

Abstract
Purpose
Determining the role of DYNC2H1 variants in nonsyndromic inherited retinal disease (IRD).
Methods
Genome and exome sequencing were performed for five unrelated cases of IRD with no identified variant. In vitro assays were developed to validate the variants identified (fibroblast assay, induced pluripotent stem cell [iPSC] derived retinal organoids, and a dynein motility assay).
Results
Four novel DYNC2H1 variants (V1, g.103327020_103327021dup; V2, g.103055779A>T; V3, g.103112272C>G; V4, g.103070104A>C) and one previously reported variant (V5, g.103339363T>G) were identified. In proband 1 (V1/V2), V1 was predicted to introduce a premature termination codon (PTC), whereas V2 disrupted the exon 41 splice donor site causing incomplete skipping of exon 41. V1 and V2 impaired dynein-2 motility in vitro and perturbed IFT88 distribution within cilia. V3, homozygous in probands 2–4, is predicted to cause a PTC in a retina-predominant transcript. Analysis of retinal organoids showed that this new transcript expression increased with organoid differentiation. V4, a novel missense variant, was in trans with V5, previously associated with Jeune asphyxiating thoracic dystrophy (JATD).
Conclusion
The DYNC2H1 variants discussed herein were either hypomorphic or affecting a retina-predominant transcript and caused nonsyndromic IRD. Dynein variants, specifically DYNC2H1 variants are reported as a cause of non syndromic IRD.
Keywords: inherited retinal disease (IRD), retinitis pigmentosa (RP), primary cilia, DYNC2H1, intraflagellar transport (IFT)
INTRODUCTION
Inherited retinal diseases (IRDs) are a clinically and genetically heterogeneous group of disorders that often lead to photoreceptor degeneration.1 Technological advances have resulted in the identification of over 300 IRD genes to date (https://sph.uth.edu/retnet/).2 Next-generation sequencing of IRD gene panels is now commonly used for clinical genetic testing and the current yield is around 66%.3 When no pathogenic variant is identified, exome sequencing or genome sequencing may help uncover the variants missed by panel-based screening. As the common IRD variants have largely been identified, exome sequencing or genome sequencing identify rare variants that require functional validation.
Biallelic variants in the cilia gene DYNC2H1 have been associated with two severe ciliopathies: Jeune asphyxiating thoracic dystrophy (JATD, MIM 613091) and short-rib polydactyly (SRP, MIM 613091) with only four documented cases of associated complex early retinal degeneration at ages 2 months, and 2, 5, and 11 years old.4–6 DYNC2H1 encodes the primary subunit of dynein-2, the motor protein that drives ciliary retrograde intraflagellar transport (IFT).7 IFT is an important bidirectional transport of multisubunit protein complexes (IFT particles) along the axonemal microtubules that is essential to normal cilia assembly, function, and maintenance.8,9 Hence, DYNC2H1 mutants show ciliary accumulation of IFT complex proteins, such as IFT88.4,10 The IFT process is important to the photoreceptor, where the connecting cilium is the major transit conduit between the inner segment and the outer segment where light is processed.11,12 A large portion of IRD genes encode for ciliary proteins.13–15 Defects in IFT have an impact on photoreceptor function and survival because poor protein transport affects outer segment health leading to photoreceptor cell death, IRD, and blindness.16 In zebrafish, variants in DYNC2H1 cause abnormal photoreceptor development.11 No mutant mice retina phenotype has been documented.
Using exome sequencing and genome sequencing, we document four novel DYNC2H1 pathogenic variants, and one variant previously reported in five unrelated cases of nonsyndromic progressive IRD. All identified variants affect either a retina-predominant transcript or are hypomorphic, suggesting a basis for a nonsyndromic retinal phenotype. Here we discuss the discovery and validation approaches used, as well as propose novel disease-causing mechanisms for DYNC2H1 in nonsyndromic IRD.
MATERIALS AND METHODS
Full methodological details
Complete methods and any associated references can be found in Supplementary Methods.
Patient recruitment and clinical evaluation
Patients were recruited and evaluated at different institutions, in different countries (Table 1, Fig. 1, Supplementary Results). Phenotypes were documented retrospectively through chart review when possible or by patient self-reporting. When possible, examinations were validated with visual field testing, fundus photography, fundus autofluorescence, optical coherence tomography (OCT), and chest/hands X-rays.
Table 1.
Summary of clinical phenotype of probands 1–5.
| Proband 1 | Proband 2 | Proband 3 | Proband 4 | Proband 5 | |
|---|---|---|---|---|---|
| Site of study | SickKids, Toronto | Leeds, England | London, England | Manchester, England | Wroclaw, Poland |
| Sequencing type | GS | ES | GS | GS | ES |
| Gender (age, years) | F (19) | F (33) | M (47) | M (32) | F (24) |
| Ethnicity | Barbados/Black and Greek | South Asian | South Asian | South Asian | Polish |
| Ocular diagnosis | ARRP | ARRP | ARRP | ARRP | ARRP |
| Onset of symptoms | Birth, photophobia 9 years, nyctalopia | 25 years, nyctalopia | 44 years, VF loss | 18 years, VF loss | 21 years |
| VA (LogMar) | 0.4, 0.5 (13 years) | 0.4, 0.4 (31 years) | 0, 0.2 (13 years) | NA | 0.1, 0 (23 years) |
| OD, OS | 0.6, 0.5 (19 years) | ||||
| Color vision | Absent | NA | NA | NA | Normal |
| ERG | Nonrecordable (19 years) | Reduced responsesa | Reduced responsesa | Reduced responsesa |
Reduced responses (23 years) |
| GVF | <10 degrees | Constricteda | Constricteda | Constricteda | Constricted |
| DYNC2H1 variants | V1: c.12605_12606dup | V3: c.9836C>G | V3: c.9836C>G | V3: c.9836C>G | V4: c.7987A>C |
| p.Asp4203Trpfs*7 | p.Ser3279* | p.Ser3279* | p.Ser3279* | p.Thr2663Pro | |
| V2: c.6632A>T | homozygous | V5: c.12716T>G | |||
| p.Glu2211Val | p.Leu4239Argb |
Normal LogMar VA is 0, legal blindness is 1.ARRP autosomal recessive retinitis pigmentosa, ERG electroretinogram, ES exome sequencing, GS genome sequencing, GVF Goldmann visual field, OD right eye, OS left eye, VA visual acuity, VF visual field.
