

Abstract
Ras-related C3 botulinum toxin substrate 1 (Rac1) is a small GTPase that is well known for its sensitivity to the environmental stress of a cell or an organism. It senses the external signals which are transmitted from membrane-bound receptors and induces downstream signaling cascades to exert its physiological functions. Rac1 is an important regulator of a variety of cellular processes, such as cytoskeletal organization, generation of oxidative products, and gene expression. In particular, Rac1 has a significant influence on certain brain functions like neuronal migration, synaptic plasticity, and memory formation via regulation of actin dynamics in neurons. Abnormal Rac1 expression and activity have been observed in multiple neurological diseases. Here, we review recent findings to delineate the role of Rac1 signaling in neurodevelopmental disorders associated with abnormal spine morphology, synaptogenesis, and synaptic plasticity. Moreover, certain novel inhibitors of Rac1 and related pathways are discussed as potential avenues toward future treatment for these diseases.
1. Introduction
As a member of the Ras-homologous (Rho) small GTPase family, Rac1 is well known for its versatility in mediating the response of cells or organisms when facing external disturbances or environmental challenges, such as heat shock [1], oxidative stress [2], mechanical stress [3], genotoxic stress [4], hypoxic stress [5], or even higher-level mental stress from social confrontation and fear [6–9]. In the last decade, Rac1 has gained increased attention in the field of neuroscience with its roles in brain structure and function becoming more widely appreciated. It is commonly accepted that Rac1 and related signaling pathways are prominently involved in the maintenance and regulation of basic nervous system functions including neurite outgrowth, neuronal migration, synaptogenesis, synaptic plasticity, and learning memory [10–13]. Moreover, Rac1 is believed to contribute to the formation of addictive behavior [14]. However, not until recently have studies revealed that Rac1 may be relevant for certain inherited neurodevelopmental disorders, likely due to its essential role in the regulation of neuronal cell structure and development [15–19]. In this review, we aim to sketch a picture of the newly identified roles of Rac1 in these diseases and to shed light on the potential of specific inhibitors for Rac1 as novel therapeutics.
2. Basic Molecular Mechanism of Rac1 Signaling
Rac1 belongs to the Rac subfamily of Rho small GTPases (~21 kDa), whose primary function is to transduce external signals to the inside of a cell. Rac proteins are among the frontline responders to external stress signals [20]. To date, three Rac proteins (Rac1–3) have been identified in vertebrates, which share a high degree of homology in amino acid sequences (88–92%) [21]. Rac1 participates in a wide spectrum of physiological processes, including actin cytoskeleton organization, cell adhesion and migration, gene expression, neurodevelopment, and synaptic plasticity [12, 22–24]. Rac1 was first identified in the human leukemia cell line HL-60 as a substrate of botulinum C3 ADP-ribosyltransferase [25, 26]. Similar to other small GTPases, Rac1 possesses a G core domain and an effector binding domain [27]. It is expressed in both the eukaryotic cytoplasm and the nucleus and cycles between the GTP-bound and GDP-bound states, marking the active and inactive forms of Rac1, respectively. To enter the active form, the bound GDP on Rac1 is replaced by GTP which is catalyzed by specific guanine nucleotide exchange factors (GEFs). Conversely, bound GTP is hydrolyzed to GDP by GTPase-activating proteins (GAPs) to produce the inactive form of Rac1 [28, 29]. Rac1 shares an identical amino acid sequence between murine, bovine, and human [30, 31]. The high degree of conservation with Rac1 protein structure and its downstream signaling cascades highlights its physiological relevance across different species. Rac1 exerts its functional impacts mainly via a downstream effector named p21-activated kinase (PAK). PAK directly phosphorylates and activates the LIM kinase (LIMK), which in turn phosphorylates and inactivates the actin-depolymerizing factor, cofilin, leading to actin depolymerization and cytoskeleton reorganization (Figure 1). In addition to the PAK-LIMK-cofilin pathway, Rac1 can also act directly through the WAVE1 and actin-related protein 2/3 (Arp2/3) complex to regulate actin nucleation and thus cellular structure, movement, and functions [32–36].
Figure 1.
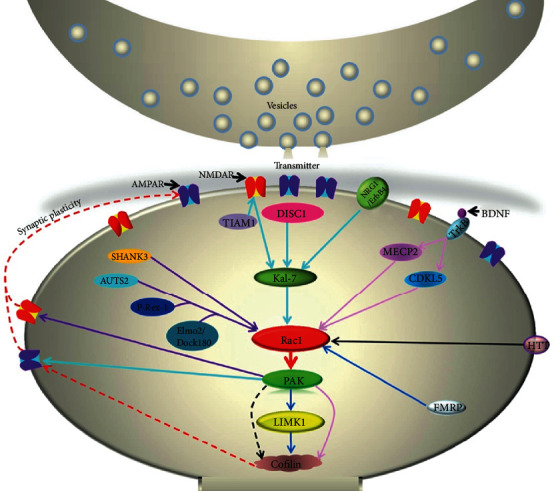
Regulation and interaction of Rac1-related signaling pathways at the postsynaptic terminal. Effectors of FXS and Huntington's disease, such as FMRP and HTT, can directly activate or inhibit Rac1 activity to modulate its downstream signaling cascades, mainly via the Rac1-PAK-cofilin pathway, which subsequently influences synaptic plasticity. In schizophrenia, NMDA receptors activate Kal-7 via TIAM1, while DISC1 and NRG1/ErbB4 interact with Kal-7 to activate or inhibit Rac1. In ASD, SHANK3 directly modulates Rac1 activity, while other effectors like AUTS2, P-Rex-1, and Elmo2/Dock180 form a complex to modulate Rac1 activity and then affect NMDA receptor activity through the PAK pathway. In Rett syndrome, BDNF activates TrkB receptors to modulate the activity of CDKL5 and MECP2 that further regulate the function of the Rac1-cofilin pathway. Abnormalities of these proteins in any pathways may affect neuroplasticity and cause neurodevelopmental disorders.
3. Rac1 and Neurodevelopmental Disorders
Given the vital role that actin dynamics plays in multiple key physiological processes in the brain, it is no surprise that Rac1 influences a wide variety of nervous system functions, including synaptogenesis, neuronal migration, neurite outgrowth, synaptic transmission and plasticity, memory, and addictive behavior formation [14, 37–41]. Aberrant Rac1 expression or activity regulation or even small alterations to its downstream signaling may lead to severe neurodevelopmental disorders (Figure 2). Here, we briefly summarize recent findings on a few hereditary neurodevelopmental disorders that involve dysregulated Rac1 expression/activity, which have not been systemically covered previously in other reviews.
Figure 2.
