


Abstract
Histones are substrates of the SUMO (small ubiquitin-like modifier) conjugation pathway. Several reports suggest histone sumoylation affects transcription negatively, but paradoxically, our genome-wide analysis shows the modification concentrated at many active genes. We find that trans-tail regulation of histone-H2B ubiquitylation and H3K4 di-methylation potentiates subsequent histone sumoylation. Consistent with the known control of the Set3 histone deacetylase complex (HDAC) by H3K4 di-methylation, histone sumoylation directly recruits the Set3 complex to both protein-coding and noncoding RNA (ncRNA) genes via a SUMO-interacting motif in the HDAC Cpr1 subunit. The altered gene expression profile caused by reducing histone sumoylation matches well to the profile in cells lacking Set3. Histone H2B sumoylation and the Set3 HDAC coordinately suppress cryptic ncRNA transcription initiation internal to mRNA genes. Our results reveal an elaborate co-transcriptional histone crosstalk pathway involving the consecutive ubiquitylation, methylation, sumoylation and deacetylation of histones, which maintains transcriptional fidelity by suppressing spurious transcription.
INTRODUCTION
Covalent attachment of the small ubiquitin-like modifier (SUMO) protein to eukaryotic proteins alters their functional properties, most commonly by changing their binding to other proteins (1,2). The C-terminus of SUMO is first activated by the heterodimeric Aos1/Uba2 SUMO-activating enzyme (E1) and then transferred to the Ubc9 SUMO-conjugating enzyme (E2). SUMO is then ligated to one or more lysine residues of the substrate, usually with the aid of a SUMO ligase (E3) (3). The modification is highly dynamic, being readily reversed by SUMO proteases (4).
SUMO modification plays key roles in many cellular processes, including mitochondrial dynamics, ribosome biogenesis and DNA repair (2). Much attention has focused on the role of SUMO in gene expression because many SUMO substrates are involved in chromatin dynamics and transcriptional regulation in both yeast and mammals (5–8). SUMO attachment can have either negative or positive effects on transcription.
All four core histones and the H2A variant H2A.Z are sumoylated in yeast (9,10), while sumoylation of histones H3, H4, H2A.X and linker histone H1 has been reported in human cells (11–14). Several SUMO modification sites have been identified, including a site in human H4 (12,15,16) and multiple sites in yeast H2B, H4, and H2A.Z (9,10); more such sites within each histone are likely to exist. Mammalian sumoylated histone H4 recruits the histone deacetylase HDAC1 and heterochromatin protein 1 (HP1), thereby attenuating transcription (11). The LSD1-CoREST repressor complex binds in vitro to nucleosomes containing sumoylated histone H4 via a SUMO-interacting motif (SIM) in CoREST, resulting in more compacted chromatin (17,18). In another example, SUMO-conjugated yeast histones appear to suppress gene expression by opposing active histone marks such as acetylation or ubiquitylation (9). Recently, our group reported a novel regulatory mechanism in which histone H2BK123 ubiquitylation stimulates histone sumoylation, which in turn impairs nucleosomal association of the Ctk1 RNA polymerase II (RNAPII) kinase during transcription elongation (19). Reversal of histone sumoylation by the Ulp2 protease is required for Ctk1 recruitment, thereby promoting RNAPII elongation.
In contrast to histone sumoylation, histone methylation has been heavily studied. Among these modifications, it is now well known that cotranscriptional methylation of histone H3 lysine-4 (H3K4) serves fundamental roles in diverse processes, including RNAPII-mediated transcription (20). Intriguingly, H2BK123 ubiquitylation is essential for H3K4 tri-methylation (me3) and di-methylation (me2) but not its mono-methylation (me1) (21,22). This histone modification crosstalk is evolutionarily conserved and involved in promoting gene transcription and telomeric silencing (22,23). Genomic studies reveal a gradient of H3K4 methylation across most active genes: H3K4me3 near the promoter, H3K4me2 in the 5′ transcribed region, and H3K4me1 more dispersed and further downstream (24,25). The degree of methylation is determined by the time the Set1/COMPASS methyltransferase spends tethered near the nucleosome (26).
These different states of methylation function distinctly in transcription (27). Whereas H3K4me3 recruits mainly positive transcriptional regulators, H3K4me2 provides a binding site for the PHD finger in Set3 (28). Set3 is a subunit of a 7-subunit deacetylase complex, SET3C, that includes two histone deacetylases, Hos2 and Hst1. Whereas SET3C acts as a meiosis-specific repressor of sporulation genes (29), it usually stimulates transcription by deacetylating histones in the 5′ ends of genes (30,31). Such deacetylation represses ‘cryptic’ internal initiation of both sense and antisense transcripts that overlap mRNA genes (30,32).
Pervasive genome-wide transcription appears to be a general feature of eukaryotic genomes, with large numbers of uncharacterized noncoding RNA (ncRNA) genes distinct from those with established functions (33). While pervasive transcripts detected in wild-type (WT) yeast cells have been named SUTs (Stable Uncharacterized Transcripts) (34), additional classes of ncRNAs were characterized upon removal of specific regulators. For example, CUTs (Cryptic Unstable Transcripts) and XUTs (Xrn1-sensitive Unstable Transcripts) were identified in mutants of Rrp6 (nuclear RNA exosome) and Xrn1 (cytoplasmic exonuclease), respectively (34–37). However, it remains unclear whether all these ncRNAs have specific functions or are byproducts of functional or spurious transcription events.
Here, we report that trans-tail crosstalk involving both H2B ubiquitylation and H3K4 methylation is required to stimulate subsequent histone sumoylation, which is enriched at numerous active genes. Histone sumoylation requires H3K4me2, but not H3K4me3. H3K4me2-marked chromatin is known to recruit SET3C to the 5′ regions of transcribed genes (32). Amplifying this effect, H3K4me2-mediated histone sumoylation provides further binding sites for SET3C via interaction with a SIM in Cpr1, a subunit of SET3C. As a result, the extent of SET3C recruitment depends on the level of histone sumoylation. Changes in ncRNA gene expression profiles correlate strongly between cells lacking Set3 and those stably expressing an H2B-4KA mutant in which H2B-SUMO conjugation is strongly impaired. Most notably, SET3C and histone sumoylation coordinately suppress ncRNAs transcribed from cryptic internal promoters.
Taken together, these data reveal a remarkably elaborate network of interdependent histone modifications in which H2B ubiquitylation promotes H3K4 di-methylation and, in turn, histone sumoylation. The latter two modifications recruit the Set3 HDAC to deacetylate histones in the 5′ ends of mRNA genes. Contrary to the idea that histone sumoylation simply represses transcription, this modification is broadly localized to active protein-coding and ncRNA genes and promotes recruitment of SET3C to these loci, preventing aberrant transcription from internal cryptic promoters. Histone sumoylation-potentiated SET3C histone deacetylation therefore maintains transcriptional fidelity by suppressing initiation of spurious transcripts.
MATERIALS AMD METHODS
Yeast strains
Yeast strains used in this study are listed in Supplementary Table S4. Standard techniques were used for strain construction. To generate MHY10883 and MHY10884, pRS314-FLAG-htb1 K6/7/16/17A and pRS314-SUMO-HTB1 CEN/TRP1 plasmids were individually transformed into MHY4027, and then the URA3-marked YCp50-HTB1 plasmid was evicted by two consecutive streakings on SD plates containing 5-fluoroorotic acid (5-FOA, 1 g/l). To make HYS37, pRS314-FLAG-HHF1 and pRS317-HHT1 were transformed into MHY4259 (hht1‐hhf1 hht2‐hhf2 [YCp50‐copyII (HHT2‐HHF2)]), and then YCp50‐copyII (HHT2‐HHF2)] was evicted by 5-FOA selection. Diploid cells (MHY606) were transformed with YCplac33-ULP2 CEN/URA3, sporulated and dissected, and then ulp2Δ haploid cells with a cover plasmid (MHY10905) were isolated.
The deletion strains were generated by replacing each ORF with kanMX4 modules constructed by PCR amplification from the corresponding strains obtained from Euroscarf. The kanMX4 disruption cassettes for set1, set2, dot1, swd1, swd3, spp1, sdc1, bre2 and shg1 were introduced into yDS050 bearing YCplac33-ULP2. The set1, set2 and dot1 deletion strains were generated by replacing each ORF in MHY10244, MHY10250, and HYS37 with the kanMX4 markers. The kanMX4 modules for set3, hos2, hst1, cpr1, snt1, sif2 and hos4 were transformed into MHY1379, MHY4027 containing pRS314-FLAG-htb1 K6/7/16/17A and MHY10244, respectively. The C-terminal tagging cassettes amplified from the SET3-TAP and HOS2-TAP locus in the tagged yeast strain from Open Biosystems were individually introduced into appropriate strains, MHY10244, MHY10248, MHY10249, MHY10879 without YCp50-HTB1, MHY10883, MHY10884, MHY10905, and HYS37. All strains were verified by PCR and/or immunoblot analysis.
Yeast growth conditions
Cells were grown at 30°C in YPD (rich) or SD (minimal) medium with suitable supplements. For plate growth assays, liquid cultures in exponential growth were normalized to 0.1 OD600 and subjected to five-fold serial dilutions. Cells were spotted onto SD-Ura medium with or without 6-azauracil (6-AU, 100 μg/ml), and the plates were incubated at 30°C for 2–4 days. For induction of GAL1 gene, cells grown in SD-Trp medium with 2% glucose to mid-log phase were shifted to SD-Trp medium containing 2% raffinose. After 2 hours, the 2% raffinose medium was replaced with SD-Trp medium containing 2% galactose and then incubated for 1 hour. Between each shift, cells were washed twice with sterile water.
