


Abstract
Pentatricopeptide repeat (PPR) motifs are α-helical structures known for their modular recognition of single-stranded RNA sequences with each motif in a tandem array binding to a single nucleotide. Protein-only RNase P 1 (PRORP1) in Arabidopsis thaliana is an endoribonuclease that uses its PPR domain to recognize precursor tRNAs (pre-tRNAs) as it catalyzes removal of the 5′-leader sequence from pre-tRNAs with its NYN metallonuclease domain. To gain insight into the mechanism by which PRORP1 recognizes tRNA, we determined a crystal structure of the PPR domain in complex with yeast tRNAPhe at 2.85 Å resolution. The PPR domain of PRORP1 bound to the structurally conserved elbow of tRNA and recognized conserved structural features of tRNAs using mechanisms that are different from the established single-stranded RNA recognition mode of PPR motifs. The PRORP1 PPR domain-tRNAPhe structure revealed a conformational change of the PPR domain upon tRNA binding and moreover demonstrated the need for pronounced overall flexibility in the PRORP1 enzyme conformation for substrate recognition and catalysis. The PRORP1 PPR motifs have evolved strategies for protein-tRNA interaction analogous to tRNA recognition by the RNA component of ribonucleoprotein RNase P and other catalytic RNAs, indicating convergence on a common solution for tRNA substrate recognition.
INTRODUCTION
Transfer RNA (tRNA) is an essential non-coding RNA required for protein translation that physically links the genetic code in mRNA to the amino acid sequence of a protein. With a classical length of 70–85 nucleotides, mature tRNAs fold into the functional 3D L-shaped structure. tRNAs undergo a maturation process whereby precursor molecules undergo a series of individual processing steps, including 5′- and 3′-sequence removal, RNA base modifications, splicing, and addition of the conserved 3′-terminal CCA sequence. Correct tRNA processing is critical for function. It is therefore not surprising that mutations in tRNA genes and tRNA processing enzymes are associated with multiple diseases (1).
Ribonuclease P (RNase P) is an endoribonuclease that catalyzes the 5′ maturation of tRNA precursors (pre-tRNA). Two forms exist in nature. In all three phylogenetic domains, RNase P occurs as a ribonucleoprotein (RNP) containing a catalytic RNA and up to 10 protein cofactors. In eukaryotes, protein-only RNase P (PRORP) forms are present in the nuclei and organelles (2,3). A minimal protein-only RNase P was also reported in some phyla of bacteria and archaea (4,5). Because RNP RNase P and PRORP catalyze identical reactions, they serve as a model system for how RNA-based and polypeptide enzymes have co-evolved (3). Since Altman and co-workers discovered in 1983 that Escherichia coli RNase P RNA (M1 RNA) is a ribozyme (6), biochemical and structural studies have focused largely on bacterial RNase P RNAs, and much information on structure-function relationships is available (2,7). The structures of bacterial, archaeal, and eukaryotic RNP RNase P in complex with tRNA elucidated the structural basis for the catalytic mechanism and tRNA recognition of RNP RNase P at the atomic level (8–11).
How 5′ pre-tRNA processing was carried out in the apparent absence of RNP RNase P in plant cells and organelles in eukaryotic cells was a mystery until the discovery of PRORP in 2008. PRORP was initially found in human mitochondria (12,13) and subsequently found in the organelles and nuclei of the model plant A. thaliana (14,15), the alga Ostreococcus tauri (16), the protozoan Trypanosoma brucei (17) and the moss Physcomitrella patens (18). A recent bioinformatics analysis described that PRORP proteins are widely present in four out of the five main eukaryotic supergroups (apparently absent in Amoebozoa), and that the occurrence of RNP RNase P and PRORP proteins seems mutually exclusive in genetic compartments of modern Eukarya (19). Human mitochondrial PRORP was the first identified (originally termed mammalian mitochondrial RNase P, MRPP3), and it requires two additional protein subunits (MRPP1 and MRPP2) for function (12). In contrast, A. thaliana PRORP requires no additional subunits to catalyze pre-tRNA maturation in vitro (although there is some evidence that plant organellar PRORP might function with other proteins in vivo (20)), and as a result, it is used as a model PRORP for comparison with RNP RNase P. A. thaliana encodes three isozymes, PRORP1, PRORP2, and PRORP3. PRORP1 is localized in mitochondria and chloroplasts, while PRORP2 and PRORP3 are in the nuclei (14). PRORP1 can catalyze the endonucleolytic maturation of pre-tRNA and substitute for RNP RNase P activity in E. coli (14) as well as in Saccharomyces cerevisiae (21). Crystal structures of PRORP1 and PRORP2 revealed that PRORP proteins comprise five tandem pentatricopeptide repeat (PPR) motifs, a central linker domain, and a metallonuclease domain belonging to the NYN family. The N-terminal PPR domain is involved in pre-tRNA binding, while the C-terminal metallonuclease domain catalyzes cleavage (22,23). The PRORP PPR domain has been shown to contact the D and TψC loops at the pre-tRNA ‘elbow’ while the NYN nuclease domain is responsible for catalysis at the 5′ end (3,22,24). No crystal structures of a PRORP in complex with pre-tRNA or tRNA have been reported, leaving open the question of how the PPR domain recognizes the structured pre-tRNA elbow.
PPR motif-containing proteins are eukaryote-specific and widely distributed among RNA-binding proteins in plants that are involved in organelle transcript processing and stability (25–27). PPR motifs are often found in tandem, and each repeat forms a helix-turn-helix fold of ∼35 amino acid residues (28). Computational and biochemical analyses have identified a putative RNA recognition code for PPR proteins (29–31). Recent crystal structures of the maize chloroplast protein PPR10 and a designed PPR protein in complex with single-stranded RNA support the proposed recognition code and revealed the molecular basis for specific and modular recognition of RNA bases (32,33).
Given the established single-stranded RNA recognition mode of PPR motifs, different models have been proposed for the PRORP1 PPR domain recognition of substrate pre-tRNA. Imai et al. proposed a model of tRNA docked onto PRORP1 with the second (PPR2) and third (PPR3) PPR motifs recognizing nucleotides C56 and A57, respectively, in the TψC loop of pre-tRNA (34). In addition, Pinker et al. reported a small angle scattering (SAXS)-based model of PRORP2 in complex with pre-tRNA, showing its PPR2 and PPR3 motifs recognizing C56 in the TψC loop and G18 in the D-loop, respectively (35). Both of these models used the rules derived for sequence-specific binding of single-stranded RNA by PPR motifs and proposed that PPR2 selects a pyrimidine while PPR3 selects a purine. In contrast, Klemm et al. demonstrated that conserved residues outside the canonical PPR RNA-interacting positions are critical for pre-tRNA recognition and tRNA sequence substitutions had little effect on PRORP1-pre-tRNA binding affinity (36). Moreover, the salt dependence of PRORP-pre-tRNA affinity indicated the involvement of ionic interactions. As a result, Klemm et al. proposed a model of the PRORP-pre-tRNA complex without sequence-specific interactions, instead showing the PPR domain interacting with backbone phosphodiester bonds of tRNA. This model is also consistent with biochemical data showing that the PPR3 motif of A. thaliana PRORP3 could not be ‘reprogrammed’ using the established single-stranded RNA-binding code (29–31) to bind to tRNA bearing a pyrimidine at nucleotide 57 (37).
To gain an atomic level understanding of how PRORP recognizes pre-tRNA and resolve the discrepancies in models of PPR motif recognition of the tRNA elbow, we determined a crystal structure of the PPR domain of A. thaliana PRORP1 in complex with yeast tRNAPhe at a resolution of 2.85 Å. The structure revealed that the PPR domain of PRORP1 recognizes the ‘elbow’ region of yeast tRNAPhe via base- and structure-specific interactions that are completely different from the established single-stranded RNA recognition by other PPR proteins. The PPR1 and PPR2 motifs recognize two invariant nucleotide residues, G19 and C56, that form a base pair between the D and TψC loops. In addition, residues within the PPR1 motif bind U17 and G20 in the D loop, and residues in the PPR3 motif interact with the TψC loop. These residues form electrostatic and stacking interactions with the tRNA and also hydrogen bond interactions that appear to be base specific. Each of these interactions contributes to the binding affinity. This tRNA elbow recognition mode is remarkably similar to that of RNP RNase P, as well as to those of 23S rRNA (38) and T-box riboswitches (39). Thus, the results presented here support the notion that PRORP proteins and functional RNAs, such as RNase P RNA, 23S rRNA and T-boxes, have converged on a similar solution to tRNA recognition (8–11,24,35,38–40). The tRNA interaction mode of the PPR domain also reveals evolution of PPR motifs to recognize structured RNA in addition to single-stranded RNA sequences.
