

Abstract
Liquid biopsy for the detection and monitoring of central nervous system (CNS) tumors is of significant clinical interest. At initial diagnosis, the majority of patients with central nervous system tumors undergo magnetic resonance imaging (MRI), followed by invasive brain biopsy to determine the molecular diagnosis of the WHO 2016 classification paradigm. Despite the importance of MRI for long-term treatment monitoring, in the majority of patients who receive chemoradiation therapy for glioblastoma (GBM), it can be challenging to distinguish between radiation treatment effects including pseudoprogression, radiation necrosis (RN) and recurrent/progressive disease (PD) based on imaging alone. Tissue biopsy-based monitoring is high risk and not always feasible. However, distinguishing these entities is of critical importance for management of patients and can significantly affect survival. Liquid biopsy strategies including circulating tumor cells (CTCs), circulating free DNA (CfDNA) and extracellular vesicles (EVs) have the potential to afford significant useful molecular information at both the stage of diagnosis and monitoring for these tumors. We review current liquid biopsy-based approaches in the context of tumor monitoring to differentiate PD from pseudoprogression and RN.
Keywords: Glioblastoma, progression, pseudoprogression, liquid biopsy, radiation necrosis, extracellular vesicles, circulating tumor cells, circulating free DNA
Introduction
Glioblastoma is the most common malignant primary central nervous system tumor. GBM is highly aggressive and the median overall survival is only 15–23 months despite aggressive treatment 1. Currently, maximal resection followed by radiation therapy with concurrent temozolomide (TMZ) and adjuvant TMZ treatment is the standard of care. Post treatment surveillance involves serial MRI. A challenge faced by clinicians is the diagnosis and management of a gadolinium enhancing lesion on a follow-up MRI post treatment. This suspicious lesion could be PD or a mere post treatment radiation effects such as pseudoprogression or radiation necrosis (RN). Pseudoprogression and RN are distinct clinical entities, which when identified and managed appropriately result in better outcomes, while PD of the tumor is often dismal. Patients with PD have a median survival of 3–6 months 2, and there is no standard of care. Systemic options include TMZ rechallenge, lomustine, and antiangiogenic therapy such as bevacizumab, but their effectiveness is limited. Re-radiation and re-resection can be considered depending on the location of the tumor and the condition of the patient 3. Conversely, antiangiogenic drugs like bevacizumab or cediranib decrease contrast enhancement by altering permeability of tumor vasculature without actual reduction in tumor burden, referred to as pseudoresponse. Distinguishing these clinical entities from PD is crucial to avoid unnecessary reoperations, premature discontinuation of adjuvant TMZ or its substitution with second line agents.
MR imaging based monitoring is the current standard of care for post-surgical monitoring. Contrast enhancement on imaging is indicative of disrupted blood brain barrier (BBB), but not tumor presence 4. Currently, MRI based Response Assessment in Neuro-Oncology (RANO) criteria is used to monitor treatment response in GBM patients.The criteria included T1 gadolinium enhancing disease, T2/FLAIR changes, new lesions, corticosteroid use, and clinical status 5. Adoption of RANO criteria for monitoring response is not without limitations. There is ambiguity in identifying radiation effects, enrolling patients into clinical trials and monitoring immunotherapy response 6. Advanced imaging modalities including diffusion-tensor imaging, perfusion imaging, MR spectroscopy (MRS), Positron Emission Tomography (PET) imaging have been used to identify true PD 7,8. Although, MRS 9 and dynamic susceptibility contrast methods 10 show promise, imaging modalities cannot establish a definitive diagnosis nor capture the heterogeneous molecular landscape of the evolving tumor which is crucial in the setting of PD. Moreover, repeated biopsies cannot be performed to monitor tumor progression due to high risk, surgical inaccessibility and life threatening complications 11. Furthermore, focal sampling cannot capture the true tumor heterogeneity.
As such, there is a great need for tools that can allow early diagnosis, molecular characterization, and assess response to therapy as well as distinguish PD from pseudoprogression and RN with higher sensitivity and specificity compared to current imaging-based technologies. Liquid biopsy refers to analysis of biofluids of patients to detect disease specific genomic or proteomic cargo for diagnostic and prognostic purposes. Liquid biopsy encompasses circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), and extracellular vesicles (EVs).A longitudinal liquid biopsy based patient monitoring could provide better perspectives into the tumor presence, molecular status, tumor evolution, response to therapy and also distinguish PD from post treatment radiation effects and ultimately strategize appropriate therapies to improve patient outcomes 12,13. In this review, we briefly discuss the clinical entities of pseudoprogression and RN in the context of various liquid biopsy platforms to distinguish PD from pseudoprogression and RN.
Pseudoprogression
Pseudoprogression is a reversible subacute post treatment radiation effect identified as an increase in the size of the contrast enhancing lesion, with or without neurological deterioration following completion of RT alone or concomitant RT-TMZ, mimicking PD 14,15. pseudoprogression most often occurs within the first 3 months following RT/RT-TMZ, but can present up to 6 months afterwards. Nearly half the patients with an enhancing lesion within 1 month post-RT have pseudoprogression 16. Unlike patients with PD, patients with pseudoprogression remain asymptomatic. Some present with complications due to transient demyelination including worsening of pre-existing symptoms, transient cognitive decline, subacute rhombencephalitis or somnolence syndrome.
Pathologically, pseudoprogression corresponds to gliosis and reactive radiation-induced changes without evidence of viable tumor tissue17. This may represent an exaggerated response to therapy involving changes to the vascular endothelium and the blood brain barrier (BBB) as well as oligodendroglial injury leading to inflammation and increased vascular permeability 11. Treatment-related cellular hypoxia could also contribute to this abnormal enhancement 11,18. Some groups suggested pseudoprogression to be an active ‘inflammatory’ response against the tumor considering the association between pseudoprogression and increased survival 19. Interestingly, patients with MGMT methylation show pseudoprogression twice as often 20. Considering the fact that MGMT methylation status is associated with response to TMZ and thus favourable prognosis 21,22, identification of MGMT status is useful in predicting pseudoprogression and differentiating it from PD 15,16,23. Conversely, patients without MGMT methylation have higher rates of PD, with rates of 60% occurrence 22. Recent studies have demonstrated a correlation between P53 overexpression and pseudoprogression 24. As such, P53 status could also be a potential biomarker for pseudoprogression. Emerging reports have suggested the association of higher expressions of interferon regulatory factor 9 (IRF9) and X ray repair cross-complementing 1 (XRCC1) in pseudoprogression 25.
Conventional MR imaging is unable to distinguish between pseudoprogression and early progression, and alternative techniques have not been validated in prospective trials 7,11,26. The current method to distinguish the two is to perform follow-up examinations of patients comparing MR images at different points in time. Asymptomatic cases of suspected pseudoprogression are followed up by serial imaging, but when there is worsening of the symptoms due to transient cerebral edema, short course of corticosteroid treatment is initiated with close clinical surveillance and serial imaging 7.
Radiation Necrosis
Radiation necrosis is a permanent post treatment radiation effect characterized by an increase in the size of the contrast enhancing lesion occurring 3 months to years after RT 11. Pseudoprogression and RN are often considered a spectrum of post treatment radiation changes. Unlike pseudoprogression, RN progresses without treatment, and has not been associated with better prognosis. With improvements in overall survival of patients with GBM, there is growing usage of reradiation, radiation surgery and hypofractionated radiotherapy adding to the cumulative dose of radiation received by a single patient contributing to the increasing incidence of RN of about 5 – 40% 27. Patients with RN can be asymptomatic or present with symptoms and signs of necrosis including stroke-like migraine attacks after radiation therapy (SMART syndrome), radiation induced cavernous malformations or aneurysms, Moya-Moya syndrome, mineralizing microangiopathy, tissue calcification, atrophy, leukoencephalomyelopathy or rarely endocrine dysfunction 28–32.
