

Abstract
Developmental methylmercury (MeHg) exposure can have lasting consequences on neural development and motor function across the lifespan. Recent evidence for MeHg targeting of myogenic pathways has drawn attention to the possibility that developing skeletal muscle plays a role in the motor deficits stemming from early life MeHg exposure. In this study we examined a potential role for muscle in influencing MeHg developmental toxicity in offspring of female mice exposed to MeHg via drinking water. Dams had access to 0, 0.5 or 5.0ppm MeHg chloride in drinking water from two weeks prior to mating through weaning. Blood, brain and muscle tissue was harvested from dams at weaning and pups at postnatal days (PND) 6, 21 and 60 for analysis of total Hg. Muscle tissue sections were examined with histological stains. Behavioral testing of offspring was conducted at PND 60 and included locomotor activity, inverted screen, grip strength and rotarod tests to assess motor function. Total Hg (tHg) levels in dam muscles at weaning were 1.7 to 3-fold higher than Hg levels in blood or brain. In PND6 male and female pups, muscle and brain tHg levels were 2 to 4-fold higher than blood tHg. Brain tHg levels decreased more rapidly than muscle tHg levels between PND 6 and 21. Premised on modeling of growth dilution, brain tissue demonstrated an elimination of tHg while muscle tissue exhibited a net uptake of tHg between PND 6 and 21. Despite overall elevated Hg levels in developing muscle, no gross morphological or cytological phenotypes were observed in muscle at PND 60. At the higher MeHg dose, grip strength was reduced in both females and males at PND 60, whereas only male specific deficits were observed in locomotor activity and inverted screen tests with marginally significant deficits on rotarod. These findings highlight a potential role for developing skeletal muscle in mediating the neuromuscular insult of early life MeHg exposure.
Keywords: methylmercury, prenatal exposure, growth dilution, myogenesis, toxicokinetics, skeletal muscle, motor behavior, neurotoxicity, myotoxicity
Introduction
Methylmercury (MeHg) continues to be a priority environmental toxicant of concern due to its well-established neurotoxic properties and bioaccumulation in fish, resulting in widespread human exposure. Since the developing fetus is exceptionally vulnerable, numerous studies have focused on adverse outcomes associated with MeHg exposure during the perinatal period. Yet, the risks of MeHg exposure remain uncertain, largely due to the variability in outcomes in prenatally exposed offspring that has been observed across several epidemiological studies [1, 2]. Such variability is also seen in controlled laboratory studies in rodents, posing experimental and statistical challenges for discerning low dose effects (reviewed in [3]). Nonetheless, it is clear that at sufficiently high doses, MeHg can elicit motor and cognitive deficits in rodent offspring previously exposed in utero (reviewed in [4]). Motor deficits have largely been attributed to MeHg targeting of the developing brain [5–7], however, recent studies have begun to demonstrate that MeHg can target the developing skeletal muscle system which could thereby contribute to altered motor function [8, 9].
Numerous studies investigating effects of developmental MeHg exposure have relied on a paradigm of controlled feeding experiments in rodent models [3, 4]. Typically, maternal dosing via food or drinking water spans a period prior to and during gestation through lactation until weaning. This is followed by assessment of behavioral and/or histological/cellular/molecular outcomes in offspring at various postnatal timepoints extending to adulthood. Such studies have revealed MeHg induced developmental traits appearing as deficits in motor behaviors (e.g. open field locomotor tests and Rotarod performance) as well as in cognitive function (e.g. reward paradigms and operant behaviors) [10, 11]. For example, Montgomery et al [12], using C57BL/6 mice and MeHg exposure during gestation showed reduced motor function in offspring as assessed by decreased activity in an open field and a shortening of time staying on the rotarod. In contrast, using a similar gestational exposure paradigm in the same strain of mice, Goulet et al [13] reported no significant reductions in locomotor functions in the offspring, whereas female offspring alone proved to be impaired in acquiring working memory. Such reports highlight the variability in MeHg toxicity outcomes that are a source of uncertainty in translating these observations to inform toxicity risk. An important consideration for interpreting such studies is the accuracy of the dose-response relationship. In pregnant rodents, dosage is intrinsically complicated by toxicokinetic processes, e.g. absorption, distribution and elimination, that ultimately dictate how much MeHg reaches the target organ, e.g. the fetal brain, and, importantly, the levels of MeHg that persist in the critical early postnatal development period. Numerous studies in mice and rats have accounted for the Hg levels in the neonatal brain that accompany the behavioral or morphological effects (reviewed in [4]) that, in general, support the observation that the dose in the fetal the brain correlates with the maternal dose during gestation. Yet, postnatally, a decrease in brain Hg, on the order of 10-fold between PND 1 and 21, is commonly observed, even in the instance where MeHg exposure to dams is continued through weaning [4, 14, 15]. The distribution of Hg in other tissues that support motor function, e.g. skeletal muscle, during the developmental time course are less well understood.
In this study we sought to examine the role of skeletal muscle as a MeHg target in mediating motor deficits that emerge in developing mice exposed during gestation and lactation. We find that both adult maternal skeletal muscle and developing neonatal muscle are prone to accumulating tHg up to 2 – 3 times that of blood or brain tissue. Furthermore, elimination of tHg in the brain during the early postnatal period (PND 6–21) is accompanied by an accumulation of tHg in the growing skeletal muscle. While no apparent gross cytological pathology is seen in the skeletal muscle, these kinetic features of early life tHg exposure are seen to foreshadow an apparent male-specific diminished performance in several motor behaviors at the adult stage. These findings highlight a potential role for developing skeletal muscle in moderating the neuromuscular insult of early life MeHg exposure.
Materials and Methods
Mice, Breeding and MeHg Exposure:
Housing and Exposure:
Male and female C57Bl6/J mice were purchased from Jackson Laboratories (Bar Harbor, ME) and allowed to acclimate in the housing room for one week prior to MeHg administration. Fourteen days prior to mating, females only began exposure to methylmercury (MeHg chloride Sigma #442534, prepared in tap water at 5ppm) via drinking water at 0.5 and 5ppm concentrations, whereas control mice and male sires received tap water alone in glass bottles. Exposures continued through mating and gestation, ending at weaning at postnatal day (PND) 21. To assure consistent dosing, we evaluated MeHg concentration in the water over time, which showed a decrease of ~15% over a 3-day period (data not shown), likely due to adsorption to the vessel. Therefore, fresh preparations of water with (and without) MeHg in clean bottles were introduced every three days. To account for background levels of Hg exposure, total Hg in the mouse chow was determined and found to be 0.012 ppm (data not shown), consistent with prior reports [16]. Males and females were group housed separately during acclimation and MeHg administration.
Breeding:
Estrous cycles of females were synchronized via pheromone induced ovulation by placing females in cages with dirty bedding from males [17]. Monogamous pairs of mice were then bred for 3 days, after which males were removed and dams remained singly housed with litters until weaning. All mice in this study were housed under a 12 h reversed light/dark cycle and temperature maintained at ~ 22 °C. A total of 50 dams were used. To preclude litter-specific effects, only single pups/sex/litter were used in all of the studies. All mice used in this study were treated humanely and with regard for alleviation of suffering and all procedures were conducted with approval of the Institutional Animal Care and Use Committees at the University of Rochester. The scheme for MeHg exposure, tissue sampling and behavior analysis is illustrated in Figure 1.
Figure 1. MeHg dosing, tissue harvesting and behavior testing scheme.