aReported.
bVariant has been previously reported in Schmidts et al.4 DYNC2H1 variants are reported according to NM_001080463.1.
Fig. 1. Phenotype documentation and family pedigrees.

(I) Proband 1: (a) Retinal imaging of the right and left eye showing some retinal pigmentary changes and narrowing of the vessels. The optic nerve is overexposed and only had mild pallor. (b) Optical coherence tomography (OCT) centered on the macula showing thinning of the outer retina. (c) MAIA™ microperimetry showing decreased retinal sensitivity (black dots) despite decent vision. This leads to fixation instability (turquoise blur from fixation points). (d) Chest X-ray, lateral and anterior–posterior (AP) view showing normal size and chest structure. The pedigree shows segregation of the variants with the phenotype. Filled symbol: affected, arrow: proband. (II) Proband 2: (a) Wide field imaging of the retina of proband 2 showing equatorial bone spiculing changes characteristic of retinitis pigmentosa (RP). (b) Wide field fundus autofluorescence showing a hyperfluorescent area that represents the limits of the functional retina. (c) OCT imaging showing thinning of the outer retina. (d) Left: normal chest X-ray (AP) and right: normal hand X-ray. This pedigree shows consanguinity with very little information about family members who were not available for segregation analysis. Diamond shape symbol with no number: uncertain number of relatives, unknown gender. (III) Proband 3: (a) Fundus autofluorescence showing retinal pigment epithelium atrophy (dark spots throughout). The right eye has a hyperfluorescent ring outlining viable retina. (b) Below is a retina photograph showing attenuated vessels, a marker of photoreceptor degeneration. (c) The OCT shows retinal thinning. Pedigree of the family is also showing an autosomal recessive inheritance pattern and consanguinity. This proband was genotyped through a large genome sequencing initiative, and family members were not available for segregation analysis. Diamond shape symbol with number: number of children, unknown gender. (IV) Proband 4: (a) Wide field fundus autofluorescence showing pigmentary changes and a hyperfluorescent ring larger than for proband 2 though they are the same age. This suggests a larger field of vision. (b) OCT view showing thinning of the outer retina. The pedigree shows pseudodominant (autosomal recessive) inheritance, seen in consanguineous families though consanguinity is not documented. The family was not available for segregation analysis. (V) Proband 5: (a) Imaging of the retina showing retinal atrophy outside the central area (macula) but the vessels were not severely constricted. bone spicules were visible only in the periphery (not shown). (b) OCT imaging showing thinning of the outer retina. c) Normal chest X-ray (AP view). The family pedigree is shown with segregation of variants with the phenotype. (VI) Normal OCT showing the different layers of the retina. Important to this work is the outer retina, which includes CL connecting cilia layer, ISL inner segment layer, OSL outer segment layer, RPE/BM retinal pigment epithelium/Bruch’s membrane, WT wild type.
Ethics statement
All studies conducted were approved by the ethics review boards of the participating institutions and met the Tenets of the Declaration of Helsinki. Informed consent and consent to publish patient photos was obtained from all subjects.
Exome sequencing and genome sequencing identified DYNC2H1 variants as a cause of nonsyndromic IRD
To identify the underlying genetic cause of retinal disease, genome sequencing of genomic DNA was used for probands 1, 3, and 4, while exome sequencing was used for probands 2 and 5 (Supplementary Methods, Fig. S1). Although the details of each approach varied between the five study groups, the general principles were similar. Briefly, for genome sequencing pooled libraries of patient DNA were sequenced and the resulting files were aligned using the GRCh37 version of the human genome. Different approaches and prediction tools were then used to analyze the coding and noncoding sequence. Specifically, under an autosomal recessive model and using control population databases, the sequences were filtered for rare variants (<1% minor allele frequency; MAF) that would be presumed pathogenic. When possible variant segregation analysis was done. Analysis of the exome used a similar approach for the coding portion of the genome (Supplementary Methods).
Transcript detection and quantification
Quantification of proband 1 DYNC2H1 transcripts was performed using MassARRAY (Agena Bioscience, San Diego, CA, USA) genotyping of complementary DNA (cDNA) reverse transcribed from patient fibroblast RNA.17 Briefly, multiplex polymerase chain reaction (PCR) amplification of the DYNC2H1 transcripts was followed by single-base extension from primers directly flanking transcript-specific single-nucleotide polymorphisms (SNPs) of interest. Primers used are listed in Table S1. Results were generated by matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectrometry using Typer 4.0 software to identify and quantify the nucleotide present in the sample at the position of interest (Supplementary Methods).