Pathologically, RN corresponds to white matter necrosis associated with calcification, fibrinoid deposition, vascular hyalinization and endothelial thickening which leads to chronic inflammatory state, oxidative stress and inhibition of neurogenesis 29,33–36. Radiation induced vascular injury initiates the process of necrosis; subsequently, increased tumor necrosis factor alpha (TNF-α) drives endothelial cell apoptosis and increased vascular permeability, and increased vascular endothelial growth factor(VEGF) induces small vessel permeability and cerebral edema 7,37–40. Conventional imaging tools cannot identify RN and alternative techniques have not yet been validated in prospective trials 7,11,26. Suspected RN can be managed with corticosteroid treatment, hyperbaric oxygen therapy, anticoagulation, anti-angiogenic agents like bevacizumab, laser interstitial thermal therapy or even surgery 7. Corticosteroids reduce radiation induced radiation induced inflammatory response, decrease BBB leakiness and reduce cerebral edema41. Hyperbaric oxygen therapy stimulates angiogenesis and restores blood supply after radiation induced vascular injury. It is even suggested as a prophylactic option in patients with high likelihood of developing RN 42,43. Anticoagulants like heparin and warfarin inhibit cytokine release, prevent platelet aggregation and coagulation 44,45. Anti-VEGF agents reduce small vessel permeability and BBB leakiness 46–49. Laser interstitial thermal therapy focuses on thermal coagulation of peri-necrotic region of abnormal angiogenesis 50. Surgery reduces mass effect, edema and decreases intracranial pressure in addition to providing true tissue diagnosis 51,52. No controlled randomized clinical trials have been performed to establish the most beneficial regimen to manage RN.
Circulating tumor cells
Circulating tumor cells are cancer cells that leave the primary tumor and enter circulation. A fraction of these CTCs have the potential to invade distant sites and progress to metastasis 53. Epithelial to mesenchymal transition (EMT) within the tumor enables some cells to gain a phenotype associated with increased motility and invasion 54,55. CTCs are found either as single cells or in clusters, the latter of which have higher metastatic potential 56–58. CTCs are hypothesized to be either randomly detached cells or metastatic tumor subclones. In either case, they contain the genomic, transcriptomic and proteomic characteristics of the primary tumor 12 and can be valuable tools to provide insight into the primary tumor 59,60. Studies in multiple cancers have shown the possibility of CTC based diagnosis 61–63, monitoring 64–73 and prognosis 74–77. These studies have also demonstrated that the presence, phenotype and the methylation status of markers within CTCs in peripheral circulation have prognostic significance 57,78,79.
GBMs rarely form clinically evident extracranial metastases 80. This is attributed to the inability of glioma cells to survive in extracranial sites, and tolerate the immune system 60,81. However, recent evidence of CTCs detected in blood of GBM patients (Table 1) 59,60,81,82 poses questions about the conventional theories of GBM dissemination, opening the field of CTC based liquid biopsy in brain tumors 59,60,82. Although the capability of the detected GBM CTCs to metastasize has not been established, they can be used as tools to diagnose and monitor GBM 59,60. Previous studies have used positive selection (surface marker based selection), negative selection (depletion of blood cells) or other novel platforms for CTC detection (Table 1).
Table 1.
Summary of studies using circulating tumor cell-based analysis for GBM.
| Author, Year | Biofluid | Methodology of CTC enrichment | Genetic cargo evaluated | Diagnostic sensitivity | Potential role |
|---|---|---|---|---|---|
| Sullivan, 2014 | Blood | CTC-iChip microfluidic technology; characterization using antibody cocktail, STEAM: SOX2, tubulin-3, EGFR, A2B5 and cMET. | SERPINE1, TGFB1,TGFBR2,VIM; EGFR amplification | 39% | Diagnosis/Prognosis |
| Muller, 2014 | Blood | Density gradient centrifugation followed by fluorescence immunocytochemistry using anti- GFAP antibody | EGFR amplification | 20% | Diagnosis/Prognosis |
| Macarthur, 2014 | Blood | Density gradient centrifugation followed TERT promotor-based CTC detection assay | TERT | 72%: pre-radiotherapy 8% post-radiotherapy | Prognosis/Monitoring |
| Malara, 2016 | Blood | Vimentin positive cell sorting and short time expansion | - | 2/2 | Prognosis/Monitoring |
| Gao, 2018* | Blood | CTCs detection based on the aneuploidy of chromosome 8 examination by CEP8-FISH | Chromosome 8 aneuploidy | 24 of 31 (77%) GBM (82%) | Diagnosis/Prognosis/Monitoring |
| Krol, 2018λ | Blood | Parsortix microfluidic system | SOX2 | 7/13(53.8%) | Diagnosis/Prognosis/ Monitoring |
Abbreviations.CEP8, Centromere Probe (CEP) 8; CTC, circulating tumor cells; EGFR, epidermal growth factor receptor; FISH, Fluorescence in situ Hybridization; GBM, Glioblastoma; GFAP, glial fibrillary acidic protein; SERPINE1, Serpin Family E Member 1; SOX2, SRY (sex determining region Y)-box 2; TERT, Telomerase reverse transcriptase; TGFB1, Transforming Growth Factor Beta 1; TGFBR2, Transforming Growth Factor Beta Receptor 2;
These described cases in this series are not limited to GBM
The study evaluates for CTC clusters
GBM-CTCs were shown to contain tumor specific molecular characteristics and invasive mesenchymal signature 59. Macarthur et al., showed an increase in CTC numbers post radiotherapy in a patient suggestive of PD, indicating the potential of CTCs in distinguishing PD from radiation effects 82. Gao et al. identified CTCs in all grades of glioma patients, and showed that CTC detection can reliably identify PD from RN 83. These studies provide a proof of principle that patients with GBM have CTCs in their peripheral blood. They demonstrate the potential of molecular characterization of these cells for minimally invasive tumor profiling and identification of PD from radiation effects. Recently, CTC clusters were also identified in the blood of GBM patients 81. Interestingly, Lui et al., demonstrated the capacity of intravenously injected CTCs to ‘reseed’ the primary site using a xenograft model and showed that CTCs also demonstrated stemness phenotype more resistant to treatments 84. This strengthens the notion that CTCs are a subset of aggressive primary GBM cells, with EMT and stemness characteristics.
Although current studies report a very high specificity, the sensitivity of CTC detection is variable, from 20.6% to 82% 59,60,82. Higher sensitivities are required to establish CTCs as a potential diagnostic modality to diagnose and monitor brain tumors. Most studies inadequately characterize CTCs, are underpowered, use limited numbers of surface markers for CTC enrichment, have samples collected at variable time points along the disease course and lack long-term followup. Furthermore, CTCs were not detectable in each of multiple samples of a given patient at a given time point. This could indicate the lack of sensitivity of current techniques in detecting CTCs or the rarity CTCs in blood (1 cell per 109 blood cells). CTC analysis requires large volumes of fresh blood, and immediate sample processing. Also, detection is currently limited by technological constraints 12,85. Several factors including localization of the primary tumor, circulation dynamics and entrapment in capillary beds limit CTC detection. Furthermore, EMT may alter the surface marker profiles, which may negatively affect CTC-assay performance 29,86. Furthermore, the role of CTCs as diagnostic screening modalities is debatable as the disease would be in an advanced stage with CTC dissemination, but it can probably be a good monitoring tool for disease progression and prognosis. Nevertheless, CTCs can provide a distant insight into the primary tumor, and analysis using complementary technology could potentially indicate the presence of a tumor, monitor disease progression, therapeutic responses, and reflect the genetic characteristics of the primary tumor.
