

Abstract
Platelet-derived growth factor is one of the major growth factors found in human and mammalian serum and tissues. Abnormal activation of platelet-derived growth factor signaling pathway through platelet-derived growth factor receptors may contribute to the development and progression of pulmonary vascular remodeling and obliterative vascular lesions in patients with pulmonary arterial hypertension. In this study, we examined the expression of platelet-derived growth factor receptor isoforms in pulmonary arterial smooth muscle and pulmonary arterial endothelial cells and investigated whether platelet-derived growth factor secreted from pulmonary arterial smooth muscle cell or pulmonary arterial endothelial cell promotes pulmonary arterial smooth muscle cell proliferation. Our results showed that the protein expression of platelet-derived growth factor receptor α and platelet-derived growth factor receptor β in pulmonary arterial smooth muscle cell was upregulated in patients with idiopathic pulmonary arterial hypertension compared to normal subjects. Platelet-derived growth factor activated platelet-derived growth factor receptor α and platelet-derived growth factor receptor β in pulmonary arterial smooth muscle cell, as determined by phosphorylation of platelet-derived growth factor receptor α and platelet-derived growth factor receptor β. The platelet-derived growth factor-mediated activation of platelet-derived growth factor receptor α/platelet-derived growth factor receptor β was enhanced in idiopathic pulmonary arterial hypertension-pulmonary arterial smooth muscle cell compared to normal cells. Expression level of platelet-derived growth factor-AA and platelet-derived growth factor-BB was greater in the conditioned media collected from idiopathic pulmonary arterial hypertension-pulmonary arterial endothelial cell than from normal pulmonary arterial endothelial cell. Furthermore, incubation of idiopathic pulmonary arterial hypertension-pulmonary arterial smooth muscle cell with conditioned culture media from normal pulmonary arterial endothelial cell induced more platelet-derived growth factor receptor α activation than in normal pulmonary arterial smooth muscle cell. Accordingly, the conditioned media from idiopathic pulmonary arterial hypertension-pulmonary arterial endothelial cell resulted in more pulmonary arterial smooth muscle cell proliferation than the media from normal pulmonary arterial endothelial cell. These data indicate that (a) the expression and activity of platelet-derived growth factor receptor are increased in idiopathic pulmonary arterial hypertension-pulmonary arterial smooth muscle cell compared to normal pulmonary arterial smooth muscle cell, and (b) pulmonary arterial endothelial cell from idiopathic pulmonary arterial hypertension patients secretes higher level of platelet-derived growth factor than pulmonary arterial endothelial cell from normal subjects. The enhanced secretion (and production) of platelet-derived growth factor from idiopathic pulmonary arterial hypertension-pulmonary arterial endothelial cell and upregulated platelet-derived growth factor receptor expression (and function) in idiopathic pulmonary arterial hypertension-pulmonary arterial smooth muscle cell may contribute to enhancing platelet-derived growth factor/platelet-derived growth factor receptor-associated pulmonary vascular remodeling in pulmonary arterial hypertension.
Keywords: platelet-derived growth factor, platelet-derived growth factorreceptor, smooth muscle cell, endothelial cell
Introduction
Pulmonary vascular remodeling characterized by concentric pulmonary arterial wall thickening and muscularization of small pulmonary arteries (PAs) and precapillary arterioles is a major cause for the elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with pulmonary arterial hypertension (PAH).1,2 Increased proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) have been implicated in the development and progression of pulmonary vascular remodeling in PAH,3,4 while the enhanced cytosolic free Ca2+ concentration ([Ca2+]cyt) is an important stimulus for PASMC proliferation and migration, contributing to pulmonary vascular remodeling.5–8 Regardless of the cause of PAH, the common denominator of the disease pathogenesis is superimposed release of some growth factors, systemic hormones, cytokines, chemokines, and resultant activation of transcription factors.8–10 The altered activation of growth factor-induced pathways drives concentric pulmonary arterial remodeling, pulmonary arteriole muscularization, and obliterative intimal lesions predominantly resulting in the elevation of pulmonary vascular resistance.11
Platelet-derived growth factor (PDGF) plays an essential role in regulating cell proliferation, differentiation, and migration.12,13 The increased level of PDGF-AA and PDGF-BB in serum and PASMC has been demonstrated in patients with PAH and animals with experimental pulmonary hypertension (PH).14–16 Inhibitors of tyrosine kinase receptors (TKRs) of PDGF (i.e. imatinib) in the pulmonary vasculature have been demonstrated to have a therapeutic effect on severe PH based on in vivo studies using animal models of experimental PH and patients with PAH by attenuating and reversing pulmonary vascular remodeling.17–20
PDGF consists of four different polypeptide chains: A, B, C, and D. The four PDGF chains can assemble into four homodimers (e.g. PDGF-AA, PDGF-BB, PDGF-CC, or PDGF-DD) and one heterodimer (e.g. PDGF-AB). Although the circulating forms of PDGF in humans are, for the most part, PDGF-BB, PDGF-AA, and PDGF-AB, among the PDGF isoforms PDGF-BB is a more potent ligand. PDGF-AA, PDGF-AB, and PDGF-BB are already activated intracellularly by furin-like proteases while PDGF-CC and PDGF-DD are secreted as inactive precursor molecules with an N-terminal CUB.12,21 The N-terminal CUB-domains need to be extracellularly cleaved before it can bind and activate its receptor. PDGF-BB is a well-known potent mitogen that stimulates mesenchymal phenotype of cells (e.g. smooth muscle cells, myofibroblasts, and fibroblasts) in the vasculature with high efficiency.22–25 PDGF receptors (PDGFRs), like most of TKRs, are composed of two members: PDGFRα and PDGFRβ. The PDGF polypeptide chains bind to the receptors with different affinities.26
The PDGF-AA and PDGF-BB polypeptide chains are functionally capable of activating PDGFR in the plasma membrane and, via various intracellular signaling pathways, stimulating PASMC proliferation and migration. Upon activation of PDGFRs, the downstream signaling cascades include, at least, two main molecular mechanisms: PI3K/AKT/mTOR and MAPK pathways. The PI3K/AKT/mTOR cascade contributes to pulmonary vascular remodeling by promoting cell proliferation, survival, and metabolic changes (Warburg effect) while MAPK signaling tightly controls cell proliferation, migration, and differentiation.12,27,28 We have previously demonstrated that PDGF-BB ligand enhances [Ca2+]cyt and promotes PASMC proliferation through AKT/mTOR pathway.7 Moreover, PDGF-BB-mediated activation of AKT/mTOR pathway and a rise in [Ca2+]cyt are also involved in switch of PASMC from quiescent (contractile) phenotype to a synthetic (proliferative) phenotype.29 PASMC phenotype switch characterized by the increased proliferation and migration has been recently linked to pulmonary vascular remodeling in PH.30
In the pulmonary circulation system, PDGF-AA and PDGF-BB are mainly synthesized and released from lung vascular endothelial cells and circulating inflammatory cells (e.g. macrophages).16,21,31 It appears that PDGF is constitutively produced in these cells and released to extracellular or intercellular space or blood to exert mitogenic and migratory effects on cells via paracrine and autocrine mechanisms. PDGF released from the endothelial cells probably helps in attracting PASMC from the media to the intima and/or fibroblasts from the adventitia to the media/intima to induce vascular remodeling; however, the direct evidence that endothelium-derived PDGF stimulates PASMC proliferation is lacking.
In this study, we aimed at investigating whether protein expression level of PDGFRs in PASMC from patients with idiopathic PAH (IPAH) was different from that in PASMC from control subjects, and whether endothelial release of PDGF was sufficient to activate the PDGF/AKT signaling in PASMC, an important pathway to stimulate PASMC proliferation, ultimately contributing to arterial wall thickening and arteriole muscularization.