Plasmids
Plasmids used in this study are listed in Supplementary Table S5. To make pRS416-GPD-SET1 and pRS416-GPD-SET1(ΔRRM), ORFs of SET1 and set1(ΔRRM) were individually amplified by PCR from the plasmid pRS414-SET1 and pRS414-SET1(ΔRRM), cleaved with SpeI and HindIII and then cloned into pRS416-GPD. pRS425-GPD-Cpr1–13Myc was generated by sequential ligation into pRS425-GPD of the CPR1 ORF without stop codon and 13Myc epitope sequence for C-terminal fusion; these sequences were PCR-amplified from yeast genomic DNA and pFA6–13Myc–kanMX6 (38), respectively. Wild-type HHT1, including 500 bp upstream and 200 bp downstream, was PCR-amplified from yeast genomic DNA, digested with ApaI and NotI and then cloned into pRS317; pRS314-FLAG-HHF1 was generated by subcloning the ApaI and BamHI fragment from pRS424-FLAG-HHF1 into pRS314. To create pRS425-GPD-Cpr1 MUT-13Myc, pRS314-FLAG-htb1-K6/7/16/17R and pRS314-hht2 R2A-HHF2, QuikChange mutagenesis was used to mutate codons 154–156 within CPR1 from IVV to NAA in pRS425-GPD-Cpr1–13Myc; the K6, 7, 16, and 17 codons in FLAG-HTB1 (histone H2B) were similarly mutated to Arg codons in pRS314-FLAG-HTB1, and the R2 codon was changed in HHT2 (histone H3) to an Ala codon in pRS314-HHT2-HHF2. All constructs were confirmed by DNA sequencing.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed as described previously (39,40). Briefly, formaldehyde was added to mid-log phase cells to a final concentration of 1% for 20 min. Cross-linking was quenched by addition of glycine to 240 mM. The cross-linked cells were collected by centrifugation, washed in TBS twice, and then lysed with glass beads in FA lysis buffer {50 mM HEPES–KOH [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na deoxycholate, 0.1% SDS, protease inhibitor cocktail (Roche, 11697498001), 1 mM PMSF (AmericanBio, AB01620)}. Chromatin sheared by sonication was incubated with Protein G-Sepharose (GE Healthcare, 17–0618-01) bound with anti-H3K4me3 (Abcam, ab8580), anti-H3K36me3 (Abcam, ab9050), anti-H3K79me3 (Abcam, ab2621), anti-H3Ac (Millipore, 07-360), anti-H3 (Abcam, ab1791) or anti-Rpb3 (BioLegend, 665004) at 4°C. TAP-, Flag- or 13Myc-tagged proteins were precipitated with IgG-Sepharose beads (GE Healthcare, 17-0969-01), anti-Flag agarose beads (Sigma, A2220) or anti-Myc agarose beads (Covance, AFC-150P), respectively. Precipitates were sequentially washed with FA lysis buffer with 275 mM NaCl, the same buffer with 500 mM NaCl, LiCl washing buffer (10 mM Tris–HCl [pH 8.0], 0.1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate) and TE (10 mM Tris–HCl [pH 8.0], 1 mM EDTA) and then eluted with elution buffer (10 mM Tris–HCl [pH 7.5], 10 mM EDTA, 1% SDS) at 65°C. Eluted chromatin fragments were treated with pronase (Roche, 11459643001), and then DNA was purified by phenol/chloroform extraction. The oligonucleotide sequences used for ChIP qPCR are listed in Supplementary Table S6. Diluted template DNA (1:8 or 1:10 dilution for IP DNA and 1:1,000 dilution for input DNA) was used in qPCR reactions using the iQ™ SYBR Green Supermix Kit (Bio-Rad, 1708884) on a LightCycler 480 II (Roche), and signals were normalized to the internal control (a fragment amplified from an untranscribed region on ChrIV, residues 1516109–1516234; SGD) and input DNA. The values of H3 modifications were further normalized by H3 levels.
Chromatin double immunoprecipitation (ChDIP)
ChDIP assays were carried out as described previously (9,19). Preparation of chromatin fragments and elution steps were performed as described above for ChIP experiments. After overnight immunoprecipitation (IP) with anti-Flag agarose beads (Sigma, A2220) at 4°C, elution was performed with triple-Flag peptide (Sigma, F3290). While 5% of the eluate was used as INPUT, the remaining 95% was incubated with anti-HA-conjugated agarose (Thermo Scientific, 26182) overnight at 4°C and then washed with FA lysis buffer four times. N-ethylmaleimide (NEM; Sigma, E3876) was added to buffers at a final concentration of 20 mM to prevent SUMO deconjugation. The oligonucleotide sequences used in ChDIP qPCR are listed in Supplementary Table S4. DNA templates were diluted 1:10 for IP and 1:50 for INPUT, respectively, for further qPCR assays.
ChIP- and ChDIP-sequencing (seq)
The libraries were constructed by protocols of the Yale Center for Genomic Analysis (YCGA) (https://medicine.yale.edu/keck/ycga/Images/8_tcm240-21598.pdf) and sequenced using 100 bp paired-end sequencing on an Illumina HiSeq 2500 according to Illumina protocols. The 10 bp dual index was read during additional sequencing reads that automatically followed the completion of read 1. Signal intensities were converted to individual base calls during a run using the system's Real-Time Analysis (RTA) Software v1.17.21.3. Reads were trimmed using Trim Galore (v0.5.0) and then aligned to the yeast reference genome (sacCer3) using BWA (v0.7.17) (41). Peaks were called using MACS2 (v2.1.1) (42) and annotated using HOMER (v4.10.4) (43) with gene models from SGD and previous ncRNA datasets (34,35). The normalized bedGraph files were generated using MACS2 (‘-B –SPMR’) and then converted to bigwig files using the bedGraphToBigWig program downloaded from the website of UCSC Genome Browser. The genome-wide profile was generated using the ‘computeMatrix scale-regions’ and plotProfile tools in the deepTools package (v3.1.3) (44).
RNA-seq
Yeast strains, WT (MHY10244), set3Δ (MHY10869) and H2B-4KA (MHY10883), were grown in SD-Trp media until reaching mid-log phase (OD600 of 0.8–1.0). One OD600 equivalent of cells was used for RNA preparation using the RNeasy Mini Kit (Qiagen, 74104). Contaminating DNA was removed from the samples using the DNA-free™ DNA Removal Kit according to the manufacturer's protocol (Ambion, AM1906).
RNA samples were submitted to YCGA, and the libraries were constructed by YCGA protocols (https://medicine.yale.edu/keck/ycga/Images/9_tcm240-21599.pdf). The libraries underwent 101-bp paired-end sequencing using an Illumina HiSeq 2500 according to Illumina protocols. Low quality reads were trimmed, and adaptor contamination was removed using Trim Galore (v0.5.0). Trimmed reads were mapped to the S. cerevisiae genome (sacCer3) using HISAT2 (v2.1.0) (45). Gene expression levels were quantified using StringTie (v 1.3.3b) (46) with gene models from SGD and previous ncRNA datasets (34,35). Differentially expressed genes were identified using DESeq2 (v 1.22.1) (47).
Immunoblotting
Preparation of yeast whole-cell extracts and immunoblotting were performed as described previously (48). Levels of TAP-, Flag- or Myc- tagged proteins, SUMO conjugates, PGK, H3 methylations or H3 were analyzed by immunoblotting with 1:2000 to 1:5000 dilution of peroxidase-anti-peroxidase complex (PAP; Sigma, P1291), anti-Flag (Sigma, F3165), anti-Myc 9E10 (Covance, MMS-150R), anti-SUMO (49), anti-PGK (Molecular Probes, 459250), anti-H3K4me3 (Abcam, ab8580), anti-H3K4me2 (Abcam, ab7766), anti-H3K4me1 (Abcam, ab8895), anti-H3K36me3 (Abcam, ab9050), anti-H3K79me3 (Abcam, ab2621) and anti-H3 (Abcam, ab1791), respectively.
Sumoylation of Set3-TAP, Hos2-TAP or Cpr1–13Myc was determined as described previously (50,51). TCA-extracted proteins from 100 OD600 equivalents of cells were resuspended in 0.6 ml of sodium dodecyl sulphate (SDS) sample buffer with 50 μl of unbuffered 2 M Tris, and then heated to 100°C for 10 min. After centrifugation, 0.4 ml of supernatants were diluted with 0.8 ml of IP buffer {50 mM Tris–HCl [pH 7.4], 5 mM EDTA, 150 mM NaCl, 1% TritonX-100, 20 mM NEM (Sigma, E3876)}, immunoprecipitated with IgG-Sepharose beads (GE Healthcare, 17-0969-01) or anti-Myc agarose beads (Covance, AFC-150P) for 4 h at 4°C, washed by IP buffer four times, and then finally eluted into SDS sample buffer by boiling for 5 min. Both IP and INPUT samples were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting as described above.
In vivo histone sumoylation assay
The relative levels of sumoylated histone H2B or H4 were analyzed as described previously (19). TCA-extracted histones from 50 OD600 equivalents of cells were resuspended in 0.6 ml of SDS sample buffer with 50 μl of unbuffered 2 M Tris, and then heated to 100°C for 10 min. After centrifugation, 0.4 ml of supernatants were diluted with 0.8 ml of IP buffer {50 mM Tris–HCl [pH 7.4], 150 mM NaCl, 0.5% NP-40, 20 mM NEM (Sigma, E3876)}, immunoprecipitated with anti-Flag agarose beads (A2220, Sigma) for 4 h at 4°C, washed four times with IP buffer and then finally eluted into SDS sample buffer by heating at 100°C for 5 min. Both IP and INPUT samples were analyzed by SDS-PAGE and immunoblotting as described in the immunoblotting section.
Chromatin association assay
Chromatin association assays were done as described previously (19,52). Forty OD600 equivalents of cells grown to mid-exponential phase were harvested, washed with water and SB buffer (20 mM Tris–HCl [pH 7.4], 1 M sorbitol), and then frozen at -80°C until ready for isolation. After washing with 1 ml of SB buffer, cells were resuspended in 1 ml of PSB buffer (20 mM Tris–HCl [pH 7.4], 2 mM EDTA, 100 mM NaCl, 10 mM β-ME). Cells were then spheroplasted with Zymolyase (MP Biomedicals, MP08320931) for 30 min at room temperature. Spheroplasts were spun down at 5,000 × g for 15 min and washed twice with LB (20 mM PIPES [pH 6.8], 0.4 M sorbitol, 150 mM potassium acetate, 2 mM magnesium acetate). Triton X-100 was added to 0.1 ml of LB (final concentration of 1%) for cell lysis, and cells were incubated on ice for 15 min. Chromatin was isolated by spinning down lysates at 10 000 × g for 15 min. The supernatant was collected and saved as the ‘soluble’ fraction. The chromatin was washed once with LB and then resuspended in a volume equal to that of the ‘soluble’ fraction. Volume equivalents were resolved by SDS-PAGE and immunoblotting as described in the immunoblotting section. SB and LB buffers contained a protease inhibitor cocktail (Roche, 11697498001), 1 mM PMSF (AmericanBio, AB01620) and 20 mM NEM (Sigma, E3876).