Circulating Tumor DNA
Circulating tumor DNA (ctDNA) is a subtype of circulating, cell-free DNA (cfDNA) that originates from tumor cells and is composed of small fragments of DNA (180–200 base pairs in length) 85,87. CtDNA is typically released during tumor cell death and rapidly cleared by phagocytic processes. As such, the concentration of cfDNA is about very low (10–100 ng/ml) in plasma in normal individuals and in early stage cancers. However, the levels could be almost 10-fold higher in patients with advanced cancers 87. The challenge in ctDNA based liquid biopsy is two-fold, in extraction and in targeted detection. At the level of extraction, optimization of methodologies would increase the chance that these markers are detected. At the level of detection sensitive technologies, including droplet digital PCR, BEAMing (beads, emulsion, amplification, and magnetics) and next generation sequencing, allow identification of targeted mutations in various biofluids 85.
Studies in multiple cancers have demonstrated the utility of cfDNA based diagnosis 88–91, monitoring and assessing response to therapy 92–96. Growing evidence also suggests that cfDNA concentration correlates with tumor burden, cancer stage, cellular turnover, and response to therapy 87,97. However, the application of this strategy to gliomas has been hindered by the relatively low abundance of detectable molecular alterations in plasma (<10% of patients) as compared to other tumor types, likely due to the BBB 87.
However, emerging studies have reported the detection of tumor specific mutations in the cfDNA of patients with glioma (Table 2) 87,98–110. Detection of glioma specific alterations such as TERT 105,111, EGFRvIII 102, IDH1108 and histone mutations 112 has shown promise in minimally invasive diagnosis, molecular profiling and classification of tumors. EGFR gene is amplified in 30–40% of GBMs and nearly 50% of them express the in-frame deleted variant of EGFR receptor, EGFRvIII and represents an aggressive subtype of GBM 113–119. IDH1 mutations occur in 10% GBMs 120. TERT promoter mutations occur in 60% of GBMs, associated with poorer outcomes. Simultaneous presence of IDH1 and TERT promoter mutations confer survival benefit for GBM patients. H3K27M mutation status has both diagnostic and prognostic significance in diffuse midline glioma 121. Furthermore, identification of prognostic markers such as MGMT can be valuable to guide therapy 98,101,107,109,110. Considering the association of MGMT promoter methylation with pseudoprogression, a positive MGMT methylation status can suggest the likelihood that a contrast enhancing lesion indicates pseudoprogression. Emerging studies also suggest the possibility of using ctDNA analysis to pursue treatment alternatives 100 as well as assess response to immunotherapy 100,122. Recent studies have shown the ability of ctDNA based longitudinal follow up in GBM patients. Miller et al. showed that CSF ctDNA based sequencing analysis can be used to track the evolution of tumors 104. Arruda and Mourliere showed that the levels of tumor specific mutation status in ctDNA fraction of CSF parallels the disease status, correlating with progressive disease 103,123,124.
Table 2.
Summary of studies using circulating free DNA-based analysis for GBM.
| Author, Year | Biofluid | Methodology of cfDNA analysis | Genetic cargo evaluated | Diagnostic sensitivity | Potential role |
|---|---|---|---|---|---|
| Balana, 2003 | Plasma | Methylation Specific PCR assay | MGMT methylation status | 81% | Prognosis; Treatment selection |
| Liu, 2010* | CSF, serum | Methylated DNA immunoprecipitation RT-PCR analysis | MGMT, p16INK4a, TIMP3, THBS1 promoter hypermethylation | CSF, 50% Serum 50% | prognosis |
| Lavon, 2010* | Serum | Methylation Specific PCR assay | MGMT promoter methylation status | 51% | Diagnosis |
| Boisselier. 2012* | Plasma | DNA amplification by COLD PCR and further characterization by digital PCR | IDH1 mutation | 60% | Diagnosis |
| Salkeni, 2013 | Plasma | Long range PCR amplification | EGFRvIII deletion variant | 23% | Monitoring |
| Majchrzak-Celińska, 2013* | Serum | Methylation Specific PCR assay | MGMT, RASSF1A, p15INK4B, p14ARF promoter methylation | 81% | Diagnosis |
| Bettegowda 2014* | Plasma | Droplet digital PCR | TP53, EGFR, PTEN | <10% | Diagnosis |
| Wang, 2015* | Serum CSF | Methylation Specific PCR assay | MGMT promotor methylation | Serum, 21% CSF, 43% | Prognosis |
| De Mattos-Arruda, 2015* | CSF, Plasma | Targeted capture massively parallel sequencing | IDH1, TP53, PTEN, EGFR, FGFR2, ERBB2 mutations | - | Monitoring |
| Schwaederle, 2016* | Plasma | Next generation sequencing | TP53, NOTCH1 | 27% | Molecular profiling, Prognosis |
| Juratli, 2018* | CSF, Plasma | Nested PCR | TERT promoter mutations | CSF, 92% Plasma, 8% | Diagnosis |
| Piccioni. 2019* | Plasma | Guardant360® cfDNA digital next generation sequencing assay | TP53, NF1, MET, APC, PDGFRA mutations MET, EGFR, ERBB2 amplifications | 55% | Molecular profiling, treatment selection |
| Miller, 2019* | CSF | Next generation sequencing | IDH1, IDH2, TP53 mutations; CDKN2A, CDKN2B deletions; EGFR amplification | 49% (posttherapy) | Prognosis, Monitoring |
| Mouliere, 2019* | CSF, Plasma Urine | Tumor-guided capture sequencing | Matched clonal and private mutations | CSF, 50% Plasma, 50% Urine, 13% | Diagnosis |
| Cordova, 2019 | Plasma | Droplet digital PCR | TERT promoter mutations | 46% | Monitoring |
Abbreviations.APC, adenomatous polyposis coli; CDKN2A, Cyclin Dependent Kinase Inhibitor 2A, CDKN2B, Cyclin Dependent Kinase Inhibitor 2B; CfDNA, circulating free DNA; CSF, cerebrospinal fluid; EGFR, epidermal growth factor receptor; ERBB2, Erb-B2 Receptor Tyrosine Kinase 2; FGFR2, Fibroblast growth factor receptor 2; IDH, isocitrate dehydrogenase; MGMT, O(6)-Methylguanine-DNA methyltransferase; NF1, neurofibromatosis type 1; PDGFRA, platelet-derived growth factor receptor alpha; PTEN, Phosphatase and tensin homolog; RASSF1A, Ras association domain family 1 isoform A; RT-PCR, real time polymerase chain reaction; TERT, Telomerase reverse transcriptase; THBS1, Thrombospondin 1; TIMP3, TIMP Metallopeptidase Inhibitor 3.
These described cases in this series are not limited to GBM.
CSF studies have consistently shown higher sensitivities in ctDNA detection compared to blood based analysis 101,105–107, however, serial monitoring may not be practical considering the invasiveness of CSF collection. While recent studies have explored the potential of alternative biofluids such as urine 123, blood based detection has shown promising sensitivity and is more practical for serial monitoring. cfDNA is shed by virtually all cells in the body; it is especially difficult to identify ctDNA within this background. Furthermore, ctDNA fragments have a very short half-life and require rapid processing 12. Most cfDNA studies in glioma have small sample sizes and have used various methods of mutant detection to allow meaningful comparisons. Lack of standardized procedures for sample collection, isolation, and analysis has been another major hurdle for the field, making it challenging to compare sensitivities across various studies. Nevertheless, development of sensitive technologies for ctDNA capture and tumor specific mutant and methylation status can provide minimally invasive diagnosis and monitoring for GBMs, providing insights into the spatiotemporal heterogeneity over time and therapy.