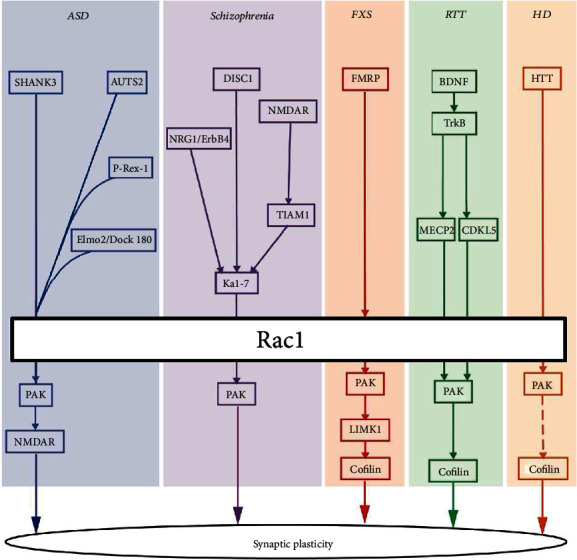
Putative schematic of the Rac1 signaling pathways involved in different neurodevelopmental disorders. The upstream effector, such as Kal-7, AUTS2, NMDAR, or FMRP, activates or inhibits Rac1 activity to modulate its downstream signaling cascades, primarily via the PAK-cofilin pathway, which orchestrates neuronal cell migration, spinogenesis, and synaptic plasticity. Abnormalities in this Rac1-related signaling complex are common features of neurodevelopmental disorders. The dashed lines indicate certain mechanisms that remain unclear and require further investigation.
3.1. Autism Spectrum Disorders
Autism spectrum disorders (ASD) refer to a group of neurodevelopmental disorders characterized by impaired social interaction and communication along with restricted or repetitive behaviors [16, 42]. ASD can be diagnosed at any age; however, they are described as developmental disorders because they become apparent mostly in the first two years of life. A number of high-risk genes linked to the etiology of ASD have been identified and characterized, including the Autism susceptibility candidate 2 (AUTS2), SH3 and multiple ankyrin repeat domains 3 (SHANK3), Ubiquitin-protein ligase E3A (UBE3A), and Methyl-CpG-binding protein 2 (MECP2) [19, 43–49]. Some of these autism-risk genes were recently found to be linked to ASD via the Rac1-associated signaling network in the brain.
The AUTS2 gene was first found to be disrupted by a de novo balanced translocation in two monozygotic twins with ASD [50]. It was later found to contribute to ASD by influencing synaptogenesis and neuron migration via Rac1. The AUTS2 protein activates Rac1 by interacting with different GEFs, such as P-Rex-1 and Elmo2/Dock180 complex, to promote the formation of lamellipodia in neurons [18, 43, 44]. Rac1 is also essential for AUTS2-mediated neuronal migration and neuritogenesis in the process of corticogenesis. At the early stages of cerebral cortex development, AUTS2 deficiency in mice results in retarded cortical neuronal migration that can be rescued by the overexpression of wild-type Rac1 [18, 44].
SHANK3 is another highly studied risk gene for ASD. The link between SHANK3 haploinsufficiency and ASD has been extensively studied in human and animal models [42, 51, 52]. SHANK3-deficient mice exhibit typical autistic cellular and behavioral phenotypes [52]. It was recently shown that the social deficits and diminished synaptic N-methyl-D-aspartate (NMDA) receptor function in SHANK3-deficient mice resulted from actin filament disorganization, caused by reduced Rac1/PAK activity and increased cofilin activity in the prefrontal cortex. To support this conclusion, it was found that both behavioral deficits and NMDA receptor malfunction were rescued by restoring Rac1/PAK activity in these mice [16, 45].
Impaired reversal learning caused by behavioral inflexibility is another hallmark symptom of ASD [53]. In a recent study with fruit flies (Drosophila melanogaster), Rac1 was a functional converging point for multiple autism risk genes, including Fragile X mental retardation 1 (FMR1), UBE3A, Neurexin-1 (Nrx-1), Neuroligin-4 (Nlg4), and Tuberous sclerosis complex 1 (TSC1). Mutations on these genes all caused similar autistic behavioral inflexibility related to Rac1-dependent memory impairment, which led to impaired reversal learning [47].
3.2. Schizophrenia
Schizophrenia is an inherited, severe psychiatric disorder that is thought to be associated with disturbances in neural network connectivity [54, 55]. An examination of postmortem brains from schizophrenia patients revealed the reduced density of dendritic spines and fewer glutamatergic synapses [56]. In the past decade, several schizophrenia risk genes have been reported, such as Disrupted-in-Schizophrenia 1 (DISC1), NMDA receptor subunit genes, Neuregulin 1/ErbB4 (NRG1/ErbB4), and Brain-derived neurotrophic factor (BDNF), which participate in the regulation of neuroplasticity and neural connectivity [15, 57–60]. Rac1 functions mostly as a downstream signaling hub molecule of these genes [11, 15, 57, 61].
Kalirin 7 (Kal-7) is a Rac1 GEF that was found to be transcriptionally downregulated in the prefrontal cortices of patients with schizophrenia [56]. Multiple lines of evidence support a critical role for Kal-7 in the modulation of spine morphology, driven mainly by the Rac1-dependent regulation of actin cytoskeleton [61–63]. Kal-7 interacts with DISC1 as a signalosome to control the duration and intensity of Rac1 activation in response to NMDA receptor activation. In rodent primary cortical neurons, DISC1 deficiency activates Rac1 and leads to rapid spine growth, while the overexpression of DISC1 suppresses Rac1 activity to reduce spine size [15]. Kal-7 also interacts with NR2B, an NMDA receptor subunit extensively involved in nervous system function and neurological diseases [10, 64–66]. It was noted that the NR2B-dependent NMDA receptor currents are diminished in neurons lacking Kal-7 [10, 61]. Additionally, Kal-7 is also involved in signaling cascades mediated by synaptic receptors like Ephrin B (EphB), Erb-B2 receptor tyrosine kinase 4 (ErbB4), and 5-Hydroxytryptamine (serotonin) receptor 2A (5HT2A), which regulate structural and functional plasticity of synapses [60, 62, 67, 68].
Like Kal-7, TIAM1 (TIAM Rac1-associated GEF 1) is a Rac1 GEF that colocalizes with the NR1 subunit of NMDA receptors. TIAM1 deficiency leads to reduced spine size, a result of TIAM1 binding to NMDA receptors and induction of local Rac1-dependent spine morphogenesis [69]. Moreover, other studies suggest that the inhibition of PAK prevents the progressive synaptic deterioration in rodent models of schizophrenia [57, 70, 71]. These studies collectively demonstrate a role for Rac1 and its regulators/effectors in the pathogenesis of schizophrenia, suggesting that Rac1 and related signaling pathways can be novel therapeutic targets.
