

Abstract
Recent advances in metagenomic sequencing have enabled discovery of diverse, distinct microbes and viruses. Bacteriophages, the most abundant biological entity on Earth, evolve rapidly, and therefore, detection of unknown bacteriophages in sequence datasets is a challenge. Most of the existing detection methods rely on sequence similarity to known bacteriophage sequences, impeding the identification and characterization of distinct, highly divergent bacteriophage families. Here we present Seeker, a deep-learning tool for alignment-free identification of phage sequences. Seeker allows rapid detection of phages in sequence datasets and differentiation of phage sequences from bacterial ones, even when those phages exhibit little sequence similarity to established phage families. We comprehensively validate Seeker's ability to identify previously unidentified phages, and employ this method to detect unknown phages, some of which are highly divergent from the known phage families. We provide a web portal (seeker.pythonanywhere.com) and a user-friendly Python package (github.com/gussow/seeker) allowing researchers to easily apply Seeker in metagenomic studies, for the detection of diverse unknown bacteriophages.
INTRODUCTION
Bacteriophages, viruses that infect bacteria (phages, for short), are ubiquitous and abundant in every type of biome, and their interactions with microbial communities heavily influence microbial ecology, impact biogeochemical cycling in various ecosystems, and to a large extent, shape the evolution of cellular organisms (1–8). Recently, the development of non-culture based, metagenomic sequencing has allowed researchers to detect numerous, diverse bacteriophages in sequence data from almost every environment, further demonstrating their broad impact on the functions of microbial communities, such as, for example, animal gut, soil, and ocean microbiomes. In particular, it has been recently shown that the human gut microbiota harbors abundant bacteriophages (9) that profoundly influence human metabolism and immunity (10–12), with clear therapeutic implications (6,12,13) for diseases such as irritable-bowel syndrome and non-alcoholic fatty liver disease (14). Yet, our understanding of the viral diversity in the majority of microbial communities is limited, given that most of the microbes from such communities have not been cultivated, complicating virus discovery (15).
Metagenomic studies using high throughput sequencing technology generate ample amounts of short read sequences from prokaryotic cells in microbial communities regardless of the cultivability of cells. Hence, multiple new viruses can be discovered from metagenomic sequencing data, substantially advancing our knowledge of the virus diversity in different types of communities (16). However, to characterize habitat-specific viromes, it is essential to efficiently extract viral sequences from complex mixtures of virus and host sequences. The existing tools for the identification of phages and prophages rely on sequence similarity (17–22), gene prediction (19,20,22) or distribution of nucleotide k-mers and specific sequence signatures (21,23). Due to this knowledge-based approach, the available methods are largely limited to the detection of viruses with sequences significantly similar to those of already known viruses. Because of the high evolution rate typical of virus genome sequences, distinct groups of viruses often have little in common with previously recognized viruses, impeding their identification by sequence similarity, even using the most sensitive of the available methods for protein sequence comparison. More recently, several deep-learning based approaches have been proposed to overcome these challenges (24,25), but these rely on millions of parameters and thus might be susceptible to similar limitations. Given that only a small minority of viruses and prokaryotes have been formally described so far (26–28), identification of previously unknown major groups of bacteriophages remains an open challenge.
Here, we introduce Seeker, an alignment-free method that leverages recent advances in deep learning to detect phages. Seeker employs Long Short-Term Memory (LSTM) models, a type of Recurrent Neural Network (RNN). By contrast to other sequence learning methods, RNNs (and specifically LSTMs) maintain a long memory of sequences, and thus can identify distant dependencies within sequences, to distinguish phages from bacteria. Seeker is unbiased, alignment-free, and is not based on pre-determined sequence features (i.e. genes, repeats, k-mers or sequence signatures), but rather, is trained to read through a complete DNA sequence, weighing the likelihood of it belonging to a phage genome. This makes Seeker a fitting choice for learning DNA sequence context including long term dependencies and subtle patterns, with more power than any method that explicitly extracts motifs or relies on direct sequence similarity. The number of parameters used by Seeker is relatively small (either 152 or 212 parameters, dependent on the model version), precluding memorization and overfitting, thus, the performance of Seeker is robust and more stable compared to other approaches. Seeker is trained on segments and thus performs better on shorter sequences, which is critical for application to metagenomic data. In addition, Seeker does not require substantive computational resources, its runtime is linear with respect to the input length and it is substantially faster than existing methods. To demonstrate the utility of Seeker on specific test cases, we used this method to identify previously unknown phages from human and sheep gut microbiomes as well as environmental metagenome data. Some of the detected phages are highly divergent from known phage families and at least one might become the founder of a distinct phage family or a higher taxon.
We provide a web portal (seeker.pythonanywhere.com) that we will maintain for at least 3 years, and a python package (github.com/gussow/seeker) for the application of Seeker and visualization of the results. This work demonstrates that, by limiting model size and conducting a careful training process, deep learning tools can overcome the limitations of alignments and gene comparisons for the identification of new phages, and has the potential to facilitate other complex sequence prediction tasks.
MATERIALS AND METHODS
Curation of phage and bacterial genomes
Phage and bacterial complete genomes were obtained for two training steps (Figure 1A). All analyzed DNA sequences were consecutively segmented into non-overlapping 1 kilobase pairs (kb) sequences as this is the recommended upper bound for input length to LSTM models (29). Smaller or overlapping segments would substantially increase both training and testing runtimes, and overlapping segments could lead to excessive sequence duplication. As bacteria have much larger genome than phages, they yield substantially more segments, and using a similar number of phage and bacteria genomes would yield an imbalanced set of segments that would bias the training toward the bacterial set. As a result, the bacterial training set size was set to match the phage training set that was used for training in each step.
Figure 1.

Seeker: a machine learning method for phage genome identification. (A) Cartoon representation of the model and training pipeline. (B) Scatter plot showing the test scores of the model trained with an embedding layer (Python model, x-axis) versus that of the model using a sequence input layer (Matlab model, y-axis). (C) ROC (Receiver Operating Characteristic) classification curves predicting phage vs. bacterial genome on the left-out test set.