Fibroblast ciliation assay
Proband 1–derived fibroblasts were used for immunofluorescence and analysis of cilia formation and function. Approximately 250,000 fibroblast cells were seeded on cleaned glass coverslips in 12-well plates. Following standard protocols detailed in the Supplementary Methods, serum starved cells were fixed with ice cold methanol and primary antibodies for IFT88, acetylated tubulin, and gamma tubulin were used for immunofluorescence experiments. A spinning disk confocal microscope (Zeiss, Oberkochen, Germany) was used for imaging and IFT88 fluorescence intensity in cilia was quantified manually using the sum intensity images in FIJI18 (Supplementary Methods).
Protein expression and purification
A H. sapiens cytoplasmic dynein-2 motor domain construct containing an N-terminal SNAPf tag19 was modified to make V1 and V2 constructs. For V1, residues 4196–4201 were changed to sequence WIALNL and residues 4202–4307 (inclusive) were deleted. For V2, residues 2160–2211 (inclusive) in AAA2 were removed, corresponding to exon skipping in V2. Variants were introduced using PCR with Q5 polymerase (New England Biolabs, Ipswich, MA, USA). Dynein-2 constructs were expressed in Spodoptera frugiperda (Sf9) insect cells (Thermo Fisher Scientific, Waltham, MA, USA) using a baculovirus system as previously described.19 Protein purifications were performed at 4 °C. Proteins of interest were eluted by resuspending the resin in TEV buffer, adding 20 μg TEV protease, and incubating the reaction overnight on a roller. TEV-cleaved proteins were separated from the resin using an empty column (Supplementary Methods).
Microtubule gliding assays
Wild type (WT), V1, and V2 dynein-2 motor protein samples were biotinylated for microtubule gliding assays via their N-terminal SNAPf tag as described19 (Supplementary Methods). V1 and V2 dynein-2 motor protein samples were immobilized on a coverslip and fluorescently labeled microtubules and adenosine triphosphate (ATP) were added. The immobilized motor domains propelled the microtubules over the coverslip, and the velocity of microtubule movement was quantified using total internal reflection fluorescence (TIRF) microscopy. As the velocity of microtubule movement depends on dynein-2 concentration,19 all constructs were normalized to a concentration of 1.6 nM. Microtubule gliding velocities were calculated from kymographs generated in FIJI.18 Graphing was performed in Prism 5 (GraphPad).
Organoid assay
Fibroblasts BJ (ATCC® CRL-2522™) were cultured and maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Labtec, Salzburg, Austria), NEAA, and penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). BJ induced pluripotent stem cells (iPSCs) made previously20 were cultured and maintained in Essential 8 Flex medium on plates coated with Geltrex (Thermo Fisher Scientific, Waltham, MA, USA). BJ iPSCs were differentiated to retinal organoids following the protocol established by Zhong and collaborators.21
For validation of DYNC2H1 microexon expression, RNA was extracted from cell lines and retinal organoids using the RNeasy Micro Kit (Qiagen, Hilden, Germany) following the manufacturer instructions. Then, 50 ng of RNA were converted to cDNA by the Tetro cDNA Synthesis Kit (Bioline, London, UK) and a mixture of oligo-dT and random hexamer primers. Next, 2.5 ng of cDNA were used to detect the DYNC2H1 gene isoforms. For control BJ-derived retinal organoid cDNA, PCR amplicons for the DYNC2H1 retinal isoform only differed by 21 bp. This caused formation of nonspecific cDNA heteroduplex. For this reason, samples were subjected to digestion by 3 units/sample of T7 Endonuclease I (New England BioLabs, Ipswich, MA, USA) at 37 °C, 60 minutes prior to loading a 2.5% agarose gel for visualization. Bands were excised and extracted from the gel using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and sent for Sanger sequencing.
RESULTS
Through an international collaboration between Canada, the UK, and European Retinal Disease Consortia (IRDC and ERDC), we have identified five DYNC2H1 variants as associated with five cases of nonsyndromic IRD from cohort members in Canada, the UK, and Poland. Proband phenotypes are summarized in Table 1, Fig. 1, and Supplementary Results. Information about the variants found is summarized in Tables S2–S6 and Figs. S1–S2.
Clinical evaluation
All probands were diagnosed with nonsyndromic retinitis pigmentosa (RP), a variant of IRD (Table 1, Fig. 1 and Supplementary Results). Probands had normal development, cognition, height, weight, and X-rays (Fig. 1). Age at onset of symptoms ranged from childhood to mid-40s (Table 1). The changes in visual acuity were slowly progressive. Generalized photoreceptor dysfunction was documented by visual fields constriction and electroretinography (ERG), which showed either nonrecordable or severely reduced responses. There was no significant refractive error. When possible, retinal examination identified the classical features of RP, i.e., spiculed pigment migration and arteriolar attenuation while outer retinal degeneration was visualized by OCT and fundus autofluorescence imaging (Fig. 1).
Identification of causative DYNC2H1 variants (V1–V5)
Genome sequencing of genomic DNA from probands 1, 3 and 4, and exome sequencing for probands 2 and 5 (Fig. S1, Supplementary Methods, Tables S2 & S3) identified presumed pathogenic variants, V1–V5, mapped to the motor domain of the DYNC2H1 protein (Fig. S2).
In proband 1 (Greek/Barbados descent) from Canada, the novel compound heterozygous DYNC2H1 variants (V1: c.12605_12606dup, p.Asp4203Trpfs*7, NM_001080463.1; V2: c.6632A>T, p.Glu2211Val, NM_001080463.1) were the only variants from the genome sequencing that met defined pathogenicity criteria (Supplementary Methods, Tables S2 & S4, Fig. S1) and segregated with the disease phenotype in the family. V1 was predicted to introduce a premature termination codon (PTC) yielding a protein lacking the C-terminal 106 amino acids. While V2 introduced a missense change, it mapped to a splice site required for incorporation of exon 41 into the protein and it was predicted in silico that V2 would cause the loss of the natural donor site of DYNC2H1 exon 41:c.6633 NM_001080463.1 (Table S5).