In vitro nucleosome binding assay
Nucleosome pull-down assays were performed largely as described (30,53) with some modifications. 100 OD600 equivalents of cells expressing Flag-tagged H2B or derivatives were harvested, washed with water twice and then frozen at –80°C until ready for extraction. Cell pellets were lysed with glass beads in IP buffer {10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% NP-40, protease inhibitor cocktail (Roche, 11697498001), 1 mM PMSF (AmericanBio, AB01620), 20 mM NEM (Sigma, E3876)} and then sonicated 20 times for 20 s in pulse mode to make soluble chromatin. After two centrifugations at 21 130 × g for 30 min at 4°C, the concentration of protein in supernatant was measured by Bradford protein assay (Bio-Rad). Two mg of protein were mixed with 20 μl of anti-Flag agarose beads (Sigma, A2220) for 2 h at 4°C. Following four washes with IP buffer, bead-bound nucleosomes were incubated with 10 mg of extract containing TAP- or 13-Myc tagged proteins for 4 h at 4°C. The complexes were washed four times with IP buffer and then eluted into SDS sample buffer by heating to 100°C for 5 min. Samples were resolved by SDS-PAGE and immunoblotting as described in immunoblotting section.
Co-immunoprecipitation (Co-IP)
Co-IP experiments were performed as described previously (19,54), with few modifications. One hundred OD600 cell equivalents were harvested, washed with water twice and then frozen at –80°C. Cell pellets were lysed with glass beads in IP buffer {50 mM HEPES [pH 7.5], 100 mM NaCl, 10% glycerol, 0.1% Triton X-100, protease inhibitor cocktail (Roche, 11697498001), 1 mM PMSF (AmericanBio, AB01620), 20 mM NEM (Sigma, E3876)}. Cell debris were removed by spinning down twice at 21 130 × g for 30 min at 4°C. The supernatant was collected, and protein concentration was determined by Bradford protein assay (Bio-Rad). 10 mg of proteins were incubated with IgG-Sepharose beads (GE Healthcare, 17-0969-01) for 4 h at 4°C. Following four washes with IP buffer, 50 μl of SDS sample buffer was added to the washed beads for elution at 100°C. Finally, eluates (IP) and INPUT were analyzed by SDS-PAGE and immunoblotting as above.
Yeast synthetic growth assays
The synthetic growth assays were carried out as described previously (19). Two type of diploid strains were prepared to determine synthetic lethality; kanMX4-based gene replacements for each gene were made in yDS050 diploid cells (ULP2/ulp2Δ::HIS3) carrying the YCplac33-ULP2 plasmid, or yDS224 (ulp2Δ::kanMX4) was crossed with MHY4259 bearing pRS314, pRS314-HHT2/HHF2, pRS314-hht2-K4R/HHF2 or pRS314-hht2-K79A/HHF2 plasmids. After tetrad dissection of sporulated cultures, double mutant haploid cells carrying URA3-marked ULP2 or HHT2-HHF2 plasmids were streaked on 5-FOA plates and grown at 30°C for 3 days. Similarly, to test synthetic growth defects, YCplac33-ULP2 was evicted by 5-FOA counter-selection in double mutants of ulp2Δ combined with deletions of genes encoding each subunit of the Set3 HDAC (MHY1379 background) or ulp2Δ set1Δ mutants with YCplac33-ULP2 and pRS414 expressing SET1 or set1(ΔRRM). MHY4027 cells carrying deletion alleles and pRS314-FLAG-htb1 K6/7/16/17A were grown on 5-FOA plates to evict YCp50-HTB1 (URA3).
Quantification and statistical analysis
We analyzed three biological replicates for all experiments of ChIP and ChDIP with triplicate qPCR reactions. Statistical parameters are reported in the Figure Legends.
RESULTS
Set1 methylase is required for histone sumoylation
The prototypical example of histone-modification crosstalk is H2BK123 ubiquitylation-dependent H3 methylation at K4 and K79 (21,22). We recently reported a novel histone-crosstalk pathway in which histone sumoylation also was dependent on H2BK123-ubiquitin during transcription (19). To examine whether histone H3 methylation might mediate this sumoylation, we first measured the level of H2B-SUMO in cells lacking the Set1, Set2 or Dot1 methyltransferases that methylate histone H3 at K4, K36 or K79, respectively (Figure 1A, upper). As expected, levels of sumoylated H2B were greatly reduced in ubc9ts cells, while loss of the Ulp2 SUMO protease led to accumulation of di- and poly-SUMO conjugated H2B with slightly lower levels of mono-SUMO-conjugated H2B. Importantly, loss of Set1, but not of Set2 or Dot1, caused strongly diminished H2B sumoylation compared with WT cells. Lack of SET1 also resulted in a steep drop in H4 sumoylation (Supplementary Figure S1A). Because global levels of SUMO conjugates were constant in the same set1Δ mutant (Figure 1A, middle), these results suggest that Set1 specifically facilitates SUMO attachment to histones.
Figure 1.

Set1 mediates histone sumoylation. (A) Anti-SUMO immunoblot analysis of immunoprecipitated Flag-tagged histone H2B (upper panel) and bulk sumoylated proteins (middle panel) from extracts of indicated strains. Anti-Flag blotting for Flag-H2B (bottom panel) was used to verify similar loading. Open circle marks a non-specific band. *, **, ***: mono-, di- and poly-sumoylated histones, respectively. (B) Diagram of PMA1, ADH1 and PYK1 genes. The TATA/promoter (Pro) and open reading frame (ORF) are represented by black and white boxes, respectively. Bars below the genes show the positions of the PCR products used in the ChIP and ChDIP analyses. 5′ and M indicate 5′ and middle region of ORF, respectively. These designations are used in all later figures. (C) ChDIP analysis of Flag-H2B-SUMO in the indicated strains expressing both Flag-H2B and HA-SUMO (Smt3). Pro and ORF PCR signals were normalized to an internal control and the input DNA. Plots show the mean ± SD of triplicates. *P <0.05; **P <0.01 using Student's t test and comparing to WT. (D) Immunoblot assay of histone H3 tri-methylation at K4, K36 and K79 in the indicated strains. Anti-H3 blotting shows similar loading. (E–G) ChIP assays of histone H3 tri-methylation at K4 (E), K36 (F) and K79 (G) in the indicated strains. H3 methylation levels were normalized to H3 levels. Plots represent mean ± SD of triplicates. *P <0.05; **P <0.01 (Student's t test comparing to WT). (H) Summary of ChDIP-seq data. The pie chart shows the distribution of H2B-SUMO peaks at TSS (transcription start site), ORF, TES (transcription end site), and intergenic regions. Numbers in parentheses indicate the number of detected sites. (I) ChDIP read-density plot for levels of H2B-SUMO in WT and set1Δ strains. A ±1.5 kb window relative to the center of peaks is shown. The dotted line indicates the peak center. The ChDIP-seq data were obtained from duplicate experiments. (J) Percentage graph of sites of H2B-SUMO altered in intensity by set1Δ. Distribution of H2B-SUMO classified in (H) was analyzed. See also Supplementary Table S1 (sheets 1–3).
To verify that these bulk H2B sumoylation changes in set1Δ cells reflected alterations within chromatin, we performed chromatin double immunoprecipitation (ChDIP) assays to investigate occupancy of SUMO-conjugated H2B at specific chromosomal loci. We selected three constitutively transcribed genes, PMA1, ADH1 and PYK1, for ChDIP (Figure 1B) as these genes are known to be regulated by trans-tail crosstalk, including histone sumoylation (19,55). Sumoylated H2B levels were evaluated by sequential ChIPs using anti-Flag antibody-conjugated beads for Flag-tagged H2B in the first round and anti-HA beads against HA-SUMO in the second round, as described previously (9,19). We observed the expected changes in H2B sumoylation levels in ubc9ts and ulp2Δ control strains at both promoter and ORF regions of the three genes (Figure 1C). Most strikingly, occupancy by sumoylated H2B, but not total H2B, at the tested loci decreased in the set1Δ strain to a degree similar to ubc9ts cells (Figure 1C and Supplementary Figure S1B). Loss of Set2 and Dot1, by contrast, had no effect on H2B-SUMO at these sites.
Reciprocally, we measured trimethylated H3K4, K36 and K79 levels in cells where H2B sumoylation was altered by either ubc9ts or ulp2Δ (Figure 1D). Unlike the strong defect in histone sumoylation noted above in cells lacking Set1, defects in the SUMO pathway did not alter bulk H3 methylation. Histone H3 tri-methylation at the PMA1 and ADH1 test loci was also not significantly changed in these SUMO pathway mutants (Figure 1E–G). This indicates that H3K4 methylation dictates histone sumoylation levels, but not the converse. When considered with earlier work, these observations suggest a dependent pathway of histone modification in which histone H2B ubiquitylation promotes methylation of histone H3K4, which in turn stimulates histone sumoylation in a multistep mechanism of chromatin regulation.
H3K4 methylation-dependent histone sumoylation at diverse genes
To investigate potential histone sumoylation-governed genes throughout the genome, we mapped H2B-SUMO occupancy by ChDIP-seq experiments (Figure 1H and Supplementary Figure S1C). Histone sumoylation was enriched at many coding sequences, following a pattern similar to the genome-wide distribution of histone ubiquitylation (56). The modification was notably enriched on large genes with high rates of transcription (see Figure 5B and C), implying a correlation of histone sumoylation with active transcription. When enrichment of H2B-SUMO derived from the ChDIP-seq data was plotted relative to transcribed regions [from the transcription start site (TSS) to transcription end site (TES)], we observed that sumoylated H2B was distributed across genes with slightly higher signals near the TSS and 5′ portions of ORFs (Supplementary Figure S1D).
Figure 5.

