

Abstract
Continuous globalization and industrialization have ensured metals are an increasing aspect of daily life. Their usefulness in manufacturing has made them vital to national commerce, security and global economy. However, excess exposure to metals, particularly as a result of environmental contamination or occupational exposures, has been detrimental to overall health. Excess exposure to several metals is considered environmental risk in the aetiology of several neurological and neurodegenerative diseases. Metal-induced neurotoxicity has been a major health concern globally with intensive research to unravel the mechanisms associated with it. Recently, greater focus has been directed at epigenetics to better characterize the underlying mechanisms of metal-induced neurotoxicity. Epigenetic changes are those modifications on the DNA that can turn genes on or off without altering the DNA sequence. This review discusses how epigenetic changes such as DNA methylation, post translational histone modification and noncoding RNA- mediated gene silencing mediate the neurotoxic effects of several metals, focusing on manganese, arsenic, nickel, cadmium, lead, and mercury.
Keywords: Metals, neurotoxicity, neurodegenerative diseases, epigenetics, environment
1. Introduction
An interplay between environmental and genetic factors have been associated with development of various forms of neurodegenerative and neurological diseases. Amongst environmental factors that have been implicated, exposure to metals is arguably the most concerning. Metals are identified by their properties which include malleability, ductility, reflectivity and conductivity. Examples are: lead (Pb), cadmium (Cd), mercury (Hg), arsenic (As), manganese (Mn) nickel (Ni) etc. Metals are innate in the surroundings, constitute majority of the periodic table and are bountiful in the atmosphere (Caito and Aschner 2015; Singh et al. 2011b; Tchounwou et al. 2012). They exist with high atomic mass and their density is usually in its minimum five times greater than the density of water (Caito and Aschner 2015). Generally, metals are useful in several aspects of life. Examples include the manufacturing industry for dry-cell batteries, planes, in production of pesticides, and ceramic manufacturing, to name a few. However, upon exposures to high amounts or form that are harmful to the body (Singh et al. 2011b), metals can exert multiple-organ toxicities. Dosage above the lowest-observed-adverse effect level (LOAEL) begets harmful consequences (Caito and Aschner 2015). Damage to cellular organelles, such as the mitochondria, cell membrane, lysosome, endoplasmic reticulum, nuclei and few catalysts that take part in absorption, detoxification and cellular rehabilitation have been reported after overexposure to metals (Tchounwou et al. 2012). However, while some of these metals such as Hg, and Pb, Mn are well-established neurotoxicants, others, such as Cd, As, and Ni are not considered “classical” neurotoxicants (see review Schofield, 2017). Occupational and environmental exposures to metals have been demonstrated to negatively impact brain function. Importantly, exposure to toxic metals has been implicated to trigger epigenetic changes (Jose et al. 2019; Yin et al. 2018). Numerous neurobehavioral diseases, neurodevelopmental diseases and neurodegenerative diseases have been known to develop due to one or more of the metal-toxicity-induced epigenetic mechanisms. Consequently, insight into the epigenetic mechanisms by which metals causes toxicity is key to modelling targeted treatments (Jose et al. 2019; see review Ryu et al. 2015). Here, we review recent evidence of epigenetic influence of metals that have been considered as environmental risk factors in the aetiology of various brain diseases, highlighting potential mechanisms by which metals overexposure might impact central nervous system function.
2. Metal neurotoxicity
Metals can be described as essential or non-essential based on their importance to the biological system. Essential metals are needed for regular body function such as acting as cofactors for enzymes and have dangerous effect on health when present in excess or dearth (Caito and Aschner 2015; see review Peres et al. 2016).
The brain is divided from the body’s circulatory system by the blood-brain barrier (BBB) which functions to prevent toxic materials from getting to the brain (Yokel 2006). However, there are specific messengers responsible for transfer of essential metals across the BBB (Caito and Aschner 2015). Non-essential metals such as mercury and lead have no established advantage to biological systems and are carried across the BBB in a manner that imitates molecule transfer (Caito and Aschner 2015; Singh et al. 2011b). These metals also get into the brain by passing the choroid plexus into the cerebrospinal fluid and build-up in specific brain regions (Yokel 2006).
Sources of these metals include: food, contaminated air, water, exposure to metals at work place. Miners and welders are at higher occupational risk of exposure to these metals (Sankhla et al. 2017). These metals can permeate the blood-brain barrier (BBB) leading to a build-up in the brain that results in neurotoxicity (Caito and Aschner 2015). Neurotoxic effects of metals can be acute or chronic with each manifesting different symptom. Acute metal toxicity is usually fast acting and serious. The symptoms include; headaches, cramping, nausea, vomiting, difficulty in breathing, radiation poisoning (from radioactive metals like uranium), convulsions, pain, cognitive, motor and language skill dysfunction, cell (neurons and glia) death, and death. Chronic toxicity is from prolonged exposure to metals and the symptoms manifested are due to build-up of these metals in tissues.
Chronic exposure to metals can be associated with neurodegeneration-linked diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD) (see Table 1). These neurodegenerative diseases all exhibit symptoms that impair the normal functioning of the nervous system and this can be explained by the minimal regeneration capability of neurons (Andrade et al. 2017; Breydo and Uversky 2011; Spencer and Lein 2014).
Table 1.
Some metals implicated as environmental risk factors in major neurological disorders
| Neurological disorders | Metals | Model | Route of exposure | Length of exposure | Mode of neurological damage | References |
|---|---|---|---|---|---|---|
| Alzheimer’s disease | Mercury | Rats pheochromcytoma cells (PC 12 cells) | Cell culture | 48hrs | Aggregation of APP (amyloid precursor protein) | (Song and Choi, 2013) |
| Manganese | Mice | oral | 5 days / week for 8 weeks | Increased levels of Ap peptides via impaired degradation. | (Tong et al., 2014) | |
| Nickel | Mice | Nasal | 3hrs | Increase in brain levels of both amyloid-P peptides Ap40andAp42. | (Kim et al., 2012) | |
| Cadmium | Mice | oral | 14 weeks | Exacerbated the toxic effect of ApoE4 | (Zhang et al., 2020) | |
| Arsenic | Rats | Oral | 10 days | Elevated level of Ap via increased expression of APP and RAGE | (Niño et al., 2018) | |
| Lead | Rats | Oral | 20 days | Upregulation in APP mRNA expression | (Basha et al., 2005) | |
| Parkinson’s disease | Cadmium | Mouse embryonic fibroblast cells | Cell culture | 24 hrs | Disruption of Ubiquitin-Proteasome System function and Upregulation of SNCA gene | (Yu et al., 2010) |
| Lead | Meriones shawi | Intraperitoneal | 3 days | Increased the levels of tyrosine hydroxylase | (Tamegart et al., 2019) | |
| Manganese | Human SK-N-MC cells | Cell culture | 72hrs | Increased a-synuclein toxicity | (Pifl et al., 2004) | |
| Mercury | Human SH-SY5Y cells | Cell culture | 24 hrs | Increased tau hyperphosphorylati on and Ap synthesis | (Olivieri et al., 2000) | |
| Arsenic | Human SH-SY5Y cells | Cell culture | 72 hrs | accumulation of a-synuclein | (Cholanians etal., 2016) | |
| Nickel | Humans (epidemiology) | Unclear | Unclear | Elevated level of Ni in the plasma and erthrocytes. | (Johansson et al., 2006) | |
| Huntington’s disease | Manganese | Mouse striatal cells | Cell culture | 26–30hrs | Defect in manganese homeostais due to mutant HTT gene | (Williams et al., 2010) |
| Amyotrophic lateral sclerosis | Arsenic | Humans (epidemiology) | Unclear | Unclear | Increased level of As in the CSF | (Patti et al., 2020) |
| Mercury | Humans (epidemiology) | Dermal | Unclear | Increased level of mercury in toenails | (Andrew et al., 2018) | |
| Autism | Mercury | Humans (epidemiology) | Prenatal | Prenatal | Increased plasma porphyrins levels | (Khaled et al., 2016) |
| Cadmium | Humans (epidemiology) | Oral, nasal. | Unclear | Increased level of blood cadmium | (Hessabi et al., 2019) | |
| Lead | Humans (epidemiology) | Oral, nasal. | Unclear | Increased level of blood mercury | (Hessabi et al., 2019) | |
| ADHD | Cadmium | Humans (epidemiology) | Oral, nasal | Unclear | Increased level of cadmium in the urine | (Lee et al., 2018) |
| Lead | Humans (epidemiology) | Oral, nasal, dermal | Unclear | Increased level of blood mercury | (El-Morsi et al., 2019) | |
| Mercury | Humans (epidemiology) | Prenatal | prenatal | (Sagiv et al., 2012) | ||
| Arsenic | Humans (epidemiology) | Oral, nasal | ≥ 1 yr | Increased level of Arsenic in the urine | (Rodríguez-Barranco et al., 2016) |
3. Mechanism of epigenetic changes
Epigenetic changes are acquirable phenotypic variations in chromatin structure that controls how genes are expressed without altering DNA sequences (Yang et al. 2016). These alterations which are activated by several environmental factors such as nutrition and heavy metal toxicity are brought about by; (i) DNA methylation (Feinberg and Fallin 2015), (ii) Posttranslational modification at the amino termini of histone proteins (Ke et al. 2008), and (iii) non-coding RNAs (ncRNAs) (Ding et al. 2011; Li et al. 2011) (Fig. 1).
Fig. 1.
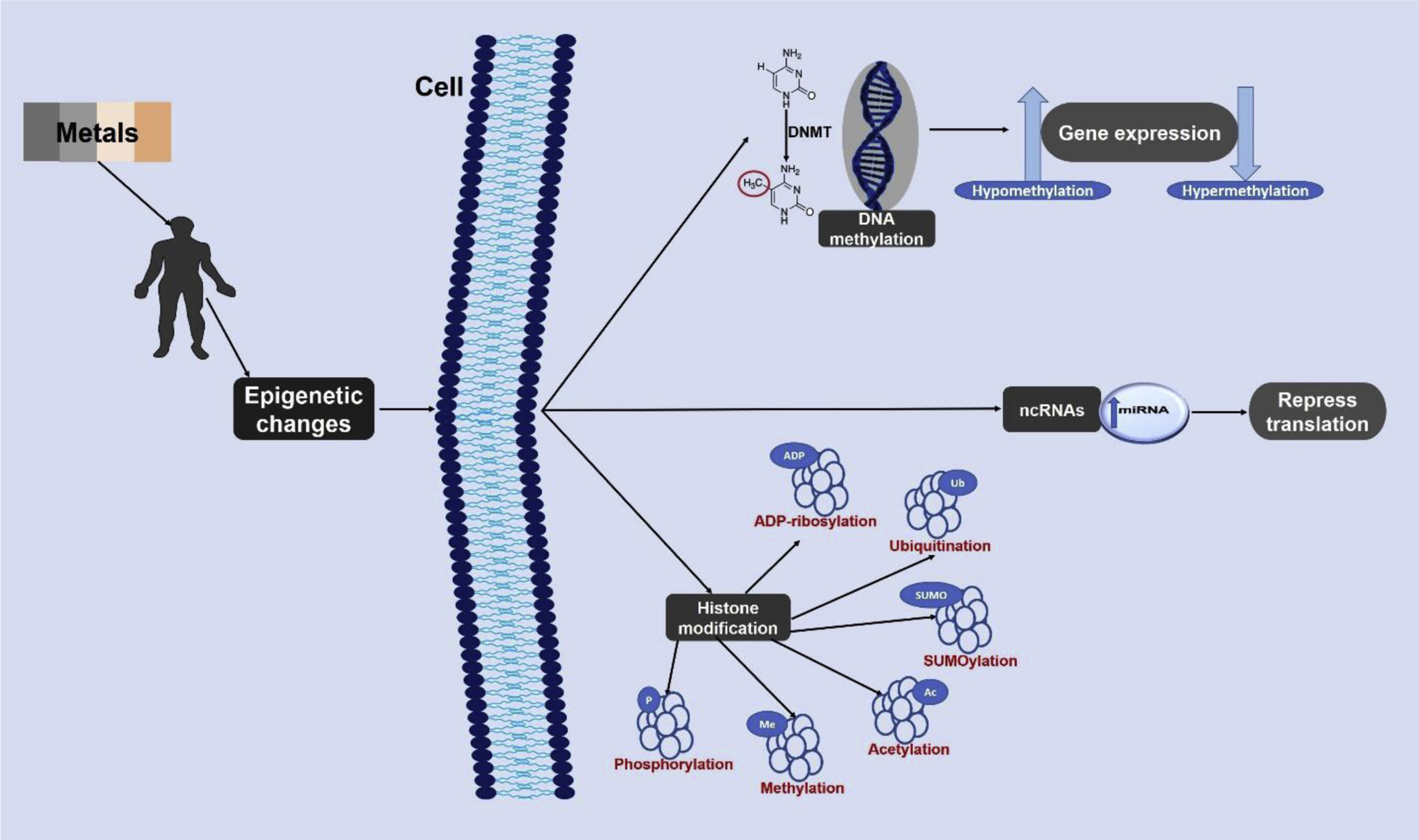
Schematic representation of the mechanism of epigenetic alterations due to exposure to neurotoxic metals. Epigenetics changes are elicited in the form of DNA methylation, histone modification, and/ or ncRNA. DNA methylation either promotes gene expression by hypomethylation or represses gene expression by hypermethylation. The histone proteins of DNA can be epigenetically modified in over 16 ways (the most common six are presented above). The ncRNA epigenetic change represses protein translation. DNMT - DNA methyltransferase; miRNA – MicroRNA;
3.1. DNA Methylation
DNA methylation is the covalent addition of a methyl group to the 5’ carbon atom in a cytosine ring. These processes mostly occur in a CpG island of the DNA, and is catalysed by the enzyme, DNA methyltransferase (Feinberg and Fallin 2015). DNA methylation, due to its contribution to numerous diseases such as; AD, diabetes, atherosclerosis, Friedrich’s ataxia, immunodeficiency, rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus is indubitably the most researched epigenetic mechanism (Heyn and Esteller 2012). It involves the enzymatic formation of a covalent bond between a methyl group from a methyl group donor, S-adenosylmethionine (SAM) and the cytosine ring of CG dinucleotides in a CpG island, resulting in 5methylcytosine (5-mC) formation (Choi et al. 2008; Doi et al. 2011). This enzymic process is regulated by the DNA methyltransferase (DNMT) family composed of DNMT1, DNMT2, DNMT3a, DNMT3b. DNMT3a and DNMT3b acts to methylate cytosine ring at the 5’ position of carbon atom, while DNMT1 helps to keep the cytosine in a methylated state even during replication (Horii et al. 2013; see review Martins-Taylor et al. 2012) (Fig.2). Methylation of the cytosine ring influences the heterochromatic and euchromatic state of the DNA. Consequently, regulating gene expression. To facilitate gene expression, there is need for the DNA to assume an euchromatic state; this is achieved via hypomethylation. In contrast, hypermethylation results in heterochromatinization. Thus, inhibiting transcription (Yang et al. 2016). Also, DNA methylation is not spontaneous process; it is however a result of several environmental triggers such as heavy metal exposure (Jordan et al. 2017; Scanlon et al. 2016).