For the first training step, we sought to curate a set of high-confidence phage and bacterial genomes. All sequences used are publicly available and were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/), with the accessions available in Supplementary Table S1. The positive set (phage genomes) was obtained from RefSeq (30) and consisted of 80% of all unique RefSeq phages (n = 2232 phages, Supplementary Table S1). The remaining 20% of phages were removed to maintain an independent set for testing that would not be used during any step of training. For the negative set (bacteria genomes), we curated a high-confidence, non-redundant set from the reference bacteria set (n = 75 bacteria, from the ncbi-genome-download project). From the latter, all instances of known phage and prophage sequences were removed using exact match of at least 100 nucleotides with any phage in our positive set or in phage sequences obtained from the PHASTER database (20). This was done to enable a high-confidence training step where the bacterial sequences were free from any prophage contamination. These data were sorted by difficulty as described below, and training was performed on the bacterial and phage segments until the phage segments were exhausted. In total, this training set consisted of n = 80000 phage and bacteria fragments. Although there may be some duplications in this set, this does not present a confounder to deep learning methods (31).
For the second training step, in order to expand the positive set, we obtained an additional larger set consisting of all annotated complete genome phages found in an exhaustive search of online databases (https://www.ncbi.nlm.nih.gov/, https://www.ebi.ac.uk/genomes/phage.html). For bacterial genomes, of which there are many more instances in the data than phage genomes, we randomly sampled a single representative per bacterial genus, and the chromosome(s) and plasmids from that representative were included (Supplementary Tables S2 and S3). The genomes were all downloaded from NCBI (n = 1269 bacteria; 13 443 phages). 240 bacterial and 7375 phage genomes were used for the second training step (yielding 250000 phage and bacteria fragments), 98 bacterial and 2155 phage genomes were used for validation, and 931 bacterial and 3931 phage genomes were left out for testing and never included in training.
Designing and training Long Short-Term Memory networks
Seeker is based on Long Short-Term Memory (LSTM) networks, a type of Recurrent Neural Network (RNN) that take a sequence as input for various prediction tasks (32). RNNs are a class of neural networks that are principally applied to sequences, as they rely on previously calculated outputs or states while computing the current hidden state. This design allows RNNs to use distant information within a sequence and learn distant dependencies that are then used for prediction. RNNs compute the hidden state of sequence position 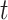 , denoted
, denoted 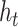 , by computing the non-linear function (usually the hyperbolic tangent) of the weight matrix
, by computing the non-linear function (usually the hyperbolic tangent) of the weight matrix 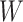 when applied to the hidden state of sequence position
when applied to the hidden state of sequence position 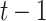 (
(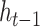 ) concatenated with the input from the current position in the sequence,
) concatenated with the input from the current position in the sequence, 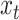 . LSTMs are a type of gated RNN that are designed to avoid the vanishing and exploding gradient problems, and thus explicitly control the contribution of the previous position in the sequence to the learning at the current position (33,34). Thus, LSTM can maintain a longer memory, pass relevant information to the next cell and forget the less relevant information in the process. The gates are termed
. LSTMs are a type of gated RNN that are designed to avoid the vanishing and exploding gradient problems, and thus explicitly control the contribution of the previous position in the sequence to the learning at the current position (33,34). Thus, LSTM can maintain a longer memory, pass relevant information to the next cell and forget the less relevant information in the process. The gates are termed 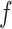 , the forget gate, designed to forget irrelevant information from previous states (sequence positions);
, the forget gate, designed to forget irrelevant information from previous states (sequence positions); 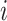 , the input gate, controlling how much of the current input
, the input gate, controlling how much of the current input 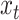 is considered;
is considered; 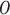 , the output gate, controlling the output that will be passed to the next hidden state; and
, the output gate, controlling the output that will be passed to the next hidden state; and 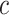 , which connects the gates to produce a cell state.
, which connects the gates to produce a cell state.
All LSTM networks used in this work are sequence-to-label LSTMs, using a single LSTM cell with 5 hidden units, with a softmax and classification layer and were trained using Adam optimizer (35), where the maximal epoch for training is set to 100. The mini batch size used for each training iteration was set to 27, with a standard gradient-clipping threshold set to 1. We maintained relatively small models with a limited number of parameters (either 152 or 212 parameters, dependent on the model version), to prevent overfitting the data, and to preclude memorization of sequences. By limiting the number of parameters to train to the weights and biases of five hidden units, the input layer (either sequence input layer or word embedding layer), and the softmax classification layer, we ensured that both models had <250 parameters to train, hence substantially reducing the risk of overfitting associated with a large number of parameters, as well as reducing the training and testing times of the models. The maximal epoch for training was initially set to 100, and was not increased because we observed that additional epochs were not improving performance on the validation set.
The 1 kb segments were used as input for two different types of layers resulting in different model architectures, which were trained to input data into the LSTM layers:
Python Keras word embedding layer, for which the DNA sequence input was transformed into integers (‘A’ = 1, ‘T’ = 2, ‘C’ = 3 and ‘G’ = 4), with the vocabulary size parameter set to 5 and the input length defined to 1000.
Matlab sequence input layer, using channel-wise normalization for zero-center normalization, for which the DNA sequence input was transformed using one-hot encoding (‘A’ = 1000, ‘T’ = 0100, ‘C’ = 0010 and ‘G’ = 0001). This model was converted into a Keras model once the training was completed.
The models performance was always assessed over complete sequences, regardless of input sequence size, by assigning each genome in a test set the average score predicted by the model across all of its 1 kb segments.
Ensuring independence between the training and test set, as well as restricting the training set and number of model parameters used, precludes memorization of strains that are used for training. We implemented these restrictions to enable a correct assessment of the approach and identification of new, divergent phages. The training, validation and test performance curves are provided in Figure 1B and in Supplementary Figure S1.
Sorting the training data by difficulty
For both the first and second training steps, the data were sorted by training difficulty (from easy to hard), to speed up the convergence of the training process and hence reduce the risk of overfitting (36). For the first training step, we approximated the difficulty of a training sample (phage or bacterial) by the average area under the curve (AUC) of the ROC obtained with LSTMs trained on its genome. We hence trained LSTMs from randomly chosen combinations of phage and bacterial genomes in the high-confidence training, such that each phage or bacterial genome was used to train five models, where in each iteration, the performance was evaluated on the rest of the training set. Then, each phage or bacterial sequence in the set was assigned a score indicating the average performance of a network trained using it; the training data was sorted by these scores, and given as input to the LSTMs for the first training phase.