MATERIALS AND METHODS
Protein expression and purification
The cDNA sequence encoding an engineered PPR domain (residues 84–292 with solubilizing substitutions Y266N, F284Q and F291Q) was subcloned into the pSMT3 vector (kindly provided by Christopher Lima, Memorial Sloan Kettering Cancer Center), which encodes an N-terminal His6-SUMO tag. The engineered PPR domain was expressed in E. coli strain BL21-CodonPlus (DE3)-RIL (Agilent Technologies) at 18°C overnight after induction with 0.5 mM IPTG. The cells were collected by centrifugation, and pellets were resuspended in lysis buffer (containing 50 mM Tris–HCl, pH 8.0, 500 mM NaCl) and stored at −80°C until use. The cells were disrupted by sonication followed by centrifugation to remove cell debris. The soluble fraction was applied to a Ni-NTA agarose column and thoroughly washed with lysis buffer containing 20 mM imidazole. The target SUMO fusion protein was eluted with lysis buffer containing 400 mM imidazole. The fusion protein was cleaved overnight with 0.2 mg of Ulp1 protease and dialyzed against a buffer containing 50 mM Tris–HCl, pH 8.0, 150 mM NaCl and 0.5 mM TCEP. The protein was then loaded onto a HiTrap Q column (GE Healthcare), which did not bind the PRORP1 PPR domain (Supplementary Figure S1A). The flow-through fractions were pooled and dialyzed against a buffer containing 25 mM HEPES–NaOH, pH 7.0, 150 mM NaCl and 0.5 mM TCEP. The sample was loaded onto a HiTrap SP column (GE Healthcare). Bound proteins were eluted using a linear gradient from 0.1 to 1 M NaCl in 25 mM HEPES–NaOH, pH 7.0. Peak fractions containing the engineered PPR domain were pooled and concentrated and reducing agent was added (final concentration of 1 mM dithiothreitol or 5 mM 2-mercaptoethanol). The protein was purified further using a Superdex 75 10/300 GL column (GE Healthcare), equilibrated with 25 mM HEPES, pH 7.0, 200 mM NaCl, and 0.5 mM TCEP. Final purified protein was concentrated to 5 mg/ml.
For crystallization, the engineered PPR domain was mixed with commercially available yeast tRNAPhe (Sigma-Aldrich). Protein and RNA were mixed at a ratio of 1:1.05, and the mixture was incubated at 4°C overnight. The protein-RNA complex was purified further using a Superdex 75 10/300 GL column (GE Healthcare), equilibrated with 25 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM MgCl2 and 0.5 mM TCEP (Supplementary Figure S1B). Peak fractions containing the complex were pooled and concentrated to A260 = 43.
Full-length PRORP1 protein for binding assays was purified as described in a previous study (22). The protein was purified by Ni-NTA agarose chromatography, followed by purification on a HiTrap SP column. Peak fractions containing the full-length protein were pooled and concentrated. The protein was purified further using a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated with 20 mM MOPS, pH 7.8, 300 mM NaCl and 0.5 mM TCEP. The peak fractions were pooled and concentrated to 76 μM. Mutant proteins were expressed at equivalent levels to WT protein, behaved similarly during the purification steps, and no differences were detected in CD spectra.
Circular dichroism analyses
To assess folding of the mutant proteins, we measured circular dichroism (CD) spectra of full-length PRORP1 wild-type, R212K, R210S and Y133Q proteins (Supplementary Figure S2). The CD spectra were measured on a Chirascan CD spectrometer (Applied Photophysics) at 20°C. For each sample (300 μl in a 0.1 cm light-path cell), three scans were accumulated in the wavelength range of 190–260 nm with a 0.2 nm step size. Protein samples were 0.13 mg/ml in 25 mM sodium phosphate, pH 7.5, 200 mM NaCl. The raw CD data were adjusted by subtracting a buffer blank. CD spectra of wild-type and mutant proteins displayed negative ellipticities at 208/222 and 215 nm, which indicate the presence of α helices and β strands, respectively.
Crystallization, data collection, structure determination and refinement
Crystallization of the purified PRORP1 PPR domain-tRNAPhe complex was performed by the sitting-drop vapor diffusion method at 4°C. Sitting drops contained 250 nl of protein–RNA complex solution mixed with 250 nl of reservoir solution (1.4–1.5 M sodium citrate). Prior to data collection, crystals were transferred to a cryoprotectant solution containing 1.6 M sodium citrate and flash cooled to −180°C. X-ray diffraction data were collected at beamline 22-ID of the Advanced Photon Source (APS) at 100 K with a wavelength of 1.000 Å. The data were processed using HKL2000 (HKL Research Inc.) (41). Phases were determined by molecular replacement using the program Phaser and search models of the PPR domain of Arabidopsis PRORP1 (PDB ID: 4G24) and yeast tRNAPhe (PDB ID: 1EHZ). Model building was carried out with the program Coot (42). The programs Refmac5 (43) and Phenix.refine (44) were used for refinement. The structures displayed good geometry when analyzed by MolProbity (45). Approximately 98% and 2% of the residues constituting the PPR domain were in the most favored and allowed regions of the Ramachandran plot, respectively. Modified bases were modeled into the structure: 2-methyl-guanosine at position 10, 5,6-dihydrouridine (D) at positions 16 and 17, N2-dimethylguanosine at position 26, O2′-methyl-cytidine at position 32, O2′-methyl-guanosine at position 34, the Y base or wybutosine at position 37, pseudouridine (ψ) at positions 39 and 55, 5-methyl-cytidine at positions 40 and 49, 7-methyl-guanosine at position 46, 5-methyl-uridine at position 54 and 1-methyl-adenosine at position 58. The average B factor for the tRNA is high due to poor electron density in regions of the tRNA that do not contact the PRORP1 protein. However, the electron density is strong at the tRNA D and TψC loops where it contacts PRORP1 (Supplementary Figure S3A).
In vitro transcription
Pre-tRNAs were synthesized as previously described (36,46) through run-off transcription from a restriction-digested (BstNI) pUC18 plasmid encoding Bacillus subtilis pre-tRNAAsp. In vitro transcription reactions were run in 5:1 excess of 5′-O-monophosphorothiate guanosine (GMPS) to GTP. The 5′-GMPS pre-tRNA product was reacted with 5-iodoaceamidofluorescein (5-IAF) in a 1:40 molar ratio (RNA:5-IAF) to produce a 5′-fluorescein labeled product. Labeling reactions were carried out in 10 mM Tris, pH 7.2 with 1 mM EDTA for 16 h at 37°C yielding 25–30% fluorescently-labeled pre-tRNA. The labeled pre-tRNA was gel purified using 12% urea-PAGE and eluted using the crush-soak method (47). Purified products were concentrated using 10 kDa MWCO Amicon® Ultra Centrifugal Filters, and ethanol precipitation. Pre-tRNA stocks were resuspended in 10 mM Tris, pH 8.0 with 1 mM EDTA, quantified by absorbance, and stored at −80°C.
The extinction coefficient for the B. subtilis pre-tRNAAsp at 260 nM was experimentally determined to be 674 390 M−1 cm−1 through alkaline hydrolysis. Concentrations of fluorescein were measured at 492 nm (extinction coefficient = 78 000 M−1 cm−1). Prior to all assays, substrates were thawed, diluted with nuclease-free water, and heated at 95°C for 90 s followed by refolding by incubating at 25°C for ≥15 min, and then incubating with metal-containing buffer for ≥15 min.