3.3. Fragile X Syndrome
Fragile X syndrome (FXS) is a hereditary neurodevelopmental disability that results from an abnormal expansion of the CGG trinucleotide repeat in the FMR1 gene on the X chromosome. The expansion leads to promoter hypermethylation and transcriptional silencing that prevents the expression of FMR1 protein (FMRP). FXS is characterized by abnormalities in dendritic spine structure, learning disabilities, and cognitive impairment. As an X-linked disorder, FXS occurs in males about two times more frequently compared to females [72–77].
Rac1 is found physically and functionally associated with FMRP. FMRP acts as a negative regulator of Rac1 synthesis [78]. Abnormally high Rac1 activity has been observed in the neocortices of FXS patients and animal models [79]. In the actin ring of murine fibroblasts, Rac1 colocalizes and interacts with FMRP and its partners, which serve as regulators of Rac1-dependent actin remodeling [80]. In developing Drosophila brains, Rac1 interacts with the homolog of FMRP to influence the cytoskeletal dynamics and neuronal outgrowth as well as synaptic morphology at neuromuscular junctions (NMJ). Loss-of-function mutations in FMRP increase the number of higher-order dendritic branches. Conversely, expression of normal FMRP dramatically decreases dendritic branching in Drosophila dendritic arborization (DA) neurons [81]. Comery et al. generated FMR1 knockout (KO) mice and observed a larger proportion of long but thin dendritic spines in the occipital cortex with elevated Rac1, similar to what was observed in humans [79]. Another study demonstrated that Rac1 expression levels are unusually high in brain stems, hippocampi, and cortices of 3-month-old mice lacking FMR1 [78].
Rac1-related signaling pathways have also been extensively investigated in FXS animal models. When Rac1 is absent in the synapses of certain brain regions, PAK activity is also downregulated. Partial inhibition of PAK by introducing dominant-negative PAK in mice results in a shift in the overall spine distribution toward shorter spines with a lower proportion of longer spines relative to wild-type neurons [82]. Opposite phenotypes have been observed in FMR1 KO mice [78]. In a separate study, the application of a small-molecule PAK inhibitor FRAX486 rescued most of the FXS phenotypes in FMR1 KO mice [83]. The activity of cofilin was also found to be suppressed in the somatosensory cortex of FMR1 KO mice, due to the hyperactivity of Rac1 [84]. These studies revealed a previously unappreciated role for impaired Rac1-PAK1-cofilin-LIMK1 signaling in abnormal spine morphology and density associated with FXS and pointed to Rac1 as a promising target for these specific abnormalities.
3.4. Rett Syndrome
Rett syndrome (RTT) is a severe progressive developmental intellectual disability that affects almost exclusively girls [85, 86]. Autopsy studies of six girls that died between the ages of 2.9 and 35 revealed excessive amounts of immature dendrites in the motor and frontal cortices [87]. Two high-risk genes linked to RTT have been identified: MECP2 and the X-linked cyclin-dependent kinase-like 5 (CDKL5). It was reported that mutations on MECP2 account for ~20% of classical RTT along with 60~80% of RTT variants [88, 89], while mutations on CDKL5 were identified in patients with the Hanefeld variant of RTT [90, 91].
In patients with MECP2 mutations, significant decreases in spine density were observed in hippocampal CA1 pyramidal neurons. Similarly, reductions of spine density were observed in the motor cortices and hippocampi of mice lacking MECP2 [92–94], which was found to be at least partially attributed to the deregulation of BDNF, a protein well known for its vital role in spine growth and synaptogenesis by binding and activating the tropomyosin-receptor kinase B (TrkB) receptor and its downstream signaling pathways [95–99]. Although more evidence is needed, Rac1 has been proposed to act as a downstream signaling effector of BDNF [100, 101]. Moreover, in the brains of RTT model mice, activation of certain Rac1 downstream proteins such as PAK and cofilin directly activates mTOR in MECP2 mutant neurons, which is responsible for the translational control of altered proteins in RTT [102].
Patients with CDKL5 mutations exhibit prominent intellectual disabilities. Knocking down CDKL5 in the rat brain results in delayed neuronal migration and severely impairs dendritic arborization. Overexpression of Rac1 rescues the dendritic growth inhibited by CDKL5 knockdown. Moreover, CDKL5 is required for the BDNF-induced activation of Rac1 [103, 104].
Taken together, Rac1 mediates the dendritic development through BDNF regulation, which may be a common mechanism in cases of RTT involving MECP2 or CDKL5 mutations. Rac1 is also responsible for the posttranslational control of altered proteins in cases of RTT involving MECP2 mutations. The modulation of Rac1-dependent spine development is potentially beneficial as a treatment for RTT.
3.5. Huntington's Disease
Huntington's disease (HD) is a hereditary and progressive nervous system disorder that is caused by a CAG trinucleotide repeat expansion in the first exon of the HTT gene, which encodes for the huntingtin protein (HTT) [105]. HD patients manifest with progressive motor, cognitive, and emotional impairments, coupled with abnormal spine morphogenesis in the cortex and striatum [106–108].
Recent studies suggest that Rac1 may contribute to the pathogenesis and symptoms of HD. In a large-scale screening study with the yeast two-hybrid system, over 2000 HTT-interacting proteins were analyzed. A few Rho GTPase signaling components, including Rac1 and PAK2, were identified as modifiers of mutant HTT toxicity, which implicates Rac1 and related signaling cascades in the onset of HD [109]. Consistent with this screen, Rac1 activity was found to be drastically enhanced in both primary human fibroblasts lacking HTT and the striatum of 1.5-month-old HD Q140/Q140 knock-in mice [110, 111]. Moreover, consistent with the conventional role of Rac1 in oxidative stress, multiple recent studies suggest that Rac1 modulates the generation of reactive oxygen species (ROS) in HD models [112, 113]. Nevertheless, even though studies suggest that Rac1 is involved in actin-dependent morphological changes of neurons during the pathogenesis of HD [110], whether Rac1 contributes to the crucial early development of HD remains unclear and requires further investigation.