For the second training step, each sample was assigned a value indicating the average performance of the LSTM networks generated from training step 1 on all its 1 kb segments. The step 2 training data was ordered by the performance score and then given as input to the LSTMs for the second training phase.
Evaluating Seeker for phages infecting bacteria of different families
We evaluated the performance of Seeker for phages infecting different bacterial families that represent different phyla. The performance of Seeker was found to be robust for most groups of hosts (Supplementary Figure S2). We additionally provide a similar table (Supplementary Table S2) showing the performance of Seeker for bacteria from different families. Seeker scores for each bacterium in the training and test data, which contain a representative of each bacterial genus, are also provided to show which bacteria are more likely to be falsely predicted as phages by Seeker.
Comparison to VirFinder, VirSorter, DeepVirFinder, PPR-Meta and VIBRANT
We compare the performance of Seeker to those of 5 previously developed methods for phage identification. The key features of these methods are summarized in Table 1.
Table 1.
Comparison of models employed for phage identification by Seeker, VirFinder, VirSorter, DeepVirFinder, PPR-Meta and VIBRANT
| Name | Model | Parameters Count | Description |
|---|---|---|---|
| Seeker | Single LSTM trained with Python or MATLAB | 157 (Python model), or 212 (MATLAB model) | Segments the genome to 1K fragments, and assigns the average score assigned by the LSTM to segments. Scores above 0.5 were considered as phage prediction. |
| VirFinder (23) | Three logistic regression models | Each model has 10890 parameters, totaling 32,670 parameters | Searches for multiple K-mer signatures that were frequently observed in known viral sequences. Scores are between 0 and 1, and scores above 0.5 were considered as phage prediction. |
| DeepVirFinder (25) | Four convolutional neural network models | Each model uses 1043001 parameters, totaling 4172004 parameters | Convolutional neural networks that extract motif intensities in sequences and then used them as features for prediction. |
| PPR-Meta (24) | Three convolutional neural network models used for phage, plasmid and chromosome | Each model uses 564632 parameters, totaling 1693896 parameters | Convolutional neural networks for different sequence lengths, for long sequences segments the genome into 1.2 kb fragments and reports average. |
| VirSorter (22) | Protein similarity | Not applicable. | Predicts proteins in sequences and detects similarity to known viral proteins. Predicted phages assigned with category scores 1 or 2 were considered as phage prediction. |
| VIBRANT (37) | Hybrid protein similarity and multi-layer perceptron approach | The multi-layer perceptron uses 63363 parameters | First extracts protein signatures based on HMM hits and then applies multi-layer perceptron to those signatures. |
In contrast to Seeker, all other approaches that are based purely on machine and deep learning (VirFinder, DeepVirFinder, PPR-meta) use between tens of thousands to millions of parameters (Table 1). The sizes of these models are larger than the number of independent phage training samples, which can lead to overfitting, and can show perfect training performance on randomly shuffled data (38). Moreover, all three approaches divide their training and test sets by date of submission. Because highly similar and even identical genomes can be submitted across numerous years, this split does not ensure independence between the training and test sets. Consequently, in practice, these methods used nearly all available data for training, and it is therefore difficult to evaluate whether these approaches can predict independent and divergent phages and bacteria, or whether they were simply overfit to the current deposited data. Given that only a small fraction of phages and bacteria have been identified and annotated so far (26–28), it is crucial for prediction methods not to be overfit to the current data, and to be able to predict novel, divergent genomes. We therefore constructed four test tests, with different levels of difficulty and divergence from existing databases of phages and bacteria, and used these sets to evaluate the performance of all approaches.
The following test sets were constructed to compare Seeker to the other approaches (Supplementary Table S3):
154574 viruses assigned with a bacterial host from the IMG/VR database were downloaded (https://img.jgi.doe.gov/cgi-bin/vr/main.cgi), and the performances of the six approaches were compared for different source environments.
Short phage and bacteria sequences ranging 1000–5000 bp were downloaded from NCBI, and the bacterial sequences were downsampled by quantile for a total of 4223 phage sequences and 6854 bacteria sequences. These sequences constitute a test set that mimics typical metagenomic data. The performances of the six approaches on these sequences were compared overall and across ranges of sequence length.
Phage sequences that were submitted to NCBI after 2018 and were not used to train Seeker. These sequences were used because more recent phage genomes may not have been available for training the other approaches, and because phages tend to evolve much faster and diverge to greater extents than bacteria, and therefore are likely to constitute a test set that is less similar to the training data used for the other approaches. We examined different families of phages, and included families with more than 5 phage sequences of length >750 bp submitted after 2018, yielding 2273 genomes from six families. All six methods were applied to these genomes, and the detection (true positive) rates were calculated.
Finally, we obtained shotgun-sequencing datasets from NCBI, considering that the other approaches were trained on full genomes as opposed to shotgun sequences, thus providing a dataset with less similarity to the sequences used for training by these methods (all annotated shotgun phage sequences [n = 419] and 1042 unclassified shotgun bacterial sequences added to NCBI after 2017 were used to reduce class imbalance in this set). We applied all six approaches to these genomes and calculated true and false positive rates and balanced accuracies.
For each phage in this divergent dataset, we additionally evaluated the sequence similarity against phages in ‘nr’ using blastn. We show that Seeker does not perform better for phages that are more similar to existing phages in nr, whereas VirFinder, VirSorter and DeepVirFinder assign significantly higher scores to the phages that are more similar to phages in nr (Supplementary Figure S4), supporting the expectation that these methods are highly reliant on sequence similarity to previously identified phages.
To compare the runtime of Seeker to those of the five other approaches, we downloaded a bacterial genome from NCBI (NC_011750.1) and created 16 segments from its nucleotide sequence, starting from the first base and continuing in steps of 250000, so that the first segment was 250 kb, the second one was 500 kb, and so forth, until the 16th segment (4000 kb). Each segment was used as input to each of the methods, and the number of CPU seconds for each run was recorded.