Methods and materials
Culture of PASMC and pulmonary arterial endothelial cell (PAEC)
Human PASMC and PAEC isolated from three normal subjects and five IPAH patients were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI, Philadelphia, PA). PASMCs were cultured in VascuLife SMC growth medium (Lifeline Cell Technologies, Frederick, MD) supplemented with 5% fetal bovine serum (FBS), 5 ng/ml human fibroblast growth factor (FGF), 5 µg/ml insulin, 50 µg/ml ascorbic acid, 10 mM l-glutamine, 5 ng/ml human epidermal growth factor (EGF), 30 mg/ml gentamicin, and 15 µg/ml amphotericin B; this is referred to as PASMC growth medium. PAECs were cultured in VascuLife vascular endothelial growth factor (VEGF) growth medium (Lifeline Cell Technologies, Frederick, MD) supplemented with 2% FBS, 5 ng/ml human EGF, 5 ng/ml human FGF, 50 µg/ml ascorbic acid, 1 µg/ml hydrocortisone hemisuccinate, 10 mM l-glutamine, 15 ng/ml human insulin-like growth factor (IGF-1), 5 ng/ml human VEGF, 0.75 U/ml heparin sulfate, 30 mg/ml gentamicin, and 15 µg/ml amphotericin B; this is referred to as PAEC growth medium. VascuLife basal medium (Lifeline Cell Technologies, Frederick, MD) without any supplements, referred to as the basal medium, was used to prepare 0.3 and 10% FBS medium for both PASMC and PAEC. The cells were cultured in an incubator under a humidified atmosphere of 5% CO2 and 95% air at 37℃. The cells of passage 5–8 were used for the experiments. The demographic information of control subjects and patients from whom we obtained PASMC and PAEC is shown in Table 1.
Table 1.
Demographic information of human subjects.
| Subjects | Gender | Age (yrs) | Race | Type of cells |
|---|---|---|---|---|
| Normal sample-1 | Male | 46 | Asian | PASMC |
| Normal sample-2 | Female | 34 | Asian | PASMC |
| Normal sample-3 | Female | 33 | White | PASMC |
| IPAH sample-1 | Female | 56 | White | PASMC |
| IPAH sample-2 | Female | 41 | White | PASMC |
| IPAH sample-3 | Male | 27 | White, non-Hispanic | PASMC |
| IPAH sample-4 | Male | 56 | White, non-Hispanic | PASMC |
| IPAH sample-5 | Female | 33 | White | PASMC |
| Normal sample-1 | Female | 50 | Non-Hispanic | PAEC |
| Normal sample-2 | Female | 36 | Non-Hispanic | PAEC |
| Normal sample-3 | Male | 47 | White | PAEC |
| IPAH sample-1 | Female | 32 | White | PAEC |
| IPAH sample-2 | Female | 16 | White | PAEC |
| IPAH sample-3 | Female | 27 | White, non-Hispanic | PAEC |
IPAH: idiopathic PAH; PAEC: pulmonary arterial endothelial cell; PASMC: pulmonary arterial smooth muscle cell.
Western blot analysis
Total protein was isolated from human PASMC and PAEC using RIPA lysis buffer (Pierce RIPA Product, Rockford, IL) supplemented with protease inhibitor cocktail (Roche; Manheim, Germany) followed by the incubation on ice for 20 min. Then the lysates were centrifuged at 12,000 rpm for 20 min at 4℃. Protein concentration was measured by bovine serum albumin (BSA) kits (Bio-Rad; Hercules, CA). Protein samples were mixed and boiled with 6 × protein loading buffer R (Bio-Rad Laemmli Sample Buffer, CA). Protein was loaded on an 8–12% odium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to 0.45 µm nitrocellulose transfer membranes (Bio-Rad), and blocked by TBST buffer containing 5% nonfat dry milk powder for 1 h at room temperature (22–24℃). Then the membranes were immunoblotted in TBST buffer containing 5% BSA with anti-rabbit PDGFRα antibody (Santa Cruz Biotechnology, CA, USA, 1:1000), anti-rabbit PDGFRβ antibody (Santa Cruz Biotechnology, CA, USA, 1:1000), anti-rabbit phosphorylated (p) PDGFRα (pPDGFRα) antibody (Santa Cruz Biotechnology, CA, USA, 1:1000), anti-rabbit pPDGFRβ antibody (Santa Cruz Biotechnology, CA, USA, 1:1000), anti-rabbit AKT antibody (Cell Signaling, Danvers, MA, 1:1000), anti-rabbit pAKT (S473) antibody (Cell Signaling, Danvers, MA, 1:1000), anti-rabbit pAKT (T308) antibody (Cell Signaling, Danvers, MA, 1:1000), and anti-mouse β-actin antibody (Santa Cruz Biotechnology, CA, USA, 1:1500) overnight at 4℃. At the next day, membranes were washed 3 times in TBST buffer for 5 min each, followed by incubation with secondary antibody in TBST buffer containing 5% milk for 2 h at RT. Signals were detected using enhanced chemiluminescence substrate (Pierce, Rockford, IL). The protein levels were quantified with Image J, normalized to β-actin control.
Immunocytochemistry
The cells grown on 25 mm cover slips in the growth medium were fixed with 4% formaldehyde for 5 min at room temperature (22–24℃) followed by the treatment with 1% Triton X-100 (Sigma-Aldrich) for 10 min at room temperature. Cells were blocked with 5% BSA in PBS for 1 h in a dark humidified chamber. Cells were then stained with anti-PDGFRα (Santa Cruz Biotechnology, CA, USA, 1:200) or anti-PDGFRβ antibodies (Santa Cruz Biotechnology, CA, USA, 1:200) overnight at 4℃ followed by the incubation with FITC-conjugated secondary antibody (Cell Signaling Technology) next day for 1 h at room temperature (22–24℃). DAPI was used for nuclear staining. Images were captured using a fluorescence microscope (Olympus, Japan) and were photographed using a digital camera.
Transfection of small interfering RNA (siRNA)
PASMCs were transiently transfected with control siRNA (10 µM; Santa Cruz Biotechnology), PDGFRα-siRNA (10 µM; Santa Cruz Biotechnology), or PDGFRβ-siRNA (10 µM; Santa Cruz Biotechnology) using Lipofectamine RNAiMAX transfection reagent protocol (Invitrogen, USA). Protein extraction or treatment with PDGF-BB using siRNA-transfected cells was performed 48 h after transfection.
Measurement of cytosolic free Ca2+ concentration ([Ca2+]cyt) in PASMC
[Ca2+]cyt in human PASMC was measured using a digital imaging fluorescent microscopy system (Nikon, Tokyo, Japan) as described previously.32 PASMCs grown on 25 mm cover slips were loaded with Fura-2/AM for 1 h at room temperature (22–24℃). The cover slips with Fura-2-loaded cells were mounted in a closed polycarbonate chamber and clamped to a heated aluminum platform (model PH-2, Warner Instruments, Hamden, CT) on the stage of an inverted microscope (Ellipse Ti-E, Nikon, Tokyo, Japan). The Fura-2 fluorescence (emitted at 510 nm) in cells excited by a xenon lamp (Hamamatsu Photonics, Hamamatsu, Japan) at 340 and 380 nm wavelength of illumination, along with the background fluorescence, was collected at room temperature (22–24℃) using a 40 × Nikon UV-fluor objective and a Nikon Eclipse Ti camera. The fluorescence signals emitted from a region of interest (5 × 5 µm) in a cell was recorded every 2 s. [Ca2+]cyt was calculated as the ratio of fluorescence intensity (F340/F380) using the following equation: [Ca2+]cyt(nM) = Kd [F − Fmin]/[Fmax − F], where F is the fluorescence ratio (F340/F380) in each measured cell; Fmin and Fmax are fluorescence values in Ca2+-free solution and Ca2+-saturated solution, respectively; and Kd (225 nM) is the dissociation constant of the Ca2+-Fura-2 complex at 37℃.
PDGF treatment
To examine PDGF-mediated effect on PASMC, we changed PASMC growth medium, when cell confluence reached 70–80%, to the basal medium without FBS or growth factor. Then, the synchronized PASMCs in the G0/G1 phase (due to 24–48 h of incubation in the basal medium) were treated with the basal medium supplemented with or without 10 ng/ml PDGF-BB for 30 min, followed by extraction of total proteins from the cells for various experiments.
Collection of conditioned medium (CM) from PAEC or PASMC
The initial growth media (with FBS and growth factors) for PASMC and PAEC were first changed to the basal media with 0.3% FBS and no growth factors, when cell confluence reached approximately 80%. Then, the 0.3% FBS basal media from PASMC or PAEC were collected after incubation for the indicated period of time. The supernatant was collected after the basal media were centrifuged at 2000 rpm for 10 min and used as the CM for experimentation. The CM were used to culture PASMC for 30 min, followed by extraction of protein from the cells for various experiments.