Extracellular vesicles
Tumor cells actively release stable membrane bound nanobodies called EVs. They carry functional genomic and proteomic cargo from their parental cells and deliver that information to surrounding and distant recipient cells to modulate their behavior. EVs are identified to modulate and reprogram the tumor microenvironment to promote tumor proliferation, reprogram metabolic activity, induce angiogenesis, escape immune surveillance, acquire drug resistance and undergo invasion 125. They can also be detected in biofluids including plasma, CSF, urine etc. Their stable configuration confers a protective niche for tumor derived mRNA, miRNA and proteins. EVs are classified according to size and biogenesis pathway: microvesicles (100–1000 nm) are formed by budding of the plasma membrane, exosomes (30–150 nm) are formed by the fusion of intracellular multivesicular bodies with the plasma membrane, apoptotic bodies (1000–5000 nm) are produced and released by dying cells, and large oncosomes ( >1 μm ) are formed by non-apoptotic blebs from plasma membrane 85,126,127. Detection of tumor specific EVs amidst the vast background of normal EVs derived from every other cell of the body is challenging. Methodologies to allow for optimal EV isolation and sensitive technologies for EV cargo analysis are being developed.
Emerging reports have demonstrated the utility of EVs as biomarkers of cancer diagnosis 3,128–130 and prognosis 131–133. Quantitative studies demonstrated that EV numbers in plasma were higher in patients with GBM patients compared to controls and the numbers dropped with therapy 134–137. Higher numbers were noted in PD compared to patients with stable disease or pseudoprogression 134,135. However, nanoparticle tracking analysis or flow cytometry based EV quantification methods are non-specific and non-representative of true tumor derived EV burden. Nevertheless, these studies indicate that the pattern of EV dynamics parallel the disease course in the broad sense.
Recent EV based mRNA studies have reported sensitivities between 28% and 82% for the detection of EGFRvIII in EVs extracted from serum of GBM patients 138,139. In addition, analysis of CSF-derived EV mRNA has shown higher sensitivities in IDH mutant detection compared to blood based EV analysis 140,141. Several protein based EV analysis methods have been used for tumor specific EV characterization 142,143. Shao et al. used micro nuclear magnetic resonance system chip based EV protein analysis and identified EGFRvIII, PDPN and IDH1 proteins in the plasma EVs of glioma patients. The sensitivities were higher for EGFRvIII and PDPN (68%) than they were for IDH1 (16%)142. Chandran et al. showed that detection of syndecan-1 in plasma (sensitivity, 71%) can differentiate high grade gliomas from low grade gliomas. Other groups have explored EV miRNAs including miR-301a144, miR-182–5p, miR328–3p, miR-339–5p, miR-340–5p, miR-485–3p, miR-486–5p and miR-543145 miR-21, miR-222, miR-124–3p146, miR-320 and miR-574–3p, as well as a small noncoding RNA, RNU6147 as diagnostic tools. Specifically, Lan et al. and Santangelo et al. showed that serum miR-301a levels 144, miR-21, miR-222 and miR-124–3p levels 146 in serum EVs were higher in GBM patients and paralleled the clinical disease course, with levels decreasing with surgical resection and increasing with recurrence 144.
Recent studies have explored the potential of fluorescent labelled EV quantification using imaging flow cytometry. Ricklefs et al. used imaging flow cytometry to show that EVs with double positive tetraspanin expression (CD63+/CD81+) are enriched in patient plasma samples 148. Galbo et al. showed that CD9+/GFAP+/SVN+ EVs can predict response to therapy 149. These studies highlight the possibility of monitoring GBM EVs using surface marker analysis. Jones et al. identified protoporphyrin positive EVs in plasma of patients with malignant glioma undergoing fluorescence guided surgery with 5-Amino levulinic acid (5-ALA) as a potential diagnostic strategy to identify and monitor malignant gliomas. As the drug is currently approved only for surgical resection, the potential of the drug in a longitudinal setting has not yet been evaluated 150.
The ability of GBM-EVs to cross the BBB has always been a topic of debate, which could be the reason for lower sensitivities of target detection in blood based EV analysis. Garcia-Romero et al. recently demonstrated that tumor specific EVs are capable of crossing intact BBB and navigating into plasma, using an orthotopic xenotransplant mouse model of human glioma-cancer stem cells featuring an intact BBB141.
Although both CSF and plasma/serum based EV analysis is promising, the superiority of a biofluid for EV based monitoring is still unclear. However, plasma/serum based monitoring is more practical for the purposes of longitudinal monitoring as repeated CSF sampling is not feasible 151. Biofluids such as urine and saliva need to be explored. Small sample sizes, variable technologies, lack of a gold standard method of EV characterization makes it difficult to make meaningful comparisons. However, these initial EV biomarker discovery studies show promise and their potential in longitudinal setting is yet to be explored.
Blood Brain Barrier
Although CSF is considered as the ideal biofluid for liquid biopsy based diagnosis and monitoring due to the anatomic proximity to the primary tumor, plasma and serum are easily accessible and minimally invasive. CSF collection is highly invasive, requires trained professionals and has several potential complications. The utility of blood based liquid biopsy depends on the inherent ability of the liquid biopsy substrates (CTCs, CtDNA, EVs) to cross the BBB and reach peripheral circulation. The BBB provides both physical and biochemical barriers with a continuous network of tight and adherens junctions between brain capillary endothelial cells preventing paracellular diffusion of hydrophilic molecules153. The most obvious path for these substrates is circumnavigation of the BBB at the regions of BBB disruption. Despite the fact that GBM is a highly aggressive and invasive brain tumor with a disruption of BBB, large sections of BBB remain intact 152. Wide scale disruptions of BBB usually occur with the progression of disease.
CTCs are large and require disrupted BBB to navigate their way into the bloodstream. This could be one of the reasons for their low abundance in blood. It is unlikely for the hydrophilic ctDNA to cross an intact BBB. CtDNA could enter the bloodstream via the sites of BBB disruption. Studies have shown higher ctDNA levels in blood in high grade gliomas than low-grade gliomas87, which can partly be attributed to the BBB disruption in high grade gliomas. A positive correlation between the extent of BBB disruption and ctDNA levels in the blood was identified by Nabavizadeh and colleagues, indicating the ability to detect ctDNA as a function of BBB disruption strengthening this notion 154. Morad et al demonstrated using in vitro and in vivo BBB models, the ability of native tumor derived EVs to breach the intact BBB and reach the circulation via transcytosis155. Kur et al showed a neuronal activity driven uptake of hematopoietic cell derived EVs by neurons across the BBB via transcytosis156. These studies provide a proof of principle that tumor specific EVs navigate through the intact BBB, and reach the peripheral circulation. However, further investigation is required to unveil the ability of liquid biopsy substrates to reach peripheral circulation as well as determine the optimal biofluid for monitoring disease progression.
Future directions
Promising developments in the field of liquid biopsy can aid clinicians making diagnostic and therapeutic decisions to manage GBMs. The potential of combining both liquid biopsy fractions, cfDNA from dying cells and actively secreted EVs from live cells might be a better representation of the ongoing tumor dynamics. Recent clinical application of liquid biopsy based diagnostics such as cobas EGFR Mutation Test version 2, which monitors T790M mutation status in plasma cfDNA in non-small cell lung cancer patients to aid the use of osimertinib 157,158, and ExoDx Prostate IntelliScore (EPI Test, Bio-Techne), a non-invasive EV based urine test measures three mRNAs considered to be important genomic RNA biomarkers that can guide urologists in determining the true need for a prostate biopsy 159,160 have shown promise of liquid biopsy for minimally invasive diagnostics and prognostics. However, there are several challenges along the pathway of blood-based biomarker development from discovery to clinical utility, and systematic approach to tackle these hurdles is critical to develop a blood based biomarker with clinical utility. These aspects are extensively reviewed elsewhere 151,161,162. Ideally, an advanced machine learning model 163,164 integrating clinical, imaging and liquid biopsy based molecular characterization could help decision making during follow-up. With the advent of sensitive technologies, liquid biopsy could be the future of tumor diagnosis, monitoring and therapy response.