Identification and characterization of unknown bacteriophages
To identify unknown bacteriophages with Seeker, we applied Seeker to unclassified metagenomic data from four projects. We searched for circular sequences (those with a direct overlap > 15 bp at the genome termini) of length >30 kb, assigned with high Seeker scores to select candidates that are most likely to be phages per Seeker's assessment (top 10% of each database and larger than 0.7), yielding 367 contigs in total (33, 61, 203 and 68 from PRJEB22623, PRJEB25190, PRJNA504765 and PRJNA577476, respectively, downloaded from NCBI, Supplementary Table S4). To quantify the proportion of unknown phages within these datasets, we ran six frame translation on each contig, ignoring stop codons, and PSI-blasted (39) the resulting proteins against CDD (40) and PVOG (41) with E-value cutoff of 0.1. in addition, we applied BlastX to each contig, with E-value cutoff of 1E-4. From these, hits to terminase, capsid and portal proteins were retained, where 311 contigs (85%) had a hit to at least one of these three. For each identified protein, the maximum percent of identity was obtained using BlastX against NR (Supplementary Table S4).
The resulting sequences were then filtered to include only those with <1% overlap with existing phage sequences (using BlastN, query coverage less than 1%). The protein sequences of these candidates were predicted using Prodigal (42) (v2.6.3) with the parameter set for metagenome mode (-p meta). The protein sequences of these candidates were compared to the phage subset of the NR protein database (accessed December 2020) using BlastP. We filtered for candidates in which fewer than 50% of the predicted proteins had BlastP hits to the proteins in this database and less than 33% of the proteins had hits to a single phage family (with E-value < 1e–6). The candidates that met these criteria were taken to represent ‘unknown’ phages and five of these were annotated and characterized (Supplementary Data 1–3). In addition to these, we selected 8 divergent phages that were not detected with VirFinder or VirSorter (some of which are highly divergent), and included annotations for some of their key protein sequences (Supplementary Data 4, 5).
Each predicted protein sequence of the candidate phages was used as a query for psi-blast (39) against the NR database (accessed December 2019) to construct a multiple sequence alignment (MSA). The resulting MSA was used as a query against the NCBI CDD database (accessed 12/2019 (40)) with an E-value cutoff of < 0.1. Additional annotations were generated with hhblits (43), using the MSAs constructed above as queries to search the PDB database clustered to 70% maximum pairwise sequence identity (downloaded from http://wwwuser.gwdg.de/∼compbiol/data/hhsuite/databases/hhsuite_dbs/, accessed December 2019).
Amber-readthrough is a genetic code in which the ribosome does not stop at the stop-codon UAG, but rather continues the process of protein synthesis. This is usually accomplished via a suppressor tRNA that recognizes UAG and allows the readthrough to occur, as documented previously for several phages from gut metagenomes (44). Two of the novel phages reported here employ amber-readthrough. For these phages, the initial prediction of protein-coding genes was performed using the standard genetic code. In both cases, the following was observed: (a) the homolog of the large terminase subunit (TerL) was small and contained a TAG stop codon but aligned to the full length of TerL sequences from other phages when the stop codon was ignored; (b) when translating with an amber-readthrough genetic code, the size of most of the genes substantially increased, enabling us to annotate genes for which otherwise no homologs were detected. Given these lines of evidence, these phages are assumed to be using an amber-readthrough genetic code, most likely, for translation of the late genes as previously demonstrated (44). The tRNAs were annotated in the amber-readthrough genomes using tRNA-scan-SE (45) (v2.0) with a bitscore cutoff of 35.
In addition, we annotated eight phages from the four metagenomic projects, which were identified by Seeker but missed by most other approaches (Supplementary Data 4, 5, Supplementary Figures S6 and S7).
For each candidate phage and its relatives, a phylogenetic tree was constructed based on the predicted terminase large subunit protein sequence. To create an alignment, terminase sequence was run against NR using PSI-Blast, and sequences with e-value <0.01 were retrieved. Sequences were then clustered at 70% identity using mmclust (46). The resulting sequences were aligned using muscle (47), and then filtered to include those with <50% gaps. The resulting alignment was used to construct a tree using FastTree, with default parameters (48).
RESULTS
Seeker: a method to differentiate phage genome sequences from bacterial ones
Seeker employs LSTM networks, a type of neural network that is structured to learn order dependence in prediction problems (32). Conceptually, LSTM networks process DNA by looking at each position in the sequence and passing information from one step in the network to the next, thus allowing information to persist. This persistence allows LSTMs to learn subtle patterns in the data which are inaccessible to other existing machine learning methods and contribute to the classification of the input sequences.
To train the model to differentiate phage from bacterial sequence, the genomes in the dataset were segmented into fragments of 1 kb which were then converted into vectors to be used as the input for the LSTM models (see Materials and Methods for details, Figure 1). We required a set of sequences from each category (phages and bacteria). Seeker was trained via two steps. When combining all training and testing data, the cumulative dataset consists of n = 15675 phages and n = 1344 bacteria, with roughly 330000 training segments for both phages and bacteria (Figure 1A). For the first training step, we curated high-confidence data of positive (phages, n = 2232) and negative (bacteria, n = 75) samples. The relatively small subset of the bacterial genomes was used to make a balanced training set, with approximately equal numbers of samples (1 kb segments) from bacteria and phages, given that bacterial genomes are much larger and hence yield many more fragments (see Materials and Methods for details; Figure 1, Supplementary Table S1). To maximally speed up the convergence of the training process, the input was ordered by training difficulty (36) (see Materials and Methods for details). Following training the models on the high-confidence set, we expanded the training to include a more diversified set of bacteria and phage strains (n = 13443 phages, n = 1269 bacteria; see Materials and Methods for details; Supplementary Table S1). At this point, a test set was set aside from these data for method assessment, and the remaining genomes were divided into training and validation sets (Figure 1). Thus, the sets used in constructing the method are training and validation, and evaluations were performed on the separate test set. We assessed the method against the test set, by assigning each genome in the test set the average score predicted by the model across all of its 1 kb segments, and found that the classification scores assigned by the two models are strongly correlated (Pearson's 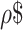 = 0.95), and can distinguish viral from bacterial sequences with high confidence (Python model AUC = 0.91, Matlab model AUC = 0.92, Figure 1, Supplementary Table S1). Bacterial plasmids were included in the bacterial test sets and showed a slightly lower performance compared to bacterial chromosomes (TNR = 0.79, Supplementary Table S1).