4. Screening Novel Rac1 Inhibitors for the Treatment of Neurodevelopmental Disorders
Due to the multifaceted roles that Rac1 plays in brain function and neurodevelopmental disease etiology, enormous efforts have been made to screen for effective Rac1 inhibitors with the hope to develop novel medications for relevant neurodevelopmental diseases (Table 1). A good number of small-molecule compounds targeting Rac1 and related signaling pathways have been developed. NSC23766 and its derivatives have been extensively investigated in multiple disease models, both in vitro and in vivo. NSC23766 inhibits Rac1 activity by disrupting its physical binding with its interacting proteins, such as GEFs, TIAM1, and triple functional domain protein (TRIO), without affecting RhoA or Cdc42 activity. Preclinically, NSC23766 has demonstrated positive effects in several disease models, including in models of cancers, cognitive disorders, brain injuries, and neurodegenerative and kidney diseases [112–118]. Based on its structure and characteristics, further optimization of NSC23766 has been performed. AZA1 is a compound structurally based on NSC23766 that has greater inhibitory potency on Rho GTPase activity, which was shown to inhibit both Rac1 and Cdc42 activity in prostate cancer cells and improve the survival rate of mice bearing human prostate cancer xenografts [119]. EHop-016 is another potent Rac inhibitor derived from NSC23766 that targets the association between Rac1 and the Rac GEF Vav2. EHop-016 is highly effective at inhibiting Rac1 activity and suppressing Rac-directed lamellipodia formation in MDA-MB-435 metastatic cancer cells and MDA-MB-231 metastatic breast cancer cells [120].
Table 1.
List of small-molecule compounds that inhibit Rac1 activity.
| Compound name | Formula | Molecular weight | Target Rac1 signaling | Target other Rho GTPases | Reference |
|---|---|---|---|---|---|
| NSC23766 | C24H35N73HCl | 530.96 | Inhibit TIAM1 and TRIO | None | [114] |
| ITX3 | C22H17N3OS | 371.45 | TRIO | RhoG and Rac1 | [122] |
| EHop-016 | C25H30N60 | 430.55 | Inhibit Vav2 | Rac3, Cdc42 | [120] |
| AZA1 | C22H20N6 | 368.43 | Rac1/PAK1 | Cdc42 | [119] |
| 1A-116 | C16H16F3N3 | 307.31 | Rac1/P-Rex-1 | None | [123, 127] |
| ETH 1864 | C25H27F3N204S2HCI | 581.47 | Rac1/TIAM1 | Rac1b, Rac2, Rac3 | [128] |
| FRAX486 | C25H23Cl2FN6O | 513.39 | PAK1-3 | None | [71, 83] |
Even though much work has been done to optimize NSC23766-based molecules, a big gap still remains with further development into clinical testing, possibly due to their low binding capacity to the target proteins.
Unraveling the crystal structures of Rac1 and its interacting proteins opened another door to identify inhibitory compounds [121]. ITX3 was identified as a selective inhibitor of TRIO and N-terminal TRIO-dependent cell structures in vitro based on the crystal structures and characteristics of the target proteins. However, the efficacy of ITX3 in animal models appears to be not ideal [122]. A thorough screening of the ZINC database containing more than 200,000 compounds led to the discovery of a number of novel Rac1 inhibitors. 1A-116 shows a robust antimetastatic effect by blocking Rac GEF P-Rex-1 (PIP3-dependent Rac exchanger 1) binding to suppress Rac1 activation [123]. In addition, ETH 1864 was identified to be a potent suppressor of Rac1a and its GEF, TIAM1, as well as the other Rac1 isoforms, Rac1b, Rac2, and Rac3. ETH 1864 has been demonstrated to decrease the NMDAR current density in rat cortical neurons and reduce spine density in the early stages of hippocampal neuronal development [124]. Consistent with this, LTP and LTD were both abolished by the treatment of ETH 1864 and NSC23766, respectively, on mouse hippocampus slices [11]. The downstream effectors of Rac1, such as PAK, have also been targets for novel inhibitor screening. Through a traditional, two-part structure-activity relationship approach, a series of PAK inhibitors were found in a library of 12,000 compounds using a FRET-based assay. Among them, FRAX486 was identified to show a potent inhibitory effect to PAK with good PK properties and brain penetration. The small molecule was demonstrated to reverse the spine abnormalities seen in animal models of FXS and schizophrenia. Behavioral phenotypes, such as hyperactivity and repetitive movements, were also rescued after treatment with FRAX486 [71, 83].
So far, although no small-molecule inhibitors have moved into clinical trials, recent alternative approaches to targeting Rac1 offer more optimism for this approach. A polypeptide that inhibits Rac1 and a few other RhoA GTPases is in preclinical testing with positive effects shown on treating neurodegenerative diseases and cancer [125]. An antisense RNAi oligonucleotide drug for Kaposi's sarcoma developed by researchers from the University of Miami has also been reported [126]. Although no drugs have been tested in humans at this moment, the application of these Rac1 inhibitors in neurodevelopmental disease models may provide new leads for diseases featuring synaptic abnormalities.
5. Conclusions
The dysregulation of Rac1 has been indicated in the processes of neuronal morphogenesis, migration, and synaptic plasticity in neurodevelopmental disorders such as schizophrenia, ASD, and FXS, as highlighted in the review. Given the lack of effective medications for these diseases, Rac1 presents an opportunity for therapeutic intervention by targeting the abnormalities in synaptic morphology and plasticity. Further exploration of the expression and modulation of specific Rac1 regulators as well as Rac1 itself under physiological and pathological conditions will be beneficial to our understanding of the underlying mechanisms of these diseases as well as the development of novel therapeutic approaches.
Acknowledgments
The work was supported by the grants of the National Science Foundation of China (Nos. 31527802 and 31871033) (S. Z.), the Beijing Natural Science Foundation (7182063), the Beijing Health System High-Level Health Technical Personnel (2014-3-058), and the Fund for Incubating Program of Capital Medical University (No. PYZ2018058) (X. H. W.).
Contributor Information
Guan Wang, Email: wangguan@bu.edu.
Shuli Zhang, Email: shulizhang@ibp.ac.cn.
Lei Zhang, Email: leizhang3030@hotmail.com.
Data Availability
No data were used to support this study.
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
The contributions of the authors involved in this study are as follows: conceptualization: Lei Zhang, Shuli Zhang, and Guan Wang; writing-original draft for the basic molecular mechanism of Rac1 and autism spectrum disorders: Xiaohui Wang; writing-original draft about Huntington's disease: Dongbin Liu; writing-original draft about Rac1 inhibitors for neurodevelopmental disease treatment: Fangzhen Wei; writing-original draft about fragile X syndrome: Linjie Li; writing-original draft for Rett syndrome: Xuefeng Wang; writing-original draft for schizophrenia: Yue Li; and supervision, figures, and editing: Lei Zhang, Shuli Zhang, and Guan Wang. Xiaohui Wang, Dongbin Liu, and Fangzhen Wei contributed equally to this work.