Presumably unrelated probands 2, 3, and 4 were found to have a novel homozygous PTC variant (V3: c.9836C>G, p.Ser3279*, NM_001080463.1) in a microexon that is specific to a noncanonical isoform of DYNC2H1 (NM_001080463.1). Organoid data indicate that this noncanonical isoform is predominant in the retina (Fig. 2). Probands 2, 3, and 4 were of South Asian descent but from different parts of the UK (Leeds, Manchester, and London, respectively), and haplotype analysis supported a founder effect for V3 in this population (Table S6).
Fig. 2. Inclusion of DYNC2H1 retinal microexon in human retinal organoid development.
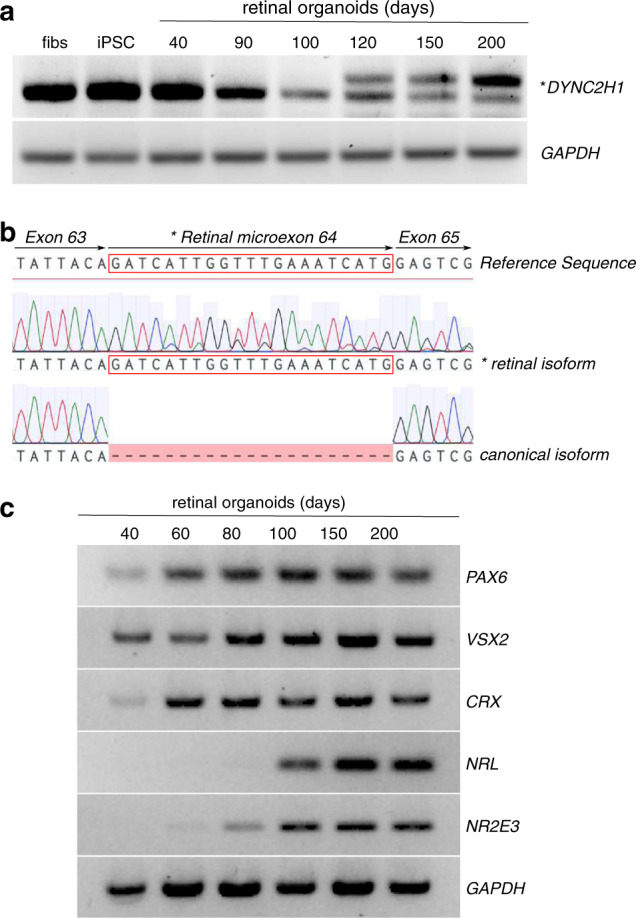
(a) Reverse-transcription polymerase chain reaction (RT-PCR) showing the expression of DYNC2H1 in BJ fibroblasts (fibs), induced pluripotent stem cells (iPSCs), and retinal organoids at 40, 90, 100, 120, 150, and 200 days of differentiation. The shorter PCR product on the gel corresponds to the canonical DYNC2H1 transcript from exons 63 and 65. The longer amplicon (*) includes the retinal-specific microexon 64 (human exon ID GT_21385 on http://ascot.cs.jhu.edu/). (b) Sequencing electropherograms of the canonical and retinal (*) isoforms with alignment to the DYNC2H1 reference sequence. The retinal microexon is boxed in red. (c) Retinal organoid differentiation was confirmed by RT-PCR for markers of retinal differentiation (PAX6, VSX2, CRX, NRL, NR2E3) at 40, 60, 80, 100, 150, and 200 days of differentiation.
Proband 5 from Poland had compound heterozygous variants (V4: c.7987A>C, p.Thr2663Pro, NM_001080463.1; V5: c.12716T>G, p.Leu4239Arg, NM_001080463.1) that segregated with the disease phenotype and were confirmed to be in trans. The missense variants V4 and V5 were predicted deleterious using in silico prediction tools available (Fig. S2, Table S4). The region affected by V4 displays high conservation, both on nucleotide (phyloP 7.29) and amino acid (up to Hawaiian sea urchin) levels (Fig. S2), which suggests its importance for the proper functioning of the protein. Indeed, Thr2263 forms part of the AAA4 nucleotide-binding pocket,22 and V4 (Thr2263Pro) is predicted to perturb nucleotide binding at AAA4 (Fig. S2). V5 maps to a β-sheet within the motor C-terminal domain (Fig. S2). Substitution of a hydrophobic leucine residue for a charged arginine residue within the β-sheet is likely to perturb the folding and stability of the protein. V5 was previously reported in HGMD (CM133150) in case of JATD, hence it is not discussed further.
Other putative pathogenic variants from the five cases were excluded based on established internal exclusion criteria (Supplementary Results, Fig. S1). Novel DYNC2H1 variants were rare (MAF < 0.001%) or not present in the Bravo23 or gnomAD24 population databases (Table S4). At least two of the predictive pathogenicity programs used predicted damaging scores for all V1–V5 (Table S4) and the amino acids involved were conserved across species (Fig. S2).
Functional validation of variants
Functional validation experiments were undertaken for V1–V3.
V1 introduces a PTC and V2 causes skipping of DYNC2H1 exon 41
Analysis of proband 1–derived fibroblast cDNA by PCR and Sanger sequencing confirmed that V1 caused a transcript with a PTC (transcript 1, Fig. 3). This PTC would yield DYNC2H1 protein lacking the 106 C-terminal amino acids. V2 caused incomplete in-frame skipping of DYNC2H1 exon 41 (transcript 2b, Fig. 3), with only 13% of the normally spliced (NS), but mutated, transcript still produced (transcript 2a, Fig. 3). Skipping of exon 41 would yield DYNC2H1 protein lacking 52 amino acids in its AAA2 module. Using Agena massARRAY genotyping, the relative abundance of each transcript was determined to be 15% transcript 1, 13% transcript 2a, and 72% transcript 2b (Fig. 3).
