

Abstract
The proximal tubule (PT) is a major target in acute kidney injury (AKI), leading to profound changes in PT cell biology. Among the genes with early and robust changes in expression are many transcription factors (TFs), which themselves account for other transcriptomic changes. Potentially important TFs are being revealed in large sequencing datasets; however, to understand whether these TFs account for adaptive or maladaptive changes requires further mechanistic studies, which may reveal novel therapeutic targets. This minireview will highlight the identification and biology of three novel TFs in AKI: Sox9, Foxml and Foxo3.
Keywords: transcription factors, AKI, proximal tubule, RNA-sequencing
Introduction
Acute kidney injury (AKI) is a major risk factor for development of renal fibrosis and chronic kidney disease (CKD). AKI can be caused by ischemic or toxic insults, and the proximal tubule (PT) is particularly susceptible due to its high oxygen demand and role in excreting drugs/toxins [1]. AKI triggers major changes in PT cellular biology, including in differentiation, morphology, cell cycle, secretome, and metabolism [2]. Successful repair and restoration of fully mature and functional PT cells is achievable, but failure of repair may stimulate differentiation of resident fibroblasts into myofibroblasts and immune cells infiltration, causing fibrosis and CKD [1]. The roles of individual signaling molecules in PT injury are partially understood, but the mechanisms by which global pathways, and the switch between successful and unsuccessful repair are regulated, are less well characterized.
Transcription factors (TFs) are master regulators of fundamental biological processes, due to their ability to regulate the expression of multiple gene targets and form feedback loops. Their expression and functions may be regulated in response to intrinsic and extrinsic changes in environment by both transcriptional and post-translational mechanisms, and even modest changes in expression can lead to significant changes in downstream pathways. In AKI, many TFs are strongly upregulated, with variable timings, durations, and magnitudes. Some, such as FOS, JUN, and P53, are well known in the setting of AKI [3]. This review will describe three recently identified novel PT TFs in AKI.
Identification of novel transcriptional actors
The reducing costs of transcriptomic analysis, improving technologies for single cell/nuclear (sc/sn) RNA-seq, and advances in tools for bioinformatic analysis and machine learning, are facilitating a rapidly expanding knowledge of the transcriptional changes that occur during AKI. Among those that occur earliest and to the greatest extent, are changes in expression of TFs that themselves likely potentiate many of the other transcriptional changes. Thus, recent transcriptomic studies have revealed many new potential transcriptional actors in AKI, in human samples and mouse models, whose biological functions remain to be elucidated [3–6]. Furthermore, scRNA-seq and translating ribosome affinity purification (TRAP)-seq (Fig. 1) technologies enable the transcriptional signatures and drivers of individual cells/cell types to be revealed [7,8]. For example, a recent unilateral ureteric obstruction (UUO) TRAP-seq analysis demonstrated both upregulation and downregulation of a number of TFs not previously studied in PT injury and fibrosis [6]. Such transcriptomics studies provide rich ‘hypothesis-generating’ datasets that will lead to identification of novel transcriptional regulators in AKI.
Fig. 1. Schematic of TRAP-sequencing.
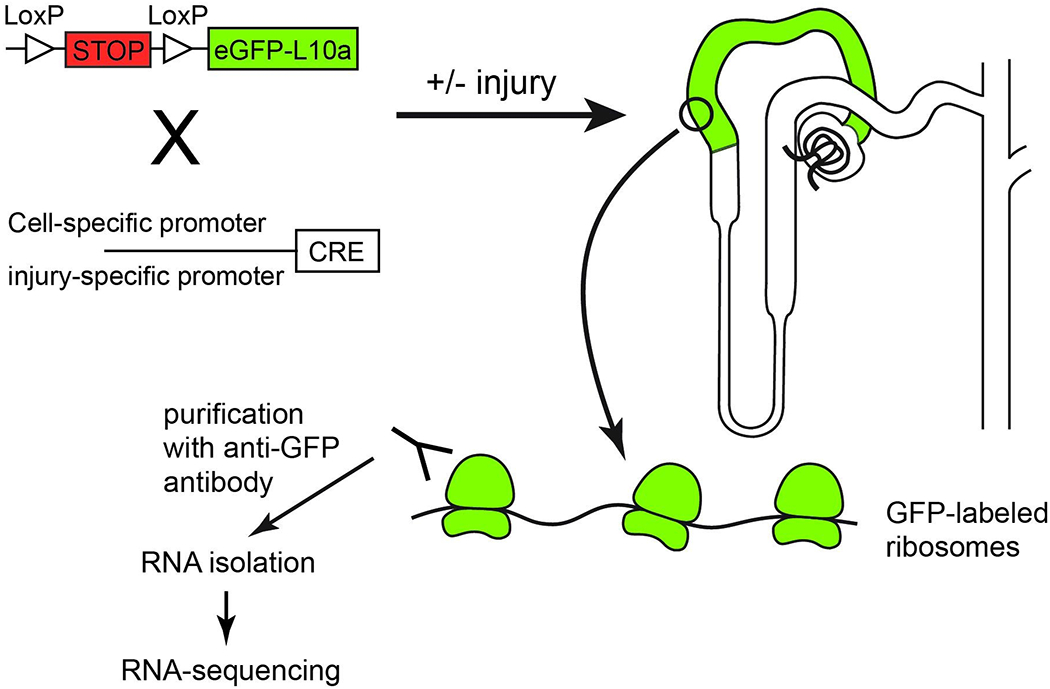