References
- 1.Han S. I., Oh S. Y., Woo S. H., et al. Implication of a small GTPase Rac1 in the activation of c-Jun N-terminal kinase and heat shock factor in response to heat shock. The Journal of Biological Chemistry. 2001;276(3):1889–1895. doi: 10.1074/jbc.M006042200. [DOI] [PubMed] [Google Scholar]
- 2.Mohammad G., Duraisamy A. J., Kowluru A., Kowluru R. A. Functional regulation of an oxidative stress mediator, Rac1, in diabetic retinopathy. Molecular Neurobiology. 2019;56(12):8643–8655. doi: 10.1007/s12035-019-01696-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang X., Shen Y., Zhang Y., et al. Rac1 mediates laminar shear stress-induced vascular endothelial cell migration. Cell Adhesion & Migration. 2013;7(6):462–468. doi: 10.4161/cam.27171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huelsenbeck S. C., Schorr A., Roos W. P., et al. Rac1 protein signaling is required for DNA damage response stimulated by topoisomerase II poisons. The Journal of Biological Chemistry. 2012;287(46):38590–38599. doi: 10.1074/jbc.M112.377903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du J., Xu R., Hu Z., et al. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1α expression in MCF-7 breast cancer cells. PLoS One. 2011;6(9, article e25213) doi: 10.1371/journal.pone.0025213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Hodes G. E., Zhang H., et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nature Communications. 2018;9(1):p. 477. doi: 10.1038/s41467-017-02794-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golden S. A., Christoffel D. J., Heshmati M., et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nature Medicine. 2013;19(3):337–344. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao Z., Tao Y., Guo X., et al. Fear conditioning downregulates Rac1 activity in the basolateral amygdala astrocytes to facilitate the formation of fear memory. Frontiers in Molecular Neuroscience. 2017;10:p. 396. doi: 10.3389/fnmol.2017.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Q., Yao W., Wang J., et al. Post-training activation of Rac1 in the basolateral amygdala is required for the formation of both short-term and long-term auditory fear memory. Frontiers in Molecular Neuroscience. 2015;8 doi: 10.3389/fnmol.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemtiri-Chlieh F., Zhao L., Kiraly D. D., Eipper B. A., Mains R. E., Levine E. S. Kalirin-7 is necessary for normal NMDA receptor-dependent synaptic plasticity. BMC Neuroscience. 2011;12(1):p. 126. doi: 10.1186/1471-2202-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez L. A., Tejada-Simon M. V. Pharmacological inactivation of the small GTPase Rac1 impairs long-term plasticity in the mouse hippocampus. Neuropharmacology. 2011;61(1-2):305–312. doi: 10.1016/j.neuropharm.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tolias K. F., Duman J. G., Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Progress in Neurobiology. 2011;94(2):133–148. doi: 10.1016/j.pneurobio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tejada-Simon M. V., Villasana L. E., Serrano F., Klann E. NMDA receptor activation induces translocation and activation of Rac in mouse hippocampal area CA1. Biochemical and Biophysical Research Communications. 2006;343(2):504–512. doi: 10.1016/j.bbrc.2006.02.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dietz D. M., Sun H., Lobo M. K., et al. Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nature Neuroscience. 2012;15(6):891–896. doi: 10.1038/nn.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi-Takagi A., Takaki M., Graziane N., et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nature Neuroscience. 2010;13(3):327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duffney L. J., Zhong P., Wei J., et al. Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Reports. 2015;11(9):1400–1413. doi: 10.1016/j.celrep.2015.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tropea D., Hardingham N., Millar K., Fox K. Mechanisms underlying the role of DISC1 in synaptic plasticity. The Journal of Physiology. 2018;596(14):2747–2771. doi: 10.1113/JP274330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hori K., Hoshino M. Neuronal migration and AUTS2 syndrome. Brain Sciences. 2017;7(12):p. 54. doi: 10.3390/brainsci7050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian C., Kay Y., Sadybekov A., Rao S., Katritch V., Herring B. E. An intellectual disability-related missense mutation in Rac1 prevents LTP induction. Frontiers in Molecular Neuroscience. 2018;11:p. 223. doi: 10.3389/fnmol.2018.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fritz G., Kaina B. Rac1 GTPase, a multifunctional player in the regulation of genotoxic stress response. Cell Cycle. 2014;12(16):2521–2522. doi: 10.4161/cc.25807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corbetta S., Gualdoni S., Albertinazzi C., et al. Generation and characterization of Rac3 knockout mice. Molecular and Cellular Biology. 2005;25(13):5763–5776. doi: 10.1128/MCB.25.13.5763-5776.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdrabou A., Wang Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells. 2018;7(12):p. 263. doi: 10.3390/cells7120263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payapilly A., Malliri A. Compartmentalisation of RAC1 signalling. Current Opinion in Cell Biology. 2018;54:50–56. doi: 10.1016/j.ceb.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Liu L., Li J., Zhang L., et al. Cofilin phosphorylation is elevated after F-actin disassembly induced by Rac1 depletion. BioFactors. 2015;41(5):352–359. doi: 10.1002/biof.1235. [DOI] [PubMed] [Google Scholar]
- 25.Didsbury J., Weber R. F., Bokoch G. M., Evans T., Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. The Journal of Biological Chemistry. 1989;264(28):16378–16382. [PubMed] [Google Scholar]
- 26.Polakis P. G., Weber R. F., Nevins B., Didsbury J. R., Evans T., Snyderman R. Identification of the ral and rac1 gene products, low molecular mass GTP-binding proteins from human platelets. The Journal of Biological Chemistry. 1989;264(28):16383–16389. [PubMed] [Google Scholar]
- 27.Yang Z. Small GTPases: versatile signaling switches in plants. Plant Cell. 2002;14(suppl 1):S375–S388. doi: 10.1105/tpc.001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marei H., Carpy A., Macek B., Malliri A. Proteomic analysis of Rac1 signaling regulation by guanine nucleotide exchange factors. Cell Cycle. 2016;15(15):1961–1974. doi: 10.1080/15384101.2016.1183852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marei H., Carpy A., Woroniuk A., et al. Differential Rac1 signalling by guanine nucleotide exchange factors implicates FLII in regulating Rac1-driven cell migration. Nature Communications. 2016;7(1) doi: 10.1038/ncomms10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia A., Zhang X. H. cDNA cloning, characterization, and expression analysis of the Rac1 gene from Scophthalmus maximus. Comparative Biochemistry and Physiology. Part B, Biochemistry & Molecular Biology. 2009;154(1):80–84. doi: 10.1016/j.cbpb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 31.García-Weber D., Millán J. Parallels between single cell migration and barrier formation: the case of RhoB and Rac1 trafficking. Small GTPases. 2016;9(4):332–338. doi: 10.1080/21541248.2016.1231655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ridley A. J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends in Cell Biology. 2006;16(10):522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Yang N., Higuchi O., Ohashi K., et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393(6687):809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 34.Edwards D. C., Sanders L. C., Bokoch G. M., Gill G. N. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nature Cell Biology. 1999;1(5):253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 35.Pollard T. D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annual Review of Biophysics and Biomolecular Structure. 2007;36(1):451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez A. M., Flamini M. I., Fu X. D., et al. Rapid signaling of estrogen to WAVE1 and moesin controls neuronal spine formation via the actin cytoskeleton. Molecular Endocrinology. 2009;23(8):1193–1202. doi: 10.1210/me.2008-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L., Zhang F., Yang T., et al. The B-cell receptor BR3 modulates cellular branching via Rac1 during neuronal migration. Journal of Molecular Cell Biology. 2016;8(4):363–365. doi: 10.1093/jmcb/mjw034. [DOI] [PubMed] [Google Scholar]