Histone sumoylation governs association of SET3C with ncRNA genes. (A) ChIP reads-density plot for recruitment of Set3-TAP in WT and H2B-4KA strains, as shown in Figure 1I. (B and C) Percentage graphs of occupancy of Set3-TAP and post-translationally sumoylated histone H2B (H2B-SUMO) in WT cells with genes classified in (B) by FPKM (Fragments Per Kilobase of Million reads mapped) reported in our earlier RNA-seq experiments (50) and in (C) by gene length. RPs, ribosomal protein genes; ND, genes that were not detected. (D) Summary of genome-wide mapping of Set3-TAP at all genes (left) and at ncRNA loci (middle) and of H2B-SUMO at ncRNA loci (right) in WT. Pie graphs show the distribution of identified sites at the indicated regions. (E) Percentage graphs of data in (D) altered by H2B-4KA or set1Δ. Significantly decreased (blue) or increased (red) signals are shown. (F) Average plot of Set3-TAP recruitment at transcribed genes in WT and H2B-4KA cells. The y-axis values indicate fold-enrichment, setting the maximum occupancy to 1 and the minimum occupancy to 0; the x-axis represents normalized distance from TSS and TES. (G and H) Pie graphs showing percentage of ORF peaks of Set3-TAP increased by H2B-4KA in (E). ChIP-seq data were subdivided into three groups (G) single exon and two ‘exons’ separated by intron or +1 frameshift, or by description of genes (H). See also Supplementary Tables S1 (sheets 4–6) and S2.
Interestingly, this profile tracked closely with the pattern of Ulp2 SUMO protease recruitment, which is required for efficient transcriptional elongation (19). This supports the inference that (poly)SUMO-modified histones are key substrates of the Ulp2 protease during RNAPII elongation. Previous reports suggested histone sumoylation has a negative role in transcription (9,11,17,19). Our data, on the other hand, have revealed that this modification is concentrated at active genes, suggesting histone sumoylation also may promote transcriptional activity.
To determine genome-wide effects of H3K4 methylation on subsequent histone sumoylation, we compared enrichment of histone sumoylation between WT and set1Δ strains throughout the genome (Figure 1I). H2B-SUMO ChDIP signals detected at the peak centers and surrounding positions were reduced in set1Δ cells, consistent with the results in Figure 1A and C. Loss of Set1 reduced H2B-SUMO occupancy by 65–83% at TSS, ORF, TES and intergenic regions; occupancy at a comparatively small portion (20–30%) of sites was either increased or unaffected in set1Δ cells (Figure 1J). These results support the inference that trans-tail crosstalk between H2B ubiquitylation and H3K4 methylation promotes subsequent histone sumoylation, suggesting this newly uncovered aspect of the histone code is a general feature of yeast chromatin regulation.
H3K4me2 is essential for cell viability in ulp2Δ cells
We previously determined that histone-H2B ubiquitylation promotes histone (poly)sumoylation, which if not reversed by Ulp2, impedes Ctk1 phosphorylation of CTD Ser2 within RNAPII during transcriptional elongation (19). Deletions of RAD6 or BRE1, encoding the E2 and E3 enzymes responsible for H2B ubiquitylation, are lethal when combined with ulp2Δ (57). To investigate whether loss of histone methylation has similar genetic interactions with ulp2Δ, we analyzed crosses between ulp2Δ [YCplac33-ULP2] cells and the methylase-deficient set1Δ, set2Δ or dot1Δ strains. Meiotic segregants were streaked on plates containing 5-fluoroorotic acid (5-FOA) to evict the WT ULP2 plasmid (Supplementary Figure S2A). Lack of SET1, but not SET2 or DOT1, was lethal with ulp2Δ, suggesting that methylation of histone H3K4 is linked with histone sumoylation, consistent with the results in Figure 1. Indeed, when cell viability was assayed in double mutants combining ulp2Δ with specific histone H3 methylation site mutants H3K4R, H3K79A or H3R2A, disruption of ULP2 was lethal only in combination with H3K4R (Supplementary Figure S2B).
Interestingly, the H3R2A mutant, which abolishes tri-methylation, but only slightly reduces di-methylation at H3K4 (58), displayed a much milder growth defect in the ulp2Δ background (Supplementary Figure S2B and C). Moreover, among the nonessential genes encoding subunits of the Set1/COMPASS complex, only deletion of SPP1 failed to show synthetic lethality or sickness when combined with ulp2Δ (Supplementary Figure S2D). Uniquely among these genes, loss of SPP1 eliminates H3K4me3 but not H3K4me2 [SHG1 is not required for H3K4 methylation (59)]. Loss of SWD1, SDC1, SWD3 or BRE2, which all affect both tri- and di-methylation at H3K4 (60), caused severe growth defects when combined with ulp2Δ.
To confirm the distinct growth effects of different methylation states at H3K4 in cells also deleted for ULP2, we compared cell growth of ulp2Δ set1Δ double mutants transformed with empty plasmid or plasmids carrying either SET1 or set1(ΔRRM). The latter allele eliminates H3K4me3 but has no effect on H3K4me2 (61). Cells carrying the set1(ΔRRM) plasmid, unlike empty vector, grew as well as the double mutant transformed with WT SET1 plasmid (Figure 2A). This again implicates H3K4me2 (and possibly H3K4me1), but not H3K4me3, as being essential for cell growth when the SUMO system is dysregulated by loss of ULP2. Taken together, these data strongly imply that H3K4me2 and SUMO have overlapping functions in the regulation of transcription.
Figure 2.

H3K4me is required for histone sumoylation. (A) Mutant set1Δ ulp2Δ strains carrying a YCplac33-ULP2 (URA3) cover plasmid and the indicated plasmids were grown on SD-Trp and SD+5-FOA at 30°C for 2–3 days. (B) Immunoblot assay of H3K4me3, me2, and me1 in the indicated strains. Anti-H3 blotting shows similar loading. (C and E) Immunoblot analysis of immunoprecipitated Flag-tagged H2B using anti-SUMO antibody in the indicated strains. Upper and lower panels show Flag-H2B-SUMO and Flag-H2B (loading control), respectively. Asterisks and open circles as in Figure 1A. (D) ChDIP analysis of Flag-H2B-SUMO for the strains in (C). Data represent mean ± SD of triplicates; *P <0.05 (Student's t test comparing to WT).
We addressed in more detail how the SUMO pathway and H3K4 methylation interact. As seen in Figure 2B, methylation of H3K4 was unchanged in ubc9ts and ulp2Δ strains. Conversely, however, plasmids bearing either WT SET1 or set1(ΔRRM) plasmids fully restored histones H2B and H4 sumoylation in set1Δ mutants (Figure 2C and D and Supplementary Figure S2E and S2F). These data indicate that elimination of H3K4me3 does not impact histone sumoylation, implying instead that H3K4me2 provides the essential methylation state of H3K4 necessary for histone sumoylation.
Histone sumoylation promotes Set3 HDAC recruitment
H3K4me2 has distinct transcriptional functions and localization patterns compared to H3K4me3. While H3K4me3 is predominantly located near promoter regions and is generally associated with transcriptional initiation, H3K4me2 is enriched in the 5′ transcribed regions of genes to create binding sites for SET3C (27). SET3C deacetylates histones via two catalytic subunits, Hos2 and Hst1, which regulate transcriptional elongation, cryptic internal initiation, or RNAPII recruitment (29,30,32,62). Since H3K4me2 also seemed closely correlated with histone sumoylation, we sought to link H3K4me2-mediated control of SET3C and SUMO-H2B.
Combining mutations in Set3 subunits with ulp2Δ or a histone H2B mutation, H2B-4KA, which is defective for H2B sumoylation (9), caused no obvious synthetic growth defects (Supplementary Figure S3A and B). Loss of SET3C subunits also had no effect on either histone sumoylation or global SUMO conjugates (Figure 2E and Supplementary Figure S3C). Therefore, SET3C does not become essential when histone sumoylation is altered, unlike H3K4me2, which is necessary for both recruitment of SET3C to chromatin and histone sumoylation. This argues that the essential shared function of histone sumoylation and H3K4 methylation is not limited to SET3C recruitment.
To determine whether histone sumoylation actually affects recruitment of SET3C, we compared levels of two components of the complex, Set3 and Hos2, at constitutively transcribed genes in WT, ubc9ts, ulp2Δ, H2B-4KA, and 2SUMO-H2B strains; the last of these is an htb1–1 htb2–1 (histone H2B null) mutant with a plasmid encoding two non-cleavable SUMO moieties fused in tandem to H2B (Figure 3A, upper and middle panels). The H2B-4KA and 2SUMO-H2B mutants mimic strains with reduced or persistent (poly)SUMO-conjugated histones, respectively. Intriguingly, we found that recruitment of Set3 and Hos2 was reduced at all three tested genes in both ubc9ts and H2B-4KA cells, but not in ulp2Δ or 2SUMO-H2B cells. We repeated the ChIP analysis with a more conservative H2B mutant in which the sumoylated Lys residues were replaced with Arg instead of Ala (H2B-4KR). As was true for H2B-4KA cells, the H2B-4KR strain also showed significantly impaired recruitment of both Set3 and Hos2 to the tested loci (Figure 3B). Together, these data suggest that histone sumoylation promotes chromatin binding of the Set3 HDAC, although other post-translational modifications of these lysines might also be important.
Figure 3.

Histone sumoylation recruits the Set3 HDAC. (A) ChIP analyses of Set3-TAP (upper panel), Hos2-TAP (middle) and H3 acetylation (bottom) in the indicated strains at 5′ ORF regions. An untagged strain (MHY10244) was used as a negative control for IP of TAP-tagged proteins. Values of H3 acetylation were normalized to H3 levels. Data represent mean ± SD of triplicates. *P <0.05; **P <0.01 (Student's t test comparing to WT). (B) ChIP analyses of Set3-TAP and Hos2-TAP in WT and H2B-4KR strains at 5′ ORF regions. Data represent mean ± SD of triplicates. *P <0.05; **P <0.01 (Student's t test comparing to WT). (C) Occupancy of Rpb3, Set3-TAP and Hos2-TAP and the ratio of H3Ac to H3 were determined by ChIP in WT and H2B 4KA strains. GAL1 OFF and ON denote uninduced and induced conditions. Data represent mean ± SD of triplicates. *P <0.05; **P <0.01 (Student's t test comparing to WT). (D) Chromatin association assays using indicated strains from (A). After fractionation into chromatin (pellet) and soluble (supernatant) components, fractions were analyzed by immunoblotting as indicated. (E) In vitro nucleosome binding assays of Set3 and Hos2. Nucleosomes were isolated by anti-Flag agarose IP from indicated strains, while a negative control strain (H2B) lacked the Flag tag. Nucleosome-immobilized beads were incubated with yeast extracts containing either Set3-TAP (MHY10885) or Hos2-TAP (MHY10886). Bound proteins were analyzed by immunoblotting. (F) Sensitivity to 6-AU of htb1–1 htb2–1 strains with pRS314 plasmids expressing Flag-H2B, Flag-H2B-4KA or 2SUMO-H2B. All strains carried pRS316 (URA3) and were spotted on SD-Ura with or without 6-AU (100 μg/ml). Plates were incubated for 2–4 days.