Lately, it has been discovered that demethylation of the DNA reflected by loss of the covalent bond between the 5’ carbon atom of a cytosine ring and the methyl group can also occur. This demethylation process is either due to lack of DNMT1 throughout reoccurring replication or by active enzymatic demethylation or modification of the methylated cytosine with the establishment of an unmethylated cytosine ring (Bhutani et al. 2011; Shen et al. 2014).
3.2. Histone Modification
The linear DNA is wrapped into several arrays of nucleosomes, the fundamental component of chromatin structure, by winding around an octamer of histone proteins which consists of two replicas each of H2A, H2B, H3 and H4. These arrays of nucleosomes are connected by the histone H5 to form chromatin which in turn condenses to forms the chromosome (see review Ryu et al. 2015). This packaging is influenced by several histone modifications and over 16 of these modifications has been identified including acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, and ADP-ribosylation (Fig. 1). These modifications which are active in more than 60 different residues of histone protein (such as lysine, arginine etc.) play an imperative role in modifying gene transcription and other chromatin-associated processes (Chen et al. 2006; Ke et al. 2008).
Histone acetylation or deacetylation are the most frequently occurring in various diseases and disorders (see review Li et al. 2014). A unique expression pattern is achieved when several modifications combines to form a histone code which alters the histone structure (see review Tran and Miyake 2017). For example, phosphorylation of histone H3 serine 10 (H3S10ph) stimulates acetylation of histone H3 lysine 14 (H3K14ac), which is a transcription-activating modification; mono-ubiquitination of histone H2B lysine 120 (H2BK120ub) stimulates methylation of histone H3 lysine 4 (H3K4me), also a transcription-activating adjustment (Choudhuri, 2011). It has become apparent that PTMs on histone proteins function in intricate combinations to regulate the various activities associated with chromatin (Gardner et al., 2011).
Various enzymes catalysing several histone modifications have been identified, and they include histone acetyltransferases and deacetylases, lysine methyltransferases and demethylases, serine/threonine/tyrosine kinases and phosphatases, and lysine ubiquitinases and deubiquitinases. These inheritable modifications are linked with several diseases (Li et al. 2014) and have different effects on gene expression. Acetylation leads to active transcription, while histone methylation leads to either active transcription or gene repression (Gatto et al. 2017; Tatton-Brown et al. 2014). Histone acetylation and methylation are mediated by enzymes that add or remove specific groups to the histone proteins. Histone acetylation is mediated by two opposing groups of enzymes, namely, histone acetyltransferases (HAT) which add acetyl groups, and histone deacetylases (HDACs) that eliminate them. Currently, the eighteen different HDACs discovered are grouped into four classes (Delcuve et al., 2012). Of all the classes, the class-1 HDACs (HDAC 1, 2, 3 and 8) play an important role in regulating gene expression. HDAC1 and HDAC2, the two most studied HDACs have been found in several co-repressor complexes such as CoREST (co-repressor for element-1- silencing transcription factor) complexes, Sin-associated protein (SAP) complex and the NuRD (nucleosome remodelling and histone deacetylation) complex. These complexes play vital role in transcriptional repression via removal of acetyl groups from histones (Kelly & Cowley, 2013). On the other hand, histone methylation is facilitated by histone methyltransferases that add methyl groups to histone residues resulting in activation or repression of transcription that can be reversed by histone demethylases (Chen et al. 2006; Golebiowski and Kasprzak 2005).
3.3. ncRNAs
The third epigenetic mechanism involves the noncoding RNAs which includes the miRNA. miRNA are small, single stranded, noncoding RNA molecules that act as translational repressors. These were first identified in Caenorhabditis elegans, however, have now been shown to be expressed ubiquitously by all species. RNAs like the DNA are important components of gene expression as they are involved in both transcription and translational process of gene synthesis. However, not all RNAs have coding function. These non-coding RNAs can either be classified as long ncRNAs or small ncRNAs. Small ncRNAs control gene expression by either repressing translation (miRNA) or degrading the mRNA (siRNA). The miRNA binds to a complementary sequence in the mRNA to trigger translational repression (Fig. 1). However, when there is an incomplete base match in the sequence, the degradation pathway driven by siRNA is triggered, resulting in mRNA degradation (Choudhuri et al. 2010). Currently, over 17,000 different miRNAs have been recognized in over 140 species (Kozomara and Griffiths-Jones 2010).
To accomplish their effector role, miRNAs are integrated into ribonucleoprotein (RNP) complexes, known as mi-RISCs (RNA-induced silencing complexes) which acts post-transcriptionally or RITS (RNA-induced transcriptional silencing complexes). The biosynthesis of miRNA and siRNA is catalysed by Endonucleases such as the Dicer complex. Dicer processes double-stranded RNA (dsRNA) into miRNA and siRNA (Jaskiewicz & Filipowicz, 2008).
4. Metal-induced epigenetic changes as a consequence of environmental toxicity
In a way, epigenetic changes could be likened to a channel through which environmental factors can impact lifelong biological changes to organisms at a molecular level (see review Martin and Fry 2016). These environmental factors can either be positive or negative (Mansuy and Mohanna 2011) and the changes they elicit can be inherited by their offspring (Waterland & Jirtle, 2003). Several studies have been able to link various neurodevelopmental disorders, resulting in neurological and cognitive dysfunction to epigenetic changes (Nagarajan et al. 2006; Schneider et al. 2013; Stansfield et al. 2012). Exposure to environmental toxins is the leading cause of alteration in the expression of genes that are responsible for several life processes. Environmental toxins can come in various forms, such as air pollutant, persistent organic pollutant, toxic metals like arsenic, mercury, lead, cadmium, nickel and manganese, exposure to polycyclic aromatic hydrocarbons, smoke from tobacco, aflatoxin B1 and so on (see review Martin and Fry 2016).
Epigenetic dysregulation has been marked as a key player in neurodegenerative disorders, such as AD, PD, HD amongst others (see Table 2) where it may facilitate interactions between genetic and environmental risk factors including metals (Bertogliat et al., 2020). However, there are few investigations into the role of metal-induced epigenetic changes in the onset of these neurological disorders. Nonetheless, all these evidences reviewed here suggest that epigenetics may be a critical pathway for metal-induced neurotoxicity. Exposure to neurotoxic metals like mercury, lead, manganese, cadmium, and many more are known to cause epigenetic changes associated with adverse health conditions. These metals and the mechanism by which they alter the expression of certain genes will be discussed in detail in the subsequent section of this review (summarized in Table 3).
Table 2.
Epigenetic changes associated with neurological diseases
| Diseases | Model | Epigenetic Modifications | References |
|---|---|---|---|
| Alzheimer’s disease | Rats | Upregulation in APP mRNA expression | (Basha et al., 2005) |
| Rats | Upregulated tau phosphorylation via the increased acetylation of H3 and hypomethylation of Cdk5 promoter region | (Li et al., 2015) | |
| Mice | Increased HDAC2 expression | (Gonzalez-Zuñiga et al., 2014) | |
| Mice | Decreased hippocampal level of 5-hydroxymethylcytosine | (Shu et al., 2016) | |
| Parkinson’s disease | Rats and Mice | Hyperacetylation of H3 and H4 | (Song et al., 2010) |
| Mice ES cells | Decreased level of miR-133b | (Kim et al., 2007) | |
| Human (postmortem brain) | Decreased expression of DNMT1 | (Desplats et al., 2011) | |
| Humans (epidemiology) | Hypomethylation of SNCA gene | (Ai et al., 2014) | |
| Huntington’s disease | STHdhQ7 and STHdhQ111 cell lines | Reduced expression of Ap-1, | (Ng et al., 2013) |
| Sox2, Pax6, and Nes genes via hypermethylation of their promoter region. | |||
| Human cells and Mice | Decreased level of 5-hydroxymethylcytosine at the 5’ end of AD0RA2A gene | (Villar-Menéndez et al., 2013) | |
| Caenorhabditis elegans | Diminished HD AC1 and HDAC2 function | (Bates et al., 2006) | |
| Amyotrophic lateral sclerosis | Mice | Upregulation of DNMT1 and DNMT3a | (Chestnut et al., 2011) |
| Mice ES cells | Dysregulation of miR-409 and miR-495 | (Capauto et al., 2018) | |
| Human PS cells | Downregulation of HDAC 6 | (Du et al., 2015) | |
| Autism | Rats | Inhibition of HDAC1 and HDAC2 | (Shimshoni et al., 2007) |
| Humans (postmortem brain) | Reduced expression of MeCP2 | (Nagarajan et al., 2006) | |
| ADHD | Humans (epidemiology) | Hypomethylation of dopamine receptor D4 gene (DRD4) and the serotonin transporter gene (SLC6A4) gene | (van Mil et al., 2014) |
Table 3.
Summary of epigenetic changes induced by neurotoxic metals
| Metal | Epigenetic changes | Consequences of epigenetic changes | References |
|---|---|---|---|
| Manganese (Mn) | Transient hypermethylation of parvalbumin | Disruption of neurogenesis and subsequent neuronal differentiation | (Wang et al., 2013) |
| Hypermethylation of Midi, Atpla3 and Nr2f1 | Disruption of neurogenesis and subsequent neuronal differentiation | (Wang et al., 2013) | |
| Sequestration of DNMT1 via a-synuclein overexpression | (Li et al. 2010; Tarale et al.,2016) | ||
| Hypo-acetylation of histone H3 and H4 via upregulation of HDAC activity and downregulation of HAT activity | Death of dopaminergic neurons | (Guo et al. 2018) | |
| Suppressed expression of miR-133b | Increased proliferation of malignant cells | (Cai et al., 2011) | |
| Downregulation of miR-7 and miR-153 | Overexpression of SNCA gene | (Tarale et al., 2018) | |
| Arsenic (As) | DNA hypomethylation leading to upregulation of ER-α | Formation of proliferative leison and hepatocellular carcinogenesis. | (see review Hussain et al. 2018) |
| Restriction of DNMT activity leading to DNA hypomethylation | Increased activation of K-ras gene in malignant RWPE-1 cells | (Reichard and Puga 2010) | |
| Hypo-acetylation of histone 3K9 | Memory deficits in mice | (Cronican et al. 2013) | |
| Downregulation of miR-199a-5p | Tumor angiogenesis in humsn bronchial epithelium | (He et al. 2014) | |
| Lead(Pb) | Decreased expression of DNMTs and MeCP2 enzymes | Increased risk of AD | (Eid et al. 2016; Sanchez-Martin et al. 2015) |
| Repressed H3K9Ac and H3K4Me2 expression and upregulation of H3K27Me3 | Increased risk of AD | (Eid et al. 2016; Sanchez-Martin et al. 2015) | |
| Inhibition of DNA methylation via alteration of IGF-1 and methionine-stimulated synthase | Impaired cognitive function | (Waly et al. 2004) | |
| Reduced DNA methylation of the PEG3 differentially methylated regions. | Impaired brain development | (Li et al. 2015) | |
| Mercury (Hg) | Decreased expression of BDNF mRNA | Behavioural alterations | (Onishchenko et al. 2008) |
| Overexpression of five major ncRNA (miR-302b, miR-367, miR-372, miR-196b, and miR-141) | Impaired learning and memory, synaptic vesicle transport, and neuronal development. | (Pallocca et al. 2013) | |
| Global DNA hypomethylation of the NSCs | Reduced neural stem cell proliferation | (Bose et al. 2012) | |
| Decreased global DNA methylation and DNMTs activity | Transgenerational inheritance of dysregulated genes | (Basu et al. 2013) | |
| Transgenerational epigenetic alterations of DNA methylation regions. | Trangenerational inheritance of phenotypes (visual deficits and hyperactivity) in zebrafish. | (Carvan et al. 2017) | |
| Nickel (Ni) | increased histone H3K9 dimethylation | transgene silencing in G12 Chinese hamster cells | (Chen et al. 2006) |
| DNA Heterochromatinization | Gpt transgene silencing in Chinese hamster V79-derived G12 and G10 cells | (Ellen et al. 2009) | |
| Dose- and time-dependent (0.2 mM) ubiquitination of H2A and H2B. | Impaired mechanism of DNA repair in cultured 1HAEo- and HPL1D human lung cells | (Karaczyn et al. 2006) | |
| Cadmium (Cd) | DNA hypermethylation and enhanced DNMT activity (chronic exposure, low dose 0–1.5 μmol/L) | Increased cellular proliferation of human embryo lung fibroblast cells. | (Jiang et al. 2008) |
| DNA hypomethylation by means of non-competitive inhibition of DNMT activity, (acute exposure in high dose 2.0μM) | Stimulation of K562 cell proliferation. | (Huang et al. 2008) | |
| Upregulation of H3K4me3, H3K27me3 and H3K9me3 occupancy at the metallothionein 3 (MT3) promoter. | Activation of MT3 gene in human urothelial cell (found in urothelial cancer) (bivalent state) | (Somji et al. 2011) | |
| Inhibition of H3 Phosphorylation via inactivity of the human vaccinia-related kinase VRK1/2. | Defective DNA repair system | (Barcia-Sanjurjo et al. 2013) |
4.1. Manganese
Mn is used in the industry for the manufacture of steel and formation of aluminium mixture, production of potassium permanganate, manufacture of glass, textile bleaching, matches and fireworks, oxidizing agent for electrode coating in welding rods, tanning of leather, oxidant in the production of hydroquinone (Bouabid et al. 2014; Caito and Aschner 2015). Manganese (Mn) is the twelfth most abundant metal with eleven oxidative forms. The most prevalent are +2, +3, +4 and +7 (Caito and Aschner 2015; Harischandra et al. 2019; Neal and Guilarte 2012). Mn is essential for several for physiologic activities such as protein and energy breakdown, bone mineralization, immunological response amongst others (Bornhorst et al. 2012). However, in toxic concentrations, it is associated with the development of several neurological disorder such as AD (Tong et al., 2014), PD (Pifl et al., 2004), and HD (Williams et al., 2010) (see Table 1).
4.1.1. Mn neurotoxicity
Mn is vital for life but can also have detrimental effect on health when present in excess (Bouabid et al. 2014). Excess intake of Mn could be from occupational exposure (mining, welding, battery manufacture etc.), methcathinone drug abuse, total parenteral nutrition (TPN) and or genetic factors like mutation of the SLC3OA10 transporter (Tuschl et al. 2012; Tuschl et al. 2013). Exposure to Mn at high levels results in a Parkinson disease look-alike syndrome called manganism. Symptoms include bradykinesia, rigidity, abnormal gait, memory and cognitive aberrations and mood disorder (Neal and Guilarte 2012).
The CNS is the prime target of excess Mn exposure. Furthermore, though the body can readily eliminate surplus Mn, mainly through the gut and liver, excess Mn deposition into brain is not easily eliminated leading to Mn build-up in the brain (Harischandra et al. 2019). Build-up of Mn occurs in the dopamine-rich basal ganglia specifically in the striatum, globus pallidus and substantia nigra (Harischandra et al. 2019; Tuschl et al. 2013). This affinity for the basal ganglia might be associated with the high level of divalent metal transporter 1 (DMT 1) (a Mn transporter), in that region (Tuschl et al. 2013). Various transporters have been recognized as carriers for Mn. Mn uptake is primarily by DMT1, with some contribution from other transporters which include transferrin, zinc transporters (ZIP8 and ZIP14), dopamine transporters, Ca channels. Conversely, Mn efflux transporters include SLC30A10, ferroportin, ATPase 13A2 (Chen et al. 2015).