Fig. 3. Analysis of the different DYNC2H1 transcripts of proband 1.

Sanger sequencing (a), Agena MassARRAY experiments (b), and in vitro microtubule gliding assays (c–f) show that proband 1 variably expresses three DYNC2H1 transcripts with impaired in vitro microtubule gliding. Variants are numbered according to the DYNC2H1 transcript NM_001080463.1. (a) The top right electropherogram shows that transcript 1 is expressed even though V1 (orange) causes a frameshift and premature termination codon (PTC) in exon 88 (brown). Top left and bottom electropherograms show that V2 (red) causes incomplete in-frame exon skipping of exon 41 (blue), resulting in the expression of transcript 2a with V2 and transcript 2b missing exon 41. Exon 40: pink. Exon 42: green. Rest of the DYNC2H1 exons and introns: black dashed lines. Location of transcript-specific identifier nucleotides used for Agena MassARRAY experiments: roman numerals. (b) MassARRAY shows product peaks for identifier nucleotides I–IV and shows that 2b is the dominant transcript. Allelotype frequency = (peak allele 1 – peak area variance allele 1) / (peak allele 1 – area variance allele 1) + (peak allele 2 – area variance allele 2). (c) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of purified wild type (WT), V2, and V1 dynein-2 motor domains. (d) Fluorescently labeled (Alexa-488) microtubules (MTs) are added and translocated by dynein in the presence of adenosine triphosphate (ATP). The movement is monitored using total internal reflection fluorescence (TIRF) microscopy. Time sequence of microtubule translocation by immobilized wild type, V2, and V1 dynein motor domains at 1.6 nM input concentration. (e) Plot of microtubule gliding velocity. Lines show mean values (± s.d.). n = 312 microtubule gliding events (WT), n = 157 (V2), n = 91 (V1). Experiments were carried out in duplicate with two different protein preparations. cDNA complementary DNA, mRNA messenger RNA, SNP single-nucleotide polymorphism.
V1/V2 perturb dynein-2 motor function
The impact of V1 and V2 on dynein-2 motor function was tested in vitro. Wild-type (WT) human dynein-2 motor domain was purified,19 along with two constructs with amino acid changes matching those caused by V1 and the V2 variant lacking exon 41 (Supplementary Methods). The V1 and V2 constructs produced protein of the expected molecular weight (Fig. 3). However, the yield of the variant proteins was substantially lower compared with WT. The activity of the variant proteins was assessed using an in vitro motility assay (Fig. 3). The WT construct propelled microtubules at an average speed of 135 nm/s (±56 nm/s). Both V1 and V2 showed severely reduced motility, with average speeds of 24 nm/s (±24 nm/s) for V2 and 12 nm/s (±9 nm/s) for V1 (Fig. 3). The proportion of microtubules that underwent any movement was also lower for V1 and V2. While 82% of microtubules were propelled by WT, only 25% and 42% of microtubules were propelled by V1 and V2, respectively. Overall, both proband 1 variants perturbed dynein-2 motor function.
V1/V2 affect IFT88 transport in fibroblasts
Proband 1–derived fibroblasts showed abnormal IFT88 protein accumulation in ciliary axonemes, which supports DYNC2H1 malfunction (Fig. 4). There was no difference in cilia incidence (data not shown).
Fig. 4. Ciliated proband 1 fibroblasts accumulate IFT88 (green) in ciliary axonemes.

(a) Wild-type (WT) (top) and proband 1 V1/V2 mutant (bottom) fibroblasts are stained with acetylated alpha tubulin (cilia axonemes, red), gamma tubulin (basal body, red), and Hoescht (nuclei, blue). Scale bars are 18 µm. Panels on the right show enlargements of boxed ciliary IFT88 staining. Scale bars are 2 µm for enlargements. (b) Boxplot represents the distribution of IFT88 fluorescence intensity in proband 1 V1/V2 mutant and WT fibroblast cells. Data points are overlaid onto the boxplot to highlight the overlapping fluorescence intensity measurements in patient and WT cells. IFT88 fluorescence intensity was measured in 100 patient’s? and 100 WT cilia. ***P < 0.0001 (Student’s unpaired t-test).
V3 is part of a retina-predominant microexon
The homozygous variant V3 is predicted to introduce a PTC in an isoform-specific 21-bp in-frame microexon (NM_001080463.1), which to date has only been described in human retinal tissue RNAseq experiments.25 RT-PCR from control retinal organoid cDNA at different stages of differentiation showed that expression of the microexon began at day 120, with expression increasing with organoid maturity to become the dominant transcript (up to day 200), correlating with photoreceptor differentiation. The microexon was not detected in cDNA from dermal fibroblasts, iPSCs, or in retinal organoids at 40, 90, or 100 days postdifferentiation (Fig. 2a). Sequencing of the resulting bands confirmed the larger product included the microexon (Fig. 2b). The microexon encodes a 7–amino acid insertion within the AAA5 module of DYNC2H1 (Fig. S2). The presence of a stop codon in the microexon would produce a severely truncated protein. The lack of a systemic phenotype in probands 2, 3, and 4 despite the presence of a homozygous stop variant in the microexon provides further support for the retinal predominance of this isoform and suggests that the canonical isoform (NM_001377.2), expressed elsewhere in the body, is unaffected.
