a:2 pair with phenylacetylene yielded a divergent result. C(sp)–H activation became evident by the presence of a distinctive hydridic 1H NMR resonance at −10.4 ppm (2JHP = 14 Hz, 1JHPt = 608 Hz). However, a single platinum complex was formed in this case, which resonates at 82.2 ppm (1JPPt = 2810 Hz) in its 31P{1H} NMR spectrum and could not be washed out using nonpolar hydrocarbon solvents. The corresponding C≡C stretching frequency rendered an infrared band shifted to lower wavenumbers (νC≡C 1982 cm–1; cf. 2048 cm–1 for 11b, 2029 cm–1 for 11c, and 2090 cm–1 for 10), while sp-hybridized carbon atoms render 13C{1H} NMR resonances at lower frequencies (91.1 and 85.9 ppm) in comparison to compounds 11b,c and 10 (116–125 ppm). These observations expose the divergent product distribution derived from using phosphines with different steric profiles. Since the recorded parameters equate with heterobimetallic σ,π-acetylide compounds 3, we assumed an analogous structure for this complex (12a), a premise that we could substantiate by X-ray diffraction analysis (Figure 4b). The bulkier nature of the phenylacetylide moiety with respect to the unsubstituted acetylide in 3a is likely the cause of a more intense distortion of the T-shaped platinum fragment, where a P–Pt–P angle of 156.71(8)° is recorded (cf. 162.76(5)° for 3a). The close proximity between the acetylide phenyl fragment and the ortho substituents of one of the flanking aryl rings of the terphenyl moiety may be responsible for the dissimilar product distributions found between PMe2ArXyl2 vs PMe2ArDipp2/PCyp2ArXyl2.
On the basis of our experimental and computational studies on acetylene activation, initial formation of a gold–alkyne adduct like 9 seems most plausible. The slightly higher acidity of phenylacetylene, as well as its higher size, may account for the prevalence of the deprotonation pathway in detriment of the 1,2-addition route toward vinylene structures, which were not detected. In turn, the dissimilar reaction products depicted in Scheme 3 might be understood on steric grounds. Thus, selective formation of compounds 11 for the bulkier PMe2ArDipp2-/PCyp2ArXyl2-based systems could be the result of a higher steric clash between the tert-butyl substituents on the platinum fragment and the terphenyl moiety of the gold-bound phosphine. In contrast, the reduced steric pressure introduced by PMe2ArXyl2 may permit easier access to the heterobimetallic compound 12a, analogous to its unsubstituted acetylide version (3a).
We thought of interest to carry out several further experiments to shed some light into the operating cooperative mechanism. In the same manner as we observed for acetylene activation, the reactivity of the individual metallic fragments starkly contrasts with that of the bimetallic pairs. Accordingly, platinum compound 2 does not exhibit any reactivity toward phenylacetylene even after heating at 80 °C for 48 h (Scheme 4a). In turn, compounds 1 promote C(sp)–H cleavage of the alkyne to form the corresponding digold σ,π-acetylide complexes 11, but at a considerably slower pace (24 h at 25 °C: 1a, 9% of 11a; 1b, 70% of 11b; 1c, 12% of 11c; NMR spectroscopic yields). To account for the origin of 10, we performed the reaction of 2 with equivalent amounts of phenylacetylene and catalytic quantities of 1. In addition, we also tested the same transformation but using 6 as the catalyst, since this presumably forms in situ after protonation of 2 by small amounts of HNTf2 derived from the reaction of 1 and phenylacetylene toward 11. Catalytic amounts of compound 6 may also derive from the cooperative Au/Pt C(sp)–H activation of phenylacetylene by a deprotonation mechanism. As anticipated, immediate conversion of 2 into 10 was observed at room temperature using either 1 or 6 in catalytic amounts as low as 2 mol % (Scheme 4b). The reaction of the independently prepared 10 with 1a leads to 3a as the major species by NMR analysis, though other unidentified side products are also formed. The reaction of the independently synthesized neutral σ-acetylide compound [(PMe2ArDipp2)Au(C≡CPh)] (13b; ORTEP diagram in Figure S4) with 1b yielded the expected digold σ,π-acetylide 11b under mild conditions (Scheme 4c). This result parallels the reactivity previously described for NHC-based gold complexes, where the latter transformation proceeds smoothly,24f as also occurs with the parent unsubstituted acetylide (C≡CH) fragment.18 Thus, terminal acetylides of type 13 may be regarded as key intermediates toward compounds 11 during the activation of PhCCH.
Scheme 4. Selected Experiments Carried out to Provide Information about the Mechanism of Phenylacetylene Activation Mediated by TMOFLPs 1:2.

On consideration of all the information discussed above, Scheme 5 contains an overall mechanistic picture to account for the phosphine-dependent product distribution during the activation of alkynes. The common gold acetylene adduct 9 is proposed to be a key intermediate. While both deprotonation and 1,2-addition mechanisms (blue and red in Scheme 5, respectively) are viable for acetylene, only the deprotonation pathway seems to be operative in the case of the more acidic phenyl-substituted alkyne. Once the latter is deprotonated to form an equimolar mixture of 13 and 6, steric factors appear to dominate the final product distribution. The combination of the more hindered terphenylphosphines with the bulkier phenylacetylene prevents formation of the corresponding Au/Pt heterobimetallic adducts likely due to a steric clash, while the latter is the only observed complex in the PMe2ArXyl2 system. In turn, terminal gold acetylides 13, although unable to react with [Pt(PtBu3)2H]+ (6), rapidly yield the corresponding digold σ,π-acetylides 11 upon combination with still unreacted triflimide complexes 1. We can also infer from our experimental observations that several of the transformations depicted in Scheme 5 constitute dynamic equilibria that are dependent on reaction conditions.
Scheme 5. Overall Representation of the Proposed Pathways to Account for Product Distribution during Alkyne Activation.