Despite a broad knowledge on Mn-induced neurotoxicity, conclusive mechanisms on Mn-induced neurotoxicity remain elusive. However, numerous determinants such as oxidative injury, mitochondrial dysfunction, protein misfolding and neuroinflammation control this process (Harischandra et al. 2019). Mn-induced neurotoxicity via oxidative stress can be ancillary to a mitochondrial dysfunction which takes part in the onset of the AD and PD. Furthermore, Mn may also directly affect mitochondrial function by impeding the electron transport chain hence an increased oxygen production, reduced ATP generation and elevated electron leakage. All these evidences considered proposes that Mn increases neurotoxicity related to AD and PD (Martins, 2019). Additionally, a study by Tong et al. evaluated Mn in whole blood and reported a positive relationship between plasma amyloid-β protein (possible risk biomarkers of AD) which proposes the probable role of Mn in amyloid-β marked AD (Tong, 2014).
4.1.2. Mn-induced epigenetic changes
Mn may cause aberrations in gene expression via epigenetics (Abel and Zukin 2008). Mn exposure leads to production of reactive oxygen species (ROS) through redox cycling which leads to oxidative stress. Oxidative stress caused by Mn leads to death of dopaminergic cells and mitochondrial malfunction (Farina et al. 2013). Evidence has shown that oxidative stress causes DNA aberrations like alterations in chromosome arrangement, base modification and deletions. This possibly alters capacity of DNA methyl transferase to use DNA as a base thereby disrupting general or gene-specific methylation (Donkena et al. 2010). Additionally, epigenetic changes that occur due to foetal exposure to Mn lead to aberrations in observable traits. Temporary hypermethylation of parvalbumin, hypermethylation of Mid1, Atp1a3 and Nr2f1 and a resultant down regulation of these genes were observed in mouse blood following prenatal exposure to Mn (Wang et al., 2013). Furthermore, there is evidence that links epigenetic alterations to neurological disorders such as PD, HD and mood swings (anxiety and depression).
Mn overexposure leads to overexpression in α-synuclein levels which has been postulated to cause PD (Li et al. 2010). This Mn induced α-synuclein overexpression has been attributed to DNA hypomethylation of α-synuclein gene promoter following the exposure to Mn (Frieling et al., 2007). Contributing to the accumulation of α-synuclein are the influence of Mn-induced oxidative stress and dopaminergic cells destruction. (Cappai et al. 2005; Cookson 2009). This accumulated of α-synuclein will form an α-synuclein oligomer that results in the fragmentation of Golgi complex. Furthermore, it negatively affects the production, maturation, and trafficking of dopamine transporter (Volles and Lansbury 2003). In the cytoplasm, α-synuclein induces the formation of proapoptotic BCL2-associated death promoter (BAD) that binds on mitochondrial membrane altering its permeability as result. Similarly, α-synuclein also causes the sequestration of DNMT1 in the cytoplasm. DNMT1 via global hypomethylation epigenetically alters PD associated genes like PARK2 (Parkin RBR E3 Ubiquitin Protein Ligase) and α-synuclein genes. On the other hand, in the nucleus, α-synuclein binds to histone protein where it results in the inhibition of histone acetyltransferase activity and consequently the inhibition of the protein kinase C (PKCδ) expression. This occurrence elicit DNA fragmentation accompanied by an attendant apoptotic cell death (Tarale et al., 2016).
Histone modification has also been shown following Mn exposure. Guo et al., (2018) observed notable suppression in the acetylation of histone H3 and H4 in PC12 cells and SHSY5Y (neuroblastoma) cells which was observed to be time-dependent. MnCl2 achieved this hypo-acetylation process by 1) increasing HDAC functioning level and protein expression level, 2) decreasing HAT functioning level and protein expression level in neurons (Guo et al. 2018). Following their investigation, Zhang et al. (2017) reported that increased histone acetylation may have a role in increased ROS level and decreased glutathione quantity in neurons. This may be due to the role of histone acetylation in enhancing the expression of Nrf2/HO-1 pathway in Mn-exposed neural cells (Zhang, 2017).
Other critical alterations due to disproportion in Mn level are downregulation of miR-7 and miR-153 and interplay with histone protein p300 (see review Wessels 2017). Concurrently, a study by demonstrated down-regulation of mi-R-7 and mi-R433 post-Mn exposure. There is no established mechanism by which this down-regulation occurs. Nevertheless, Mn is identified to cause neuronal cell death via oxidative stress hence they postulated that Mn effect on redox balance and its transcription factors resulting from oxidative stress may cause a dysregulated miRNA expression (Tarale et al., 2018). The relationship between ncRNAs and Mn exposed neural cells remain unclear however, the expression pattern of ncRNAs were notably altered in Mn-exposed hippocampal cells (Ma, 2016). Mn has been established to cause death of dopaminergic neurons which is a pathologic hallmark for PD. The midbrain tissues of PD victims are found to be lacking miR-133b yet it is enriched in dopaminergic neurons. Using rat midbrain cultures, miR-133b was found to target Pitx3 (Kim, 2007). A case-control study using laser dissected post-mortem dopaminergic neurons further affirmed the lack of miR-133b in PD patients (Sonntag, 2010; Zovoilis et al. 2011). The presence of miRNA in the plasma of victims of PD has also been observed by Khoo and colleagues. They identified increased expression of miR-626, miR-505 and k-TSP1 as biomarkers for PD (Khoo et al. 2012).
4.2. Arsenic
Arsenic (As) is a prevalent element that is classified as a metalloid. Metalloids are elements that have properties in between that of metals and non-metals (Caito and Aschner 2015; Singh et al. 2011a). It is also categorized as a non-essential metal because it has no known physiologic role (Tchounwou et al. 2012). Arsenic is found in both organic and inorganic form in the environment (Singh et al. 2011a). Organic arsenic exists as mono-methylarsonic acid (MMA), di-methylarsonic acid (DMA) and tri-methylarsonic acid, which are collectively methylated metabolites (Tchounwou et al. 2012), whereas inorganic arsenic exists as arsenides (−3), arsenites (+3) and arsenate (+5). Arsenic plays no role in normal bodily activity, but has industrial functions. Since ancient times, arsenic has been adopted for strengthening of alloys of copper and lead. Recently, it’s use includes production of automobile batteries, pyrotechnics, laser diodes and LEDs, as semiconductors and also in agricultural activities (as wood preservative, pesticides, fungicides, herbicides etc.) (Caito and Aschner 2015; Mochizuki et al. 2019). These industrial functions serve as a means of exposure (inhalation) to arsenic, which has deleterious effect to health. Other means of exposure include contaminated food and water, skin contact, and possibly the parenteral route (Caito and Aschner 2015; Singh et al. 2011a; Tchounwou et al. 2012). In the nervous system, it accumulates and could contribute to development of several neurologic disorders which includes AD (Niño et al., 2018), PD (Cholanians et al., 2016), ALS (Patti et al., 2020), and ADHD (Rodríguez-Barranco et al., 2016) (see Table 1).
4.2.1. As neurotoxicity
Arsenic is a non-essential metal that is associated with toxic effect in the biological system. The harmful effect of arsenic has long been acknowledged and it has been used as a homicidal agent because it is odourless, highly poisonous and present symptoms related to common diseases (Caito and Aschner 2015; Hughes et al. 2011). Arsenic readily crosses the BBB; hence the brain is easily affected upon overexposure to the metal (Singh et al. 2011a). Inorganic arsenic crosses the BBB using several distinct transporters. Arsenite (AsIII) is transported via aqua(glycerol)porins (AQP), organic union transporters and glucose transporter (GLUT) whereas arsenate (AsV) is transferred via phosphate transporters where it is reduced to AsIII (Calatayud et al. 2012; Torres-Avila et al. 2010). According to WHO the mean daily intake of arsenic from drinking-water will generally be less than 10μg/l except in those areas where drinking water contains elevated concentration. Mild exposure to arsenic (≥2mg As/kg/day) results in arsenic encephalopathy with symptoms such as headache, confusion, seizure, coma and death whereas severe exposure to arsenic at lower concentration has a negative effect on the peripheral nervous system and also leads to a defective motor activity (Caito and Aschner 2015; Naujokas et al. 2013). Several reports have also linked exposure to arsenic in humans with chromosomal alterations and micronuclei formation (Ghosh et al. 2006). The mechanism for arsenic-induced neurotoxicity has not been fully unveiled. However, several hypotheses have been made, the possible mechanisms reported include; mitochondrial dysfunction, lipid peroxidation, apoptosis, increased calpain and thiamine deficiency (Dwivedi and Flora 2011; Singh et al. 2011a).
4.2.2. As-induced epigenetic changes
Metabolism of arsenic is vital for its toxic effect. This process occurs by reduction and methylation reactions by gluthathione-S-transferase omega 1 (GSTO1) and arsenic (III) methyltransferase (AS3MT). As is methylated through one-carbon metabolism with S-adenosyl methionine as the methyl donor and reduction reaction occurs with reduced glutathione as electron donor (Singh et al. 2011a). In liver cells, exposure to arsenic results in hepatic DNA hypomethylation which causes abnormal gene expression (example; over expression of ER-α) and cancerous growth in the liver (Hussain et al. 2018). Arsenic is methylated majorly in the parts of the brain where AS3MT is expressed by disparate hypothesized mechanisms. One mechanism is by methyltransferase competition for S-adenosylmethionine (SAM). A reduced SAM level due to arsenic breakdown possibly causes a cofactor restriction on DNA methyltransferase (DNMT) enzymes. Another way through which reduction in SAM level can occur is through the transfer of homocysteine to the trans-sulfuration pathway amid oxidative stress. Restriction of DNMT functioning bolsters DNA hypomethylation and has been said to be another mechanism through which DNA methylation occur (Reichard and Puga 2010).
Aside DNA methylation, few reports have linked arsenic with histone modifications. One of the reports was by Jo et al., (2009). They showed that AsIII cause reduced histone 4 lysine 16 acetyltransferase (H4K16Ac) action resulting in toxicity in the urothelial cell (Bailey et al. 2013; Jo et al. 2009; Sánchez-Peña et al. 2010). Arsenic has also been reported to cause histone acetylation, one of the identified processes of modification of histones. It alters pyruvate dehydrogenase function which is an enzyme for the conversion of pyruvate to Acetyl-CoA, needed for histone acetylation. A research on foetal exposure of mice to arsenic recorded hypo-acetylation at histone 3K9 (H3K9) associated with defective spatial and episodic memory (Cronican et al. 2013).
There is little work on the effect of arsenic on ncRNAs expression compared to DNA methylation and post translational histone modification. Evidence shows that arsenite exposure to human lymphoblast resulted in a meaningful change in five miRNAs (miR-210, miR-22, miR-34a, miR-221 and miR-222) which was not followed by any alteration in general DNA methylation (Marsit et al. 2006). Another study implicated arsenic exposure to BEAS-2B cells to result in production of reactive oxygen species and potentiate tumorigenesis through the downregulation of miR-199a-5p (He et al. 2014). Changes in miRNA expression has also been associated with foetal exposure to arsenic. A research carried out by Rage et al in northern Mexico’s populace after intake of contaminated water with arsenic levels over 50mg/L showed a disruption in miRNA expression levels in infant cord blood (Rager et al. 2014).
4.3. Lead
Lead (Pb) is an abundant metal found on earth. Over the years, it has been considered as a neurotoxin due to its detrimental effect on the nervous system. It has high bioavailability and can readily cross the blood-brain barrier (Eid et al. 2016). Lead toxicity makes up 0.6% of the global burden of disease (Parada et al. 2014). The major route of exposure to Pb is through ingestion. Kids between the ages of 1 to 3 are more susceptible to exposure. They can pick up lead dust via inhalation and/or ingestion, from leaded paint, wearing off, when playing on the floor or their toys (Parada et al. 2014). Bioaccumulation of lead is implicated in several neurological disorders such as AD (Basha et al., 2005), PD (Tamegart et al., 2019), and Autism (Hessabi et al., 2019) (see Table 1).
4.3.1. Pb neurotoxicity
Generally, an excessive exposure to Pb in early life can result in growth retardation, impaired cognitive development in infancy, attention deficits hyperactivity disorder, neurotoxicity, neurobehavioral alterations, difficulties in executive functions and decrease in brain volume (Li et al. 2015). There is also an increased risk of developing kidney disease, PD and AD (Parada et al. 2014).
Pb is a neurotoxin that is harmful to the central nervous system and affecting processes like hearing, cognitive functions and posture in children (Lin et al. 2020). Studies have shown that exposure to Pb in early life results in latent overexpression of AD-related proteins (tau, APP, and amyloid-beta proteins) later in life. The alterations in these proteins are associated with epigenetic changes (Basha et al. 2005; Bihaqi et al. 2014; Bihaqi and Zawia 2013; Wu et al. 2008). Growing evidence shows that males are more susceptible to a high level of Pb exposure than females, with respect to increased risk of developing neurological and behavioural disorders (Lin et al. 2020). Exposure to Pb can cause brain damage by altering the homeostasis of calcium and zinc ions in the brain, disrupting the synthesis and secretion of neurotransmitters as well as the receptor density. It also results in increased oxidative stress, damage to the BBB thereby abnormally increasing its permeability (Basha et al. 2005). Furthermore, on entering the brain, Pb alters heme synthesis, and consequently contributes to loss of oligodendrocytes density, decreased cortical synaptogenesis, blockage of voltage-sensitive calcium channels, interference of protein kinase, dysfunction of neurotransmitter (see review Ray, 2016).
4.3.2. Pb-induced epigenetic Changes
The neurotoxicity of Pb has been associated with epigenetic changes like DNA methylation that control transcriptional pathways involved in synapse function, neurogenesis and memory-related gene expressions (Sánchez-Martín et al. 2015). Study shows that exposure to Pb in early life led to a significant decrease in the level of DNMT1, DNMT3a, and MeCP2 expression (Bihaqi et al. 2014; Schneider et al. 2013). DNMT1 and DNMT3a play a vital role in the development, maturation and function of the nervous system. During development DNMT1 and DNMT3a are responsible for neuronal maturation (in addition to synaptic transmission) and neural development respectively (Schneider et al. 2013). Evidence from Schneider et al. experiment showed that a deficiency of DNMT1 in the hippocampus of perinatal females and early postnatal males exposed to Pb may result in a defect synaptic plasticity in these animals. A similar occurrence is also found following an alteration in DNMT3a expression in the hippocampus of perinatal males and early postnatal females exposed to Pb. The downregulation of DNMT1 and DNMT3a expression in the forebrain neurons, due to exposure to Pb, result in impaired synaptic plasticity, memory formation and learning (Schneider et al. 2013). The resultant reduction in the activity of DNMT1 due to early life exposure to Pb also alter the regulation of APP (amyloid precursor protein) and BACE-1 (beta secretase 1) genes, that are involved in AD pathway. Also contributing to the dysregulation is the This early life (from post-natal day 1 through post-natal day 20) exposure to Pb causes an overexpression of APP mRNA, as well as increase the level Aβ later in life (Basha et al. 2005). Similarly, a high level of cerebral 8-hydroxy-2-deoxyguanosine (8-oxo-dG) associated with hypomethylation (Mastroeni et al., 2009). This suggests that the exposure to lead early in life is a strong risk factor for late onset AD. Epigenetic changes on the expression and/or methylation of MeCP2 protein can result in Rett syndrome, and different types of cognitive and behavioural disorders such as mild learning disability, autism, mental retardation, and attention deficit hyperactivity disorder (Nagarajan et al. 2006). The degree of cognitive impairment experienced as a result of MeCP2 alterations depends on the concentration of Pb exposure. Thus, animals with compromised but yet a little MeCP2 protein function exhibited a high level of anxiety, poor social interactions, cerebellar learning dysfunction, poor nest building and diurnal activities and cognitive as well as behavioural dysfunctions (Schneider et al. 2013).