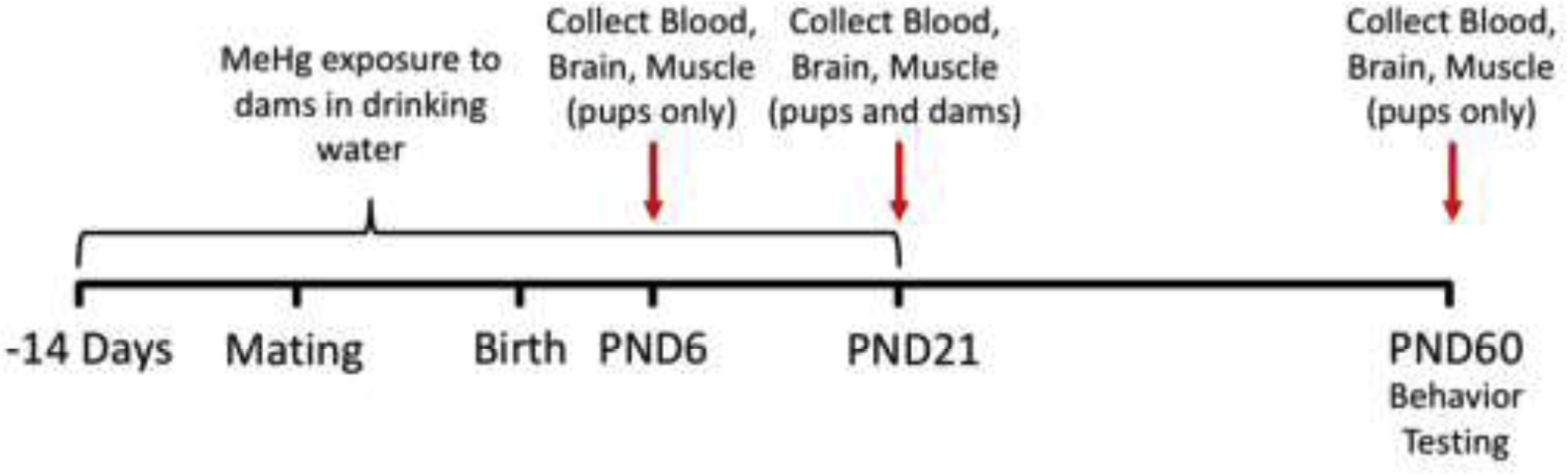
See text for description.
Behavioral Testing:
Behavioral tests were selected to emphasize analyses of motor function, including locomotor activity, hind- and forelimb grip strength, Rotarod (balance and endurance) and inverted screen (strength and endurance). Behavioral testing began at postnatal day 60.
Locomotor activity was measured across 60-min test sessions in automated chambers equipped with 48-channel infrared photobeams (Med Associates). Photobeam breaks were recorded in 5-min bins to assess horizontal, vertical, and ambulatory movements across time (resulting in 12 bins for a 1-h locomotor activity session). Ambulatory counts were defined as the number of beam breaks in ambulatory movement; ambulatory episodes were defined by a minimum number of ambulatory counts (at least three successive photobeam breaks). The total ambulatory time refers to the time in ambulatory movement status, whereas distance represents the Euclidean distance of all ambulatory episodes, which together can also be used to determine speed (average velocity). Vertical activity was defined as movement that broke photobeams placed in the z-axis with breaks in the x and y-axes as well. Resting time was defined as time spent with no new photobeam breaks. Stereotypic counts and stereotypic time measure activity within a 2 photobeam by 2 photobeam square in the arena. Jump counts are measured as the number of time periods that no x or y-axis beam breaks were detected, with a break in the z-axis, whereas jump time reflects the total duration in seconds of the jump counts.
For the inverted screen test, the mouse was placed in the middle of a mesh screen square. The mesh screen square was then slowly rotated 180 degrees placing the mouse upside down. The time until the subject has righted itself was then recorded in a 3-minute trial; failure to achieve righting was scored as 3 minutes. The subject is considered to have righted itself when it is securely on the top side of the screen (defined as all four feet placed on the top of the screen). Three trials were collected for every subject; where the mouse did not right itself or dropped off the screen, a maximum trial time of 3 minutes was recorded.
For rotarod performance, an acceleration protocol was utilized, with a starting speed of 4 mph, which was then increased by 4 mph at 15 sec intervals to a final value of 40 mph. The time for the mouse to fall from the rotarod and the speed at which this occurred were recorded. A total of 3 trials were carried out for each mouse in each session, with a 120 second inter-trial interval between each trial. Sessions were run for 5 consecutive days, with data from the first two sessions (training phase) collected to determine whether there were any differences between groups in ability to learn the task.
Grip strength was analyzed for both forelimbs and hind limbs using a digital force meter equipped with precision force gauges to retain the peak force applied on a digital display and with a grid or wire system that allows measurement of mouse grip strength by either or both paws. Body weight was first collected for each mouse. Each mouse was then tested 3 times (3 trials) on both the hindlimbs and forelimbs with a minimum 60 second inter-trial interval. For forelimb grip strength, the mouse was lifted by the tail to a height where the front paws are at the same height as the bar and then moved horizontally towards the bar until it became within reach, producing a symmetric, tight grip with both paws, at which point the mouse was gently pulled at a constant speed until its grasp was broken and the resistance measured. For hind-limb grip strength, rear limbs were similarly directed toward the grasp bar. Measurements were discarded if the animal used only one paw or also used its hind paws, turned backwards during the pull, or left the bar without resistance.
Tissue harvesting:
Mice at PND 6, 21 and 60 were weighed and then sacrificed by cervical dislocation without the use of sedatives and blood, skeletal muscle and brains removed. Skeletal muscle (quadriceps and gastrocnemius) was further dissected to capture a fresh sample to freeze for Hg analyses and a sample processed for histology (see below). Brains were further dissected to capture cortex, cerebellum, hippocampus, olfactory bulb and rest of brain (R.O.B.), the latter used for the bulk of the Hg analyses, and the former preserved for future histological analyses. Blood and brain tissues from dams was collected upon sacrifice after weaning of pups (PND 21). Randomization across groups was used during sacrifice to prohibit circadian variation occurring in a single group.
Hg measurements:
For offspring, tissue total Hg (tHg) concentrations were determined in blood, brain (rest of brain) and muscle (quadricep) on a wet weight basis. Samples of cortex, cerebellum, hippocampus and olfactory bulb of three dams treated at the 5 ppm MeHg level were also analyzed for comparison of Hg levels across brain regions. Speciation of MeHg and Hg2+ was not done as discerning the Hg species related to the toxicity was not an aim of the study. Tissue wet weights were determined using a Mettler MX5 microbalance. Total Hg was analyzed using ICP-mass spectrometry on a Perkin-Elmer Nexion 2000 instrument. Tissue samples were digested in high purity nitric acid with heating at 90°C for two hours to achieve complete hydrolysis. Sample diluted 1:50 in trace element free water were introduced to the ICP-MS and Hg was determined relative to a calibration curve established with element standard solutions (Perkin Elmer). Limit of quantitation and limit of detection for 202Hg for the ICP-MS was 2.0 part per trillion (ppt) and limit of quantification of 20 ppt, respectively.
Histology:
Muscle histology was assessed in 39 pairs of quadriceps and gastrocnemius muscles spanning control and MeHg exposures across PND6, 21 and 60. Following dissection muscles were frozen in isopentane cooled in liquid nitrogen and 10 μm cryosections were obtained for the following histochemical stains: Hematoxylin and Eosin (H&E), Trichrome (TRI) and NADH using standard protocols [18]. Histologic assessment and image capture was performed by light microscopy at 10X and 20X magnification with the evaluator blinded to the mouse’s MeHg exposure.
Muscle fiber size was determined by a modified method of the lesser fiber diameter [18]. Briefly, 20X images of H&E stained sections were analyzed in Image J (NIH, Image J). Section areas analyzed were selected at random and the evaluator was blinded to the mouse’s MeHg exposure. For each fiber cross-section a line was drawn parallel to the longest axis. A perpendicular line was then drawn at the widest dimension of the fiber diameter. This latter line length, the lesser fiber diameter, was recorded in pixels and converted to μm using a calibrated image of a ruler. Quadriceps and gastrocnemius muscle from 4–5 animals of each sex were measure and mean fiber diameter determined on 78–105 fibers in each sample. The mean and standard error for diameter was calculated for each sex and each treatment. The data was also expressed as a histogram of percent of fiber bundles that fall within 5μm increments of bundle diameter between 15–70 μm.
Statistical Analyses:
Total Hg measurements in tissues (brain, muscle, blood) and offspring weights were analyzed by one way analysis of variance (ANOVA) and carried out separately for each sex by MeHg dose and developmental time using Graphpad Prism 6. Multiple comparisons of mean values were performed by Tukey’s method and multiplicity adjusted p values of HSD post-hoc tests reported where p≤ 0.05 was considered significant. Behavioral outcomes were analyzed using either repeated measures ANOVA or one factor ANOVA (MeHg treatment: 0, 0.5 and 5.0 ppm) and carried out separately for each sex. Post-hoc ANOVAs or t-tests were then carried out dependent upon findings of main effects or interactions as appropriate, using JMP14. A p value of ≤ 0.05 was considered statistically significant.
Results
Dosimetry: Dose dependent concentrations of total Hg in dam tissues.