Conclusions
In summary, we provide evidence for the potential of a cooperative Au(I)/Pt(0) bimetallic system that behaves as a metal-only frustrated Lewis pair for the activation of small molecules. We have demonstrated that subtle modifications of the phosphine ligands bound to gold have a strong effect on the regioselectivity of acetylene activation. Thus, simply by adjusting the steric hindrance of those phosphines, we have been able to select the operating mechanism (deprotonation vs 1,2-addition) during acetylene activation with full specificity. This is possible without the need to alter the basicity of the platinum(0) moiety, thus contrasting with main-group FLP systems. Moreover, ligand modification also permitted us to diverge from the product distribution that results from C–H bond cleavage in phenylacetylene. While the less congested system based on PMe2ArXyl2 yields a heterobimetallic σ,π-acetylide compound, the more hindered pairs constructed around PMe2ArDipp2 and PCyp2ArXyl2 resulted in the formation of equimolar mixtures of digold σ,π-acetylide compounds, [Pt(PtBu3)2(H)]+, and [Pt(PtBu3)2(H)(C≡CPh)]. These results highlight one of the advantages of incorporating transition metals into frustrated designs, namely the ability to easily tune the stereoelectronic properties of the acidic site, in turn a challenging task in traditional FLPs that rely on the electrophilicity of fluorinated boranes. The straightforward control of these properties in TMFLPs may be exploited for the development of more selective catalytic transformations drawn on the concept of frustration.
Experimental Section
General Considerations
All preparations and manipulations were carried out using standard Schlenk and glovebox techniques, under an atmosphere of argon and of high-purity nitrogen, respectively. All solvents were dried, stored over 4 Å molecular sieves, and degassed prior to use. Toluene (C7H8) and n-pentane (C5H12) were distilled under nitrogen over sodium. Tetrahydrofuran (THF) and diethyl ether were distilled under nitrogen over sodium/benzophenone. [D6]Benzene was dried over molecular sieves (4 Å) and CD2Cl2 over CaH2 and distilled under argon. [AuCl(THT)] (THT = tetrahydrothiophene)25 and compounds 1a,71b,181c,72,26 and 6(16) were prepared as described previously. Other chemicals were commercially available and were used as received. Solution NMR spectra were recorded on Bruker AMX-300, DRX-400, and DRX-500 spectrometers. Spectra were referenced to external SiMe4 (δ 0 ppm) using the residual proton solvent peaks as internal standards (1H NMR experiments) or the characteristic resonances of the solvent nuclei (13C NMR experiments), while 31P was referenced to H3PO4. Spectral assignments were made by routine one- and two-dimensional NMR experiments where appropriate (Figure 5). Infrared spectra were recorded on a Bruker Vector 22 spectrometer, and sampling preparation was carried out in Nujol. For elemental analyses a LECO TruSpec CHN elementary analyzer was utilized. 1965525, 1965526, and 1986501–1986504 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Figure 5.

General labeling scheme used for 1H and 13C{1H} NMR assignments.
[(PMe2ArXyl2)Au(μ-η2:η1-C≡CH)Pt(H)(PtBu3)2]NTf2 (3a)
A solid mixture of 1a (100 mg, 0.121 mmol) and 2 (73 mg, 0.121 mmol) was dissolved in 5 mL of toluene and stirred at room temperature for 12 h under a C2H2 atmosphere (0.5 bar). The solution was layered with pentane (10 mL) and stored at −30 °C overnight to yield compound 3a as colorless crystals (115 mg, 66%). Anal. Calcd for C52H83AuF6NO4P3PtS2: C, 43.1; H, 5.8; N, 1.0; S, 4.4. Found: C, 43.1; H, 5.5; N, 1.1; S, 4.4. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.26 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.18 (m, 1 H, Hd), 7.15 (d, 4 H, 3JHH = 7.6 Hz, Ha), 6.71 (dd, 2 H, 3JHH = 7.5 Hz, 4JHP = 3.5 Hz, Hc), 2.98 (m, 1 H, C≡CH), 2.15 (s, 12 H, MeXyl), 1.52 (d, 6 H, 2JHP = 9.5 Hz, PMe2), 1.49 (vt, 54 H, 3JHP = 6.4 Hz, tBu), −10.27 (m, 1H, 2JHP = 14 Hz, 1JHPt = 571 Hz, Pt–H). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 146.8 (d, 2JCP = 10 Hz, C3), 141.1 (d, 3JCP = 4 Hz, C2), 136.8 (C1), 132.4 (CHd), 131.4 (d, 3JCP = 8 Hz, CHc), 129.3 (CHb), 128.8 (CHa), 127.4 (d, 1JCP = 57 Hz, C4), 126.6 (d, 2JCP = 79 Hz, C≡CH), 121.4 (q, 1JCF = 323 Hz, CF3), 108.2 (d, 2JCP = 20 Hz, C≡CH), 41.2 (vt, 1JCP = 8 Hz, Pt-C(CH3)3), 33.1 (Pt-(C(CH3)3)), 21.8 (MeXyl), 17.0 (d, 1JCP = 38 Hz, PMe2). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 82.9 (1JPPt = 2768 Hz, P(tBu)3), 0.8 (Au–P). IR (Nujol): ν(≡C–H) 3171, ν(C≡C) 1843 cm–1.
[(PCyp2ArXyl2)Au(μ-η1:η1-HC=CH)Pt(PtBu3)2]NTf2 (4c)
A mixture of compounds 1c (100 mg, 0.107 mmol) and 2 (64 mg, 0.107 mmol) was dissolved in toluene (5 mL), and the argon atmosphere was replaced by C2H2 (0.5 bar), upon which the bright yellow solution changed to an intense orange. The solution was then filtered, layered with pentane, and stored at −20 °C overnight to yield compound 4c as orange crystals (68 mg, 41%). Anal. Calcd for C60H95AuF6NO4P3PtS2: C, 46.3; H, 6.1; N, 1.0; S, 4.1. Found: C, 45.9; H, 5.7; N, 1.0; S, 3.8. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.19–7.08 (m, 3 H, Hb, Hd), 7.01 (d, 4 H, 3JHH = 7.6 Hz, Ha), 6.66 (dd, 2 H, 3JHH = 7.7 Hz, 4JHP = 2.7 Hz, Hc), 4.51 (dt, 1 H, 3JHH = 7.6 Hz, 3JHP = 3.4 Hz, 2JHPt = 110 Hz, Hβ), 4.20 (dd, 1 H, 3JHH = 7.6 Hz, 3JHP = 6.4 Hz, 3JHPt = 194 Hz, Hα), 2.22–2.09 (m, 2 H, CH), 1.97 (s, 24 H, MeXyl), 1.71–1.47 (m, 12 H, CH2), 1.36 (vt, 54 H, 3JHP = 6.5 Hz, tBu), 1.24–1.09 (m, 4H, CH2). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 159.4 (d, 2JCP = 113, 1JCH = 198 Hz, CHα), 148.9 (d, 2JCP = 10 Hz, C3), 142.4 (d, 3JCP = 5 Hz, C2), 137.9 (C1), 137.0 (d, 3JCP = 6 Hz, CHc), 132.4 (d, 4JCP = 8 Hz, CHd), 131.6 (d, 1JCP = 40 Hz, C4), 129.3 (CHa), 125.7 (CHb), 121.7 (q, 1JCF = 322 Hz, CF3), 115.5 (d, 3JCP = 24, 1JCH = 193 Hz, CHβ), 41.2 (vt, 1JCP = 8 Hz, Pt-C(CH3)3), 38.0 (d, 1JCP = 28 Hz, PCH), 35.0 (d, 2JCP = 10 Hz, CH2), 33.1 (Pt-(C(CH3)3)), 32.5 (d, 2JCP = 10 Hz, CH2), 26.0 (d, 2JCP = 10 Hz, CH2), 25.7 (d, 2JCP = 10 Hz, CH2), 21.4 (MeXyl). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 70.6 (1JPPt = 3323 Hz, P(tBu)3), 51.7 (4JPPt = 277 Hz, Au–P). IR (Nujol): ν(=C–H) 3172, ν(C=C) 1645 cm–1.