To test the predicted locally increased histone acetylation when sumoylation is impaired (30,31), we analyzed acetylated histone H3 levels in the same strains (Figure 3A, bottom panel). Consistent with the inverse correlation between SET3C recruitment and histone acetylation, H2B-4KA cells displayed a sharp increase in histone H3 acetylation. However, contrary to prediction, H3 acetylation levels were greatly repressed in ubc9ts cells. Ubc9-mediated SUMO modification serves diverse roles in transcription (63–65), and many proteins involved in chromatin organization, transcription, and RNA metabolism are Ubc9 substrates (7,66,67). Hence, hypo-acetylation of H3 in ubc9ts cells is likely caused by other sequelae of reduced cellular SUMO conjugation.
It was previously reported that GAL genes are targets of Set3- and Hos2-mediated transcriptional activation (31). We therefore examined the GAL1 gene to determine whether lack of histone sumoylation limits SET3C recruitment when transcription is induced by galactose (Figure 3C). Increased 5′ GAL1 occupancy by the RNAPII subunit Rpb3 and TAP-tagged Set3 and Hos2 proteins was observed in WT cells under inducing conditions. By contrast, binding of these factors was significantly reduced in H2B-4KA cells. In accord with reduced HDAC recruitment, the H2B-4KA mutation led to increase H3 acetylation at the 5′ end of GAL1 following gene induction. These data support a model in which local histone sumoylation enhances chromatin binding of SET3C during transcriptional induction.
To assess the effect of histone sumoylation on the global loading of Set3 and Hos2 onto chromatin, we carried out a chromatin-association assay that measures overall changes in the levels of these proteins in chromatin obtained from yeast spheroplasts (Figure 3D). Separation of soluble from chromatin-bound proteins was verified by immunoblotting using anti-PGK (soluble) and anti-H3 (chromatin) antibodies. Both Set3 and Hos2 partitioned predominantly into the chromatin fraction from WT as well as 2SUMO-H2B cells. By contrast, in H2B-4KA cells, the chromatin localization of both proteins was strongly reduced, paralleling the ChIP results in Figure 3A and C. These results suggest that the impact of histone sumoylation on SET3C recruitment is not limited to a few specific genes but rather, is affecting much of the genome.
To exclude possible indirect effects due to reduced histone sumoylation, we monitored direct interaction between isolated nucleosomes harboring histone mutants and SET3C (Figure 3E). Nucleosomes were immunoprecipitated via Flag-tagged H2B from WT, H2B-4KA or ubc9ts cell lysates and then incubated with cell extracts from yeast expressing Set3-TAP or Hos2-TAP. Nucleosomes containing histone H2B-4KA interacted more poorly with Set3 and Hos2 compared to WT nucleosomes. Association of these SET3C subunits with nucleosomes isolated from ubc9ts cells was reduced even further. Because the nucleosomal histone octamer has additional SUMO attachments beyond the four mutated lysines in H2B-4KA (9), this result is consistent with sumoylation of other histone residues (or chromatin factors) being able to enhance nucleosome binding by SET3C. Taken together, our data indicate that histone sumoylation broadly promotes chromatin binding of SET3C, regulating local histone acetylation.
Both set3Δ and hos2Δ strains are sensitive to MPA (mycophenolic acid), which inhibits transcriptional elongation by reducing nucleotide pools (30). Similarly, 6-azauracil (6-AU) also inhibits transcriptional elongation, and 2SUMO-H2B cells, in which histone H2B is persistently sumoylated, are strongly growth impaired on 6-AU (19), as are set1 mutants (68). We therefore tested whether cells with the opposite defect, diminished histone sumoylation, also causes sensitivity to 6-AU (Figure 3F). Although the H2B-4KA mutant exhibited slow growth on normal medium at least as severe as the 2SUMO-H2B defect at 30°C, it showed little additional sensitivity to 6-AU at this temperature. However, when grown at 34°C on 6-AU plates, H2B-4KA cell growth was strongly impaired, suggesting that SUMO-histone conjugation, like SET3C-catalyzed histone deacetylation, promotes transcriptional elongation.
A SUMO-interacting motif in the Cpr1 subunit of SET3C enables chromatin binding
Given that histone sumoylation resulted in recruitment of SET3C to actively transcribed genes, we next sought to determine how such SUMO modifications stimulate chromatin association of the complex. Intriguingly, the multifunctional Cpr1 protein, a subunit of SET3C, was found to associate with SUMO in two screens using affinity capture and mass spectrometry (69,70). The Cpr1-SUMO interaction has not been verified, and its cellular role remains obscure. Therefore, we tested for covalent sumoylation of Cpr1 protein by IP of 13Myc-tagged Cpr1 from denatured lysates from WT or ulp2Δ cells (Figure 4A, last two lanes). Cpr1 was not detectably conjugated to SUMO.
Figure 4.

Mutation of the SET3C Cpr1 subunit SIM blocks chromatin binding. (A) IP of Set3-TAP and Hos2-TAP with IgG-Sepharose and Cpr1–13Myc with anti-Myc agarose from denatured yeast extracts in ulp2Δ strains with or without YCplac33-ULP2. Precipitated proteins were followed by immunoblotting. Anti-PGK was used as a loading control. The top panels show overexposure of SUMO blot. Potentially polySUMO-conjugated Set3-TAP is seen in the stacker in lane 2. Arrowheads, unmodified TAP-tagged proteins. (B) Mutation of SIM within Cpr1. Putative SIM residues 154–158 are bolded and underlined. (C) Chromatin association assays using htb1–1 htb2–1 cpr1Δ strains with pRS314 plasmids expressing Flag-H2B or Flag-H2B-4KA and pRS425-GPD-Cpr1–13Myc or pRS425-GPD-Cpr1 MUT-13Myc. Chromatin and soluble fractions were analyzed by immunoblotting. (D) In vitro nucleosome binding assays of Cpr1 WT and MUT. Nucleosomes isolated on anti-Flag agarose from the indicated strains were incubated with extracts from cpr1Δ cells (MHY10887) carrying pRS425-GPD-borne Cpr1–13Myc or Cpr1-MUT-13Myc, precipitated, and then immunoblotting with the indicated antibodies. (E) ChIP assays at the indicated 5′ ORF regions using strains in (C). An untagged strain (MHY10244) was used as a negative control. Data represented mean ± SD of triplicates. *P <0.05; **P <0.01 (Student's t test between indicated pairs of values).
Based on the GPS-SUMO tool for predicting SUMO-interacting motifs (SIMs) (71), Cpr1 has a potential SIM at residues 154–158 (IVVAK). We therefore generated a mutant, Cpr1 MUT, in which the residues IVV were replaced with NAA (Figure 4B). Co-IP experiments showed the Cpr1 SIM did not affect Set3 complex assembly or stability (Supplementary Figure S4). The effect of the Cpr1 SIM mutation on SET3C binding to chromatin was tested with the chromatin association assay used above. Similar to the partitioning of the Set3 and Hos2 subunits of SET3C (Figure 3E), the Cpr1 subunit also concentrated in the chromatin fraction (Figure 4C). Critically, Cpr1 MUT was found in lower amounts in the chromatin fraction and correspondingly higher levels in the soluble fraction, suggesting the SIM is needed for Cpr1 localization to chromatin. Chromatin association of Cpr1 was also impaired in the H2B-4KA mutant (Figure 4C), similar to the results with Set3 and Hos2 (Figure 3E). Combining Cpr1 with the mutated SIM with H2B-4KA exacerbated the chromatin binding defects of the single mutants, suggesting that both the SIM in Cpr1 and histone sumoylation are required for chromatin localization of Cpr1 and that each mutation by itself only partially inactivates SUMO-dependent Cpr1–chromatin interaction.
We also tested the effect of mutating the Cpr1 SIM on the direct interaction of SET3C with nucleosomes in vitro (Figure 4D). Consistent with the above chromatin association data, nucleosomal binding of Cpr1 was dependent on histone sumoylation, and binding was also reduced by mutation of the Cpr1 SIM. The interaction of the Cpr1 SIM mutant with nucleosomes was further impaired by concomitant hypo-sumoylation of histones due to the H2B-4KA mutation. This also suggests that other SUMO-conjugated factors, including potentially other histones, may be recognized by the Cpr1 SIM to promote SET3C loading onto nucleosomes.
To determine if the Cpr1 SIM-mediated association with nucleosomes depends on histone sumoylation at specific loci, we carried out ChIP assays in WT and H2B-4KA cells with 13Myc-tagged WT or SIM-mutant Cpr1 (Figure 4E). Consonant with the results in Figure 4C and D, ChIP analysis revealed that recruitment of Cpr1 at the SET3C-regulated PMA1, ADH1 and PYK1 genes was significantly reduced by either the H2B-4KA or Cpr1 SIM mutation. Mutation of the Cpr1 SIM further suppressed its association with the tested genes in H2B-4KA strains, implying that the Cpr1 SIM mediates recognition of diverse sumoylated chromatin proteins; however, there was no significant change in recruitment of the Cpr1 SIM mutant between WT and H2B-4KA strains (Figure 4E). This strongly suggests that association of sumoylated histones with the Cpr1 SIM is essential for Cpr1 recruitment to specific chromatin loci. Overall, these data demonstrate the importance of Cpr1 recognition of SUMO-conjugated histones via the Cpr1 SIM motif.