Twenty seven of the 50 dams used for the study were selected to perform total mercury (tHg) analyses on tissues collected at weaning, which occurred after approximately 56 days of continuous exposure to MeHg via the drinking water (Figure 1). Among these 27 dams, analyses of tHg in blood was determined for representatives of each MeHg treatment group: 0.0ppm (n=6), 0.5ppm (n=11), 5.0 ppm (n=10) (Suppl. Table 1). Additionally, three dams from each treatment group were randomly selected to perform additional analyses of tHg in brain and muscle tissue (Table 1). A dose dependent accumulation of tHg was seen in all three tissues (Table 1 and Suppl. Table 1). Notably, a wide range of blood tHg values, varying by more than 2.5-fold, was seen within each treatment group (Suppl. Table 1). Detectable levels of Hg were observed in the control group receiving no MeHg in the water, which also demonstrated a wide range of values. This latter Hg likely resulted from MeHg contained in the fish meal constituent of the mouse chow. Total Hg in the mouse chow was determined to be 0.012ppm (data not shown). For the 0.5 and 5.0ppm MeHg treatments via the water, tHg levels in the dam’s blood and brain were seen to parallel that in the water being provided. In contrast, tHg levels in muscle showed to be 1.7 to 3-fold greater than the blood and the brain tissues at both treatment levels (Table 1). Since rest of brain (R.O.B.) tissue was used in the analysis, we compared tHg levels across four additional brain regions (cortex, cerebellum, hippocampus and olfactory bulb) in three dams receiving the 5 ppm MeHg treatment. No significant difference was found between ROB and the other brain regions (Suppl. Table 2), whereas cerebellum showed significantly less Hg accumulation relative to cortex, hippocampus and olfactory bulb (Suppl. Table 2). With the trace level MeHg exposure in the control (0 ppm) group no significant difference in tHg levels between blood, brain and muscle tissue was seen (Table 1).
Table 1 :
Total mercury concentration (ppm) in dam tissue at weaning
| MeHg | Blood | Brain | Muscle | p value |
|---|---|---|---|---|
| 0 ppm | 0.011 ± 0.007 | 0.006 ± 0.001 | 0.006 ± 0.002 | n.s. |
| 0.5 ppm | 0.474 ± 0.067 | 0.674 ± 0.035 | 1.436 ± 0.042 | a, c, e |
| 5 ppm | 4.777 ± 1.049 | 4.996 ± 0.499 | 8.412 ± 0.923 | b, d, f |
| p value | g | h | h |
Mean ± SEM; n = 3
a–b, one-way ANOVA for tissues;
p<0.0001;
p<0.05
c – d, Post-hoc, Blood × Muscle; one-way ANOVA, Tukey’s HSD;
p<0.0001;
p=0.054
e – f, Post-hoc, Brain × Muscle; one-way ANOVA, Tukey’s HSD;
p<0.0001;
p=0.068
g–h, one-way ANOVA for dose;
p<0.005;
p<0.0001
Dosimetry: Dose and time dependent concentrations of total Hg in pup tissues.
Total Hg was determined in blood, brain and muscle of pups at postnatal day (PND) 6, 21 and 60 subsequent to MeHg exposure to the dams as outlined in Figure 1. PND 6 pups demonstrated a dose dependent increase in tHg in all three tissues of both male and females (Table 2). Interestingly, at both 0.5 and 5.0 ppm MeHg dosage levels, tHg in both brain and muscle showed to be 2 to 4-fold higher than that of blood in PND 6 pups (Table 2), indicating preferential distribution of Hg to these peripheral organs relative to blood at this early stage. Interestingly, in PND 6 pups from dams exposed to the trace level MeHg via the mouse chow (0 ppm), only in muscle was there a greater accumulation of tHg relative to blood (Table 2). This pattern of higher levels of Hg in muscle relative to brain and/or blood appeared to persist over developmental time (Table 2). At PND 6 and 21, there was no significant differences in Hg concentrations between females and males in each of the respective tissues at the 5.0 ppm dose (Unpaired t-test, p>0.2). At PND 60, at the 5.0 ppm treatment level, the remaining Hg in male muscle was significantly lower than female muscle (p=0.012, Unpaired t-test), while Hg levels in brain and blood were not statistically different between males and females.
Table 2.
Total mercury concentration (ppm) in pup tissue at postnatal day (PND) 6, 21, or 60
| Female | Male | |||||||
|---|---|---|---|---|---|---|---|---|
| MeHg | Blood | Brain | Muscle | P | Blood | Brain | Muscle | P |
| PND 6 | ||||||||
| 0 ppm | 0.005 ± 0.003 | 0.010 ± 0.003 | 0.103 ± 0.018 | a, f, g | 0.001 ± 0.001 | 0.004 ± 0.000 | 0.083 ± 0.026 | c, f, g |
| 0.5 ppm | 0.114 ± 0.037 | 0.218 ± 0.015 | 0.447 ± 0.044 | a, f, g | 0.078 ± 0.016 | 0.200 ± 0.009 | 0.446 ± 0.040 | a, e, f, g |
| 5 ppm | 1.267 ± 0.103 | 3.175 ± 0.348 | 3.466 ± 0.380 | d, g | 1.080 ± 0.175 | 2.720 ± 0.350 | 2.966 ± 0.139 | b, e, g |
| PND 21 | ||||||||
| 0 ppm | 0.008 ± 0.003 | 0.000 ± 0.000 | 0.014 ± 0.006 | ns | 0.006 ± 0.001 | 0.001 ± 0.000 | 0.005 ± 0.002 | d, e, g |
| 0.5 ppm | 0.150 ± 0.006 | 0.069 ± 0.005 | 0.193 ± 0.010 | a, e, f, g | 0.093 ± 0.009 | 0.077 ± 0.005 | 0.171 ± 0.014 | a, f, g |
| 5 ppm | 0.897 ± 0.150 | 0.695 ± 0.047 | 1.519 ± 0.158 | c, f, g | 0.791 ± 0.120 | 0.614 ± 0.055 | 1.425 ± 0.136 | b, f, g |
| PND 60 | ||||||||
| 0 ppm | 0.003 ± 0.000 | 0.004 ± 0.000 | 0.009 ± 0.001 | a, f, g | 0.001 ± 0.000 | 0.003 ± 0.000 | 0.005 ± 0.001 | a, f, g |
| 0.5 ppm | 0.005 ± 0.001 | 0.010 ± 0.001 | 0.015 ± 0.002 | b, f, g | 0.002 ± 0.000 | 0.006 ± 0.000 | 0.007 ± 0.001 | a, f, g |
| 5 ppm | 0.017 ± 0.004 | 0.083 ± 0.022 | 0.059 ± 0.013 | d, e | 0.013 ± 0.002 | 0.040 ± 0.002 | 0.020 ± 0.002 | a, e, g |
| P (5ppm) | h, j | h, k | i, j | h, j | h, j | h, j | ||
Mean ± SEM; n ≥ 4
a–d, one-way ANOVA for tissues,
p< or = 0.0001;
p<0.001;
p<0.01;
p<0.05.