[(PCyp2ArXyl2)Au(C≡CH)] (5c)
A suspension of (PCyp2ArXyl2)AuCl7 (200 mg, 0.29 mmol) in toluene (10 mL) was cooled to −40 °C, and a toluene solution containing a small excess (1.2 equiv) of Mg(C≡CH)Br was added dropwise. The mixture was stirred for an additional 2 h at −40 °C. The volatiles were removed in vacuo, and the residue was extracted with pentane. Evaporation of the solvent led to compound 5c as a white powder (35 mg, 18%). Gold acetylides are potentially explosive and should be handled with caution. Anal. Calcd for C34H40AuP: C, 60.4; H, 6.0. Found: C, 60.4; H, 5.8. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.29 (t, 2 H, 3JHH = 7.6 Hz, Hb), 7.09 (d, 4 H, 3JHH = 7.56 Hz, Ha), 6.94 (td, 1 H, 3JHH = 7.6 Hz, 5JHP = 1.6 Hz, Hd), 6.64 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 2.7 Hz, Hc), 2.17–2.05 (m, 2 H, PCH), 1.97 (s, 12 H, MeXyl), 1.78 (d, 1H, 4JHP = 5.5 Hz, AuC≡CH), 1.7–1.1 (m, 16 H, CH2). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 148.8 (d, 2JCP = 10 Hz, C3), 141.9 (d, 3JCP = 5 Hz, C2), 136.3 (C1), 132.0 (d, 3JCP = 7 Hz, CHc), 130.8 (d, 4JCP = 5 Hz, CHd), 130.2 (d, 2JCP = 158 Hz, AuC≡CH), 128.9 (d, 1JCP = 48 Hz, C4), 128.7 (CHb), 128.4 (CHa), 86.6 (d, 3JCP = 25 Hz, 1JCH = 233 Hz, AuC≡CH), 38.3 (d, 1JCP = 31 Hz, PCH), 35.1 (d, 2JCP = 10 Hz, CH2), 32.4 (d, 2JCP = 10 Hz, CH2), 25.4 (d, 2JCP = 11 Hz, CH2), 25.3 (d, 2JCP = 13 Hz, CH2), 21.6 (MeXyl). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 55.5. IR (Nujol): ν(≡C–H) 3286, ν(C≡C) 1944 cm–1.
[Pt(PtBu3)2(H)(C≡CPh)] (10)
A toluene (5 mL) solution of 2 (64 mg, 0.107 mmol), phenylacetylene (11 μL, 0.107 mmol), and 1b or 1c (2 mg, 0.002 mmol) was stirred at room temperature for 15 min. The volatiles were removed in vacuo, and the residue was extracted with pentane (3 × 5 mL). Evaporation of the solvent yielded compound 10 as a colorless oil (26 mg, 35%). Suitable crystals for X-ray diffraction studies can be obtained by slow pentane evaporation at room temperature. Anal. Calcd for C32H60P2Pt: C, 54.8; H, 8.6. Found: C, 54.9; H, 8.4. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.64 (d, 2 H, 3JHH = 7.3 Hz, o-C6H5), 7.21 (t, 2 H, 3JHH = 8.2 Hz, m-C6H5), 7.02 (t, 1 H, 3JHH = 7.3 Hz, p-C6H5), 1.58 (vt, 54 H, 3JHP = 6.3 Hz, tBu), −9.46 (t, 1 H, 2JHP = 15.4, 1JHPt = 532.9 Hz, Pt-H). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 131.3 (CH-Ar), 130.7 (CH-Ar), 129.3 (C–Ar), 124.2 (CH-Ar), 118.8 (C≡CPh), 117.6 (C≡CPh), 40.4 (vt, 1JCP = 8 Hz, 2JCPt = 35 Hz, Pt-P(C(CH3)3), 33.1 (Pt-P(C(CH3)3). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 81.3 (1JPPt = 2880 Hz). IR (Nujol): ν(C≡C) 2090 cm–1.