Fluorescence anisotropy binding assays
Binding assays were performed in Corning black polystyrene half-area 96-well plates (Product number 3686), as previously described (22,46). In short, PRORP1 variants were serially diluted from 20 μM to 9 nM and equal volumes of enzyme and 20 nM 5′-fluorescein-pre-tRNA substrate were mixed; a minimum of 12 concentrations was analyzed. Enzyme-substrate mixtures were incubated at 25 ± 1°C in 30 mM MOPS, pH 7.8, 330 mM NaCl, 1 mM TCEP and 20 mM CaCl2. Anisotropy readings of the 5′-fluorescein-pre-tRNA tag were measured with a Tecan Ultra plate reader using an excitation wavelength of 485 nm, and emission wavelength of 535 nm. The anisotropy measurements at each enzyme concentration were observed 5 times over 15–20 min to ensure complete equilibration. The concentration dependence of the anisotropy changes was well described by a single binding isotherm (Equation 1) (where FA is the fluorescence anisotropy, FA0 is the initial anisotropy, ΔFA is the total change in anisotropy, P is the concentration of PRORP and KD is the dissociation constant). The KD values and standard error for KD values were calculated by fitting Equation (1) to the data points from a single experimental trial using GraphPad Prism to carry out non-linear regression analysis.
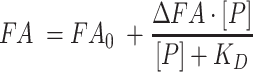 |
(1) |
Single-turnover kinetic assays
Single-turnover assay reactions were initiated through addition of 5–45 μM enzyme to an equal volume of 30 nM 5′-fluorescein-pre-tRNA substrate. Reactions were carried out at 25 ± 1°C in 30 mM MOPS, pH 7.8, 330 mM NaCl, 1 mM TCEP and 20 mM MgCl2. At specific time points (0–4800 s), 4 μl aliquots of the reaction were quenched with an equal volume of 100 mM EDTA, 8 M urea, 0.05% bromophenol blue, 0.05% xylene cyanol and 2 μg/μl yeast tRNA. Fluorescently labeled pre-tRNA substrate and 5′ leader product were separated by electrophoresis on 22.5% denaturing urea–PAGE gel. Gels were visualized using an Amersham Typhoon Biomolecular Imager, and the fraction of product was quantified using ImageJ software. A minimum of 10 time points was analyzed for each mutant. Observed single-turnover rate constants and standard errors were obtained by fitting a single exponential to the data points from a single experimental trial using GraphPad Prism 8 (Equation 2).
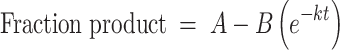 |
(2) |
Electrophoretic mobility shift assays
tRNAs were radiolabeled at the 5′ end using [γ-32P] ATP and T4 polynucleotide kinase, then purified using an Illustra MicroSpin G-25 column (GE Healthcare). RNA-binding reactions included 0.9 nM radiolabeled RNA and protein serially diluted (2-fold) from 25 μM to 3 nM. Binding reactions were incubated for 1 h at 20°C in 20 mM HEPES, pH 7.9, 100 mM NaCl, 1 mM TCEP, 10 mM CaCl2, 0.02% (v/v) Tween 20, 0.1 mg/ml poly r(U), and 2.5% (v/v) glycerol and separated by electrophoresis on 10% TBE polyacrylamide gels (Invitrogen). Gels were dried and exposed to storage phosphor screens for 6–20 h, scanned with a Typhoon 8600 Imager, and the band intensities were quantified with ImageQuant 5.2. The data for three technical replicates were analyzed and KD values were calculated via non-linear regression analysis for one-site binding with GraphPad Prism 7.
RESULTS
Engineering a PRORP1 PPR domain for crystallization
To understand substrate recognition by PRORP enzymes, we sought to determine a crystal structure of a PRORP in complex with tRNA. Through protein engineering, we obtained crystals suitable for structure determination of the PPR domain of A. thaliana PRORP1 in complex with tRNAPhe. Our attempts to crystallize a PRORP containing both PPR and catalytic domains in complex with tRNA were unsuccessful. We therefore focused on the PRORP1 PPR domain as the module that drives tRNA recognition (24,34–36). We engineered three regions of the PPR domain to promote crystallization. First, we noted that in the crystal structure of Arabidopsis PRORP1 (residues 76–572, PDB ID: 4G24) helices α10 and α11 of the PPR domain form a hydrophobic interface with the central zinc-finger domain (22). Three aromatic residues within the PPR domain (Tyr266, Phe284 and Phe291) are located at the interface. To increase the solubility of the PPR domain, we substituted these aromatic residues with hydrophilic residues (Y266N, F284Q and F291Q). Second, previous studies demonstrated that the N-terminal flexible region is involved in tRNA binding and lysine residues in this region might contact tRNA (34,40), although residues 76–94 were disordered in the crystal structure of PRORP1. We defined the minimal N-terminal region required for tRNA binding by measuring binding of N-terminal deletions of the PPR domain (named N76, N83 or N86 to indicate the N-terminal residue with all fragments extending to the C-terminal residue 294) to yeast tRNAPhe by electrophoretic mobility shift assay (EMSA). The affinity of the N86 variant for tRNA was considerably weaker than that of the N76 and N83 variants (Table 1), indicating that the N83 variant is optimal for tRNA binding affinity. Third, a long loop (LAEAATESSP) between the two α-helices of the PPR2 motif is shorter in the other Arabidopsis isoforms, PRORP2 and PRORP3. Substituting this loop with a shorter loop (LASASS) had little effect on tRNA binding affinity (Table 1, PRORP1 PPR N83 ΔPPR2 loop). The engineered PRORP1 PPR domain (84–294) with three solubilizing substitutions (Y266N, F284Q and F291Q) and a shorter loop in the PPR2 motif produced well behaved protein that retained tRNA recognition and was used successfully for crystallization in complex with yeast tRNAPhe. For simplicity, we will refer to this as the PRORP1 PPR domain.
Table 1.
Binding affinities of PRORP1 PPR domain variants
| PRORP1 PPR domain variant | tRNA | K D (nM)a |
|---|---|---|
| PRORP1 PPR N76 | Yeast tRNAPhe | 627 ± 55 |
| PRORP1 PPR N83 | Yeast tRNAPhe | 281 ± 44 |
| PRORP1 PPR N83 ΔPPR2 loop | Yeast tRNAPhe | 317 ± 92 |
| PRORP1 PPR N86 | Yeast tRNAPhe | 3405 ± 338 |
| PRORP1 PPR N83 | Arabidopsis mito pre-tRNACys | 257 ± 54 |
aMeasured using electrophoretic mobility shift assays in 20 mM HEPES, pH 7.9, 100 mM NaCl, 1 mM TCEP, 10 mM CaCl2, 0.02% (v/v) Tween 20, 0.1 mg/ml poly r(U) and 2.5% (v/v) glycerol at room temperature. The mean and standard error of the mean values for three technical replicates are reported. Representative data are shown in Supplementary Figure S9.
Structure description
We determined a crystal structure of the Arabidopsis PRORP1 PPR domain in complex with yeast tRNAPhe at 2.85 Å resolution by molecular replacement (Supplementary Table S1). Two independent crystallographic complexes were present in an asymmetric unit. The N-terminal 11 residues of the PPR domain (SRKAKKKAIQQ) are disordered in the crystal structure in complex with tRNAPhe, despite their importance for tRNA binding (Table 1). The basic residues of the N-terminal flexible region could interact non-specifically with negatively-charged phosphate groups of the tRNA backbone to enhance substrate binding affinity. The two complexes include chain A (PPR domain)-chain B (tRNAPhe) and chain C (PPR domain)-chain D (tRNAPhe) and are highly similar overall (root mean square deviation [rmsd] value of 1.00 Å over 192 CA atoms in the protein, 1.36 Å over 1569 atoms in the tRNA, and 1.94 Å over 1352 main chain atoms in the complex). However, each PPR domain in the asymmetric unit binds its corresponding tRNAPhe in a slightly different manner. The chain D tRNA molecule appears to be influenced by crystal packing forces, resulting in the chain C-chain D complex lacking some interactions. Hence, we focus on the chain A-chain B complex to describe the PPR domain–tRNA interaction.