Post-hoc tests:
Blood × Brain; one-way ANOVA, Tukey’s HSD; p < 0.05,
Blood × Muscle; one-way ANOVA, Tukey’s HSD; p < 0.05
Brain × Muscle; one-way ANOVA, Tukey’s HSD; p < 0.05
h, I, one-way ANOVA for development time (5.0ppm dose),
p< or = 0.0001;
p<0.001
Post hoc:
PND6 × PND21; one-way ANOVA, Tukey’s HSD; p < 0.05,
PND21 × PND60; one-way ANOVA, Tukey’s HSD; p < 0.05
Among the MeHg exposed pups, from PND 6 to 60 tHg levels showed an overall decrease in all three tissues in the offspring (Table 2, Figure 2, Suppl Figure 1) with a similar overall pattern seen in male and females. However, the decrease in tHg levels between PND 6 and 21 appeared variable between tissues. As seen with pups exposed to the 5.0 ppm maternal dose (Table 2, Figure 2), tHg in the brain and muscle showed a sharp decline from PND6 to PND21, with more than a four-fold drop in brain Hg levels and a two-fold drop in muscle Hg levels. The corresponding decrease in blood tHg from PND6 to PND21 was more moderate with only ~30% drop (Figure 2). To examine this discrepancy more closely we considered that the decrease in tissue Hg is expected to occur not only by elimination but by growth dilution. In addition, organs are known to grow at different rates over early postnatal development [19]. Growth dilution factors (fold-change in volume) were therefore determined based on values for the increase in organ volume over the PND 6–21 and PND 21–60 intervals extracted from the data of Siddiqui et al [19] and shown in Figure 2 (panel G). We then plotted predicted levels of tHg at PND 21 and 60 resulting solely from growth dilution from the PND6 time point together with the measured values of tHg in each tissue at these time points (Figure 2, Suppl. Fig. 1). In blood, the measured Hg levels tracked closely with that predicted from growth dilution in the blood compartment (Fig. 2A,B). In brain, the tHg decrease observed at PND21 was far below the level predicted from growth dilution alone (Fig. 2C, D). In contrast, in muscle, the tHg levels observed at PND21 were 2 to 3-fold higher than is predicted from growth dilution (Fig. 2E,F). At PND 60 the observed tHg levels approached baseline in all three tissues. An overall similar profile was seen at the 0.5 ppm MeHg dose treatments (Suppl. Fig. 1). In the control exposed pups baseline tHg levels in blood were 1/20th to 1/70th of the 0.5 ppm treated pups. This baseline tHg level was seen to increase from PND 6 to 21. However, at PND 60 blood tHg levels were not different than at PND 6, indicating no gross accumulation of Hg from the chow in the pups once they reached feeding age.
Figure 2. Hg elimination and growth dilution.
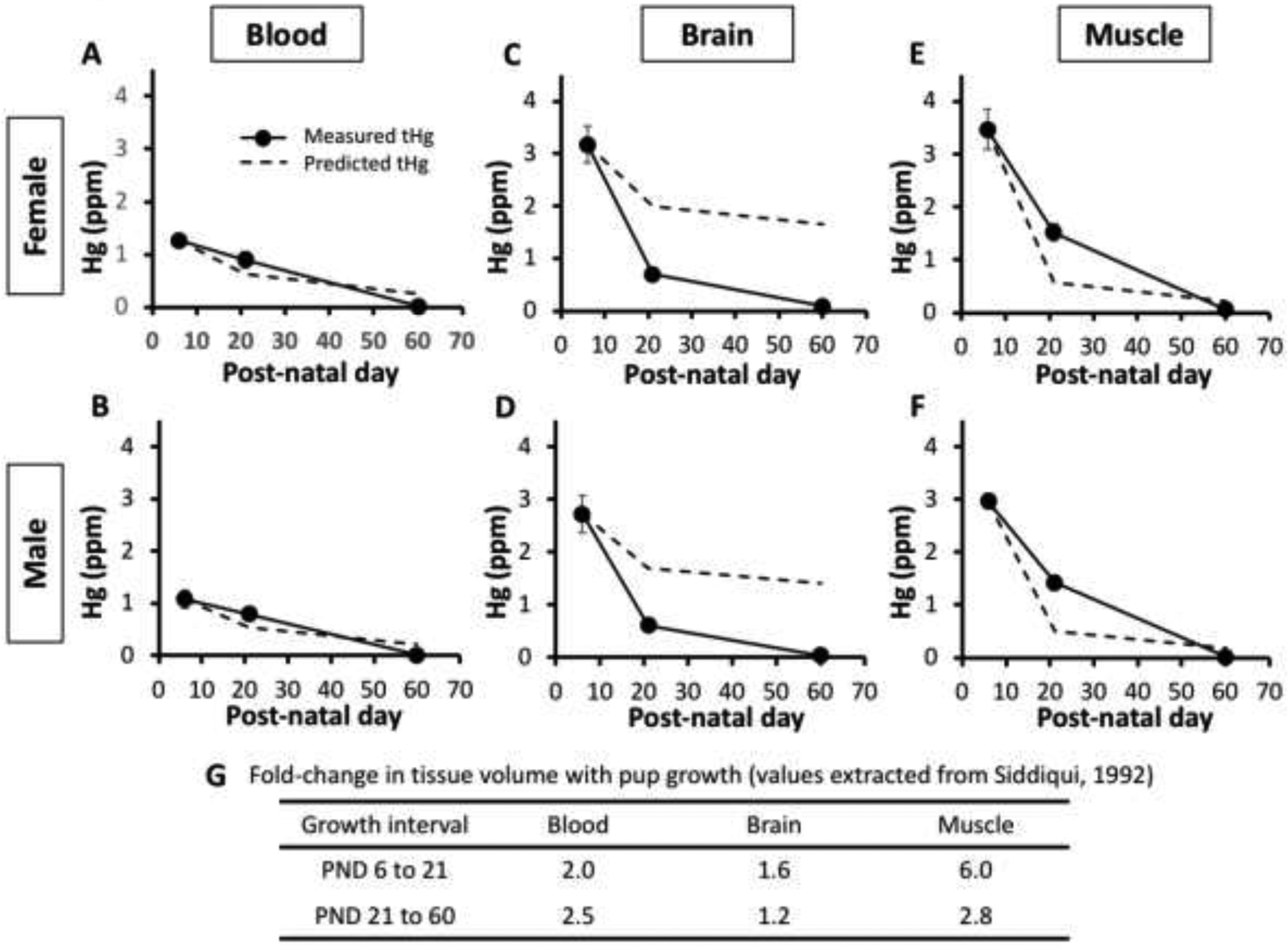
Time course of tHg levels (in ppm) in blood (A,B) , brain (C,D) and muscle (E,F) from PND6, 21 and 60 mice. Measured tHg values (solid line) are plotted together with predicted values of tissue tHg at PND 21 and 60 based on growth dilution within the respective organs (dashed lines). Measured tHg values are from the 5.0ppm maternal dosage experiments. Table (G) shows values for volume changes between PND growth intervals for the respective organs used in growth dilution calculations. (Values extracted from the data of [19]).
Effects of developmental MeHg exposure on body weight and muscle morphology.
Prenatal MeHg exposure at 5.0 ppm in the dam’s drinking water significantly decreased PND 0 pup birth weight by 7% and 5% in both female and male offspring, respectively (Table 3). At PND 60, male offspring trended toward a 5% lower weight which did not reach significance. Muscle histology was assessed with Hematoxylin and Eosin (H&E), Trichrome (TRI) and NADH stains focusing on the p21 and p60 developmental time points. No remarkable phenotypes arising from MeHg treatment were observed, whereby with H&E and TRI staining all sections showed normal muscle histology with polygonal fibers with normal variability in fiber size, absence of central nucleation or angulated atrophic fibers (Suppl Figure 2A and data not shown). No muscle fiber necrosis or regeneration and no increase in interstitial connective tissue was seen (data not shown). Similarly, NADH staining showed a normal intermyofibrillar staining pattern (data not shown). Images of H+E stains were then used to quantify muscle fiber diameter in PND 60 quadricep and gastrocnemius muscles using the lesser fiber diameter method [18]. Despite a trend toward reduced mean bundle diameter in males with MeHg, no significant difference in fiber diameter distribution was seen across all treatments within both sexes (Suppl Figure 2B).
Table 3.
Offspring weights (grams) across treatment and developmental time
| MeHg | Female | Male |
|---|---|---|
| PND 0 | ||
| 0 ppm | 1.32 ± 0.03 | 1.35 ± 0.02 |
| 0.5 ppm | 1.29 ± 0.02 | 1.30 ± 0.01 |
| 5 ppm | 1.22 ± 0.02a | 1.28 ± 0.02b |
| PND 60 | ||
| 0 ppm | 21.23 ± 0.32 | 27.95 ± 0.58 |
| 0.5 ppm | 21.71 ± 0.37 | 27.39 ± 0.62 |
| 5 ppm | 21.55 ± 0.61c | 26.39 ± 0.54c |
Mean ± SEM; PND 0 n ≥ 18; PND 60 n=10
MeHg dose effect one-way ANOVA,
F(2,62)=4.008, p<0.0231;
F(2, 29)=3.552, p<0.0351;
non-significant.
Effects of developmental MeHg exposure on motor behavior
Behavioral assessments were done to investigate potential motor disturbances across an array of motor functions. First, we examined changes in locomotor activity using an automated open field paradigm. Cumulative changes in vertical activity, ambulatory distance and total resting time are shown for individual females (top row) and males (bottom row) in Figure 3. MeHg-induced changes in locomotor activity differed by sex and MeHg exposure concentration. In females, no statistically significant differences were seen in any of these outcome measures. In contrast, males showed a significant reduction in vertical counts that was restricted to the 5.0 ppm MeHg exposure (F(2,27)=4.44, p=0.0215), with marginal reductions in ambulatory distance travelled (F(2,27)=2.94, p=0.0684), and a marginal increase in resting time (F92,27)=3.02, p=0.066).