Histone sumoylation governs association of SET3C with ncRNA genes
To investigate genome-wide effects of histone sumoylation on SET3C recruitment (Supplementary Figure S5), we performed ChIP-seq analysis in WT and H2B-4KA strains expressing TAP-tagged Set3. When enrichment of Set3-TAP was compared between WT and H2B-4KA strains (Figure 5A), peak read densities were significantly reduced in H2B-4KA cells, in line with the single gene analyses in Figure 3. In WT cells, Set3 proteins were predominantly enriched on long genes with high expression (Figure 5B and C), showing a similar pattern with the genome-wide location of sumoylated histone H2B (H2B-SUMO) in ChDIP-seq experiments. Set3 was recruited to diverse sites around TSS (999), ORF (139), TES (172), intron (8) and intergenic (25) regions (Figure 5D, left panel) that suggested a preferred function in the early stages of transcription. This is congruent with previous reports that SET3C preferentially modulates histone acetylation in the 5′ regions of genes (30,32).
SET3C represses cryptic internal initiation and anti-sense transcription at Set3-regulated genes that overlap with ncRNA transcripts (32). We analyzed occupancy of Set3 and H2B-SUMO at previously mapped locations of SUT and CUT ncRNAs (34,35) (Figure 5D, middle and right panels). Notably, both Set3 and H2B-SUMO were generally observed at these loci, consistent with both histone sumoylation and SET3C regulating the transcription of ncRNA genes. Although peaks signals for Set3-TAP and H2B-SUMO were also found at some intergenic sites, most peaks overlapped coding sequences.
In agreement with the idea that H2B sumoylation globally promotes SET3C association in the 5′ regions of actively transcribed genes, ChIP signals of Set3-TAP were significantly diminished by the H2B-4KA mutation at ∼80% of sites in Set3-enriched protein-coding genes (Figure 5E, left panel). While we also found increased levels of Set3-TAP at a small number (46) of ORF regions (Figure 5E, G and H), comparing global Set3 binding on normalized TSS to TES plots clearly showed that the H2B-4KA mutation generally impedes recruitment of Set3 to transcribed genes (Figure 5F).
The H2B-4KA strain showed a significant decline in Set3 recruitment at ∼80% of ncRNA loci (Figure 5E, middle panel). Notably, at ∼70% of these loci, SUMO-conjugated H2B was significantly decreased in cells lacking the Set1 methyltransferase (Figure 5E, right panel). These results suggest that ncRNA gene loci are targets of both SET3C regulation and H3K4 methylation-sensitive histone sumoylation. By contrast, peak signals of Set3-TAP or H2B-SUMO in ∼20% or ∼40% of intergenic regions were higher in H2B-4KA or set1 mutants, respectively, implying that histone sumoylation and SET3C have distinct roles in chromatin regulation at these sites. Taken together, our results strongly suggest that sequential H3K4 methylation and histone sumoylation stimulate recruitment of SET3C to ncRNA as well as mRNA genes.
Histone sumoylation suppresses cryptic initiation of transcription
Earlier genome-wide microarray analysis of set3Δ cells revealed 119 genes that were down-regulated >1.7-fold relative to WT in response to carbon source shifts, indicating a positive role for local histone deacetylation by SET3C (32). Derepressed ncRNA expression that overlapped these genes was found to delay or attenuate induction of the latter mRNAs. Because our ChIP-seq data showed histone sumoylation promotes Set3 binding to both protein-coding and ncRNA genes (Figure 5), we asked if histone sumoylation-stimulated Set3 binding drove parallel transcriptome changes.
For this we derived genome-wide gene expression profiles of set3Δ and H2B-4KA cells using RNA-seq and then compared the results with publicly available datasets (Figure 6A and Supplementary Figure S6). The mutant profiles included many genes with at least a 1.5-fold change in expression levels relative to WT (Supplementary Figure S7A and B). Interestingly, both up- and down-regulated gene groups were substantially overlapping between the set3Δ and H2B-4KA strains with between half to three-quarters of protein-coding or ncRNA genes changing in parallel (Figure 6A). In both mutants, more genes exhibited an increase rather than a decrease in expression, and the changes were greater for ncRNA than mRNA genes. These data suggest that a major role for histone sumoylation is to promote SET3C-catalyzed deacetylation and transcriptional repression, especially of ncRNA genes.
Figure 6.

Histone sumoylation suppresses cryptic initiation of transcription. (A) Transcriptome analysis of set3Δ and H2B-4KA strains compared with WT (MHY10244). Venn diagrams show significantly down- or up-regulated genes or ncRNAs (>1.5-fold). The RNA-seq data were obtained from duplicate samples. (B) Classification of ncRNA clusters according to start site and direction of transcription. Protein-coding ORF and orientation of ncRNA transcription are represented by white box and arrow, respectively. ncRNA initiation sites either do or do not overlap with known ORFs, either in the same orientation (A and C groups) or in opposite orientation (B and D groups) to the coding transcript. E group comprise ncRNA transcripts that are do not overlap with an ORF. (C) Percentage graphs of ncRNA expression altered by set3Δ or H2B-4KA in the groups shown in (A). Significantly decreased or increased signals are represented as bar graphs. (D) Venn diagrams of ncRNAs whose levels were increased by set3Δ or H2B-4KA shown in (C). Numbers on the graph refer to the number of ncRNAs increased in expression in either mutant; the percentage of the total ncRNAs that increased in level in H2B-4KA cells that also increased in set3Δ is shown in parentheses. (E) Percentage graphs of the expression changes for genes overlapping those ncRNAs with increased expression in set3Δ (left panel) or H2B-4KA (right panel). See also Supplementary Table S3.
In S. cerevisiae, in addition to well-known ncRNAs such as rRNA, tRNA or snoRNA, pervasive transcription of ncRNAs has been reported in WT or exosome-deficient cells (34–37). Most of the newly identified ncRNAs have not been functionally validated. To determine which ncRNAs are governed by the histone sumoylation-SET3C pathway, we classified ncRNA genes belonging to the SUT (stable uncharacterized transcripts) and CUT (cryptic unstable transcripts) groups into five classes according to their initiation points and transcriptional orientation (Figure 6B). The A and B ncRNAs start within a protein-coding gene body in either the same or opposite direction of the primary mRNA transcript, respectively. The C and D groups comprise sense and anti-sense types of ncRNA initiated outside of any ORFs. The E group includes ncRNA genes that do not overlap any ORF but potentially still run into the 5′ or 3′ untranslated regions of mRNAs (Supplementary Figure S7C). This grouping revealed that either set3Δ or H2B-4KA mutation led to increased expression of a large fraction of group A ncRNAs (∼70%) and possibly a small increase in group B (Figure 6C). Furthermore, almost all the ncRNA genes in group A (96.6%) increased in expression in response to both mutations, and expression levels of ∼57–65% of all the other ncRNA groups were also observed to increase in both mutants (Figure 6D). These results suggest that histone sumoylation and SET3C regulate the majority of these ncRNAs, and the two factors have particularly strong suppressive effects on spurious transcription of sense ncRNAs initiated from sites internal to mRNA ORFs.
Notably, our results indicated that expression levels of mRNAs from genes that overlap with ncRNAs were not generally altered in either mutant (Figure 6E). This supports the hypothesis that the principal function of histone sumoylation and histone deacetylation by SET3C is to prevent spurious or cryptic ncRNA transcription, especially when these initiate within mRNA genes.
DISCUSSION
Histone-SUMO conjugates were first reported in human cells in 2003 (11), yet functional characterization of such conjugates in chromatin dynamics has remained largely unexplored. Here we report that histone sumoylation depends on both H2B ubiquitylation and H3K4 methylation and such sumoylation enhances gene expression by distinct mechanisms during different phases of transcription (Figure 7 and Supplementary Figure S8). The degree of H3K4 methylation generally shows a gradient on transcribed genes, with mostly tri-methylation near active promoters and di-methylation in the 5′-transcribed regions (24,25). At the promoter during the transition from transcriptional initiation to elongation, the form of RNAPII with CTD-S5 phosphorylated interacts with the H2B ubiquitylation enzymes Rad6 and Bre1, and the Set1/COMPASS histone methyltransferase through the PAF complex (72) (Figure 7A). H2B ubiquitylation and H3K4 methylation triggers recruitment of Ubc9 and presumably an E3 ligase to catalyze histone poly-sumoylation. The presence of both histone ubiquitylation and sumoylation inhibits the recruitment of Ctk1 kinase to nucleosomes, preventing its phosphorylation of CTD-S2 in RNAPII (19,73). The proteases Ulp2 and Ubp8 (within the SAGA DUB module) are recruited to the transcription machinery to remove polySUMO and ubiquitin, respectively, from histones. This facilitates Ctk1-mediated CTD-S2 phosphorylation and transcript elongation (19,73) (Figure 7B). Gcn5 histone acetyltransferase (HAT), another SAGA subunit, along with other HATs mediates hyperacetylation of histones in 5′ gene regions (74). Finally, H3K4me2- and SUMO-conjugated histones provide binding sites for SET3C through the Set3 PHD finger and Cpr1 SIM, leading to histone deacetylation and a more compact chromatin structure that prevents initiation of spurious ncRNAs from cryptic internal sites [(30) and our data] (Figure 7C).
Figure 7.