Histone modification has been observed following early life exposure to Pb. The levels of H3K9Ac and H3K4Me2 proteins which are associated with gene activation, decreases. Contrastingly, the level of H3K27Me3 protein, associated gene repression, increases. Thus, following the early life exposure to Pb, the histone repressive marks were upregulated (Eid et al. 2016). This supports the hypothesis that early-life exposure to Pb compromises the developmental processes in the brain with increased risk of developing neurodegenerative diseases (Parada et al. 2014; Sánchez-Martín et al. 2015).
Even the lowest level of exposure to Pb is harmful to the foetus because of the ease with which Pb crosses the placental barrier during development. The developing brain compared to other organs is unfortunately much more vulnerable to the exposure of Pb. it affects early brain development by altering the genomic DNA methylation in cord blood that results in long-term effects, such as behavioural and cognitive impairment, later in life (see review Ceccatelli et al. 2013). The perinatal exposure to a low concentration of Pb has been shown to disrupt the activity of synaptic protein phosphatases, which controls synaptic plasticity (Gąssowska et al. 2016). A study, by Waly et al., of the human neuroblastoma culture exposed to Pb shows that Pb can alter the IGF-1 stimulated methionine synthase, a DNA methylation regulation enzyme (Waly et al. 2004).
The brain neurotrophic factor (BDNF) is an important protein that is involved in neuronal and synaptic development. BDNF-tropomyosin-related kinase B (TrkB) trans-synaptic signalling is responsible for maintaining the bidirectional communication between neurons in the process of synaptogenesis (Stansfield et al. 2012). Exposure to Pb has been found to cause an alteration in the BDNF-TrkB signalling pathway as well as in the synaptic proteins, which are associated with impaired synaptic development and function that may result in disruption synaptic maturation and neurodevelopmental process. These alterations caused by Pb exposure are associated with cognitive deficits and behavioural disorders in children (Stansfield et al. 2012).
Studies have been done to investigate the effect of Pb exposure on the expression of miRNAs. The result of the study showed an over-expression of miRNA-222, which is associated with the regulation of tumour suppressor and CDK-inhibitor 1b (p27Kp1), and an under-expression miRNA-146a which is associated with the immune response (Bose et al. 2012; Li et al. 2015).
4.4. Mercury
Mercury (Hg) is among the toxic elements in the periodic table. It exists in three (metallic/elemental, inorganic, and organic) forms with variable toxicity. It is among those chemicals considered to be of major public health concern (Fisher and WHO 2003). The metallic mercury exists as a volatile liquid at room temperature, which is detrimental to the CNS if inhaled (Aschner et al., 2013). Mercury in its inorganic form is found richly in the environment mainly as cinnabar and metacinnabar minerals. It may also be present in other minerals as impurities. Some other examples of inorganic mercury compounds include mercuric chloride, mercuric sulphide, mercuric acetate, and mercurous chloride (Fisher & WHO 2003).
MeHg is absorbed in the GI tract and enters the bloodstream where it is transported to all organs, the brain inclusive which it enters freely via its interaction with endothelial cysteine sulphhydral group of the BBB and forming a methionine analogue (a neutral amino acid) which enter the brain by imitating the structural form of methionine for entering the brain (Clarkson 2002; Hintelmann 2010; Karri et al. 2016). Upon entrance into the brain, Hg can induce several deleterious effects such as aggregation of APP in AD (Song and Choi, 2013), and increased tau hyperphosphorylation in PD (Zahir et al., 2005). It has also been associated with ALS and autism in several epidemiologic studies (Andrew et al., 2018; Khaled et al., 2016) (see Table 1).
4.4.1. Hg neurotoxicity
The neurotoxic consequence of mercury can still be traced down to the event of the Minamata Bay, on the western coast of Japan between the late 1950s and early 1960s. Mercury poisoning from the consumption of MeHg-contaminated seafood was recorded to have affected thousands of people, killing several hundreds of people (see reviews Ceccatelli et al. 2010; Ceccatelli et al. 2013). Those affected showed neurological deficits including paralysis, ataxia, deafness, speech impairment, mental disorders, sensory disturbances in the distal part of the limbs, tremor, nystagmus, anosmia, ageusia, hyposmia, and hypogeusia (see review Ceccatelli et al. 2013). Prenatal exposure to a low dose of mercury can result in the disruption of some of the critical developmental processes which would subsequently lead to neurodevelopment disorders later on. Neuropathological examination on brains exposed to MeHg prenatally exhibits several developmental anomalies such as; dysplasia of cerebral and cerebellar cortices, neuronal ectopia and several others. Similarly, exposure to low concentration of some metals, mercury inclusive, can directly induce the formation of α-synuclein in dopaminergic neurons of the substantia nigra of the basal ganglia resulting in PD (see review Zahir et al. 2005). In addition, according to the investigation of Olivieri and colleagues, showed that cobalt and mercury were able to induce oxidative stress and cytotoxicity, as well as increasing the secretion of β-amyloid 1–40 and 1–42 on the SHSY5Y neuroblastoma cells which had been exposed to these metals (Olivieri et al., 2002). This occurrence may result in neurodegenerative diseases, like PD and ADs (see review Zahir et al. 2005). Mercury can also bind to the sulphydryl groups of proteins, as well as on the disulphide groups in amino acids thereby leading to the inhibition of sulphur oxidation and blocks related enzymes. The inhibition of cellular sulphur oxidation is evident in several neurodegenerative diseases, such as; AD, PD, ALS, Autism (Wilkinson & Waring, 2002; Zahir et al. 2005).
Exposure to a concentration of 0.05 mg/m3 or lower of mercury can result in memory disturbance, tiredness, subclinical finger tremor, aberrant EEG by computerized analysis and poor performance in neurobehavioral/ neuropsychological tests. Another investigation carried out on workers exposed to low concentrations of mercury showed that depression and short term-auditory memory impairment were common in these workers (Branco et al., 2017; Zahir et al. 2005).
4.4.2. Hg-induced epigenetic Changes
Several studies have been able to link early developmental exposure to low doses of MeHg to long-term behavioural impairment like learning capabilities and depression-like behaviour (Johansson et al. 2007; Onishchenko et al. 2008; Onishchenko et al. 2007).
Brain-derived neurotrophic factor (BDNF) is a protein that is encoded by the BDNF gene (Binder and Scharfman 2004). Studies showed that BDNF expression is low in depressed patients as well as in mice models of depression. The hippocampus is the part of the brain responsible for modulating emotions such as depression (Onishchenko et al. 2008). The exposure to methylmercury results in a decrease in the level of BDNF in the hippocampus and the dentate gyrus and such a decrease is said to be associated decreased neurogenesis (Onishchenko et al. 2008), neuroinflammation, alterations in the serotonergic system as well as the cyclic AMP response element-binding protein (CREB) activity in the hippocampus (Chen et al. 2019). MeHg also alters BDNF gene expression which is associated with epigenetic changes in the chromatin structure of the BDNF gene. MeHg has a suppressive effect on the BDNF promoter region which is attributed to the DNA hypermethylation, increasing histone H3–K27 trimethylation and decreasing H3 acetylation at promoter IV facilitated by exposure to MeHg. In like manner, the level of BDNF mRNA in the dentate gyrus decreased as a result of the effect of MeHg (Onishchenko et al. 2008). Fortunately, these epigenetic changes resulting in depression can be reversed by treatment with anti-depressant like fluorexine which up-regulate the expression of BDNF genes in the hippocampal formation, therefore, reversing the repressed chromatin state and re-establishing the BDNF mRNA level (Onishchenko et al. 2008).
Additional evidence shows that exposure to a nanomolar concentration of MeHg results in inheritable consequences such as a decrease in neural stem cells (NSCs) proliferation, changes in the expression of genes involved in regulating cell cycle, mitochondrial dysfunction and alterations in cellular senescence (Bose et al. 2012). It was found that there was an overexpression of p16 and p21 gene, at the mRNA and protein level, in the brain of mice model exposed to a micromolar concentration of MeHg. The p21, as well as p16 genes are cell cycle regulating genes that code for the inhibitor proteins for cyclin-dependent kinases (CDK). Their overexpression halts the progression of the cell cycle. Similarly, MeHg exposure also results in the inhibition of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) phosphorylation, which alters the transition from G1 to S phase in the cell cycle. Inhibition of ERK1/2 phosphorylation have been shown to be caused by the inhibition of DNMTs (So et al. 2011) The overexpression of p16 and p21 genes and the inhibition of ERK1/2 phosphorylation are associated with the induction of premature cellular senescence (Bose et al. 2012), which is a state of irreversible cell growth arrest (Aird and Zhang 2013).
The neural stem cells were used as a model by Bose et al., in 2012, to study the direct and long-lasting inherited changes that occurred in the brain due to low-dose exposure to MeHg. The study shows that MeHg decreases the proliferation of NSCs both as a result of direct exposure and as an inherited consequence to the exposure. In addition, global DNA hypomethylation (a biomarker for environmental toxicity) was observed in the NSCs after the exposure to a nanomolar dose of MeHg (Bose et al. 2012). The consequence of this MeHg exposure on the NSCs is its long-lasting morphological and functional alterations to the neural stem cells, which increase the risk of neurodegeneration and/or neurodevelopmental disorders later in life. Furthermore, the rate of proliferation of neural progenitor cells in the sub granule region of the dentate gyrus decreases due to exposure to MeHg (Bose et al. 2012). In another study, Basu and colleagues reported reduction in global DNA methylation and DNMT activity in minks exposed to MeHg (Basu et al. 2013). A recent study by Carvan et al. (2017) shows MeHg is capable of eliciting a transgenerational effect on unexposed offspring in zebrafish. MeHg elicits its epigenetic alterations on the differential DNA methylation regions (DMRs) (existing in CpG deserts with low CpG density) up on till the F2. These epigenetically altered DMR associated genes observed in the F2 generation give rise to some behavioral phenotypic changes (such as hyperactivity and visual deficit), which are due to dysregulation of metabolic pathways, neuroactive receptor-ligand interactions, actin cytoskeleton pathway, and MAPK (Mitogen-activated protein kinase) pathway (Carvan III et al. 2017).
An investigation by Maccani and colleagues demonstrated a possible novel mechanism by which in-utero exposure to Hg could potentiate aberrant infant neurobehavioral outcomes. MeHg crosses the placenta, causing an epigenetic alteration by disrupting the placental DNA methylation patterns (known to be detrimental to foetal development). 339 loci of genes already known to be affected by Hg exposure-induced neurodevelopmental disorders were examined, 10 of which were associated with high-risk NICU Network Neurobehavioral Scale (NNNS) profile. Interestingly, 6 of these loci found to be located in the EMID2 COL26A1 (collagen type xxvi α−1 chain) gene were hypomethylated due to Hg exposure. Although, the precise role of EMID2 modulation on neurodevelopment is still under investigation, however its hypomethylation is suggested to likely result in adverse neurobehavioral outcome in infants (Maccani et al. 2015). Furthermore, exposure to MeHgCl (methylmercury chloride) lead to over-expression of miR-302b, miR-367, miR-372, miR-196b, and miR-141. These five miRNAs (which are ncRNAs) are pivotal in the manner by which cells respond to stress and in the developmental process. Therefore, these alterations caused by MeHgCl affect neurological development via the disruption of the signalling pathways involved in both learning and memory formation (Pallocca et al. 2013).
4.5. Nickel
Nickel (Ni) is an abundant essential element that has an extensive range of application. It is the twenty-fourth most abundant element in the Earth’s crust. Nickel is used industrially in steel and alloy production, electroplating, nickel-cadmium battery production (nickel hydroxide), chemical catalysis, the manufacture of electronic components such as vacuum tubes and transistors (nickel carbonate), and in the manufacture of metal items such as ships, jet turbines, armaments, spark plugs, dental tools, factory tools, and household utensils. (Klein and Costa 2015). The processing of Ni has led to occupational exposure and an elevated level of contamination and the pollution of water, soil and air (Muñoz and Costa 2012). Although present in the human body, its functional significance in humans remains unidentified, with no enzymes employing Ni having been established in mammals (Huang et al. 2013; Maroney and Ciurli 2013). Also, it has been implicated in several neurological diseases including AD (Kim et al., 2012) and PD (Johansson et al., 2006) (see Table 1).
4.5.1. Ni neurotoxicity
Ni overexposure either via oral, nasal and dermal routes induces toxicity to the nervous system and other body systems (Afridi et al. 2011; Ijomone et al. 2018b). The rate of absorption of Nickel is reliant on the route of exposure; orally consumed nickel is absorbed in a diminutive quantity (about 1%) via the intestinal tract. 20% of those from inhaled sources are absorbed by the respiratory tract (Klein and Costa 2015). The absorbed Ni is distributed to several organs. The most targeted site of distribution is the kidney, which is the primary site of nickel accumulation, followed by the lung, brain, and pancreas. The absorbed Nickel reaches the intracellular compartment of the brain via various transporter systems, which includes (i) competitive transport by transferrin which is usually involved in cellular uptake of iron, (ii) competitive transport by divalent metal transporter 1 (DMT-1), also involved Mn, cobalt, copper, and zinc transport. Due to the shared transport system, Ni has the ability to disrupt the homeostasis of iron, Mn, cobalt, copper, and zinc uptake (Chen et al. 2006; Das and Büchner 2007). The extent of cellular damage following Ni-induced neurotoxicity is dependent on the route of exposure. Nickel also has a dose-dependent effect on some antioxidant enzymes. It surges LPO levels, and decreases GSH, GST, and glutathione peroxidase (GSH-Px) level (Ijomone et al. 2018a).
There are relatively limited literatures on Ni-induced neurotoxicity. However, an independent work by Topal et al., and He et al. demonstrated the neurotoxic effect of Ni. Topal et al., demonstrated using a fish, the behavioural change and altered antioxidant and cholinergic activities induced by Ni exposure, (Topal et al. 2015). He et al., also noticed similar alterations while experimenting on mice (He et al. 2013). Ni toxicity also affects tissue morphology by causing degeneration of hippocampal and striatal cells and subsequent alteration in cognition and locomotion (Ijomone et al. 2018a). Furthermore, another study showed Ni treated rat cells showed electron dense and heterochromatic neurons with loss of Golgi apparatus and distension of endoplasmic reticulum. The study also demonstrated the region-dependent effect of Ni toxicity in the brain, with the cortex and striatum showing less ultrastructural damage compared to the hippocampus at the same dose. It was also established that Ni increased the expression of caspase-3 (a protein involved in the apoptotic pathway) in the striatum, CA3 and DG of the hippocampus, and also the possibility of perturbed α-synuclein expression following Ni treatment (Ijomone et al. 2018b).