Measurement level of PDGF in CM
PASMCs or PAECs were plated in Petri dishes at the density of 5 × 104 cells/dish and cultured in the growth medium for 24 h. The growth medium was then replaced by the basal medium with 0.3% FBS followed by medium collection at the indicated period of time. The CM samples collected at different times were immediately frozen at −80℃ before experimentation. The level of PDGF-AA and PDGF-BB in the collected CM samples was measured by ELISA kit (Sigma, St Louis, MO).
MTT assay
PASMCs from normal subjects and IPAH patients were plated in 96-well plates at the density of 5 × 103 cells/well and cultured in the growth media for 24 h. Then, cells were incubated in (a) the basal medium with 0.3% FBS, (b) the basal medium with 10% FBS, (c) the normal PAEC-CM, and (d) the IPAH PAEC-CM. After 48 h of culture in the media, the cells were incubated in an incubation medium containing 0.5 mg/ml 3 -(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT). After 4 h, the incubation medium was removed and the MTT solubilization solution (including 10% Triton-X 100 in acidic isopropanol) was used to extract the blue MTT-formazan product. The absorbance of the formazan solution was measured at 540 nm using a spectrophotometer to assess cell viability.
Drugs and chemicals
Imatinib Mesylate (Selleckchem, Houston, TX) was dissolved in distilled water to make a stock solution. Recombinant human PDGF-BB protein (Sigma, St Louis, MO) was dissolved in dimethyl sulfoxide. The stock solutions were aliquoted and kept frozen at −20℃ until use.
Statistics
Summarized data are shown as means ± standard error (SE). The statistical significance between two groups was determined using a standardized unpaired Student’s t-test or ANOVA and post hoc tests (Student–Newman–Keuls). Significant differences were expressed in the results or figures as p < 0.05.
Results
Upregulated expression of PDGFRα and PDGFRβ in PASMC from patients with IPAH
PDGF induces PASMC proliferation and contributes to the progression of PAH through binding with their cognate receptors (PDGFR). We first compared the protein expression level of PDGFR in PASMC isolated form three normal subject and five IPAH patients. We found that PDGFRα and PDGFRβ were both upregulated in IPAH-PASMC in comparison to normal PASMC. The upregulation of PDGFRα and PDGFRβ in IPAH-PASMC was associated with increased phosphorylation (p) of AKT at S473 (pAKT (S473)) and T308 (pAKT (T308)) sites (Fig. 1A). Using specific siRNA for PDGFRα or PDGFRβ, we were able to knockdown the expression of PDGFRα or PDGFRβ in IPAH-PASMC. Interestingly, we found that downregulation of PDGFRβ with siRNA resulted in a compensatory increase in the protein level of PDGFRα, whereas downregulation of PDGFRα with siRNA slightly decreased PDGFRβ in IPAH-PASMC (Fig. 1B). These results indicate that protein expression of PDGFRα and PDGFRβ is upregulated in PASMC from patients with IPAH.
Fig. 1.

Upregulated protein expression of PDGFRα and PDGFRβ in PASMC from patients with IPAH. (A) Representative Western blot images (a) and summarized data (b, means ± SE) showing the protein expression of PDGFRα, PDGFRβ, pAKT at S473 (pAKT-S473) and at T308 (pAKT-T308), and total AKT in PASMC isolated from normal subjects (blue bars, n = 3 subjects) and IPAH patients (red bars, n = 5 patients). *p < 0.05, ***p < 0.001 versus normal. (B) Representative Western bolt images (a) and summarized data (b, means ± SE) showing the protein expression of PDGFRα and PDGFRβ in IPAH-PASMC transfected with control siRNA (si-Control) and siRNA specifically targeting PDGFRα (si-PDGFRα) and PDGFRβ (si-PDGFRβ). ***p < 0.001 versus si-Control. (C) Representative Western bolt images (a) and summarized data (b, means ± SE) showing the protein expression of PDGFRα, PDGFRβ, pAKT (S473), and total AKT in PASMC and PAEC isolated from normal subjects and IPAH patients. **p < 0.001 versus normal PASMC. (D) Representative images showing immunofluorescence in PASMC (upper panels) and PAEC (lower panels) isolated from normal subjects and IPAH patients stained for PDGFRα (a) and PDGFRβ (b) (green). Nuclei counterstained with DAPI (blue). AKT: protein kinase B; DAPI: 4′,6-diamidino-2-phenylindole; IPAH: idiopathic PAH; PAEC: pulmonary arterial endothelial cell; pAKT: phosphorylated AKT; PASMC: pulmonary arterial smooth muscle cell; PDGFRα: platelet-derived growth factor receptor α; PDGFRβ: platelet-derived growth factor receptor β; siRNA: small interfering RNA.
Furthermore, we found that upregulation of PDGFRα and PDGFRβ only occurred in PASMC from IPAH patients, but not in PAEC from IPAH patients (Fig. 1C). In PAEC isolated from normal subjects and patients with IPAH, both PDGFRα and PDGFRβ expression levels were too low to be detected (ND) in our WB experiments (Fig. 1C). The immunohistochemical staining of PDGFRα and PDGFRβ in PASMC and PAEC showed similar results. The expression level of PDGFRα and PDGFRβ was higher in IPAH-PASMC than in normal PASMC (Fig. 1D, upper panels), whereas the expression of PDGFRα and PDGFRβ was hardly detected in normal and IPAH PAEC (Fig. 1D, lower panels). These results imply that the protein expression pattern is different in PASMC and PAEC from patients with IPAH. Further studies are needed to specify the potential different mechanisms involved in transcriptional and translational regulation of PDGFRα and PDGFRβ in PASMC and PAEC from normal subjects and IPAH patients.
Increased activation of PDGFRα and PDGFRβ in PASMC from patients with IPAH
PDGFR is a family of receptor tyrosine kinases, which can be activated by binding to its ligand, PDGF. After ligand binding, PDGFR is autophosphorylated at tyrosine residue sites, and then recruits and activates its downstream signaling pathway, for example PI3K/AKT. As the expression of PDGFR is upregulated in IPAH-PASMC (see Fig. 1A), we then examined and compared PDGF-mediated phosphorylation of PDGFR in normal and IPAH PASMC incubated in serum-free basal medium. In this experiment, we used PDGF-BB33 (10 ng/ml for 30 min) as the ligand to induce phosphorylation of PDGFR and AKT in order to evaluate PDGF-BB-mediated PDGFR activation, determined by the levels of phosphorylated PDGFR (pPDGFR) and phosphorylated AKT (pAKT). As shown in Fig. 2Aa and b, PDGF-BB-induced phosphorylation of PDGFRα and PDGFRβ was significantly enhanced in IPAH-PASMC in comparison to normal PASMC. However, the PDGF-BB-induced increase in pAKT (at S473 and T308) was not significantly different in normal and IPAH PASMC due likely to the significant increase in the basal phosphorylation level of AKT at S473 and T308 (Fig. 2Ac). PDGF-BB-mediated increases in pPDGFRα, pPDGFRβ, and pAKT (at S473 and T308) were significantly inhibited in both normal and IPAH PASMC pre-incubated with a low dose (0.3–1 µM) of pharmacological blocker of PDGFR, imatinib,20,34 a TKR blocker, for 30 min (Fig. 2Aa and b); the imatinib-mediated inhibition of pAKT at S473 seemed to be greater in IPAH-PASMC than in normal PASMC (Fig. 2Ac, third panel).
Fig. 2.