The crystal structure of the PRORP1 PPR domain-tRNA complex revealed that the PPR domain undergoes conformational changes that place PPR motifs 1–4 in position to interact with the tRNA. The PRORP1 PPR domain comprises five consecutive PPR repeats and one additional C-terminal helix (Figure 1A). The PPR5 motif does not interact with the tRNA. Instead it may aid in positioning the PPR1–4 motifs for tRNA elbow recognition relative to the nuclease active site. As noted in the crystal structure of full-length PRORP1, the central linker domain interacts with the PPR5 motif and the terminal α-helix of the PPR domain (22). PPR5 together with the central linker domain bridges the tRNA elbow recognition and catalytic domains. It also serves as a C-terminal cap to the PPR domain, which stabilizes the terminal α-helices in the PPR4 motif (48). When compared with the PPR domain in the structure of full-length apo PRORP1 (PDB ID: 4G24), the first three PPR repeats (PPR1, PPR2 and PPR3) are different in their configurational details. The tRNAPhe-bound PPR domain exhibits a more curved conformation (Supplementary Figure S3B). With the PPR4 and PPR5 motifs aligned, the PPR3 motif in the complex is shifted away from the tRNA, whereas PPR1 is closer to the tRNA molecule. These changes allow PPRs 1–4 to interact with the tRNA, inducing a more extensive interaction surface than had been predicted. Conformational flexibility is a common feature of PPR proteins. Previous studies show that PPR proteins utilize considerable structural adaptability to bind to single-stranded RNA (32,49). In contrast, the overall structure of yeast tRNAPhe is unaltered by the binding of the PPR domain. The structure of yeast tRNAPhe is highly similar to the previously determined structure of the tRNA alone (PDB ID: 1EHZ, rmsd value of 1.56 Å over 1568 atoms).
Figure 1.
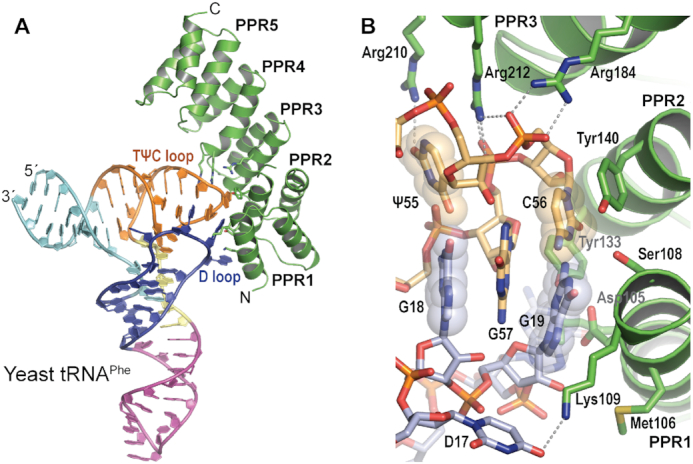
The PRORP1 PPR domain recognizes the tRNA D and TψC loops. (A) Ribbon diagram of the crystal structure of the PRORP1 PPR-tRNAPhe complex. PRORP1 PPR is shown in green with tRNA-interacting residues displayed as stick models. The tRNA is shown as a cartoon colored by region: acceptor stem (cyan), D stem loop (blue), anticodon stem loop (magenta), variable region (yellow), and TψC stem loop (orange). (B) Close-up view of the G19–C56 base pair accommodation pocket of the PRORP1 PPR domain. PRORP1 and tRNA are colored as in (A) with atom colors: oxygen (red), nitrogen (blue), phosphorus (orange) and sulfur (yellow). Dashed lines indicate interactions between PRORP1 and tRNA, and the G19–C56 and G18–ψ55 base pairs are indicated by transparent spheres.
The PRORP1 PPR domain nestles the tRNA elbow in a pocket formed by PPRs 1–4
The Arabidopsis PRORP1 PPR domain recognizes conserved features of the ‘elbow’ of the L-shaped tRNA, formed by the D and TψC loops (Figure 1). A ubiquitous G19–C56 tertiary interaction between the D and TψC loops is located at the tip of the tRNA elbow, and the PPR domain forms a pocket that accommodates the G19–C56 base pair (Figure 1B). Residues that contact the TψC loop are more conserved than those that contact the D loop (Supplementary Figure S4). The G19 base is surrounded by residues from the PPR1 motif (Asp105, Met106, Ser108 and Lys109) and Tyr133 from the PPR2 motif (Figures 1B and 2). The OH-group of Tyr133 is hydrogen bonded to the N2 atom of the G19 base, potentially a base-specific interaction (Figure 2). The C56 base forms a stacking interaction with the phenol ring of Tyr140 in the PPR2 motif (Figures 1B and 2). Together these interactions appear to recognize the structure as well as the sequence of the conserved base pair. In addition to recognizing the G19-C56 base pair, PRORP1 interacts with the base pair between G18 (D loop) and the pseudouridine, ψ55 (TψC loop). The tRNAPhe in our structure was obtained from yeast, so it has 14 post-transcriptional modification sites (50). The guanidinium group of Arg210 in the PPR3 motif is hydrogen bonded to the O2 atom of the ψ55 base (Figures 1B and 2). The intercalation of G57 between these two base pairs forms the tRNA elbow's structural core whose sequence and structure are probed by PRORP1.
Figure 2.

PRORP1 PPR–tRNA interactions. Schematic representation of interactions between the PRORP1 PPR domain and yeast tRNAPhe. PRORP1 PPR residues are green, tRNA TψC-loop nucleotides are orange, and tRNA D-loop nucleotides are light blue. Circles represent tRNA phosphate groups (P). Dotted and double lines indicate hydrophilic and stacking interactions, respectively.
The PRORP1 PPR domain forms a variety of additional tRNA interactions using basic side chains (Figure 2). Two unpaired nucleotides in the D loop are recognized by lysine side chains. Lys101 in the PPR1 motif stacks with the G20 base (Figure 2). The ϵ-amino group of Lys109 in the PPR1 motif makes a hydrogen bond with the O4 atom of the 5,6-dihydrouridine (D) base, D17 (Figures 1B and 2). This appears to be a base-specific contact recognizing a modified nucleotide but it is also capable of recognizing the O4 atom of an unmodified uracil. The phosphate backbone of the TψC loop is contacted by two arginine residues. The guanidino groups of Arg184 in the PPR3 motif and Arg212 in the PPR4 motif make salt–bridge interactions with the phosphate moieties of C56 and G57, respectively (Figures 1B and 2). These interactions are consistent with the salt dependence of PRORP-pre-tRNA affinity, which suggested up to three direct contacts with substrate backbone phosphodiester bonds by the PPR domain (36).
As described above, we observed PPR domain base-specific interactions with two modified bases in tRNA, D17 and ψ55. To gain insight into the importance of base modifications for tRNA affinity, we used EMSAs to compare the KD values for binding of the isolated PPR domain to in vitro transcribed Arabidopsis mitochondrial pre-tRNACys lacking modified nucleotides to that of modified tRNAPhe. These KD values of 281 ± 44 nM for yeast tRNAPhe and 257 ± 54 nM for Arabidopsis mitochondrial pre-tRNACys are comparable (Table 1), suggesting that the post-transcriptional modifications are dispensable for tRNA recognition by the Arabidopsis PRORP1 PPR domain. The contacts between Lys109–D17 and Arg210–ψ55 that we observed in the crystal structure could be substituted by interaction of the side chains with the O4 atom of an unmodified uracil.
Our crystal structure illustrating that the PRORP1 PPR domain specifically binds the tRNA elbow fully explains previous mutagenesis experiments that identified amino acids residues in the PRORP1 PPR domain that are critical for tRNA binding (36,40). Klemm et al. generated a number of PRORP1 mutations and analyzed the binding affinity of full-length PRORP1 to B. subtilis pre-tRNAAsp (36). This analysis identified Tyr133, Tyr140, Arg184 and Arg212 as essential residues for tRNA binding (Supplementary Table S2). Mutation of each of these residues to alanine substantially reduced binding affinity. Our crystal structure is in agreement with these results, as each of these amino acid residues is located at the interface between the PPR domain and tRNAPhe (Figures 1B and 2). Furthermore, it appears that the interaction of Arg212 requires specific contacts that cannot be satisfied by a lysine at this position; we found that the R212K mutation in PRORP1 also dramatically reduced binding affinity of pre-tRNAAsp, as measured by fluorescence anisotropy (Table 2). The purified R212K mutant protein expressed similarly to WT protein and maintains protein structure as determined by CD analysis. Chen et al. combined chemical modification of lysines with multiple-reaction monitoring mass spectrometry and identified Lys101 and Lys109 as putative tRNA-contacting residues (40). They further showed mutation of these lysine residues to alanine had a small to moderate effect on pre-tRNA binding by full-length PRORP1 (Supplementary Table S2). Our crystal structure also agrees with this finding, as these two residues recognize D-loop nucleotides through stacking and hydrogen bond interactions, respectively (Figure 2). The consistency between our crystal structure of the PRORP1 PPR domain bound to a tRNA and activity assays with mutant full-length proteins indicates that the structure accurately depicts interactions that are critical for enzyme-substrate recognition.