In summary, we have identified five variants in DYNC2H1 that cause nonsyndromic IRD, either because they only affect a retina-predominant transcript or because they are hypomorphic, resulting in a milder phenotype.
DISCUSSION
This work reports the association of mutant dyneins and DYNC2H1 variants as a cause of nonsyndromic IRD, an important cause of blindness worldwide. This not only highlights the new role of dyneins in IRD but also extends the range of severity associated with pathogenic variants in DYNC2H1 and shows how interpretation of rare variants in a different phenotype requires thorough validation.
Thus far, human biallelic pathogenic variants in DYNC2H1 have been associated exclusively with JATD or SRPs, occasionally lethal conditions.4,26 JATD features include various skeletal anomalies such as short ribs, narrow thorax, various bone anomalies, renal or liver disease with only four cases of associated early retinal degeneration.4–6 SRP is more severe and often leads to early prenatal lethality.26 The manifestation of JATD and SRP may present with varying degrees of severity.4,26 Variability in disease presentation is not fully understood but includes genetic influences.27,28 The role of modifier alleles has been suggested in a case of SRP type II with double heterozygous variants in DYNC2H1 and NEK1.29 In addition to genetic influences, other possible explanations for the variability in disease manifestation include environmental influences, epigenetic factors, and varying impact of variants on DYNC2H1 protein structure and function.4,29 Retinal involvement in DYNC2H1 JATD and SRP cases is rare, but since these ciliopathies can be quite severe and some affected individuals die early, the retinal involvement could be underestimated.4 The association of nonsyndromic IRD with syndromic ciliopathy-related disease genes is increasingly documented.30–32 Furthermore, variants in more than 80 cilia-related genes have been associated with nonsyndromic IRD.14,15,33 Some of the mechanisms through which mutated syndromic ciliopathy genes lead to nonsyndromic IRD include variants in amino acids uniquely important for visual function, variants in retina-specific transcripts, variants causing reduced expression of syndromic ciliopathy genes, and enhanced aberrant splicing in photoreceptors.31,32,34,35 Documented DYNC2H1 disease-associated variants are spread throughout the length of the gene.4 These variants may lead to impaired ciliary protein localization, reduced motility, and decreased IFT.8,9 The four novel pathogenic variants identified here all map within the motor domain of dynein-2 and all variants fall on residues conserved in vertebrates (Fig. S2).
The in vitro mobility assays indicate that both V1 and V2 variants perturb the motor function of dynein-2 (Fig. 3). This suggests that dynein-2 activity in proband 1 would be a fraction of wild-type protein; however, the residual activity in most tissues is evidently sufficient to prevent syndromic disease.
V2 affects the penultimate nucleotide of DYNC2H1 exon 41 and changes the existing AG 5’ donor splice site to TG. Although V2 causes in-frame exon skipping, the translated protein would be missing 52 amino acids from the AAA2 module of the motor domain. We showed that V2 leads to incomplete aberrant splicing, and that the nonskipped (NS) transcript (which still harbors V2) remains expressed at approximately 13% (Fig. 3). This NS transcript may well retain some function, as it would only contain one variant (V2) that maps to a residue on the surface of the protein that is not strictly conserved. Hence it would be less likely to affect folding/function (Fig. S2). The level of NS transcript could be sufficient to ensure DYNC2H1 function in most tissues, with the exception of the retina, which is highly reliant on functional IFT. Our work suggests the mobility of IFT88 is impaired. (Fig. 4). IFT88 is critical for photoreceptor formation and human variants in IFT88 have been associated with photoreceptor degeneration.16,36,37 Another possible explanation for the isolated retinal phenotype in proband 1 could be that V2 causes different proportions of transcripts in the retina, resulting in even more aberrant splicing.
In this study, V3, a homozygous novel PTC variant, was identified in three nonsyndromic probands. This variant was detected in a noncanonical isoform, which includes a 21-bp microexon and appears to have a founder effect in South Asians in the UK. Based on RT-PCR from retinal organoids and RNAseq data25 we hypothesized that that this affected isoform could be the major isoform expressed in the photoreceptors. The noncanonical isoform was not detected in differentiated fibroblasts and was only detected in matured retinal organoids from day 120 onward, which coincided with photoreceptor differentiation. As the organoids matured beyond 200 days, this noncanonical transcript became the dominant transcript (Fig. 2). This isoform is not expressed in any other body tissues according to expression data from GTEx.38 This provides another example of a variant in a retina-predominant transcript as a cause of nonsyndromic disease, a phenomenon previously documented.20 It will be interesting to investigate how this microexon affects DYNC2H1 function and if it is tailored to a specific function in photoreceptor retrograde IFT.
In contrast to V1-V3 variants, the V4 and V5 variants were both missense, causing substitution of threonine 2663 (NM_001080463.1) with proline, and leucine 4239 (NM_001080463.1) for arginine, respectively. V4 was located within the AAA4 module of DYNC2H1, proximal to a nucleotide-binding site. While the variant itself has not been reported previously, a nearby arginine residue (c.2662, NM_001080463.1) has been found mutated in two instances in SRP Majewski type and SRP type I (unpublished data, ClinVar RCV000515850.1).29 The V4 residue displayed high conservation, both at the nucleotide and amino acid level (Fig. S2). For proband 5 no functional work could be done, but we hypothesize a situation similar to proband 1 in that V4 could represent a hypomorphic allele, although additional studies would be needed to prove this. It is not known if V4 could possibly affect a retina-specific isoform. V4 may be assumed to be essential for the proper functioning of the protein as proper nucleotide binding at AAA4 is thought to play an important structural role in dynein-2.22 V4 is also immediately adjacent to one of the critical conserved nucleotide-binding motifs in DYNC2H1.22 Substitution to a proline, a radically different amino acid, is extremely likely to perturb nucleotide binding at this site and hence perturb the activity of the protein, as shown for similar variants in DYNC1H1.39 V5 occurs in the DYNC2H1 C-terminal tail domain. This variant was reported in JATD and the leucine residue, as well as thymine nucleotide, are highly conserved.4 The presumed milder impact of these variants may explain the milder disease course and later age of IRD onset (21 years old) for this individual.