- 38.Li L. Z., Yin N., Li X. Y., et al. Rac1 modulates excitatory synaptic transmission in mouse retinal ganglion cells. Neuroscience Bulletin. 2019;35(4):673–687. doi: 10.1007/s12264-019-00353-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costa J. F., Dines M., Lamprecht R. The role of Rac GTPase in dendritic spine morphogenesis and memory. Front Synaptic Neurosci. 2020;12 doi: 10.3389/fnsyn.2020.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pennucci R., Gucciardi I., de Curtis I. Rac1 and Rac3 GTPases differently influence the morphological maturation of dendritic spines in hippocampal neurons. PLoS One. 2019;14(8, article e0220496) doi: 10.1371/journal.pone.0220496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lv L., Liu Y., Xie J., et al. Interplay between α2-chimaerin and Rac1 activity determines dynamic maintenance of long-term memory. Nature Communications. 2019;10(1):p. 5313. doi: 10.1038/s41467-019-13236-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Rubeis S., Study T. D. D. D., He X., et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hori K., Nagai T., Shan W., et al. Heterozygous disruption of autism susceptibility candidate 2 causes impaired emotional control and cognitive memory. PLoS One. 2015;10(12):p. e0145979. doi: 10.1371/journal.pone.0145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hori K., Nagai T., Shan W., et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Reports. 2014;9(6):2166–2179. doi: 10.1016/j.celrep.2014.11.045. [DOI] [PubMed] [Google Scholar]
- 45.Duffney L. J., Wei J., Cheng J., et al. Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. The Journal of Neuroscience. 2013;33(40):15767–15778. doi: 10.1523/JNEUROSCI.1175-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J. H., Espinera A. R., Chen D., et al. Neonatal inflammatory pain and systemic inflammatory responses as possible environmental factors in the development of autism spectrum disorder of juvenile rats. Journal of Neuroinflammation. 2016;13(1):p. 109. doi: 10.1186/s12974-016-0575-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong T., He J., Wang S., Wang L., Cheng Y., Zhong Y. Inability to activate Rac1-dependent forgetting contributes to behavioral inflexibility in mutants of multiple autism-risk genes. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(27):7644–7649. doi: 10.1073/pnas.1602152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Astrinidis A., Cash T. P., Hunter D. S., Walker C. L., Chernoff J., Henske E. P. Tuberin, the tuberous sclerosis complex 2 tumor suppressor gene product, regulates Rho activation, cell adhesion and migration. Oncogene. 2002;21(55):8470–8476. doi: 10.1038/sj.onc.1205962. [DOI] [PubMed] [Google Scholar]
- 49.Sadybekov A., Tian C., Arnesano C., Katritch V., Herring B. E. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nature Communications. 2017;8(1):p. 601. doi: 10.1038/s41467-017-00472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sultana R., Yu C. E., Yu J., et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics. 2002;80(2):129–134. doi: 10.1006/geno.2002.6810. [DOI] [PubMed] [Google Scholar]
- 51.Betancur C., Buxbaum J. D. SHANK3 haploinsufficiency: a "common" but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Molecular Autism. 2013;4(1):p. 17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kouser M., Speed H. E., Dewey C. M., et al. Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. The Journal of Neuroscience. 2013;33(47):18448–18468. doi: 10.1523/JNEUROSCI.3017-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dajani D. R., Uddin L. Q. Demystifying cognitive flexibility: implications for clinical and developmental neuroscience. Trends in Neurosciences. 2015;38(9):571–578. doi: 10.1016/j.tins.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramos-Miguel A., Barr A. M., Honer W. G. Spines, synapses, and schizophrenia. Biological Psychiatry. 2015;78(11):741–743. doi: 10.1016/j.biopsych.2015.08.035. [DOI] [PubMed] [Google Scholar]
- 55.Datta D., Arion D., Corradi J. P., Lewis D. A. Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biological Psychiatry. 2015;78(11):775–785. doi: 10.1016/j.biopsych.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glantz L. A., Lewis D. A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Archives of General Psychiatry. 2000;57(1):65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 57.Chen S. Y., Huang P. H., Cheng H. J. Disrupted-in-Schizophrenia 1-mediated axon guidance involves TRIO-RAC-PAK small GTPase pathway signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5861–5866. doi: 10.1073/pnas.1018128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moutin E., Nikonenko I., Stefanelli T., et al. Palmitoylation of cdc42 promotes spine stabilization and rescues spine density deficit in a mouse model of 22q11.2 deletion syndrome. Cerebral Cortex. 2017;27(7):3618–3629. doi: 10.1093/cercor/bhw183. [DOI] [PubMed] [Google Scholar]
- 59.Han D., Xu L., Xiao H., Prado Schmidt G. C., Shi S. Dizocilpine reduces head diameter of dendritic spines in the hippocampus of adolescent rats. Psychiatry Research. 2013;210(1):351–356. doi: 10.1016/j.psychres.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 60.Cahill M., Penzes P. Colocalization between endogenous erbB4 and endogenous kalirin-7 in interneuronal dendrites. Molecular Psychiatry. 2012;17(1):p. 1. doi: 10.1038/mp.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kiraly D. D., Lemtiri-Chlieh F., Levine E. S., Mains R. E., Eipper B. A. Kalirin binds the NR2B subunit of the NMDA receptor, altering its synaptic localization and function. The Journal of Neuroscience. 2011;31(35):12554–12565. doi: 10.1523/JNEUROSCI.3143-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie Z., Srivastava D. P., Photowala H., et al. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56(4):640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Penzes P., Johnson R. C., Kambampati V., Mains R. E., Eipper B. A. Distinct roles for the two Rho GDP/GTP exchange factor domains of kalirin in regulation of neurite growth and neuronal morphology. The Journal of Neuroscience. 2001;21(21):8426–8434. doi: 10.1523/JNEUROSCI.21-21-08426.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berberich S., Punnakkal P., Jensen V., et al. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. The Journal of Neuroscience. 2005;25(29):6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sze C.-I., Bi H., Kleinschmidt-DeMasters B. K., Filley C. M., Martin L. J. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. Journal of the Neurological Sciences. 2001;182(2):151–159. doi: 10.1016/S0022-510X(00)00467-6. [DOI] [PubMed] [Google Scholar]