Figure 3. Locomotor activity.
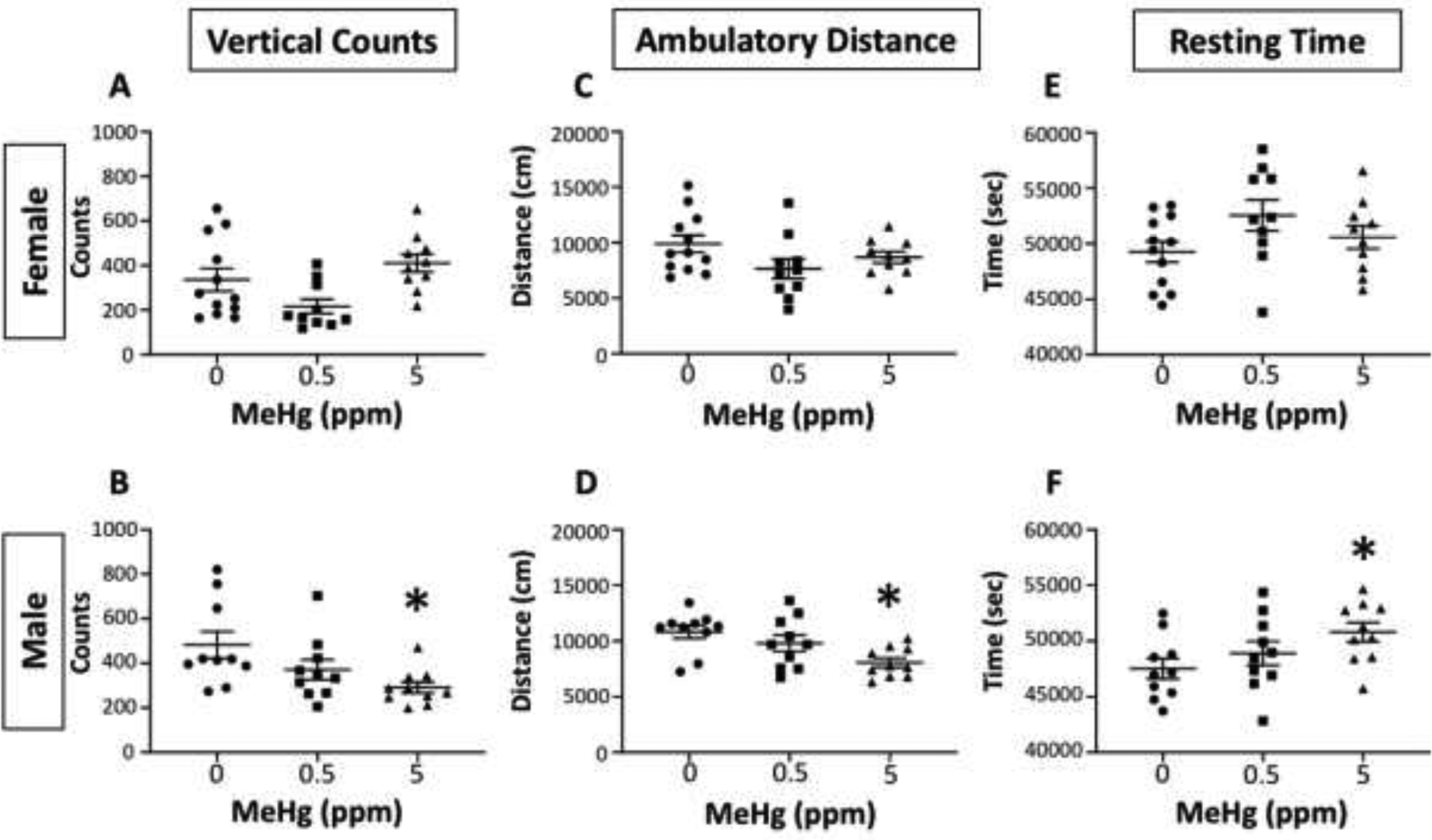
Individual values for vertical counts (counts; left column), ambulatory distance (cm; middle column) and resting time (secs; right column) of female (top row) and male (bottom row) offspring following maternal exposure to 0, 0.5 or 5.0 ppm MeHg as indicated. *=significantly different from 0 ppm control in post-hoc testing following significant main effect in ANOVA.
The inverted screen paradigm required mice to lift themselves up on a wire mesh screen. The time required for righting is shown in Figure 4 for females (left) and males. No systematic effects of MeHg were seen across the 3 sessions for females, whereas a main effect of group was found for males, which post-hoc testing confirmed was due to longer times for the 5.0 MeHg group (F(1,28)=5.04, p=0.0328) which were seen in session 2 (p=0.0009). Grip strength was measured using a strain gauge and force/weight (i.e., corrected for body weight) for forelimb and hindlimb grip strength are shown for females (top row) and males (bottom row) in Figure 5. With respect to forelimb grip strength, significant reductions were seen at the 5.0 ppm MeHg exposure concentration in both males (F(2,27)=3.496, p=0.0445) and females (F(2,29)=3.9, p=0.0315). In contrast, no changes in hindlimb grip strength were found in MeHg-exposed females, while slight reductions were found in males at the 0.5 ppm exposure (F(2,27)=3.99, p=0.03).
Figure 4. Inverted screen righting behavior.
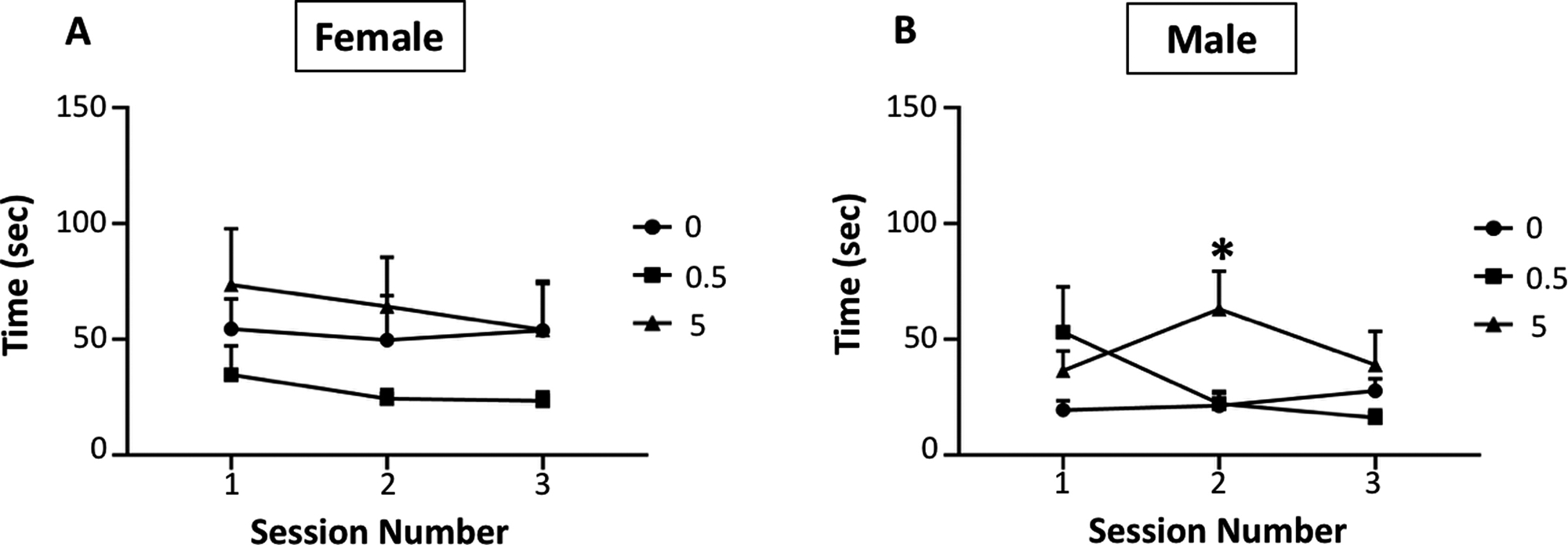
Group mean ± S.E. values for time (sec) spent righting for female (left) and male (right) offspring exposed to 0, 0.5 or 5.0 ppm MeHg, as indicated by symbols, during pregnancy and lactation. Main effect of group was found for males (F(1,28)=5.04, p=0.0328).