= 0.95), and can distinguish viral from bacterial sequences with high confidence (Python model AUC = 0.91, Matlab model AUC = 0.92, Figure 1, Supplementary Table S1). Bacterial plasmids were included in the bacterial test sets and showed a slightly lower performance compared to bacterial chromosomes (TNR = 0.79, Supplementary Table S1).
We next compared Seeker to existing approaches for phage genome identification VirSorter (22), VirFinder (23), DeepVirFinder (25), PPR-Meta (24) and VIBRANT (37). For the purpose of training, other approaches divided the genomes into training and test sets based on the year of deposition in NCBI databases. Because closely related, highly similar viruses and bacteria are deposited across different years, this does not ensure independence between training and test sets. Thus, neither the test sets used by other approaches nor the test set used to formally evaluate Seeker (Figure 1C) would be appropriate for comparison. To produce an unbiased comparison of these methods, we evaluated their performances across four different test sets, in order to compare the strengths and weaknesses of each method. First, to compare the ability of all approaches to recover environmental phages that are highly similar to previously identified ones, we used phage genomes from the IMG/VR database, across different environment categories. Because the IMG/VR dataset was composed by searching for explicit genome similarity to known phages, this is an example of a collection of familiar phages and their close relatives. Therefore, unsurprisingly, all approaches perform well on this data, but the most recent approaches, namely, Seeker, PPR-Meta, DeepVirFinder and VIBRANT, show the best performances (Figure 2A and B). Second, to evaluate the ability of these methods to distinguish phage sequences from bacterial ones in short contigs, which typically comprise metagenomic data, we downloaded short sequences of phages and bacteria (1K–5K of length) from NCBI (Supplementary Table S3). Seeker shows the best accuracy on this dataset (Figure 2C), whereas approaches that utilize sequence similarity (VirSorter and VIBRANT) predictably are the least accurate on short sequences due to their low true positive rate on this type of data (Figure 2C). Seeker is also the most stable across different ranges of sequence length (Figure 2D), whereas the rest of the approaches tend to show better performance for longer sequences, especially, those methods that utilize sequence similarity (VirSorter and VIBRANT, Figure 2D).
Figure 2.

Comparison of the performance of Seeker to those of other approaches for the IMG/VR dataset and for short phage and bacterial sequences. (A) True positive rates of Seeker (yellow), VirFinder (Blue), VirSorter (red), DeepVirFinder (green), PPR-Meta (purple) and VIBRANT (brown) for phages in the IMG/VR data set. (B) Overall true positive rates of the nine data points in panel (A); the error bars show standard deviations of the rates; the color code is the same as in panel A. (C) Balanced accuracy, true positive rate (TPR), precision (positive predictive rate) and true negative rate (TNR) of the six methods, for short sequences mimicking metagenomics data; the color code is the same as in panel A. (D) Balanced accuracy of the six methods for short sequences mimicking metagenome projects, for three ranges of sequence lengths; the color code is the same as in panel A.
Third, we required a test set that was not seen during the training of any of the methods. Thus, we obtained environmental sequences from six phage families that were added to the NCBI databases after 2018, and therefore, were not represented in the training datasets used to train any of the previous methods (Supplementary Table S3). Applied to these phage sequences, Seeker performed better than any of the other five approaches (Seeker overall True Positive rate (TPR) = 0.90, VirFinder TPR = 0.79; VirSorter TPR = 0.57; DeepVirFinder TPR = 0.78; PPR-Meta TPR = 0.7; VIBRANT TPR = 0.52, Figure 3A and B). Furthermore, Seeker showed a more stable performance with less variance compared to other approaches (Figure 3B).
Figure 3.

Performance of Seeker compared to those of other approaches on divergent sequence datasets. (A) The true positive rates of Seeker (yellow), VirFinder (Blue), VirSorter (red), DeepVirFinder (green), PPR-Meta (purple) and VIBRANT (brown) for environmental sequences from six bacteriophage families deposited in NCBI databases after 2018. (B) The overall true positive rates for the six data points in panel (A); the error bars show the standard deviations of the rates; the color code is the same as in panel A. (C) The balanced accuracy, true positive rate (TPR), precision (positive predictive rate) and true negative rate (TNR) for the six approaches, on the shotgun sequencing test set; the color code is the same as in panel A. (D) The CPU times (seconds, y-axis) for Seeker (yellow), VirFinder (Blue), VirSorter (red), DeepVirFinder (green), PPR-meta (purple) and VIBRANT (brown) depending on the input size (x-axis).
Fourth, to obtain a test set with even less similarity to the sequences used for training, we tested all three methods on shotgun-sequencing datasets from the NCBI (n = 419 phages, n = 1042 bacteria, see Methods, Supplementary Table S3d). We found that Seeker outperformed the other methods (Figure 3C). In addition, Seeker scores on phages in this dataset were not correlated with contig length, in contrast to VirFinder, VirSorter and DeepVirFinder (Supplementary Figure S3). As this is the most divergent dataset examined, we further evaluated whether the performance of different approaches was better for phages with higher sequence similarity to known ones. We found that the scores assigned by Seeker were not higher for phages that are more similar to those in existing databases, whereas the scores obtained with the other approaches that had non-random true positive rate (VirFinder, VirSorter DeepVirFinder) were significantly higher for more familiar phages (Supplementary Figure S4). Together, these results indicate that Seeker is stable and not confounded by contig length, and is able to detect phages that are divergent from those that were seen during training. In addition, Seeker is substantially faster than all existing approaches, and its runtime is linear with respect to the input length (Figure 3D).