Increased activation of PDGFRα and PDGFRβ in PASMC from patients with IPAH. (A) Representative Western blot images (a) and summarized data (b, means ± SE) showing the levels of phosphorylated (p) PDGFRα (pPDGFRα), PDGFRβ (pPDGFRβ), AKT (pAKT-S473 and pAKT-T308) in normal and IPAH PASMC pre-treated with (0.3 or 1 µM) or without (0 µM) imatinib for 30 min, followed by the treatment with vehicle (−, control) or 10 ng/ml PDGF-BB (+, PDGF) for 30 min. *p < 0.05, **p < 0.01 versus control; #p < 0.05, ##p < 0.01 versus PDGF; §p < 0.05, §§p < 0.01, §§§p < 0.001 versus normal–control. Summarized data (c, means ± SE) showing the PDGF-BB-induced changes in protein levels of pPDGFRα, pPDGFRβ, pAKT-S473, and pAKT-T308 in normal and IPAH PASMC pre-treated with (PDGF + Imatinib) or without (PDGF) imatinib (1 µM). *p < 0.05, **p < 0.01, ***p < 0.001 versus PDGF; §p < 0.05, §§p < 0.01 versus normal PASMC. The data shown in (c) are constructed from the data shown in (b). (B) Representative fluorescent images (a) and summarized data (b, means ± SE) showing [Ca2+]cyt in normal and IPAH PASMC pre-treated with or without imatinib (1 µM) for 30 min, followed by the treatment with vehicle (control) or 10 ng/ml PDGF-BB (PDGF) for 30 min. ***p < 0.001 versus control; ###p < 0.001 versus PDGF; §§§P < 0.001 versus normal control. Summarized data (c, means ± SE) showing PDGF-BB-induced changes in [Ca2+]cyt in normal or IPAH PASMC pre-treated with (PDGF + Imatinib) or without (PDGF) imatinib (1 µM). **p < 0.01, ***p < 0.001 versus PDGF. The data shown in (c) are constructed from the data shown in (b). PDGF was applied to cells in the serum-free media. AKT: protein kinase B; IPAH: idiopathic PAH; pAKT: phosphorylated AKT; PASMC: pulmonary arterial smooth muscle cell; PDGFRα: platelet-derived growth factor receptor α; PDGFRβ: platelet-derived growth factor receptor β.
PDGF-BB-mediated activation of PDGFR can also increase cytosolic free Ca2+ concentration ([Ca2+]cyt). A rise in [Ca2+]cyt in PASMC is a major trigger for pulmonary vasoconstriction and an important stimulus for PASMC proliferation and migration contributing to the development of pulmonary vascular remodeling. We found that the basal [Ca2+]cyt was higher in IPAH-PASMC than in normal PASMC; however, the PDGF-BB-mediated increase in [Ca2+]cyt was not significantly different in normal and IPAH PASMC incubated in serum-free basal medium (Fig. 2Ba and b). Pharmacological blockade of PDGFR with imatinib (1 µM for 30 min) resulted in a significant inhibition on PDGF-BB-mediated increases in [Ca2+]cyt in both normal and IPAH PASMC (Fig. 2Bc). These results indicate that blockade of PDGFR with imatinib exerts efficient inhibitory effect on PDGFR-associated phosphorylation of AKT and increase in [Ca2+]cyt in PASMC from patients with IPAH.
Downregulation of PDGFRα inhibits PDGF-induced AKT phosphorylation in PASMC
There are two isoforms of PDGFR, PDGFRα and PDGFRβ, which form hetero- and homodimers and differ in their ability to respond to isoforms of PDGF. We used siRNA to knockdown PDGFRα or PDGFRβ to evaluate the respective effect of PDGFRα and PDGFRβ on pAKT. When PASMCs were incubated in the serum-free basal medium, the expression levels of PDGFRα and PDGFRβ were both higher in IPAH PASMC than in normal PASMC, but the basal level of pAKT was not significantly different between normal and IPAH PASMC (Fig. 3A and B). PDGF-BB treatment (10 ng/ml for 30 min) had no effect on the protein expression levels of PDGFRα and PDGFRβ, but significantly increased phosphorylation of AKT at S473 (Fig. 3). Downregulation of PDGFRα with siRNA significantly inhibited the PDGF-BB-induced increase in pAKT (Fig. 3A), whereas downregulation of PDGFRβ with siRNA had negligible effect on the PDGF-BB-induced increase in pAKT (at S473), in both normal and IPAH PASMC (Fig. 3B). Since knockdown of PDGFRβ resulted in compensatory increase in the expression of PDGFRα (see Fig. 1B), we also used siRNAs for both PDGFRα and PDGFRβ to double knockdown PDGFRα and PDGFRβ in PASMC to see their effects on pAKT. We found that downregulation of both PDGFRα and PDGFRβ significantly inhibited the PDGF-BB-induced increase in pAKT in normal and IPAH PASMC; the effect of double knockdown of PDGFRα and PDGFRβ was similar to the effect of downregulation of PDGFRα alone (Fig. 3C). These results imply that PDGFRα is a predominant PDGFR that mediates PDGF-mediated phosphorylation of pAKT in PASMC.
Fig. 3.

Knockdown of PDGFRα inhibits PDGF-induced phosphorylation of AKT in PASMC. (A) Representative Western blot images (a) and summarized data (b, means ± SE) showing protein expression levels of PDGFRα, pAKT (S473), and total AKT in normal and IPAH PASMC transfected with control siRNA (siRNA-Control) and siRNA specifically targeting PDGFRα (siRNA-PDGFRα) for 48 h, followed by the treatment with (+) or without (−) PDGF-BB (PDGF, 10 ng/ml) for 30 min. ***p < 0.001 versus siRNA-Control; #p < 0.05, ###p < 0.001 versus siRNA-Control + PDGF; §§p < 0.01 versus normal PASMC. (B) Representative Western blot images (a) and summarized data (b, means ± SE) showing protein expression levels of PDGFRβ, pAKT (S473) and total AKT in normal and IPAH PASMC transfected with control siRNA (siRNA-Control) and siRNA specifically targeting PDGFRβ (siRNA-PDGFRβ) for 48 h, followed by the treatment with (+) or without (−) PDGF-BB (PDGF, 10 ng/ml) for 30 min. **p < 0.01, ***p < 0.001 versus siRNA-Control; ###p < 0.001 versus siRNA-Control + PDGF; §p < 0.05 versus normal PASMC. (C) Representative Western blot images (a) and summarized data (b, means ± SE) showing protein expression levels of PDGFRα, PDGFRβ, pAKT (S473), and total AKT in normal and IPAH PASMC transfected with control siRNA (siRNA-Control) and siRNAs specifically targeting PDGFRα and PDGFRβ (siRNA-PDGFRα/β) for 48 h, followed by the treatment with (+) or without (−) PDGF-BB (PDGF, 10 ng/ml) for 30 min. *p < 0.05, **p < 0.01, ***p < 0.001 versus siRNA-Control; #p < 0.05, ##p < 0.01, ###p < 0.001 versus siRNA-Control + PDGF; §p < 0.05, §§p < 0.01 versus normal PASMC. PDGF was applied to cells in the serum-free media. AKT: protein kinase B; IPAH: idiopathic PAH; pAKT: phosphorylated AKT; PASMC: pulmonary arterial smooth muscle cell; PDGFRα: platelet-derived growth factor receptor α; PDGFRβ: platelet-derived growth factor receptor β; siRNA: small interfering RNA.
PAECs from patients with IPAH secrete high levels of PDGF-AA and PDGF-BB
Functional communication or interaction between PAEC and PASMC via a paracrine mechanism plays an important role in the development and progression of pulmonary vasculopathy in patients with pulmonary vascular disease. In the next set of experiments, we examined whether PDGF is produced in PAEC and secreted into the extracellular medium and compared the expression level of PDGF (PDGF-AA and PDGF-BB) in PAEC from normal subjects and IPAH patients. We cultured PAEC in the 0.3% FBS basal medium and then collected the culture medium (PAEC-CM) at 6, 12, 24, and 48 h (Fig. 4Aa). We then used ELISA assay to measure the concentration of PDGF-AA and PDGF-BB in the CM collected at different times from normal and IPAH PAEC. As shown in Fig. 4Ab (left panel), the concentration of PDGF-AA was increased in the PAEC-CM in a time-dependent manner. In comparison to the CM from normal PAEC, the PDGF-AA level was slightly, but with statistical significance, greater in the CM from IPAH PAEC (Fig. 4Ab, left panel). Although the level of PDGF-BB in the CM was much less than that of PDGF-AA, IPAH-PAEC seemed to produce more PDGF-BB to the medium than normal PAEC after 24 and 48 h of incubation in the 0.3% FBS basal medium (Fig. 4Ab, right panel and inset). Nevertheless, the negligible amount of PDGF-AA or PDGF-BB was determined in the medium used to incubate or culture normal PASMC (Fig. 4B) or IPAH PASMC (data not shown). These data imply that (a) PAEC, but not PASMC, actively produce and secret PDGF (predominantly PDGF-AA) to the extracellular medium, and (b) PAEC from IPAH patients produces and secretes more PDGF-AA and PDGF-BB to the medium than normal PAEC.
Fig. 4.