Figure 5. Grip strength tests.
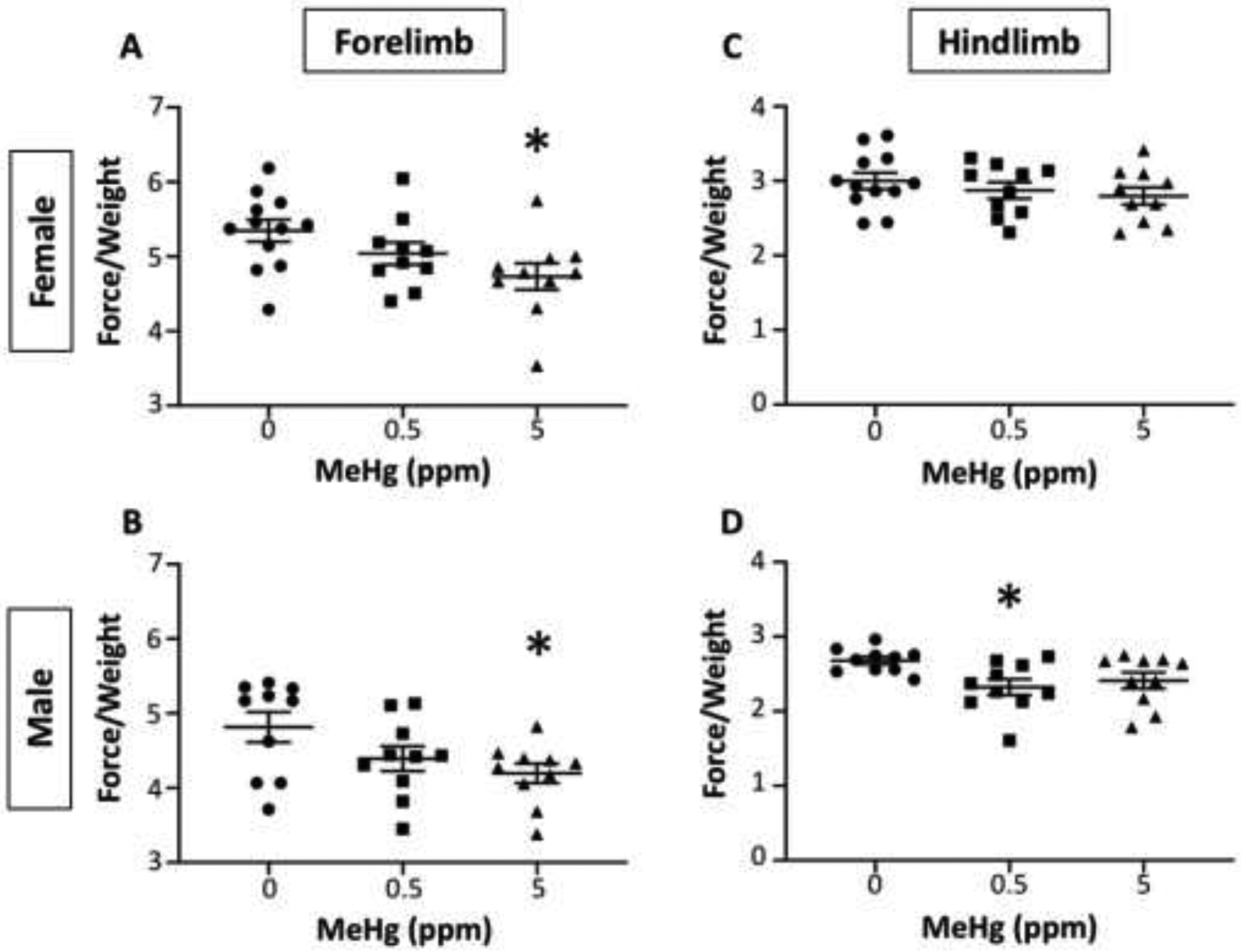
Individual values for forelimb (left column) and hindlimb (right column) grip strength in force normalized to the individual animal’s body weight for female (top row) and male (bottom row) offspring following exposure during pregnancy and lactation to 0, 0.5 or 5.0 ppm MeHg. *=significantly different from 0 ppm control in post-hoc testing following significant main effect in ANOVA.
Average time (left column) and average speed (right column) on the Rotarod across 3 sessions is shown for females (top row) and males (bottom row) of Figure 6. No effects were seen in females. In males, MeHg had no significant effect on the average time spent on the Rotarod, and marginally reduced the average speed (F(2,25)=2.6, p=0.094).
Figure 6. Rotarod tests.
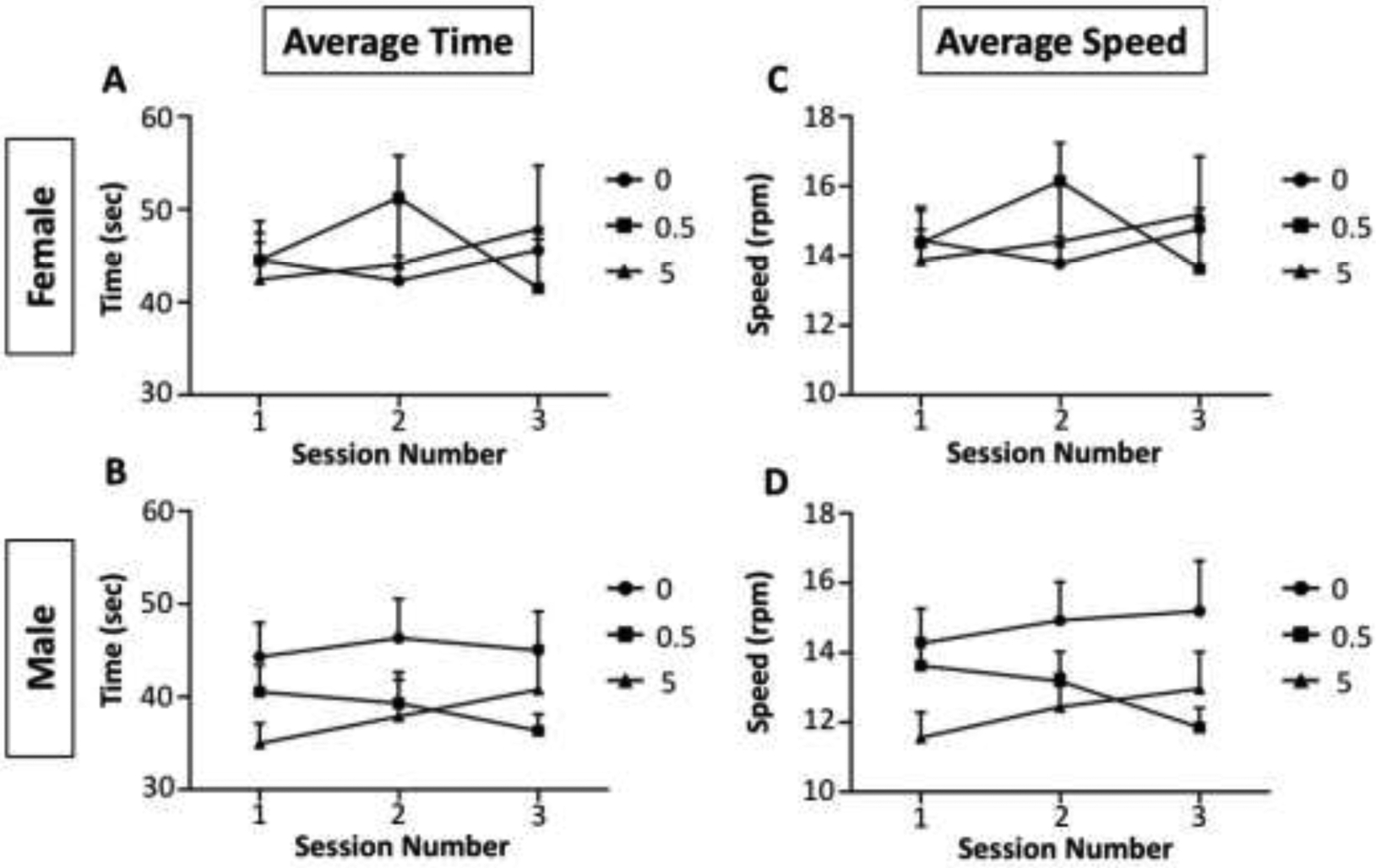
Group mean ± S.E. values for time (sec) spent on the Rotarod (left column) and average speed (rpm) on the rotarod (right column) for females (top row) and males (bottom row) following exposure during pregnancy and lactation to 0, 0.5 or 5.0 ppm MeHg. (Males marginally reduced the average speed (F(2,25)=2.6, p=0.094))
Discussion
We have investigated the potential for MeHg to affect developing skeletal muscle and its possible contribution to later life locomotor deficits. To do this, we first accounted for the accumulation of total Hg in the blood, brain and muscle tissues of both the dam and the developing pups subsequent to maternal exposure via the drinking water. Firstly, in the dams, we observe an overall correlation of the external dose (0.5 and 5.0 ppm in the water) with the mean blood tHg concentrations which were correspondingly an order of magnitude difference between the 0.5 to the 5 ppm treatment group. However, the internal dose (as measured in blood) across the dam’s varied more than 2.5-fold between individual dams receiving the same external MeHg dose. This variability presents a unique problem for dose response studies, as elevated variability at the high dose, can limit significant statistical detection at the low dose (Suppl. Table 1). This variability presents both a challenge and opportunity to explain variation in developmental MeHg toxicity. Consistently and regardless of this high degree of individual variability, we see that Hg accumulates to higher levels in muscle than in blood, in both the dams and the offspring. At PND6, Hg level in the pup muscle is twice that in the brain at the 0.5ppm dose, yet these are nearly equivalent at the 5.0ppm dose. However, over development to PND21, the Hg in pup brains is seen to drop to significantly lower levels than in muscle. Remarkably, at PND 21 Hg levels in muscle appear to exceed that predicted from growth dilution alone, albeit in the absence of known variance of the predicted values, suggesting a net uptake of Hg to muscle over this developmental period. While this profile indicates that developing muscle experiences higher internal dose levels than brain, no gross morphological or cytological phenotypes were detected in the muscles by standard histological staining methods. Nonetheless, with several motor behaviors a male-biased deficit was seen in the PND 60 mice that had experienced the higher MeHg dose including reduced grip strength with the lower MeHg dose.