Conclusion
Liquid biopsy strategies offer minimally invasive tools for diagnosis as well as monitoring brain tumors for response to therapy and for predicting treatment related changes. Despite recent advances in liquid biopsy based biomarking brain tumors, the sensitivity of detection in brain tumors have been low. As of now, there are no clinically applicable circulating biomarkers for the diagnosis and monitoring of GBMs, but promising developments in the field with complimentary sensitive technologies have moved the needle closer to a clinical assay. Biobanking and appropriate sample collection and handling protocols are needed to allow the field to harvest and save biofluids for development and validation of biomarkers and technologies. Ideally, a three-pronged monitoring approach correlating clinical status, imaging characteristics and liquid biopsy based molecular characterization, to provide a comprehensive clinical and molecular snapshot of the tumor in space and time, to assess the evolution of the tumor, and identify true PD from radiation effects could be a potential solution to the current challenge.
Figure 1.
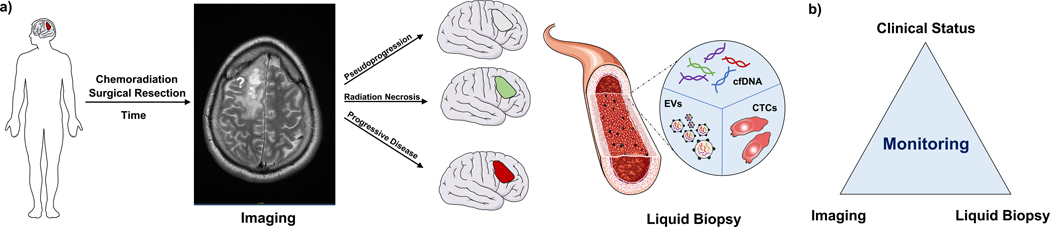
a. Patients with glioblastoma, post-surgical resection and chemoradiation, are monitored using serial MR imaging. A gadolinium enhancing lesion on contrast enhanced magnetic resonance imaging (MRI) could either be true progressive disease (PD), or radiation effects such as pseudoprogression or radiation necrosis (RN). Liquid biopsy strategies including circulating tumor cells (CTCs), circulating free DNA (CfDNA), extracellular vesicles (EVs) can provide minimally invasive modalities of monitoring brain tumors. b. Three-pronged monitoring approach using integrating clinical status, imaging modalities and liquid biopsy strategies could be a potential solution for tracking the tumor evolution over time and therapy. Blood vessel image from Smart Servier.
Table 3.
Summary of studies using extracellular vesicle EV-based analysis for GBM.
| Author, Year | Biofluid | Methodology of EVanalysis | Genetic cargo evaluated | Diagnostic sensitivity | Potential role |
|---|---|---|---|---|---|
| Skog, 2008 | Serum | Nested RT-PCR EGFRvIII mRNA | EGFRvIII | 28% | Diagnosis |
| Shao, 2012 | Plasma | Micro nuclear magnetic resonance system chip based EV protein analysis | EGFRvIII, IDH1, PDPN proteins | 68% (EGFRvIII, PDPN) 16% IDH1 | Diagnosis |
| Chen & Balaj, 2013 | CSF, Serum | BEAMing (beads, emulsion, amplification, magnetics) RT-PCR and ddPCR | IDH1 mutation | 62.5% 0% | Diagnosis |
| Akers, 2013 | CSF | RT-PCR | miR-21 | 85% initial cohort, 87% validation cohort | Diagnosis/Monitoring |
| Manterola, 2014 | Serum | RT-PCR | miR-320, miR-574–3p, RNU6–1 expression | miR-320, 65%, miR-574–3p, 59%, RNU6–1, 73% | Diagnosis |
| Koch, 2014 | Plasma | Flow cytometry: size of 300 nm or greater and Annexin V positivity | - | - | Monitoring |
| Evans, 2016 | Plasma | Flow cytometry: Annexin V positivity | - | - | Monitoring/ Prognosis |
| Garcia-Romero, 2017* | Plasma | Fast Cold-PCR | IDH1 mutation | 48% | Diagnosis |
| Galbo, 2017* | Serum | Imaging flow cytometry- fluorescent labelled antibodies | CD9+/GFAP+/SVN+ EVs | - | Monitoring |
| Andre-Gregoire, 2018 | Plasma | Tunable resistive pulse sensing analysis (TRPS) | - | - | - |
| Ricklefs, 2018* | Plasma | Droplet PCR | PD-L1 DNA | 67% | Monitoring |
| Manda, 2018* | Serum | Semi-nested RT-PCR | EGFRvIII mRNA | 82% | Diagnosis |
| Lan, 2018* | Serum | RT-PCR | miR-301A | _ | Prognosis/Monitoring |
| Ebrahimkhani, 2018* | Serum | Deep sequencing | miR-182–5p, miR-328–3p, miR-339–5p, miR-340–5p, miR-485–3p, miR-486–5p and miR-543 | 92% λ | Diagnosis |
| Santangelo, 2018* | Serum | RT-PCR | miR-21, miR-222, miR-124–3p | miR-21, 84%, miR-222, 80% miR-124–3p 78% | Diagnosis/Monitoring |
| Osti, 2019* | Plasma | Nanoparticle tracking analysis, Mass spectrometry | Proteins: vWF, APCS, C4B, AMBP, APOD, AZGP1, C4BPB, Serpin3, FTL, C3, and APOE | - | Monitoring |
| Jones & Yekula, 2019 | Plasma | Imaging flow cytometry based monitoring of PpIX positive EVs pre and post 5-ALA based fluorescent guided surgery | PpIX positive EVs | 4 out of 4 | Diagnosis/Monitoring |
| Chandran, 2019 | Plasma | Mass spectrometry, Nanoparticle tracking analysis, Electron microscopy | Levels of Syndecan 1 | 71% | Diagnosis/Classification |
| Ricklefs, 2019* | Plasma | Imaging flow cytometry- fluorescent labelled antibodies | CD63+/CD81+ EVs | - | - |
Abbreviations.AMBP, Alpha-1-Microglobulin/ Bikunin Precursor; APCS, Serum amyloid P component; APOD, Apolipoprotein D; APOE, Apolipoprotein E; AZGP1, Alpha-2-Glycoprotein 1; C3, complement C3; C4B, Complement C4B; C4BPB, Complement Component 4 Binding Protein Beta; CSF, cerebrospinal fluid; ddPCT, droplet digital PCR; EGFR, epidermal growth factor receptor; FTL, Ferritin Light Chain; IDH, isocitrate dehydrogenase; PDPN, podoplanin; PpIX, Protoporphyrin; RT-PCR, reverse transcriptase polymerase chain reaction; vWF, von Willebrand factor; 5-ALA, 5 Aminolaevulinic acid.
These described cases in this series are not limited to GBM.
Acknowledgments
Funding sources
This work is supported by grants U01 CA230697 (BSC, LB), UH3 TR000931 (BSC), P01 CA069246 (BSC). The funding sources had no role in the writing the manuscript or decision to submit the manuscript for publication. The authors have not been paid to write this article by any entity. The corresponding author has full access to the manuscript and assumes final responsibility for the decision to submit for publication.
Footnotes
Declaration of competing interests
None of the other authors declare any conflicts of interest.