- 66.Ma J., Choi B. R., Chung C. H., Min S., Jeon W., Han J. S. Chronic brain inflammation causes a reduction in GluN2A and GluN2B subunits of NMDA receptors and an increase in the phosphorylation of mitogen-activated protein kinases in the hippocampus. Molecular Brain. 2014;7(1):p. 33. doi: 10.1186/1756-6606-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones K. A., Srivastava D. P., Allen J. A., Strachan R. T., Roth B. L., Penzes P. Rapid modulation of spine morphology by the 5-HT2A serotonin receptor through kalirin-7 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(46):19575–19580. doi: 10.1073/pnas.0905884106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Penzes P., Jones K. A. Dendritic spine dynamics--a key role for kalirin-7. Trends in Neurosciences. 2008;31(8):419–427. doi: 10.1016/j.tins.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tolias K. F., Bikoff J. B., Burette A., et al. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45(4):525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 70.Xie Z., Photowala H., Cahill M. E., et al. Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. The Journal of Neuroscience. 2008;28(24):6079–6091. doi: 10.1523/JNEUROSCI.1170-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayashi-Takagi A., Araki Y., Nakamura M., et al. PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(17):6461–6466. doi: 10.1073/pnas.1321109111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stefanovic S., DeMarco B. A., Underwood A., Williams K. R., Bassell G. J., Mihailescu M. R. Fragile X mental retardation protein interactions with a G quadruplex structure in the 3'-untranslated region of NR2B mRNA. Molecular BioSystems. 2015;11(12):3222–3230. doi: 10.1039/C5MB00423C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonaccorso C. M., Spatuzza M., di Marco B., et al. Fragile X mental retardation protein (FMRP) interacting proteins exhibit different expression patterns during development. International Journal of Developmental Neuroscience. 2015;42(1):15–23. doi: 10.1016/j.ijdevneu.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 74.Heulens I. Fragile X syndrome: from gene discovery to therapy. Frontiers in Bioscience. 2011;16(1):p. 1211. doi: 10.2741/3785. [DOI] [PubMed] [Google Scholar]
- 75.Bolduc F. Fragile X mental retardation 1 and filamin A interact genetically in Drosophila long-term memory. Front Neural Circuits. 2010;3 doi: 10.3389/neuro.04.022.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hagerman R. J., Berry-Kravis E., Hazlett H. C., et al. Fragile X syndrome. Nature Reviews. Disease Primers. 2017;3(1) doi: 10.1038/nrdp.2017.65. [DOI] [PubMed] [Google Scholar]
- 77.Rajaratnam A., Shergill J., Salcedo-Arellano M., Saldarriaga W., Duan X., Hagerman R. Fragile X syndrome and fragile X-associated disorders. F1000Res. 2017;6:p. 2112. doi: 10.12688/f1000research.11885.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bongmba O. Y. N., Martinez L. A., Elhardt M. E., Butler K., Tejada-Simon M. V. Modulation of dendritic spines and synaptic function by Rac1: a possible link to fragile X syndrome pathology. Brain Research. 2011;1399:79–95. doi: 10.1016/j.brainres.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Comery T. A., Harris J. B., Willems P. J., et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(10):5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castets M., Schaeffer C.´., Bechara E., et al. FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Human Molecular Genetics. 2005;14(6):835–844. doi: 10.1093/hmg/ddi077. [DOI] [PubMed] [Google Scholar]
- 81.Lee A., Li W., Xu K., Bogert B. A., Su K., Gao F. B. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development. 2003;130(22):5543–5552. doi: 10.1242/dev.00792. [DOI] [PubMed] [Google Scholar]
- 82.Hayashi M. L., Choi S. Y., Rao B. S. S., et al. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron. 2004;42(5):773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 83.Dolan B. M., Duron S. G., Campbell D. A., et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(14):5671–5676. doi: 10.1073/pnas.1219383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pyronneau A., He Q., Hwang J. Y., Porch M., Contractor A., Zukin R. S. Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Science Signaling. 2017;10(504, article eaan0852) doi: 10.1126/scisignal.aan0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Filippis B., Ricceri L., Fuso A., Laviola G. Neonatal exposure to low dose corticosterone persistently modulates hippocampal mineralocorticoid receptor expression and improves locomotor/exploratory behaviour in a mouse model of Rett syndrome. Neuropharmacology. 2013;68:174–183. doi: 10.1016/j.neuropharm.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 86.Hagberg B. Rett's syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatrica Scandinavica. 1985;74(3):405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- 87.Cianfaglione R., Clarke A., Kerr M., et al. A national survey of Rett syndrome: behavioural characteristics. Journal of Neurodevelopmental Disorders. 2015;7(1):p. 11. doi: 10.1186/s11689-015-9104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheadle J. P., Gill H., Fleming N., et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Human Molecular Genetics. 2000;9(7):1119–1129. doi: 10.1093/hmg/9.7.1119. [DOI] [PubMed] [Google Scholar]
- 89.Gill H., Cheadle J. P., Maynard J., et al. Mutation analysis in the MECP2 gene and genetic counselling for Rett syndrome. Journal of Medical Genetics. 2003;40(5):380–384. doi: 10.1136/jmg.40.5.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Keogh C., Pini G., Dyer A. H., et al. Clinical and genetic Rett syndrome variants are defined by stable electrophysiological profiles. BMC Pediatrics. 2018;18(1):p. 333. doi: 10.1186/s12887-018-1304-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pini G., Bigoni S., Engerström I. W., et al. Variant of Rett syndrome and CDKL5 gene: clinical and autonomic description of 10 cases. Neuropediatrics. 2012;43(1):37–43. doi: 10.1055/s-0032-1308856. [DOI] [PubMed] [Google Scholar]