Mice carrying a transgene composed of a stop cassette flanked by LoxP sites upstream of cDNA encoding enhanced green fluorescent protein (eGFP) tagged ribosomal protein subunit L10a, are bred with mice containing Cre recombinase controlled by a cell-specific or injury-specific promoter. Upon excision of the stop cassette, ribosomes within the cell-type of interest can be purified using anti-GFP antibodies, the RNA isolated, and RNA-sequencing undertaken.
Novel transcription factors in PT injury and repair
Molecular changes occurring in AKI frequently resemble reactivation of developmental pathways. One highly upregulated developmental TF is SRY homeobox 9 (SOX9), which is expressed in the tips of ureteric branches during development, but is not widely expressed in uninjured adult kidney [5]. Nephron-specific TRAP-microarray identified Sox9 as a highly upregulated TF in PT cells after ischemia-reperfusion injury (IRI) [5] and Sox9 was similarly upregulated after unilateral ureteric obstruction (UUO) [5], unilateral partial nephrectomy [9], and in folic acid nephropathy (FAN) [10]. Sox9 expression preceded PT cell dedifferentiation, and a large proportion of SOX9+ cells were proliferating 48 hours after IRI, suggesting a relationship between de novo SOX9 expression and re-entry of PT cells into the cell cycle [5]. Fate-tracing studies confirmed that SOX9+ cells proliferated in both IRI and FAN, followed by re-differentiation of the newly generated PT cells [5,10]. Interestingly, 28 days after IRI, some PT cells with persistent Sox9 expression had failed to re-differentiate, whereas fully differentiated PT cells no longer expressed Sox9 [5]. Mice lacking Sox9 in the nephron had reduced tubular cell proliferation and more severe fibrosis after FAN, indicating the important role of Sox9 in tubular regeneration after AKI [5].
Another important TF in proliferation and regeneration after AKI, Forkhead box M1 (Foxm1), was identified using TRAP-seq in an inducible Kim1 -reporter mouse, a strategy to detect actively transcribing genes specifically in injured PT cells [4]. In contrast to Foxj1, which was upregulated 14 days after unilateral IRI, Foxm1 was strongly upregulated after 2 days, and returned to baseline after 14 days, and similar upregulation was found in a human AKI sample. Foxm1 upregulation was found to be downstream of epidermal growth factor receptor (EGFR) signaling, and in vitro, knockdown of Foxm1 in primary human PT cells led to reduced proliferation [4].
Another member of the FOX family, FoxO3, is also strongly upregulated after cisplatin, UUO and IRI. However, in contrast to Foxm1 expression which appears to be transcriptionally regulated, Foxo3 gene expression may be downregulated by miR-122 (which is downregulated in AKI) [11], and FoxO3 protein post-translationally activated by acetylation/deacetylation [11,12], and by inhibition of proline hydroxylation, which would usually promote FoxO3 protein degradation [13,14]. Activated FoxO3 protein interacts with P53, which may contribute to tubular cell death [11,12], and increases autophagic capacity and autophagosome formation [13,14], whilst inducible knockdown of Foxo3 initiated after IRI led to exacerbated injury, and increased fibrosis [13]. The seemingly contradictory outcomes of FoxO3 activation in upregulating both apoptosis and autophagy may be a product of the balance in post-translational modifications (e.g. acetylation versus hydroxylation), duration of FoxO3 induction (e.g. early versus sustained), and/or levels of regulators and binding partners of FoxO3, in any given injury type or cell; however these details remain to be elucidated.