References
- 1.Ostrom QT et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro-Oncology vol. 18 v1–v75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gruber ML & Buster WP Temozolomide in Combination With Irinotecan for Treatment of Recurrent Malignant Glioma. Am. J. Clin. Oncol 27, 33–38 (2004). [DOI] [PubMed] [Google Scholar]
- 3.van Linde ME et al. Treatment outcome of patients with recurrent glioblastoma multiforme: a retrospective multicenter analysis. J. Neurooncol 135, 183–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambady P, Bettegowda C. & Holdhoff M. Emerging methods for disease monitoring in malignant gliomas. CNS Oncol 2, 511–522 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen PY et al. Updated Response Assessment Criteria for High-Grade Gliomas: Response Assessment in Neuro-Oncology Working Group. J. Clin. Oncol 28, 1963–1972 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Chukwueke UN & Wen PY Use of the Response Assessment in Neuro-Oncology (RANO) criteria in clinical trials and clinical practice. CNS Oncol 8, CNS28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parvez K, Parvez A. & Zadeh G. The diagnosis and treatment of pseudoprogression, radiation necrosis and brain tumor recurrence. Int. J. Mol. Sci 15, 11832–11846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thust SC, van den Bent MJ & Smits M. Pseudoprogression of brain tumors. Journal of Magnetic Resonance Imaging vol. 48 571–589 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiang GC et al. Magnetic Resonance Spectroscopy, Positron Emission Tomography and Radiogenomics-Relevance to Glioma. Front. Neurol 9, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen VA, Simonsen HJ, Law I, Larsson HBW & Hansen AE Evaluation of dynamic contrast-enhanced T1-weighted perfusion MRI in the differentiation of tumor recurrence from radiation necrosis. Neuroradiology 55, 361–369 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Brandsma D, Stalpers L, Taal W, Sminia P. & van den Bent MJ Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 9, 453–461 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Shankar GM, Balaj L, Stott SL, Nahed B. & Carter BS Liquid biopsy for brain tumors. Expert Review of Molecular Diagnostics vol. 17 943–947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bark JM, Kulasinghe A, Chua B, Day BW & Punyadeera C. Circulating biomarkers in patients with glioblastoma. British Journal of Cancer vol. 122 295–305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruz LCH da et al. Pseudoprogression and Pseudoresponse: Imaging Challenges in the Assessment of Posttreatment Glioma. American Journal of Neuroradiology vol. 32 1978–1985 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandes AA et al. Disease progression or pseudoprogression after concomitant radiochemotherapy treatment: Pitfalls in neurooncology. Neuro. Oncol 10, 361–367 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellingson BM, Wen PY, van den Bent MJ & Cloughesy TF Pros and cons of current brain tumor imaging. Neuro. Oncol 16 Suppl 7, vii2–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsien C. et al. Parametric Response Map As an Imaging Biomarker to Distinguish Progression From Pseudoprogression in High-Grade Glioma. J. Clin. Oncol 28, 2293–2299 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gahramanov S. et al. Potential for differentiation of pseudoprogression from true tumor progression with dynamic susceptibility-weighted contrast-enhanced magnetic resonance imaging using ferumoxytol vs. gadoteridol: a pilot study. Int. J. Radiat. Oncol. Biol. Phys 79, 514–523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yaman E. et al. Radiation induced early necrosis in patients with malignant gliomas receiving temozolomide. Clin. Neurol. Neurosurg 112, 662–667 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Brandes AA et al. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation With MGMT promoter methylation status. J. Clin. Oncol 27, 1275–1279 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Esteller M. et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med 343, 1350–1354 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Hegi ME et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Zikou A. et al. Radiation Necrosis, Pseudoprogression, Pseudoresponse, and Tumor Recurrence: Imaging Challenges for the Evaluation of Treated Gliomas. Contrast Media Mol. Imaging 2018, 6828396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang H-C et al. Pseudoprogression in patients with malignant gliomas treated with concurrent temozolomide and radiotherapy: potential role of p53. Journal of Neuro-Oncology vol. 102 157–162 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Qian X. et al. Identification of biomarkers for pseudo and true progression of GBM based on radiogenomics study. Oncotarget 7, 55377–55394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandes AA et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J. Clin. Oncol 26, 2192–2197 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Langleben DD & Segall GM PET in differentiation of recurrent brain tumor from radiation injury. J. Nucl. Med 41, 1861–1867 (2000). [PubMed] [Google Scholar]
- 28.Fink J, Born D. & Chamberlain MC Radiation necrosis: relevance with respect to treatment of primary and secondary brain tumors. Curr. Neurol. Neurosci. Rep 12, 276–285 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Belka C, Budach W, Kortmann RD & Bamberg M. Radiation induced CNS toxicity-molecular and cellular mechanisms. Br. J. Cancer 85, 1233–1239 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheline GE, Wara WM & Smith V. Therapeutic irradiation and brain injury. Int. J. Radiat. Oncol. Biol. Phys 6, 1215–1228 (1980). [DOI] [PubMed] [Google Scholar]
- 31.Giglio P. & Gilbert MR Cerebral radiation necrosis. Neurologist 9, 180–188 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Johannesen TB, Lien HH, Hole KH & Lote K. Radiological and clinical assessment of long-term brain tumour survivors after radiotherapy. Radiother. Oncol 69, 169–176 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Reinhold HS, Calvo W, Hopewell JW & van der Berg AP Development of blood vessel-related radiation damage in the fimbria of the central nervous system. Int. J. Radiat. Oncol. Biol. Phys 18, 37–42 (1990). [DOI] [PubMed] [Google Scholar]
- 34.Schultheiss TE, Kun LE, Ang KK & Stephens LC Radiation response of the central nervous system. Int. J. Radiat. Oncol. Biol. Phys 31, 1093–1112 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Yoshii Y. Pathological review of late cerebral radionecrosis. Brain Tumor Pathol. 25, 51–58 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Zhao W. & Robbins MEC Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Curr. Med. Chem 16, 130–143 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Ansari R. et al. Anti-TNFA (TNF-α) Treatment Abrogates Radiation-Induced Changes in Vascular Density and Tissue Oxygenation. Radiation Research vol. 167 80–86 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Mayhan WG Cellular mechanisms by which tumor necrosis factor-alpha produces disruption of the blood-brain barrier. Brain Res. 927, 144–152 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Wilson CM, Gaber MW, Sabek OM, Zawaski JA & Merchant TE Radiation-induced astrogliosis and blood-brain barrier damage can be abrogated using anti-TNF treatment. Int. J. Radiat. Oncol. Biol. Phys 74, 934–941 (2009). [DOI] [PubMed] [Google Scholar]
- 40.van Bruggen N. et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J. Clin. Invest 104, 1613–1620 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaw PJ & Bates D. Conservative treatment of delayed cerebral radiation necrosis. J. Neurol. Neurosurg. Psychiatry 47, 1338–1341 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohshi K. et al. Successful treatment of radiation-induced brain necrosis by hyperbaric oxygen therapy. J. Neurol. Sci 209, 115–117 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Leber KA, Eder HG, Kovac H, Anegg U. & Pendl G. Treatment of cerebral radionecrosis by hyperbaric oxygen therapy. Stereotact. Funct. Neurosurg 70 Suppl 1, 229–236 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Glantz MJ et al. Treatment of radiation-induced nervous system injury with heparin and warfarin. Neurology 44, 2020–2027 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Rizzoli HV & Pagnanelli DM Treatment of delayed radiation necrosis of the brain. Journal of Neurosurgery vol. 60 589–594 (1984). [DOI] [PubMed] [Google Scholar]