4.5.2. Ni-induced epigenetics changes
A number of studies have suggested that epigenetic alterations underlie nickel pathogenesis (Scanlon et al. 2017; Yin et al. 2018). Ni induces suppression of the DNA repair gene, O6-methylguanine DNA methyltransferase (MGMT) by some epigenetic modifications. These modifications include (i) promote hypermethylation by DNMT, MeCP2 and MBD2 recruitment, (ii) increased histone methylation and reduced acetylation indicated by high level of H3K9me2 and low levels of H4ac and H3K9ac respectively (Chen et al. 2006). It has been reported that silencing of the tumour suppressor genes, ELF4 (encoding ETS-related transcription factor Elf-4) following Ni-induced methylation. There was a similar occurrence in the Cdkn2a/p16, Rarb, and Rassf1a gene which are related to carcinogenesis (Klein and Costa 2015). Hypermethylation of the promoter for the tumour suppressor gene p53 due to Ni exposure have also been examined in wild type C57BL/6 mice and mice heterozygous for the tumour suppressor p53 gene (Melnikova and Ananthaswamy 2005).
Ni also brings about alterations in chromatin structure by modifying histone tails via methylation, acetylation, phosphorylation, or ubiquitination. Dose- and time dependent, inhibition of histone H2A, H2B, H3 and H4 via acetylation, and substantial increases of H3K9 dimethylation and ubiquitination of H2A and H2B are commonly found and they generally result in heterochromatinization and subsequent suppression of global gene transcription of tumour suppressor gene (Ellen et al. 2009; Karaczyn et al. 2006). This was demonstrated when Ni induced tumorigenesis caused heterochromatinization-associated with gene repression-of p16 gene following hypermethylation and subsequent gene repression (see review Ryu et al. 2015). Ni ions impede histone acetyltransferases causing extensive deacetylation. It also impedes H3K9 demethylases, ensuing increased levels of H3K9me2 (Jose et al. 2019). Ni toxicity also results in increased levels of H2A and 2B ubiquitination leading to alterations in chromatin structure, repression of gene expression, and subsequent carcinogenesis when the tumour suppressor genes are involved (Ke et al. 2008).
Furthermore, pre-treatment of mouse PW cells and human cells with the histone deacetylase inhibitor trichostatin A (TSA) significantly inhibited the ability of Ni to induce cell transformation suggesting that gene silencing mediated by histone alteration may be involved in Ni-induced cell alteration (Li et al. 2009). Inhibition of histone acetyltransferase activity has been stated as the mechanism by which Ni exposure lowers histone acetylation (Drukteinis et al. 2005). In an additional study carried out by Chen et al., there was an increase in H3K9 demethylation at the gpt locus in G12 Chinese hamster cells, resulting in the suppression of the transgene expression. This action was attributed to the inhibition of histone H3K9 demethylase (JHDM2A) by Ni, and the effect was reversed by treatment with 5-aza-2´-deoxycytidine, a DNA-demethylating agent (Chen et al. 2006). Additionally, Ni-induced histone modification is time and dose dependent; reduced Ni dose caused increase in H2B ubiquitination, while increased dose >0.25 Mm resulted in decreased H2B ubiquitination resulting in gene silencing and activation, respectively (Karaczyn et al. 2006).
Generally, it has been shown that Ni increases DNA methylation, has a time and dose dependent effect on histone ubiquitination, leads to either increase or decrease in histone methylation and acetylation through modulation of the activity of relevant enzymes, such as the HMT, acetyltransferase, or demethylase. Although Ni has a low mutagenic activity, it is implicated in a number of cases where exposure has led to the formation of cancer. It is therefore believed that alteration in the expression of certain genes (tumour suppressor genes, oncogenes, DNMTs, and HMTs) due to Ni toxicity has the potential to cause cancer, either directly or indirectly (Salnikow and Zhitkovich 2007).
4.6. Cadmium
Cadmium (Cd) is a non-essential transitional metal, typically present as a divalent cation, complexed with other elements (e.g. CdCl2). It is used in various processes, for example in the manufacture of laser batteries, in certain electroplating methods, one of the components of tobacco, in the production of metal (see review Bernhoft 2013; see review Wang and Du 2013). Exposure to Cd usually occurs through inhalation and ingestion of contaminated food or water. Cigarette smoking has been regarded as the most substantial source of human Cd exposure. Absorption through skin is also possible but insignificant (Abernethy et al. 2010). The ingested or inhaled Cd are subsequently absorbed for distribution through several transporters (Nordberg 2009). Due to similarity of the transport system, Cd absorption is largely dependent upon particle size, and level of other metals such as iron, calcium and zinc. Deficiency of one of these metals has been linked to increase in Cd absorption (see review Bernhoft 2013). The absorbed Cd is usually transported in the body bound to sulfhydryl group-containing protein e.g. metallothionein. About 30% of absorbed Cd are deposited in the liver, same goes for the kidney. The rest are distributed to other body organs including the brain (Abernethy et al. 2010). In the brain, Cd could contribute to disorders such AD by exacerbating the toxic effect of ApoE4 (Zhang et al., 2020), and PD via the disruption of Ubiquitin-Proteasome System (Yu et al., 2010). It has also been associated with autism and ADHD by several epidemiological studies (Hessabi et al., 2019; Lee et al., 2018) (see Table 1).
4.6.1. Cd neurotoxicity
The CNS is generally susceptible to Cd overexposure, and the risk is higher in neonates due to underdeveloped BBB (see review Wang and Du 2013). In the CNS, Cd preferentially stores in the choroid plexus and cerebral cortical neurons (Xu et al. 2011) than in other brain structures. This was shown by a post-mortem human study where Cd level in the choroids plexus was approximately 2–3 folds of that in the cerebral cortex (see review Wang and Du 2013). Also, another study showed Cd-induced neuronal death in cortical neurons via a collective mechanism of apoptosis and necrosis that involves reactive oxygen species generation and lipid peroxidation (Lopez et al. 2006). Cd induces changes in the histomorphology of BBB leading to increased permeability and subsequent intracellular accumulation and toxicity (Goncalves et al. 2010). Olfactory transportation of cadmium through the olfactory nerve to the olfactory bulb helps Cd bypass the BBB. Thereby implicating occupational inhalation of cadmium as an olfactory toxicant (Bondier et al. 2008; Czarnecki et al. 2011).
In the CNS, cadmium induces tissues injury by (i) oxidative stress leading to apoptosis (Lopez et al. 2006; Matovíc et al. 2011), (ii) free intracellular calcium (Ca2+) homeostasis disruption, resulting in apoptosis. Cd may block the influx of Ca2+ through membrane channels into the nerve terminal following the action potential; these decreases in calcium influx caused by Cd would be associated with an altered transmitter release (Bodereau-Dubois et al. 2012; Xu et al. 2011), (iii) creating epigenetic changes in DNA that alters gene expression (Chen et al. 2011; Lopez et al. 2006). Additional pathologic mechanism include competitive interference with the physiologic action of Zn or Mg. Cd has been shown to hamper Zn uptake, by using transport systems that usually function to control Zn levels in brain. Reduction in Zn and Cu level were observed following treatment with Cd in 15- and 21-days old animals. This early Cd exposure may produce alteration in the synthesis of several lipids, leading to CNS dysfunctions and a possibility of being manifested in later life (Moulis 2010) and impairment of mitochondrial function and subsequently inducing apoptosis (Cannino et al. 2009). Cd also induces several biochemical variations in the CNS, it inhibits the discharge of acetylcholine by interfering with calcium metabolism, upsurges serotonin sensibility (Goncalves et al. 2010), as well as the antioxidant level (Méndez-Armenta and Ríos 2007).
Cd exposure alters behaviours and reduces learning ability (Méndez-Armenta and Ríos 2007). Behavioural deficits, neurochemical alterations, and brain lesions were described in experimental animals, while in humans acute Cd poisoning produced Parkinsonism symptoms (Bao et al. 2009). Cd accumulation prior to and at birth, might cause irreversible or lasting changes in the brain, which in turn leads to the altered gene expression (Ishitobi et al. 2007; Ishitobi and Watanabe 2005).
4.6.2. Cd-induced epigenetic changes
Several studies have demonstrated the epigenetic effect of Cd-induced toxicity and has identifying DNA methylation as a reoccurring mechanism (Doi et al. 2011; Jiang et al. 2008). Cd induces changes to the DNA by epigenetics rather that alteration to DNA sequence because it binds weakly to the DNA. Cd may well act as an epigenetic or indirectly genotoxic carcinogen since it is, in general, poorly mutagenic. These epigenetic changes are achieved through DNA methylation, histone modification and miRNA (Doi et al. 2011; Jiang et al. 2008).
Cd exposure affects the methylation pattern of DNA by interfering with DNMT-DNA interaction and resulting in either hypermethylation or hypomethylation depending on the duration of exposure. In a cultured cell, chronic myelogenous leukaemia K562, Short time exposure (for about 24 hours to 1 week) led to DNA hypomethylation by means of non-competitive inhibition of DNMT activity by Cd. To the contrary, global DNA hypermethylation and enhanced DNMT activity has been noted in human embryo lung fibroblast cells (HLF) following prolonged Cd exposure for about 8 to 10 weeks (Huang et al. 2008; Jiang et al. 2008).
The process of cytosine methylation is driven by the DNA methyltransferase family which includes DNMT1, DNMT2, DNMT3a, DNMT3b. DNMT3a and DNMT3b methylate cytosine, this methylation state is maintained by DNMT1 even during replication (Horii et al. 2013; Martins-Taylor et al. 2012). The level of expression of these proteins is affected by time-dependent Cd exposure. DNMT3b overexpression is demonstrated following chronic Cd-induced toxicity in human prostate (Benbrahim-Tallaa et al. 2007), while downregulation of DNMT activity was found in rat livers after acute Cd overexposure, while upregulation of DMNT activity and subsequent hypermethylation was observed in the same cells following chronic exposure (Wang et al. 2012). This is also consistent with observations in human embryo lung fibroblast cell (Jiang et al. 2008), myelogenous leukaemia K562 cell (Huang et al. 2008).
Valinluck et al., (2004) have implicated oxidative stress as an indirect mechanism by which Cd overexposure influences DNA methylation. The upsurge in the level of reactive oxygen species results in low methyltransferases-DNA interaction (Valinluck et al. 2004), and subsequent cytosine hypomethylation at CpG sites. This could be also due to a possible direct interaction of Cd with the methyltransferase DNA binding domain (see review Wang and Du 2013).
Histone modification is another mechanism by which Cd induces toxicity. Several studies have connected Cd overexposure to several histone modifications (see review Wang et al. 2012). Cd-induced histone modifications have mainly been described in in-vitro studies. The levels of H3K4me3, H3K27me3 and H3K9me3 occupancy at the metallothionein 3 (MT3) promoter increased in Cd treated cells (Somji et al. 2011). Apart from modifying the methylation status of histone H3 lysine residues, Cd also decreases the phosphorylation of H3 via inhibition of the human vaccinia-related kinase VRK1/2 (Barcia-Sanjurjo et al. 2013). Additionally, Cd-induced epigenetic change has been suggested as mechanism for the pathogenesis of AD. (Zhou et al., 2020) demonstrated that Cd exposure upregulated HDAC2 expression and also impaired learning and memory. The aberrant up-regulation of HDAC2 have been identified as an etiological factor in the onset of AD in a mice model (Qing et al., 2008). This demonstrates the ability of Cd to modulate histones through inhibition of histone modifying enzymes.
5. Concluding remarks
The underlying cause of many neurologic and neurodegenerative diseases are still unresolved. However, it is now widely accepted that the environmental toxicants contribute to the development of many of these disorders. Metals, appear to be the most implicated environment toxicants resulting in CNS disorders. Metal neurotoxicity has been implicated in various brain diseases, including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Data from epidemiologic studies have continuously linked these diseases to metal exposure. Several mechanisms have been implicated in metals-induced neurotoxicity; however, oxidative stress appears a common feature in most metal neurotoxicity. The impact of metals on epigenetics as mechanism for the pathogenesis of CNS disorders is a focus of recent researches. Because epigenetic changes are inheritable, the precise process by which metals induce these changes are of considerable importance. Thus, researches have been focused on identifying the epigenetic changes associated with metal-induced neurotoxicity and its role in the development of brain diseases. These epigenetic changes are also inducible by metals at low concentration. Here we have reviewed recent evidence highlighting the possible influence of metal overexposure on overall brain by mediating epigenetic changes. Our review has summarized epigenetic changes including DNA methylation, post-translational histone modification and ncRNAs, induced by metals could lead to upregulation or downregulation of specific genes and consequently resulting in CNS disorders. It is noteworthy that the epigenetic changes through endpoints of DNA methylation and histone modification are common to all metals reviewed here. Additionally, we observed that epigenetic changes via DNA methylation mostly involve inhibition or reduction of DNA methyltransferase activities for all metals. On the other hand, epigenetic changes through ncRNAs were identified Mn, Pb, Hg, As but not Ni and Cd. Nevertheless, reports of changes through ncRNAs are limited and further studies are needed to delineate involvement ncRNAs in metal-induced epigenetic changes. Understanding metal-induced epigenetic changes could provide better insight on the mechanism through which neurotoxicity occurs and can help model targeted therapies for the diseases of the brain.