The profile of Hg accumulation in muscle we observe in early mouse development suggests that developing muscle may play an important toxicokinetic role in moderating MeHg effects in other tissues. Between PND 6 and 21, the growing muscle compartment is apparently accumulating Hg while brain tissue is actively eliminating Hg. Considering that excretory mechanisms of the liver and kidney are not fully mature in this same developmental window, the growing muscle may serve as an important reservoir for Hg. The notion that skeletal muscle may influence MeHg toxicokinetics by influencing MeHg distribution can be extended to the adult mouse. We see that, in dams, the muscle harbors 1.7 to 3 times the level of Hg than is in blood or brain. Since skeletal muscle approaches 35–40% of the body mass in the mature animal [20], and considering a selective partitioning of up to 3-fold of Hg to muscle relative to blood, the muscle compartment is a major determinant of the volume of distribution of MeHg. In fundamental terms, where the biological half-life (t1/2) is directly influenced by the volume of distribution , muscle can therefore influence steady state level of Hg in the animal under conditions of chronic repeated exposures. This notion is supported by the extensive toxicokinetic analyses done with adult mice receiving a bolus dose of MeHg [21, 22], where it was seen that Hg in the muscle was 3 to 5-fold higher than in the brain and ~2-fold higher than in blood. Interestingly, Nielsen et al. [22] observed significant sex-specific differences in half-life and in organ distribution of MeHg between female and male mice. We did not see significant differences in Hg levels in between male and female offspring at the 5.0 ppm dose level at the PND6 and PND 21 timepoints. Thus, sex differences in MeHg distribution may be a trait of adult mice. On this note, we see that female mice exhibited slightly higher Hg in muscle and brain at PND60 compared to males. It is also important to consider that extrapolation of these toxicokinetic properties in mice, and other lab animals, to humans must account for the species-specific differences in tissue distributions that exist. For example, the brain:blood ratio of Hg in rats can be 0.06–0.08 while in humans it is typically >3 [23]. These factors highlight that the relationship of skeletal muscle mass to the internal dose of MeHg across critical organs over the dynamic period of fetal and neonatal growth warrants further investigation.
Accumulation of MeHg in the growing skeletal muscle suggest that either selective transport or additional binding sites are emerging in muscle tissue as it is growing. One possible explanation resides in the well documented MeHg transport mechanism that engages amino acid transporters. MeHg conjugated to cysteine is actively taken up by L-type large neutral amino acid transporters (LAT1) in a process of molecular mimicry, whereby MeHg-cysteine mimics methionine [24, 25]. Since muscle undergoes a 6-fold expansion in the PND 6 to 21 growth period it follows that MeHg uptake likely accompanies the large import of amino acids that is required to support protein synthesis in developing muscle. It can be postulated that maturation of the blood brain barrier over the same time period will also limit MeHg uptake in the brain [26]. These complementary mechanisms present the possibility that developing muscle could serve a protective role with respect to the brain by serving as a MeHg sink.
Across several behaviors that are especially reliant on motor function, we observe decreased performance primarily in male mice at both MeHg concentrations. Deficits in motor behavior with developmental MeHg exposure have long been attributed to effects on the CNS, and correlated in particular with cerebellar lesions [27] [6, 28]. However, the overall elevated Hg level in muscle, relative to brain, raises the question whether or not aberrant muscle development contributes to these motor function deficits. Myopathic effects are difficult to distinguish from neuropathic effects with behavioral assays. Nonetheless, reduced performance, predominantly in males, in locomotor activity, righting time on the inverted screen and, in particular, grip strength, are suggestive that some aspect of compromised muscle physiology could be a contributing factor. Rotarod performance was not significantly altered, however, performance was marginally affected with MeHg exposed males. These marginal effects, given the variance in performance, may explain prior inconsistencies for this test in revealing MeHg effects [13, 29–31].
The extent to which MeHg can induce muscle pathology, either through developmental exposure or acutely in adult muscle, has received little attention. Usuki et al. [32] have shown muscle pathology in adult rats (9 weeks old) receiving MeHg (20 ppm ad libitum in water) that present with reduced fiber bundle size, centralized nuclei and non-grouping atrophic angulated fibers. In the present study, despite a reduced body weight in MeHg exposed pups at birth, histological examination of muscle tissue at PND 60 did not demonstrate any gross morphological or cytological effects associated with the muscle bundles. Future studies should examine gross morphological effects following behavioral experiences to assess if usage or functional strain would act as a secondary insult on muscle bundles. Furthermore, in preliminary analysis of gene expression by quantitative PCR, no significant effect of MeHg on expression of the myogenic genes MyoD, Myogenin and Pax7, as well as the Notch receptor and Notch target genes, Hes1, Hey1 and HeyL, was observed, even in PND 6 muscle tissue (data not shown). These results suggest that early myogenic signaling was not grossly perturbed. It follows that if muscle-specific deficits exist they are likely to stem from alterations in differentiated structures such as the neuromuscular junction, the myotendinous junction, the composition or assembly of myofilaments or mitochondrial abundance and/or fitness [33, 34]. It is also likely that defects may lie in neural structures of the motor unit, yet could be the consequence aberrant developmental cues that arise from muscle. Future studies aimed at more extensive analysis of neuromuscular phenotypes stemming from early life MeHg exposure at the physiological and subcellular level are needed.
Although these findings point to an important role for muscle in moderating developmental neuromuscular toxicity of MeHg, our study has limitations for interpreting a specific mechanism. Firstly, it is difficult to ascertain from the motor behaviors employed here whether deficits lie in aberrant neural or muscle function, or some combination of both. Future studies will require evaluation of electrophysiological properties of isolated muscle fibers to begin to make this distinction. In addition, Hg distribution between brain and muscle tissues may not be fully represented by the tissue subsampling employed here, (i.e. ‘rest of brain’ regions and quadriceps). Finally, statistically, our data found limited low dose behavior effects, despite multiple linear dose response relationships for Hg levels (e.g lower grip strength with 0.5 ppm was not significant but with 5.0 ppm reaches significance). Given the large amount of variability observed in the tHg concentrations of dams, it is worth noting that ANOVA analysis averages variation across all groups which limits the ability to discern statistically significant low dose effects. More advanced statistical methods are needed to address the unequal variance contributions at high doses that may be prohibiting identification of low dose effects. Nonetheless, the different tissue profiles of the change in Hg content over time are indicative of intrinsic toxicokinetic differences that could potentially affect the internal dose in the brain at critical postnatal time points.