In summary, the biallelic DYNC2H1 variants are reported as a cause of non syndromic IRD in five cases. These data result from either reduced stability or function of DYNC2H1 or in loss of a retina-predominant isoform to which the photoreceptors are uniquely susceptible. Although IRD is a rare feature of JATD, this report highlights the extreme variability of ciliopathy-related phenotypes. Considering the tight relationship of dynein with IFT, which is essential to photoreceptor function, genes encoding dynein-2 should be considered as candidates for the unsolved cases of IRD. Variants in syndromic disease genes associated with nonsyndromic phenotypes are emerging. We highlight the different proposed disease mechanisms for these nonsyndromic cases: variants in retina-predominant isoforms, tissue-specific mis-splicing, and hypomorphic alleles.
Supplementary information
Acknowledgements
Funding was provided by Fighting Blindness Canada (E.H.); Vision Science Research Program (A. Vig); University of Toronto McLaughlin Centre Whole Genome Sequence Initiative, Henry Brent Chair in Innovative Pediatric Ophthalmology Research (E.H.); Career Development Award (A. vincent) Foundation Fighting Blindness, USA Career Development Award, Brenda Eye Research Fund (Novartis, E.H., A. Vincent); Fight For Sight (UK) Early Career Investigator Award (G.A.); National Institute For Health Research–Biomedical Research Centre at Great Ormond Street Hospital Institute for Child Health (G.A.); Moorfields Eye Charity, National Institute For Health Research–Biomedical Research Centre at Moorfields Eye Hospital and UCL Institute of Ophthalmology (G.A.). This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. M.E.C. and D.O. received funds from Retina UK, and The Wellcome Trust. IRDC funding for Leeds, Manchester, 100 REtina UK and Fight for Sight. A.M.T. received funds from National Science Center (Poland), UMO-2015/19/D/NZ2/03193. The authors acknowledge the European Rare Disease Consortium (ERDC) colleagues for being part of a safe environment within which to share data. A.J.R. thanks M. Mladenov for expert technical assistance and the following agencies for grant support: Wellcome Trust and Royal Society (104196/Z/14/Z), BBSRC (BB/P008348/1), and Royal Society (RG170260). The authors also thank Bhooma Thiruvahindrapuram of The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, Canada for assistance with data analysis.
Disclosure
G.W. received a Wellcome Trust Seed Award in Science 204378/Z/16/Z. K.N.K. reports advisory board fees from MeiraGTx and Roche. M.M. reports educational travel grants from Novartis, Bayer, and Allergan; lecture or advisory board fees from Novartis and Bayer; and personal research funding from Alcon and Roche. A. Vincent is a consultant for Adverum Technologies. (Novartis E.H.) is a consultant for Sanofi (DSMB) and Deep Genomics. The other authors declare no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Anjali Vig, James A. Poulter.
A list of authors and their affiliations appears at the end of the paper.
Contributor Information
Anthony Roberts, Email: a.roberts@mail.cryst.bbk.ac.uk.
Elise Heon, Email: elise.heon@sickkids.ca.
Genomics England Research Consortium:
J. C. Ambrose, P. Arumugam, E. L. Baple, M. Bleda, F. Boardman-Pretty, J. M. Boissiere, C. R. Boustred, H. Brittain, M. J. Caulfield, G. C. Chan, C. E. H. Craig, L. C. Daugherty, A. de Burca, A. Devereau, G. Elgar, R. E. Foulger, T. Fowler, P. Furió-Tarí, J. M. Hackett, D. Halai, A. Hamblin, S. Henderson, J. E. Holman, T. J. P. Hubbard, K. Ibáñez, R. Jackson, L. J. Jones, D. Kasperaviciute, M. Kayikci, L. Lahnstein, K. Lawson, S. E. A. Leigh, I. U. S. Leong, F. J. Lopez, F. Maleady-Crowe, J. Mason, E. M. McDonagh, L. Moutsianas, M. Mueller, N. Murugaesu, A. C. Need, C. A. Odhams, C. Patch, D. Perez-Gil, D. Polychronopoulos, J. Pullinger, T. Rahim, A. Rendon, P. Riesgo-Ferreiro, T. Rogers, M. Ryten, K. Savage, K. Sawant, R. H. Scott, A. Siddiq, A. Sieghart, D. Smedley, K. R. Smith, A. Sosinsky, W. Spooner, H. E. Stevens, A. Stuckey, R. Sultana, E. R. A. Thomas, S. R. Thompson, C. Tregidgo, A. Tucci, E. Walsh, S. A. Watters, M. J. Welland, E. Williams, K. Witkowska, S. M. Wood, and M. Zarowiecki
Supplementary information
The online version of this article (10.1038/s41436-020-0915-1) contains supplementary material, which is available to authorized users.