[(PR2Ar)2Au2(μ-η1:η2-C≡CPh)]NTf2 (11)
To a solution of compounds 1 (100 mg, 0.107 mmol) and 2 (64 mg, 0.107 mmol) in toluene (5 mL) was added 1 equiv of phenylacetylene (11 μL, 0.107 mmol) and the mixture stirred at room temperature for 15 min. The volatiles were removed in vacuo, and the residue was washed with pentane (3 × 5 mL). The resulting fine white powder (60 mg, 11b; 65 mg, 11c) contains a mixture of compounds 11 and 6, the latter of which could not be separated by common methods but whose spectroscopic features did not hamper full characterization of compounds 11. A single crystal of 11b suitable for X-ray diffraction studies was grown by slow diffusion of pentane into a toluenes solution (2:1 v/v) at −30 °C. Alternatively, compound 11b could be synthesized free of 6: to a solution of 1b (100 mg, 0.107 mmol) in toluene (5 mL) was added 1 equiv of phenylacetylene (11 μL, 0.107 mmol) and the mixture stirred at room temperature for 18 h. The volatiles were removed in vacuo, and the residue was washed with pentane (3 × 5 mL) to yield 11b as a white powder (90 mg, 50%). Data for compound 11b are as follows. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.55 (d, 2 H, 3JHH = 7.6 Hz, o-C6H5), 7.35 (m, 2 H, m-C6H5), 7.26 (t, 4 H, 3JHH = 7.6 Hz, Hb), 7.09 (d, 8 H, 3JHH = 7.8 Hz, Ha), 7.02 (t, 1 H, 3JHH = 8.4 Hz, p-C6H5), 6.93 (m, 6 H, Hc, Hd), 2.54 (sept, 8 H, 3JHH = 6.7 Hz, iPr(CH)), 1.57 (d, 12 H, 2JHP = 9.8 Hz, PMe2), 1.29 (d, 24 H, 3JHH = 6.7 Hz, iPr(CH3)), 0.90 (d, 24 H, 3JHH = 6.7 Hz, iPr(CH3)). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 146.6 (C1), 146.0 (d, 2JCP = 10 Hz, C3), 138.6 (d, 5JCP = 3 Hz, C2), 134.5 (d, 3JCP = 9 Hz, CHc), 133.0 (CH-Ar), 131.5 (d, 1JCP = 43 Hz, C4), 130.7 (CH-Ar), 130.1 (CHb), 129.7 (C-Ar), 129.3 (CHd), 125.7 (CH-Ar), 124.0 (CHa), 120.9 (q, 1JCF = 323 Hz, CF3), 119.9 (C≡CPh), 116.4 (C≡CPh), 31.6 (iPr(CH), 25.5 (iPr(CH3), 23.1 (iPr(CH3), 17.2 (d, 1JCP = 38 Hz, PMe2). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 0.4. IR (Nujol): ν(C≡C) 2048 cm–1. Data for compound 11c are as follows. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.42 (d, 2 H, 3JHH = 8 Hz, o-C6H5), 7.24 (t, 4 H, 3JHH = 7.6 Hz, Hb), 7.08 (d, 8 H, 3JHH = 7.6 Hz, Ha), 7.05 (m, 5 H, 3JHH = 7.6 Hz, Hd, m-C6H5,p-C6H5), 6.63 (dd, 4 H, 3JHH = 7.6 Hz, 4JHP = 3.3 Hz, Hc), 1.94 (s, 24 H, MeXyl), 2.25–2.13 (m, 4 H, PCH), 1.87–1.22 (m, 32 H, CH2). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 148.1 (d, 2JCP = 10 Hz, C3), 141.5 (d, 3JCP = 5 Hz, C2), 136.8 (C1), 133.6 (d, 3JCP = 7 Hz, CHc), 132.5 (d, 4JCP = 2 Hz, CHd), 131.9 (CH-Ar), 130.6 (CH-Ar), 129.3 (CHb), 129.1 (CHa), 128.8 (C–Ar), 126.5 (d, 1JCP = 46 Hz, C4), 125.7 (CH-Ar), 124.2 (C≡CPh), 121.6 (C≡CPh), 120.1 (q, 1JCF = 323 Hz, CF3), 40.2 (d, 1JCP = 36 Hz, PCH), 38.7 (d, 3JCP = 7 Hz, CH2), 38.4 (d, 3JCP = 6.6 Hz, CH2), 35.8 (d, 2JCP = 12 Hz, CH2), 32.9 (d, 2JCP = 13 Hz, CH2), 21.6 (MeXyl). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 53.9. IR (Nujol): ν(C≡C) 2029 cm–1.
[(PMe2ArXyl2)Au(μ-η2:η1-C≡CPh)Pt(H)(PtBu3)2]NTf2 (12a)
To a solution of 1a (100 mg, 0.121 mmol) and 2 (73 mg, 0.121 mmol) in toluene (5 mL) was added 1 equiv of phenylacetylene (13 μL, 0.121 mmol) and the mixture stirred at room temperature for 15 min. The volatiles were removed in vacuo, and the residue was washed with pentane (3 × 5 mL) to yield compound 12a as a white powder (70 mg, 38%). Anal. Calcd for C58H87AuF6NO4P3PtS2: C, 45.7; H, 5.8; N, 0.9; S, 4.2. Found: C, 45.9; H, 5.6; N, 1.1; S, 4.3. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.39 (d, 2 H, 3JHH = 7.3 Hz, o-C6H5), 7.15 (m, 3 H, m-C6H5, p-C6H5), 7.09 (m, 5 H, Hb, Hd), 6.99 (d, 4 H, 3JHH = 7.6 Hz, Ha), 6.58 (dd, 2 H, 3JHH = 7.6 Hz, 4JHP = 3.3 Hz, Hc), 2.06 (s, 12 H, MeXyl), 1.38 (vt, 54 H, 3JHP = 6.3 Hz, tBu), 1.33 (d, 6 H, 2JHP = 10 Hz, PMe2), −10.40 (m, 1H, 2JHP = 14 Hz, 1JHPt = 608 Hz, Pt-H). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 145.9 (d, 2JCP = 10 Hz, C3), 140.5 (d, 3JCP = 4 Hz, C2), 137.9 (C1), 136.6 (CH-Ar), 132.2 (d, 4JCP = 2 Hz, CHd), 131.2 (CH-Ar), 129.3 (d, 3JCP = 9 Hz, CHc), 129.2 (C-Ar), 128.9 (CHb), 128.6 (CHa), 125.7 (d, 1JCP = 60 Hz, C4), 124.2 (CH-Ar), 121.3 (q, 1JCF = 323 Hz, CF3), 91.1 (C≡CPh), 85.9 (C≡CPh), 41.0 (vt, 1JCP = 8 Hz, 2JCPt = 35 Hz, Pt-P(C(CH3)3), 33.1 (Pt-P(C(CH3)3), 32.2 (MeXyl), 21.9 (d, 1JCP = 36 Hz, PMe2). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 82.2 (1JPPt = 2810 Hz, P(tBu)3), 3.3 (Au–P). IR (Nujol): ν(C≡C) 1982 cm–1.