- 46.Levin VA et al. Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int. J. Radiat. Oncol. Biol. Phys 79, 1487–1495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong ET, Huberman M, Lu X-Q & Mahadevan A. Bevacizumab reverses cerebral radiation necrosis. J. Clin. Oncol 26, 5649–5650 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez J, Kumar AJ, Conrad CA & Levin VA Effect of bevacizumab on radiation necrosis of the brain. Int. J. Radiat. Oncol. Biol. Phys 67, 323–326 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Furuse M, Kawabata S, Kuroiwa T. & Miyatake S-I Repeated treatments with bevacizumab for recurrent radiation necrosis in patients with malignant brain tumors: a report of 2 cases. J. Neurooncol 102, 471–475 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Rahmathulla G, Recinos PF, Valerio JE, Chao S. & Barnett GH Laser interstitial thermal therapy for focal cerebral radiation necrosis: a case report and literature review. Stereotact. Funct. Neurosurg 90, 192–200 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Siu A. et al. Radiation necrosis following treatment of high grade glioma--a review of the literature and current understanding. Acta Neurochir. 154, 191–201; discussion 201 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Mou Y-G et al. Surgical management of radiation-induced temporal lobe necrosis in patients with nasopharyngeal carcinoma: report of 14 cases. Head Neck 33, 1493–1500 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Dasgupta A, Lim AR & Ghajar CM Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol. Oncol 11, 40–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joosse SA, Gorges TM & Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Molecular Medicine vol. 7 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J. & Weinberg RA Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Aceto N. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gkountela S. et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 176, 98–112.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ignatiadis M, Lee M. & Jeffrey SS Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin. Cancer Res 21, 4786–4800 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Sullivan JP et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discovery vol. 4 1299–1309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Müller C. et al. Hematogenous dissemination of glioblastoma multiforme. Sci. Transl. Med 6, 247ra101 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Cristofanilli M. et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med 351, 781–791 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Krebs MG et al. Molecular analysis of circulating tumour cells-biology and biomarkers. Nat. Rev. Clin. Oncol 11, 129–144 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Ilie M. et al. ‘Sentinel’ Circulating Tumor Cells Allow Early Diagnosis of Lung Cancer in Patients with Chronic Obstructive Pulmonary Disease. PLoS ONE vol. 9 e111597 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan W-T et al. Circulating tumor cell status monitors the treatment responses in breast cancer patients: a meta-analysis. Sci. Rep 7, 43464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paoletti C. et al. Development of Circulating Tumor Cell-Endocrine Therapy Index in Patients with Hormone Receptor–Positive Breast Cancer. Clinical Cancer Research vol. 21 2487–2498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smerage JB et al. Circulating Tumor Cells and Response to Chemotherapy in Metastatic Breast Cancer: SWOG S0500. Journal of Clinical Oncology vol. 32 3483–3489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyamoto DT et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov. 2, 995–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antonarakis ES et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo X. et al. Isolation and molecular characterization of circulating melanoma cells. Cell Rep. 7, 645–653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakaizawa K. et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. British Journal of Cancer vol. 106 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maheswaran S. et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med 359, 366–377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gasch C. et al. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem 59, 252–260 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Kondo Y. et al. KRAS mutation analysis of single circulating tumor cells from patients with metastatic colorectal cancer. BMC Cancer 17, 311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bono J. S. de et al. Circulating Tumor Cells Predict Survival Benefit from Treatment in Metastatic Castration-Resistant Prostate Cancer. Clinical Cancer Research vol. 14 6302–6309 (2008). [DOI] [PubMed] [Google Scholar]
- 75.Giuliano M. et al. Circulating tumor cells as early predictors of metastatic spread in breast cancer patients with limited metastatic dissemination. Breast Cancer Research vol. 16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Normanno N, De Luca A, Gallo M, Chicchinelli N. & Rossi A. The prognostic role of circulating tumor cells in lung cancer. Expert Rev. Anticancer Ther 16, 859–867 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Tsai W-S et al. Circulating Tumor Cell Count Correlates with Colorectal Neoplasm Progression and Is a Prognostic Marker for Distant Metastasis in Non-Metastatic Patients. Sci. Rep 6, 24517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lorente D. et al. Circulating tumour cell increase as a biomarker of disease progression in metastatic castration-resistant prostate cancer patients with low baseline CTC counts. Annals of Oncology vol. 29 1554–1560 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Tong B. et al. Prognostic significance of circulating tumor cells in non-small cell lung cancer patients undergoing chemotherapy. Oncotarget vol. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith DR, Hardman JM & Earle KM Contiguous glioblastoma multiforme and fibrosarcoma with extracranial metastasis. Cancer 24, 270–276 (1969). [DOI] [PubMed] [Google Scholar]
- 81.Krol I. et al. Detection of circulating tumour cell clusters in human glioblastoma. British Journal of Cancer vol. 119 487–491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Macarthur KM et al. Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Res. 74, 2152–2159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gao F. et al. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget 7, 71330–71340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu T. et al. Circulating Glioma Cells Exhibit Stem Cell-like Properties. Cancer Res. 78, 6632–6642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zachariah MA, Oliveira-Costa JP, Carter BS, Stott SL & Nahed BV Blood-based biomarkers for the diagnosis and monitoring of gliomas. Neuro. Oncol 20, 1155–1161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aktas B. et al. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Research vol. 11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bettegowda C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med 6, 224ra24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forshew T. et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med 4, 136ra68 (2012). [DOI] [PubMed] [Google Scholar]
- 89.Kinde I, Wu J, Papadopoulos N, Kinzler KW & Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. U. S. A 108, 9530–9535 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wen L. et al. Erratum: Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell Research vol. 25 1376–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mao L, Hruban RH, Boyle JO, Tockman M. & Sidransky D. Detection of oncogene mutations in sputum precedes diagnosis of lung cancer. Cancer Res. 54, 1634–1637 (1994). [PubMed] [Google Scholar]
- 92.Gormally E. et al. TP53 and KRAS2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: a prospective study. Cancer Res. 66, 6871–6876 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Fernandez-Cuesta L. et al. Identification of Circulating Tumor DNA for the Early Detection of Small-cell Lung Cancer. EBioMedicine vol. 10 117–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fackler MJ et al. Novel Methylated Biomarkers and a Robust Assay to Detect Circulating Tumor DNA in Metastatic Breast Cancer. Cancer Research vol. 74 2160–2170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Genovese G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sozzi G. et al. O-297 Quantification of free circulating DNA as a diagnostic marker in lung cancer. Lung Cancer vol. 41 S86–S87 (2003). [DOI] [PubMed] [Google Scholar]
- 97.Palmirotta R. et al. Liquid biopsy of cancer: a multimodal diagnostic tool in clinical oncology. Therapeutic Advances in Medical Oncology vol. 10 175883591879463 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Balaña C. et al. O6-methyl-guanine-DNA methyltransferase methylation in serum and tumor DNA predicts response to 1,3-bis(2-chloroethyl)-1-nitrosourea but not to temozolamide plus cisplatin in glioblastoma multiforme. Clin. Cancer Res 9, 1461–1468 (2003). [PubMed] [Google Scholar]
- 99.Schwaederle M. et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget vol. 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Piccioni DE et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol 8, CNS34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Z. et al. promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. Biomed Rep 3, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salkeni MA et al. Detection of EGFRvIII mutant DNA in the peripheral blood of brain tumor patients. J. Neurooncol 115, 27–35 (2013). [DOI] [PubMed] [Google Scholar]
- 103.De Mattos-Arruda L. et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun 6, 8839 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miller AM et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565, 654–658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Juratli TA et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clinical Cancer Research vol. 24 5282–5291 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Mouliere F. et al. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu B-L et al. Quantitative detection of multiple gene promoter hypermethylation in tumor tissue, serum, and cerebrospinal fluid predicts prognosis of malignant gliomas. Neuro. Oncol 12, 540–548 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boisselier B. et al. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology 79, 1693–1698 (2012). [DOI] [PubMed] [Google Scholar]