Amongst other developmental TFs whose biology and roles remain to be explored are members of the Krüppel-like factor (KLF) family. For example, Klf4, Klf5, and Klf6 were all strongly upregulated, and Klf15 downregulated, in IRI and UUO kidneys [3,6]. The role of this family of TFs has not yet been explored in the PT in AKI.
Conclusions
Large-scale sequencing studies are providing a wealth of data on transcriptional control of successful and unsuccessful repair in AKI. However, altered expression alone does not reveal whether a given TF may have an adaptive or maladaptive role. Understanding the role of each TF and their interactions with other TFs, whether cooperative or opposing, requires extensive studies to determine the biological outcomes and mechanisms of action. In turn, new pathways and potential therapeutic targets in PT cell repair may be elucidated. Targeting of TFs for therapeutic benefit has the attraction of being able to modify multiple downstream target pathways using a single drug; however, this same feature may also be detrimental and lead to undesirable effects through the TF regulating other pathways whose modulation may be unwanted. Thus, design of drugs targeting TFs may require a more subtle approach than complete blocking of function, such as disrupting interactions with specific binding partners.
Acknowledgments
Funding sources
This work was supported by funds from the UAB-UCSD O’Brien Center (SEP), NIH/NIDDK (1R01DK112984) (SKM), VA Merit (1I01BX003698) (SKM), and Dialysis Clinic Inc. (SKM).
Footnotes
Disclosure statement
The authors have no conflicts of interest to declare.
References
- 1.Gewin LS: Renal fibrosis: Primacy of the proximal tubule. Matrix Biol 2018;68–69:248–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu BC, Tang TT, Lv LL, Lan HY: Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int 2018;93:568–579. [DOI] [PubMed] [Google Scholar]
- 3.Cippa PE, Sun B, Liu J, Chen L, Naesens M, McMahon AP: Transcriptional trajectories of human kidney injury progression. JCI Insight 2018;3:e123151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang-Panesso M, Kadyrov FF, Lalli M, Wu H, Ikeda S, Kefaloyianni E, Abdelmageed MM, Herrlich A, Kobayashi A, Humphreys BD: FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J Clin Invest 2019;129:5501–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar S, Liu J, Pang P, Krautzberger AM, Reginensi A, Akiyama H, Schedl A, Humphreys BD, McMahon AP: Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep 2015;12:1325–1338. [DOI] [PubMed] [Google Scholar]
- 6.Wu H, Lai CF, Chang-Panesso M, Humphreys BD: Proximal Tubule Translational Profiling during Kidney Fibrosis Reveals Proinflammatory and Long Noncoding RNA Expression Patterns with Sexual Dimorphism. J Am Soc Nephrol 2020;31:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, Grgic I, Kumar S, Humphreys BD, Hide WA, McMahon AP: Cell-specific translational profiling in acute kidney injury. J Clin Invest 2014;124:1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H, Kirita Y, Donnelly EL, Humphreys BD: Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J Am Soc Nephrol 2019;30:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Q, Wang Y, Zhang T, Zuo W: Notch-mediated Sox9(+) cell activation contributes to kidney repair after partial nephrectomy. Life Sci 2018;193:104–109. [DOI] [PubMed] [Google Scholar]
- 10.Kang HM, Huang S, Reidy K, Han SH, Chinga F, Susztak K: Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep 2016;14:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CG, Kim JG, Kim HJ, Kwon HK, Cho IJ, Choi DW, Lee WH, Kim WD, Hwang SJ, Choi S, Kim SG: Discovery of an integrative network of microRNAs and transcriptomics changes for acute kidney injury. Kidney Int 2014;86:943–953. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Mu Y, Zhou X, Ji H, Gao X, Cai WW, Guan Q, Xu T: SIRT2-mediated FOXO3a deacetylation drives its nuclear translocation triggering FasL-induced cell apoptosis during renal ischemia reperfusion. Apoptosis 2017;22:519–530. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Kang H, Zhang Q, D’Agati VD, Al-Awqati Q, Lin F: FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest 2019;129:2374–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Zviti R, Ha C, Wang ZV, Hill JA, Lin F: Forkhead box O3 (FoxO3) regulates kidney tubular autophagy following urinary tract obstruction. J Biol Chem 2017;292:13774–13783. [DOI] [PMC free article] [PubMed] [Google Scholar]