Soluble PDGF in the CM collected from IPAH-PAEC is higher than in the media from normal PAEC. (A) Schematic diagram (a) showing how the CM was collected for ELISA experiments and summarized data (b, means ± SE) showing the levels of PDGF-AA (left panel) and PDGF-BB (right panels) in CM collected 6, 12, 24, or 48 h after incubation with normal PAEC (normal EC-CM) or IPAH-PAEC (IPAH EC-CM). Inset: increased scale of Y-axis to see difference of PDGF-BB levels between normal EC-CM and IPAH EC-CM. ***p < 0.001 versus normal EC-CM (ANOVA). (B) Schematic diagram (a) showing how the CM was collected for ELISA experiments and summarized data (b, means ± SE) showing the levels of PDGF-AA (left panel) and PDGF-BB (right panel) in CM collected 48 h after incubation with (PASMC-CM) or without (cell-free) normal PASMC. 0.3% FBS basal medium was used for the collection. ELISA: enzyme-linked immunosorbent assay; FBS: fetal bovine serum; IPAH: idiopathic PAH; PAEC: pulmonary arterial endothelial cell; PASMC: pulmonary arterial smooth muscle cell.
PAEC-CM activates PDGFRα and PDGFRβ in PASMC
We then tested whether PAEC-produced PDGF in the medium can activate PDGFR in PASMC. We collected 0.3% FBS basal medium from normal PAEC (PAEC-CM) after 48 h of incubation; we also collected medium from a cell-free Petri dish (cell-free-CM) as a control (Fig. 5A). The cell-free-CM and PAEC-CM were used to incubate normal and IPAH PASMC for 30 min and then the phosphorylation of PDGFRα (pPDGFRα) and PDGFRβ (pPDGFRβ) was determined using antibodies specifically against pPDGFRα and pPDGFβ. As shown in Fig. 5B and C, pPDGFRα but not pPDGFRβ in PASMC was significantly increased by incubating PASMC in the PAEC-CM for 30 min compared with cell-free-CM. The PDGFRα phosphorylation induced by PAEC-CM was significantly greater in IPAH-PASMC than in normal PASMC (Fig. 5C). Interestingly, the PAEC-CM also induced phosphorylation of AKT (pAKT), but the PAEC-CM-mediated pAKT increase was not significantly different in normal and IPAH PASMC (Fig. 5C). These results suggest that PDGF, both PDGF-AA and PDGF-BB, produced and secreted from PAEC, is able to activate PDGFRα in PASMC and that PDGF-mediated activation or phosphorylation of PDGFRα is significantly enhanced in PASMC from IPAH patients due apparently to the upregulated PDGFRα in these cells.
Fig. 5.

CM collected from PAEC activates PDGFRα in PASMC. (A) Schematic diagram showing how the CM was collected for Western blot experiments from cell-free controls and normal PAEC and applied to normal and IPAH PASMC to test the effects of CM on PDGFR. (B) Representative Western blot images on phosphorylated (p) PDGFRα (pPDGFRα), PDGFRβ (pPDGFRβ), and AKT (pAKT-S473) in normal and IPAH PASMC incubated for 30 min with the CM collected from cell-free controls (cell-free CM) or normal PAEC (PAEC-CM) after 48 h. (C) Summarized data (means ± SE) showing the protein expression of pPDGFRα (left panel), pPDGFRβ (middle panel), and pAKT-S473 (right panel) in normal and IPAH-PASMC incubated with cell-free CM or PAEC-CM for 30 min. *p < 0.05, **p < 0.01 versus cell-free CM; #P < 0.05 versus normal PASMC. 0.3% FBS basal medium was used for the collection. FBS: fetal bovine serum; IPAH: idiopathic PAH; PAEC: pulmonary arterial endothelial cell; pAKT: phosphorylated AKT; PASMC: pulmonary arterial smooth muscle cell; PDGFRα: platelet-derived growth factor receptor α; PDGFRβ: platelet-derived growth factor receptor β.
PAEC-CM increases PASMC proliferation
Abnormal PASMC proliferation is an important contributor to the development and progression of concentric pulmonary vascular remodeling in PAH and other forms of PH. As PAEC-CM increased phosphorylation of PDGFR and AKT in PASMC, we then investigated if the PAEC-CM (or PDGF released from PAEC) can stimulate PASMC proliferation determined by MTT assay. We first collected the CM used to incubate normal and IPAH PAEC for 24 or 48 h. Then, we cultured normal and IPAH PASMC in the PAEC-CM for 48 h before cell proliferation was evaluated by MTT assay (Fig. 6Aa). The normal PAEC-CM collected at 24 h of incubation had negligible effect on PASMC proliferation, while the normal PAEC-CM collected at 48 h significantly increased proliferation of normal and IPAH PASMC compared with control. The normal PAEC-CM collected at 48 h caused more proliferation in IPAH-PASMC than in normal PASMC (Fig. 6Ab). The IPAH-PAEC-CM collected at 24 or 48 h of incubation increased normal and IPAH PASMC proliferation. The IPAH-PAEC-CM collected at 48 h resulted in more proliferation of IPAH PASMC than of normal PASMC (Fig. 6Ab). Interestingly, the IPAH-PAEC-CM induced higher proliferation in IPAH-PASMC compared with the normal PAEC-CM collected at 24 or 48 h. Furthermore, we found that 10% FBS basal medium could significantly increase MTT activity in PASMC, and 10% FBS basal medium-mediated cell proliferation in IPAH-PASMC was greater than in normal PASMC (Fig. 6B). These data suggest that the normal PAEC-CM due likely to its containing PAEC-released PDGF (a large amount of PDGF-AA and small amount of PDGF-BB) can efficiently increase PASMC proliferation; the proliferative effect is much greater in IPAH PASMC than in normal PASMC because of, potentially, upregulated PDGFR in IPAH PASMC. Furthermore, the proliferative effect of the IPAH-PAEC-CM on normal and IPAH PASMC is greater than of the normal PAEC-CM because of, potentially, more PDGF is produced and released from IPAH PAEC than normal PAEC.
Fig. 6.
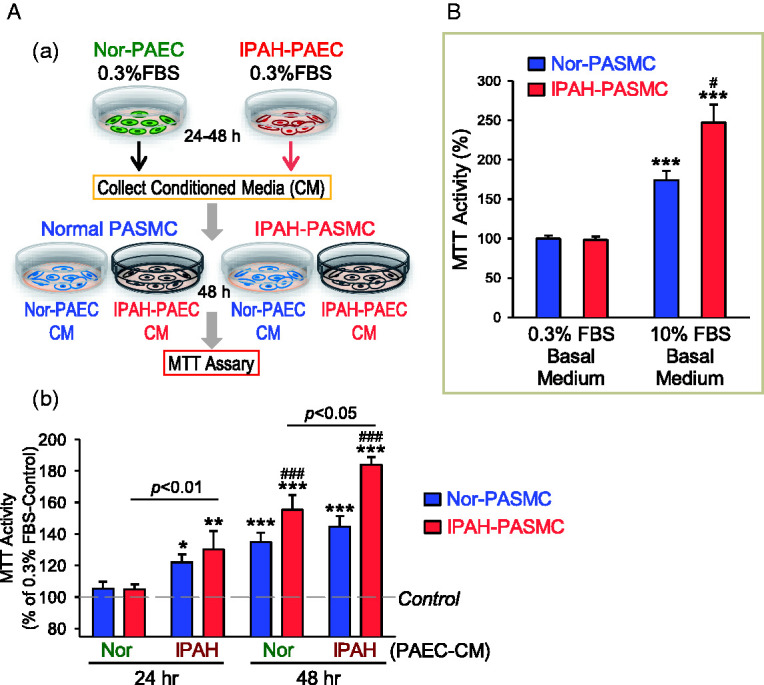
CM collected from normal and IPAH PAEC stimulates PASMC proliferation. (A) Schematic diagram (a) showing how the CM was collected for MTT experiments from normal (Nor-PAEC) and IPAH PAEC (IPAH-PAEC) and applied to PASMC isolated from normal subjects (normal PASMC) and IPAH patients (IPAH-PASMC) to test the effects of PAEC-CM on PASMC proliferation. Summarized data (b, means ± SE) showing MTT activity in normal PASMC (Nor-PASMC) and IPAH-PASMC cultured for 48 h in Nor-PAEC-CM or IPAH-PAEC-CM collected 24 or 48 h after incubation with normal or IPAH PAEC. *p < 0.05, **p < 0.01, ***p < 0.001 versus control (0.3% FBS basal medium, the dotted line); ###p < 0.001 versus Nor-PASMC. 0.3% FBS basal medium was used for the collection. (B) Summarized data (means ± SE) showing MTT activity in Nor-PASMC and IPAH-PASMC cultured for 48 h in 0.3% FBS basal medium or 10% FBS basal medium. ***p < 0.001 versus 0.3% FBS basal medium; #p < 0.05 versus Nor-PASMC. FBS: fetal bovine serum; IPAH: idiopathic PAH; MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide; PAEC: pulmonary arterial endothelial cell; PASMC: pulmonary arterial smooth muscle cell.