Using Seeker for phage discovery
Encouraged by the results of Seeker testing against diverse sets of known phages, we used this method to search metagenomic sequence datasets for previously undetected phage genomes. At this stage, we sought to practically demonstrate Seeker's utility to detect novel phages with limited similarity to known ones in metagenomic data sets, which we envision as the most common use for Seeker. We filtered four metagenomic sequencing projects for circular contigs with a high Seeker score, for a total of 367 candidate phage genomes (Supplementary Table S4). Each candidate was then searched for the protein sequences of three phage markers, i.e. protein-coding genes that are represented in all known tailed phages (41), namely, terminase (large subunit), capsid and portal proteins (see Methods for details). We found that, for 311 of the candidates (85%), we were able to detect at least one of these markers (Figure 4A, Supplementary Table S4), most often, the terminase, the most conserved of the three protein markers sequence-wise. The remaining candidates are either not phages and therefore false positives, or contain extremely divergent forms of these markers. In the majority of the candidates where the markers were detected, the sequences of the marker proteins are substantially dissimilar from their closest known homologs, with <50% identity (Figure 4B and C), further indicating the novelty of these phages detected by Seeker. A similar analysis applied to these four metagenomic projects without filtering for circular contigs resulted in equivalent performances (Supplementary Table S5, Figure S5).
Figure 4.

Terminase, capsid and portal proteins in candidate novel bacteriophages. (A) The Venn diagram shows, for each metagenomic project analyzed (marked with different shapes), the percentage of circular contigs identified by Seeker with different combinations of the three phage markers detected (color-coded). (B) Maximum percent identity across all three markers (terminase, capsid, and portal) and their closest known homologs, and that of the terminase separately, for each project. (C) The proportion of proteins with less than 50% identity to their closest known homologs for each of the three markers, and across all of the three markers combined (terminase, capsid and portal), shown separately for the four analyzed projects.
We explored in detail five of the unknown phages discovered by Seeker in this set, with an explicit focus on the phages that bore the least sequence similarity to known phages (see Materials and Methods for details), starting with two phages detected in the gut metagenomes. The first of these phages (OLNE01000568.1), which we refer to as Flitwick, was detected in a human gut metagenome. Flitwick has a 33716 bp circular genome, with 25 predicted genes, and uses an alternative genetic code, with readthrough of amber stop codons. This could, in part, explain why this phage has not been previously identified. We annotated 13 of Flitwick's genes (52%, Figure 5A, Supplementary Data 1–3), in particular, several encoding structural proteins including the major capsid protein and the large terminase subunit. We additionally detected four tRNA genes in the phage genome one of which is predicted to be the suppressor of the amber stop codon. The position of Flitwick in the phylogenetic tree of the large terminase subunit shows that this is a distinct member of the Siphoviridae family (Figure 5B).
Figure 5.

Novel bacteriophages identified by Seeker in gut metagenomes. (A) Annotated gene map of the Flitwick phage. (B) Phylogenetic tree of large terminase subunit for the relatives of the Flitwick phage. (C) Annotated gene map of the Regulus phage. (D) Phylogenetic tree of large terminase subunits for the relatives of the Regulus phage.
The second phage (ODAI012083904.1), which we refer to as Regulus, was detected in sheep rumen metagenome (Methods). Regulus has a 432079 bp circular genome and is thus a previously unknown ‘jumbo’ phage (49), with 554 predicted genes, of which we were able to annotate 127 (23%). Regulus also uses an amber-readthrough genetic code. In the phylogenetic tree of the large terminase subunits, Regulus forms a distinct branch in the Myoviridae family (Figure 5D).
Identification of these phages with Seeker illustrates its ability to detect phage sequences that are distantly related to phages that were seen during training, and additionally, demonstrates that Seeker does not depend on the genetic code used by a phage.
We next explored in detail 3 of the environmental metagenome phages detected by Seeker, all of which are divergent from any known phage family. The first of these (SDBT01001083.1), which we named Ignotus, has a 46652 bp circular genome with 88 predicted genes, of which 17 (19%) could be annotated (Figure 6A). The predicted terminase and capsid protein are too divergent to be reliably aligned with the other phage terminase or capsid proteins (although recognized at a statistically significant level), and therefore, we were unable to reconstruct a phylogenetic tree (Supplementary Data 1–3). Thus, Ignotus will, probably, become the founder of a distinct phage family or a higher taxon.
Figure 6.

Novel bacteriophages identified by Seeker in environmental metagenomes. (A–C) Annotated gene maps of the phages Ignotus, Alastor and Wulfric, respectively. (D, E) Phylogenetic trees constructed from the large terminase subunits of the phages Alastor and Wulfric, respectively.
The second phage in this set (WNFG01000004.1), named Alastor, has a 164 887 bp circular genome with 223 predicted genes, of which 68 (31%) could be annotated (Figure 6B, Supplementary Data 1–3). The major capsid and portal proteins of this phage are moderately similar to proteins in a subset of the phages in the family Herelleviridae, but its terminase is highly divergent from known terminases (Figure 6D). The last phage we analyzed from this set (WNGI01000014.1), named Wulfric, has a 103078 bp circular genome with 133 predicted genes, of which 43 (32%) could be annotated (Figure 6C, Supplementary Data 1–3). Phylogenetic analysis of the large terminase subunits shows that Wulfric is a distinct member of the family Podoviridae (Figure 6E).
Additionally, we discovered eight new phages from the four metagenomic projects that were identified by Seeker but not by most of the other approaches (Supplementary Figures S6 and S7 and Supplementary Data 4,5). Phylogenetic trees for the predicted terminase sequences of this set indicated that five phages formed deep branches within Caudovirales (namely, WNGN01000162.1, SDBU01000213.1, SDBT01001087.1, WNGL01000548.1 and OLNE01000281.1). Of the remaining three, for one, we were able to identify the portal and tail proteins, but not the terminase (SDBT01001081.1). The terminases of the remaining two phages (SDBT01000149.1 and SDBT01000023.1) were too divergent to be reliably aligned with other phage terminase, and therefore, we were unable to reconstruct a phylogenetic tree for these cases. Conceivably, these highly diverged phages represent new phage groups, perhaps, with a family rank.
DISCUSSION
Bacteriophages play vital roles in nearly every ecosystem on earth and, through their presence in microbiomes, directly impact human health. Metagenomic sequencing has brought about a new era of bacteriophage discovery, where the crucial hurdle is the ability to extract viral sequences and discover unknown bacteriophages from a large pool of metagenomic sequences. Existing methods, which mostly detect phage sequences based on direct similarity to the phages present in the current databases, are often inadequate for detection of phages distantly related to the known ones, and are slow when applied to long sequences and large datasets.