Acknowledgements
OMI acknowledges the Young IBRO Regions Connecting Awards towards collaborative activities between The Neuro- Lab, Federal University of Technology Akure, Nigeria, and Aschner’s Lab, Albert Einstein College of Medicine, USA. MA is supported by National Institute of Health (NIH), USA grants; NIEHS R01 10563, NIEHS R01 07331 and NIEHS R01 020852.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abel T, Zukin RS, 2008. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Current opinion in pharmacology 8(1), 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abernethy DR, DeStefano AJ, Cecil TL, Zaidi K, Williams RL, 2010. Metal impurities in food and drugs. Pharmaceutical Research 27(5), 750–755. [DOI] [PubMed] [Google Scholar]
- Afridi HI, Kazi TG, Kazi N, Kandhro GA, Baig JA, Shah AQ, Wadhwa SK, Khan S, Kolachi NF, Shah F, 2011. Evaluation of status of cadmium, lead, and nickel levels in biological samples of normal and night blindness children of age groups 3–7 and 8–12 years. Biological trace element research 142(3), 350–361. [DOI] [PubMed] [Google Scholar]
- Ai S. x., Xu Q, Hu Y. c., Song C. y., Guo J. f., Shen L, Wang C. r., Yu R. l., Yan X. x., Tang B. s., 2014. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. Journal of the neurological sciences 337(1–2), 123–128. [DOI] [PubMed] [Google Scholar]
- Aird KM, Zhang R, 2013. Detection of senescence-associated heterochromatin foci (SAHF), Cell Senescence. Springer, pp. 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade V, Aschner M, Dos Santos AM, 2017. Neurotoxicity of metal mixtures, Neurotoxicity of Metals. Springer, pp. 227–265. [DOI] [PubMed] [Google Scholar]
- Andrew AS, Chen CY, Caller TA, Tandan R, Henegan PL, Jackson BP, Hall BP, Bradley WG, Stommel EW, 2018. Toenail mercury Levels are associated with amyotrophic lateral sclerosis risk. Muscle & nerve 58(1), 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin DW, Spolding B, Gondalia S, Shandley K, Palombo EA, Knowles S, Walder K, 2014. Genetic variation associated with hypersensitivity to mercury. Toxicology international 21(3), 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey KA, Wu MC, Ward WO, Smeester L, Rager JE, García-Vargas G, Del Razo LM, Drobná Z, Stýblo M, Fry RC, 2013. Arsenic and the epigenome: interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. Journal of biochemical and molecular toxicology 27(2), 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Q-S, Lu C-Y, Song H, Wang M, Ling W, Chen W-Q, Deng X-Q, Hao Y-T, Rao S, 2009. Behavioural development of school-aged children who live around a multi-metal sulphide mine in Guangdong province, China: a cross-sectional study. BMC public health 9(1), 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia-Sanjurjo I, Vázquez-Cedeira M, Barcia R, Lazo PA, 2013. Sensitivity of the kinase activity of human vaccinia-related kinase proteins to toxic metals. JBIC Journal of Biological Inorganic Chemistry 18(4), 473–482. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge Y-W, Lahiri DK, Zawia NH, 2005. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and β-amyloid in the aging brain. Journal of Neuroscience 25(4), 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu N, Head J, Nam D-H, Pilsner JR, Carvan MJ, Chan HM, Goetz FW, Murphy CA, Rouvinen-Watt K, Scheuhammer AM, 2013. Effects of methylmercury on epigenetic markers in three model species: mink, chicken and yellow perch. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology 157(3), 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates EA, Victor M, Jones AK, Shi Y, Hart AC, 2006. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. Journal of Neuroscience 26(10), 2830–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Waterland RA, Dill AL, Webber MM, Waalkes MP, 2007. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environmental health perspectives 115(10), 1454–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhoft RA, 2013. Cadmium toxicity and treatment. The Scientific World Journal 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertogliat MJ, Morris-Blanco KC, Vemuganti R, 2020. Epigenetic mechanisms of neurodegenerative diseases and acute brain injury. Neurochemistry international 133, 104642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N, Burns DM, Blau HM, 2011. DNA demethylation dynamics. cell. 146(6), 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihaqi SW, Bahmani A, Subaiea GM, Zawia NH, 2014. Infantile exposure to lead and late-age cognitive decline: relevance to AD. Alzheimer’s & Dementia 10(2), 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihaqi SW, Zawia NH, 2013. Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 39, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bind M-A, Baccarelli A, Zanobetti A, Tarantini L, Suh H, Vokonas P, Schwartz J, 2012. Air pollution and markers of coagulation, inflammation and endothelial function: Associations and epigene-environment interactions in an elderly cohort. Epidemiology (Cambridge, Mass.) 23(2), 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Scharfman HE, 2004. Brain-derived neurotrophic factor. Growth factors (Chur, Switzerland) 22(3), 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodereau-Dubois B, List O, Calas-List D, Marques O, Communal P-Y, Thany SH, Lapied B, 2012. Transmembrane potential polarization, calcium influx, and receptor conformational state modulate the sensitivity of the imidacloprid-insensitive neuronal insect nicotinic acetylcholine receptor to neonicotinoid insecticides. Journal of Pharmacology and Experimental Therapeutics 341(2), 326–339. [DOI] [PubMed] [Google Scholar]
- Bondier JR, Michel V, Propper A, Badot PM, 2008. Harmful effects of cadmium on olfactory system in mice. Inhalation Toxicology 20(13), 1169–1177. [DOI] [PubMed] [Google Scholar]
- Bornhorst J, Wehe CA, Hüwel S, Karst U, Galla H-J, Schwerdtle T, 2012. Impact of manganese on and transfer across blood-brain and blood-cerebrospinal fluid barrier in vitro. Journal of Biological Chemistry 287(21), 17140–17151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Onishchenko N, Edoff K, Janson Lang AM, Ceccatelli S, 2012. Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicological Sciences 130(2), 383–390. [DOI] [PubMed] [Google Scholar]
- Bouabid S, Delaville C, De Deurwaerdère P, Lakhdar-Ghazal N, Benazzouz A, 2014. Manganese-induced atypical parkinsonism is associated with altered basal ganglia activity and changes in tissue levels of monoamines in the rat. PloS one 9(6), e98952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L, Uversky VN, 2011. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 3(11), 1163–1180. [DOI] [PubMed] [Google Scholar]
- Cai F, Zhong W, Zhang G, 2011. Effect of Manganese Chloride on Generation of Reactive Oxygen Species and Expression of miR-133b in Pheochro-mocytoma Cells. Journal of Environment and Health(12), 6. [Google Scholar]
- Caito S, Aschner M, 2015. Neurotoxicity of metals, Handbook of clinical neurology. Elsevier, pp. 169–189. [DOI] [PubMed] [Google Scholar]
- Calatayud M, Velez D, Devesa V, 2012. Metabolism of inorganic arsenic in intestinal epithelial cell lines. Chemical research in toxicology 25(11), 2402–2411. [DOI] [PubMed] [Google Scholar]
- Cannino G, Ferruggia E, Luparello C, Rinaldi AM, 2009. Cadmium and mitochondria. Mitochondrion 9(6), 377–384. [DOI] [PubMed] [Google Scholar]
- Capauto D, Colantoni A, Lu L, Santini T, Peruzzi G, Biscarini S, Morlando M, Shneider NA, Caffarelli E, Laneve P, 2018. A regulatory circuitry between Gria2, miR-409, and miR-495 is affected by ALS FUS mutation in ESC-derived motor neurons. Molecular neurobiology 55(10), 7635–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccatelli S, Bose R, Edoff K, Onishchenko N, Spulber S, 2013. Long-lasting neurotoxic effects of exposure to methylmercury during development. Journal of internal medicine 273(5), 490–497. [DOI] [PubMed] [Google Scholar]
- Ceccatelli S, Daré E, Moors M, 2010. Methylmercury-induced neurotoxicity and apoptosis. Chemico-biological interactions 188(2), 301–308. [DOI] [PubMed] [Google Scholar]
- Chen H, Ke Q, Kluz T, Yan Y, Costa M, 2006. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Molecular and cellular biology 26(10), 3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Chakraborty S, Mukhopadhyay S, Lee E, Paoliello MM, Bowman AB, Aschner M, 2015. Manganese homeostasis in the nervous system. Journal of neurochemistry 134(4), 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xu Y, Xu B, Guo M, Zhang Z, Liu L, Ma H, Chen Z, Luo Y, Huang S, 2011. CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. Journal of neurochemistry 119(5), 1108–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W-Y, Zhang H, Gatta E, Glover EJ, Pandey SC, Lasek AW, 2019. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol 78, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ, 2011. Epigenetic regulation of motor neuron cell death through DNA methylation. Journal of Neuroscience 31(46), 16619–16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AO, Brown SE, Szyf M, Maysinger D, 2008. Quantum dot-induced epigenetic and genotoxic changes in human breast cancer cells. Journal of molecular medicine 86(3), 291–302. [DOI] [PubMed] [Google Scholar]
- Cholanians AB, Phan AV, Ditzel EJ, Camenisch TD, Lau SS, Monks TJ, 2016. From the cover: Arsenic induces accumulation of α-synuclein: Implications for synucleinopathies and neurodegeneration. Toxicological Sciences 153(2), 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri S, Cui Y, Klaassen CD, 2010. Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicology and applied pharmacology 245(3), 378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, 2002. The three modern faces of mercury. Environmental health perspectives 110(suppl 1), 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consales C, Toft G, Leter G, Bonde JPE, Uccelli R, Pacchierotti F, Eleuteri P, Jönsson BA, Giwercman A, Pedersen HS, 2016. Exposure to persistent organic pollutants and sperm DNA methylation changes in Arctic and European populations. Environmental and molecular mutagenesis 57(3), 200–209. [DOI] [PubMed] [Google Scholar]
- Cronican AA, Fitz NF, Carter A, Saleem M, Shiva S, Barchowsky A, Koldamova R, Schug J, Lefterov I, 2013. Genome-wide alteration of histone H3K9 acetylation pattern in mouse offspring prenatally exposed to arsenic. PloS one 8(2), e53478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki LA, Moberly AH, Rubinstein T, Turkel DJ, Pottackal J, McGann JP, 2011. Invivovisualization of olfactory pathophysiology induced by intranasal cadmium instillation in mice. NeuroToxicology 32(4), 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao T, Hong X, Wang X, Tang W-Y, 2015. Aberrant 5’-CpG methylation of cord blood TNFα associated with maternal exposure to polybrominated diphenyl ethers. PloS one 10(9), e0138815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KK, Büchner V, 2007. Effect of nickel exposure on peripheral tissues: role of oxidative stress in toxicity and possible protection by ascorbic acid. Reviews on environmental health 22(2), 157–173. [DOI] [PubMed] [Google Scholar]
- Delcuve GP, Khan DH, Davie JR, 2012. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clinical epigenetics 4(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E, 2011. α-Synuclein sequesters Dnmt1 from the nucleus a novel mechanism for epigenetic alterations in lewy body diseases. Journal of Biological Chemistry 286(11), 9031–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Chen Z, Zhu C, 2011. Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). Journal of experimental botany 62(10), 3563–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Puri P, McCann A, Bannigan J, Thompson J, 2011. Epigenetic effect of cadmium on global de novo DNA hypomethylation in the cadmium-induced ventral body wall defect (VBWD) in the chick model. Toxicological Sciences 120(2), 475–480. [DOI] [PubMed] [Google Scholar]
- Donkena KV, Young CY, Tindall DJ, 2010. Oxidative stress and DNA methylation in prostate cancer. Obstetrics and gynecology international 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drukteinis J, Medrano T, Ablordeppey E, Kitzman J, Shiverick K, 2005. Benzo [a] pyrene, but not 2, 3, 7, 8-TCDD, induces G2/M cell cycle arrest, p21CIP1 and p53 phosphorylation in human choriocarcinoma JEG-3 cells: a distinct signaling pathway. Placenta 26, S87–S95. [DOI] [PubMed] [Google Scholar]
- Du Z-W, Chen H, Liu H, Lu J, Qian K, Huang C-L, Zhong X, Fan F, Zhang S-C, 2015. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nature communications 6(1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaway KW, Islam MS, Coulson RL, Lopez SJ, Ciernia AV, Chu RG, Yasui DH, Pessah IN, Lott P, Mordaunt C, 2016. Cumulative impact of polychlorinated biphenyl and large chromosomal duplications on DNA methylation, chromatin, and expression of autism candidate genes. Cell reports 17(11), 3035–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi N, Flora SJ, 2011. Concomitant exposure to arsenic and organophosphates on tissue oxidative stress in rats. Food and chemical toxicology 49(5), 1152–1159. [DOI] [PubMed] [Google Scholar]
- Eid A, Bihaqi SW, Renehan WE, Zawia NH, 2016. Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellen TP, Kluz T, Harder ME, Xiong J, Costa M, 2009. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 48(21), 4626–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Morsi D, El-Bakary A, Hasaneen B, Elatta H, 2019. Lead and cadmium hair levels in a sample of Egyptian children with attention deficit hyperactivity disorder. J. Clin. Toxicol 9(1), 1–8. [Google Scholar]
- Farina M, Avila DS, Da Rocha JBT, Aschner M, 2013. Metals, oxidative stress and neurodegeneration: a focus on iron, manganese and mercury. Neurochemistry international 62(5), 575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Fallin MD, 2015. Epigenetics at the crossroads of genes and the environment. Jama 314(11), 1129–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gąssowska M, Baranowska-Bosiacka I, Moczydłowska J, Frontczak-Baniewicz M, Gewartowska M, Strużyńska L, Gutowska I, Chlubek D, Adamczyk A, 2016. Perinatal exposure to lead (Pb) induces ultrastructural and molecular alterations in synapses of rat offspring. Toxicology 373, 13–29. [DOI] [PubMed] [Google Scholar]
- Gatto S, Gagliardi M, Franzese M, 2017. ICF-specific DNMT3B dysfunction interferes with intragenic regulation of mRNA transcription and alternative splicing. Nucleic Acids Research Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Basu A, Mahata J, Basu S, Sengupta M, Das JK, Mukherjee A, Sarkar AK, Mondal L, Ray K, 2006. Cytogenetic damage and genetic variants in the individuals susceptible to arsenic-induced cancer through drinking water. International journal of cancer 118(10), 2470–2478. [DOI] [PubMed] [Google Scholar]
- Golebiowski F, Kasprzak KS, 2005. Inhibition of core histones acetylation by carcinogenic nickel (II). Molecular and cellular biochemistry 279(1–2), 133–139. [DOI] [PubMed] [Google Scholar]
- Goncalves JF, Fiorenza AM, Spanevello RM, 2010. N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chemico-Biological Interactions 186(1), 53–60. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zuñiga M, Contreras PS, Estrada LD, Chamorro D, Villagra A, Zanlungo S, Seto E, Alvarez AR, 2014. c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer’s disease. Molecular cell 56(1), 163–173. [DOI] [PubMed] [Google Scholar]
- Guo L, Byun HM, Zhong J, Motta V, Barupal J, Zheng Y, Dou C, Zhang F, McCracken JP, Diaz A, 2014. Effects of short-term exposure to inhalable particulate matter on DNA methylation of tandem repeats. Environmental and molecular mutagenesis 55(4), 322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zhang Z, Wang Q, Zhang J, Wang L, Zhang Q, Li H, Wu S, 2018. Manganese chloride induces histone acetylation changes in neuronal cells: Its role in manganese-induced damage. Neurotoxicology 65, 255–263. [DOI] [PubMed] [Google Scholar]
- Harischandra DS, Ghaisas S, Zenitsky G, Jin H, Kanthasamy A, Anantharam V, Kanthasamy A, 2019. Manganese-induced Neurotoxicity: New Insights into Protein Misfolding, Mitochondrial Impairment and Neuroinflammation. Frontiers in neuroscience 13, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Kobayashi Y, Cui X, Hirano S, 2005. A new metabolic pathway of arsenite: arsenic–glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Archives of toxicology 79(4), 183–191. [DOI] [PubMed] [Google Scholar]
- He J, Wang M, Jiang Y, Chen Q, Xu S, Xu Q, Jiang B-H, Liu L-Z, 2014. Chronic arsenic exposure and angiogenesis in human bronchial epithelial cells via the ROS/miR-199a-5p/HIF-1 α/COX-2 pathway. Environmental health perspectives 122(3), 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M-D, Xu S-C, Zhang X, Wang Y, Xiong J-C, Zhang X, Lu Y-H, Zhang L, Yu ZP, Zhou Z, 2013. Disturbance of aerobic metabolism accompanies neurobehavioral changes induced by nickel in mice. Neurotoxicology 38, 9–16. [DOI] [PubMed] [Google Scholar]
- Hessabi M, Rahbar MH, Dobrescu I, Bach MA, Kobylinska L, Bressler J, Grove ML, Loveland KA, Mihailescu I, Nedelcu MC, 2019. Concentrations of lead, mercury, arsenic, cadmium, manganese, and aluminum in blood of Romanian children suspected of having Autism spectrum disorder. International Journal of Environmental Research and Public Health 16(13), 2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Esteller M, 2012. DNA methylation profiling in the clinic: applications and challenges. Nat. Rev. Genet 13, 679–692. [DOI] [PubMed] [Google Scholar]
- Hintelmann H, 2010. Organomercurials. Their formation and pathways in the environment, Organometallics in environment and toxicology. Royal Society of Chemistry Cambridge, UK, pp. 365–401. [DOI] [PubMed] [Google Scholar]
- Horii T, Tamura D, Morita S, Kimura M, Hatada I, 2013. Generation of an ICF syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. International Journal of Molecular Sciences 14(10), 19774–19781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Zhang Y, Qi Y, Chen C, Ji W, 2008. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicology letters 179(1), 43–47. [DOI] [PubMed] [Google Scholar]
- Huang J, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wu B, 2013. The association between splenocyte apoptosis and alterations of Bax, Bcl-2 and caspase-3 mRNA expression, and oxidative stress induced by dietary nickel chloride in broilers. International journal of environmental research and public health 10(12), 7310–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen K, Yousefi P, Bradman A, Yan L, Harley KG, Kogut K, Eskenazi B, Holland N, 2014. Effects of age, sex, and persistent organic pollutants on DNA methylation in children. Environmental and molecular mutagenesis 55(3), 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ, 2011. Arsenic exposure and toxicology: a historical perspective. Toxicological Sciences 123(2), 305–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A, Raveendran VA, Kundu S, Samanta T, Shunmugam R, Pal D, Sarma JD, 2018. Mechanisms of Arsenic-Induced Toxicity with Special Emphasis on Arsenic-Binding Proteins. Arsenic-Analytical and Toxicological Studies, 1st ed.; Stoytcheva M, Zlatev R, Eds, 57–80. [Google Scholar]
- Idring S, Magnusson C, Lundberg M, Ek M, Rai D, Svensson AC, Dalman C, Karlsson H, Lee BK, 2014. Parental age and the risk of autism spectrum disorders: findings from a Swedish population-based cohort. International journal of epidemiology 43(1), 107–115. [DOI] [PubMed] [Google Scholar]
- Ijomone OM, Okori SO, Ijomone OK, Ebokaiwe AP, 2018a. Sub-acute nickel exposure impairs behavior, alters neuronal microarchitecture, and induces oxidative stress in rats’ brain. Drug and chemical toxicology 41(4), 377–384. [DOI] [PubMed] [Google Scholar]
- Ijomone OM, Olatunji SY, Owolabi JO, Naicker T, Aschner M, 2018b. Nickel-induced neurodegeneration in the hippocampus, striatum and cortex; an ultrastructural insight, and the role of caspase-3 and α-synuclein. Journal of Trace Elements in Medicine and Biology 50, 16–23. [DOI] [PubMed] [Google Scholar]
- Ishitobi H, Mori K, Yoshida K, Watanabe C, 2007. Effectsof perinatal exposure to low-dose cadmium on thyroid hormonerelated and sex hormone receptor gene expressions in brain of offspring,” NeuroToxicology 28(4), 790–797. [DOI] [PubMed] [Google Scholar]
- Ishitobi H, Watanabe C, 2005. Effects of low-dose perinatal cadmium exposure on tissue zinc and copper concentrations in neonatal mice and on the reproductive development of female offspring. Toxicology letters 159(1), 38–46. [DOI] [PubMed] [Google Scholar]
- Ivorra C, Fraga MF, Bayón GF, Fernández AF, Garcia-Vicent C, Chaves FJ, Redon J, Lurbe E, 2015. DNA methylation patterns in newborns exposed to tobacco in utero. Journal of translational medicine 13(1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskiewicz L, Filipowicz W, 2008. Role of Dicer in posttranscriptional RNA silencing, Rna Interference. Springer, pp. 77–97. [DOI] [PubMed] [Google Scholar]
- Jiang G, Xu L, Song S, Zhu C, Wu Q, Zhang L, Wu L, 2008. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology 244(1), 49–55. [DOI] [PubMed] [Google Scholar]
- Jo WJ, Ren X, Chu F, Aleshin M, Wintz H, Burlingame A, Smith MT, Vulpe CD, Zhang L, 2009. Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicology and applied pharmacology 241(3), 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, Ceccatelli S, 2007. Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotoxicity research 11(3–4), 241–260. [DOI] [PubMed] [Google Scholar]
- Johansson E, Westermarck T, Hasan M, Nilsson B, Stephen S, Adem A, 2006. Alterations in nickel and cadmium concentrations in erythrocytes and plasma of patients with Parkinson’s disease. Вестник Оренбургского государственного университета(12–2).