Discussion
PDGF/PDGFR/AKT signaling is an important signaling cascade for cell migration and proliferation.27,35 PAECs functionally interact with PASMC via multiple mechanisms to regulate PASMC contraction, migration, and proliferation. It has been well documented that PAECs produce and release the endothelium-derived constricting (e.g. endothelin and thromboxane A2) and relaxing (e.g. nitric oxide and prostacyclin) factors to regulate PASMC contraction and relaxation, and, subsequently, pulmonary vasoconstriction and vasodilation. PAECs may also produce and release various mitogenic factors and cytokines that regulate PASMC proliferation via a paracrine mechanism and ultimately contribute to the development and progression of concentric pulmonary arterial wall thickening in patients with PAH and animals with experimental PH.
This study shows that (i) the protein expression and phosphorylation of PDGFRα and PDGFRβ are significantly increased in PASMC isolated from IPAH patients compared to PASMC isolated from normal subjects; (ii) PAECs in culture produce and release PDGF, mainly PDGF-AA (but also small amount of PDGF-BB), to the medium, while PAECs from IPAH patients secret more PDGF-AA and PDGF-BB than PAEC from normal subjects; (iii) the CM (0.3% FBS basal medium) used to incubate normal PAEC for 48 h results in significant phosphorylation of PDGFRα (pPDGFRα) and AKT (pAKT) in normal PASMC, while the CM from normal PAEC causes more increase in pPDGFRα and proliferation in IPAH-PASMC than in normal PASMC; (iv) the CM from normal or IPAH PAEC causes more proliferation in IPAH-PASMC compared with normal PASMC while the proliferation of IPAH-PASMC was higher in IPAH-PAEC-CM than in normal PAEC-CM. These data suggest that the paracrine mechanism by which PAEC-released PDGF (PDGF-AA and PDGF-BB) activates PDGFRα in PASMC and stimulates PASMC proliferation via the AKT signaling cascade is an important mechanism involved, at least in part, in the development of pulmonary vascular remodeling in PAH. Inhibition of endothelial production and secretion of PDGF (PDGF-BB and PDGF-AA) and blockade of smooth muscle PDGFRα may be an efficient combination therapy for PAH and other forms of severe PH.
It has been shown that PDGF and PDGFR are upregulated or increased in small remodeled human PAs with plexiform lesions in patients with PAH and animals with severe experimental PH with predominant expression in PASMC.16,35 Upregulation of PDGFR is one of the potential mechanisms for PDGF-mediated pulmonary vascular remodeling.36,37 Although PDGFRα is used as a fibroblast marker,38 its expression has been detected in both normal and IPAH PASMC.7,16,22 It is possible that PDGFRα may be a marker for fibroblasts under normal conditions because of its high expression level, but it is likely that PDGFRα is upregulated in PASMC under pathological conditions. A recent study provided strong evidence that PDGFRα+ and α-SMA+ cells represent two distinct mesenchymal cell populations involved in the development of lung fibrosis,39 while another group, in contrast, reported that only 45% of myofibroblasts (α-SMA+ cells) are PDGFRα+40 in an adult pulmonary fibrosis model. Lineage tracing study on human and murine specimens revealed that PDGFRα+ cells were expanded, but they were in majority not α-SMA+ within the normal and remodeled vessel wall in patients with IPAH and animals with experimental PH.41 Sub-phenotype of cells42 and/or partially reprogrammed cells43,44 may exist in the diseased tissues and affected cell population. Normal PASMC may become more “fibroblast-like SMC” or myofibroblasts (or SMC-like fibroblasts) in the remodeled pulmonary arterioles and/or intimal and plexiform lesions in patients with IPAH patients. A higher level of PDGFRα in IPAH-PASMC than in normal PASMC could also be due to PASMC dedifferentiation from contractile to proliferative phenotype30,45,46 which can contribute to the population of SMC-like (mesenchymal or myofibroblasts) cells identified in remodeled vessels, muscularized arterioles, and plexiform lesions in IPAH patients.47,48 Taken together, we speculate that (a) phenotypical alterations of PASMC, (b) sub-phenotype of PASMC, and (c) endothelial-to-mesenchymal transition may all take place and contribute to the development and progression of pulmonary vascular remodeling and obliterative vascular lesions in patients with IPAH and animals with experimental PH. Further characterization of heterogeneous SMC (and other cell) population in the remodeled PAs, muscularized arterioles and capillaries, and obliterative intimal lesions in patients with IPAH would clarify these questions and specify the subtype of cells that highly express PDGFRα (and PDGFRβ).
We determined the protein expression of PDGFR in human PASMC but were unable to detect any PDGFR in both normal or IPAH PAEC probably due to lack or very low level of expression. However, the expression of PDGFRα has been identified in microvascular endothelial cells and liver endothelial cells49,50 while PDGFRβ is expressed in mouse capillary endothelial cells.51 Moreover, ten Freyhaus et al.35 reported that PDGF-BB failed to activate PDGFRs in human coronary endothelial cells suggesting that the activated PDGF/PDGFR signaling is likely unique to PASMC or vascular SMC, myofibroblasts, fibroblasts, and pericytes.
Although the exact upstream mechanisms underlying the increased PDGF/PDGFR in PASMC isolated from IPAH patients are unclear, published reports indicate that the promoters of PDGF and PDGFRs contain the binding consensus for many different transcription factors (e.g. GATA, p53, Oct4, c-Jun, Mixl1)52–57 that are implicated in the development and progression of PAH and PH. Thus, PDGFRα promoter has unique putative binding sites for transcription factors GATA, Oct4, and Mixl152,55,57 while TGF-β, p53, and c-Jun were found to increase the promoter activity of PDGFβ gene.53,54,56 Furthermore, Yu et al.58 demonstrated that FoxM1 regulates PDGF-A transcription by directly binding to two sites of the PDGFA promoter. Increased FoxM1 promotes cell proliferation in PDGF/AKT-dependent manner and forms a positive feedback loop to maintain the upregulated FoxM1 level. Our previous study showed that c-Jun, a transcription factor belonging to the AP-1 family, was involved in PDGF-mediated Ca2+-dependent upregulation of TRPC6 channels in PASMC.59 PDGF-mediated increase in c-Jun may contribute to the upregulation of PDGF and PDGFRs in PASMC by an autocrine mechanism. These observations suggest that transcriptional activation through the TGF-β/GATA4/5 and spermine/CaSR/Ca2+/CaM/c-Jun signaling cascades, as well as the MDM2-associated posttranscriptional modification of p53 protein pathway, may potentially be involved in the upregulation of PDGFRα and PDGFβ genes in PASMC from patients with IPAH.
The findings from this study showed that due to the upregulated PDGFRα and PDGFRβ in IPAH-PASMC, PDGF-induced phosphorylation of AKT (at S473 and T308) was enhanced in IPAH-PASMC compared to normal PASMC. However, we were not able to demonstrate that PDGF induces more increases in pAKT at S473 and T308 in IPAH-PASMC, due likely to the increased basal phosphorylation level of AKT at these two sites. As expected, PDGF also increased [Ca2+]cyt in PASMC, but the PDGF-mediated rise in [Ca2+]cyt was not different between normal and IPAH PASMC. These data suggest that the expression and phosphorylation of PDGFR were both increased in IPAH-PASMC compared to normal PASMC, which may serve as an important pathogenic mechanism in the development and progression of pulmonary vascular remodeling due to enhanced PASMC proliferation in PAH.