Neural networks are often described as black boxes. This is due to their structure, which includes a large number of parameters, and their use of non-linear functions that transform the input into an uninterpretable numeric form. Nevertheless, recent advances in deep learning have demonstrated the enormous power these approaches can wield in detecting otherwise opaque patterns and trends in complex datasets (50). Here, we utilize LSTM neural networks that, to our knowledge, have not been previously employed to detect the origin of a DNA sequence, to enable alignment-free detection of viral sequences from large-scale sequencing data. These LSTM networks, despite a limited number of parameters and training data, are able to detect implicit, long-range patterns within the data. This feature is further demonstrated by the detection of previously unknown phages using Seeker. Some of the discovered phages are highly divergent from known phage families and might become the founders of distinct phage families (Figures 5 and 6, Supplementary Figures S6 and S7).
Like any computational approach, Seeker is not devoid of limitations. First, although the overall performance is reliable and robust, it does not perfectly distinguish phages from bacterial genomes, and some bacteria and phages are misclassified. To facilitate estimation of where Seeker could fail, we provide the complete table of Seeker scores for bacteria in the training and test sets which contain a representative from every bacterial genus (Supplementary Table S1d). In addition, we analyzed Seeker's performance across phages infecting multiple bacterial families representing different phyla and found that the performance of Seeker is robust with respect to the phage and host diversity (Supplementary Figure S2, Table S2). Seeker was not trained on eukaryotic DNA and cannot be used to detect eukaryotic contamination in metagenomic sequence data. Eukaryotic sequences might be misidentified as phages, therefore users suspecting eukaryotic contamination should take appropriate steps to filter out potential eukaryotic sequences. Neither was Seeker trained to identify prophages within bacterial genomes, and its ability to do so has not been tested. Developing a fast and reliable method to detect prophages is desirable, but expanding Seeker into an alignment-free approach for prophage identification would be highly challenging. Such an expansion would require explicit training on prophage sequences, along with the development of a sophisticated method to scan bacterial genomes and to accurately define the threshold scores to distinguish prophage sequences from the surrounding bacterial sequences, with good true positive and true negative rates. For these reasons, we expect a follow-up to this work, incorporating a version of Seeker as the first filtering step, with a second, reference-based step, will be valuable to enhance the speed and accuracy of prophage identification.
A comparison of Seeker with five methods for phage identification in sequence databases, VirSorter (22), VirFinder (23), DeepVirFinder (25), PPR-Meta (24) and VIBRANT (37), demonstrated a more robust, more reliable and much faster performance of Seeker on diverse test sets. Our comparisons demonstrate that most approaches perform well on datasets containing familiar phages with high levels of similarity to known phages (Figure 2A and B). However, for large metagenomic sequence data, the long runtime and computational requirements of some approaches (Figure 3D) lead to practical limitations, as it may take days or even weeks along with considerable computational resources to analyze large data sets. In addition to speed, a major advantage of Seeker is that it maintains a high level of performance when applied to viral sequences with little similarity to those seen during its training and thus is well suited to discover new groups of phages (Figure 3A–C). Importantly, VirSorter and VIBRANT offer annotation of virus genomes, in addition to prediction, which is valuable and can compensate for the long runtime for some cases.
Seeker is freely and publicly available, as a webtool (seeker.pythonanywhere.com), a command-line tool and a Python package (github.com/gussow/seeker). Researchers can easily utilize any of these options to process their metagenomic datasets and rapidly discover previously unknown phages. Given its ability to detect a wide diversity of bacteriophages, we expect that widespread application of Seeker leads to the discovery of numerous phages, some of which would represent distinct families or even higher taxa in the forthcoming new phage taxonomy (51). More generally, this work demonstrates that LSTM neural networks can learn long-term dependencies within DNA sequences and thus can efficiently tackle tasks that are not easily amenable to standard techniques based on explicit sequence similarity. Future studies are warranted to evaluate the approach developed here for other sequence categories.
DATA AVAILABILITY
The code for the Seeker Python package and for training Seeker is publicly available (https://github.com/gussow/seeker). The training and testing data accession are available in Supplementary Table S1.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Kira S. Makarova for helpful discussions. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Contributor Information
Noam Auslander, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
Ayal B Gussow, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
Sean Benler, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
Yuri I Wolf, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
Eugene V Koonin, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Library of Medicine at the NIH. Funding for open access charge: US Department of Health and Human Services, Intramural fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Fuhrman J.A. Marine viruses and their biogeochemical and ecological effects. Nature. 1999; 399:541–548. [DOI] [PubMed] [Google Scholar]
- 2. Wommack K.E., Colwell R.R.. Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000; 64:69–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Edwards R.A., Rohwer F.. Viral metagenomics. Nat. Rev. Microbiol. 2005; 3:504–510. [DOI] [PubMed] [Google Scholar]
- 4. Rohwer F., Thurber R.V.. Viruses manipulate the marine environment. Nature. 2009; 459:207–212. [DOI] [PubMed] [Google Scholar]
- 5. Rodriguez-Valera F., Martin-Cuadrado A.B., Rodriguez-Brito B., Pašić L., Thingstad T.F., Rohwer F., Mira A.. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009; 7:828–836. [DOI] [PubMed] [Google Scholar]
- 6. Reyes A., Semenkovich N.P., Whiteson K., Rohwer F., Gordon J.I.. Going viral: next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012; 10:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gilbert J.A., Blaser M.J., Caporaso J.G., Jansson J.K., Lynch S. V., Knight R.. Current understanding of the human microbiome. Nat. Med. 2018; 24:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Busby B., Kristensen D.M., Koonin E. V.. Contribution of phage-derived genomic islands to the virulence of facultative bacterial pathogens. Environ. Microbiol. 2013; 15:307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hurwitz B.L., U’Ren J.M., Youens-Clark K.. Computational prospecting the great viral unknown. FEMS Microbiol. Lett. 2016; 363:fnw077. [DOI] [PubMed] [Google Scholar]