[(PMe2ArDipp2)Au(C≡CPh)] (13b)
Following a previously reported method,27 to a solution of phenylacetylene (36 μL, 0.325 mmol) and KOH (18 mg, 0.325 mmol) in 15 mL of methanol was added a suspension of (PMe2ArDipp2)AuCl (150 mg, 0.216 mmol). The solution was stirred for 20 h at room temperature, and the solid was filtered and washed with diethyl ether (2 × 5 mL) to yield compound 13b as a white solid (139 mg, 85%). Gold acetylides are potentially explosive and should be handled with caution. Anal. Calcd for C40H48AuP: C, 63.5; H, 6.4. Found: C, 63.2; H, 6.7. 1H NMR (400 MHz, C6D6, 25 °C): δ 7.74 (d, 2 H, 3JHH = 7.3 Hz, o-C6H5), 7.36 (t, 2 H, 3JHH = 7.8 Hz, m-C6H5), 7.17 (d, 4 H, 3JHH = 7.8 Hz, Ha), 7.06 (t, 1 H, 3JHH = 8.3 Hz, p-C6H5), 6.99 (m, 5 H, Hb, Hc, Hd), 2.64 (sept, 4 H, 3JHH = 6.5 Hz, iPr(CH)), 1.34 (d, 12 H, 3JHH = 6.5 Hz, iPr(CH3)), 0.92 (d, 12 H, 3JHH = 6.5 Hz, iPr(CH3)), 0.90 (d, 6 H, 2JHP = 10 Hz, PMe2). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ 146.6 (CH-Ar), 145.7 (d, 2JCP = 11 Hz, C3), 139.0 (d, 3JCP = 4 Hz, C2), 135.8 (C1), 134.3 (CH-Ar), 132.8 (d, 3JCP = 7 Hz, CHc), 132.6 (d, 4JCP = 2 Hz, CHd), 130.4 (d, 1JCP = 47 Hz, C4), 129.8 (CHb), 128.9 (CHa), 125.9 (C-Ar), 123.8 (CH-Ar), 123.0 (C≡CPh), 102.4 (C≡CPh), 31.6 (iPr(CH)), 25.7 (iPr(CH3)), 23.2 (iPr(CH3)), 16.9 (d, 1JCP = 35 Hz, PMe2). 31P{1H} NMR (160 MHz, C6D6, 25 °C): δ 9.7. IR (Nujol): ν(C≡C) 2115 cm–1. MS-ESI (m/z): calcd for C40H48AuPt, 756.76; found, [M + H] 757.3, [M + Na] 779.3, [M + K] 795.3.
Computational Details
Geometry optimization of minima and transition states was carried out with the Gaussian software package.28 Optimizations were carried out without symmetry restrictions using the ωB97xD functional,29 which includes empirical dispersion corrections.30 The 6-31g(d,p) basis set31 was used for nonmetal atoms, and Au and Pt atoms were described with the SDD basis and associated electron core potential (ECP).32 Bulk solvent effects (dichloromethane) were included during optimization with the SMD continuum model.33 Vibrational analysis was carried out on the stationary points to characterize them as minima or transition states as well as to calculate the zero-point corrections, and thermal corrections were made to the enthalpy and free energy. Free energies were corrected (ΔGqh) to account for errors associated with the harmonic oscillator approximation. Thus, according to Truhlar’s quasi harmonic approximation, all vibrational frequencies below 100 cm–1 were set to this value so that the entropy contribution was not overestimated.34 These anharmonic corrections were calculated with the Goodvibes code.35 The CYLview visualization software has been used to create some of the figures.36
Acknowledgments
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575) and by the Spanish Ministry of Economy and Competitiveness (Project CTQ2016-75193-P [AEI/FEDER, UE]). J.J.M. and M.P.-J. thank the University of Sevilla and the Ministry of Science, Innovation and Universities, respectively, for Ph.D. fellowships. The use of computational facilities at the Supercomputing Centre of Galicia (CESGA) and the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, are acknowledged. We gratefully acknowledge Prof. Ernesto Carmona for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.0c00330.
Accession Codes
CCDC 1965525, 1965526 and 1986501–1986504 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]; b Stephan D. W.; Erker G. Frustrated Lewis Pairs II. Top. Curr. Chem. 2013, 334, 1. [DOI] [PubMed] [Google Scholar]; c Stephan D. W.; Erker G. Frustrated Lewis Pairs I. Top. Curr. Chem. 2012, 332, 1. 10.1007/128_2012_381. [DOI] [PubMed] [Google Scholar]; d Stephan D. W.; Erker G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem., Int. Ed. 2010, 49, 46–76. 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]; e Stephan D. W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. 10.1021/jacs.5b06794. [DOI] [PubMed] [Google Scholar]; f Stephan D. W. The Broadening Reach of Frustrated Lewis Pair Chemistry. Science 2016, 354, aaf7229 10.1126/science.aaf7229. [DOI] [PubMed] [Google Scholar]; g Jupp A. R.; Stephan D. W. New Directions for Frustrated Lewis Pair Chemistry. Trends in Chemistry 2019, 1, 35–48. 10.1016/j.trechm.2019.01.006. [DOI] [Google Scholar]
- Flynn S. R.; Wass D. F. Transition Metal Frustrated Lewis Pairs. ACS Catal. 2013, 3, 2574–2581. 10.1021/cs400754w. [DOI] [Google Scholar]