Our data also showed that PDGF-AA concentration was much higher than PDGF-BB in the CM from normal PAEC, while PDGF-AA and PDGF-BB were both significantly higher in the CM from IPAH PAEC than in normal PAEC. PDGF could be released as free and bounded isoforms or as constituents of extracellular vesicles.60 We will continue our research to clarify the form of PDGF-AA and PDGF-BB in PAEC-CM. Incubation of PASMC with the CM collected from IPAH-PAEC resulted in higher proliferative rate probably due to higher concentration of released PDGF-AA and/or PDGF-BB. It is worth noting that the highest proliferation was shown in IPAH-PASMC in response to the CM from IPAH-PAEC. We were able to detect the negligible amount of PDGF secreted by PASMC but it was not enough to promote PASMC proliferation (data not shown). These findings led us to conclude that, in patients with IPAH, the increased level of PAEC-secreted PDGF-AA and PDGF-BB and the upregulated PDGFR in PASMC coordinate with each other contributing to enhance PASMC proliferation and pulmonary vascular remodeling.
Imatinib, a 2-phenyl amino pyrimidine derivative that specifically inhibits TKRs like PDGFR and c-Kit, efficiently inhibited PDGF-induced increases in pAKT and [Ca2+[cyt in our experiments. Large doses of imatinib also efficiently inhibit EGFR, ErbB2, insulin receptor, and IGF-I receptor (with an IC50 >100 µM) as well as Flt-3, cFms/v-Fms, and vSrc (with an IC50 > 10 µM).21,61 Imatinib has been approved by the FDA for the treatment of inoperable and metastatic malignant gastrointestinal stromal tumors. In recent years, imatinib has also been found to attenuate experimental PH.18,20,62,63 In clinical studies, imatinib could improve exercise capacity and hemodynamics in patients with advanced PAH.17,64,65 However, it has been reported that treatment with imatinib is accompanied by severe adverse effects, including subdural hematomas, as well as poor tolerability.66 A possible reason for the imatinib-associated advert effects (e.g. cardiac toxicity, abnormal bone, mineral metabolism, and immune function disorder)67 is that it inhibits not only PDGFR, but also other TKRs important for cell survival.61,68 Taken together, inhibition of endothelial PDGF synthesis and secretion, and blockade of smooth muscle PDGFR would be a good therapeutic strategy to inhibit PASMC proliferation and migration, attenuate pulmonary vascular remodeling, and eventually prevent the development of PH or reverse established PH.
In summary, the pathogenic paracrine interaction of increased endothelial PDGF and upregulated smooth muscle PDGFR is enhanced in patients with PAH and animals with experimental PH. Given the adverse effects of conventional PDGFR inhibitors (e.g. imatinib), it is valid and promising to develop specific transcriptional and translational inhibitors of endothelial PDGF and smooth muscle PDGFR as therapeutic approach for PAH and other forms of severe PH.
Acknowledgments
The human lung cells for this study were provided by the CMREF-PHBI (Cardiovascular Medical Research and Education Fund/Pulmonary Hypertension Breakthrough Initiative) Research Network.
Authors’ contributions
KW, HT, AB, AM, and JXJY conceived and designed research; KW, HT, RL, SGC, ZW, RJA, PPJ, MX, MR, Shamin R, FB, Shayan R performed experiments: KW, HT, RL, AB, RJA, PPJ, MX, MR, Shamin R, FB, Shayan R, DV-J, TSS, AAD, JGNG, JYJS, PAT, JW, AM, and JXJY analyzed data; KW, HT, AB, PPJ, MX, DV-J, TSS, AAD, JGNG, JYJS, PAT, AM, and JXJY interpreted results of experiments; KW, HT, AB, and JXJY prepared figures; KW, HT, AB, and JXJY drafted manuscript; KW, HT, AB, PPJ, MX, MR, Shamin R, FB, Shayan R, DV-J, TSS, AAD, JGNG, JYJS, PAT, JW, AM, and JXJY edited and revised manuscript; KW, HT, RL, SGC, ZW, AB, RJA, PPJ, MX, MR, Shamin R, FB, Shayan R, DV-J, TSS, AAD, JGNG, JYJS, PAT, JW, AM, and JXJY approved final version of manuscript.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This work was supported in part by the grants from the National Heart, Lung and Blood Institute of the National Institutes of Health (R35HL135807 and R01HL146764). AB was supported by the American Heart Association Postdoctoral Fellowship (20POST35210959).
ORCID iDs
Joe G.N. Garcia https://orcid.org/0000-0002-6934-0420
Ayako Makino https://orcid.org/0000-0003-1259-8604
References
- 1.Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54: S20–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res 2017; 367: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenmark KR, Frid MG, Graham BB, et al. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res 2018; 114: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tajsic T, Morrell NW. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr Physiol 2011; 1: 295–317. [DOI] [PubMed] [Google Scholar]
- 5.Li C, Qin F, Xue M, et al. miR-429 and miR-424-5p inhibit cell proliferation and Ca2+ influx by downregulating CaSR in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 2019; 316: C111–C120. [DOI] [PubMed] [Google Scholar]
- 6.Kuhr FK, Smith KA, Song MY, et al. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 2012; 302: H1546–H1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogawa A, Firth AL, Smith KA, et al. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 2012; 302: C405–C411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med 2013; 91: 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaillancourt M, Ruffenach G, Meloche J, et al. Adaptation and remodelling of the pulmonary circulation in pulmonary hypertension. Can J Cardiol 2015; 31: 407–415. [DOI] [PubMed] [Google Scholar]
- 10.Humbert M, Montani D, Perros F, et al. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol 2008; 49: 113–118. [DOI] [PubMed] [Google Scholar]
- 11.Spiekerkoetter E, Goncharova EA, Guignabert C, et al. Hot topics in the mechanisms of pulmonary arterial hypertension disease: cancer-like pathobiology, the role of the adventitia, systemic involvement, and right ventricular failure. Pulm Circ 2019; 9: 2045894019889775. [DOI] [PMC free article] [PubMed]
- 12.Noskovicova N, Petrek M, Eickelberg O, et al. Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am J Respir Cell Mol Biol 2015; 52: 263–284. [DOI] [PubMed] [Google Scholar]
- 13.Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest 2005; 115: 2691–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng CC, Chi PL, Shen MC, et al. Caffeic acid phenethyl ester rescues pulmonary arterial hypertension through the inhibition of AKT/ERK-dependent PDGF/HIF-1α in vitro and in vivo. Int J Mol Sci 2019; 20: pii: E1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selimovic N, Bergh CH, Andersson B, et al. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur Respir J 2009; 34: 662–668. [DOI] [PubMed] [Google Scholar]
- 16.Perros F, Montani D, Dorfmuller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2008; 178: 81–88. [DOI] [PubMed] [Google Scholar]
- 17.Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013; 127: 1128–1138. [DOI] [PubMed] [Google Scholar]
- 18.Pankey EA, Thammasiboon S, Lasker GF, et al. Imatinib attenuates monocrotaline pulmonary hypertension and has potent vasodilator activity in pulmonary and systemic vascular beds in the rat. Am J Physiol Heart Circ Physiol 2013; 305: H1288–H1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tu L, De Man FS, Girerd B, et al. A critical role for p130Cas in the progression of pulmonary hypertension in humans and rodents. Am J Respir Crit Care Med 2012; 186: 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 2005; 115: 2811–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ten Freyhaus H, Dumitrescu D, Berghausen E, et al. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs 2012; 21: 119–134. [DOI] [PubMed] [Google Scholar]
- 22.Yamamura A, Nayeem MJ, Al Mamun A, et al. Platelet-derived growth factor up-regulates Ca2+-sensing receptors in idiopathic pulmonary arterial hypertension. FASEB J 2019; 33: 7363–7374. [DOI] [PubMed] [Google Scholar]
- 23.Sysol JR, Natarajan V, Machado RF. PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 2016; 310: C983–C992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Q, Liu L, Mandal J, et al. PDGF-BB induces PRMT1 expression through ERK1/2 dependent STAT1 activation and regulates remodeling in primary human lung fibroblasts. Cell Signal 2016; 28: 307–315. [DOI] [PubMed] [Google Scholar]
- 25.Lambers C, Roth M, Jaksch P, et al. Treprostinil inhibits proliferation and extracellular matrix deposition by fibroblasts through cAMP activation. Sci Rep 2018; 8: 1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antoniu SA. Targeting PDGF pathway in pulmonary arterial hypertension. Expert Opin Ther Targets 2012; 16: 1055–1063. [DOI] [PubMed] [Google Scholar]