- 109.Lavon I, Refael M, Zelikovitch B, Shalom E. & Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro. Oncol 12, 173–180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Majchrzak-Celińska A. et al. Detection of MGMT, RASSF1A, p15INK4B, and p14ARF promoter methylation in circulating tumor-derived DNA of central nervous system cancer patients. J. Appl. Genet 54, 335–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cordova C. et al. Plasma cell-free circulating tumor DNA (ctDNA) detection in longitudinally followed glioblastoma patients using TERT promoter mutation-specific droplet digital PCR assays. Journal of Clinical Oncology vol. 37 2026–2026 (2019). [Google Scholar]
- 112.Azad TD, Jin MC, Bernhardt LJ & Bettegowda C. Liquid biopsy for pediatric diffuse midline glioma: a review of circulating tumor DNA and cerebrospinal fluid tumor DNA. Neurosurg. Focus 48, E9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sugawa N, Ekstrand AJ, James CD & Collins VP Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. U. S. A 87, 8602–8606 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ekstrand AJ, Sugawa N, James CD & Collins VP Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proceedings of the National Academy of Sciences 89, 4309–4313 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frederick L, Wang XY, Eley G. & James CD Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 60, 1383–1387 (2000). [PubMed] [Google Scholar]
- 116.Aldape KD et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol 63, 700–707 (2004). [DOI] [PubMed] [Google Scholar]
- 117.Ohgaki H. & Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol 64, 479–489 (2005). [DOI] [PubMed] [Google Scholar]
- 118.Parsons DW et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Louis DN et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Huang TY et al. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol Commun 5, 28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao J. et al. Author Correction: Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med 25, 1022 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Mouliere F. et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cell-free DNA in newly diagnosed patients with glioblastoma – a clinical prospective feasibility study. Oncotarget vol. 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yekula A. et al. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Frontiers in Immunology vol. 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roy S. et al. Navigating the Landscape of Tumor Extracellular Vesicle Heterogeneity. Int. J. Mol. Sci 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yekula A. et al. Large and small extracellular vesicles released by glioma cells in vitro and in vivo. Journal of Extracellular Vesicles vol. 9 1689784 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Park YH et al. Author Correction: Prostate-specific extracellular vesicles as a novel biomarker in human prostate cancer. Sci. Rep 9, 6051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.García-Silva S. et al. Correction: Use of extracellular vesicles from lymphatic drainage as surrogate markers of melanoma progression and BRAFV600E mutation. The Journal of Experimental Medicine vol. 216 1230–1230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van der Mijn JC et al. Analysis of AKT and ERK1/2 protein kinases in extracellular vesicles isolated from blood of patients with cancer. J Extracell Vesicles 3, 25657 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goto T. et al. An elevated expression of serum exosomal microRNA-191, - 21, −451a of pancreatic neoplasm is considered to be efficient diagnostic marker. BMC Cancer 18, 116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fu F, Jiang W, Zhou L. & Chen Z. Circulating Exosomal miR-17–5p and miR-92a-3p Predict Pathologic Stage and Grade of Colorectal Cancer. Transl. Oncol 11, 221–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu Q. et al. Circulating exosomal microRNAs as prognostic biomarkers for non-small-cell lung cancer. Oncotarget 8, 13048–13058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Koch CJ et al. Microvesicles as a Biomarker for Tumor Progression versus Treatment Effect in Radiation/Temozolomide-Treated Glioblastoma Patients. Transl. Oncol 7, 752–758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Evans SM et al. Initial evidence that blood-borne microvesicles are biomarkers for recurrence and survival in newly diagnosed glioblastoma patients. J. Neurooncol 127, 391–400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Osti D. et al. Clinical Significance of Extracellular Vesicles in Plasma from Glioblastoma Patients. Clin. Cancer Res 25, 266–276 (2019). [DOI] [PubMed] [Google Scholar]
- 137.André-Grégoire G, Bidère N. & Gavard J. Temozolomide affects Extracellular Vesicles Released by Glioblastoma Cells. Biochimie vol. 155 11–15 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Skog J. et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10, 1470–1476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Manda SV et al. Exosomes as a biomarker platform for detecting epidermal growth factor receptor-positive high-grade gliomas. J. Neurosurg 128, 1091–1101 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Chen WW et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 mRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol. Ther. Nucleic Acids 2, e109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.García-Romero N. et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget vol. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shao H. et al. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med 18, 1835–1840 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chandran VI et al. Ultrasensitive Immunoprofiling of Plasma Extracellular Vesicles Identifies Syndecan-1 as a Potential Tool for Minimally Invasive Diagnosis of Glioma. Clinical Cancer Research vol. 25 3115–3127 (2019). [DOI] [PubMed] [Google Scholar]
- 144.Lan F. et al. Serum exosomal miR-301a as a potential diagnostic and prognostic biomarker for human glioma. Cell. Oncol 41, 25–33 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Ebrahimkhani S. et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis Oncol 2, 28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Santangelo A. et al. A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. J. Neurooncol 136, 51–62 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Manterola L. et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro. Oncol 16, 520–527 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ricklefs FL et al. Imaging flow cytometry facilitates multiparametric characterization of extracellular vesicles in malignant brain tumours. J Extracell Vesicles 8, 1588555 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Galbo PM et al. Circulating CD9 /GFAP /survivin exosomes in malignant glioma patients following survivin vaccination. Oncotarget vol. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jones PS et al. Characterization of plasma-derived protoporphyrin-IX-positive extracellular vesicles following 5-ALA use in patients with malignant glioma. EBioMedicine 48, 23–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yekula A. et al. From laboratory to clinic: Translation of extracellular vesicle based cancer biomarkers. Methods (2020) doi: 10.1016/j.ymeth.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sarkaria JN et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro. Oncol 20, 184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cardoso FL, Brites D. & Brito MA Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res. Rev 64, 328–363 (2010). [DOI] [PubMed] [Google Scholar]
- 154.Mair R. et al. Measurement of Plasma Cell-Free Mitochondrial Tumor DNA Improves Detection of Glioblastoma in Patient-Derived Orthotopic Xenograft Models. Cancer Res. 79, 220–230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morad G. et al. Tumor-Derived Extracellular Vesicles Breach the Intact Blood–Brain Barrier via Transcytosis. ACS Nano vol. 13 13853–13865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kur I-M et al. Neuronal activity triggers uptake of hematopoietic extracellular vesicles in vivo. PLoS Biol. 18, e3000643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jenkins S, Cross D. & Scudder SA Osimertinib (TAGRISSOTM) and the cobas® EGFR Mutation Test v2. Companion and Complementary Diagnostics 429–443 (2019) doi: 10.1016/b978-0-12-813539-6.00023-7. [DOI] [Google Scholar]
- 158.Brown P. The Cobas® EGFR Mutation Test v2 assay. Future Oncology vol. 12 451–452 (2016). [DOI] [PubMed] [Google Scholar]
- 159.McKiernan J. et al. A Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer at Initial Biopsy. JAMA Oncol 2, 882–889 (2016). [DOI] [PubMed] [Google Scholar]
- 160.McKiernan J. et al. A Prospective Adaptive Utility Trial to Validate Performance of a Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer in Patients with Prostate-specific Antigen 2–10 ng/ml at Initial Biopsy. European Urology vol. 74 731–738 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Ioannidis JPA & Bossuyt PMM Waste, Leaks, and Failures in the Biomarker Pipeline. Clin. Chem 63, 963–972 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Zhao Z. et al. Extracellular vesicles as cancer liquid biopsies: from discovery, validation, to clinical application. Lab Chip 19, 1114–1140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jang B-S, Jeon SH, Kim IH & Kim IA Prediction of Pseudoprogression versus Progression using Machine Learning Algorithm in Glioblastoma. Sci. Rep 8, 12516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Akbari H. et al. NIMG-70. QUANTITATIVE IMAGE ANALYSIS AND MACHINE LEARNING TECHNIQUES FOR DISTINGUISHING TRUE PROGRESSION FROM PSEUDOPROGRESSION IN PATIENTS WITH GLIOBLASTOMA. Neuro-Oncology vol. 20 vi191–vi192 (2018). [Google Scholar]