Table 2.
Binding affinities and kinetic constants of full-length PRORP1 to 5′-fluorescein-pre- tRNAAsp
| PRORP1 Variant | k obs (min−1)a | k obs relative to WT | K D (nM)b | K D relative to WT | ΔΔG°binding (kcal/mol) |
|---|---|---|---|---|---|
| Δ76 WT | 2.62 ± 0.28 | 1 | 390 ± 30 | 1 | 0 |
| Y133D | 1.17 ± 0.22 | 0.45 | 11100 ± 1600 | 28.4 | 1.9 |
| Y133Q | 2.76 ± 0.34 | 1.05 | 3200 ± 600 | 8.2 | 1.2 |
| R210A | 1.59 ± 0.20 | 0.61 | 4200 ± 400 | 10.8 | 1.4 |
| R210S | 0.49 ± 0.09 | 0.19 | 10600 ± 1000 | 27.2 | 1.9 |
| R212K | n.d.c | n.d. | ≥ 50000d | ≥ 130 | ≥ 2.9 |
aMeasured using single turnover cleavage assays in 30 mM MOPS, pH 7.8, 330 mM NaCl, 1 mM TCEP and 20 mM CaCl2 at 25 ± 1°C. The kobs and standard error values are determined from nonlinear least squares regression of a single exponential equation to the data points from a single experimental trial using GraphPad Prism.
bMeasured using fluorescence anisotropy in 30 mM MOPS, pH 7.8, 330 mM NaCl, 1 mM TCEP and 20 mM CaCl2 at 25 ± 1°C. The KD and standard error values are determined from non-linear least squares regression of a single binding isotherm to the data points from a single experimental trial using GraphPad Prism.
cBased on the binding affinity data, we were unable to measure single turnover kinetics for the R212K mutant under saturating enzyme concentration.
dThe lower limit for the KD for R212K was estimated from the observation of <10% change in anisotropy at 20 μM R212K mutant, assuming that the total change in anisotropy upon binding Fl-pre-tRNA was similar to the that of the other PRORP1 variants.
PRORP1 PPR motifs have evolved to recognize conserved tRNA bases and structural features
Our crystal structure confirms that the PRORP1 PPR motifs recognize tRNA using different mechanisms than observed for single-stranded RNA recognition. PPR motifs are often used for single-stranded RNA binding and recognize specific RNA bases modularly, using amino acid side chains at positions 2, 5 and 35 (alternatively 1, 4 and ii) of the PPR motif (29,30). Based on the RNA recognition code of PPR motifs, it was predicted that Asn136 and Asn175 of the PPR2 motif of PRORP1 would recognize a pyrimidine, and Thr180 and Arg210 of PPR3 would recognize a purine (34,35). Contrary to this prediction, our crystal structure reveals that Asn136, Asn175 and Thr180 do not contact the tRNA (Figure 3A, B). Residues at position 35 of PRORP1 PPR3 (Arg210) and position 2 of PPR4 (Arg212) do interact with the ψ55 base and RNA backbone, but in a completely different manner than RNA base recognition by other PPR proteins (Figure 3A, B). Arg210 at position 35 of PPR3 recognizes the ψ55 base and Arg212 at position 2 of PPR4 interacts with backbone phosphate groups (Figure 3A). In contrast, residues at positions 2, 5 and 35 of repeat 2 of PPR10 recognize a uracil base using stacking and hydrogen bond interactions (Figure 3B). Y133 at position 2 of PRORP1 PPR2 also interacts with the tRNA, but it is turned away from the prospective PPR base recognition pocket to hydrogen bond with G19 (Figure 3C).
Figure 3.

PRORP1 PPR motifs use distinct mechanisms for tRNA recognition. (A) Recognition of ψ55 by PRORP1 repeat 3. (B) Recognition of U2 by PPR10 repeat 2 (PDB ID: 4M59). (C) Recognition of G19 by PRORP1 repeat 2 (PDB ID: 6LVR). (D) Differences in specific RNA recognition by PRORP1 and PPR10. RNA-interacting residues in PPR motifs 1–4 of PRORP1 and PPR10 are shown. Nucleotides recognized are shown below and connected by a yellow line. Individual recognition modules are boxed with PRORP1 colored green and PPR10 colored blue.
We identified several PRORP1-tRNA contacts that suggest base-specific recognition and found that Tyr133 and Arg210 are critically important for PRORP1 binding affinity and catalytic activity. No tRNA sequence substitution has yet been shown to affect PRORP recognition, but prior experiments to look for base recognition were based on the single-stranded RNA PPR recognition code and the corresponding premise that PRORP1 PPR2 interacts with a pyrimidine in the tRNA. In our crystal structure, Tyr133 interacts with G19 and together with Tyr140 appears to recognize the G19–C56 base pair (Figure 2). Binding by Tyr133 and Tyr140 assures selection of this conserved feature. Mutation of Tyr133 to Phe strongly affects tRNA recognition (Supplementary Table S2) (36), which suggests the importance of the interaction between the Tyr133 OH and G19 N2 groups. Many of the tRNA-contacting residues identified by our crystal structure are conserved in the PRORP2 and PRORP3 isoforms but some, including Tyr133, are different. Tyr133 is Gln in PRORP2 and PRORP3 and in the more distantly related human mitochondrial MRPP3, it is Asp (Supplementary Figure S5). To investigate the importance of these interactions we measured the binding affinity of the PRORP1 Y133Q and Y133D mutations for 5′-fluorescein-pre-tRNA substrate using fluorescence anisotropy analysis (Figure 4A, Table 2). These substitutions decreased binding affinity by 8- and 28-fold relative to wild-type protein, equivalent to a loss of 1.2 and 1.9 kcal/mol, and these losses are similar to the decreased binding affinities of the Y133A (17-fold) and Y133F (30-fold) mutants that were reported previously (36).
Figure 4.

Fluorescence anisotropy binding curves for PRORP1 variants binding to B. subtilis 5′-fluorescein-pre-tRNAAsp substrate. (A) Binding curves for PRORP1 Y133 and R212 variants. (B) Binding curves for PRORP1 R210 variants. WT binding curves are shown in black. The assays were carried out in 30 mM MOPS, pH 7.8, 330 mM NaCl, 1 mM TCEP and 20 mM CaCl2 at 25 ± 1°C. A hyperbola (Equation 1, Materials and Methods) was fit to the fraction change in anisotropy measured from a single experimental trial using GraphPad Prism to derive values for the dissociation constant (KD) (Table 2).
In addition to Tyr133, hydrogen bond interactions between Arg210 and Lys109 and the tRNA appeared to be base specific. Arg210 interacts with the O2 group of ψ55 in our crystal structure (Figures 1B and 2) and it is conserved in PRORPs 1–3 (Supplementary Figure S5). To evaluate its involvement in tRNA recognition, we generated two mutants, R210A and R210S, and measured their pre-tRNA binding affinity by fluorescence anisotropy analysis. We found that the R210A and R210S mutations increased the pre-tRNA KD values by 11- and 27-fold, respectively, compared to that of wild-type PRORP1 (Figure 4A, Table 2). These losses in binding affinity correspond to decreases of 1.4 and 1.9 kcal/mol, respectively. This result demonstrates that the base-specific interaction of Arg210 in the PRORP1 PPR3 plays a crucial role in tRNA recognition. Unfortunately, we could not directly test the effect of a base substitution in the tRNA because ψ55 also interacts with G18 and the phosphate backbone such that base substitutions would also affect the tRNA structure. Lys109 contacts the O4 atom of D17, a modified nucleotide (Figure 1B), however mutation to alanine was shown previously to have little effect on RNA-binding affinity (Supplementary Table S2) (40). Lys109 is conserved among PRORPs 1–3 (Supplementary Figure S5), but the nucleotide at position 17 in the tRNA D loop is variable (Supplementary Figure S6). The modest effect of the K109A mutation appears consistent with the need to tolerate tRNA substitutions.