In summary, we show that gestational and lactational MeHg exposure in mice results in greater accumulation of Hg in developing muscle, relative to brain and blood, in early postnatal development of offspring, which foreshadows adult motor behavior deficits in male mice. These findings open up the possibility that prolonged MeHg exposure in developing muscle contributes to later life motor deficits.
Supplementary Material
Highlights.
Methylmercury accumulates in muscle tissue to higher levels than in brain and blood in dams and maternally exposed offspring.
Growing muscle potentially serves as a reservoir for methylmercury that leaves the brain in early postnatal development.
Early life methylmercury exposure can lead to male-biased adult motor deficits.
Acknowledgements
We thank Tom Scrimale for technical assistance in ICP-MS Hg analyses.
Funding
This research was supported by the National Institute of Health, NIH R01ES025721 (M.D.R., PI), and the NIH supported University of Rochester Environmental Health Science Center P30ES001247.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oken E and Bellinger DC, Fish consumption, methylmercury and child neurodevelopment. Curr Opin Pediatr, 2008. 20(2): p. 178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karagas MR, et al. , Evidence on the human health effects of low-level methylmercury exposure. Environ Health Perspect, 2012. 120(6): p. 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisen-Hersh EB, et al. , Behavioral effects of developmental methylmercury drinking water exposure in rodents. J Trace Elem Med Biol, 2014. 28(2): p. 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castoldi AF, et al. , Neurodevelopmental toxicity of methylmercury: Laboratory animal data and their contribution to human risk assessment. Regul Toxicol Pharmacol, 2008. 51(2): p. 215–29. [DOI] [PubMed] [Google Scholar]
- 5.Patel E and Reynolds M, Methylmercury impairs motor function in early development and induces oxidative stress in cerebellar granule cells. Toxicol Lett, 2013. 222(3): p. 265–72. [DOI] [PubMed] [Google Scholar]
- 6.Sager PR, Aschner M, and Rodier PM, Persistent, differential alterations in developing cerebellar cortex of male and female mice after methylmercury exposure. Brain Res, 1984. 314(1): p. 1–11. [DOI] [PubMed] [Google Scholar]
- 7.Rodier PM, Aschner M, and Sager PR, Mitotic arrest in the developing CNS after prenatal exposure to methylmercury. Neurobehav Toxicol Teratol, 1984. 6(5): p. 379–85. [PubMed] [Google Scholar]
- 8.Montgomery SL, et al. , Genome-Wide Association Analysis of Tolerance to Methylmercury Toxicity in Drosophila implicates myogenic and neuromuscular developmental pathways. PLOS One, 2014. 9(10):e110375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prince LM and Rand MD, Notch Target Gene E(spl)mdelta Is a Mediator of Methylmercury-Induced Myotoxicity in Drosophila. Front Genet, 2017. 8: p. 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paletz EM, Craig-Schmidt MC, and Newland MC, Gestational exposure to methylmercury and n-3 fatty acids: effects on high- and low-rate operant behavior in adulthood. Neurotoxicol Teratol, 2006. 28(1): p. 59–73. [DOI] [PubMed] [Google Scholar]
- 11.Weston HI, et al. , Sex-dependent and non-monotonic enhancement and unmasking of methylmercury neurotoxicity by prenatal stress. Neurotoxicology, 2014. 41: p. 123–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montgomery KS, et al. , Chronic, low-dose prenatal exposure to methylmercury impairs motor and mnemonic function in adult C57/B6 mice. Behav Brain Res, 2008. 191(1): p. 55–61. [DOI] [PubMed] [Google Scholar]
- 13.Goulet S, Dore FY, and Mirault ME, Neurobehavioral changes in mice chronically exposed to methylmercury during fetal and early postnatal development. Neurotoxicol Teratol, 2003. 25(3): p. 335–47. [DOI] [PubMed] [Google Scholar]
- 14.Stern S, et al. , Perinatal and lifetime exposure to methylmercury in the mouse: blood and brain concentrations of mercury to 26 months of age. Neurotoxicology, 2001. 22(4): p. 467–77. [DOI] [PubMed] [Google Scholar]
- 15.Newland MC and Reile PA, Blood and brain mercury levels after chronic gestational exposure to methylmercury in rats. Toxicol Sci, 1999. 50(1): p. 106–16. [DOI] [PubMed] [Google Scholar]
- 16.Weiss B, et al. , Methylmercury contamination of laboratory animal diets. Environ Health Perspect, 2005. 113(9): p. 1120–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitten MK, Effect of exteroceptive factors on the oestrous cycle of mice. Nature, 1957. 180(4599): p. 1436. [DOI] [PubMed] [Google Scholar]
- 18.Dubowitz V and Sewry C, Muscle Biopsy: A Practical Approach. 2007, England: Bailliere Tindall; 720. [Google Scholar]
- 19.Siddiqui RA, et al. , Growth allometry of organs, muscles and bones in mice from lines divergently selected on the basis of plasma insulin-like growth factor-I. Growth Dev Aging, 1992. 56(1): p. 53–60. [PubMed] [Google Scholar]
- 20.Brown RP, et al. , Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health, 1997. 13(4): p. 407–84. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen JB and Andersen O, Methyl mercuric chloride toxicokinetics in mice. I: Effects of strain, sex, route of administration and dose. Pharmacol Toxicol, 1991. 68(3): p. 201–7. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen JB and Andersen O, Methyl mercuric chloride toxicokinetics in mice. II: Sexual differences in whole-body retention and deposition in blood, hair, skin, muscles and fat. Pharmacol Toxicol, 1991. 68(3): p. 208–11. [DOI] [PubMed] [Google Scholar]
- 23.Omata S, et al. , Species difference between rat and hamster in tissue accumulation of mercury after administration of methylmercury. Arch Toxicol, 1986. 59(4): p. 249–54. [DOI] [PubMed] [Google Scholar]
- 24.Aschner M and Clarkson TW, Methyl mercury uptake across bovine brain capillary endothelial cells in vitro: the role of amino acids. Pharmacol Toxicol, 1989. 64(3): p. 293–7. [DOI] [PubMed] [Google Scholar]
- 25.Yin Z, et al. , The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J Neurochem, 2008. 107(4): p. 1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, et al. , Establishment and Dysfunction of the Blood-Brain Barrier. Cell, 2015. 163(5): p. 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimura M, Cheng J, and Zhao W, Perinatal exposure to low-dose methylmercury induces dysfunction of motor coordination with decreases in synaptophysin expression in the cerebellar granule cells of rats. Brain Res, 2012. 1464: p. 1–7. [DOI] [PubMed] [Google Scholar]
- 28.Choi BH, Kudo M, and Lapham LW, A Golgi and electron-microscopic study of cerebellum in methylmercury-poisoned neonatal mice. Acta Neuropathol, 1981. 54(3): p. 233–7. [DOI] [PubMed] [Google Scholar]
- 29.Onishchenko N, et al. , Developmental exposure to methylmercury alters learning and induces depression-like behavior in male mice. Toxicol Sci, 2007. 97(2): p. 428–37. [DOI] [PubMed] [Google Scholar]
- 30.Vitalone A, et al. , Neurobehavioral assessment of rats exposed to low doses of PCB126 and methyl mercury during development. Environ Toxicol Pharmacol, 2008. 25(1): p. 103–13. [DOI] [PubMed] [Google Scholar]
- 31.Cheng J, et al. , Neurobehavioral effects, c-Fos/Jun expression and tissue distribution in rat offspring prenatally co-exposed to MeHg and PFOA: PFOA impairs Hg retention. Chemosphere, 2013. 91(6): p. 758–64. [DOI] [PubMed] [Google Scholar]
- 32.Usuki F, et al. , Beneficial effects of mild lifelong dietary restriction on skeletal muscle: prevention of age-related mitochondrial damage, morphological changes, and vulnerability to a chemical toxin. Acta Neuropathol, 2004. 108(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 33.Larsson L, et al. , Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol Rev, 2019. 99(1): p. 427–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gan Z, et al. , Skeletal muscle mitochondrial remodeling in exercise and diseases. Cell Res, 2018. 28(10): p. 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.