- Jordan A, Zhang X, Li J, Laulicht-Glick F, Sun H, Costa M, 2017. Nickel and cadmium-induced SLBP depletion: A potential pathway to metal mediated cellular transformation. PloS one 12(3), e0173624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose CC, Wang Z, Tanwar VS, Zhang X, Zang C, Cuddapah S, 2019. Nickel induced transcriptional changes persist post exposure through epigenetic reprograming. bioRxiv, 806588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaczyn AA, Golebiowski F, Kasprzak KS, 2006. Ni (II) affects ubiquitination of core histones H2B and H2A. Experimental cell research 312(17), 3252–3259. [DOI] [PubMed] [Google Scholar]
- Karri V, Schuhmacher M, Kumar V, 2016. Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Environmental toxicology and pharmacology 48, 203–213. [DOI] [PubMed] [Google Scholar]
- Ke Q, Ellen TP, Costa M, 2008. Nickel compounds induce histone ubiquitination by inhibiting histone deubiquitinating enzyme activity. Toxicology and applied pharmacology 228(2), 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RD, Cowley SM, 2013. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Portland Press Ltd. [DOI] [PubMed] [Google Scholar]
- Khaled EM, Meguid NA, Bjørklund G, Gouda A, Bahary MH, Hashish A, Sallam NM, Chirumbolo S, El-Bana MA, 2016. Altered urinary porphyrins and mercury exposure as biomarkers for autism severity in Egyptian children with autism spectrum disorder. Metabolic brain disease 31(6), 1419–1426. [DOI] [PubMed] [Google Scholar]
- Khoo SK, Petillo D, Kang UJ, Resau JH, Berryhill B, Linder J, Forsgren L, Neuman LA, Tan AC, 2012. Plasma-based circulating MicroRNA biomarkers for Parkinson’s disease. Journal of Parkinson’s disease 2(4), 321–331. [DOI] [PubMed] [Google Scholar]
- Kile ML, Fang S, Baccarelli AA, Tarantini L, Cavallari J, Christiani DC, 2013. A panel study of occupational exposure to fine particulate matter and changes in DNA methylation over a single workday and years worked in boilermaker welders. Environmental Health 12(1), 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A, 2007. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317(5842), 1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Knight EM, Saunders EL, Cuevas AK, Popovech M, Chen L-C, Gandy S, 2012. Rapid doubling of Alzheimer’s amyloid-β40 and 42 levels in brains of mice exposed to a nickel nanoparticle model of air pollution. F1000Research 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Costa M, 2015. Nickel, Handbook on the Toxicology of Metals. Elsevier, pp. 1091–1111. [Google Scholar]
- Kozomara A, Griffiths-Jones S, 2010. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic acids research 39(suppl_1), D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-J, Chou M-C, Chou W-J, Huang C-W, Kuo H-C, Lee S-Y, Wang L-J, 2018. Heavy metals’ effect on susceptibility to attention-deficit/hyperactivity disorder: implication of lead, cadmium, and antimony. International journal of environmental research and public health 15(6), 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang C, Zi X, Tu Q, Guo K, 2015. Epigenetic modulation of Cdk5 contributes to memory deficiency induced by amyloid fibrils. Experimental brain research 233(1), 165–173. [DOI] [PubMed] [Google Scholar]
- Li R, Shugart YY, Zhou W, An Y, Yang Y, Zhou Y, Zhang B, Lu D, Wang H, Qian J, 2009. Common genetic variations of the cytochrome P450 1A1 gene and risk of hepatocellular carcinoma in a Chinese population. European Journal of Cancer 45(7), 1239–1247. [DOI] [PubMed] [Google Scholar]
- Li S, Wang Y, Wang H, Bai Y, Liang G, Wang Y, Huang N, Xiao Z, 2011. MicroRNAs as participants in cytotoxicity of CdTe quantum dots in NIH/3T3 cells. Biomaterials 32(15), 3807–3814. [DOI] [PubMed] [Google Scholar]
- Li Y, Sun L, Cai T, Zhang Y, Lv S, Wang Y, Ye L, 2010. α-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain research bulletin 81(4–5), 428–433. [DOI] [PubMed] [Google Scholar]
- Li Y, Xie C, Murphy SK, Skaar D, Nye M, Vidal AC, Cecil KM, Dietrich KN, Puga A, Jirtle RL, 2015. Lead exposure during early human development and DNA methylation of imprinted gene regulatory elements in adulthood. Environmental health perspectives 124(5), 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-C, Tsai M-S, Chen M-H, Chen P-C, 2020. Mercury, Lead, Manganese, and Hazardous Metals, Health Impacts of Developmental Exposure to Environmental Chemicals. Springer, pp. 247–277. [Google Scholar]
- Lopez E, Arce C, Oset-Gasque M, Canadas S, Gonzalez M, 2006. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radical Biology and Medicine 40(6), 940–951. [DOI] [PubMed] [Google Scholar]
- Ma S, Qing L, Yang X, Liang G, Zhang L.e., Li Q, Xiong F, Peng S, Ma Y, Huang X, 2016. Expression Profiles of Long Noncoding RNAs and Messenger RNAs in Mn-Exposed Hippocampal Neurons of Sprague–Dawley Rats Ascertained by Microarray: Implications for Mn-Induced Neurotoxicity. PloS one 11(1), e0145856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccani JZ, Koestler DC, Lester B, Houseman EA, Armstrong DA, Kelsey KT, Marsit CJ, 2015. Placental DNA methylation related to both infant toenail mercury and adverse neurobehavioral outcomes. Environmental health perspectives 123(7), 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, Tarantini L, Schwartz J, 2011. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environmental health perspectives 119(7), 977–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy IM, Mohanna S, 2011. Epigenetics and the human brain: where nurture meets nature, Cerebrum: the Dana forum on brain science. Dana Foundation. [PMC free article] [PubMed] [Google Scholar]
- Maroney MJ, Ciurli S, 2013. Nonredox nickel enzymes. Chemical reviews 114(8), 4206–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit CJ, Eddy K, Kelsey KT, 2006. MicroRNA responses to cellular stress. Cancer research 66(22), 10843–10848. [DOI] [PubMed] [Google Scholar]
- Martin EM, Fry RC, 2016. A cross-study analysis of prenatal exposures to environmental contaminants and the epigenome: support for stress-responsive transcription factor occupancy as a mediator of gene-specific CpG methylation patterning. Environmental epigenetics 2(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Taylor K, Schroeder DI, LaSalle JM, Lalande M, Xu RH, 2012. Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics 7(1), 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matović V, Buha A, Bulat Z, Dukić-Ćosić D, 2011. Cadmium toxicity revisited: focus on oxidative stress induction and interactions with zinc and magnesium. Arhiv za Higijenu Rada iToksikologiju 16(1), 65–76. [DOI] [PubMed] [Google Scholar]
- Melnikova VO, Ananthaswamy HN, 2005. Cellular and molecular events leading to the development of skin cancer. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 571(1–2), 91–106. [DOI] [PubMed] [Google Scholar]
- Méndez-Armenta M, Ríos C, 2007. Cadmium neurotoxicity. Environmental Toxicology and Pharmacology 23(3), 350–358. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Phyu KP, Aung MN, Zin PW, Yano Y, Myint MZ, Thit WM, Yamamoto Y, Hishikawa Y, Thant KZ, 2019. Peripheral neuropathy induced by drinking water contaminated with low-dose arsenic in Myanmar. Environmental health and preventive medicine 24(1), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulis JM, 2010. Cellular mechanisms ofcadmium toxicity related to the homeostasis ofessential metals. BioMetals 23(5), 877–896. [DOI] [PubMed] [Google Scholar]
- Muñoz A, Costa M, 2012. Elucidating the mechanisms of nickel compound uptake: a review of particulate and nano-nickel endocytosis and toxicity. Toxicology and applied pharmacology 260(1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan R, Hogart A, Gwye Y, Martin MR, LaSalle JM, 2006. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 1(4), 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, Suk WA, 2013. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environmental health perspectives 121(3), 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal A, Guilarte T, 2012. Mechanisms of heavy metal neurotoxicity: lead and manganese. J Drug Metab Toxicol S 5(2), 1–13. [Google Scholar]
- Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, Labadorf A, Housman DE, Fraenkel E, 2013. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proceedings of the National Academy of Sciences 110(6), 2354–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niño SA, Martel-Gallegos G, Castro-Zavala A, Ortega-Berlanga B, Delgado JM, Hernández-Mendoza H.c., Romero-Guzmán E, Ríos-Lugo J, Rosales-Mendoza S, Jimenez-Capdeville ME, 2018. Chronic Arsenic Exposure Increases Aβ (1–42) Production and Receptor for Advanced Glycation End Products Expression in Rat Brain. Chemical research in toxicology 31(1), 13–21. [DOI] [PubMed] [Google Scholar]
- Nordberg GF, 2009. Historical perspectives on cadmium toxicology. Toxicology and applied pharmacology 238(3), 192–200. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Brack C, Müller-Spahn F, Stähelin H, Herrmann M, Renard P, Brockhaus M, Hock C, 2000. Mercury induces cell cytotoxicity and oxidative stress and increases β-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. Journal of Neurochemistry 74(1), 231–236. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Novakovic M, Savaskan E, Meier F, Baysang G, Brockhaus M, Müller-Spahn F, 2002. The effects of β-estradiol on SHSY5Y neuroblastoma cells during heavy metal induced oxidative stress, neurotoxicity and β-amyloid secretion. Neuroscience 113(4), 849–855. [DOI] [PubMed] [Google Scholar]
- Onishchenko N, Karpova N, Sabri F, Castrén E, Ceccatelli S, 2008. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. Journal of neurochemistry 106(3), 1378–1387. [DOI] [PubMed] [Google Scholar]
- Onishchenko N, Tamm C, Vahter M, Hökfelt T, Johnson JA, Johnson DA, Ceccatelli S, 2007. Developmental exposure to methylmercury alters learning and induces depression-like behavior in male mice. Toxicological sciences 97(2), 428–437. [DOI] [PubMed] [Google Scholar]
- Organization, W.H., 2003. Elemental mercury and inorganic mercury compounds: human health aspects, Elemental mercury and inorganic mercury compounds: Human health aspects.
- Pallocca G, Fabbri M, Sacco MG, Gribaldo L, Pamies D, Laurenza I, Bal-Price A, 2013. miRNA expression profiling in a human stem cell-based model as a tool for developmental neurotoxicity testing. Cell biology and toxicology 29(4), 239–257. [DOI] [PubMed] [Google Scholar]
- Parada B, Kimberly A, Jay S, 2014. Lead Toxicity: What is it and How is it Happening?