- For recent examples see:; a Hamilton H. B.; King A. M.; Sparkes H. A.; Pridmore N. E.; Wass D. F. Zirconium-Nitrogen Intermolecular Frustrated Lewis Pairs. Inorg. Chem. 2019, 58, 6399–6409. 10.1021/acs.inorgchem.9b00569. [DOI] [PubMed] [Google Scholar]; b Jian Z.; Daniliuc C. G.; Kehra G.; Erker G. Frustrated Lewis Pair vs Metal-Carbon σ-Bond Insertion Chemistry at an o-Phenylene-Bridged Cp2Zr+/PPh2 System. Organometallics 2017, 36, 424–434. 10.1021/acs.organomet.6b00828. [DOI] [Google Scholar]; c Jiang Y.; Blacque O.; Fox T.; Berke H. Catalytic CO2 Activation Assisted by Rhenium Hydride/B(C6F5)3 Frustrated Lewis Pairs–Metal Hydrides Functioning as FLP Bases. J. Am. Chem. Soc. 2013, 135, 7751–7760. 10.1021/ja402381d. [DOI] [PubMed] [Google Scholar]; d Simonneau A.; Turrel R.; Vendier L.; Etienne M. Group 6 Transition-Metal/Boron Frustrated Lewis Pair Templates Activate N2 and Allow its Facile Borylation and Silylation. Angew. Chem., Int. Ed. 2017, 56, 12268–12272. 10.1002/anie.201706226. [DOI] [PubMed] [Google Scholar]; e Rahman M. M.; Smith M. D.; Amaya J. A.; Makris T. M.; Peryshkov D. V. Activation of C-H Bonds of Alkyl-and Arylnitriles by the TaCl5-PPh3 Lewis Pair. Inorg. Chem. 2017, 56, 11798–11803. 10.1021/acs.inorgchem.7b01800. [DOI] [PubMed] [Google Scholar]; f Cui P.; Comanescu C. C.; Iluc V. M. Frustrated Lewis Pair-like Reactions of Nucleophilic Palladium Carbenes with B(C6F5)3. Chem. Commun. 2015, 51, 6206–6209. 10.1039/C5CC00868A. [DOI] [PubMed] [Google Scholar]; g Chang K.; Xu X. Frustrated Lewis Pair Behavior of a Neutral Scandium Complex. Dalton Trans. 2017, 46, 4514–4517. 10.1039/C7DT00676D. [DOI] [PubMed] [Google Scholar]; h Wang Z.; Ying A.; Fan Z.; Hervieu C.; Zhang L. Tertiary Amino Group in Cationic Gold Catalyst: Tethered Frustrated Lewis Pairs That Enable Ligand-Controlled Regiodivergent and Stereoselective Isomerizations of Propargylic Esters. ACS Catal. 2017, 7, 3676–3680. 10.1021/acscatal.7b00626. [DOI] [Google Scholar]; i Barnett B. R.; Neville M. L.; Moore C. E.; Rheingold A. L.; Figueroa J. S. Oxidative-Insertion Reactivity Across a Geometrically Constrained Metal→Borane Interaction. Angew. Chem., Int. Ed. 2017, 56, 7195–7199. 10.1002/anie.201702151. [DOI] [PubMed] [Google Scholar]; j Zhang S.; Appel A. M.; Bullock M. Reversible Heterolytic Cleavage of the H-H Bond by Molybdenum Complexes: Controlling the Dynamics of Exchange Between Proton and Hydride. J. Am. Chem. Soc. 2017, 139, 7376–7387. 10.1021/jacs.7b03053. [DOI] [PubMed] [Google Scholar]; k Arndt S.; Rudolph M.; Hashmi A. S. K. Gold-based Frustrated Lewis Acid/Base Pairs (FLPs). Gold Bull. 2017, 50, 267–282. 10.1007/s13404-017-0219-7. [DOI] [Google Scholar]; l Zwettler N.; Walg S. P.; Belaj F.; Mösch-Zanetti N. C. Heterolytic Si-H Bond Cleavage at a Molybdenum-Oxido-Based Lewis Pair. Chem. - Eur. J. 2018, 24, 7149–7160. 10.1002/chem.201800226. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Chang K.; Wang X.; Fan Z.; Xu X. Reactions of Neutral Scandium/Phosphorus Lewis Pairs with Small Molecules. Inorg. Chem. 2018, 57, 8568–8580. 10.1021/acs.inorgchem.8b01292. [DOI] [PubMed] [Google Scholar]
- Chapman A. M.; Haddow M. F.; Wass D. F. Frustrated Lewis Pairs Beyond the Main Group: Synthesis, Reactivity, and Small Molecule Activation with Cationic Zirconocene-Phosphinoaryloxide Complexes. J. Am. Chem. Soc. 2011, 133, 18463–18478. 10.1021/ja207936p. [DOI] [PubMed] [Google Scholar]
- Metters O. J.; Forrest S. J. K.; SparkesIan H. A.; Manners I.; Wass D. F. Small Molecule Activation by Intermolecular Zr(IV)-Phosphine Frustrated Lewis Pairs. J. Am. Chem. Soc. 2016, 138, 1994–2003. 10.1021/jacs.5b12536. [DOI] [PubMed] [Google Scholar]
- a Campos J. Dihydrogen and Acetylene Activation by a Gold(I)/Platinum(0) Transition Metal Only Frustrated Lewis Pair. J. Am. Chem. Soc. 2017, 139, 2944–2947. 10.1021/jacs.7b00491. [DOI] [PubMed] [Google Scholar]; b Hidalgo N.; Bajo S.; Moreno J. J.; Navarro-Gilabert C.; Mercado B.; Campos J. Reactivity of a Gold(I)/Platinum(0) Frustrated Lewis Pair with Germanium and Tin Dihalides. Dalton Trans. 2019, 48, 9127–9138. 10.1039/C9DT00702D. [DOI] [PubMed] [Google Scholar]
- Hidalgo N.; Moreno J. J.; Pérez-Jiménez M.; Maya C.; López-Serrano J.; Campos J. Evidence for Genuine Bimetallic Frustrated Lewis Pair Activation of Dihydrogen with Gold(I)/Platinum(0) Systems. Chem. - Eur. J. 2020, 26, 5982–5993. 10.1002/chem.201905793. [DOI] [PubMed] [Google Scholar]
- a Rokob T. A.; Pápai I. Hydrogen Activation by Frustrated Lewis Pairs: Insights from Computational Studies. Top. Curr. Chem. 2013, 332, 157–211. 10.1007/128_2012_399. [DOI] [PubMed] [Google Scholar]; b Paradies J. Mechanisms in Frustrated Lewis Pair-Catalyzed Reactions. Eur. J. Org. Chem. 2019, 2019, 283–294. 10.1002/ejoc.201800944. [DOI] [Google Scholar]; c Rocchigiani L. Experimental Insights into the Structure and Reactivity of Frustrated Lewis Pairs. Isr. J. Chem. 2015, 55, 134–149. 10.1002/ijch.201400139. [DOI] [Google Scholar]; d Liu L.; Lukose B.; Jaque P.; Ensing B. Reaction Mechanism of Hydrogen Activation by Frustrated Lewis pairs. Green Energy Environ 2019, 4, 20–28. 10.1016/j.gee.2018.06.001. [DOI] [Google Scholar]
- Rokob T. A.; Hamza A.; Stirling A.; Pápai I. On the Mechanism of B(C6F5)3-Catalyzed Direct Hydrogenation of Imines: Inherent and Thermally Induced Frustration. J. Am. Chem. Soc. 2009, 131, 2029–2036. 10.1021/ja809125r. [DOI] [PubMed] [Google Scholar]