To further understand the role that the newly identified tRNA-interacting residues (Tyr133, Arg210 and Arg212) play in catalysis, single-turnover kinetic rate constants (kobs) were determined under enzyme saturating conditions with limiting (30 nM) 5′-fluorescein-pre-tRNAAsp substrate (Table 2 and Figure 5). Under saturating conditions, kobs measures the reaction of the enzyme-bound substrate, removing the effects of altering the binding affinity of the substrate. Therefore, significant changes in kobs compared to wild-type PRORP1 indicate that the mutated residue plays a role in more than just the initial binding step. Of the isoforms measured, Y133Q maintained wild-type level kobs (Table 2), indicating that the Y133Q mutation interfered with equilibrium association but not catalytic competence. R212K displayed appreciable loss in binding affinity (KD ≥ 50 μM), suggesting that this residue is important for tRNA recognition. However, because we were unable to achieve enzyme saturating conditions for R212K, we cannot accurately determine the single-turnover activity for this mutant enzyme to compare with wild-type (Figure 5). In contrast, Y133D and R210A exhibited ∼2-fold decreases in single-turnover activity while R210S displayed a ∼5-fold decrease in single-turnover activity (Table 2 and Figure 5). These decreases indicate that cleavage of substrate in the enzyme-substrate complex was compromised. We suggest that the specific interactions between the pre-tRNA ‘elbow’ and the PRORP PPR domain mediated by Tyr133 and Arg210 play a role in optimally positioning the 5′ leader cleavage site at the metallonuclease domain active site. Mutation of these residues interferes with the formation of a catalytically competent PRORP-pre-tRNA complex as well as decreasing substrate binding affinity. Similarly, the absence of the substrate D-loop, which is recognized by Tyr133, decreases the single-turnover activity and affinity of PRORP3 (37).
Figure 5.
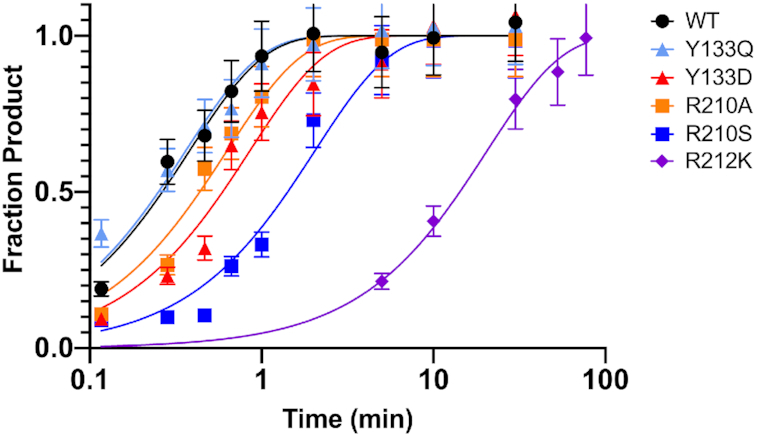
Time courses for the single-turnover cleavage of 5′-fluorescein pre-tRNA substrate catalyzed by PRORP1 variants, measured in 30 mM MOPS, pH 7.8, 330 mM NaCl, 1 mM TCEP and 20 mM MgCl2 at 25 ± 1°C. The enzyme concentration was 30 μM. A single exponential (Equation 2, Materials and Methods) was fit to the time dependence of the fraction of product formation measured from a single experimental trial using GraphPad Prism to determine a single-turnover rate constant (kobs) (Table 2). The R212K mutant was evaluated under sub-saturating enzyme concentration. Representative data are shown in Supplementary Figure S10.
DISCUSSION
Our crystal structure of the PPR domain of PRORP1 in complex with tRNA illustrates its substrate recognition strategy emphasizing interactions with conserved nucleotide and structural elements of the tRNA elbow region and employing new mechanisms of PPR motif–RNA interaction. This specific recognition of the pre-tRNA elbow is essential for catalytic activity. By bringing together tRNA nucleotide and structural recognition, it requires the expansion of RNA recognition modes by PPR motifs beyond the established RNA base recognition mechanism.
The new PPR motif recognition modes in PRORP1 confirm one of the distinct proposals from the models of PPR motif-tRNA interaction proposed previously (24,36,37): that the PPR domain in PRORP does not use the canonical base-selection model, but rather utilizes a strategy like recognition of the G19–C56 base pair by the RNP RNase P ribozyme (Figure 6) (9). Evolution of the RNA recognition modes of PPR motifs to allow recognition of structured RNA in addition to single-stranded RNA sequences is reminiscent of the PUF (Pumilio/fem-3 binding factor) family of proteins. Both types of α-helical repeat proteins are well known for their modular recognition of RNA sequences (51). In addition, these α-helical repeat scaffolds can be modified for structured RNA recognition (52,53). Many other families of α-helical repeat proteins are known for their roles in RNA metabolism (54), presenting the possibility that they, too, serve as versatile scaffolds for RNA recognition.
Figure 6.
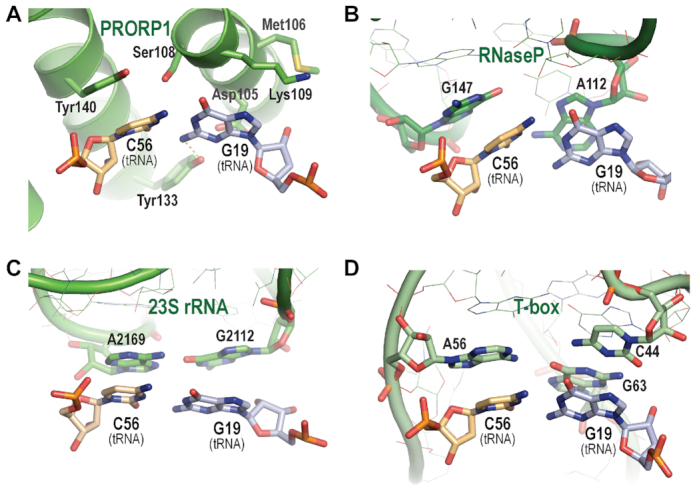
Evolutionary convergence of G19–C56 tRNA base pair recognition. (A) PRORP1 PPR domain recognition of the G19–C56 base pair. (B) Bacterial RNP RNase P RNA recognition of the G19–C56 base pair (PDB ID: 3Q1Q). (C) 23S rRNA recognition of the G19–C56 base pair (PDB ID: 4V4I). (D) T-box riboswitch recognition of the G19–C56 base pair (PDB ID: 4LCK). PRORP1 PPR domain, RNase P RNA, 23S rRNA and T-box riboswitch in green are shown as cartoons with interacting residues displayed as stick models. Amino acid side chains and tRNA D-loop (light blue) and TψC-loop (orange) nucleotides are shown with atom colors: oxygen (red), nitrogen (blue), phosphorus (orange) and sulfur (yellow). Non-tRNA interacting nucleotides of bacterial RNase P, 23S rRNA and T-box riboswitch RNAs are shown as thin stick models.
The central features of recognition of the tRNA elbow are a stacking interaction between PRORP1 Tyr140 and C56 in the G19–C56 base pair and electrostatic interactions of Arg184 and Arg212 with phosphate groups important for the positioning of G57. These highly conserved tRNA structural elements are recognized by residues that are retained in Arabidopsis PRORPs and the human MRPP3 (Supplementary Figure S5). Mutation of these residues has severe effects on pre-tRNA binding affinity (Supplementary Table S2). Mutation of Tyr140 to alanine in full-length PRORP1 severely diminished pre-tRNA binding affinity (∼200-fold weaker) and mutation to phenylalanine decreased pre-tRNA binding affinity ∼6-fold (36). Similarly, mutation of Arg184 and Arg212 to alanine dramatically reduced pre-tRNA binding affinity. In addition, base-specific interactions of PRORP1 Tyr133 with the N2 atom of G19 and Arg210 with the O4 atom of ψ55 appear to be important for pre-tRNA recognition. Mutation of Tyr133 or Arg210 decreases pre-tRNA binding affinity, indicating their important contribution to recognition. Despite their importance in PRORP1, Tyr133 is not conserved in Arabidopsis PRORP2 and PRORP3, and neither Tyr133 nor Arg210 is conserved in human MRPP3 (Supplementary Figure S5). PRORP2 and PRORP3, which process pre-tRNAs retaining the G19-C56 base pair between the D and TψC loops, may have evolved alternative mechanisms for base-specific binding consistent with their set of pre-tRNA substrates. For MRPP3, mammalian mitochondrial tRNAs retain the overall L-shape, but their sequences are degenerate, especially in the D and TψC loops. Consequently, MRPP3 may recognize only the structure of pre-tRNA substrates.