- Patti F, Fiore M, Chisari CG, D’Amico E, Fermo SL, Toscano S, Copat C, Ferrante M, Zappia M, 2020. CSF neurotoxic metals/metalloids levels in amyotrophic lateral sclerosis patients: comparison between bulbar and spinal onset. Environmental Research, 109820. [DOI] [PubMed] [Google Scholar]
- Peres TV, Schettinger MRC, Chen P, Carvalho F, Avila DS, Bowman AB, Aschner M, 2016. Manganese-induced neurotoxicity: a review of its behavioral consequences and neuroprotective strategies. BMC Pharmacology and Toxicology 17(1), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifl C, Khorchide M, Kattinger A, Reither H, Hardy J, Hornykiewicz O, 2004. α-Synuclein selectively increases manganese-induced viability loss in SK-N-MC neuroblastoma cells expressing the human dopamine transporter. Neuroscience letters 354(1), 34–37. [DOI] [PubMed] [Google Scholar]
- Pinyayev TS, Kohan MJ, Herbin-Davis K, Creed JT, Thomas DJ, 2011. Preabsorptive metabolism of sodium arsenate by anaerobic microbiota of mouse cecum forms a variety of methylated and thiolated arsenicals. Chemical research in toxicology 24(4), 475–477. [DOI] [PubMed] [Google Scholar]
- Rager JE, Bailey KA, Smeester L, Miller SK, Parker JS, Laine JE, Drobná Z, Currier J, Douillet C, Olshan AF, 2014. Prenatal arsenic exposure and the epigenome: altered microRNAs associated with innate and adaptive immune signaling in newborn cord blood. Environmental and molecular mutagenesis 55(3), 196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard JF, Puga A, 2010. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2(1), 87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Barranco M, Gil F, Hernández AF, Alguacil J, Lorca A, Mendoza R, Gómez I, Molina-Villalba I, González-Alzaga B, Aguilar-Garduño C, 2016. Postnatal arsenic exposure and attention impairment in school children. Cortex 74, 370–382. [DOI] [PubMed] [Google Scholar]
- Ryu H-W, Lee DH, Won H-R, Kim KH, Seong YJ, Kwon SH, 2015. Influence of toxicologically relevant metals on human epigenetic regulation. Toxicological research 31(1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv SK, Thurston SW, Bellinger DC, Amarasiriwardena C, Korrick SA, 2012. Prenatal exposure to mercury and fish consumption during pregnancy and attention-deficit/hyperactivity disorder–related behavior in children. Archives of pediatrics & adolescent medicine 166(12), 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salam MT, Byun H-M, Lurmann F, Breton CV, Wang X, Eckel SP, Gilliland FD, 2012. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. Journal of Allergy and Clinical Immunology 129(1), 232–239. e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K, Zhitkovich A, 2007. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chemical research in toxicology 21(1), 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Martín FJ, Lindquist DM, Landero-Figueroa J, Zhang X, Chen J, Cecil KM, Medvedovic M, Puga A, 2015. Sex-and tissue-specific methylome changes in brains of mice perinatally exposed to lead. Neurotoxicology 46, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Peña LC, Petrosyan P, Morales M, González NB, Gutiérrez-Ospina G, Del Razo LM, Gonsebatt ME, 2010. Arsenic species, AS3MT amount, and AS3MT gen expression in different brain regions of mouse exposed to arsenite. Environmental research 110(5), 428–434. [DOI] [PubMed] [Google Scholar]
- Sankhla MS, Sharma K, Kumar R, 2017. Heavy Metal Causing Neurotoxicity in Human Health. Int J Innov Res Sci, Eng Technol 6(5), 7721–7726. [Google Scholar]
- Scanlon SE, Scanlon CD, Hegan DC, Sulkowski P, Glazer PM, 2016. Abstract LB-029: Negative transcriptional and epigenetic regulation of DNA repair pathways by the heavy metals nickel and arsenic. AACR. [Google Scholar]
- Scanlon SE, Scanlon CD, Hegan DC, Sulkowski PL, Glazer PM, 2017. Nickel induces transcriptional down-regulation of DNA repair pathways in tumorigenic and non-tumorigenic lung cells. Carcinogenesis 38(6), 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Kidd S, Anderson D, 2013. Influence of developmental lead exposure on expression of DNA methyltransferases and methyl cytosine-binding proteins in hippocampus. Toxicology letters 217(1), 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield K, 2017. The metal neurotoxins: an important role in current human neural epidemics? International journal of environmental research and public health 14(12), 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Song C, He C, Zhang Y, 2014. Mechanism and function of oxidative reversal ofDNA and RNA methylation. Annu Rev Biochem 83, 585–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimshoni JA, Dalton EC, Jenkins A, Eyal S, Ewan K, Williams RS, Pessah N, Yagen B, Harwood AJ, Bialer M, 2007. The effects of central nervous system-active valproic acid constitutional isomers, cyclopropyl analogs, and amide derivatives on neuronal growth cone behavior. Molecular pharmacology 71(3), 884–892. [DOI] [PubMed] [Google Scholar]
- Shu L, Sun W, Li L, Xu Z, Lin L, Xie P, Shen H, Huang L, Xu Q, Jin P, 2016. Genome-wide alteration of 5-hydroxymenthylcytosine in a mouse model of Alzheimer’s disease. BMC genomics 17(1), 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Goel RK, Kaur T, 2011. Mechanisms pertaining to arsenic toxicity. Toxicology international 18(2), 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Gautam N, Mishra A, Gupta R, 2011. Heavy metals and living systems: An overview. Indian journal of pharmacology 43(3), 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A-Y, Jung J-W, Lee S, Kim H-S, Kang K-S, 2011. DNA methyltransferase controls stem cell aging by regulating BMI1 and EZH2 through microRNAs. PloS one 6(5), e19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somineni HK, Zhang X, Myers JMB, Kovacic MB, Ulm A, Jurcak N, Ryan PH, Hershey GKK, Ji H, 2016. Ten-eleven translocation 1 (TET1) methylation is associated with childhood asthma and traffic-related air pollution. Journal of Allergy and Clinical Immunology 137(3), 797–805. e795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somji S, Garrett SH, Toni C, Zhou XD, Zheng Y, Ajjimaporn A, Sens MA, Sens DA, 2011. Differences in the epigenetic regulation of MT-3 gene expression between parental and Cd+ 2 or As+ 3 transformed human urothelial cells. Cancer cell international 11(1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Kanthasamy A, Anantharam V, Sun F, Kanthasamy AG, 2010. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Molecular pharmacology 77(4), 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J-W, Choi B-S, 2013. Mercury induced the accumulation of amyloid beta (Aβ) in PC12 cells: the role of production and degradation of Aβ. Toxicological research 29(4), 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag K-C, 2010. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain research 1338, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer PS, Lein PJ, 2014. Neurotoxicity, Encyclopedia of Toxicology: Third Edition. Elsevier, pp. 489–500. [Google Scholar]
- Sriram K, Lin GX, Jefferson AM, Stone S, Afshari A, Keane MJ, McKinney W, Jackson M, Chen BT, Schwegler-Berry D, 2015. Modifying welding process parameters can reduce the neurotoxic potential of manganese-containing welding fumes. Toxicology 328, 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield KH, Pilsner JR, Lu Q, Wright RO, Guilarte TR, 2012. Dysregulation of BDNF-TrkB signaling in developing hippocampal neurons by Pb2+: Implications for an environmental basis of neurodevelopmental disorders. Toxicological Sciences 127(1), 277–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamegart L, Abbaoui A, Makbal R, Zroudi M, Bouizgarne B, Bouyatas MM, Gamrani H, 2019. Crocus sativus restores dopaminergic and noradrenergic damages induced by lead in Meriones shawi: A possible link with Parkinson’s disease. Acta histochemica 121(2), 171–181. [DOI] [PubMed] [Google Scholar]
- Tarale P, Daiwile AP, Sivanesan S, Stöger R, Bafana A, Naoghare PK, Parmar D, Chakrabarti T, Krishnamurthi K, 2018. Manganese exposure: Linking down-regulation of miRNA-7 and miRNA-433 with α-synuclein overexpression and risk of idiopathic Parkinson’s disease. Toxicology in Vitro 46, 94–101. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Seal S, Ruark E, 2014. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nature Genetics 46(4), 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ, 2012. Heavy metal toxicity and the environment, Molecular, clinical and environmental toxicology. Springer, pp. 133–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Yang H, Tian X, Wang H, Zhou T, Zhang S, Yu J, Zhang T, Fan D, Guo X, 2014. High manganese, a risk for Alzheimer’s disease: High manganese induces amyloid-β related cognitive impairment. Journal of Alzheimer’s disease 42(3), 865–878. [DOI] [PubMed] [Google Scholar]
- Topal A, Atamanalp M, Oruç E, Halıcı MB, Şişecioğlu M, Erol HS, Gergit A, Yılmaz B, 2015. Neurotoxic effects of nickel chloride in the rainbow trout brain: assessment of c-Fos activity, antioxidant responses, acetylcholinesterase activity, and histopathological changes. Fish physiology and biochemistry 41(3), 625–634. [DOI] [PubMed] [Google Scholar]
- Torres-Avila M, Leal-Galicia P, Sánchez-Peña LC, Del Razo LM, Gonsebatt ME, 2010. Arsenite induces aquaglyceroporin 9 expression in murine livers. Environmental research 110(5), 443–447. [DOI] [PubMed] [Google Scholar]
- Tran NQV, Miyake K, 2017. Neurodevelopmental disorders and environmental toxicants: epigenetics as an underlying mechanism. International journal of genomics 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, 2012. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. The American Journal of Human Genetics 90(3), 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschl K, Mills PB, Clayton PT, 2013. Manganese and the brain, International review of neurobiology. Elsevier, pp. 277–312. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Tsai H-H, Rogstad DK, Burdzy A, Bird A, Sowers LC, 2004. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic acids research 32(14), 4100–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mil NH, Steegers-Theunissen RP, Bouwland-Both MI, Verbiest MM, Rijlaarsdam J, Hofman A, Steegers EA, Heijmans BT, Jaddoe VW, Verhulst FC, 2014. DNA methylation profiles at birth and child ADHD symptoms. Journal of psychiatric research 49, 51–59. [DOI] [PubMed] [Google Scholar]
- Villar-Menéndez I, Blanch M, Tyebji S, Pereira-Veiga T, Albasanz JL, Martín M, Ferrer I, Pérez-Navarro E, Barrachina M, 2013. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A 2A R levels in Huntington’s disease. Neuromolecular medicine 15(2), 295–309. [DOI] [PubMed] [Google Scholar]
- Waly M, Olteanu H, Banerjee R, Choi S, Mason J, Parker B, Sukumar S, Shim S, Sharma A, Benzecry J, 2004. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Molecular psychiatry 9(4), 358–370. [DOI] [PubMed] [Google Scholar]
- Wang B, Du Y, 2013. Cadmium and its neurotoxic effects. Oxidative medicine and cellular longevity 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li Y, Shao C, Tan Y, Cai L, 2012. Cadmium and its epigenetic effects. Current medicinal chemistry 19(16), 2611–2620. [DOI] [PubMed] [Google Scholar]
- Wang L, Shiraki A, Itahashi M, Akane H, Abe H, Mitsumori K, Shibutani M, 2013. Aberration in epigenetic gene regulation in hippocampal neurogenesis by developmental exposure to manganese chloride in mice. toxicological sciences 136(1), 154–165. [DOI] [PubMed] [Google Scholar]
- Wessels I, 2017. Epigenetics and Minerals: An Overview. Handbook of Nutrition, Diet, and Epigenetics, 1–19. [Google Scholar]
- White AJ, Chen J, McCullough LE, Xu X, Cho YH, Teitelbaum SL, Neugut AI, Terry MB, Hibshoosh H, Santella RM, 2015. Polycyclic aromatic hydrocarbon (PAH)–DNA adducts and breast cancer: modification by gene promoter methylation in a population-based study. Cancer Causes & Control 26(12), 1791–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AJ, Chen J, Teitelbaum SL, McCullough LE, Xu X, Cho YH, Conway K, Beyea J, Stellman SD, Steck SE, 2016. Sources of polycyclic aromatic hydrocarbons are associated with gene-specific promoter methylation in women with breast cancer. Environmental research 145, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L, Waring R, 2002. Cysteine dioxygenase: modulation of expression in human cell lines by cytokines and control of sulphate production. Toxicology in vitro 16(4), 481–483. [DOI] [PubMed] [Google Scholar]
- Williams BB, Li D, Wegrzynowicz M, Vadodaria BK, Anderson JG, Kwakye GF, Aschner M, Erikson KM, Bowman AB, 2010. Disease-toxicant screen reveals a neuroprotective interaction between Huntington’s disease and manganese exposure. Journal of neurochemistry 112(1), 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M, Todd G, Roney N, Crawford J, Coles C, McClure P, Garey J, Zaccaria K, Citra M, 2012. Toxicological Profile for Manganese (Atlanta, GA: Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles). [PubMed]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, 2008. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. Journal of Neuroscience 28(1), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Chen S, Luo Y, 2011. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction ofreactive oxygen species and activation ofMAPK/mTOR network. PLoS One 6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Mas A, Diamond MP, Al-Hendy A, 2016. The mechanism and function of epigenetics in uterine leiomyoma development. Reproductive Sciences 23(2), 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, Mo J, Dai J, Wang H, 2018. Nickel (ii) inhibits the oxidation of DNA 5-methylcytosine in mammalian somatic cells and embryonic stem cells. Metallomics 10(3), 504–512. [DOI] [PubMed] [Google Scholar]
- Yokel RA, 2006. Blood-brain barrier flux of aluminum, manganese, iron and other metals suspected to contribute to metal-induced neurodegeneration. Journal of Alzheimer’s Disease 10(2–3), 223–253. [DOI] [PubMed] [Google Scholar]
- Yu X, Robinson JF, Sidhu JS, Hong S, Faustman EM, 2010. A system-based comparison of gene expression reveals alterations in oxidative stress, disruption of ubiquitin-proteasome system and altered cell cycle regulation after exposure to cadmium and methylmercury in mouse embryonic fibroblast. Toxicological sciences 114(2), 356–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahir F, Rizwi SJ, Haq SK, Khan RH, 2005. Low dose mercury toxicity and human health. Environmental toxicology and pharmacology 20(2), 351–360. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang H, Abel GM, Storm DR, Xia Z, 2020. The Effects of Gene-Environment Interactions Between Cadmium Exposure and Apolipoprotein E4 on Memory in a Mouse Model of Alzheimer’s Disease. Toxicological Sciences 173(1), 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Liu P, Wang J, Xiao X, Meng X, Zhang Y, 2016. Umbilical cord blood PBDEs concentrations are associated with placental DNA methylation. Environment international 97, 1–6. [DOI] [PubMed] [Google Scholar]
- Zhou R, Zhao J, Li D, Chen Y, Xiao Y, Fan A, Chen X-T, Wang H-L, 2020. Combined exposure of lead and cadmium leads to the aggravated neurotoxicity through regulating the expression of histone deacetylase 2. Chemosphere, 126589. [DOI] [PubMed] [Google Scholar]
- Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, Rao P, Farinelli L, Delalle I, Schmitt A, Falkai P, 2011. microRNA-34c is a novel target to treat dementias. The EMBO journal 30(20), 4299–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]