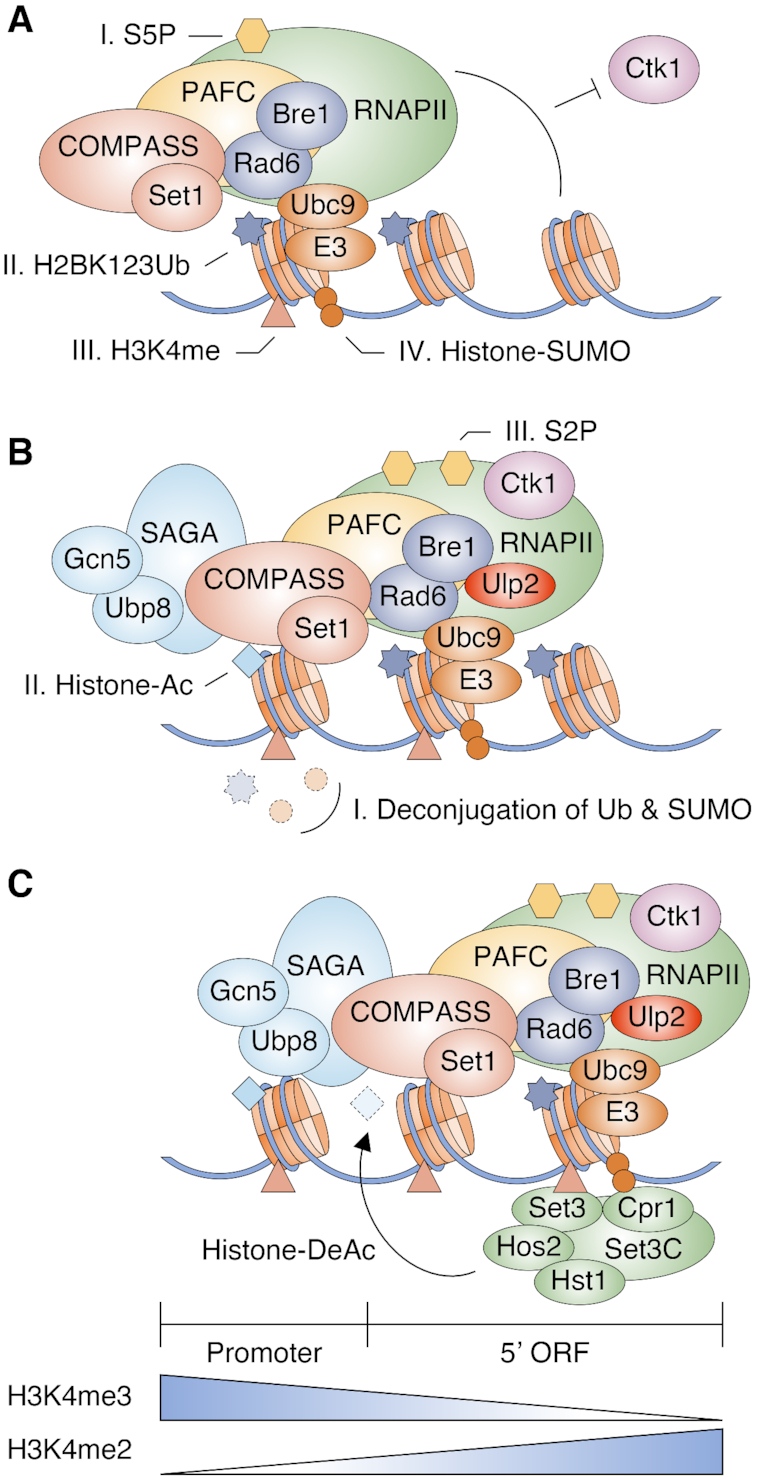
Model for the functions of histone sumoylation in transcription. Illustration reflects the relevant components but not precise physical association or order of events.
Nucleosome binding assays comparing ubc9ts and H2B-K4A mutants suggest sumoylation of other residues in histones or other chromatin components is required for full nucleosomal binding of SET3C (Figures 3E and 4D). Since SUMO can form branched chains and is subject to other modifications such as phosphorylation, acetylation and ubiquitylation (3), complex signaling codes involving SUMO could regulate transcription in various ways. We suggest SUMO attachment to diverse histone sites creates many binding sites for Cpr1 and also locally decompacts chromatin to facilitate SET3C association. This would be consistent with the known binding of the LSD1–CoREST repressor complex to sumoylated histones via a CoREST SIM and less tightly folded chromatin (17,18).
Multiple histone modifications and their combinations are proposed to constitute codes for distinct functional states of chromatin (75). Our data provide a striking example of the potential complexity of such coding, with a dependent series of at least four different histone modifications acting on transcription. Set3 protein may itself be (poly)sumoylated (Figure 4A). This would be analogous to the ability of Rad6/Bre1 to promote H3K4 methylation by two possible pathways, ubiquitylation of H2B and COMPASS component Swd2 (76).
Unlike a previous report (32), our data show SET3C suppresses transcription (sense) from internal cryptic promoters more widely than that of overlapping antisense transcripts. However, the earlier study used microarray experiments in yeast cells responding to carbon source shifts; cells may have a greater need to modulate antisense transcription under rapidly changing growth conditions. Antisense-mediated transcriptional interference is able to repress expression of associated mRNAs in specific conditions (77). However, since most yeast ncRNAs are likely to result from leaky transcription initiation under most conditions (78), a primary role for SET3C may be to prevent inappropriate internal ncRNA transcription.
Components of yeast SET3C show strong sequence similarities to the mammalian HDAC3/SMRT complex (29). In addition, fusion of SUMO to histone H4 promotes the local binding of HDAC1 along with HP1 or with the LSD1-CoREST complex in human cells (11,17). Therefore, histone sumoylation-stimulated HDAC recruitment may be an evolutionarily conserved mechanism to control pervasive transcription. In eukaryotes, ncRNA cellular levels are established by multiple means, including inhibition of inappropriate RNA polymerase initiation and exosome-mediated RNA decay (78). Our findings indicate that histone sumoylation is central to both controlling elongation of mRNA transcripts and inhibiting spurious transcriptional initiation of ncRNAs within the bodies of protein-coding genes. Both functions are likely to be conserved in higher eukaryotes as well.
DATA AND AVAILABILITY
Data generated or used in this publication have been deposited in Gene Expression Omnibus (GEO) with accession numbers:
Supplementary Material
ACKNOWLEDGEMENTS
We thank D.E. Gottschling, I.M. Fingerman, P. Hieter, S. Buratowski and S.L. Berger for supplying plasmids, and S.H. Ahn for comments on the manuscript.
Notes
Present address: Dan Su, Alexion Pharmaceuticals, 100 College St., New Haven, CT 06510, USA.
Contributor Information
Hong-Yeoul Ryu, School of Life Sciences, BK21 Plus KNU Creative BioResearch Group, College of National Sciences, Kyungpook National University, Daegu 41566, Republic of Korea.
Dejian Zhao, Yale Center for Genome Analysis, Yale University, New Haven, CT 06520, USA.
Jianhui Li, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520, USA.
Dan Su, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520, USA.
Mark Hochstrasser, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [GM053756, GM136325 to M.H.]; National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) [2020R1C1C1009367 to H.Y.R.]. Funding for open access charge: NIH [GM136325 to M.H.]; National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) [2020R1C1C1009367 to H.Y.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009; 458:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flotho A., Melchior F.. Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013; 82:357–385. [DOI] [PubMed] [Google Scholar]
- 3. Hendriks I.A., Vertegaal A.C.. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016; 17:581–595. [DOI] [PubMed] [Google Scholar]
- 4. Hickey C.M., Wilson N.R., Hochstrasser M.. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012; 13:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gill G. Something about SUMO inhibits transcription. Curr. Opin. Genet. Dev. 2005; 15:536–541. [DOI] [PubMed] [Google Scholar]
- 6. Zhao J. Sumoylation regulates diverse biological processes. Cell. Mol. Life Sci. 2007; 64:3017–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Makhnevych T., Sydorskyy Y., Xin X., Srikumar T., Vizeacoumar F.J., Jeram S.M., Li Z., Bahr S., Andrews B.J., Boone C. et al.. Global map of SUMO function revealed by protein-protein interaction and genetic networks. Mol. Cell. 2009; 33:124–135. [DOI] [PubMed] [Google Scholar]
- 8. Chymkowitch P., Nguea P.A., Enserink J.M.. SUMO-regulated transcription: Challenging the dogma. Bioessays. 2015; 37:1095–1105. [DOI] [PubMed] [Google Scholar]
- 9. Nathan D., Ingvarsdottir K., Sterner D.E., Bylebyl G.R., Dokmanovic M., Dorsey J.A., Whelan K.A., Krsmanovic M., Lane W.S., Meluh P.B. et al.. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006; 20:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kalocsay M., Hiller N.J., Jentsch S.. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell. 2009; 33:335–343. [DOI] [PubMed] [Google Scholar]
- 11. Shiio Y., Eisenman R.N.. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:13225–13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hendriks I.A., D'Souza R.C., Yang B., Verlaan-de Vries M., Mann M., Vertegaal A.C.. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014; 21:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matafora V., D’Amato A., Mori S., Blasi F., Bachi A.. Proteomics analysis of nucleolar SUMO-1 target proteins upon proteasome inhibition. Mol. Cell. Proteomics. 2009; 8:2243–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen W.T., Alpert A., Leiter C., Gong F., Jackson S.P., Miller K.M.. Systematic identification of functional residues in mammalian histone H2AX. Mol. Cell. Biol. 2013; 33:111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamoliatte F., McManus F.P., Maarifi G., Chelbi-Alix M.K., Thibault P.. Uncovering the SUMOylation and ubiquitylation crosstalk in human cells using sequential peptide immunopurification. Nat. Commun. 2017; 8:14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galisson F., Mahrouche L., Courcelles M., Bonneil E., Meloche S., Chelbi-Alix M.K., Thibault P.. A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol. Cell. Proteomics. 2011; 10:M110 004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dhall A., Weller C.E., Chu A., Shelton P.M.M., Chatterjee C.. Chemically sumoylated histone H4 stimulates intranucleosomal demethylation by the LSD1-CoREST complex. ACS Chem. Biol. 2017; 12:2275–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dhall A., Wei S., Fierz B., Woodcock C.L., Lee T.H., Chatterjee C.. Sumoylated human histone H4 prevents chromatin compaction by inhibiting long-range internucleosomal interactions. J. Biol. Chem. 2014; 289:33827–33837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ryu H.Y., Su D., Wilson-Eisele N.R., Zhao D., Lopez-Giraldez F., Hochstrasser M.. The Ulp2 SUMO protease promotes transcription elongation through regulation of histone sumoylation. EMBO J. 2019; 38:e102003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012; 81:65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shahbazian M.D., Zhang K., Grunstein M.. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol. Cell. 2005; 19:271–277. [DOI] [PubMed] [Google Scholar]
- 22. Sun Z.-W., Allis C.D.. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002; 418:104–108. [DOI] [PubMed] [Google Scholar]
- 23. Henry K.W., Berger S.L.. Trans-tail histone modifications: wedge or bridge. Nat. Struct. Biol. 2002; 9:565–566. [DOI] [PubMed] [Google Scholar]
- 24. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 25. Pokholok D.K., Harbison C.T., Levine S., Cole M., Hannett N.M., Lee T.I., Bell G.W., Walker K., Rolfe P.A., Herbolsheimer E. et al.. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005; 122:517–527. [DOI] [PubMed] [Google Scholar]
- 26. Soares L.M., He P.C., Chun Y., Suh H., Kim T., Buratowski S.. Determinants of histone H3K4 methylation patterns. Mol. Cell. 2017; 68:773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buratowski S., Kim T.. The role of cotranscriptional histone methylations. Cold Spring Harb. Symp. Quant. Biol. 2010; 75:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pinskaya M., Morillon A.. Histone H3 lysine 4 di-methylation: a novel mark for transcriptional fidelity. Epigenetics. 2009; 4:302–306. [DOI] [PubMed] [Google Scholar]