References
- 1.Wert KJ, Lin JH, Tsang SH. General pathophysiology in retinal degeneration. Dev Ophthalmol. 2014;53:33–43. doi: 10.1159/000357294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanany M, Rivolta C, Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci USA. 2020;117:2710. doi: 10.1073/pnas.1913179117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Resta C, Spiga I, Presi S, et al. Integration of multigene panels for the diagnosis of hereditary retinal disorders using next generation sequencing and bioinformatics approaches. EJIFCC. 2018;29:15–25. [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidts M, Arts HH, Bongers EMHF, et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J Med Genet. 2013;50:309. doi: 10.1136/jmedgenet-2012-101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baujat G, Huber C, El Hokayem J, et al. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J Med Genet. 2013;50:91–98. doi: 10.1136/jmedgenet-2012-101282. [DOI] [PubMed] [Google Scholar]
- 6.Cossu C, Incani F, Serra ML, et al. New mutations in DYNC2H1 and WDR60 genes revealed by whole-exome sequencing in two unrelated Sardinian families with Jeune asphyxiating thoracic dystrophy. Clin Chim Acta. 2016;455:172–180. doi: 10.1016/j.cca.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Roberts AJ. Emerging mechanisms of dynein transport in the cytoplasm versus the cilium. Biochem Soc Trans. 2018;46:967–982. doi: 10.1042/BST20170568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ocbina PJR, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn. 2008;237:2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi P, Li W-J, Dong M-Q, Ou G. Dynein-driven retrograde intraflagellar transport is triphasic in C. elegans sensory cilia. Curr Biol. 2017;27:1448–1461. doi: 10.1016/j.cub.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Ocbina PJR, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krock BL, Mills-Henry I, Perkins BD. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest Ophthalmol Vis Sci. 2009;50:5463–5471. doi: 10.1167/iovs.09-3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Insinna C, Besharse JC. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev Dyn. 2008;237:1982–1992. doi: 10.1002/dvdy.21554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 14.Bujakowska KM, Liu Q, Pierce EA. Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb Perspect Biol. 2017;9:a028274. doi: 10.1101/cshperspect.a028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanna H, Baehr W. Retina ciliopathies: from genes to mechanisms and treatment. Vis Res. 2012;75:1. doi: 10.1016/j.visres.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Pazour GJ, Baker SA, Deane JA, et al. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradić M, Costa J, Chelo IM. Genotyping with Sequenom. In: Orgogozo V, Rockman MV, editors. Molecular methods for evolutionary genetics. Totowa, NJ: Humana Press; 2011. pp. 193–210. [DOI] [PubMed] [Google Scholar]
- 18.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toropova K, Mladenov M, Roberts AJ. Intraflagellar transport dynein is autoinhibited by trapping of its mechanical and track-binding elements. Nat Struct Mol Biol. 2017;24:461. doi: 10.1038/nsmb.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arno G, Agrawal Smriti A, Eblimit A, et al. Mutations in REEP6 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2016;99:1305–1315. doi: 10.1016/j.ajhg.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong X, Gutierrez C, Xue T, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. 2014;5:4047–4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt H, Zalyte R, Urnavicius L, Carter AP. Structure of human cytoplasmic dynein-2 primed for its power stroke. Nature. 2015;518:435–438. doi: 10.1038/nature14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The NHLBI Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program. BRAVO variant browser: University of Michigan and NHLBI. 2018. https://bravo.sph.umich.edu/freeze5/hg38/.
- 24.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farkas MH, Grant GR, White JA, Sousa ME, Consugar MB, Pierce EA. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics. 2013;14:486. doi: 10.1186/1471-2164-14-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dagoneau N, Goulet M, Geneviève D, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu Y, Arbogast T, Lorenzo SM, et al. Oligogenic effects of 16p11.2 copy-number variation on craniofacial development. Cell Rep. 2019;28:3320–3328. doi: 10.1016/j.celrep.2019.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kousi M, Katsanis N. Genetic modifiers and oligogenic inheritance. Cold Spring Harb Perspect Med. 2015;5:a017145. doi: 10.1101/cshperspect.a017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El Hokayem J, Huber C, Couvé A, et al. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. J Med Genet. 2012;49:227. doi: 10.1136/jmedgenet-2011-100717. [DOI] [PubMed] [Google Scholar]
- 30.Xu M, Yang L, Wang F, et al. Mutations in human IFT140 cause nonsyndromic retinal degeneration. Hum Genet. 2015;134:1069–1078. doi: 10.1007/s00439-015-1586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb TR, Parfitt DA, Gardner JC, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum Mol Genet. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pretorius PR, Aldahmesh MA, Alkuraya FS, Sheffield VC, Slusarski DC. Functional analysis of BBS3 A89V that results in nonsyndromic retinal degeneration. Hum Mol Genet. 2011;20:1625–1632. doi: 10.1093/hmg/ddr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali MU, Rahman MSU, Cao J, Yuan PX. Genetic characterization and disease mechanism of retinitis pigmentosa; current scenario. 3 Biotech. 2017;7:251–251. doi: 10.1007/s13205-017-0878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parfitt David A, Lane A, Ramsden Conor M, et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell. 2016;18:769–781. doi: 10.1016/j.stem.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riazuddin SA, Iqbal M, Wang Y, et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am J Hum Genet. 2010;86:805–812. doi: 10.1016/j.ajhg.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sukumaran S, Perkins BD. Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 intraflagellar transport mutants. Vis Res. 2009;49:479–489. doi: 10.1016/j.visres.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chekuri A, Guru AA, Biswas P, et al. IFT88 mutations identified in individuals with nonsyndromic recessive retinal degeneration result in abnormal ciliogenesis. Hum Genet. 2018;137:447–458. doi: 10.1007/s00439-018-1897-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kon T, Nishiura M, Ohkura R, Toyoshima YY, Sutoh K. Distinct functions of nucleotide-binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry. 2004;43:11266–11274. doi: 10.1021/bi048985a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.