Our crystal structure indicates conformational changes in PRORP enzymes that are required for recognition and 5′ cleavage of pre-tRNA. Previous biochemical and biophysical analyses suggested that PRORPs adopt multiple conformations (35). A hinge between the NYN metallonuclease and central zinc-binding domains allows reorientation of the nuclease domain with respect to the PPR domain. Superposition of our tRNA-bound PPR domain with the PPR domains in crystal structures of Arabidopsis PRORP1 or PRORP2 shows that without a conformational change the position of the nuclease domain would overlap the acceptor-stem of the tRNA (Supplementary Figure S7). Therefore, the relative positions of the nuclease and PPR domains must be more open to recognize the pre-tRNA elbow and place the nuclease active site at the pre-tRNA 5′ end. Molecular dynamics simulations have illustrated the ranges of conformational flexibility in PRORP enyzmes (35,55). In addition to this requirement for overall conformational change, we observed flexibility within the PRORP PPR domain (Supplementary Figure S3) that may be crucial to recognize pre-tRNA and to release mature tRNA product.
PRORP enzymes must process a set of diverse pre-tRNA substrates, and our data suggest multiple mechanisms to promote recognition plasticity. In addition to tRNA sequence diversity, 5′ leader and 3′ trailer processing occur prior to structure-stabilizing base modifications (56,57). Therefore, PRORP enzymes might recognize and bind partially folded or dynamic pre-tRNA tertiary structures during the early stages of maturation, and in turn they might aid in achieving the final tRNA tertiary structure. Recognition of a highly conserved pre-tRNA structural feature like the G19–C56 base pair is an excellent strategy to capture a range of pre-tRNA conformations. Our data indicating that the PRORP1 PPR domain binds equally well to modified and unmodified tRNA also allows binding as the tRNA matures. Flexibility of the PPR domain and overall PRORP1 conformation permits optimization of binding to different pre-tRNA structures.
Recognition of the G19–C56 base pair at the tRNA elbow is a core element of PRORP PPR interactions, and it is striking that this mode of recognition by a protein is analogous to recognition of the same feature by three functional RNAs (Figure 6). The crystal structure of Thermotoga maritima RNP RNase P in complex with tRNA showed that highly conserved nucleotides A112 and G147 in single-stranded loops of the RNase P RNA form a binding pocket and interact via stacking interactions with G19 and C56 (Figure 6B) (9). This interaction is conserved in structures of RNP RNase P from bacteria, archaea, yeast, and human (Supplementary Figure S8) (8–11). Furthermore, this tRNA-binding mode is like that between the L1 stalk of the 23S rRNA and tRNA at the E-site in the 50S ribosome (38) and two single-stranded loops of T-box riboswitches and their cognate tRNAs (39) (Figure 6C, D). Since these functional RNAs share no common ancestor, it is thus postulated that the specific binding to the tRNA elbow with stacking interaction has evolved independently at least three times (39). Evolution of the recognition α-helices in the PPR1 and PPR2 motifs mimics the binding pocket for the tRNA elbow formed by the two single-stranded chains of these functional RNAs. Hence, PRORPs add a fourth molecule to this evolutionarily convergent solution for tRNA elbow recognition.
DATA AVAILABILITY
Atomic coordinates and structure factors for the PRORP1 PPR domain-yeast tRNAPhe complex have been deposited with the Protein Data Bank under accession number 6LVR.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to our colleagues B. Klemm and L. Pedersen for critical reading of our manuscript. We thank John Gonczy for assistance with data collection at SER-CAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory and Lars Pedersen and Juno Krahn for crystallographic and data collection support at NIEHS. We also thank Takuo Minato and Ki-Soek Yoon for assistance with CD spectra measurement at Kyushu University.
Contributor Information
Takamasa Teramoto, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA; Department of Bioscience and Biotechnology, Faculty of Agriculture, Kyushu University, Fukuoka 819-0395, Japan.
Kipchumba J Kaitany, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Yoshimitsu Kakuta, Department of Bioscience and Biotechnology, Faculty of Agriculture, Kyushu University, Fukuoka 819-0395, Japan.
Makoto Kimura, Department of Bioscience and Biotechnology, Faculty of Agriculture, Kyushu University, Fukuoka 819-0395, Japan.
Carol A Fierke, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Departments of Chemistry and Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA.
Traci M Tanaka Hall, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institutes of Health; National Institute of Environmental Health Sciences [1ZIA50165 to T.M.T.H.] (in part); National Institutes of Health [GM55387 to C.A.F.]; Robert A. Welch Foundation [1904939 to C.A.F.]; the Advanced Photon Source used for this study was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences [W-31-109-Eng-38]. Funding for open access charge: National Institutes of Health, National Institute of Environmental Health Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1. Abbott J.A., Francklyn C.S., Robey-Bond S.M.. Transfer RNA and human disease. Front Genet. 2014; 5:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Esakova O., Krasilnikov A.S.. Of proteins and RNA: the RNase P/MRP family. RNA. 2010; 16:1725–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schelcher C., Sauter C., Giege P.. Mechanistic and structural studies of protein-only RNase P compared to ribonucleoproteins reveal the two faces of the same enzymatic activity. Biomolecules. 2016; 6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daniels C.J., Lai L.B., Chen T.H., Gopalan V.. Both kinds of RNase P in all domains of life: surprises galore. RNA. 2019; 25:286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nickel A.I., Waber N.B., Gossringer M., Lechner M., Linne U., Toth U., Rossmanith W., Hartmann R.K.. Minimal and RNA-free RNase P in Aquifex aeolicus. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guerrier-Takada C., Gardiner K., Marsh T., Pace N., Altman S.. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983; 35:849–857. [DOI] [PubMed] [Google Scholar]
- 7. Kazantsev A.V., Pace N.R.. Bacterial RNase P: a new view of an ancient enzyme. Nat. Rev. Microbiol. 2006; 4:729–740. [DOI] [PubMed] [Google Scholar]
- 8. Lan P., Tan M., Zhang Y., Niu S., Chen J., Shi S., Qiu S., Wang X., Peng X., Cai G. et al.. Structural insight into precursor tRNA processing by yeast ribonuclease P. Science. 2018; 362:eaat6678. [DOI] [PubMed] [Google Scholar]
- 9. Reiter N.J., Osterman A., Torres-Larios A., Swinger K.K., Pan T., Mondragon A.. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature. 2010; 468:784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wan F., Wang Q., Tan J., Tan M., Chen J., Shi S., Lan P., Wu J., Lei M.. Cryo-electron microscopy structure of an archaeal ribonuclease P holoenzyme. Nat. Commun. 2019; 10:2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu J., Niu S., Tan M., Huang C., Li M., Song Y., Wang Q., Chen J., Shi S., Lan P. et al.. Cryo-EM structure of the human ribonuclease P holoenzyme. Cell. 2018; 175:1393–1404. [DOI] [PubMed] [Google Scholar]
- 12. Holzmann J., Frank P., Loffler E., Bennett K.L., Gerner C., Rossmanith W.. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008; 135:462–474. [DOI] [PubMed] [Google Scholar]
- 13. Vilardo E., Nachbagauer C., Buzet A., Taschner A., Holzmann J., Rossmanith W.. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012; 40:11583–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gobert A., Gutmann B., Taschner A., Gossringer M., Holzmann J., Hartmann R.K., Rossmanith W., Giege P.. A single Arabidopsis organellar protein has RNase P activity. Nat. Struct. Mol. Biol. 2010; 17:740–744. [DOI] [PubMed] [Google Scholar]
- 15. Gutmann B., Gobert A., Giege P.. PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev. 2012; 26:1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai L.B., Bernal-Bayard P., Mohannath G., Lai S.M., Gopalan V., Vioque A.. A functional RNase P protein subunit of bacterial origin in some eukaryotes. Mol. Genet. Genomics. 2011; 286:359–369. [DOI] [PubMed] [Google Scholar]