- 29. Pijnappel W.W., Schaft D., Roguev A., Shevchenko A., Tekotte H., Wilm M., Rigaut G., Seraphin B., Aasland R., Stewart A.F.. The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev. 2001; 15:2991–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim T., Buratowski S.. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell. 2009; 137:259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang A., Kurdistani S.K., Grunstein M.. Requirement of Hos2 histone deacetylase for gene activity in yeast. Science. 2002; 298:1412–1414. [DOI] [PubMed] [Google Scholar]
- 32. Kim T., Xu Z., Clauder-Münster S., Steinmetz L.M., Buratowski S.. Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell. 2012; 150:1158–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tisseur M., Kwapisz M., Morillon A.. Pervasive transcription - lessons from yeast. Biochimie. 2011; 93:1889–1896. [DOI] [PubMed] [Google Scholar]
- 34. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Munster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M.. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009; 457:1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neil H., Malabat C., d’Aubenton-Carafa Y., Xu Z., Steinmetz L.M., Jacquier A.. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009; 457:1038–1042. [DOI] [PubMed] [Google Scholar]
- 36. van Dijk E.L., Chen C.L., d’Aubenton-Carafa Y., Gourvennec S., Kwapisz M., Roche V., Bertrand C., Silvain M., Legoix-Ne P., Loeillet S. et al.. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011; 475:114–117. [DOI] [PubMed] [Google Scholar]
- 37. Wery M., Descrimes M., Vogt N., Dallongeville A.S., Gautheret D., Morillon A.. Nonsense-mediated decay restricts LncRNA levels in yeast unless blocked by double-stranded RNA structure. Mol. Cell. 2016; 61:379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Longtine M.S., McKenzie A. III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R.. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998; 14:953–961. [DOI] [PubMed] [Google Scholar]
- 39. Ryu H.Y., Wilson N.R., Mehta S., Hwang S.S., Hochstrasser M.. Loss of the SUMO protease Ulp2 triggers a specific multichromosome aneuploidy. Genes Dev. 2016; 30:1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ryu H.Y., Ahn S.. Yeast histone H3 lysine 4 demethylase Jhd2 regulates mitotic rDNA condensation. BMC Biol. 2014; 12:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 42. Feng J., Liu T., Qin B., Zhang Y., Liu X.S.. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kroetz M.B., Su D., Hochstrasser M.. Essential role of nuclear localization for yeast Ulp2 SUMO protease function. Mol. Biol. Cell. 2009; 20:2196–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li S.J., Hochstrasser M.. A new protease required for cell-cycle progression in yeast. Nature. 1999; 398:246–251. [DOI] [PubMed] [Google Scholar]
- 50. Ryu H.Y., Lopez-Giraldez F., Knight J., Hwang S.S., Renner C., Kreft S.G., Hochstrasser M.. Distinct adaptive mechanisms drive recovery from aneuploidy caused by loss of the Ulp2 SUMO protease. Nat. Commun. 2018; 9:5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gillies J., Hickey C.M., Su D., Wu Z., Peng J., Hochstrasser M.. SUMO pathway modulation of regulatory protein binding at the ribosomal DNA locus in saccharomyces cerevisiae. Genetics. 2016; 202:1377–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gilbert T.M., McDaniel S.L., Byrum S.D., Cades J.A., Dancy B.C., Wade H., Tackett A.J., Strahl B.D., Taverna S.D.. A PWWP domain-containing protein targets the NuA3 acetyltransferase complex via histone H3 lysine 36 trimethylation to coordinate transcriptional elongation at coding regions. Mol. Cell. Proteomics. 2014; 13:2883–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim T., Buratowski S.. Two Saccharomyces cerevisiae JmjC domain proteins demethylate histone H3 Lys36 in transcribed regions to promote elongation. J. Biol. Chem. 2007; 282:20827–20835. [DOI] [PubMed] [Google Scholar]
- 54. Sa-Moura B., Funakoshi M., Tomko R.J. Jr., Dohmen R.J., Wu Z., Peng J., Hochstrasser M.. A conserved protein with AN1 zinc finger and ubiquitin-like domains modulates Cdc48 (p97) function in the ubiquitin-proteasome pathway. J. Biol. Chem. 2013; 288:33682–33696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xiao T., Kao C.F., Krogan N.J., Sun Z.W., Greenblatt J.F., Osley M.A., Strahl B.D.. Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol. Cell. Biol. 2005; 25:637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shieh G.S., Pan C.H., Wu J.H., Sun Y.J., Wang C.C., Hsiao W.C., Lin C.Y., Tung L., Chang T.H., Fleming A.B. et al.. H2B ubiquitylation is part of chromatin architecture that marks exon-intron structure in budding yeast. BMC Genomics. 2011; 12:627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lewis A., Felberbaum R., Hochstrasser M.. A nuclear envelope protein linking nuclear pore basket assembly, SUMO protease regulation, and mRNA surveillance. J. Cell Biol. 2007; 178:813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kirmizis A., Santos-Rosa H., Penkett C.J., Singer M.A., Vermeulen M., Mann M., Bahler J., Green R.D., Kouzarides T.. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007; 449:928–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roguev A., Schaft D., Shevchenko A., Pijnappel W.W., Wilm M., Aasland R., Stewart A.F.. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001; 20:7137–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schneider J., Wood A., Lee J.S., Schuster R., Dueker J., Maguire C., Swanson S.K., Florens L., Washburn M.P., Shilatifard A.. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol. Cell. 2005; 19:849–856. [DOI] [PubMed] [Google Scholar]
- 61. Fingerman I.M., Wu C.-L., Wilson B.D., Briggs S.D.. Global loss of Set1-mediated H3 Lys4 trimethylation is associated with silencing defects in Saccharomyces cerevisiae. J. Biol. Chem. 2005; 280:28761–28765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Govind C.K., Qiu H., Ginsburg D.S., Ruan C., Hofmeyer K., Hu C., Swaminathan V., Workman J.L., Li B., Hinnebusch A.G.. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell. 2010; 39:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Srikumar T., Lewicki M.C., Costanzo M., Tkach J.M., van Bakel H., Tsui K., Johnson E.S., Brown G.W., Andrews B.J., Boone C. et al.. Global analysis of SUMO chain function reveals multiple roles in chromatin regulation. J. Cell Biol. 2013; 201:145–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rosonina E., Duncan S.M., Manley J.L.. SUMO functions in constitutive transcription and during activation of inducible genes in yeast. Genes Dev. 2010; 24:1242–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chymkowitch P., Nguea P.A., Aanes H., Koehler C.J., Thiede B., Lorenz S., Meza-Zepeda L.A., Klungland A., Enserink J.M.. Sumoylation of Rap1 mediates the recruitment of TFIID to promote transcription of ribosomal protein genes. Genome Res. 2015; 25:897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhou W.D., Ryan J.J., Zhou H.L.. Global analyses of sumoylated proteins in Saccharomyces cerevisiae - Induction of protein sumoylation by cellular stresses. J. Biol. Chem. 2004; 279:32262–32268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wykoff D.D., O'Shea E.K.. Identification of sumoylated proteins by systematic immunoprecipitation of the budding yeast proteome. Mol. Cell. Proteomics. 2005; 4:73–83. [DOI] [PubMed] [Google Scholar]
- 68. Soares L.M., Radman-Livaja M., Lin S.G., Rando O.J., Buratowski S.. Feedback control of Set1 protein levels is important for proper H3K4 methylation patterns. Cell Rep. 2014; 6:961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hannich J.T., Lewis A., Kroetz M.B., Li S.J., Heide H., Emili A., Hochstrasser M.. Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J. Biol. Chem. 2005; 280:4102–4110. [DOI] [PubMed] [Google Scholar]
- 70. Ho Y., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K. et al.. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002; 415:180–183. [DOI] [PubMed] [Google Scholar]
- 71. Zhao Q., Xie Y., Zheng Y., Jiang S., Liu W., Mu W., Liu Z., Zhao Y., Xue Y., Ren J.. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014; 42:W325–W330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wood A., Schneider J., Dover J., Johnston M., Shilatifard A.. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J. Biol. Chem. 2003; 278:34739–34742. [DOI] [PubMed] [Google Scholar]
- 73. Wyce A., Xiao T., Whelan K.A., Kosman C., Walter W., Eick D., Hughes T.R., Krogan N.J., Strahl B.D., Berger S.L.. H2B ubiquitylation acts as a barrier to Ctk1 nucleosomal recruitment prior to removal by Ubp8 within a SAGA-related complex. Mol. Cell. 2007; 27:275–288. [DOI] [PubMed] [Google Scholar]
- 74. Brownell J.E., Zhou J., Ranalli T., Kobayashi R., Edmondson D.G., Roth S.Y., Allis C.D.. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996; 84:843–851. [DOI] [PubMed] [Google Scholar]
- 75. Jenuwein T., Allis C.D.. Translating the histone code. Science. 2001; 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 76. Vitaliano-Prunier A., Menant A., Hobeika M., Geli V., Gwizdek C., Dargemont C.. Ubiquitylation of the COMPASS component Swd2 links H2B ubiquitylation to H3K4 trimethylation. Nat. Cell Biol. 2008; 10:1365–1371. [DOI] [PubMed] [Google Scholar]
- 77. Nevers A., Doyen A., Malabat C., Neron B., Kergrohen T., Jacquier A., Badis G.. Antisense transcriptional interference mediates condition-specific gene repression in budding yeast. Nucleic Acids Res. 2018; 46:6009–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jensen T.H., Jacquier A., Libri D.. Dealing with pervasive transcription. Mol. Cell. 2013; 52:473–484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.