- a Mahdi T.; Stephan D. W. Enabling Catalytic Ketone Hydrogenation by Frustrated Lewis Pairs. J. Am. Chem. Soc. 2014, 136, 15809–15812. 10.1021/ja508829x. [DOI] [PubMed] [Google Scholar]; b Scott D. J.; Fuchter M. J.; Ashley A. E. Nonmetal Catalyzed Hydrogenation of Carbonyl Compounds. J. Am. Chem. Soc. 2014, 136, 15813–15816. 10.1021/ja5088979. [DOI] [PubMed] [Google Scholar]; c Légaré M. A.; Rochette E.; Lavergne J. L.; Bouchard N.; Fontaine F. G. Bench-Stable Frustrated Lewis Pair Chemistry: Fluoroborate Salts as Precatalysts for the C-H Borylation of Heteroarenes. Chem. Commun. 2016, 52, 5387–5390. 10.1039/C6CC01267A. [DOI] [PubMed] [Google Scholar]
- Legare Lavergne J.; Jayaraman A.; Misal Castro L. C.; Rochette E.; Fontaine F.-G. Metal-Free Borylation of Heteroarenes Using Ambiphilic Aminoboranes: On the Importance of Sterics in Frustrated Lewis Pair C-H Bond Activation. J. Am. Chem. Soc. 2017, 139, 14714–14723. 10.1021/jacs.7b08143. [DOI] [PubMed] [Google Scholar]
- a Jiang C.; Blacque O.; Berke H. Activation of Terminal Alkynes by Frustrated Lewis Pairs. Organometallics 2010, 29, 125–133. 10.1021/om9008636. [DOI] [Google Scholar]; b Dureen M. A.; Brown C. C.; Stephan D. W. Deprotonation and Addition Reactions of Frustrated Lewis Pairs with Alkynes. Organometallics 2010, 29, 6594–6607. 10.1021/om1009044. [DOI] [Google Scholar]
- Lu Z.; Ye H.; Wang H. New Organoboranes in “Frustrated Lewis Pair” Chemistry. Top. Curr. Chem. 2012, 334, 59–80. 10.1007/128_2012_382. [DOI] [PubMed] [Google Scholar]
- See for example:; a Dureen M. A.; Stephan D. W. Terminal Alkyne Activation by Frustrated and Classical Lewis Acid/Phosphine Pairs. J. Am. Chem. Soc. 2009, 131, 8396–8397. 10.1021/ja903650w. [DOI] [PubMed] [Google Scholar]; b Fukazawa A.; Yamada H.; Yamaguchi S. Phosphonium-and borate-bridged zwitterionic ladder stilbene and its extended analogues. Angew. Chem., Int. Ed. 2008, 47, 5582–5585. 10.1002/anie.200801834. [DOI] [PubMed] [Google Scholar]; c Geier S. J.; Dureen M. A.; Ouyang E. Y.; Stephan D. W. New Strategies to Phosphino-Phosphonium Cations and Zwitterions. Chem. - Eur. J. 2010, 16, 988–993. 10.1002/chem.200902369. [DOI] [PubMed] [Google Scholar]
- Marín M.; Moreno J. J.; Alcaide M. M.; Álvarez E.; López-Serrano J.; Campos J.; Nicasio M. C.; Carmona E. Evaluating Stereoelectronic Properties of Bulky Dialkylterphenylphosphine Ligands. J. Organomet. Chem. 2019, 896, 120–128. 10.1016/j.jorganchem.2019.06.003. [DOI] [Google Scholar]
- Goel R. G.; Srivastava R. C. Preparation, Characterization, and Some Reactions of Hydridobis(tri-tert-butyl)phosphineplatinum(II) Cation Containing Three-Coordinate Platinum. Can. J. Chem. 1983, 61, 1352–1359. 10.1139/v83-238. [DOI] [Google Scholar]
- Ortega-Moreno L.; Fernández-Espada M.; Moreno J. J.; Navarro C.; Campos J.; Conejero S.; López-Serrano J.; Maya C.; Peloso R.; Carmona E. Synthesis, Properties, and Some Rhodium, Iridium, and Platinum Complexes of a Series of Bulky m-Terphenylposphine Ligands. Polyhedron 2016, 116, 170–181. 10.1016/j.poly.2016.04.023. [DOI] [Google Scholar]
- Espada M. F.; Campos J.; López-Serrano J.; Poveda M. L.; Carmona E. Methyl-, Ethenyl-, and Ethynyl-Bridged Cationic Digold Complexes Stabilized by Coordination to a Bulky Terphenylphosphine Ligand. Angew. Chem., Int. Ed. 2015, 54, 15379–15384. 10.1002/anie.201508931. [DOI] [PubMed] [Google Scholar]
- a Steinborn D.; Aisa A. M. A.; Heinemann F. W.; Lehmann S. Synthesis and Structure of a μ-(E)-vinylene-bis[dimethylglyoximato(1-)-dimethylglyoximato(2-)-(triphenylphosphine)rhodate] Complex a Vinylene-Bridged Dinuclear Rhodium Complex. J. Organomet. Chem. 1997, 527, 239–245. 10.1016/S0022-328X(96)06648-X. [DOI] [Google Scholar]; b Wieteck M.; Vilhelmsen M. H. L.; Nösel P.; Schulmeister J.; Rominger F.; Rudolph M.; Pernpointner M.; Hashmi A. S. K. Conjugated Vinylgold(I)-Vinylideneruthenium(II) Complexes and Related Organoruthenium Compounds: Stable Analogues of Intermediates Proposed in Dual Gold Catalysis. Adv. Synth. Catal. 2016, 358, 1449–1462. 10.1002/adsc.201600255. [DOI] [Google Scholar]
- a Arndt S.; Hansmann M. M.; Motloch P.; Rudolph M.; Rominger F.; Hashmi A. S. K. Intramolecular anti-Phosphinoauration of Alkynes: An FLP-Motivated Approach to Stable Aurated Phosphindolium Complexes. Chem. - Eur. J. 2017, 23, 2542–2547. 10.1002/chem.201605914. [DOI] [PubMed] [Google Scholar]; b Arndt S.; Hansmann M. M.; Rominger F.; Rudolph M.; Hashmi A. S. K. Direct Access to π-Extended Phosphindolium Salts by Simple Proton-Induced Cyclization of (o-Alkynylphenyl)phosphanes. Chem. - Eur. J. 2017, 23, 5429–5433. 10.1002/chem.201700889. [DOI] [PubMed] [Google Scholar]; c Arndt S.; Borstelmann J.; Saatlo R. E.; Antoni P. W.; Rominger F.; Rudolph M.; An Q.; Vaynzof Y.; Hashmi A. S. K. The Gold(I)-Mediated Domino Reaction to Fused Diphenyl Phosphoniumfluorenes: Mechanistic Consequences for Gold-Catalyzed Hydroarylations and Application in Solar Cells. Chem. - Eur. J. 2018, 24, 7882–7889. 10.1002/chem.201800460. [DOI] [PubMed] [Google Scholar]