- 17. Taschner A., Weber C., Buzet A., Hartmann R.K., Hartig A., Rossmanith W.. Nuclear RNase P of Trypanosoma brucei: a single protein in place of the multicomponent RNA-protein complex. Cell Rep. 2012; 2:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sugita C., Komura Y., Tanaka K., Kometani K., Satoh H., Sugita M.. Molecular characterization of three PRORP proteins in the moss Physcomitrella patens: nuclear PRORP protein is not essential for moss viability. PLoS One. 2014; 9:e108962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lechner M., Rossmanith W., Hartmann R.K., Tholken C., Gutmann B., Giege P., Gobert A.. Distribution of ribonucleoprotein and protein-only RNase P in Eukarya. Mol. Biol. Evol. 2015; 32:3186–3193. [DOI] [PubMed] [Google Scholar]
- 20. Bouchoucha A., Waltz F., Bonnard G., Arrive M., Hammann P., Kuhn L., Schelcher C., Zuber H., Gobert A., Giege P.. Determination of protein-only RNase P interactome in Arabidopsis mitochondria and chloroplasts identifies a complex between PRORP1 and another NYN domain nuclease. Plant J. 2019; 100:549–561. [DOI] [PubMed] [Google Scholar]
- 21. Weber C., Hartig A., Hartmann R.K., Rossmanith W.. Playing RNase P evolution: swapping the RNA catalyst for a protein reveals functional uniformity of highly divergent enzyme forms. PLos Genet. 2014; 10:e1004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Howard M.J., Lim W.H., Fierke C.A., Koutmos M.. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16149–16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karasik A., Shanmuganathan A., Howard M.J., Fierke C.A., Koutmos M.. Nuclear protein-only ribonuclease P2 structure and biochemical characterization provide insight into the conserved properties of tRNA 5′ end processing enzymes. J. Mol. Biol. 2016; 428:26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gobert A., Pinker F., Fuchsbauer O., Gutmann B., Boutin R., Roblin P., Sauter C., Giege P.. Structural insights into protein-only RNase P complexed with tRNA. Nat. Commun. 2013; 4:1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujii S., Small I.. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 2011; 191:37–47. [DOI] [PubMed] [Google Scholar]
- 26. Kotera E., Tasaka M., Shikanai T.. A pentatricopeptide repeat protein is essential for RNA editing in chloroplasts. Nature. 2005; 433:326–330. [DOI] [PubMed] [Google Scholar]
- 27. Nakamura T., Yagi Y., Kobayashi K.. Mechanistic insight into pentatricopeptide repeat proteins as sequence-specific RNA-binding proteins for organellar RNAs in plants. Plant Cell Physiol. 2012; 53:1171–1179. [DOI] [PubMed] [Google Scholar]
- 28. Ringel R., Sologub M., Morozov Y.I., Litonin D., Cramer P., Temiakov D.. Structure of human mitochondrial RNA polymerase. Nature. 2011; 478:269–273. [DOI] [PubMed] [Google Scholar]
- 29. Barkan A., Rojas M., Fujii S., Yap A., Chong Y.S., Bond C.S., Small I.. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLos Genet. 2012; 8:e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yagi Y., Hayashi S., Kobayashi K., Hirayama T., Nakamura T.. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One. 2013; 8:e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yagi Y., Tachikawa M., Noguchi H., Satoh S., Obokata J., Nakamura T.. Pentatricopeptide repeat proteins involved in plant organellar RNA editing. RNA Biol. 2013; 10:1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen C., Zhang D., Guan Z., Liu Y., Yang Z., Yang Y., Wang X., Wang Q., Zhang Q., Fan S. et al.. Structural basis for specific single-stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat. Commun. 2016; 7:11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yin P., Li Q., Yan C., Liu Y., Liu J., Yu F., Wang Z., Long J., He J., Wang H.W. et al.. Structural basis for the modular recognition of single-stranded RNA by PPR proteins. Nature. 2013; 504:168–171. [DOI] [PubMed] [Google Scholar]
- 34. Imai T., Nakamura T., Maeda T., Nakayama K., Gao X., Nakashima T., Kakuta Y., Kimura M.. Pentatricopeptide repeat motifs in the processing enzyme PRORP1 in Arabidopsis thaliana play a crucial role in recognition of nucleotide bases at TpsiC loop in precursor tRNAs. Biochem. Biophys. Res. Commun. 2014; 450:1541–1546. [DOI] [PubMed] [Google Scholar]
- 35. Pinker F., Schelcher C., Fernandez-Millan P., Gobert A., Birck C., Thureau A., Roblin P., Giege P., Sauter C.. Biophysical analysis of Arabidopsis protein-only RNase P alone and in complex with tRNA provides a refined model of tRNA binding. J. Biol. Chem. 2017; 292:13904–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klemm B.P., Karasik A., Kaitany K.J., Shanmuganathan A., Henley M.J., Thelen A.Z., Dewar A.J.L., Jackson N.D., Koutmos M., Fierke C.A.. Molecular recognition of pre-tRNA by Arabidopsis protein-only ribonuclease P. RNA. 2017; 23:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brillante N., Gossringer M., Lindenhofer D., Toth U., Rossmanith W., Hartmann R.K.. Substrate recognition and cleavage-site selection by a single-subunit protein-only RNase P. Nucleic Acids Res. 2016; 44:2323–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Korostelev A., Trakhanov S., Laurberg M., Noller H.F.. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006; 126:1065–1077. [DOI] [PubMed] [Google Scholar]
- 39. Zhang J., Ferre-D’Amare A.R.. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013; 500:363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen T.H., Tanimoto A., Shkriabai N., Kvaratskhelia M., Wysocki V., Gopalan V.. Use of chemical modification and mass spectrometry to identify substrate-contacting sites in proteinaceous RNase P, a tRNA processing enzyme. Nucleic Acids Res. 2016; 44:5344–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 42. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 43. Murshudov G.N., Vagin A.A., Dodson E.J.. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr. 1997; 53:240–255. [DOI] [PubMed] [Google Scholar]
- 44. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Howard M.J., Klemm B.P., Fierke C.A.. Mechanistic studies reveal similar catalytic strategies for phosphodiester bond hydrolysis by protein-only and RNA-dependent ribonuclease P. J. Biol. Chem. 2015; 290:13454–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milligan J.F., Uhlenbeck O.C.. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989; 180:51–62. [DOI] [PubMed] [Google Scholar]
- 48. Grove T.Z., Cortajarena A.L., Regan L.. Ligand binding by repeat proteins: natural and designed. Curr. Opin. Struct. Biol. 2008; 18:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coquille S., Filipovska A., Chia T., Rajappa L., Lingford J.P., Razif M.F., Thore S., Rackham O.. An artificial PPR scaffold for programmable RNA recognition. Nat. Commun. 2014; 5:5729. [DOI] [PubMed] [Google Scholar]
- 50. Shi H., Moore P.B.. The crystal structure of yeast phenylalanine tRNA at 1.93 A resolution: a classic structure revisited. RNA. 2000; 6:1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hall T.M.T. De-coding and re-coding RNA recognition by PUF and PPR repeat proteins. Curr. Opin. Struct. Biol. 2016; 36:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qiu C., McCann K.L., Wine R.N., Baserga S.J., Hall T.M.T.. A divergent Pumilio repeat protein family for pre-rRNA processing and mRNA localization. Proc. Natl. Acad. Sci. USA. 2014; 111:18554–18559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J., McCann K.L., Qiu C., Gonzalez L.E., Baserga S.J., Hall T.M.T.. Nop9 is a PUF-like protein that prevents premature cleavage to correctly process pre-18S rRNA. Nat. Commun. 2016; 7:13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hammani K., Bonnard G., Bouchoucha A., Gobert A., Pinker F., Salinas T., Giege P.. Helical repeats modular proteins are major players for organelle gene expression. Biochimie. 2014; 100:141–150. [DOI] [PubMed] [Google Scholar]
- 55. Chen T.H., Sotomayor M., Gopalan V.. Biochemical studies provide insights into the necessity for multiple arabidopsis thaliana protein-only RNase P isoenzymes. J. Mol. Biol. 2019; 431:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Binder S., Stoll K., Stoll B.. Maturation of 5′ ends of plant mitochondrial RNAs. Physiol. Plant. 2016; 157:280–288. [DOI] [PubMed] [Google Scholar]
- 57. Rackham O., Busch J.D., Matic S., Siira S.J., Kuznetsova I., Atanassov I., Ermer J.A., Shearwood A.M., Richman T.R., Stewart J.B. et al.. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 2016; 16:1874–1890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the PRORP1 PPR domain-yeast tRNAPhe complex have been deposited with the Protein Data Bank under accession number 6LVR.