- 10. Kernbauer E., Ding Y., Cadwell K.. An enteric virus can replace the beneficial function of commensal bacteria. Nature. 2014; 516:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cani P.D., Possemiers S., Van De Wiele T., Guiot Y., Everard A., Rottier O., Geurts L., Naslain D., Neyrinck A., Lambert D.M. et al.. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009; 58:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Norman J.M., Handley S.A., Baldridge M.T., Droit L., Liu C.Y., Keller B.C., Kambal A., Monaco C.L., Zhao G., Fleshner P. et al.. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015; 160:447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumarasamy K.K., Toleman M.A., Walsh T.R., Bagaria J., Butt F., Balakrishnan R., Chaudhary U., Doumith M., Giske C.G., Irfan S. et al.. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010; 10:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tripathi A., Debelius J., Brenner D.A., Karin M., Loomba R., Schnabl B., Knight R.. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018; 15:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delwart E.L. Viral metagenomics. Rev. Med. Virol. 2007; 17:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simmonds P., Adams M.J., Benkő M., Breitbart M., Brister J.R., Carstens E.B., Davison A.J., Delwart E., Gorbalenya A.E., Harrach B. et al.. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017; 15:161–168. [DOI] [PubMed] [Google Scholar]
- 17. Fouts D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006; 34:5839–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lima-Mendez G., Van Helden J., Toussaint A., Leplae R.. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics. 2008; 24:863–865. [DOI] [PubMed] [Google Scholar]
- 19. Zhou Y., Liang Y., Lynch K.H., Dennis J.J., Wishart D.S.. PHAST: a fast phage search tool. Nucleic Acids Res. 2011; 39:W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Akhter S., Aziz R.K., Edwards R.A.. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity-and composition-based strategies. Nucleic Acids Res. 2012; 40:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roux S., Enault F., Hurwitz B.L., Sullivan M.B.. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015; 3:e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F.. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017; 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang Z., Tan J., Wu S., Li M., Xu C., Xie Z., Zhu H.. PPR-Meta: a tool for identifying phages and plasmids from metagenomic fragments using deep learning. Gigascience. 2019; 8:doi:10.1093/gigascience/giz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ren J., Song K., Deng C., Ahlgren N.A., Fuhrman J.A., Li Y., Xie X., Poplin R., Sun F.. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020; 8:64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pace N.R. A molecular view of microbial diversity and the biosphere. Science. 1997; 276:734–740. [DOI] [PubMed] [Google Scholar]
- 27. Kellenberger E. Exploring the unknown. EMBO Rep. 2001; 2:5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anthony S.J., Epstein J.H., Murray K.A., Navarrete-Macias I., Zambrana-Torrelio C.M., Solovyov A., Ojeda-Flores R., Arrigo N.C., Islam A., Khan S.A. et al.. A strategy to estimate unknown viral diversity in mammals. MBio. 2013; 4:e00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hochreiter S., Schmidhuber J.. Long Short-Term memory. Neural Comput. 1997; 9:1735–1780. [DOI] [PubMed] [Google Scholar]
- 30. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu F., Seff A., Zhang Y., Song S., Funkhouser T., Xiao J.. LSUN: construction of a large-scale image dataset using deep learning with humans in the loop. 2015; arXiv doi:04 June 2016, preprint: not peer reviewedhttps://arxiv.org/abs/1506.03365.
- 32. Hochreiter S., Schmidhuber J.. Long short-term memory. Neural Comput. 1997; 9:1735–1780. [DOI] [PubMed] [Google Scholar]
- 33. Hakkani-Tür D., Tur G., Celikyilmaz A., Chen Y.N., Gao J., Deng L., Wang Y.Y.. Multi-domain joint semantic frame parsing using bi-directional RNN-LSTM. Proceedings of the Annual Conference of the International Speech Communication Association, INTERSPEECH. 2016; [Google Scholar]
- 34. Sak H., Senior A., Beaufays F.. Long short-term memory recurrent neural network architectures for large scale acoustic modeling. Proceedings of the Annual Conference of the International Speech Communication Association, INTERSPEECH. 2014; [Google Scholar]
- 35. Kingma D.P., Ba J.L.. Adam: a method for stochastic optimization. Int.Conf.Learn.Represent.2015. 2015; http://doi.acm.org.ezproxy.lib.ucf.edu/10.1145/1830483.1830503. [Google Scholar]
- 36. Bengio Y., Louradour J., Collobert R., Weston J.. Curriculum learning. ACM International Conference Proceeding Series. 2009; [Google Scholar]
- 37. Kieft K., Zhou Z., Anantharaman K.. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020; 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chiyuan Z., Bengio S., Hardt M., Recht B., Vinyals O.. Understanding deep learning requires re- thinking generalization. 2017; arXiv doi:26 February 2017,preprint: not peer reviewedhttps://arxiv.org/abs/1611.03530.
- 39. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu S., Wang J., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R., Gwadz M., Hurwitz D.I., Marchler G.H., Song J.S. et al.. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020; 48:D265–D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grazziotin A.L., Koonin E. V., Kristensen D.M.. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017; 45:D491–D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hyatt D., Chen G.L., LoCascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Remmert M., Biegert A., Hauser A., Söding J.. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012; 9:173–175. [DOI] [PubMed] [Google Scholar]
- 44. Ivanova N.N., Schwientek P., Tripp H.J., Rinke C., Pati A., Huntemann M., Visel A., Woyke T., Kyrpides N.C., Rubin E.M.. Stop codon reassignments in the wild. Science. 2014; 344:909–913. [DOI] [PubMed] [Google Scholar]
- 45. Lowe T.M., Eddy S.R.. TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1996; 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steinegger M., Söding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 47. Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Price M.N., Dehal P.S., Arkin A.P.. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009; 26:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan Y., Gao M.. Jumbo bacteriophages: an overview. Front. Microbiol. 2017; 8:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eraslan G., Avsec Ž., Gagneur J., Theis F.J.. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 2019; 20:389–403. [DOI] [PubMed] [Google Scholar]
- 51. Adriaenssens E.M., Sullivan M.B., Knezevic P., van Zyl L.J., Sarkar B.L., Dutilh B.E., Alfenas-Zerbini P., Łobocka M., Tong Y., Brister J.R. et al.. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2020; 165:1253–1260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code for the Seeker Python package and for training Seeker is publicly available (https://github.com/gussow/seeker). The training and testing data accession are available in Supplementary Table S1.