- a Brown T. J.; Widenhoefer R. A. Cationic Gold(I) π-Complexes of Terminal Alkynes and Their Conversion to Dinuclear σ,π-Acetylide Complexes. Organometallics 2011, 30, 6003–6009. 10.1021/om200840g. [DOI] [Google Scholar]; b Hashmi A. S. K; Lauterbach T.; Nösel P.; Vilhelmsen M. H.; Rudolph M.; Rominger F. Dual Gold Catalysis: σ,π-Propyne Acetylide and Hydroxyl-Bridged Digold Complexes as Easy-To-Prepare and Easy-To-Handle Precatalysts. Chem. - Eur. J. 2013, 19, 1058–1065. 10.1002/chem.201203010. [DOI] [PubMed] [Google Scholar]; c Zhao X.; Rudolph M.; Hashmi A. S. K. Dual gold catalysis-an update. Chem. Commun. 2019, 55, 12127–12135. 10.1039/C9CC06078B. [DOI] [PubMed] [Google Scholar]; d Asiri A. M.; Hashmi A. S. K. Gold-catalysed reactions of diynes. Chem. Soc. Rev. 2016, 45, 4471–4503. 10.1039/C6CS00023A. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Li S. A Novel Addition Mechanism for the Reaction of “Frustrated Lewis Pairs” with Olefins. Eur. J. Inorg. Chem. 2008, 2008, 2501–2505. 10.1002/ejic.200800281. [DOI] [Google Scholar]
- a Debrouwer W.; Heugebaert T. S. A.; Roman B. I.; Stevens C. V. Homogeneous Gold-Catalyzed Cyclization Reactions of Alkynes with N-and S-Nucleophiles. Adv. Synth. Catal. 2015, 357, 2975–3006. 10.1002/adsc.201500520. [DOI] [Google Scholar]; b Couce-Rios A.; Lledós A.; Fernández I.; Ujaque G. Origin of the Anti-Markovnikov Hydroamination of Alkenes Catalyzed by L-Au(I) Complexes: Coordination Mode Determines Regioselectivity. ACS Catal. 2019, 9, 848–858. 10.1021/acscatal.8b03843. [DOI] [Google Scholar]
- See for example:; a Gimeno A.; Cuenca A. B.; Suárez-Pantiga S.; De Arellano C. R.; Medio-Simýn M.; Asensio G. Competitive Gold-Activation Modes in Terminal Alkynes: An Experimental and Mechanistic Study. Chem. - Eur. J. 2014, 20, 683–688. 10.1002/chem.201304087. [DOI] [PubMed] [Google Scholar]; b Hansmann M. M.; Rominger F.; Boone M. P.; Stephan D. W.; Hashmi A. S. K. Reactivity of Organogold Compounds with B(C6F5)3: Gold-Boron Transmetalation via σ-B/π-Au Species. Organometallics 2014, 33, 4461–4470. 10.1021/om5006885. [DOI] [Google Scholar]; c Grirrane A.; García H.; Corma A.; Álvarez E. Air-Stable, Dinuclear and Tetranuclears σ,π-Acetylide Gold(I) Complexes and Their Catalytic Implications. Chem. - Eur. J. 2013, 19, 12239–12244. 10.1002/chem.201301623. [DOI] [PubMed] [Google Scholar]; d Obradors C.; Echavarren A. M. Intermolecular Gold-Catalyzed Cycloaddition of Alkynes with Oxoalkenes. Chem. - Eur. J. 2013, 19, 3547–3551. 10.1002/chem.201300131. [DOI] [PubMed] [Google Scholar]; e Grirrane A.; García H.; Corma A.; Álvarez E. Intermolecular [2 + 2] Cycloaddition of Alkyne-Alkene Catalyzed by Au(I) Complexes. What Are the Catalytic Sites Involved?. ACS Catal. 2011, 1, 1647–1653. 10.1021/cs2004278. [DOI] [Google Scholar]; f Gómez-Suárez A.; Dupuy S.; Slawin A. M. Z.; Nolan S. P. Straightforward Synthetic Access to gem-Diaurated and Digold σ,π-Acetylide Species. Angew. Chem., Int. Ed. 2013, 52, 938–942. 10.1002/anie.201208234. [DOI] [PubMed] [Google Scholar]
- Uson R.; Laguna A.; Laguna M.; Briggs D. A.; Murray H. H.; Fackler J. P. (Tetrahydrothiophene)gold(I) or gold(III) Complexes. Inorg. Synth. 2007, 26, 85–91. 10.1002/9780470132579.ch17. [DOI] [Google Scholar]
- Jaw H. R. C.; Mason W. R. Electronic Absorption and MCD Spectra for Isoelectronic Linear Two-Coordinate Bis(tri-tert-butylphosphine)metal Complexes of Platinum(0) and Gold(I). Inorg. Chem. 1989, 28, 4370–4373. 10.1021/ic00323a017. [DOI] [Google Scholar]
- Wan X.-K.; Cheng X.-L.; Tang Q.; Han Y.-Z.; Hu G.; Jiang D.; Wang Q.-M. Atomically Precise Bimetallic Au19Cu30 Nanocluster with an Icosidodecahedral Cu30 Shell and an Alkynyl-Cu Interface. J. Am. Chem. Soc. 2017, 139, 9451–9554. 10.1021/jacs.7b04622. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Rev. B.01; Gaussian, Inc.: Wallingford CT, 2010.
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- a Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]; b Hehre W. J.; Ditchfield R.; Pople J. A. Self–Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]; c Hariharan P. C.; Pople J. A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]; d Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. 10.1063/1.444267. [DOI] [Google Scholar]
- Andrae D.; Haeussermann U.; Dolg M.; Stoll H.; Preuss H. Energy-Adjusted Ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Funes-Ardoiz I.; Paton R. S.. Goodvibes 2.0.2, 2016; 10.5281/zenodo.595246. [DOI]
- Legault C. Y.CYLview, 1.0b; Université de Sherbrooke: 2009; http://www.cylview.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.