- 92.Sun Y., Gao Y., Tidei J. J., et al. Loss of MeCP2 in immature neurons leads to impaired network integration. Human Molecular Genetics. 2019;28(2):245–257. doi: 10.1093/hmg/ddy338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Balakrishnan S., Mironov S. L. CA1 neurons acquire Rett syndrome phenotype after brief activation of glutamatergic receptors: specific role of mGluR1/5. Frontiers in Cellular Neuroscience. 2018;12:p. 363. doi: 10.3389/fncel.2018.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gulmez Karaca K., Brito D. V. C., Zeuch B., Oliveira A. M. M. Adult hippocampal MeCP2 preserves the genomic responsiveness to learning required for long-term memory formation. Neurobiology of Learning and Memory. 2018;149:84–97. doi: 10.1016/j.nlm.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 95.McFarland K. N., Huizenga M. N., Darnell S. B., et al. MeCP2: a novel huntingtin interactor. Human Molecular Genetics. 2014;23(4):1036–1044. doi: 10.1093/hmg/ddt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li W., Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology. 2014;76:737–746. doi: 10.1016/j.neuropharm.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klein R., Nanduri V., Jing S., et al. The Trkb tyrosine protein-kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66(2):395–403. doi: 10.1016/0092-8674(91)90628-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu B., Pang P. T., Woo N. H. The yin and yang of neurotrophin action. Nature Reviews. Neuroscience. 2005;6(8):603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 99.Zuccato C., Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nature Reviews. Neurology. 2009;5(6):311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- 100.Bellot A., Guivernau B., Tajes M., Bosch-Morató M., Valls-Comamala V., Muñoz F. J. The structure and function of actin cytoskeleton in mature glutamatergic dendritic spines. Brain Research. 2014;1573:1–16. doi: 10.1016/j.brainres.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 101.Chen Q., Zhu Y. C., Yu J., et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. The Journal of Neuroscience. 2010;30(38):12777–12786. doi: 10.1523/JNEUROSCI.1102-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Filippis B., Nativio P., Fabbri A., et al. Pharmacological stimulation of the brain serotonin receptor 7 as a novel therapeutic approach for Rett syndrome. Neuropsychopharmacology. 2014;39(11):2506–2518. doi: 10.1038/npp.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fuchs C., Medici G., Trazzi S., et al. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathology. 2019;29(5):658–674. doi: 10.1111/bpa.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fuchs C., Gennaccaro L., Trazzi S., et al. Heterozygous CDKL5 knockout female mice are a valuable animal model for CDKL5 disorder. Neural Plasticity. 2018;2018:18. doi: 10.1155/2018/9726950.9726950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tang B. L. Unconventional secretion and intercellular transfer of mutant huntingtin. Cell. 2018;7(6):p. 59. doi: 10.3390/cells7060059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Graham R. K., Pouladi M. A., Joshi P., et al. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. The Journal of Neuroscience. 2009;29(7):2193–2204. doi: 10.1523/JNEUROSCI.5473-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmidt M. E., Buren C., Mackay J. P., et al. Altering cortical input unmasks synaptic phenotypes in the YAC128 cortico-striatal co-culture model of Huntington disease. BMC Biology. 2018;16(1):p. 58. doi: 10.1186/s12915-018-0526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reiner A., Albin R. L., Anderson K. D., D'Amato C. J., Penney J. B., Young A. B. Differential loss of striatal projection neurons in Huntington disease. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(15):5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tourette C., Li B., Bell R., et al. A large scale huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. The Journal of Biological Chemistry. 2014;289(10):6709–6726. doi: 10.1074/jbc.M113.523696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tousley A., Iuliano M., Weisman E., et al. Rac1 activity is modulated by huntingtin and dysregulated in models of Huntington's disease. Journal of Huntington's Disease. 2019;8(1):53–69. doi: 10.3233/JHD-180311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Valencia A., Sapp E., Kimm J. S., et al. Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington's disease. Human Molecular Genetics. 2013;22(6):1112–1131. doi: 10.1093/hmg/dds516. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112.Marei H., Malliri A. GEFs: dual regulation of Rac1 signaling. Small GTPases. 2016;8(2):90–99. doi: 10.1080/21541248.2016.1202635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marei H., Malliri A. Rac1 in human diseases: the therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases. 2016;8(3):139–163. doi: 10.1080/21541248.2016.1211398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gao Y., Dickerson J. B., Guo F., Zheng J., Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(20):7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Q. G., Wang R., Han D., Dong Y., Brann D. W. Role of Rac1 GTPase in JNK signaling and delayed neuronal cell death following global cerebral ischemia. Brain Research. 2009;1265:138–147. doi: 10.1016/j.brainres.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou Z., Hu J., Passafaro M., Xie W., Jia Z. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. The Journal of Neuroscience. 2011;31(3):819–833. doi: 10.1523/JNEUROSCI.3869-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim I. H., Park S. K., Hong S. T., et al. Inositol 1,4,5-trisphosphate 3-kinase A functions as a scaffold for synaptic Rac signaling. The Journal of Neuroscience. 2009;29(44):14039–14049. doi: 10.1523/JNEUROSCI.2483-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wu P., Ding Z. B., Meng S. Q., et al. Differential role of Rac in the basolateral amygdala and cornu ammonis 1 in the reconsolidation of auditory and contextual Pavlovian fear memory in rats. Psychopharmacology. 2014;231(15):2909–2919. doi: 10.1007/s00213-014-3462-0. [DOI] [PubMed] [Google Scholar]
- 119.Zins K., Lucas T., Reichl P., Abraham D., Aharinejad S. A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS One. 2013;8(9, article e74924) doi: 10.1371/journal.pone.0074924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Montalvo-Ortiz B. L., Castillo-Pichardo L., Hernández E., et al. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. The Journal of Biological Chemistry. 2012;287(16):13228–13238. doi: 10.1074/jbc.M111.334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Worthylake D. K., Rossman K. L., Sondek J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature. 2000;408(6813):682–688. doi: 10.1038/35047014. [DOI] [PubMed] [Google Scholar]
- 122.Bouquier N., Vignal E., Charrasse S., et al. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chemistry & Biology. 2009;16(6):657–666. doi: 10.1016/j.chembiol.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 123.Cardama G., Comin M., Hornos L., et al. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anti-Cancer Agents in Medicinal Chemistry. 2014;14(6):840–851. doi: 10.2174/18715206113136660334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Onesto C., Shutes A., Picard V., Schweighoffer F., der C. J. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods in Enzymology. 2008;439:111–129. doi: 10.1016/S0076-6879(07)00409-0. [DOI] [PubMed] [Google Scholar]
- 125.Klaus A., et al. A polypeptide capable of glycosylating tyrosine residues of target proteins. WO; 2014. [Google Scholar]
- 126.Dong C., et al. Use of Rac1 inhibitors to treat Kaposi's sarcoma. 2009. US2008/011916.
- 127.Gonzalez N., Cardama G. A., Comin M. J., et al. Pharmacological inhibition of Rac1-PAK1 axis restores tamoxifen sensitivity in human resistant breast cancer cells. Cellular Signalling. 2017;30:154–161. doi: 10.1016/j.cellsig.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 128.Raynaud F., Moutin E., Schmidt S., et al. Rho-GTPase-activating protein interacting with Cdc-42-interacting protein 4 homolog 2 (Rich2): a new Ras-related C3 botulinum toxin substrate 1 (Rac1) GTPase-activating protein that controls dendritic spine morphogenesis. The Journal of Biological Chemistry. 2014;289(5):2600–2609. doi: 10.1074/jbc.M113.534636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used to support this study.