- 27.Xiao Y, Peng H, Hong C, et al. PDGF promotes the Warburg effect in pulmonary arterial smooth muscle cells via activation of the PI3K/AKT/mTOR/HIF-1α signaling pathway. Cell Physiol Biochem 2017; 42: 1603–1613. [DOI] [PubMed] [Google Scholar]
- 28.Wilson JL, Yu J, Taylor L, et al. Hyperplastic growth of pulmonary artery smooth muscle cells from subjects with pulmonary arterial hypertension is activated through JNK and p38 MAPK. PLoS One 2015; 10: e0123662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goncharova EA, Simon MA, Yuan JX. mTORC1 in pulmonary arterial hypertension: at the crossroads between vasoconstriction and vascular remodeling? Am J Respir Crit Care Med 2020; 201: 1177–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez RA, Wan J, Song S, et al. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol 2015; 308: C581–C593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassoun PM, Mouthon L, Barbera JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 2009; 54: S10–S19. [DOI] [PubMed] [Google Scholar]
- 32.Song S, Carr SG, McDermott KM, et al. STIM2 (stromal interaction molecule 2)-mediated increase in resting cytosolic free Ca2+ concentration stimulates PASMC proliferation in pulmonary arterial hypertension. Hypertension 2018; 71: 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qian Z, Li Y, Yang H, et al. PDGF-BB promotes proliferation and migration via regulating miR-1181/STAT3 axis in human pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2018; 315: L965–L976. [DOI] [PubMed] [Google Scholar]
- 34.Sommer N, Ghofrani HA, Pak O, et al. Current and future treatments of pulmonary arterial hypertension. Br J Pharmacol: 1-25. Epub ahead of print 7 February 2020. DOI: 10.1111/bph.15016. [DOI] [PubMed]
- 35.Ten Freyhaus H, Berghausen EM, Janssen W, et al. Genetic ablation of PDGF-dependent signaling pathways abolishes vascular remodeling and experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol 2015; 35: 1236–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig J, Mikhailenko I, Noyes N, et al. The LDL receptor-related protein 1 (LRP1) regulates the PDGF signaling pathway by binding the protein phosphatase SHP-2 and modulating SHP-2-mediated PDGF signaling events. PLoS One 2013; 8: e70432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashino T, Yamamoto M, Yoshida T, et al. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler Thromb Vasc Biol 2013; 33: 760–768. [DOI] [PubMed] [Google Scholar]
- 38.Saygin D, Tabib T, Bittar HET, et al. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm Circ 2020; 10: 2045894020908782. [DOI] [PMC free article] [PubMed]
- 39.Biasin V, Crnkovic S, Sahu-Osen A, et al. PDGFRα and αSMA mark two distinct mesenchymal cell populations involved in parenchymal and vascular remodeling in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2020; 318: L684–L697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li R, Bernau K, Sandbo N, et al. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. Elife 2018; 7: e36865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crnkovic S, Marsh LM, El Agha E, et al. Resident cell lineages are preserved in pulmonary vascular remodeling. J Pathol 2018; 244: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wytock TP, Motter AE. Distinguishing cell phenotype using cell epigenotype. Sci Adv 2020; 6: eaax7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JS, Choi HW, Hong YJ, et al. Generation of partially reprogrammed cells and fully reprogrammed iPS cells by plasmid transfection. Methods Mol Biol 2016; 1357: 85–95. [DOI] [PubMed] [Google Scholar]
- 44.Ocampo A, Reddy P, Martinez-Redondo P, et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016; 167: 1719–1733.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas JA, Deaton RA, Hastings NE, et al. PDGF-DD, a novel mediator of smooth muscle cell phenotypic modulation, is upregulated in endothelial cells exposed to atherosclerosis-prone flow patterns. Am J Physiol Heart Circ Physiol 2009; 296: H442–H452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garat CV, Majka SM, Sullivan TM, et al. CREB depletion in smooth muscle cells promotes medial thickening, adventitial fibrosis and elicits pulmonary hypertension. Pulm Circ 2020; 10: 2045894019898374. [DOI] [PMC free article] [PubMed]
- 47.Sheikh AQ, Saddouk FZ, Ntokou A, et al. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep 2018; 23: 1152–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheikh AQ, Misra A, Rosas IO, et al. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 2015; 7: 308ra159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marx M, Perlmutter RA, Madri JA. Modulation of platelet-derived growth factor receptor expression in microvascular endothelial cells during in vitro angiogenesis. J Clin Invest 1994; 93: 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heldin P, Pertoft H, Nordlinder H, et al. Differential expression of platelet-derived growth factor α- and β-receptors on fat-storing cells and endothelial cells of rat liver. Exp Cell Res 1991; 193: 364–369. [DOI] [PubMed] [Google Scholar]
- 51.Smits A, Hermansson M, Nister M, et al. Rat brain capillary endothelial cells express functional PDGF B-type receptors. Growth Factors 1989; 2: 1–8. [DOI] [PubMed] [Google Scholar]
- 52.Minato Y, Kuwahara-Otani S, Maeda S, et al. Platelet-derived growth factor receptor α gene is regulated by multiple first exons. Biochem Biophys Res Commun 2019; 510: 489–494. [DOI] [PubMed] [Google Scholar]
- 53.Peng Y, Yan S, Chen D, et al. Pdgfrb is a direct regulatory target of TGFβ signaling in atrioventricular cushion mesenchymal cells. PLoS One 2017; 12: e0175791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uramoto H, Hackzell A, Wetterskog D, et al. pRb, Myc and p53 are critically involved in SV40 large T antigen repression of PDGF β-receptor transcription. J Cell Sci 2004; 117: 3855–3865. [DOI] [PubMed] [Google Scholar]
- 55.Kraft HJ, Mosselman S, Smits HA, et al. Oct-4 regulates alternative platelet-derived growth factor α receptor gene promoter in human embryonal carcinoma cells. J Biol Chem 1996; 271: 12873–12878. [DOI] [PubMed] [Google Scholar]
- 56.Shimizu H, Hagio M, Iwaya H, et al. Deoxycholic acid is involved in the proliferation and migration of vascular smooth muscle cells. J Nutr Sci Vitaminol 2014; 60: 450–454. [DOI] [PubMed] [Google Scholar]
- 57.Pereira LA, Wong MS, Mossman AK, et al. Pdgfrα and Flk1 are direct target genes of Mixl1 in differentiating embryonic stem cells. Stem Cell Res 2012; 8: 165–179. [DOI] [PubMed] [Google Scholar]
- 58.Yu G, Zhou A, Xue J, et al. FoxM1 promotes breast tumorigenesis by activating PDGF-A and forming a positive feedback loop with the PDGF/AKT signaling pathway. Oncotarget 2015; 6: 11281–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu Y, Sweeney M, Zhang S, et al. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 2003; 284: C316–C330. [DOI] [PubMed] [Google Scholar]
- 60.Ostman A, Thyberg J, Westermark B, et al. PDGF-AA and PDGF-BB biosynthesis: proprotein processing in the Golgi complex and lysosomal degradation of PDGF-BB retained intracellularly. J Cell Biol 1992; 118: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996; 2: 561–566. [DOI] [PubMed] [Google Scholar]
- 62.Medarametla V, Festin S, Sugarragchaa C, et al. PK10453, a nonselective platelet-derived growth factor receptor inhibitor, prevents the progression of pulmonary arterial hypertension. Pulm Circ 2014; 4: 82–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nayyar D, Muthiah K, Kumarasinghe G, et al. Imatinib for the treatment of pulmonary arterial hypertension and pulmonary capillary hemangiomatosis. Pulm Circ 2014; 4: 342–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frost AE, Barst RJ, Hoeper MM, et al. Long-term safety and efficacy of imatinib in pulmonary arterial hypertension. J Heart Lung Transplant 2015; 34: 1366–1375. [DOI] [PubMed] [Google Scholar]
- 65.Farha S, Dweik R, Rahaghi F, et al. Imatinib in pulmonary arterial hypertension: c-Kit inhibition. Pulm Circ 2014; 4: 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Dabh A and Acharya D. EXPRESS: pulmonary hypertension with dasatinib and other tyrosine kinase inhibitors. Pulm Circ 2019; 9: 2045894019865704. [DOI] [PMC free article] [PubMed]
- 67.Kerkela R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 2006; 12: 908–916. [DOI] [PubMed] [Google Scholar]
- 68.Gambaryan N, Perros F, Montani D, et al. Imatinib inhibits bone marrow-derived c-kit+ cell mobilisation in hypoxic pulmonary hypertension. Eur Respir J 2010; 36: 1209–1211. [DOI] [PubMed] [Google Scholar]